
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of natural Lachnophyllum methyl ester, Lachnophyllum lactone and their synthetic analogs†
Kodjo
Adande
ab,
Oudjaniyobi
Simalou
 b,
Juline
Ardanuy
a,
Kodjo
Eloh
c,
Chérine
Mehalla
d,
Patricia
Constant
d,
Isabelle
Fabing
a,
Yves
Génisson
*a and
Stéphanie
Ballereau
*a
b,
Juline
Ardanuy
a,
Kodjo
Eloh
c,
Chérine
Mehalla
d,
Patricia
Constant
d,
Isabelle
Fabing
a,
Yves
Génisson
*a and
Stéphanie
Ballereau
*a
aLaboratoire de Synthèse et Physico-Chimie de Molécules d'Intérêt Biologique (SPCMIB), Université de Toulouse, CNRS, Université Toulouse III – Paul Sabatier (UT3), 118 route de Narbonne, 31062, Toulouse Cedex 9, France. E-mail: stephanie.ballereau@univ-tlse3.fr; yves.genisson@univ-tlse3.fr
bLaboratoire de Chimie Organique et Des Substances Naturelles (Lab COSNat), Département de Chimie, Faculté Des Sciences, Université de Lomé, Lomé, Togo
cLaboratoire de Chimie Organique et des Sciences de l'Environnement (LaCOSE), Département de Chimie, Faculté Des Sciences et Techniques, Université de Kara, Kara, Togo
dInstitut de Pharmacologie et de Biologie Structurale (IPBS), Université de Toulouse, CNRS, Université Toulouse III - Paul Sabatier (UT3), Toulouse, France
First published on 4th September 2024
Abstract
(2Z)-Lachnophyllum methyl ester and (4Z)-Lachnophyllum lactone were recently identified as major components in essential oils and extracts of Conyza bonariensis from Togo. Extended biological evaluation of these acetylenic compounds was however hampered by the reduced amounts isolated. A synthetic route was designed providing access to larger quantities of these two natural products as well as to original non-natural analogs with the prospect of exploring for the first time the structure–activity relationships in this series. Using LC/MS analysis, synthetic samples allowed confirming the presence of the two previously isolated natural products in plant extracts obtained by the accelerated solvent extraction technique. The nematocidal activity of the synthesized compounds confirmed the potency of the natural products, which remain the most active among all analogs tested. The synthesized compounds were also assessed against Leishmania infantum axenic amastigotes and the Mycobacterium tuberculosis H37Rv pathogenic strain. (2Z)-Lachnophyllum methyl ester, (4Z)-Lachnophyllum lactone and lactone analogs exhibited the strongest antileishmanial potency. As expected, a longer alkyl chain was necessary to observe significant antimycobacterial activity. The lactone analog bearing a C10 lipophilic appendage displayed the highest antimycobacterial potency. The notable activities of lactones, naturally occurring or analogs, either nematicidal, antileishmanial or antimycobacterial, were compared to their cytotoxicity for mammalian cells and revealed moderate selectivity index values. In this regard, the innocuous (2Z)-Lachnophyllum methyl ester and its analogs open up more promising perspectives for the discovery of bioactive agents to protect both agricultural crops and human health.
Introduction
Among the numerous plants traditionally used in herbal medicine, Conyza plant species of the Asteraceae family are known for their various pharmacological applications ranging from the treatment of malaria to toothache.1 The chemical compositions of essential oils obtained from Conyza species collected in various regions of the world such as Africa, South America or the Mediterranean Sea area have been extensively studied.2 In addition to known flavonoids or terpenoids, enyne derivatives were also identified as components of Conyza species essential oil extracts.3–6We recently isolated (2Z)-Lachnophyllum methyl ester and (4Z)-Lachnophyllum lactone (Fig. 1) as major components in extracts of Conyza bonariensis.7 The quantities obtained either from essential oils or solvent extraction were however too low to explore further the biological activities of these acetylenic compounds. To overcome this limitation, we developed a synthetic route to these natural compounds. The designed synthetic route affords original analogs also to extend the structure–activity relationship of this family of compounds.
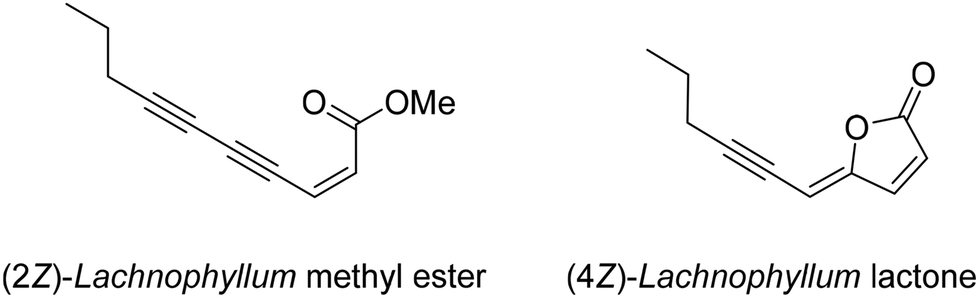 | ||
| Fig. 1 Structures of (2Z)-Lachnophyllum methyl ester and (4Z)-Lachnophyllum lactone. | ||
Results and discussion
Synthesis
Despite the numerous publications describing the isolation of Lachnophyllum esters and lactones from plants, mostly from Conyza species,2 there were, until the work of Soriano et al. published during this study,8 only two articles reporting the chemical synthesis of these natural compounds. The synthesis of the Lachnophyllum lactone as a mixture of E and Z stereoisomers starting from 2-trimethylsiloxyfuran was described in 19819 whereas the (2E) Lachnophyllum ethyl ester was mentioned as an intermediate in a synthetic study published in 1987.10 We thus designed a synthetic route allowing us not only to obtain larger quantities of natural Lachnophyllum methyl ester and lactone but also to access non-natural analogs for biological evaluation. | ||
| Scheme 1 Retrosynthetic route to Lachnophyllum methyl esters and their analogs. | ||
According to route A, a Cadiot–Chodkiewicz coupling between methyl pent-2-en-4-ynoate (2) and an alkyne bromide leads to the methyl hepta-2-en-4,6-diynoate motif, characteristic of the Lachnophyllum esters. This sequence has already been validated for the synthesis of natural epoxypolyynes.11,12 Alternatively, the more convergent route B relies on a Sonogashira coupling of methyl 3-iodoacrylate (3) with a terminal 1,3-diyne. These two synthetic pathways potentially allow for the synthesis of the two stereoisomers (Z or E) of Lachnophyllum esters as well as their synthetic analogs.
The commercially available methyl 3-iodoacrylates (Z-3 and E-3) can also be readily prepared in the laboratory.13,14 The methyl pent-2-en-4-ynoates (Z-2 and E-2) were obtained from the corresponding vinyl iodides (Z-3 and E-3 respectively) by means of a Sonogashira coupling with TMS-acetylene followed by the cleavage of the trimethylsilyl protecting group (Scheme 2).13
