
Catalyst-free reactions of anilines with β-chloroenones: synthesis of α-chloroenaminones and 1,4-benzodiazepines†
Sundaram
Suresh‡
,
Sowndarya
Palla‡
,
Dai-Ru
Chung
,
Hung-Sheng
Chien
,
Bo-Xun
Du
,
Jivan
Shinde
,
Veerababurao
Kavala
and
Ching-Fa
Yao
 *
*
Department of Chemistry, National Taiwan Normal University, No. 88, Sec. 4, Ting-Zhou Rd, Taipei-11677, Taiwan, Republic of China. E-mail: cheyaocf@ntnu.edu.tw
First published on 27th August 2024
Abstract
The Michael addition of anilines to β-chloroenones gives enaminones by the elimination of hydrochloric acid (HCl). These enaminones are transformed into α-chloroenaminones via in situ sp2 C–H functionalization. Anilines that are attached to an electron-donating group react more readily with β-chloroenone to give the corresponding products in excellent yields. A highly atom-economical method has been developed using dimethyl sulfoxide (DMSO) as a green oxidant and solvent. The desired α-functionalized enaminones are formed in good yields with excellent Z-selectivity. We have established the generality of this reaction with many substrates, and scaled-up reactions have been performed to showcase the practical applications. A catalyst-free double annulation of β-chloroenones with o-phenylenediamine has also been demonstrated for the synthesis of 1,4-benzodiazepine derivatives in moderate yields under mild reaction conditions.
Introduction
Enaminones with many reactive sites exhibit a wide range of reactivities such as functionalization of the sp2 C–H bond, reactions of the amino group, and 1,2- and 1,4-addition reactions.1 Reactions of the sp2 C–H bond have been largely studied for C–C, C–O, C–N, C–X, and C–S bond formation in the recent past.1 Recently, a highly regioselective method was reported for the site-selective acyoxylation of either the α- or β-site of enaminones by changing the solvent system.2 In 2016, an elegant metal-free reaction of enaminone with thiophenols was developed for the synthesis of polyfunctionalized aminothioalkenes in the presence of KIO3.3 Over the past few years, several other methods have been established for the production of SCN-containing enaminones.4 Enaminones have been previously utilized for sp2 C–H halogenation using either a transition metal catalyst or metal-free reaction conditions (Scheme 1a).5 The C–H functionalization of enaminone has gained considerable research attention and is widely used to produce a variety of functionalized enaminone derivatives.6 | ||
| Scheme 1 Synthesis of enaminones and benzodiazepines. | ||
Some reactions involving N,N-dimethylenaminones have been outlined in which the N,N-dimethyl group acts as a leaving group.7 Due to the presence of both nucleophilic and electrophilic sites, the enaminones are an important starting material for producing molecular diversity and complexity. In addition, enaminone derivatives have been utilized as key intermediates to construct heterocyclic compounds.8 Moreover, they are also found in many bioactive compounds and exhibit antitumor, anti-inflammatory, antibiotic, and antidepressant activities.9
Synthetic methods have been documented to readily access enaminones because of their wide range of applications in interdisciplinary fields.10 Relatively few methods have been reported in the literature that did not use the corresponding enaminones as the starting materials for the synthesis of α-functionalized enaminones. For example, Murakami and co-workers described a transition metal-catalyzed reaction of N-sulfonyl-1,2,3-triazoles with formamides that directly gave α-functionalized enaminones.11
Recently, a metal-free strategy was reported for the one-pot synthesis of α-bromoenaminones by reacting 3-bromopropenals with anilines in the presence of a catalytic amount of TsOH·H2O (Scheme 1b).12 One-pot methods involving the direct synthesis of α-functionalized compounds are quite rare, and therefore, the development of such methods can be highly useful to readily access enaminone intermediates for applications in synthetic chemistry.
Benzodiazepines are ubiquitous and an important structural unit found in many marketed drugs and natural products (Fig. 1).13 The synthesis of benzodiazepines has been accomplished via condensation of o-phenylenediamines with an α,β-unsaturated carbonyl compound under acidic conditions (Scheme 1c).14 The synthesis of bioactive benzodiazepine derivatives can also be achieved using a carbonyl compound as a coupling partner with o-phenylenediamines.15 There are no methods in the literature that utilize β-chloroenones as a reaction partner in the synthesis of benzodiazepine derivatives. Herein, we disclose a catalyst-free divergent method for the synthesis of α-chloroenaminones and 1,4-benzodiazepines using DMSO as a green solvent and reagent (Scheme 1d).
 | ||
| Fig. 1 Representative examples of bioactive enaminones and benzodiazepines. | ||
Results and discussion
An atom-economical synthesis of α-functionalized enaminones was anticipated from the reaction of anilines with halopropenones in DMSO. Synthesis of β-bromoenone was previously reported by reacting phenylpropynone with aluminium chloride (AlCl3) and sodium bromide (NaBr).16 We decided to prepare β-bromoenone using this strategy for application in the one-pot synthesis of α-functionalized enaminone derivatives. Later, reactions were performed, and the results are presented in Table 1.|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time (h) | Yield | Yield |
3aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
4aab (%) |
||||
| a All reactions were carried out using 1a (0.3 mmol, 50 mg), 2a (0.25 mmol, 23 mg), and solvent (2 mL). b The yields were determined by 1H NMR analyses of the crude reaction mixtures using CH2Br2 as the internal standard. c Isolated yield. DMF = dimethylformamide; DCE = 1,2-dichloroethane. | |||||
| 1 | DMSO | rt | 12 | — | 64 (58)c |
| 2 | DMSO | 50 | 4 | 28 | 41 |
| 3 | DMSO | 70 | 4 | 75 | 6 |
| 4 | DMSO | 70 | 6 | 84 | 5 |
| 5 | DMSO | 90 | 4 | 97 (92)c | Trace |
| 6 | DMF | 90 | 4 | — | 56 |
| 7 | Toluene | 90 | 4 | — | 51 |
| 8 | CH3CN | 90 | 4 | — | 49 |
| 9 | 1,4-Dioxane | 90 | 4 | — | 31 |
| 10 | DCE | 90 | 4 | — | 82 |

First, a reaction was carried out at room temperature, and enaminone 4aa was obtained in 58% isolated yield (entry 1). When the reaction was carried out at 50 °C, the desired α-functionalized enaminone 3aa was formed in 28% yield along with 4aa in 41% yield (entry 2). The single crystal analysis of 3aa revealed that the product is a (Z)-α-chloroenaminone and not the expected bromoenaminone (see the ESI†). This outcome suggests that the starting material 1a used here should be a β-chloroenone.
The reaction temperature was further increased to 70 °C, resulting in the formation of product 3aa in 75% and 84% yields (entries 3 and 4), respectively. However, the reaction at 90 °C gave the (Z)-α-chloroenaminone 3aa in excellent yield of 92% (entry 5). The solvent screening revealed that DMSO can act as an oxidant, and is crucial for the in situ chlorination of enaminone 4aa (entries 6–10). Thus, atom-economical synthesis was accomplished using DMSO as a solvent system in which DMSO can also act as a green oxidizing agent.
We performed reactions using different β-chloroenones and anilines to establish the generality of this method (Table 2). The reactions were carried out under the conditions that afforded the product (Z)-3aa in 92% yield. We found that ortho-toluidine reacted with 2a and gave the corresponding enaminone 3ab in 68% yield, suggesting that the steric hindrance of the ortho-methyl group decreased the rate of this reaction to some extent. The reactions using meta- and para-toluidines as reaction partners resulted in the formation of the desired α-functionalized products (3ac and 3ad) in good yields. These observations further indicated that the steric effect of a substituent attached at the ortho-position can influence the rate of this reaction.

A smooth reaction was observed of 4-ethylaniline 2e with 1a under the reaction conditions to give α-chloroenaminone 3ae in 80% yield. The product 3af was obtained in an excellent yield of 87% when para-isopropylaniline 2f was used as a reaction partner with 1a. In the case of 2,6-dimethylaniline, the corresponding enaminone 3ag was isolated in only 65% yield. The analysis of the crude reaction mixture revealed that a minor product 4ag, which was not isolated in a pure form, was formed in 15% yield. Due to the presence of two ortho-methyl groups, the reactivity of aniline derivative 2g was decreased by a considerable amount. Nevertheless, the reaction of 2,4-dimethylaniline with 1a resulted in the formation of product 3ah in an 80% yield.
Anilines bearing an electron-donating group reacted more efficiently and afforded the corresponding products 3ai and 3aj in 90% and 94% yields, respectively. This reaction was found to be sluggish when ortho-nitroaniline was reacted with 1a, and the desired product 3ak was isolated in only 40% yield after 8 h. Combined steric and electron-withdrawing effects of the ortho-nitro group greatly retarded this reaction. However, the product 3al was formed in 79% yield using meta-nitroaniline as a reaction partner with 1a.
