
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Propargyl alcohol as an acrolein equivalent: synthesis of β-(3-indolyl)acroleins and β-(imidazo[1,2-a]pyridin-3-yl)acroleins†
Bhawani
a,
Sonam
a,
Vikki N.
Shinde
a,
Prakash N.
Swami
a,
Krishnan
Rangan
 b and
Anil
Kumar
*a
b and
Anil
Kumar
*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Pilani Campus, Rajasthan 333031, India. E-mail: anilkumar@pilani.bits-pilani.ac.in
bDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Hyderabad Campus, Telangana 500078, India
First published on 24th September 2024
Abstract
A simple and straightforward method has been developed to access distinctly substituted β-(3-indolyl)acroleins and β-(imidazo[1,2-a]pyridin-3-yl)acroleins using propargyl alcohol as an acrolein equivalent. A broad substrate scope, good yields, easily accessible substrates, and metal-free conditions are the salient features of the developed methodology. This work contributes to a significant advancement in the sustainable synthesis of functionalized acroleins.
Among the nitrogen-containing heterocycles, indoles and imidazo[1,2-a]pyridines are well-known scaffolds widely found in many naturally occurring and synthetic compounds.1 A myriad of indole and imidazopyridine-based compounds have been studied for a broad range of biological and pharmacological activities such as antimicrobial, antiviral, antiprotozoal, anti-inflammatory, anti-ulcer, and anticancer activities.2 These heterocycles are essential pharmacophores of several marketed drugs such as indomethacin, atevirdine, delavirdine, arbidol, panobinostat, alpidem, zolpidem, olprinone, and minodronic acids.1c,3 Owing to the therapeutic applications of these heterocyclic motifs, the synthesis and functionalization of indoles4 and imidazo[1,2-a]pyridines5 has been a major area of focus and continues to be an important research area for chemists.
On the other hand, the acrolein moiety has been used as a highly active linker to design cytotoxic compounds.6 3-(Coumarin-3-yl)acrolein derivatives have been evaluated as novel anticancer chemotherapeutic candidates with remarkable antiproliferative activity against various cancer cell lines.7 3-(3-(4-Fluorophenyl)-1-isopropyl-1H-indol-2-yl)acrolein is a key intermediate of the anti-cholesterol fluvastatin drug.8 β-Substituted acroleins are also important intermediates for the construction of complex molecular architectures.9 The presence of acrolein functionality on indoles and imidazo[1,2-a]pyridines would allow their easy transformations. For example, they could be key building blocks for the synthesis of various fused nitrogen heterocycles employing C–H functionalization10 and [4 + 2]-cycloaddition reactions.11 The synthesis of 3-(indol-3-yl)acroleins is generally achieved by the Wittig reaction of indole-3-carbaldehydes and Wittig ylides.12 It is notable that the synthesis of 3-(indol-3-yl)acroleins is quite challenging and there are only a few methods starting from indole (Scheme 1). In early days, 3-(indol-3-yl)acrolein was obtained employing an extended Vilsmeier reaction of indole with 3-methyl-3-phenylaminopropenal (Scheme 1a). Vanderwal and co-workers reported an interesting method for the preparation of 3-(indol-3-yl)acroleins by a ring-opening reaction of pyridinium salts, but the use of toxic BrCN and a narrow substrate scope limit its broad application.13 Jiao's group synthesized 3-(indol-3-yl)acroleins by morpholine-catalysed addition of indoles with α,β-unsaturated aldehydes followed by oxidation with DDQ (Scheme 1b).14 To the best of our knowledge, there is no report on the synthesis of β-(imidazo[1,2-a]pyridin-3-yl)acroleins.