 | ||
Scheme 2 Synthesis of methyl 3-iodoacrylates Z-3 and E-3 and methyl pent-2-en-4-ynoates Z-2 and E-2. Reagents and conditions: (a) NaI, AcOH, 70 °C, 84%; (b) (i) TMSC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, PdCl2(PPh3)2, CuI, Et3N, THF, 0 °C to rt; (ii) TBAF, THF, 0 °C to rt, 68% over two steps for Z-2, 76% over two steps for E-2; (c) (i) HI, H2O, reflux, (ii) H2SO4, MeOH, reflux, 74% over two steps. CH, PdCl2(PPh3)2, CuI, Et3N, THF, 0 °C to rt; (ii) TBAF, THF, 0 °C to rt, 68% over two steps for Z-2, 76% over two steps for E-2; (c) (i) HI, H2O, reflux, (ii) H2SO4, MeOH, reflux, 74% over two steps. | ||
In order to compare the two envisioned alternative reaction sequences, we initially synthesized the naturally occurring (2Z)-Lachnophyllum methyl ester and its (2E) isomer according to routes A and B (Scheme 3). Thus, 1-bromopent-1-yne was engaged in a Cadiot–Chodkiewicz coupling with methyl (Z)-pent-2-en-4-ynoate (Z-2) to deliver (2Z)-Lachnophyllum methyl ester (Z-1a) in 38% yield (Table 1, entry 1). The same reaction with methyl (E)-pent-2-en-4-ynoate (E-2) gave (2E)-Lachnophyllum methyl ester (E-1a) in 25% yield (Table 1, entry 2). According to route B, the (2Z)-Lachnophyllum methyl ester (Z-1a) was obtained from the Sonogashira coupling of hepta-1,3-diyne15 with methyl (Z)-3-iodoacrylate (Z-3) in 70% yield (Table 1, entry 5). Similarly, (2E)-Lachnophyllum methyl ester (E-1a) resulted from the same reaction with methyl (E)-3-iodoacrylate (E-3) in a 54% yield (Table 1, entry 6).
 | ||
| Scheme 3 Synthesis of Lachnophyllum methyl esters Z-1a and E-1a and their analogs Z-1b and E-1b. Reagents and conditions: (i) Cadiot–Chodkiewicz coupling: CuCl, NH2OH·HCl, nBuNH2, THF, 0 °C to rt; (ii) Sonogashira coupling: PdCl2(PPh3)2, CuI, Et3N, THF, 40 °C overnight. | ||
| Entry | Transformation (Route) | Substrates | Product | Yield (%) | |
|---|---|---|---|---|---|
| 1 | Cadiot–Chodkiewicz coupling (Route A) | C3H7CCBr |
Z-2 | Z-1a | 38 |
| 2 | E-2 | E-1a | 25 | ||
| 3 | C10H21CCBr |
Z-2 | Z-1b | 40 | |
| 4 | E-2 | E-1b | 55 | ||
| 5 | Sonogashira coupling (Route B) | C3H7CC–CCH |
Z-3 | Z-1a | 71 |
| 6 | E-3 | E-1a | 54 | ||
| 7 | C10H21CC–CCH |
Z-3 | Z-1b | 82 | |
| 8 | E-3 | E-1b | 81 | ||
In addition to being more convergent, route B, relying on the Sonogashira coupling, is thus more efficient than the Cadiot–Chodkiewicz coupling-based route A for the synthesis of both (Z) and (E) isomers of Lachnophyllum methyl esters. This trend was confirmed during the synthesis of a long chain analog of natural Lachnophyllum methyl esters. Compound Z-1b was thus obtained in 40% yield from 1-bromododec-1-yne16 according to route A (Table 1, entry 3) versus 82% yield for the reaction with tetradeca-1,3-diyne17 in route B (Table 1, entry 7). The same gap in efficiency was observed for the synthesis of E-1b (Table 1, entry 7 vs. entry 8).
In addition to being the most effective tested route for the synthesis of natural Lachnophyllum methyl esters, the Sonogashira coupling with building block 3 also provides efficient access to various analogs. Indeed, it can be used not only with diynes as depicted in Scheme 1, but also with other alkynes such as alkyl or aromatic ones. To exemplify this flexibility, the synthesis of saturated analog Z-1c (Scheme 4) and analogs Z-1d and E-1d, where one of the triple bonds is replaced by an aromatic ring, was considered (Scheme 5). This replacement of a triple bond by a bioisosteric aromatic ring proved to be advantageous in another polyacetylenic lipid family.18 Methyl (Z)-dec-2-en-4-ynoate (Z-1c) was obtained by the coupling of (Z)-3-iodoacrylate (Z-3) with hept-1-yne with 67% yield (Scheme 4).
 | ||
| Scheme 4 Synthesis of analog Z-1c. Reagents and conditions: (i) PdCl2(PPh3)2, CuI, Et3N, THF, 0 °C to rt, 67%. | ||
 | ||
| Scheme 5 Synthesis of analogs Z-1d and E-1d. Reagents and conditions: (i) PdCl2(PPh3)2, CuI, Et3N, THF, 40 °C overnight, Z-1d, 89%, E-1d, 89%. | ||
Coupling of (Z)-3-iodoacrylate (Z-3) (or (E)-3-iodoacrylate (E-3)) with 1-ethynyl-4-propylbenzene furnished compound Z-1d in 89% yield (respectively E-1d with the same yield) (Scheme 5).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 or silver carbonate at room temperature.22
20 or silver carbonate at room temperature.22
 | ||
| Scheme 6 Preparation and subsequent lactonization of (Z)-dec-2-en-4-ynoic acid. Reagents and conditions: (i) NaOH, MeOH/H2O (8:2), 80 °C, 88%; (ii) AgI (0.1 eq.), DMF, 100 °C, 4c:5 = 6:4, 62%; (iii) Ag2CO3 (0.1 eq.), DMF, rt, 4c:5 = 8:2, 55%. | ||
This route, lacking selectivity in our hands, was anticipated to reveal even more intricate with (Z)-alk-2-en-4,6-diynoic acid. We thus chose to explore a potentially more robust pathway based on a palladium-catalyzed tandem alkyne–alkene cross coupling/lactonization process (Scheme 7).23 This one-pot access to γ-ylidene butenolides was first described starting from (Z)-3-bromopropenoic acid24,25 to notably provide access to (Z)-5-benzylidenefuran-2(5H)-one24 (Scheme 7, R = Ph) or the naturally occurring (+)-goniobutenolide A.25 It was later developed with (Z)-3-iodopropenoic acid for the synthesis of natural products xerulin15,26 and freelingyne27 as well as related synthetic compounds.28,29
 | ||
| Scheme 7 Retrosynthetic route to Lachnophyllum lactone and its analogs. | ||
As a model reaction, (Z)-3-iodoacrylic acid30 was engaged in the Pd-catalyzed tandem coupling/cyclization reaction with 1-heptyne (Scheme 8 and Table 2, entry 3). (Z)-5-Hexylidenefuran-2(5H)-one (4c) was obtained as the sole product in a 66% yield upon smooth heating overnight in the presence of PdCl2(PPh3)2, CuI and Et3N in CH3CN.
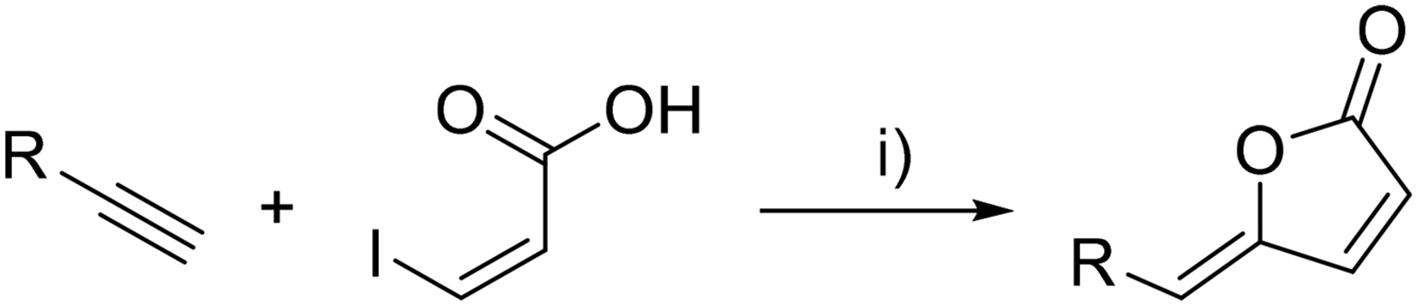 | ||
| Scheme 8 Synthesis of (4Z)-Lachnophyllum lactone and its analogs. Reagents and conditions: (i) PdCl2(PPh3)2, CuI, PPh3 (when R was an aromatic ring), Et3N, CH3CN, 40 °C overnight. | ||
| Entry | Alkyne | Lactone | Yield (%) | |
|---|---|---|---|---|
| 1 |
|
4a |