The nitro group at the meta-position can withdraw electrons only by an inductive effect. The result obtained from the reaction of para-nitroaniline with 1a suggested that both inductive and mesomeric effects are operating in this case. It was established from the above observations that the efficiency of this reaction is diminished when an aniline bearing a sterically hindered and electron-withdrawing group was used for the synthesis of enaminones.
To increase our understanding, we then used halo-substituted aniline derivatives. Using ortho-chloroaniline, the enaminone product 3an was obtained in a 51% isolated yield. The reactions of meta- and para-chloroaniline offered the corresponding products 3ao and 3ap in 63% and 82% yields, respectively. The impact of the electronic effect on the outcomes of these reactions was further established from these results. When para-bromoaniline was reacted with 1a, the desired product was formed in 85% yield.
Next, we employed benzylamine as a coupling partner and observed the formation of product 3ar in 50% yield. Furthermore, we also observed that the enaminone product 3as can be formed with only a 36% yield under the reaction conditions and using phenylethylamine 2s as a reaction partner. With these observations, we determined that amines with the aliphatic unit are not a good reaction partner. Reactions were also performed to produce α-functionalized enaminones containing an indole moiety using 5-amino-N-benzylindole as a Michael donor. Nevertheless, a mixture of inseparable products was only formed in small quantities.
We then studied the effect of substituents that are attached to the aromatic ring of β-chloroenones. The β-chloroenones containing either an electron-donating or an electron-withdrawing group were subjected to reactions under optimized reaction conditions. The reactions were initially carried out using β-chloroenone 1b and anilines with an electron-donating group. As observed earlier, anilines that contain an electron-donating group reacted more efficiently than anilines with an electron-withdrawing group. A similar trend of reactivity was observed using β-chloroenone 1c with a para-methoxy group as a Michael acceptor in this atom-economical synthesis.
Subsequently, reactions of β-chloroenone 1d with anilines were studied to evaluate a chloro-substituent effect on the outcomes of reactions. The desired products (3da, 3dj, and 3dp) were obtained in moderate to good yields. However, enaminone product 3dl was obtained with only a 47% yield from the reaction of 1d with meta-nitroaniline 2l under the reaction conditions. Finally, we used β-chloroenone bearing a bromo group at the para-position to understand the effect of this group on this reaction. The corresponding α-chloroenaminones (3ea, 3ej, 3el, and 3ep) were obtained in good yields when the corresponding anilines were reacted with 1e under catalyst-free conditions.
To demonstrate the practical applications of this strategy, scaled-up reactions were examined under optimal reaction conditions (Scheme 2). A reaction of β-chloroenone 1a with aniline 2a was carried out on a 3.5 mmol scale, and the desired product 3aa was obtained in 85% yield. We also tested the efficiency of this strategy using aniline derivative 2d as a coupling partner with 1a. In this case, the corresponding product 3ad was isolated in 87% yield when the reaction was carried out at 90 °C for 8 h.
 | ||
| Scheme 2 Scaled-up reactions. | ||
When o-phenylenediamine 5a was used as an amine partner at 90 °C, this reaction did not give fruitful results. However, we observed the formation of 1,4-benzodiazepine 6aa in 40% yield under ambient conditions (see the ESI†). A similar result was also obtained when 5b was reacted with 1a (Table 3). The desired products (6ba, 6bb, 6ca, and 6cb) were isolated in moderate yields using β-chloroenones attached to an electron-donating group. Moreover, the efficiency of this reaction decreased to a considerable extent when 1d was employed with a chloro substituent attached at the para-position.

In these cases, the expected 1,4-benzodiazepine derivatives 6da and 6db were only formed in 10–15% yields. The reactions of o-phenylenediamines 5a and 5b with 1e offered the corresponding 1,4-benzodiazepine derivatives 6ea and 6eb in 20% and 27% yields, respectively. Subsequently, reactions of 1a with 4,5-dichloro-o-phenylenediamine 5c were investigated, and it was found that this reaction was sluggish when this substrate was used. This result revealed that o-phenylenediamines bearing electron-withdrawing groups are not a suitable coupling partner.
The reaction between β-chloroenone 1a and aniline 2a delivered enaminone 4aa as the product in DMF at 90 °C (Scheme 3). Moreover, the intermediate 4aa formed only as the product in reactions that were carried out using other solvent systems (Table 1, entries 6–10). Hence, we established that DMSO as a solvent system is required for the generation of α-chlorinated enaminone derivatives.
 | ||
| Scheme 3 Control experiment. | ||
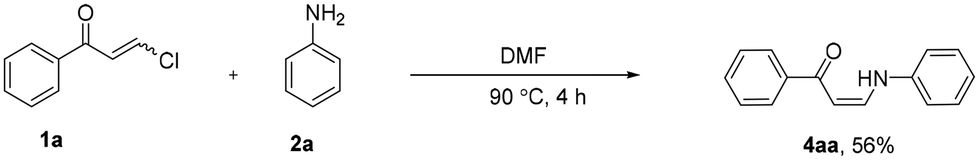
To explain the formation of α-chloroenaminone in an atom-economical manner, a suitable reaction mechanism is proposed in Scheme 4. The 1,4-addition of aniline 2a to 1a gives enaminone 4aa with the elimination of hydrochloric acid (HCl). The in situ oxidation of HCl produces chlorine molecules (Cl2). Reaction of 4aa with Cl2 can give imine intermediate A, which then undergoes isomerization to give a thermodynamically more stable product (Z)-3aa under the reaction conditions. Similarly, o-phenylenediamine 5a reacts with two molecules of 1a to give dienaminone B as an intermediate. Next, this intermediate is transformed into another intermediate Cvia an intramolecular cyclization. The more stable product 6aa is finally formed under ambient reaction conditions.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
A catalyst-free method was developed for the synthesis of a variety of (Z)-α-chloroenaminones. The corresponding enaminone products were obtained in excellent yields when anilines bearing an electron-donating group were employed as a reaction partner. The reactions were accomplished with high atom economy (>99%) using DMSO as the solvent system. We also found that anilines attached to an electron-withdrawing group required a longer reaction time.The reactions using o-phenylenediamine as an amine partner gave benzodiazepine derivatives in moderate yields under catalyst-free conditions. Therefore, molecular diversity was readily achieved by using these methods. Because enaminones have been widely used as reactive intermediates, this method could find significant applications in synthetic chemistry.
Experimental section
General information
All of the reagents and solvents were purchased from commercial suppliers and were directly used without any further purification. DMSO that was obtained from a commercial supplier was used in this study. Prior to use, 10 mL round bottom flasks were dried in an oven and cooled to room temperature. All reactions were carried out under a nitrogen atmosphere.Flash column chromatography was performed on 63–200 mesh silica gel using hexane and ethyl acetate as eluents. 1H and 13C NMR spectra were recorded on a Bruker Ascend spectrometer at 400 and 100 MHz, respectively. Chemical shifts are reported in parts per million (ppm) on the δ scale using CDCl3 as an internal standard. Multiplicities are indicated using abbreviations: s = singlet; d = doublet; t = triplet; q = quartet; dd = doublet of doublets; m = multiplet, br = broad signal. Coupling constants are expressed in Hertz (Hz). High-resolution mass spectrometry (HRMS) was performed, and spectra were recorded in electrospray ionization-time-of-flight (ESI-TOF) mode. The melting points (mp) were obtained using an Electro-Thermal FARGO MP-2D capillary melting point apparatus.
General procedure for the synthesis of β-chloroenones 1
To an oven-dried 50 mL reaction flask cooled under a N2 atmosphere was added the corresponding propargyl ketone (3 mmol), CH3CN (20 mL), tert-butanol (2 mL), AlCl3 (4.5 mmol, 599 mg), and NaBr (6 mmol, 617 mg). The reaction mixture was heated at 80 °C for 16 h, and was subsequently cooled to room temperature and quenched by the addition of water (5–10 mL). Then, the reaction mixture was extracted with ethyl acetate (30 mL), and the aqueous layer was again extracted using ethyl acetate (20 mL). The combined organic layer was washed with brine solution and dried over MgSO4. The organic layer was evaporated under reduced pressure, and the crude product was purified by silica gel column chromatography using an ethyl acetate/hexane (1:99) mixture as the eluent.
:1 (v/v) as the eluent. Yield 85% (424 mg); colorless liquid. 1H NMR (400 MHz, CDCl3) δ 7.95–7.92 (m, 2.1H), 7.70 (d, J = 12.0 Hz, 0.08H), 7.62–7.58 (m, 1.1H), 7.52–7.45 (m, 3.1H), 7.32 (d, J = 12.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 187.8, 138.2, 136.8, 133.4, 128.8, 128.6, 128.5; HRMS (EI) m/z calcd for C9H7ClO [M]+ 166.0185, found 166.0181.