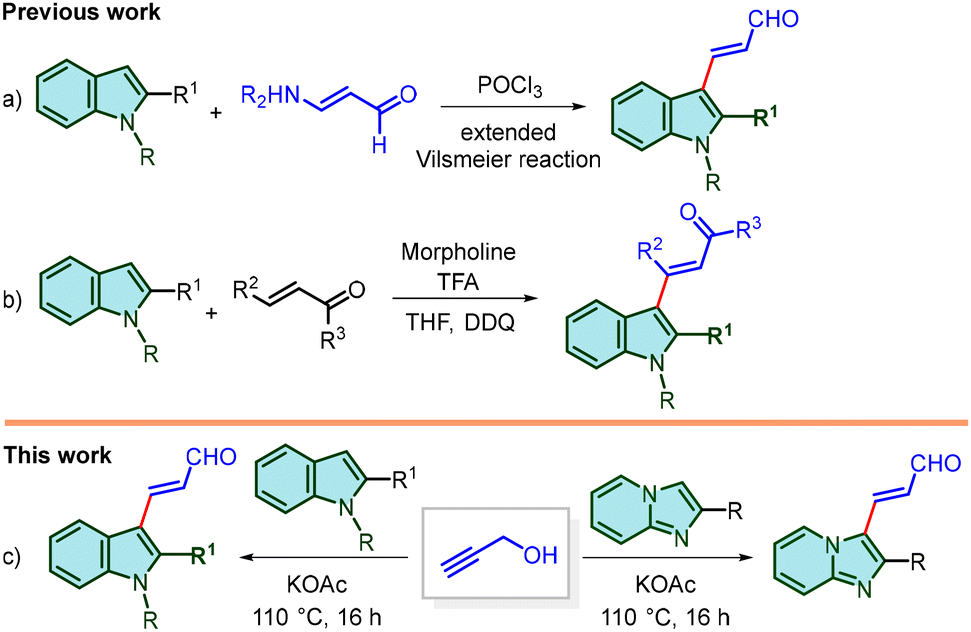 | ||
| Scheme 1 Synthesis of β-(3-indolyl)acroleins and β-(imidazo[1,2-a]pyridin-3-yl)acroleins. | ||
During our investigation on manganese-mediated ortho-alkenylation of 2-phenylimidazo[1,2-a]pyridine (1a) with propargyl alcohol (2), β-(2-phenylimidazo[1,2-a]pyridin-3-yl)acrolein (3a) was obtained in 15% yield as a side product (Table 1, entry 1). With this interesting result, we decided to optimize the reaction conditions to improve the yield of this acrolein derivative 3a (Table 1). Initially, we varied the catalyst and 3a was obtained in 18% yield along with 3a′ (33%) on using Mn(OAc)2, while the reaction failed with Mn2(CO)10 (Table 1, entries 2 and 3). Interestingly, when the reaction was performed in the absence of THF using propargyl alcohol (2) as a solvent, 3a (60%) was obtained as the major product (Table 1, entry 4). Moreover, the yield of 3a increased slightly to 63% in the absence of the catalyst, indicating that the manganese catalyst has no role in the formation of 3a (Table 1, entry 5). Among different acetates, the use of KOAc produced 3a in 73% yield which further increased to 77% on increasing the amount of KOAc to two equivalents (Table 1, entries 5–8). No significant change in yield was noticed upon increasing the reaction temperature up to 150 °C; however, lowering the reaction temperature to 80 °C resulted in a decreased yield (59%) of 3a (Table 1, entries 9 and 10). Finally, the reaction carried out in the absence of KOAc did not produce any product (Table 1, entry 11). Based on the above experimental results, KOAc (2 equiv.) in propargyl alcohol for 16 h at 120 °C were chosen as the optimum reaction conditions (Table 1, entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | t (°C) | % Yieldb | |
| 3a′ | 3a | |||||
| a Reaction conditions: 1a (0.52 mmol), 2 (2 mL), catalyst (10 mol%), additive (1.03 mmol, 2 equiv.), 16 h. b Isolated yield. | ||||||
| 1 | MnBr(CO)5 | NaOAc | THF | 120 | 48 | 15 |
| 2 | Mn(OAc)2 | NaOAc | THF | 120 | 33 | 18 |
| 3 | Mn2(CO)10 | NaOAc | THF | 120 | Trace | — |
| 4 | MnBr(CO)5 | NaOAc | 2 | 120 | Trace | 60 |
| 5 | — | NaOAc | 2 | 120 | — | 63 |
| 6 | — | KOAc | 2 | 120 | 73 | |
| 7 | — | CsOAc | 2 | 120 | 58 | |
| 8 | — | KOAc (2 equiv.) | 2 | 120 | — | 77 |
| 9 | — | KOAc (2 equiv.) | 2 | 0 | — | 59 |
| 10 | — | KOAc (2 equiv.) | 2 | 150 | — | 74 |
| 11 | — | — | 2 | 120 | — | — |

Having the optimal reaction conditions in hand, we shifted our focus to evaluate the scope and limitations of this method (Table 2). Imidazo[1,2-a]pyridines substituted with electron-donating groups (Me and OMe), halogens (F, Cl, and Br) and an electron-withdrawing group (CN) at the para-position of the C2-phenyl ring reacted smoothly to produce the corresponding substituted acrolein derivatives (3a–3g) in moderate to very good yields (41–83%). Higher yields of products were obtained from imidazopyridines with electron-donating groups on the C2-phenyl ring as compared with those having electron-withdrawing groups. This was further established by a one-pot competitive reaction of 1b and 1e with 2 which produced the corresponding products 3b and 3e in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratios. Subsequently, reactions of 2 with ortho-substituted 2-arylimidazo[1,2-a]pyridines (1h and 1i), meta-substituted 2-arylimidazo[1,2-a]pyridines (1j and 1k), 2-(naphthalen-2-yl)imidazo[1,2-a]pyridine (1l) and 2-(thiophen-3-yl)imidazo[1,2-a]pyridine (1m) produced the desired acrolein derivatives (3h–3m) in good yields (60–75%). Notably, imidazo[1,2-a]pyridines (1n–1v) with substituents (Me and Br) on various positions of the imidazopyridine nucleus also reacted smoothly furnishing the expected acrolein derivatives (3n–3v) in moderate to very good yields (56–85%). The reaction of but-3-yn-2-ol (2b) with 1r under standard conditions was sluggish and produced the corresponding alkenylated derivative 3w only in 21% yield. Unfortunately, the reaction of 3-phenylprop-2-yn-1-ol (2c) with 1a failed to produce the corresponding product under standard conditions. The structures of all the β-(imidazo[1,2-a]pyridin-3-yl)acrolein derivatives (3) were ascertained by IR, 1H NMR, 13C NMR and HRMS analyses. Furthermore, the structures of 3a (CCDC no. 2266949†) and 3m (CCDC no. 2266961†) were unambiguously confirmed by single crystal X-ray diffractometer analysis.