|
46 |
| 2 |

|
4b |

|
73 |
| 3 |

|
4c |

|
66 |
| 4 |

|
4d |

|
41 |
| 5 |

|
4e |

|
33 |
Encouraged by this result, we envisioned targeting the (4Z)-Lachnophyllum lactone using this palladium-catalyzed tandem process. Several precedents were described for the preparation of (poly)unsaturated γ-alkylidene butenolides from enyne precursors,15,26–29 but no example making use of a terminal diyne was known. Gratifyingly, the reaction of (Z)-3-iodopropenoic acid with hepta-1,3-diyne under the same conditions delivered the natural unsaturated lactone 4a (Table 2, entry 1) in 46% yield. Simultaneously to our work, this pathway was used for the gram scale synthesis of (4Z)-Lachnophyllum lactone.8
As for Lachnophyllum esters, we applied this reaction to the synthesis of analogs of natural lactone 4a. The longer alkyl chain analog 4b (Table 2, entry 2) was obtained from tetradeca-1,3-diyne in a 73% yield. Reaction between 1-ethynyl-4-propylbenzene and (Z)-3-iodopropenoic acid gave analog 4d (Table 2, entry 4) wherein the triple bond is replaced by an aromatic ring. In this case, the yield could be optimized by adding 0.1 equivalent of triphenylphosphine to the reaction mixture.19 This synthetic pathway also gave access to a functionalized aromatic analog 4e (Table 2, entry 5) with 5-ethynyl-1,3-difluoro-2-(heptyloxy)benzene31 in a 33% yield. Finally, we also varied the iodopropenoic acid using (Z)-3-iodobut-2-enoic acid,32 which upon reaction under the same coupling/cyclisation conditions with tetradeca-1,3-diyne gave compound 4f in a 35% yield (Scheme 9).
 | ||
| Scheme 9 Synthesis of (4Z)-Lachnophyllum lactone analog 4f. Reagents and conditions: (i) PdCl2(PPh3)2, CuI, Et3N, CH3CN, 40 °C overnight, 35%. | ||
Extraction optimization and HPLC analyses
The extraction methods used previously to isolate (2Z)-Lachnophyllum methyl ester and (4Z)-Lachnophyllum lactone from extracts of Conyza bonariensis7 are very solvent consuming, require the use of toxic solvents such as chloroform, hexane and methanol, and lead to very low extraction yields (0.34%). We thus tried to optimize the extraction method to improve the yield, the environmental impact and the safety of the process. Traditional Soxhlet, ultrasonic and supercritical carbon dioxide extraction techniques were carried out on the roots of Conyza bonariensis using different solvents or co-solvents such as heptane, methyl tert-butyl ether and ethanol. However, the masses extracted were low, with extraction yields remaining less than 1%. Moreover, these techniques require a long extraction time (between 10 and 12 h) and hundreds of milliliters of solvent. To overcome this problem, accelerated solvent extraction (ASE) was therefore preferred. Automating the extraction of 7 g of powdered plant virtually eliminates the variability of manual sample preparation, guaranteeing uniform and reproducible chromatographic analyses. The ASE system (Dionex™ ASE™ 350) automates extraction and filtration (10 μm) steps, requiring only 15 minutes (3 cycles of 5 minutes) and 100 mL of solvent. The yield of this ASE extraction was 0.55% for methyl tert-butyl ether (MTBE), 1.22% for heptane and 4.61% for ethanol.Analyses of the three fractions were carried out in UHPLC-PDA-MS on a reverse-phase Acquity Premier BEH C18 1.7 μm (2.1 × 100) mm column (see the ESI†). The heptanoic fraction appeared to be the richest in terms of compounds extracted and contained (4Z)-Lachnophyllum lactone and (2Z)-Lachnophyllum methyl ester (retention times of 7.048 and 8.160 min, respectively, Fig. 2B) as the hexanoic fraction previously obtained from the classical liquid extraction (Fig. 2A).7 Despite many efforts, attempts to fractionate this heptanoic extract on a semi-preparative scale were unsuccessful. This strengthens the relevance of our synthesis route to access these natural compounds.
 | ||
| Fig. 2 Analytical chromatograms obtained using an Acquity Premier BEH C18 1.7 μm (2.1 × 100) mm column, H2O/CH3CN, 0.1% HCOOH (95:5 to 0:100, 0.3 mL min−1, 40 °C, UV detection at 254 nm). (A) Hexanoic extract obtained according to Adande et al.;7 (B) heptanoic extract obtained by ASE; (C) compound 4a; (D) compound Z-1a; (E) compound E-1a. | ||
In addition to NMR analyses, UHPLC-PDA-MS was also used to confirm the purity of the synthesized compounds Z-1a and 4a and their identicalness with the natural compounds found in the plant extracts. The same analytical method was used for all samples and the chromatograms were compared (Fig. 2). Compounds 4a (Fig. 2C) and Z-1a (Fig. 2D) emerge at retention times of 7.014 min and 8.050 min, respectively, identical to those present in the extracts as confirmed by extracted MS. In addition, despite similar retention times, the major product at 8.894 min in the heptanoic fraction (Fig. 2B) or at 8.883 in the hexanoic fraction (Fig. 2A) is not (2E)-Lachnophyllum methyl ester E-1a (a retention time of 8.793 min, Fig. 2E) according to MS analysis (see the ESI†).
Biological evaluation
Many biological evaluations have been carried out on plant extracts or essential oils containing Lachnophyllum ester or lactone. These studies identified a wide range of biological activities including antitumor33,34 and antimicrobial35–37 for the (2Z)-methyl ester and allelopathic4,38–40 and antifungal8,41–44 for the lactone. We also recently evidenced insecticidal and nematicidal activities of different extracts of Conyza bonariensis, of which (2Z)-Lachnophyllum methyl ester and (4Z)-Lachnophyllum lactone were the major components.7The literature indicates that the two closely related natural products Matricaria esters and lactone (Δ8,9 unsaturated analogs, Fig. 3) possess weak antifungal42,43 and antimycobacterial activities45 and that (2E,8Z)-Matricaria methyl ester exhibits promising antileishmanial potency.46
 | ||
| Fig. 3 Structures of (4Z,8Z)-Matricaria lactone and (2E,8Z)-Matricaria methyl ester. | ||
Despite the numerous biological activities demonstrated for the Lachnophyllum ester or lactone, to the best of our knowledge, no studies on their antitubercular or antileishmanial activities have been reported so far. This prompted us to evaluate the in vitro potency of naturally occurring Lachnophyllum methyl esters and lactones against the associated pathogens Mycobacterium tuberculosis and Leishmania. The cytotoxicity towards African Green Monkey kidney mammalian cell lines (VERO) was also evaluated in order to determine their selectivity in favor of the mycobacterial or protozoan organisms. All the results of these different biological evaluations are brought together in Table 3.
| Compound | Nematicidal activity | Antileishmanial activity | Antimycobacterial activity | Cytotoxicity (VERO cells) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Structure | IC50 (mg L−1) | EC50 (μM) | EC50 (mg L−1) | MIC (μM) | IC50 (μM) | IC50 (mg L−1) | CC50 (μM) | CC50 (mg L−1) | |
| Z-1a |

|
75.3 ± 18.9 | 22.4 ± 2.7 | 4.0 ± 0.5 | >10 | ND | >250 | >44 | |
| E-1a |

|
56.9 ± 16.2 | 133 ± 6.8 | 23.4 ± 2.1 | ND | ND | ND | ||
| Z-1b |
|
>200 | 61.2 ± 2.1 | 16.8 ± 0.7 | 2.5 | 1.11 | 0.30 | >250 | >68 |
| E-1b |

|
>200 | ND | 5 | 1.48 | 0.41 | >250 | >68 | |
| Z-1c |

|
152 ± 47.3 | 77.8 ± 3.6 | 14.0 ± 0.9 | >10 | ND | >250 | >45 | |
| Z-1d |
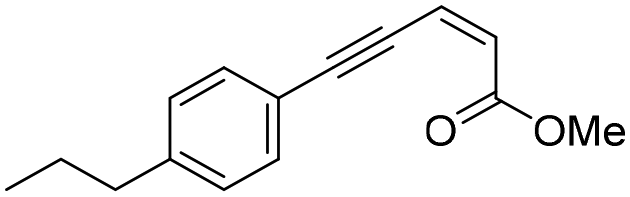
|
>200 | 56.1 ± 2.3 | 12.8 ± 0.7 | ND | ND | >250 | >57 | |
| 4a |

|
18.9 ± 12.3 | 2.8 ± 0.6 | 0.4 ± 0.1 | >10 | ND | 23.1 ± 0.9 | 3.7 ± 0.1 | |
| 4b |

|
>200 | 9.7 ± 1.4 | 2.5 ± 0.4 | 1.25 | 0.44 | 0.11 | 18.5 ± 1.2 | 4.8 ± 0.3 |
| 4c |

|
110 ± 42.2 | 9.6 ± 1.0 | 1.6 ± 0.2 | >10 | ND | 43.9 ± 2.5 | 7.3 ± 0.4 | |
| 4d |