:1 (v/v) as the eluent. Yield 82% (443 mg); colorless liquid. 1H NMR (400 MHz, CDCl3) δ 7.85–7.82 (m, 2.5H), 7.67 (d, J = 12.0 Hz, 0.17H), 7.57 (d, J = 16.0 Hz, 0.17H), 7.45 (d, J = 12.0 Hz, 1H), 7.33–7.28 (m, 3.6H), 2.43 (s, 3.6H); 13C NMR (100 MHz, CDCl3) δ 187.3, 144.5, 137.7, 134.3, 129.5, 128.8, 128.7, 128.5, 21.7; HRMS (EI) m/z calcd for C10H9ClO [M]+ 180.0342, found 180.0338.
:1 (v/v) as the eluent. Yield 75% (441 mg); white solid, 53.5–55 °C. 1H NMR (400 MHz, CDCl3) δ 7.95–7.91 (m, 2.3H), 7.66 (d, J = 12.0 Hz, 0.14H), 7.57 (d, J = 16.0 Hz, 0.14 Hz), 7.43 (d, J = 16.0 Hz, 1H), 7.31 (d, J = 16.0 Hz, 1H), 6.98–6.94 (m, 2.3H), 3.88 (s, 3.4H); 13C NMR (100 MHz, CDCl3) δ 186.1, 163.9, 137.3, 132.5, 131.0, 130.9, 129.7, 128.3, 114.0, 55.5; HRMS (EI) m/z calcd for C10H9ClO2 [M]+ 196.0291, found 196.0284.
:1 (v/v) as the eluent. Yield 52% (312 mg); white solid, melting point: 37.5–39 °C. 1H NMR (400 MHz, CDCl3) δ 7.91–7.88 (m, 2.2H), 7.74 (d, J = 16.0 Hz, 0.19H), 7.56 (d, J = 16.0 Hz, 0.19H), 7.52–7.47 (m, 3H), 7.29 (d, J = 12.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 186.5, 186.4, 140.0, 138.8, 135.1, 134.8, 132.2, 130.0, 129.9, 128.2, 128.0; HRMS (ESI) m/z calcd for C9H7Cl2O [M + H]+ 200.9874, found 200.9874.
:1 (v/v) as the eluent. Yield 64% (469 mg); white solid, melting point: 41–42 °C. 1H NMR (400 MHz, CDCl3) δ 7.83–7.80 (m, 2.2H), 7.75 (d, J = 12.0 Hz, 0.04H), 7.67–7.64 (m, 2.2H), 7.55 (d, J = 16.0 Hz, 0.06H), 7.50 (d, J = 12.0 Hz, 1H), 7.29 (d, J = 16.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 186.6, 138.8, 135.6, 132.2, 130.1, 130.0, 128.7, 128.0; HRMS (EI) m/z calcd for C9H6ClBrO [M]+ 243.9291, found 243.9287.
General procedure for the synthesis of α-chloroenaminones 3
Aniline (0.25 mmol), β-chloroenone (0.3 mmol), and DMSO (2 mL) were added to a 10 mL reaction flask that was dried in an oven and cooled to room temperature under a N2 atmosphere. The reaction mixture was heated at 90 °C using an oil bath for 4–8 h. The reaction mixture was then cooled to room temperature and transferred to a separatory funnel using ethyl acetate (30 mL). Water (30 mL) was added, and the organic layer was separated. The aqueous layer was extracted again with ethyl acetate (20 mL). The combined organic layer was washed with brine solution and dried over MgSO4. The solvent was evaporated under reduced pressure using a rotary evaporator. The crude product was purified by silica gel column chromatography using an ethyl acetate/hexane mixture as the eluent.:15 (v/v) as the eluent. Yield 92% (59 mg); yellow solid, melting point: 144–145 °C. 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 12 Hz, 1H), 7.63–7.60 (m, 2H), 7.55–7.50 (m, 1H), 7.48–7.44 (m, 2H), 7.33–7.29 (m, 2H), 7.24 (d, J = 12 Hz, 1H), 7.10–7.06 (m, 1H), 6.93–6.90 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 188.1, 141.2, 139.2, 138.8, 131.1, 130.0, 128.5, 128.4, 128.1, 124.1, 116.4, 109.8; HRMS (ESI) m/z calcd for C15H13NOCl [M + H]+ 258.0686, found 258.0691.
:15 (v/v) as the eluent. Yield 58% (32 mg); yellow solid, melting point: 138–139 °C. 1H NMR (400 MHz, CDCl3) δ 12.18 (d, J = 12 Hz, 1H), 7.98–7.96 (m, 2H), 7.57–7.45 (m, 4H), 7.39–7.34 (m, 2H), 7.14–7.08 (m, 3H), 6.06 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 191.0, 144.9, 140.2, 139.2, 131.6, 129.7, 128.5, 127.3, 123.7, 116.3, 93.7; HRMS (ESI) m/z calcd for C15H14NO [M + H]+ 224.1075, found 224.1081.
:10 (v/v) as the eluent. Yield 68% (46 mg); yellow solid, melting point: 128–130 °C. 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 12.0 Hz, 1H), 7.63–7.61 (m, 2H), 7.54–7.50 (m, 1H), 7.47–7.44 (m, 2H), 7.20–7.14 (m, 2H), 7.07 (d, J = 12.0 Hz, 1H), 7.01 (t, J = 8.0 Hz, 1H), 6.84 (d, J = 8.0 Hz, 1H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.9, 141.8, 138.8, 137.5, 131.4, 131.0, 128.5, 128.4, 127.5, 126.2, 124.2, 115.2, 110.1, 17.2; HRMS (EI) m/z calcd for C16H14ClNO [M]+ 271.0764, found 271.0758.
:15 (v/v) as the eluent. Yield 84% (57 mg); yellow solid, melting point: 104–105.5 °C. 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 12.0 Hz, 1H), 7.62–7.61 (d, J = 8.0 Hz, 2H), 7.54–7.51 (m, 1H), 7.46 (t, J = 8.0 Hz, 2H), 7.20–7.16 (m, 2H), 6.89 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 188.1, 141.3, 140.1, 139.1, 138.8, 131.0, 129.7, 128.5, 128.4, 125.0, 117.2, 113.3, 109.5, 21.4; HRMS (EI) m/z calcd for C16H14ClNO [M]+ 271.0764, found 271.0780.
:15 (v/v) as the eluent. Yield 93% (63 mg); yellow solid, melting point: 161–163 °C. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 12.0 Hz, 1H), 7.62–7.59 (m, 2H), 7.53–7.49 (m, 1H), 7.46–7.42 (m, 2H), 7.17 (d, J = 16.0 Hz, 1H), 7.10 (d, J = 8.0 Hz, 2H), 6.83–6.80 (m, 2H), 2.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.0, 141.7, 138.9, 136.8, 133.9, 130.9, 130.4, 128.5, 128.4, 116.5, 109.3, 20.7; HRMS (EI) m/z calcd for C16H14ClNO [M]+ 271.0764, found 271.0755.
:15 (v/v) as the eluent. Yield 80% (57 mg); yellow solid, melting point: 146–147 °C. 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 12.0 Hz, 1H), 7.65–7.62 (m, 2H), 7.56–7.52 (m, 1H), 7.49–7.45 (m, 2H), 7.20–7.15 (m, 3H), 6.88–6.85 (m, 2H), 2.63 (q, J = 8.0 Hz, 2H), 1.23 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 188.0, 141.7, 140.4, 138.9, 136.9, 130.9, 129.2, 128.5, 128.4, 116.5, 109.3, 28.1, 15.6; HRMS (EI) m/z calcd for C17H16ClNO [M]+ 285.0920, found 285.0913.
:10 (v/v) as the eluent. Yield 87% (65 mg); yellow solid, melting point: 160–161 °C. 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 12.0 Hz, 1H), 7.62–7.59 (m, 2H), 7.54–7.49 (m, 1H), 7.47–7.42 (m, 2H), 7.19–7.13 (m, 3H), 6.87–6.84 (m, 2H), 2.86 (sep, J = 8.0 Hz, 1H), 1.21 (d, J = 8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 188.0, 145.1, 141.8, 138.9, 137.0, 130.9, 128.5, 128.4, 127.8, 116.5, 109.3, 33.4, 23.9; HRMS (EI) m/z calcd for C18H18ClNO [M]+ 299.1077, found 299.1074.