:3 ratios. Subsequently, reactions of 2 with ortho-substituted 2-arylimidazo[1,2-a]pyridines (1h and 1i), meta-substituted 2-arylimidazo[1,2-a]pyridines (1j and 1k), 2-(naphthalen-2-yl)imidazo[1,2-a]pyridine (1l) and 2-(thiophen-3-yl)imidazo[1,2-a]pyridine (1m) produced the desired acrolein derivatives (3h–3m) in good yields (60–75%). Notably, imidazo[1,2-a]pyridines (1n–1v) with substituents (Me and Br) on various positions of the imidazopyridine nucleus also reacted smoothly furnishing the expected acrolein derivatives (3n–3v) in moderate to very good yields (56–85%). The reaction of but-3-yn-2-ol (2b) with 1r under standard conditions was sluggish and produced the corresponding alkenylated derivative 3w only in 21% yield. Unfortunately, the reaction of 3-phenylprop-2-yn-1-ol (2c) with 1a failed to produce the corresponding product under standard conditions. The structures of all the β-(imidazo[1,2-a]pyridin-3-yl)acrolein derivatives (3) were ascertained by IR, 1H NMR, 13C NMR and HRMS analyses. Furthermore, the structures of 3a (CCDC no. 2266949†) and 3m (CCDC no. 2266961†) were unambiguously confirmed by single crystal X-ray diffractometer analysis.

Next, we extended the scope of the method to indoles. As can be noted from Table 3, the reaction of 2 with 2-arylindoles (4a–i) substituted with electron-donating groups (Me, OMe, and OH) and halogens (F, Cl and Br) at the para-position, a methyl group at the ortho-position and a CF3 group at the meta-position of the C2–phenyl ring produced the corresponding β-(indol-3-yl)acroleins (5a–i) in moderate to good yields (52–77%). 2-(Thiophen-3-yl)indole (4j) on reaction with 2 produced the corresponding acrolein derivative 5j with 69% yield. Furthermore, indoles with substitutions at the C5-position (4k) and the C5, C6-position (4l) were found to be suitable substrates, leading to the formation of acrolein derivatives 5k and 5l in 61% and 63% yields, respectively. Additionally, N-methylindoles (4m–o) also reacted smoothly with 2 to produce the corresponding acrolein derivatives (5m–o) in moderate to good yields (44–72%). The structures of all the β-(indol-3-yl)acrolein derivatives (5) were ascertained by IR, 1H NMR, 13C NMR and HRMS analyses and the structure of 5c (CCDC no. 2266943†) was unambiguously confirmed by single crystal X-ray diffractometer analysis.

The synthetic utility of the developed protocol is demonstrated by gram-scale synthesis and synthetic transformation of the product (Scheme 2). Keeping the ratios of the reagents consistent with the standard reaction, the reaction of 2 with 1a (1.0 g) and 4a (1.0 g) afforded 0.96 g (76%) of 3a and 0.92 g (72%) of 5a, respectively (Scheme 2a). Furthermore, reduction of 3b with NaBH4 produced γ-(2-(p-tolyl)imidazo[1,2-a]pyridin-3-yl)allyl alcohol (6) in 92% yield (Scheme 2b). Similarly, the reaction of 3a with phosphorus ylide (7) in the presence of NaH in dry THF for 5 h at 0 °C–RT afforded the corresponding conjugated alkene product 8 in 68% yield (Scheme 2b).
 | ||
| Scheme 2 Gram-scale experiment and synthetic transformation of products. | ||
The presence of radical scavengers TEMPO and BHT (3 equivalents each) in the reaction of 1a and 2 did not suppress the yield of 3a (Scheme 3). The result obtained from the above experiment suggests that the reaction does not follow a radical pathway. It is believed that the reaction proceeds via hydroarylation of propargyl alcohol followed by oxidation. More work on understanding the mechanism of this reaction is under progress in our lab.