|
ND | 36.2 ± 1.3 | 7.8 ± 0.3 | ND | ND | 85.6 ± 3.2 | 18.3 ± 0.7 | |
| 4e |

|
ND | ND | 5 | 1.6 | 0.52 | 18.4 ± 1.6 | 5.9 ± 0.5 | |
| 4f |
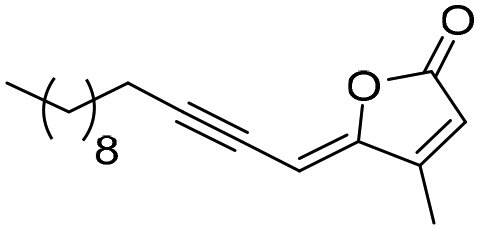
|
ND | ND | 5 | 2.3 | 0.63 | 124.4 ± 2.4 | 34.1 ± 0.7 | |
The nematicidal activity of natural compounds (Z-1a, E-1a, 4a) and their analogs (Z-1b, E-1b, Z-1c, Z-1d, 4b, 4c) was evaluated on second stage juveniles J2 of Meloidogyne incognita. Natural compounds, Lachnophyllum lactone (4a), (2E)-Lachnophyllum methyl ester (E-1a) and (2Z)-Lachnophyllum methyl ester (Z-1a), showed nematicidal activity with IC50 values under 100 mg mL−1 (Table 3), with lactone 4a being the most effective with an IC50 = 18.9 mg L−1. Lachnophyllum lactone (4a) has been widely identified in Asteraceae family plants, and several studies report its herbicidal activity on Lemna paucicostata,38 a bio-fungicide and bio-preservative on orange fruits.8,44 A diethyl ether extract from the roots of C. bonariensis containing a high level of Lachnophyllum lactone also demonstrated nematicidal activity7 on M. incognita. Somehow, the Michael acceptor behavior of the lactone, likely to be associated with its bioactivity, should be more pronounced than that of the esters. E-1a and Z-1a isomers were approximately 3 times less potent than the lactone, with the trans isomer being slightly more effective than the cis one. The same trend was reported when (4Z)- and (4E)-Matricaria lactones were tested against weed growth.40 Furthermore, the nematicidal activity of the synthetic sample of Z-1a (IC50 = 75.3 mg L−1) was similar to that of the sample of the natural product isolated from C. bonariensis (IC50 = 78.6 mg L−1),7 highlighting the relevance of the synthetic approach for confirming the nematicidal activity of these compounds.
The nematicidal activities of the natural products and their synthetic analogs were assessed to explore the structure–activity relationship in this series. Three groups of compounds were evaluated, namely long-chain, saturated and aromatic analogs. None of the long-chain analogs (Z-1b, E-1b and 4b) showed any biological activity on the phytoparasitic nematodes M. incognita with IC50 > 200 mg L−1. Short chain organic compounds may induce more damage to cell membranes that allow leakage of K+ ions causing osmotic stress to nematodes.47,48 In addition to the impact of chain length on nematicidal activity, the influence of compound unsaturation was also observed. Saturated analogs Z-1c and 4c were approximately 2 to 6 times less effective against nematodes than their unsaturated analogs, with IC50 values of 151.9 mg L−1 and 110.0 mg L−1, respectively. It can be suggested that conjugation of the Michael acceptor system of Lachnophyllum natural products could enhance its electrophilicity. The conjugation of acetylenic bonds with other functions and unsaturations could also increase the biological activity of compounds.49 The aromatic analog Z-1d of Lachnophyllum ester did not show significant nematicidal activity with an IC50 > 200 mg L−1. Overall, the most potent compounds tested in this study demonstrated nematicidal activities on phytoparasitic nematodes in the same range as that of commercial synthetic nematocides such as abamectin (IC50 = 2.1 mg L−1).50 Interestingly, some SAR trends observed in this nematicidal evaluation, highlighting the importance of bioactivity of the lactone moiety and the conjugated triple acetylenic units, were also shared with other biological studies of this work.
As mentioned earlier, the (4Z)-Lachnophyllum lactone (4a) was 8 times more potent against Leishmania infantum axenic amastigotes than the corresponding methyl ester Z-1a. This trend was also observed for the other tested lactones: lactone 4b (EC50 = 9.7 μM) was more than 6 times more potent than ester Z-1b while lactone 1c (EC50 = 9.6 μM) was almost 8 times more potent than ester Z-1c. The gap in potency between the lactone 4d (EC50 = 36.2 μM) and the corresponding ester Z-1d embedding a phenyl group was less prominent with only a 1.5-fold difference in favor of the lactone.
The naturally occurring (4Z)-Lachnophyllum lactone (4a) demonstrated the best antileishmanial activity among the synthetic samples of natural products and their analogs tested. Even if it remained lower than that of the reference molecule amphotericin B (EC50 = 0.07 μM), this activity was better than the one reported for (2E,8Z)-Matricaria methyl ester (EC50 = 61.2 μM).46 However, the selectivity index with respect to the cytotoxicity on mammalian cells was modest (SI = 8) as otherwise observed for (2E,8Z)-Matricaria methyl ester.46 From this point of view, the methyl esters Z-1a, Z-1b, Z-1c and Z-1d are more promising for developing new anti-leishmanial drugs as they do not present any detectable cytotoxicity against VERO cells at up to 250 μM concentration while their anti-leishmanial activity remains significant.
In the lactone series, the same trend was observed. The naturally occurring lactone 4a and its partially saturated analog 4c were inactive with MIC > 10 μM. Considering the favorable influence of the chain length on antimycobacterial activity, we also evaluated the lipophilic lactones 4b, 4e and 4f. Compound 4b proved to be the most active of all tested compounds with a MIC down to 1.25 μM. This potency compared favorably with that of ciprofloxacin, a drug used as a reference in the assay (MIC = 2.5 μM). Adding a methyl group to the lactone led to a 4-fold decrease in activity with compound 4f exhibiting a MIC of 5 μM. These data are in agreement with the putative Michael acceptor behavior of these compounds. Besides, the insertion in the alkyl chain of a fluorinated aromatic ring gives a similar loss of activity with compound 4e presenting a MIC = 5 μM.
Considering the cytotoxicity towards mammalian cells (vide infra), lactone 4b with a CC50 = 18.5 μM presented a moderate selectivity index of 15. Nevertheless, the methyl esters Z-1b and E-1b, devoid of cytotoxicity at a concentration up to 250 μM, were more promising compounds despite not having the highest activity against Mycobacterium tuberculosis.
Conclusion
In the present paper, we have developed a synthetic route allowing to access two naturally occurring compounds, (2Z)-Lachnophyllum methyl ester and (4Z)-Lachnophyllum lactone, that we recently identified as major components in essential oils and extracts of Conyza bonariensis. A nematicidal activity was evidenced for the plant extracts, suggesting that these two naturally occurring compounds could be responsible for this biological property but the quantities obtained from the plant were too low to validate this hypothesis.Lachnophyllum methyl esters (Z and E) were synthesized through a Sonogashira coupling between methyl 3-iodoacrylates and hepta-1,3-diyne whereas access to (4Z)-Lachnophyllum lactone resulted from a palladium-catalyzed tandem alkyne–alkene cross coupling/lactonization process with (Z)-3-iodoacrylic acid and hepta-1,3-diyne. Varying the alkyne partner in these two reactions yielded analogs of the naturally occurring compounds.
The synthesis allowed us to obtain sufficient quantities to evaluate not only nematicidal but also antileishmanial and antimycobacterial activities of naturally occurring compounds and their analogs. We could thus confirm the nematicidal activity of (4Z)-Lachnophyllum lactone, which remains the most active compound among all tested analogs. Compared to the synthetic analogs, the naturally occurring compounds (Z-1a, E-1a, and 4a) were more active against nematodes. Regarding the antileishmanial activity, naturally occurring compounds Z-1a and 4a proved to be the most active and, overall, lactone analogs showed lower EC50 values than the corresponding esters. As expected, longer alkyl chains (compared to the naturally occurring compounds) were necessary to observe significant antimycobacterial activity. The lactone analog 4b presenting a C10 lipophilic chain was the most antimycobacterial compound. The notable activities of lactones, naturally occurring or analogs, either nematicidal, antileishmanial or antimycobacterial should be compared to the relatively high cytotoxicity of these compounds for mammalian cells, conferring them a low selectivity index. From this point of view, (2Z)-Lachnophyllum methyl ester and its analogs are more promising since, despite more moderate biological activities, their innocuity toward mammalian cells suggests greater potential in this series. These results open up perspectives in the discovery of new bioactive agents to protect both agricultural crops and human health.
Experimental section
General synthetic methods