:10 (v/v) as the eluent. Yield 65% (46 mg); yellow solid, melting point: 153–154 °C. 1H NMR (400 MHz, CDCl3) δ 7.56–7.53 (m, 2H), 7.47–7.42 (m, 1H), 7.40–7.36 (m, 2H), 7.31 (d, J = 12.0 Hz, 1H), 7.07 (s, 3H), 6.66 (d, J = 12.0 Hz, 1H), 2.27 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 187.9, 148.2, 139.0, 137.0, 132.7, 130.7, 129.1, 128.3, 126.9, 107.9, 18.5; HRMS (EI) m/z calcd for C17H16ClNO [M]+ 285.0920, found 285.0910.
:10 (v/v) as the eluent. Yield 80% (57 mg); yellow solid, melting point: 149–150 °C. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 12.0 Hz, 1H), 7.65–7.62 (m, 2H), 7.56–7.51 (m, 1H), 7.49–7.45 (m, 2H), 7.09–6.97 (m, 3H), 6.77 (d, J = 8.0 Hz, 1H), 2.33 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.9, 142.4, 138.9, 135.2, 134.1, 132.0, 130.9, 128.5, 128.4, 127.9, 126.4, 115.7, 109.6, 20.6, 17.1; HRMS (EI) m/z calcd for C17H16ClNO [M]+ 285.0920, found 285.0910.
:20 (v/v) as the eluent. Yield 90% (65 mg); yellow solid, melting point: 103–104 °C. 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 12.0 Hz, 1H), 7.73 (d, J = 12.0 Hz, 1H), 7.62–7.60 (m, 2H), 7.54–7.50 (m, 1H), 7.48–7.44 (m, 2H), 7.04–7.00 (m, 1H), 6.92–6.86 (m, 2H), 6.80 (dd, J = 1.2, 8.0 Hz, 1H), 3.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.1, 148.1, 140.4, 138.9, 130.9, 128.6, 128.5, 128.4, 123.7, 121.2, 113.4, 111.1, 110.1, 55.9; HRMS (ESI) m/z calcd for C16H15NO2Cl [M + H]+ 288.0791, found 288.0803.
:20 (v/v) as the eluent. Yield 94% (68 mg); yellow solid, melting point: 147–148 °C. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 12.0 Hz, 1H), 7.63–7.61 (m, 2H), 7.55–7.45 (m, 3H), 7.19 (d, J = 12.0 Hz, 1H), 6.92–6.87 (m, 4H), 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.9, 156.6, 142.4, 138.9, 132.7, 130.9, 128.4, 128.4, 118.2, 115.1, 108.8, 55.6; HRMS (ESI) m/z calcd for C16H15NO2Cl [M + H]+ 288.0791, found 288.0790.
:20 (v/v) as the eluent. Yield 40% (30 mg); yellow solid, melting point: 137–138 °C. 1H NMR (400 MHz, CDCl3) δ 10.56 (d, J = 12.0 Hz, 1H), 8.29 (dd, J = 8.0, 1.2 Hz, 1H), 7.98 (d, J = 12.0 Hz, 1H), 7.72–7.69 (m, 2H), 7.63–7.57 (m, 2H), 7.53–7.49 (m, 2H), 7.16–7.12 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 188.4, 138.1, 137.4, 136.5, 136.3, 135.7, 131.7, 128.7, 128.5, 127.1, 122.2, 115.7, 114.3; HRMS (EI) m/z calcd for C15H12N2O3Cl [M + H]+ 303.0536, found 303.0542.
:20 (v/v) as the eluent. Yield 79% (60 mg); yellow solid, melting point: 188–189 °C. 1H NMR (400 MHz, CDCl3) δ 9.90 (d, J = 12.0 Hz, 1H), 8.13–8.12 (m, 1H), 7.95 (d, J = 12.0 Hz, 1H), 7.87–7.83 (m, 1H), 7.68–7.65 (m, 2H), 7.61–7.50 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 187.8, 148.9, 142.9, 142.2, 139.1, 131.6, 131.2, 129.0, 128.9, 123.0, 117.8, 112.4, 109.3; HRMS (EI) m/z calcd for C15H12N2O3Cl [M + H]+ 303.0536, found 303.0539.
:20 (v/v) as the eluent. Yield 62% (47 mg); yellow solid, melting point: 255–256 °C. 1H NMR (400 MHz, CDCl3) δ 10.04 (brs, 1H), 8.20–8.16 (m, 2H), 7.96 (brs, 1H), 7.69–7.66 (m, 2H), 7.63–7.59 (m, 1H), 7.54–7.51 (m, 2H), 7.41–7.38 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 188.0, 146.9, 142.5, 141.8, 138.8, 131.8, 129.0, 129.0, 126.0, 117.1, 1108; HRMS (EI) m/z calcd for C15H12N2O3Cl [M + H]+ 303.0536, found 303.0541.
:15 (v/v) as the eluent. Yield 51% (37 mg); yellow solid, melting point: 128–129 °C. 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 12.0 Hz, 1H), 7.71–7.65 (m, 3H), 7.59–7.54 (m, 1H), 7.51–7.47 (m, 2H), 7.42 (dd, J = 8.0, 1.2 Hz, 1H), 7.26–7.21 (m, 1H), 7.02 (dd, J = 8.0 1.2 Hz, 1H), 6.92 (dd, J = 8.0, 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 188.1, 139.5, 138.5, 135.8, 131.3, 130.2, 128.6, 128.5, 128.3, 124.0, 122.5, 114.8, 111.5; HRMS (EI) m/z calcd for C15H12NOCl2 [M + H]+ 292.0296, found 292.0284.
:15 (v/v) as the eluent. Yield 63% (46 mg); yellow solid, melting point: 123–124 °C. 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 12.0 Hz, 1H), 7.57–7.55 (m, 2H), 7.50–7.46 (m, 1H), 7.43–7.38 (m, 2H), 7.15 (d, J = 8.0 Hz, 1H), 7.06 (d, J = 12.0 Hz, 1H), 6.99–6.96 (m, 1H), 6.85 (t, J = 2.0 Hz, 1H), 6.74–6.71 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 188.1, 140.4, 140.1, 138.4, 135.7, 131.3, 130.9, 128.56, 128.53, 124.0, 116.4, 114.3, 110.7; HRMS (EI) m/z calcd for C15H12NOCl2 [M + H]+ 292.0296, found 292.0305.
:15 (v/v) as the eluent. Yield 82% (60 mg); yellow solid, melting point: 179–180 °C. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 12.0 Hz, 1H), 7.65–7.62 (m, 2H), 7.58–7.54 (m, 1H), 7.50–7.46 (m, 2H), 7.30–7.24 (m, 3H), 6.90–6.87 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 188.1, 140.7, 138.6, 137.9, 131.2, 129.9, 129.2, 128.53, 128.50, 117.5, 110.2; HRMS (EI) m/z calcd for C15H12NOCl2 [M + H]+ 292.0296, found 292.0299.
:15 (v/v) as the eluent. Yield 85% (71 mg); yellow solid, melting point: 192–193 °C. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 12.0 Hz, 1H), 7.65–7.62 (m, 2H), 7.58–7.54 (m, 1H), 7.50–7.47 (m, 2H), 7.45–7.41 (m, 2H), 7.21 (d, J = 12.0 Hz, 1H), 6.85–6.81 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 188.1, 140.5, 138.5, 138.3, 132.9, 131.2, 128.54, 128.50, 117.8, 116.6, 110.3; HRMS (EI) m/z calcd for C15H12NOClBr [M + H]+ 335.9791, found 335.9792.
:10 (v/v) as the eluent. Yield 50% (34 mg); yellow solid, melting point: 133–134 °C. 1H NMR (400 MHz, CDCl3) δ 7.53–7.50 (m, 2H), 7.49–7.45 (m, 1H), 7.42–7.33 (m, 6H), 7.26–7.23 (m, 2H), 5.71 (t, J = 4.0 Hz, 1H), 4.44 (d, J = 4.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 187.6, 149.6, 139.2, 136.9, 130.4, 129.1, 128.3, 128.29, 128.26, 127.3, 106.5, 51.9; HRMS (EI) m/z calcd for C16H15NOCl [M + H]+ 272.0842, found 272.0849.
:10 (v/v) as the eluent. Yield 36% (26 mg); yellow solid, melting point: 169–170 °C. 1H NMR (400 MHz, CDCl3) δ 7.42 (t, J = 8.0 Hz, 1H), 7.35–7.25 (m, 6H), 7.14 (d, J = 8.0 Hz, 2H), 7.01 (d, J = 16.0 Hz, 1H), 5.45 (t, J = 8.0 Hz, 1H), 3.50 (q, J = 8.0 Hz, 2H), 2.84 (t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 187.4, 150.1, 139.2, 137.4, 130.2, 128.99, 128.95, 128.26, 128.19, 127.0, 105.9, 49.8, 37.6; HRMS (EI) m/z calcd for C17H16NOCl [M]+ 285.0920, found 285.0912.