 | ||
| Scheme 3 Radical trapping experiment. | ||
Conclusions
In conclusion, we have developed a simple and straightforward method to access distinctly substituted β-(3-indolyl)acrolein and β-(imidazo[1,2-a]pyridin-3-yl)acrolein derivatives using propargyl alcohol as an acrolein equivalent. The salient features of this method are the use of readily available and easy-to-handle propargyl alcohol as an acrolein equivalent, good to high yields, a wide substrate scope, metal-free reaction conditions, and simple workup procedures. A wide range of synthetically useful functional groups such as Cl, Br, F, OMe, OH, CF3 NO2, and CN were tolerated well under these conditions. Furthermore, gram-scale synthesis also demonstrates that the method can be applied to bulk processes.Author contributions
B. optimized the reaction conditions and prepared β-(imidazo[1,2-a]pyridin-3-yl)acroleins. S. prepared β-(indol-3-yl)acroleins. B., S., P. N. S. and V. N. S. recorded data, analysed and discussed the results with A. K. K. R. recorded and analysed single crystal data. A. K. supervised the project and wrote the manuscript. All authors contributed to the manuscript and have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank BITS Pilani and DST New Delhi for the NMR and HRMS facilities, respectively. B. thanks BITS Pilani for the institute fellowship.References
- (a) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed; (b) A. Garrido, G. Vera, P.-O. Delaye and C. Enguehard-Gueiffier, Eur. J. Med. Chem., 2021, 226, 113867 CrossRef CAS PubMed; (c) T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed.
- N. Devi, D. Singh, K. R. Rawal, J. Bariwal and V. Singh, Curr. Top. Med. Chem., 2016, 16, 2963–2994 CrossRef CAS PubMed.
- S. Samanta, S. Kumar, E. K. Aratikatla, S. R. Ghorpade and V. Singh, RSC Med. Chem., 2023, 14, 644–657 RSC.
- (a) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed; (b) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed; (c) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Adv. Synth. Catal., 2021, 363, 2723–2739 CrossRef CAS; (d) P. Kumar, P. J. Nagtilak and M. Kapur, New J. Chem., 2021, 45, 13692–13746 RSC.
- (a) C.-H. Ma, M. Chen, Z.-W. Feng, Y. Zhang, J. Wang, Y.-Q. Jiang and B. Yu, New J. Chem., 2021, 45, 9302–9314 RSC; (b) J. A. Tali, G. Kumar, B. K. Sharma, Y. Rasool, Y. Sharma and R. Shankar, Org. Biomol. Chem., 2023, 21, 7267–7289 RSC; (c) A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555–1575 RSC.
- M.-S. Tang, H.-T. Wang, Y. Hu, W.-S. Chen, M. Akao, Z. Feng and W. Hu, Mol. Nutr. Food Res., 2011, 55, 1291–1300 CrossRef CAS PubMed.
- L. Chen, Q. Lv, J. Cai, J. Liang, Z. Liang, J. Lin, Y. Xiao, R. Chen, Z. Zhang, Y. Hong and H. Ji, Front. Pharmacol., 2023, 14, 1141121 CrossRef CAS PubMed.
- (a) J. T. Zacharia, T. Tanaka and M. Hayashi, J. Org. Chem., 2010, 75, 7514–7518 CrossRef CAS PubMed; (b) V. K. A. Kalalbandi, J. Seetharamappa and U. Katrahalli, RSC Adv., 2015, 5, 38748–38759 RSC.
- A. R. Pradipta and K. Tanaka, Chem. Rec., 2021, 21, 646–662 CrossRef CAS PubMed.
- R. K. Saunthwal, M. Patel, S. Kumar, A. K. Danodia and A. K. Verma, Chem. – Eur. J., 2015, 21, 18601–18605 CrossRef CAS PubMed.
- H. Zheng, P. He, Y. Liu, Y. Zhang, X. Liu, L. Lin and X. Feng, Chem. Commun., 2014, 50, 8794–8796 RSC.
- M. Šíša, D. Pla, M. Altuna, A. Francesch, C. Cuevas, F. Albericio and M. Álvarez, J. Med. Chem., 2009, 52, 6217–6223 CrossRef PubMed.
- A. M. Kearney and C. D. Vanderwal, Angew. Chem., Int. Ed., 2006, 45, 7803–7806 CrossRef CAS PubMed.
- S.-K. Xiang, B. Zhang, L.-H. Zhang, Y. Cui and N. Jiao, Chem. Commun., 2011, 47, 8097–8099 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, XRD data and copies of NMR (1H & 13C). CCDC 2266943, 2266949 and 2266961. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01168f |
| This journal is © The Royal Society of Chemistry 2024 |