All reagents were obtained from commercial suppliers and used without any further purification. If not specified, reactions were run under a nitrogen atmosphere in oven-dried glassware. Standard inert atmosphere techniques were used in handling all air and moisture sensitive reagents. Toluene, dichloromethane (DCM), tetrahydrofuran (THF), dimethylformamide (DMF) and diethyl ether (Et2O) were obtained by filtration through a drying column on a filtration system. Thin-layer chromatography (TLC) analyses were performed on precoated, aluminum-backed silica gel (Merck 60 F254). Visualization of the developed chromatogram was performed under UV light (254 nm) and using 10% phosphomolybdic acid in EtOH or an aqueous potassium permanganate (KMnO4) stain. Flash chromatography was performed using flash silica gel (SDS 35–70 μm). Nuclear magnetic resonance spectra were recorded on a Bruker Avance 300 or 400 or 500 MHz spectrometer. Chemical shifts for 1H NMR spectra are given in parts per million (ppm) with the residual solvent resonance peak of CHCl3 as the reference (δ = 7.26 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet and br = broad), coupling constant in Hz and integration. Chemical shifts for 13C NMR spectra are given in ppm using the central peak of CDCl3 (δ = 77.16 ppm) as the reference. All 13C NMR spectra were obtained with complete proton decoupling. Infrared analyses were run on a Thermo-Nicolet Diamond ATR (4 cm−1 of resolution, 16 scans) equipped with a DTGS detector and are reported in reciprocal centimeters (cm−1). High-resolution mass spectrometry (HRMS) was performed on a Thermo-Finnigan MAT 95 XL instrument. Mass spectrometry m/z values are given in Dalton units.Synthesis of Lachnophyllum esters and their analogs (1)
(2Z)-Lachnophyllum methyl ester (Z-1a) (or methyl (Z)-deca-2-en-4,6-diynoate). General procedure A was followed using terminal alkyne Z-213 (0.25 g, 2.31 mmol), 1-bromopent-1-yne (0.4 g, 2.72 mmol), CuCl (6.9 mg, 0.069 mmol), hydroxylamine hydrochloride (3.4 mg, 0.048 mmol), and 30% of n-butylamine (5 mL). The resulting residue was purified by silica gel chromatography using 5% of diethyl ether in pentane to give Z-1a as a yellow solid (157.4 mg, 38% yield).
General procedure B was followed using iodoalkene Z-313 (50 mg, 0.24 mmol), hepta-1,3-diyne (32.5 mg, 0.35 mmol), PdCl2(PPh3)2 (8.5 mg, 0.012 mmol), and CuI (2.3 mg, 0.012 mmol). The crude product was purified using 5% diethyl ether in pentane to give Z-1a as a yellow solid (29.7 mg, 70% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 6.23 (d, J = 11.4 Hz, 1H); 6.20 (dt, J = 11.7, 1.0 Hz, 1H); 3.79 (s, 3H); 2.36 (td, J = 7.0, 0.9 Hz, 2H); 1.69–1.53 (m, 2H); 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 164.8, 130.7, 122.6, 90.1, 86.6, 70.9, 65.2, 51.6, 21.8, 21.6, 13.5. HRMS (DCI-CH4): calcd for C11H13O2 [M + H]+: 177.0916 m/z, found: 177.0910 m/z. FTIR: 3091.3, 3026.1, 2959.1, 2933.4, 2904.9, 2873.3, 2226.0, 1714.8, 1600.1.
(2E)-Lachnophyllum methyl ester (E-1a) (or methyl (E)-deca-2-en-4,6-diynoate). General procedure A was followed using terminal alkyne E-213 (100 mg, 0.91 mmol), 1-bromopent-1-yne (160 mg, 1.09 mmol), CuCl (2.67 mg, 0.03 mmol), hydroxylamine hydrochloride (1.32 mg, 0.019 mmol), and 30% of n-butylamine (1.5 mL). The resulting residue was purified by silica gel chromatography using 5% of diethyl ether in pentane to give E-1a as yellow solid (40.7 mg, 25% yield).
General procedure B was followed using iodoalkene E-314 (150 mg, 0.71 mmol), hepta-1,3-diyne (98 mg, 1.06 mmol), PdCl2(PPh3)2 (25 mg, 0.036 mmol), and CuI (6.8 mg, 0.036 mmol). The crude product was purified using 5% diethyl ether in pentane to give E-1a as a yellow solid (67 mg, 54% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 6.78 (dt, J = 15.8, 1.1 Hz, 1H); 6.32 (d, J = 15.9 Hz, 1H); 3.78 (s, 3H); 2.36 (td, J = 7.0, 1.1 Hz, 2H); 1.61 (h, J = 7.3 Hz, 2H); 1.03 (t, J = 7.4 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ (ppm) 166.0, 132.0, 124.6, 89.1, 83.3, 71.3, 64.9, 51.9, 21.62, 13.5. HRMS (DCI-CH4): calcd for C11H13O2 [M + H]+: 177.0916 m/z, found: 177.0913 m/z. FTIR: 3069.3, 2965.2, 2934.6, 2232.9, 2141.3, 1725.1, 1614.0.
Methyl (Z)-heptadeca-2-en-4,6-diynoate (Z-1b). General procedure A was followed using terminal alkyne Z-213 (125 mg, 1.13 mmol), 1-bromododec-1-yne (330 mg, 1.36 mmol), CuCl (3.4 mg, 0.034 mmol), hydroxylamine hydrochloride (1.58 mg, 0.023 mmol), and 30% of n-butylamine (2.5 mL). The resulting residue was purified by silica gel chromatography using 5% of diethyl ether in pentane to give Z-1b as a yellow solid (126 mg, 40% yield).
General procedure B was followed using iodoalkene Z-313 (50 mg, 0.24 mmol), tetradeca-1,3-diyne (68 mg, 0.36 mmol), PdCl2(PPh3)2 (8.4 mg, 0.012 mmol), and CuI (2.3 mg, 0.012 mmol). The crude product was purified using 5% diethyl ether in pentane to give Z-1b (54 mg, 82% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 6.24 (d, J = 11.4 Hz, 1H); 6.19 (dt, J = 11.3, 0.9 Hz, 1H); 3.80 (s, 1H); 2.38 (td, J = 7.0, 1.0 Hz, 2H); 1.63–1.51 (m, 2H); 1.48–1.19 (m, 14H); 0.96–0.85 (m, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 164.8, 130.7, 122.6, 90.4, 86.7, 70.8, 65.1, 51.6, 31.9, 29.6, 29.5, 29.3, 29.1, 28.9, 28.1, 22.7, 19.8, 14.1. HRMS (DCI-CH4): calcd for C18H27O2 [M + H]+: 275.2011 m/z, found: 275.2008 m/z. FTIR: 3096.0, 3037.5, 2953.8, 2922.6, 2852.4, 2223.3, 1716.8, 1600.4.
Methyl (E)-heptadeca-2-en-4,6-diynoate (E-1b). General procedure A was followed using terminal alkyne E-213 (73 mg, 0.64 mmol), 1-bromododec-1-yne (188 mg, 0.77 mmol), CuCl (1.9 mg, 0.02 mmol), hydroxylamine hydrochloride (0.9 mg, 0.013 mmol), and 30% of n-butylamine (1.1 mL). The resulting residue was purified by silica gel chromatography using 5% of diethyl ether in pentane to give E-1b as a yellow solid (99 mg, 55% yield).
General procedure B was followed using iodoalkene E-314 (50 mg, 0.24 mmol), tetradeca-1,3-diyne (68 mg, 0.36 mmol), PdCl2(PPh3)2 (8.3 mg, 0.012 mmol), and CuI (2.3 mg, 0.012 mmol). The crude product was purified using 5% diethyl ether in pentane to give E-1b (53 mg, 81% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 6.78 (dt, J = 15.8, 1.1 Hz, 1H); 6.32 (d, J = 15.9 Hz, 1H); 3.78 (s, 3H); 2.38 (td, J = 7.0, 1.1 Hz, 2H); 1.67–1.50 (m, 2H); 1.47–1.15 (m, 14H); 0.92–0.80 (m, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 166.0, 132.0, 124.5, 89.3, 83.4, 71.3, 64.8, 51.9, 31.9, 29.5, 29.4, 29.3, 29.1, 28.8, 28.0, 22.7, 19.7, 14.1. HRMS (DCI-CH4): calcd for C18H27O2 [M + H]+: 275.2011 m/z, found: 275.2014 m/z. FTIR: 2957.8, 2926.0, 2854.6, 2231.3, 2141.5, 1786.6, 1727.0, 1614.0.
Methyl (Z)-dec-2-en-4-ynoate (Z-1c). General procedure B was followed at 0 °C to rt using iodoalkene Z-313 (500 mg, 2.35 mmol), hept-1-yne (720 μL, 2.82 mmol), PdCl2(PPh3)2 (30 mg, 0.042 mmol, 0.017 eq.), and CuI (8 mg, 0.042 mmol, 0.017 eq.). The crude product was purified using 10% diethyl ether in pentane to give Z-1c (282 mg, 67% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 6.18 (dt, J = 11.4, 2.4 Hz, 1H); 6.05 (dt, J = 11.4, 0.7 Hz, 1H); 3.77 (s, 3H), 2.46 (tdd, J = 7.2, 2.4, 0.7 Hz, 2H); 1.70–1.52 (m, 2H); 1.62 (q, J = 7.6 Hz, 2H); 1.51–1.18 (m, 4H); 0.92 (t, J = 6.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 165.4, 126.9, 124.3, 104.5, 51.4, 31.1, 28.1, 22.2, 20.1, 13.9. HRMS (DCI-CH4): calcd for C11H17O2 [M + H]+: 181.1229 m/z, found: 181.1233 m/z. FTIR: 2954.7, 2932.3, 2860.1, 2207.6, 1731.6, 1716.9, 1610.7.
Methyl (Z)-5-(4-propylphenyl)pent-2-en-4-ynoate (Z-1d). General procedure B was followed using iodoalkene Z-313 (83 mg, 0.390 mmol,), 1-ethynyl-4-propylbenzene (84 mg, 0.581 mmol), PdCl2(PPh3)2 (13.6 mg, 0.019 mmol), and CuI (3.6 mg, 0.019 mmol). The crude product was purified using 5% diethyl ether in pentane to give ester Z-1d (79 mg, 89% yield) as a yellow oil.
1 H NMR (300 MHz, CDCl3) δ (ppm) 7.45 (d, J = 8.2 Hz, 2H); 7.15 (d, J = 8.2 Hz, 2H); 6.37 (d, J = 11.4 Hz, 1H); 6.12 (d, J = 11.4 Hz, 1H); 3.80 (s, 3H); 2.59 (d, J = 7.6 Hz, 2H); 1.71–1.55 (m, 2H); 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 165.3, 144.4, 132.1, 128.6, 127.2, 123.4, 119.8, 102.0, 86.0, 51.5, 38.0, 24.3, 13.7. HRMS (DCI-CH4): calcd for C15H17O2 [M + H]+: 229.1229 m/z, found: 229.1216 m/z. FTIR: 3028.0, 2958.5, 2931.0, 2871.5, 2199.8, 2180.2, 1727.3, 1713.7, 1613.1, 1599.3.
Methyl (E)-5-(4-propylphenyl)pent-2-en-4-ynoate (E-1d). General procedure B was followed using iodoalkene E-314 (49.0 mg, 0.231 mmol), 1-ethynyl-4-propylbenzene (50 mg, 0.347 mmol), PdCl2(PPh3)2 (8.1 mg, 0.011 mmol), and CuI (2.2 mg, 0.011 mmol). The crude product was purified using 5% diethyl ether in pentane to give ester E-1d (46.9 mg, 89% yield) as a yellow solid.
1 H NMR (300 MHz, CDCl3) δ (ppm) 7.39 (d, J = 8.2 Hz, 2H); 7.15 (d, J = 8.1 Hz, 2H); 6.99 (d, J = 15.8 Hz, 1H); 6.29 (d, J = 15.8 Hz, 1H); 3.78 (s, 3H); 2.59 (t, J = 7.6 Hz, 2H); 1.72–1.56 (m, 2H); 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 166.5, 144.5, 131.9, 129.1, 128.7, 125.6, 119.3, 99, 85.9, 51.8, 38, 24.3, 13.7. HRMS (DCI-CH4): calcd for C15H17O2 [M + H]+: 229.1229 m/z, found: 229.1226 m/z. FTIR: 3077.9, 3033.1, 2955.3, 2928.4, 2869.2, 2195.0, 1710.84, 1621.7, 1601.7.