:15 (v/v) as the eluent. Yield 75% (51 mg); yellow solid, melting point: 142–143 °C. 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 12.0 Hz, 1H), 7.54–7.52 (m, 2H), 7.33–7.31 (m, 2H), 7.29–7.25 (m, 2H), 7.18 (d, J = 12.0 Hz, 1H), 7.10–7.05 (m, 1H), 6.94–6.91 (m, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.0, 141.6, 140.8, 139.3, 135.9, 129.9, 129.1, 128.7, 123.9, 116.3, 109.8, 21.5; HRMS (EI) m/z calcd for C16H15NOCl [M + H]+ 272.0842, found 272.0833.
:15 (v/v) as the eluent. Yield 70% (53 mg); yellow solid, melting point: 160–161 °C. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 12.0 Hz, 1H), 7.52–7.50 (m, 2H), 7.26–7.24 (m, 2H), 7.12 (d, J = 12.0 Hz, 1H), 6.91–6.84 (m, 4H), 3.78 (s, 3H), 2.42 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.8, 156.5, 142.0, 141.3, 136.1, 132.8, 129.0, 128.6, 118.1, 115.1, 108.9, 55.6, 21.5; HRMS (EI) m/z calcd for C17H17NO2Cl [M + H]+ 302.0948, found 302.0947.
:15 (v/v) as the eluent. Yield 60% (47 mg); yellow solid, melting point: 223–224 °C. 1H NMR (400 MHz, CDCl3) δ 9.84 (d, J = 12.0 Hz, 1H), 815–8.14 (m, 1H), 7.97 (d, J = 12.0 Hz, 1H), 7.85 (dt, J = 8.0, 4.0 Hz, 1H), 7.61–7.56 (m, 4H), 7.32 (d, J = 8.0 Hz, 2H), 2.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.6, 148.9, 142.4, 142.3, 141.6, 136.3, 131.3, 129.4, 129.2, 122.9, 117.7, 112.4, 109.4, 21.5; HRMS (EI) m/z calcd for C16H14N2O3Cl [M + H]+ 317.0693, found 317.0696.
:15 (v/v) as the eluent. Yield 63% (48 mg); yellow solid, melting point: 189–190 °C. 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 16.0 Hz, 1H), 7.55–7.52 (m, 2H), 7.28–7.25 (m, 4H), 7.17 (d, J = 12.0 Hz, 1H), 6.89–6.86 (m, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.0, 141.8, 140.2, 138.0, 135.7, 129.9, 129.1, 129.0, 128.7, 117.4, 110.3, 21.5; HRMS (EI) m/z calcd for C16H14NOCl2 [M + H]+ 306.0452, found 306.0450.
:20 (v/v) as the eluent. Yield 78% (56 mg); yellow solid, melting point: 125–127 °C. 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 12.0 Hz, 1H), 7.66–7.62 (m, 2H), 7.34–7.29 (m, 2H), 7.14 (d, J = 12.0 Hz, 1H), 7.09–7.05 (m, 1H), 6.98–6.92 (m, 4H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.3, 162.1, 140.4, 139.3, 131.0, 130.7, 129.9, 123.9, 116.2, 113.7, 109.6, 55.4; HRMS (EI) m/z calcd for C16H15NO2Cl [M + H]+ 288.0791, found 288.0800.
:25 (v/v) as the eluent. Yield 72% (57 mg); yellow solid, melting point: 162–163 °C. 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 12.0 Hz, 1H), 7.66–7.62 (m, 2H), 7.10 (d, J = 12.0 Hz, 1H), 6.99–6.95 (m, 2H), 6.94–6.89 (m, 4H), 3.89 (s, 3H), 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.1, 161.9, 156.5, 141.5, 132.9, 131.3, 130.6, 118.1, 115.1, 113.7, 108.7, 55.6, 55.4; HRMS (EI) m/z calcd for C17H16NO3Cl [M]+ 317.0819, found 317.0812.
:20 (v/v) as the eluent. Yield 55% (46 mg); yellow solid, melting point: 169–170 °C. 1H NMR (400 MHz, CDCl3) δ 7.94–7.88 (m, 2H), 7.82 (t. J = 4.0 Hz, 1H), 7.73–7.69 (m, 2H), 7.52 (t, J = 8.0 Hz, 1H), 7.34–7.27 (m, 2H), 7.03–6.99 (m, 2H), 3.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.3, 162.6, 149.4, 140.8, 138.2, 130.9, 130.8, 130.4, 121.5, 117.9, 113.9, 111.5, 110.7, 55.5; HRMS (EI) m/z calcd for C16H14N2O4Cl [M + H]+ 333.0642, found 333.0645.
:20 (v/v) as the eluent. Yield 65% (52 mg); yellow solid, melting point: 174–175 °C. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 12.0 Hz, 1H), 7.68–7.65 (m, 2H), 7.32–7.28 (m, 2H), 7.12 (d, J = 16.0 Hz, 1H), 7.00–6.96 (m, 2H), 6.92–6.88 (m, 2H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.2, 162.3, 139.5, 138.1, 130.8, 130.7, 129.9, 128.9, 117.3, 113.7, 110.1, 55.4; HRMS (EI) m/z calcd for C16H13NO2Cl2 [M]+ 321.0323, found 321.0318.
:15 (v/v) as the eluent. Yield 80% (58 mg); yellow solid, melting point: 170–171 °C. 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 12.0 Hz, 1H), 7.61–7.57 (m, 2H), 7.48–7.44 (m, 2H), 7.37–7.30 (m, 3H), 7.15–7.11 (m, 1H), 6.98–6.95 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 186.8, 141.1, 139.0, 137.3, 137.1, 129.99, 129.97, 128.7, 124.3, 116.4, 109.3; HRMS (EI) m/z calcd for C15H12NOCl2 [M + H]+ 292.0296, found 292.0301.
:15 (v/v) as the eluent. Yield 75% (60 mg); yellow solid, melting point: 173–174 °C. 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 16.0 Hz, 1H), 7.59–7.57 (m, 2H), 7.46–7.43 (m, 2H), 7.17 (d, J = 12.0 Hz, 1H), 6.93–6.88 (m, 4H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 186.6, 156.8, 142.3, 137.3, 137.1, 132.5, 129.9, 128.7, 118.3, 115.2, 108.5, 55.6; HRMS (EI) m/z calcd for C16H14NO2Cl2 [M + H]+ 322.0402, found 322.0405.
:15 (v/v) as the eluent. Yield 47% (39 mg); yellow solid, melting point: 200–201 °C. 1H NMR (400 MHz, CDCl3) δ 9.90 (d, J = 12.0 Hz, 1H), 8.19 (t, J = 4.0 Hz, 1H), 7.88–7.85 (m, 1H), 7.71–7.65 (m, 3H), 7.60–7.55 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 186.6, 148.9, 143.2, 142.1, 137.9, 136.3, 131.2, 130.9, 128.9, 123.2, 117.9, 112.7, 109.1; HRMS (EI) m/z calcd for C15H11N2O3Cl2 [M + H]+ 337.0147, found 337.0147.
:15 (v/v) as the eluent. Yield 69% (56 mg); yellow solid, melting point: 199–200.5 °C. 1H NMR (400 MHz, CDCl3) δ 9.69 (d, J = 12.0 Hz, 1H), 7.84 (d, J = 12.0 Hz, 1H), 7.66–7.63 (m, 2H), 7.56–7.53 (m, 2H), 7.38–7.34 (m, 2H), 7.28–7.24 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 186.3, 143.8, 139.8, 138.1, 136.0, 130.8, 129.7, 128.9, 127.8, 119.4, 108.1; HRMS (EI) m/z calcd for C15H11NOCl3 [M + H]+ 325.9906, found 325.9912.
:15 (v/v) as the eluent. Yield 91% (76 mg); yellow solid, melting point: 179–180 °C. 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 12.0 Hz, 1H), 7.62–7.59 (m, 2H), 7.52–7.48 (m, 2H), 7.36–7.32 (m, 2H), 7.23 (d, J = 12.0 Hz, 1H), 6.95–6.93 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 186.9, 141.1, 138.9, 137.5, 131.7, 130.1, 125.7, 124.4, 116.4, 109.4; HRMS (EI) m/z calcd for C15H11NOClBr [M]+ 334.9713, found 334.9703.
:20 (v/v) as the eluent. Yield 87% (79 mg); yellow solid, melting point: 188–189 °C. 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 12.0 Hz, 1H), 7.60–7.57 (m, 2H), 7.50–7.46 (m, 2H), 7.18 (d, J = 12.0 Hz, 1H), 6.91–6.85 (m, 4H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 186.7, 156.8, 142.3, 137.7, 132.5, 131.6, 130.1, 125.5, 118.3, 115.2, 108.5, 55.6; HRMS (EI) m/z calcd for C16H13NO2ClBr [M]+ 364.9818, found 364.9812.
:20 (v/v) as the eluent. Yield 72% (68 mg); yellow solid, melting point: 197–198 °C. 1H NMR (400 MHz, CDCl3) δ 9.91 (s, 1H), 8.20 (t, J = 2.0 Hz, 1H), 7.99 (s, 1H), 7.88–7.85 (m, 1H), 7.72–7.56 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 186.7, 148.9, 143.2, 142.1, 138.2, 131.9, 131.2, 131.1, 125.2, 123.2, 117.9, 112.7, 109.0; HRMS (EI) m/z calcd for C15H10N2O3ClBr [M]+ 379.9563, found 379.9554.
:15 (v/v) as the eluent. Yield 76% (70 mg); yellow solid, melting point: 209–210 °C. 1H NMR (400 MHz, CDCl3) δ 9.69 (d, J = 12.0 Hz, 1H), 7.84 (d, J = 16.0 Hz, 1H), 7.70–7.67 (m, 2H), 7.59–7.56 (m, 2H), 7.38–7.34 (m, 2H), 7.28–7.24 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 186.4, 143.7, 139.8, 138.5, 131.9, 130.9, 129.7, 127.8, 124.9, 119.4, 108.1; HRMS (EI) m/z calcd for C15H11NOCl2Br [M + H]+ 369.9401, found 369.9404.
General procedure for the scaled-up synthesis of 3aa and 3ad
Aniline 2a or 2d (3.5 mmol), β-chloroenone 1a (4.2 mmol, 0.697 g), and DMSO (20 mL) were added to a 100 mL reaction flask that was dried in an oven and cooled to room temperature under an N2 atmosphere. The reaction mixture was heated at 90 °C using an oil bath for 8 h. Then, the reaction mixture was cooled to room temperature and transferred to a separatory funnel using ethyl acetate (60 mL). Water (50 mL) was added, and the organic layer was separated. The aqueous layer was extracted again with ethyl acetate (30 mL). The combined organic layer was washed with brine solution and dried over MgSO4. The solvent was evaporated under reduced pressure using a rotary evaporator. The crude product was purified by silica gel column chromatography using an ethyl acetate/hexane mixture as the eluent.General procedure for the synthesis of 1,4-benzodiazepine derivatives
o-Phenylenediamine (0.5 mmol), β-chloroenone (0.5 mmol), and DMSO (3 mL) were added to an oven-dried 10 mL reaction flask cooled under a N2 atmosphere. The reaction mixture was stirred for 12 h at room temperature. Then, the reaction mixture was transferred to a separatory funnel using ethyl acetate (30 mL). Water (30 mL) was added, and the organic layer was separated. The aqueous layer was extracted again with ethyl acetate (20 mL). The combined organic layer was washed with brine solution and dried over MgSO4. The solvent was evaporated under reduced pressure using a rotary evaporator. The crude product was purified by silica gel column chromatography using an ethyl acetate/hexane mixture as the eluent.:20 (v/v) as the eluent. Yield 40% (37 mg); yellow solid, melting point: 102–103 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.45 (d, J = 8.0 Hz, 1H), 7.94–7.91 (m, 2H), 7.60 (t, J = 8.0 Hz, 1H), 7.50–7.39 (m, 7H), 7.01 (d, J = 8.0 Hz, 1H), 6.89–6.85 (m, 2H), 6.78–6.72 (m, 2H), 6.69–6.66 (m, 2H), 5.72 (d, J = 4.0 Hz, 1H), 5.11–5.06 (m, 1H), 3.11–2.98 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 199.2, 192.4, 146.7, 141.0, 137.7, 137.0, 133.6, 131.4, 129.9, 129.0, 128.5, 128.4, 123.8, 122.1, 121.0, 120.3, 116.0, 51.5, 45.6; HRMS (EI) m/z calcd for C24H20N2O2 [M]+ 368.1525, found 368.1537.
:20 (v/v) as the eluent. Yield 42% (44 mg); yellow solid, melting point: 113–114 °C. 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 4.0 Hz, 2H), 7.50 (t, J = 8.0 Hz, 1H), 7.46–7.34 (m, 6H), 7.30–7.26 (m, 2H), 7.05 (d, J = 8.0 Hz, 1H), 6.45 (s, 1H), 6.37 (s, 1H), 5.17 (dd, J = 8.0, 4.0 Hz, 1H), 4.98 (brs, 1H), 3.32 (dd, J = 16.0, 4.0 Hz, 1H), 2.87 (dd, J = 12.0, 16.0 Hz, 1H), 2.00 (s, 3H), 1.96 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.0, 193.9, 146.9, 140.3, 136.8, 134.0, 133.1, 132.9, 130.0, 129.7, 128.5, 128.4, 128.3, 128.0, 123.4, 121.1, 115.7, 52.2, 43.8, 18.8, 18.7; HRMS (EI) m/z calcd for C26H24N2O2 [M]+ 396.1838, found 396.1834.
:20 (v/v) as the eluent. Yield 39% (39 mg); yellow solid, melting point: 67–68 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.38 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.32–7.23 (m, 6H), 7.02 (d, J = 8.0 Hz, 1H), 6.87–6.85 (m, 2H), 6.76–6.70 (m, 2H), 6.66–6.63 (m, 1H), 5.67 (d, J = 4.0 Hz, 1H), 5.05 (quint, J = 4.0 Hz, 1H), 3.06–2.95 (m, 2H), 2.35 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 198.7, 192.4, 146.4, 143.9, 139.6, 138.2, 137.7, 134.6, 131.5, 129.6, 128.9, 128.6, 123.7, 122.1, 120.9, 120.2, 116.1, 51.5, 45.4, 21.6, 21.4; HRMS (EI) m/z calcd for C26H24N2O2 [M]+ 396.1838, found 396.1850.
:20 (v/v) as the eluent. Yield 37% (39 mg); yellow solid, melting point: 108–109 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.23 (d, J = 8.0 Hz, 1H), 7.83–7.80 (m, 2H), 7.28–7.22 (m, 6H), 6.97 (d, J = 8.0 Hz, 1H), 6.61 (s, 1H), 6.41 (s, 1H), 5.41 (brs, 1H), 5.1–4.96 (m, 1H), 3.05–2.90 (m, 2H), 2.36 (s, 6H), 2.05 (s, 3H), 1.97 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 198.9, 192.2, 146.4, 143.8, 139.4, 138.4, 135.2, 134.7, 131.5, 130.1, 129.5, 128.9, 128.6, 128.5, 128.4, 123.1, 121.2, 115.7, 51.6, 45.4, 21.6, 21.4, 19.0, 18.8; HRMS (EI) m/z calcd for C28H28N2O2 [M]+ 424.2151, found 424.2136.
:40 (v/v) as the eluent. Yield 41% (44 mg); yellow solid, melting point: 110–111 °C. 1H NMR (400 MHz, CDCl3) δ 7.85–7.83 (m, 2H), 7.65–7.52 (m, 1H), 7.45–7.43 (m, 2H), 7.13 (d, J = 8.0 Hz, 1H), 6.84–6.81 (m, 4H), 6.76–6.69 (m, 3H), 6.62–6.61 (m, 1H), 5.16 (dd, J = 1.7, 12.0 Hz, 1H), 4.98 (brs, 1H), 3.80 (s, 6H), 3.33 (d, J = 12.0 Hz, 1H), 2.82 (dd, J = 12.0, 16.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 198.3, 193.4, 163.5, 161.2, 146.0, 136.4, 132.4, 131.2, 130.6, 130.3, 129.8, 124.1, 122.4, 121.7, 119.9, 116.0, 113.6, 113.4, 55.4, 55.3, 52.5, 43.7; HRMS (EI) m/z calcd for C26H24N2O4 [M]+ 428.1736, found 428.1731.
:40 (v/v) as the eluent. Yield 38% (44 mg); yellow solid, melting point: 115–116 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.23 (d, J = 8.0 Hz, 1H), 7.93–7.89 (m, 2H), 7.42–7.38 (m, 2H), 7.03 (d, J = 8.0 Hz, 1H), 7.00–6.96 (m, 4H), 6.63 (s, 1H), 6.41 (s, 1H), 5.42 (brs, 1H), 4.97 (d, J = 8.0 Hz, 1H), 3.82 (s, 3H), 3.81 (s, 3H), 3.03 (dd, J = 4.0, 16.0 Hz, 1H), 2.89 (dd, J = 12.0, 16.0 Hz), 2.05 (s, 3H), 1.97 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 197.7, 191.6, 163.5, 160.8, 145.9, 135.1, 133.3, 131.3, 130.8, 130.5, 130.1, 129.0, 128.4, 123.1, 121.1, 115.6, 114.2, 113.7, 55.9, 55.7, 45.1, 19.0, 18.8; HRMS (EI) m/z calcd for C28H28N2O4 [M]+ 456.2049, found 456.2033.