Synthesis of (4Z)-Lachnophyllum lactone and its analogs (4)
(4Z)-Lachnophyllum lactone (4a) (or (Z)-5-(hex-2-yn-1-ylidene)furan-2(5H)-one). General procedure C was followed using hepta-1,3-diyne (75 mg, 0.76 mmol), (Z)-3-iodoacrylic acid (100 mg, 0.505 mmol), PdCl2P(Ph3)2 (8.9 mg, 0.013 mmol), CuI (4.8 mg, 0.025 mmol), and Et3N (2 mL, 13.6 mmol). Lactone 4a was obtained as a yellow oil (37.2 mg, 46% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 7.37 (d, J = 5.4 Hz, 1H); 6.20 (dd, J = 5.4, 0.9 Hz, 1H); 5.30 (td, J = 2.6, 0.9 Hz, 1H); 2.40 (td, J = 7.0, 2.5 Hz, 2H); 1.59 (h, J = 7.2 Hz, 2H); 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 168.9, 156.1, 142.7, 120.1, 104.6, 95.1, 74.8, 22.1, 21.8, 13.5. HRMS (DCI-CH4): calcd for C10H11O2 [M + H]+: 163.0759 m/z, found: 163.0752 m/z. FTIR: 3138.5, 3106.6, 3046.3, 2964.8, 2934.3, 2873.4, 2211.0, 1780.9, 1752.3.
(Z)-5-(Tridec-2-yn-1-ylidene)furan-2(5H)-one (4b). General procedure C was followed using tetradeca-1,3-diyne (115 mg, 0.616 mmol), (Z)-3-iodoacrylic acid (80 mg, 0.404 mmol), PdCl2P(Ph3)2 (7.1 mg, 0.01 mmol), CuI (3.8 mg, 0.02 mmol), and Et3N (1.5 mL, 10.9 mmol). Lactone 4b was obtained as a yellow oil (76.9 mg, 73% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 7.36 (d, J = 5.2 Hz, 1H); 6.21 (dd, J = 5.4, 0.8 Hz, 1H); 5.30 (t, J = 2.6 Hz, 1H); 2.44 (td, J = 7.1, 2.5 Hz, 2H); 1.57 (q, J = 7.1 Hz, 2H); 1.47–1.34 (m, 2H); 1.34–1.20 (m, 14H); 0.92–0.83 (m, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 169.0, 156.2, 142.8, 120.3, 105.0, 95.3, 74.8, 32.0, 29.7, 29.6, 29.4, 29.2, 29.1, 28.5, 22.8, 20.3, 14.2. HRMS (DCI-CH4): calcd for C17H25O2 [M + H]+: 261.1855 m/z, found: 261.1848 m/z. FTIR: 3066.3, 3049.1, 2953.6, 2922.9, 2849.9, 2868.9, 2209.2, 1787.3, 1759.1.
(Z)-5-Hexylidenefuran-2(5H)-one (4c). General procedure C was followed using hept-1-yne (170 mg, 1.33 mmol), (Z)-3-iodoacrylic acid (176 mg, 0.89 mmol), PdCl2P(Ph3)2 (15.6 mg, 0.022 mmol), CuI (8.5 mg, 0.045 mmol), and Et3N (3.4 mL, 24.03 mmol). Lactone 4c was obtained as a yellow oil (98 mg, 66% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 7.32 (d, J = 5.4 Hz, 1H); 6.12 (d, J = 5.4 Hz, 1H); 5.29 (t, J = 8.0 Hz, 1H); 2.38 (q, J = 7.6 Hz, 2H); 1.54–1.38 (m, 2H); 1.36–1.26 (m, 4H); 0.92–0.80 (m, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 170.2, 149.7, 143.7, 118.9, 117.9, 31.4, 28.6, 26.4, 22.4, 14.0. HRMS (DCI-CH4): calcd for C10H15O2 [M + H]+: 167.1072 m/z, found: 167.1070 m/z. FTIR: 3110.0, 2860.3, 2957.3, 2930.2, 2873.5, 1780.1, 1745.8.
(Z)-5-(4-Propylbenzylidene)furan-2(5H)-one (4d). General procedure C was followed using 1-ethynyl-4-propylbenzene (81.8 mg, 5.68 mmol), (Z)-3-iodoacrylic acid (75 mg, 0.38 mmol), PdCl2P(Ph3)2 (13.3 mg, 0.019 mmol), CuI (3.6 mg, 0.019 mmol), PPh3 (9.9 mg, 0.038 mmol), and Et3N (1.4 mL, 10.26 mmol). Lactone 4d was obtained as a yellow solid (76.9 mg, 41% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 7.71 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 5.4 Hz, 1H), 7.21 (d, J = 8.1 Hz, 2H), 6.19 (dd, J = 5.4, 0.8 Hz, 1H), 6.02 (s, 1H), 2.61 (t, J = 7.6 Hz, 2H), 1.65 (h, J = 7.4 Hz, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 170.5, 148.1, 145.4, 144.7, 130.9, 130.5, 129.2, 117.7, 114.6, 38.1, 24.4, 13.9. HRMS (DCI-CH4): calcd for C14H15O2 [M + H]+: 215.1072 m/z, found: 215.1064 m/z. FTIR: 3106.5, 3022.2, 2960.2, 2930.5, 1786.6, 1748.1, 1608.8, 1550.0.
(Z)-5-(3,5-Difluoro-4-(heptyloxy)benzylidene)furan-2(5H)-one (4e). General procedure C was followed using 5-ethynyl-1,3-difluoro-2-(heptyloxy)benzene (see the ESI†) (50 mg, 0.251 mmol), (Z)-3-iodoacrylic acid (95.6 mg, 0.38 mmol), PdCl2P(Ph3)2 (8.8 mg, 0.012 mmol), CuI (2.4 mg, 0.012 mmol), PPh3 (6.6 mg, 0.025 mmol), and Et3N (0.94 mL, 6.78 mmol). Lactone 4e was obtained as a yellow solid (27 mg, 33% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 7.47 (d, J = 5.4 Hz, 1H), 7.41–7.29 (m, 2H), 6.24 (dd, J = 5.4, 0.8 Hz, 1H), 5.87 (s, 1H), 4.19 (t, J = 6.7 Hz, 2H), 1.76 (p, J = 6.8 Hz, 2H), 1.53–1.40 (m, 2H), 1.40–1.19 (m, 6H), 0.92–0.85 (m, 2H). 13C NMR (75 MHz, CDCl3) δ (ppm) 169.7, 155.8 (dd, J = 247.9, 6.6 Hz), 148.9, 145.1, 136.7 (t, J = 14.1 Hz), 127.4 (t, J = 9.7 Hz), 118.8, 114.6–114.1 (m), 111.8 (t, J = 3.0 Hz), 74.9 (t, J = 3.3 Hz), 31.9, 30.1, 29.1, 25.7, 22.7, 14.2. 19F {1H} NMR (282 MHz, CDCl3) δ (ppm) −127.5. HRMS (DCI-CH4): calcd for C18H21O3F2 [M + H]+: 323.1459 m/z, found: 323.1451 m/z. FTIR: 3127.3, 3112.4, 2952.3, 2927.1, 2896.8, 1773.4, 1744.1, 1515.9, 1440.6.
(Z)-4-Methyl-5-(tridec-2-yn-1-ylidene)furan-2(5H)-one (4f). General procedure C was followed using tetradeca-1,3-diyne (134.4 mg, 0.707 mmol), (Z)-3-iodobut-2-enoic acid32 (100 mg, 0.471 mmol), PdCl2P(Ph3)2 (16.6 mg, 0.024 mmol), CuI (4.5 mg, 0.024 mmol), and Et3N (3 mL, 19.09 mmol). Lactone 4f was obtained as a yellow powder (58.6 mg, 35% yield).
1 H NMR (300 MHz, CDCl3) δ (ppm) 5.95 (s, 1H), 5.31 (dt, J = 2.6, 1.5 Hz, 1H), 2.43 (td, J = 7.1, 2.5 Hz, 2H), 2.14 (d, J = 1.4 Hz, 3H), 1.68–1.49 (m, 2H), 1.49–1.39 (m, 2H), 1.37–1.16 (m, 14H), 0.92–0.81 (m, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 168.4, 157.3, 153.9, 117.0, 104.0, 91.6, 74.4, 32.0, 29.7, 29.6, 29.4, 29.2, 29.0, 28.6, 22.8, 20.2, 14.2, 11.6. HRMS (DCI-CH4): calcd for C18H27O2 [M + H]+: 275.2011 m/z, found: 275.2011 m/z. FTIR: 3046.9, 2925.4, 2854.3, 2212.2, 1782.5, 1754.2, 1641.0, 1605.7.
Accelerated solvent extraction ASE conditions
Accelerated solvent extraction (ASE) has been done on 7 g of powdered roots of the plant. The accelerated solvent extraction system (Dionex™ ASE™ 350) automates extraction and filtration (10 μm). Each sample was extracted for 15 minutes (3 cycles of 5 minutes) with 3 × 34 mL of solvent at 100 °C and 117 bars. Samples were extracted with MTBE, heptane or ethanol. Each fraction was analyzed by UHPLC-PDA-MS.UHPLC-PDA-MS analysis conditions
All analyses of samples were carried out by reverse-phase UHPLC-PDA-MS on an Acquity Premier BEH C18 1.7 μm (2.1 × 100) mm column. Water with 0.1% HCOOH and acetonitrile with 0.1% HCOOH were used as solvents A and B respectively at a flow rate of 0.3 mL min−1 at 40 °C. The gradient was generic, starting at 5% B and remaining there for the first minute, gradually increasing to 100% B over 10 minutes, then remaining there for 2 minutes before returning to the initial conditions. Two detection modes were used: the UV detector at 254 nm and the single quadrupole positive and negative electrospray mass detector between 100 and 950 Daltons.Nematicidal assay
Nematicidal tests were conducted according to Adande, K. et al.7 and references therein. Meloidogyne incognita nematode species used for the test were obtained from tomato (Solanum lycopersicum L.) roots harvested in a greenhouse. The highly susceptible root-knot nematodes of cultivar cv Belladonna were infested by this nematode population. Experimental plants maintained in the greenhouse at a temperature of 25 to 28 °C, 60% humidity, and a 16-hour photoperiod were matured in plastic pots (18 cm diameter) filled with a 10:1 (v/v) mixture of peat and perlite. The 40 days plants were uprooted, while the free-soiled roots after the washing process were cut into 2 cm pieces. A procedure using sodium hypochlorite facilitated nematode egg extraction,51 and second-stage larvae (J2) were obtained using the modified Baermann method at 28 °C. All J2 that hatched within the first 3 days were discarded, and subsequent generations were collected and used for biological tests.52
The nematicidal activities of the synthesized natural compounds and their analogues were assessed by IC50 values, which indicated the loss of mobility in second-stage juveniles (J2). Stock solutions of the test samples were prepared in dimethyl sulfoxide (DMSO). The final test solutions were obtained by diluting the stock solutions with water containing the surfactant Polysorbate 20 (Tween-20). After preliminary tests, the DMSO and Tween-20 concentrations in each test well never exceeded 1.0% v/v and 0.3% v/v respectively. At these concentrations, the mobility of nematodes was preserved compared to nematodes maintained in pure water.53 Moreover, a 96-well microplate was used for the tests. Each test well contained 200 μL of solution with 25–30 J2s of the nematode. After 72 hours, the mortality of the nematodes was examined under a reversed microscope (Zeiss, Germany). Each test was repeated 4 times while the whole experiment was repeated at least twice at different time points. The naturally dead nematodes (less than 5% of the total number of J2s) observed in the control group consisting of water, Tween 20, and DMSO were discarded for dead J2 percentage calculation. The correction was made according to the Schneider–Orelli formula54 given below:

The corrected rates of mortality of J2 treated with the tested compounds were subjected to nonlinear analysis using the log-logistic equation proposed by Seefeldt et al.55

Antileishmanial activity on L. infantum axenic amastigotes
L. infantum promastigotes (MHOM/MA/67/ITMAP-263, CNR Leishmania, Montpellier, France, expressing luciferase activity) were cultivated in RPMI 1640 medium supplemented with 10% foetal calf serum (FCS), 2 mM L-glutamine and antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin) and harvested in the logarithmic phase of growth by centrifugation at 900g for 10 min. The supernatant was removed carefully and was replaced by the same volume of RPMI 1640 complete medium at pH 5.4 and incubated for 24 h at 24 °C. The acidified promastigotes were then incubated for 24 h at 37 °C in a ventilated flask to transform acidified promastigotes into axenic amastigotes. The effects of the tested compounds on the growth of L. infantum axenic amastigotes were assessed as follows: L. infantum amastigotes were incubated at a density of 2 × 106 parasites per mL in sterile 96-well plates with various concentrations of compounds dissolved in DMSO (final concentration 0.5% v/v), in duplicate. Appropriate controls, DMSO and amphotericin B, were added to each set of experiments. After a 48 h incubation period at 37 °C, each plate-well was then microscopically examined to detect any precipitate formation. To estimate the luciferase activity of axenic amastigotes, 80 μL of Steady Glow® reagent (Promega) was added to each white 96-well plates, according to the manufacturer's instructions, and the plates were incubated for 2 min. The luminescence was measured using a FLUOstar Omega microplate reader (BMG Labtech). Efficient concentration 50% (EC50) was defined as the concentration of drug required to inhibit the metabolic activity of L. infantum amastigotes by 50% compared to the control. EC50 values were calculated by non-linear regression analysis performed on dose–response curves using TableCurve 2D V5 software.Mycobacterial growth inhibition assays
The susceptibility of Mycobacterium tuberculosis strain H37Rv to all synthesized compounds was evaluated using a colorimetric microassay based on the reduction of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma) to formazan by metabolically active bacteria. Briefly, serial twofold dilutions of each compound solubilized in DMSO were prepared in 7H9 broth (Difco Middlebrook 7H9 broth, Becton Dickinson and Company, complemented with glycerol (4 g L−1)) using 96-well microtiter plates, and 100 μL of M. tuberculosis H37Rv suspension in 7H9 broth were added to each well (OD600 0.05). After 6 days of incubation, MTT was added (50 μL, 1 mg mL−1 in 7H9 broth). After 24 h incubation, 50 μL of SDS 20% were added to each well. The optical densities were measured at 570 nm with a microplate reader Expert Plus (ASYS HITECH, Austria). The absorbance value for untreated bacilli was set as 100% growth control. The MIC from at least three independent experiments was determined as the lowest concentration of the compound that inhibited bacterial growth; dose–response curves based on a non-linear regression were fitted using the GraphPad Prism software. When indicated, the selectivity index defined as the ratio between CC50 on VERO cells and MIC obtained on Mycobacterium tuberculosis was calculated.Cytotoxicity evaluation on the VERO cell line
The evaluation of the tested molecules’ cytotoxicity by the MTT assay was done on the VERO cell line. Briefly, cells (5 × 104 cells per mL) in 100 μL of complete medium [MEM supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine and antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin)] were seeded into each well of 96-well plates and incubated at 37 °C under a humidified 5% CO2 with 95% air atmosphere. After 24 h incubation, 100 μL of medium with various product concentrations and appropriate controls were added and the plates were incubated for 72 h at 37 °C. Each plate-well was then microscope examined for detecting possible precipitate formation before the medium was aspirated from the wells. 100 μL of MTT solution (0.5 mg mL−1 in complete MEM) were then added to each well. The cells were incubated for 1 h at 37 °C. After this time, the MTT solution was removed and DMSO (100 μL per well) was added to dissolve the resulting formazan crystals. The plates were shaken vigorously (300 rpm) for 5 min. The absorbance was measured at 570 nm with a microplate spectrophotometer (BIOTEK Eon). DMSO was used as the blank. CC50 values were calculated by non-linear regression analysis performed on dose–response curves using TableCurve 2D V5 software.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Erasmus+ program KA171 – Mobility of higher education students and staff for financing the mobility of Kodjo Adande. We thank Ms. Margaux Bossuat for the synthesis of 1,3-difluoro-2-(heptyloxy)-5-iodobenzene. We thank the Integrated Screening Platform of Toulouse (PICT, IBISA) for providing access to UPLC Acquity equipment from Waters and Institut de Chime de Toulouse ICT-UAR 2599 (Université de Toulouse, CNRS, Toulouse, France, https://ict.cnrs.fr) for providing access to Autopurification preparative HPLC equipment from Waters. We also thank Emmanuel Desmartin of Toulouse White Biotechnology (UMS INRAE/INSA/CNRS) for his valuable help with the accelerated solvent extractor and Audrey Brault from ThermoFisher Scientific. The antileishmanial activities and cytotoxicity on VERO cells assays were performed by Ms Sandra Bourgeade-Delmas, Manager of the “Plateau technique de Biologie Cellulaire” at Pharma-Dev, UMR152 IRD/UT3.References
- S. Opiyo, IOSR J. Appl. Chem., 2023, 16, 36–48 CAS.
- S. Opiyo, P. Njoroge and K. Muna, IOSR J. Appl. Chem., 2023, 16, 61–71 CAS.
- J. F. Sanz and J. A. Marco, Liebigs Ann. Chem., 1991, 1991, 399–400 CrossRef.
- O. Tzakou, A. Gani, G. Economou and A. Yannitsaros, J. Essent. Oil Res., 2004, 16, 425–428 CrossRef CAS.
- L. C. A. Barbosa, V. F. Paula, A. S. Azevedo, E. A. M. Silva and E. A. Nascimento, Flavour Fragrance J., 2005, 20, 39–41 CrossRef CAS.
- O. Tzakou, C. Vagias, A. Gani and A. Yannitsaros, Flavour Fragrance J., 2005, 20, 425–428 CrossRef CAS.
- K. Adande, K. Eloh, O. Simalou, M.-F. Bakaï and P. Caboni, Am. J. Anal. Chem., 2023, 14, 95–120 CrossRef CAS.
- G. Soriano, D. Arnodo, M. Masi, M. Fernández-Aparicio, B. B. Landa, C. Olivares-García, A. Cimmino and C. Prandi, J. Agric. Food Chem., 2024, 72, 4737–4746 CrossRef CAS PubMed.
- M. Asaoka, N. Yanagida, K. Ishibashi and H. Takei, Tetrahedron Lett., 1981, 22, 4269–4270 CrossRef CAS.
- A. Carpita, D. Neri and R. Rossi, Gazz. Chim. Ital., 1987, 117, 481–489 CAS.
- D. Grandjean, P. Pale and J. Chuche, Tetrahedron Lett., 1992, 33, 5355–5358 CrossRef CAS.
- D. Grandjean, P. Pale and J. Chuche, Tetrahedron, 1993, 49, 5225–5236 CrossRef CAS.
- S. Garrais, J. Turkington and W. P. D. Goldring, Tetrahedron, 2009, 65, 8418–8427 CrossRef CAS.
- K. S. Madden, H. R. E. Jokhoo, F. D. Conradi, J. P. Knowles, C. W. Mullineaux and A. Whiting, Org. Biomol. Chem., 2019, 17, 3752–3759 RSC.
- V. Fiandanese, D. Bottalico, G. Marchese and A. Punzi, Tetrahedron, 2004, 60, 11421–11425 CrossRef CAS.
- V. Le Fouler, G. Duret, P. Bisseret and N. Blanchard, Tetrahedron Lett., 2018, 59, 3349–3352 CrossRef CAS.
- M. Bourkhis, H. Gaspard, P. Rullière, D. K. C. de Almeida, D. Listunov, E. Joly, R. Abderrahim, M. C. de Mattos, M. C. F. de Oliveira, V. Maraval, R. Chauvin and Y. Génisson, ChemMedChem, 2018, 13, 1124–1130 CrossRef CAS PubMed.
- M. Bossuat, P. Rullière, N. Preuilh, A. Peixoto, E. Joly, J.-G. Gomez, M. Bourkhis, F. Rodriguez, F. Gonçalves, I. Fabing, H. Gaspard, V. Bernardes-Génisson, V. Maraval, S. Ballereau, R. Chauvin, S. Britton and Y. Génisson, J. Med. Chem., 2023, 66, 13918–13945 CrossRef CAS PubMed.
- M. Kotora and E.-i. Negishi, Synthesis, 1997, 121–128 CrossRef CAS.
- Y. Ogawa, M. Maruno and T. Wakamatsu, Heterocycles, 1995, 41, 2587–2599 CrossRef CAS.
- R. Rossi, F. Bellina, A. Catanese, L. Mannina and D. Valensin, Tetrahedron, 2000, 56, 479–487 CrossRef CAS.
- L. Anastasia, C. Xu and E.-i. Negishi, Tetrahedron Lett., 2002, 43, 5673–5676 CrossRef CAS.
- M. V. N. De Souza, Mini-Rev. Org. Chem., 2005, 2, 139–145 CrossRef CAS.
- X. Lu, X. Huang and S. Ma, Tetrahedron Lett., 1993, 34, 5963–5966 CrossRef CAS.
- M. Kotora and E.-i. Negishi, Tetrahedron Lett., 1996, 37, 9041–9042 CrossRef CAS.
- E.-i. Negishi, A. Alimardanov and C. Xu, Org. Lett., 2000, 2, 65–67 CrossRef CAS PubMed.
- F. Liu and E.-i. Negishi, J. Org. Chem., 1997, 62, 8591–8594 CrossRef CAS PubMed.
- V. Fiandanese, D. Bottalico and G. Marchese, Tetrahedron, 2001, 57, 10213–10218 CrossRef CAS.
- D. Rambabu, S. Bhavani, K. S. Nalivela, S. Mukherjee, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2013, 54, 2151–2155 CrossRef CAS.
- T. Zoller and D. Uguen, Tetrahedron Lett., 1998, 39, 6719–6720 CrossRef CAS.
- Q. Zhang, P. Prins, S. C. Jones, S. Barlow, T. Kondo, Z. An, L. D. A. Siebbeles and S. R. Marder, Org. Lett., 2005, 7, 5019–5022 CrossRef CAS PubMed.
- S. Inack-Ngi, R. Rahmani, L. Commeiras, G. Chouraqui, J. Thibonnet, A. Duchêne, M. Abarbri and J.-L. Parrain, Adv. Synth. Catal., 2009, 351, 779–788 CrossRef CAS.
- R. C. Ferreira, S. S. Duarte, V. M. de Sousa, R. R. M. de Souza, K. K. G. Marques, R. A. de Abrantes, Y. M. do Nascimento, N. F. de Sousa, M. T. Scotti, L. Scotti, J. F. Tavares, J. C. R. Gonçalves, M. S. da Silva and M. V. Sobral, Pharmaceuticals, 2023, 16, 1553 CrossRef CAS PubMed.
- R. C. Ferreira, Y. M. do Nascimento, P. B. de Araújo Loureiro, R. X. Martins, M. E. de Souza Maia, D. F. Farias, J. F. Tavares, J. C. R. Gonçalves, M. S. da Silva and M. V. Sobral, Biomolecules, 2023, 13, 1439 CrossRef CAS PubMed.
- P. Satyal, B. K. Chhetri, N. S. Dosoky, S. Shrestha, A. Poudel and W. N. Setzer, Nat. Prod. Commun., 2015, 10, 1934578X1501001028 CAS.
- F. Ayaz, N. Küçükboyacı and B. Demirci, J. Essent. Oil Res., 2017, 29, 336–343 CrossRef CAS.
- V. Kumar, C. S. Mathela, G. Tewari, A. Panwar and V. Pandey, Indian J. Nat. Prod. Resour., 2017, 8, 63–68 CAS.
- S. C. N. Queiroz, C. L. Cantrell, S. O. Duke, D. E. Wedge, V. K. Nandula, R. M. Moraes and A. L. Cerdeira, J. Agric. Food Chem., 2012, 60, 5893–5898 CrossRef CAS PubMed.
- M. Fernández-Aparicio, G. Soriano, M. Masi, P. Carretero, S. Vilariño-Rodríguez and A. Cimmino, Agriculture, 2022, 12, 790 CrossRef.
- A. C. Peralta, G. Soriano, J. G. Zorrilla, M. Masi, A. Cimmino and M. Fernández-Aparicio, Molecules, 2022, 27, 7421 CrossRef CAS PubMed.
- L. Rahalison, M. Benathan, M. Monod, E. Frenk, M. P. Gupta, P. N. Solis, N. Fuzzati and K. Hostettmann, Planta Med., 1995, 61, 360–362 CrossRef CAS PubMed.
- G. Vidari, S. Abdo, G. Gilardoni, A. Ciapessoni, M. Gusmeroli and G. Zanoni, Fitoterapia, 2006, 77, 318–320 CrossRef CAS PubMed.
- R. S. Porto, S. Rath and S. C. N. Queiroz, J. Braz. Chem. Soc., 2017, 28, 913–919 CAS.
- D. Terao, S. C. N. Queiroz and A. d. H. N. Maia, J. Phytopathol., 2022, 170, 158–166 CrossRef CAS.
- T. S. Lu, C. L. Cantrell, S. L. Robbs, S. G. Franzblau and N. H. Fischer, Planta Med., 1998, 64, 665–667 CrossRef CAS PubMed.
- S. C. Pandey, D. S. Dhami, A. Jha, G. C. Shah, A. Kumar and M. Samant, ACS Omega, 2019, 4, 14640–14649 CrossRef PubMed.
- N. Togashi, A. Shiraishi, M. Nishizaka, K. Matsuoka, K. Endo, H. Hamashima and Y. Inoue, Molecules, 2007, 12, 139–148 CrossRef CAS PubMed.
- K. Eloh, M. Demurtas, A. Deplano, A. Ngoutane Mfopa, A. Murgia, A. Maxia, V. Onnis and P. Caboni, J. Agric. Food Chem., 2015, 63, 9970–9976 CrossRef CAS PubMed.
- D. A. Konovalov, Pharm. Chem. J., 2014, 48, 613–631 CrossRef CAS.
- K. Eloh, M. Demurtas, M. G. Mura, A. Deplano, V. Onnis, N. Sasanelli, A. Maxia and P. Caboni, J. Agric. Food Chem., 2016, 64, 4876–4881 CrossRef CAS PubMed.
- R. S. Hussey and K. R. Barker, Plant Dis. Rep., 1973, 57, 1025–1028 Search PubMed.
- D. L. Coyne, J. M. Nicol and B. Claudius-Cole, Les nématodes des plantes: Un guide pratique des techniques de terrain et de laboratoire, Secrétariat SP-IPM, Institut International d'Agriculture Tropicale (IITA), Cotonou, Benin, 2010 Search PubMed.
- P. Caboni, N. G. Ntalli, N. Aissani, I. Cavoski and A. Angioni, J. Agric. Food Chem., 2012, 60, 1146–1151 CrossRef CAS PubMed.
- W. Püntener and O. Zahner, Manual for Field Trials in Plant Protection, rev.enl., in CIBA-GEIGY documenta, Ciba-Geigy, Basle, Switzerland, 2nd edn, 1981 Search PubMed.
- S. S. Seefeldt, J. E. Jensen and E. P. Fuerst, Weed Technol., 1995, 9, 218–227 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01224k |
| This journal is © The Royal Society of Chemistry 2024 |