:20 (v/v) as the eluent. Yield 10% (11 mg); yellow solid, melting point: 151–152 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.51 (d, J = 8.0 Hz, 1H), 7.94–7.91 (m, 2H), 7.56–7.52 (m, 2H), 7.51–7.49 (m, 2H), 7.43–7.40 (m, 2H), 6.97 (d, J = 8.0 Hz, 1H), 6.90–6.86 (m, 1H), 6.78–6.74 (m, 2H), 6.70–6.64 (m, 1H), 5.77 (brs, 1H), 5.02 (t, J = 8.0 Hz, 1H), 3.06–2.98 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 198.1, 190.9, 146.8, 139.7, 138.5, 137.8, 135.7, 134.6, 131.2, 130.4, 130.3, 129.1, 128.5, 123.9, 122.1, 121.0, 120.5, 115.8, 51.5, 45.9; HRMS (EI) m/z calcd for C24H18N2O2Cl2 [M]+ 436.0745, found 436.0736.
:20 (v/v) as the eluent. Yield 15% (18 mg); yellow solid, melting point: 141–142 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.37 (d, J = 8.0 Hz, 1H), 7.94–7.91 (m, 2H), 7.56–7.52 (m, 2H), 7.51–7.48 (m, 2H), 7.40–7.37 (m, 2H), 6.93 (d, J = 8.0 Hz, 1H), 6.64 (s, 1H), 6.43 (s, 1H), 5.57 (s, 1H), 4.99 (t, J = 8.0 Hz, 1H), 3.05–2.95 (m, 2H), 2.06 (s, 3H), 1.98 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 198.3, 190.8, 146.8, 139.9, 138.4, 135.8, 135.2, 134.5, 131.8, 130.4, 130.2, 129.1, 128.8, 128.6, 128.5, 123.2, 121.4, 115.5, 51.7, 45.8, 19.0, 18.8; HRMS (EI) m/z calcd for C26H22N2O2Cl2 [M]+ 464.1058, found 464.1051.
:15 (v/v) as the eluent. Yield 20% (26 mg); yellow solid, melting point: 203–204 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.48 (d, J = 8.0 Hz, 1H), 7.86–7.83 (m, 2H), 7.70–7.66 (m, 2H), 7.65–7.62 (m, 2H), 7.36–7.32 (m, 2H), 6.96 (d, J = 8.0 Hz, 1H), 6.89–6.86 (m, 1H), 6.77–6.73 (m, 2H), 6.69–6.65 (m, 1H), 5.76 (d, J = 4.0 Hz, 1H), 5.05–5.00 (m, 1H), 3.06–2.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 198.3, 191.0, 146.8, 140.1, 137.8, 136.1, 132.1, 131.4, 131.2, 130.5, 127.7, 124.0, 123.3, 122.1, 121.0, 120.5, 115.8, 51.5, 45.8; HRMS (EI) m/z calcd for C24H18N2O2Br2 [M]+ 523.9735, found 523.9738.
:15 (v/v) as the eluent. Yield 27% (37 mg); yellow solid, melting point: 167–169 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.35 (d, J = 8.0 Hz, 1H), 7.85–7.82 (m, 2H), 7.68–7.66 (m, 2H), 7.64–7.61 (m, 2H), 7.33–7.30 (m, 2H), 6.93 (d, J = 8.0 Hz, 1H), 6.63 (s, 1H), 6.42 (s, 1H), 5.52 (d, J = 8.0 Hz, 1H), 5.05–4.96 (m, 1H), 3.05–2.95 (m, 2H), 2.05 (s, 3H), 1.98 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 198.5, 190.8, 146.9, 140.2, 136.1, 135.4, 132.0, 131.8, 131.4, 130.5, 130.4, 128.7, 128.5, 127.6, 123.2, 123.1, 121.4, 115.5, 51.6, 45.8, 19.1, 18.8; HRMS (EI) m/z calcd for C26H22N2O2Br2 [M + 2]+ 554.0028, found 554.0021.
:15 (v/v) as the eluent. Yield 28% (33 mg); the product was obtained as a mixture of stereoisomers in a 1:0.7 ratio. Yellow solid, melting point: 145–147 °C. 1H NMR (400 MHz, CDCl3) δ 12.20 (d, J = 12.0 Hz, 0.7H), 12.11 (d, J = 12.0 Hz, 1H), 7.95–7.93 (m, 3.3H), 7.49–7.38 (m, 7H), 7.17–6.96 (m, 3.5H), 6.85–6.79 (m, 2H), 6.12 (d, J = 8.0 Hz, 0.7H), 6.05 (d, J = 8.0 Hz, 1H), 3.56 (brs, 2.7H); 13C NMR (100 MHz, CDCl3) δ 191.3, 190.9, 147.3, 146.4, 139.3, 139.2, 137.2, 132.2, 131.5, 131.4, 128.6, 128.5, 128.3, 127.5, 127.3, 125.3, 125.2, 119.9, 119.0, 117.8, 117.3, 95.5, 93.8; HRMS (EI) m/z calcd for C15H14N2O [M]+ 238.1106, found 238.1102.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Ministry of Science and Technology ROC (MOST 111-2133-M-003-016) is gratefully acknowledged for its financial support. The authors are grateful to the instrumentation center at National Taiwan Normal University for providing the analytical NMR (Ms Chiu-Hui He) and HRMS (Ms Hsiu-Min Huan) data presented in this paper.References
- (a) Y. Han, L. Zhou, C. Wang, S. Feng, R. Ma and J.-P. Wan, Chin. Chem. Lett., 2024, 35, 108977 CrossRef CAS; (b) Z. Wang, B. Zhao, Y. Liu and J.-P. Wan, Adv. Synth. Catal., 2022, 364, 1508 CrossRef CAS; (c) M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479 CrossRef CAS; (d) I. J. Amaye, R. D. Haywood, E. M. Mandzo, J. J. Wirick and P. L. Jackson-Ayotunde, Tetrahedron, 2021, 83, 131984 CrossRef CAS.
- F. Wang, W. Sun, Y. Wang, Y. Jiang and T.-P. Loh, Org. Lett., 2018, 20, 1256 CrossRef CAS.
- J.-P. Wan, S. Zhong, L. Xie, X. Cao, Y. Liu and L. Wei, Org. Lett., 2016, 18, 584 CrossRef CAS PubMed.
- (a) Y. Gao, Y. Liu and J.-P. Wan, J. Org. Chem., 2019, 84, 2243 CrossRef CAS; (b) Z. Yang, L. Hu, T. Cao, L. An, L. Li, T. Yang and C. Zhou, New J. Chem., 2019, 43, 16441 RSC; (c) X. Duan, X. Liu, X. Cuan, L. Wang, K. Liu, H. Zhou, X. Chen, H. Li and J. Wang, J. Org. Chem., 2019, 84, 12366 CrossRef CAS PubMed.
- (a) Y. Xie, Z. Zhang, B. Zhang, N. He, M. Peng, S. Song, B. Wang and F. Yu, J. Org. Chem., 2024, 89, 8521 CrossRef CAS; (b) G. S. Sorabad and M. R. Maddani, New J. Chem., 2019, 43, 6563 RSC; (c) H. Xu, P. Zhou, R. Hang, J. Zhou, L.-L. Lu, Y. Shen and F.-C. Yu, Tetrahedron Lett., 2016, 57, 4965 CrossRef CAS; (d) N. G. Ramesh, E. H. Heijne, A. J. H. Klunder and B. Zwanenburg, Tetrahedron, 2002, 58, 1361 CrossRef CAS.
- (a) T. Zhang, W. Yao, J.-P. Wan and Y. Liu, Adv. Synth. Catal., 2021, 363, 4811 CrossRef CAS; (b) B. Zhang, D. Liu, Y. Sun, Y. Zhang, J. Feng and F. Yu, Org. Lett., 2021, 23, 3076 CrossRef CAS PubMed; (c) Y. Lin, J. Jin, C. Wang, J.-P. Wan and Y. Liu, J. Org. Chem., 2021, 86, 12378 CrossRef CAS PubMed; (d) T. Luo, J.-P. Wan and Y. Liu, Org. Chem. Front., 2020, 7, 1107 RSC; (e) Y. Siddaraju and K. R. Prabhu, J. Org. Chem., 2017, 82, 3084 CrossRef CAS; (f) Y. Yuan, W. Hou, D. Zhang-Negrerie, K. Zhao and Y. Du, Org. Lett., 2014, 16, 5410 CrossRef CAS PubMed.
- (a) J.-i. Yamaguchi, Y. Akagi, R. Koike, K. Katou, T. Katakura, T. Hanase and T. Homma, Tetrahedron Lett., 2024, 135, 154876 CrossRef CAS; (b) X. Li, Z. Chen, Y. Liu, N. Luo, W. Chen, C. Liu, F. Yu and J. Huang, J. Org. Chem., 2022, 87, 10349 CrossRef PubMed; (c) H. Wu, T. Luo, J.-P. Wan, J. Jiang and Y. Liu, Eur. J. Org. Chem., 2022, e202200552 CrossRef CAS; (d) T. Liu, L. Wei, B. Zhao, Y. Liu and J.-P. Wan, J. Org. Chem., 2021, 86, 9861 CrossRef CAS PubMed; (e) Z. Jiang, J. Zhou, H. Zhu, H. Liu and Y. Zhou, Org. Lett., 2021, 23, 4406 CrossRef CAS PubMed; (f) J. Chen, P. Guo, J. Zhang, J. Rong, W. Sun, Y. Jiang and T.-P. Loh, Angew. Chem., Int. Ed., 2019, 58, 12674 CrossRef CAS PubMed; (g) J. Sun, X. Cheng, J. K. Mansaray, W. Fei, J. Wan and W. Yao, Chem. Commun., 2018, 54, 13953 RSC.
- (a) M. Baidya, D. Maiti, L. Roy and S. D. Sarkar, Angew. Chem., Int. Ed., 2022, 61, e202111679 CrossRef CAS PubMed; (b) L. Fu, J.-P. Wan, L. Zhou and Y. Liu, Chem. Commun., 2022, 58, 1808 RSC; (c) H. Guo, L. Tian, Y. Liu and J.-P. Wan, Org. Lett., 2022, 24, 228 CrossRef CAS PubMed; (d) L. Fu, Y. Liu and J.-P. Wan, Org. Lett., 2021, 23, 4363 CrossRef CAS PubMed; (e) S. Mkrtchyan and V. O. Iaroshenko, Chem. Commun., 2020, 56, 2606 RSC; (f) S. Zhou, D.-Y. Liu, S. Wang, J.-S. Tian and T.-P. Loh, Chem. Commun., 2020, 56, 15020 RSC; (g) M.-N. Zhao, Z.-J. Zhang, Z.-H. Ren, D.-S. Yang and Z.-H. Guan, Org. Lett., 2018, 20, 3088 CrossRef CAS; (h) Y.-X. Jiao, L.-S. Wei, C.-Y. Zhao, K. Wei, D.-L. Mo, C.-X. Pan and G.-F. Su, Adv. Synth. Catal., 2018, 360, 4446 CrossRef CAS; (i) S. Tang, X. Gao and A. Lei, Chem. Commun., 2017, 53, 3354 RSC; (j) J. Liu, W. Wei, T. Zhao, X. Liu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2016, 81, 9326 CrossRef CAS; (k) C.-J. Wu, Q.-Y. Meng, T. Lei, J.-J. Zhong, W.-Q. Liu, L.-M. Zhao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Catal., 2016, 6, 4635 CrossRef CAS.
- (a) R. Kumar, N. Saha, P. Purohit, S. K. Garg, K. Seth, V. S. Meena, S. Dubey, K. Dave, R. Goyal, S. S. Sharma, U. C. Banerjee and A. K. Chakraborti, Eur. J. Med. Chem., 2019, 182, 111601 CrossRef CAS PubMed; (b) M. M. Ghorab, F. A. Ragab, H. I. Heiba, M. G. Ei-Gazzar and M. G. M. Ei-Gazzar, Bioorg. Med. Chem. Lett., 2018, 28, 1464 CrossRef CAS; (c) M. Cindric, M. Rubcic, T. Hrenar, J. Pisk, D. Cvijanovic, J. Lovric and V. Vrdoljak, J. Mol. Struct., 2018, 1154, 636 CrossRef CAS; (d) P. V. Sowmya, B. Poojary, B. C. Revanasiddappa, M. Vijayakumar, P. Nikil and V. Kumar, Res. Chem. Intermed., 2017, 43, 7399 CrossRef CAS.
- (a) K. Keskar, C. Zepeda-Velazquez, C. B. Dokuburra, H. A. Jenkins and J. McNulty, Chem. Commun., 2019, 55, 10868 RSC; (b) M. J. Gonzalez-Soria and F. Alonso, Adv. Synth. Catal., 2019, 361, 5005 CrossRef CAS; (c) A. Pal and S. R. Hussaini, ACS Omega, 2019, 4, 269 CrossRef CAS; (d) Y.-P. Liu, C.-J. Zhu, C.-C. Yu, A.-E. Wang and P.-Q. Huang, Eur. J. Chem., 2019, 7169 CrossRef CAS; (e) X. Duan, S. Yang, C. Yao, F. Jia, L. Wang, W. Li, Z. Meng, Y. Zhou and X. Li, ChemistrySelect, 2019, 4, 12992 CrossRef CAS; (f) Z. Zheng, Q. Tao, Y. Ao, M. Xu and Y. Li, Org. Lett., 2018, 20, 3907 CrossRef CAS; (g) A. Leggio, A. Comande, E. L. Belsito, M. Greco, L. L. Feudo and A. Liguori, Org. Biomol. Chem., 2018, 16, 5677 RSC; (h) Y.-W. Kang, Y. J. Cho, S. J. Han and H.-Y. Jang, Org. Lett., 2016, 18, 272 CrossRef CAS; (i) W. Shi, S. Sun, M. Wu, B. Catano, W. Li, J. Wang, H. Guo and Y. Xing, Tetrahedron Lett., 2015, 56, 468 CrossRef CAS; (j) S. M. Kim, D. Lee and S. H. Hong, Org. Lett., 2014, 16, 6168 CrossRef CAS PubMed.
- T. Miura, Y. Funakoshi, T. Tanaka and M. Murakami, Org. Lett., 2014, 16, 2760 CrossRef CAS PubMed.
- S. Suresh, P. B. Patil, P.-H. Yu, C.-C. Fang, Y.-Z. Weng, V. Kavala and C.-F. Yao, Adv. Synth. Catal., 2021, 363, 4915 CrossRef CAS.
- (a) S. A. Dhabale, S. Kumar, N. Bhanwala and G. L. Khatik, Curr. Org. Chem., 2023, 27, 1471 CrossRef CAS; (b) J. Schimer, P. Cígler, J. Veselý, K. G. Šašková, M. Lepšík, J. Brynda, P. Řezáčová, M. Kožíšek, I. Císařová, H. Oberwinkler, H.-G. Kraeusslich and J. Konvalinka, J. Med. Chem., 2012, 55, 10130 CrossRef CAS PubMed; (c) L.-Z. Wang, X.-Q. Li and Y.-S. An, Org. Biomol. Chem., 2015, 13, 5497 RSC.
- (a) S. Farooq and Z. Ngaini, J. Heterocycl. Chem., 2021, 58, 1914 CrossRef CAS; (b) R. Jamatia, A. Gupta, B. Dam, M. Saha and A. K. Pal, Green Chem., 2017, 19, 1576 RSC; (c) M. Kodomari, T. Noguchi and T. Aoyama, Synth. Commun., 2004, 34, 1783 CrossRef CAS.
- (a) F. K. Sarkar, A. Gupta, R. Jamatia, J. M. H. Anal and A. K. Pal, New J. Chem., 2021, 45, 19553 RSC; (b) K. J. Tamuli and M. Bordoloi, ChemistrySelect, 2020, 5, 1353 CrossRef CAS; (c) M. Jeganathan and K. Pitchumani, ACS Sustainable Chem. Eng., 2014, 2, 1169 CrossRef CAS; (d) C.-W. Kuo, C.-C. Wang, V. Kavala and C.-F. Yao, Molecules, 2008, 13, 2313 CrossRef CAS PubMed; (e) S. D. Sharma, P. Gogoi and D. Konwar, Green Chem., 2007, 9, 153 RSC; (f) R. Kumar, P. Chaudhary, S. Nimesh, A. K. Verma and R. Chandra, Green Chem., 2006, 8, 519 RSC.
- L. Feray, P. Perfetti and M. Bertrand, Tetrahedron, 2009, 65, 8733 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectra copies of all the compounds and mass spectrometric data. CCDC 2334143 (3aa), 2370736 (3af) and 2360356 (6ea). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00954a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |