
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating H2S release from self-assembled peptide H2S-donor conjugates using cysteine derivatives†
Zhao
Li‡
ab,
Marius
Thomas‡
c,
Christian M.
Berač
c,
Oliver S.
Stach
c,
Pol
Besenius
 *c and
John B.
Matson
*ab
*c and
John B.
Matson
*ab
aDepartment of Chemistry, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: jbmatson@vt.edu
bMacromolecules Innovation Institute, Virginia Tech, Blacksburg, Virginia 24061, USA
cDepartment of Chemistry, Johannes Gutenberg-University Mainz, Duesbergweg 10-14, D-55128 Mainz, Germany. E-mail: besenius@uni-mainz.de
First published on 18th September 2024
Abstract
Self-assembled peptides provide a modular and diverse platform for drug delivery, and innovative delivery methods are needed for delivery of hydrogen sulfide (H2S), an endogenous signaling molecule (gasotransmitter) with significant therapeutic potential. Of the available types of H2S donors, peptide/protein H2S donor conjugates (PHDCs) offer significant versatility. Here we discuss the design, synthesis, and in-depth study of a PHDC containing three covalently linked components: a thiol-triggered H2S donor based on an S-aroylthiooxime (SATO), a GFFF tetrapeptide, and a tetraethylene glycol (TEG) dendron. Conventional transmission electron microscopy showed that the PHDC self-assembled into spherical structures without heat or stirring, but it formed nanofibers with gentle heat (37 °C) and stirring. Circular dichroism (CD) spectroscopy data collected during self-assembly under nanofiber-forming conditions suggested an increase in β-sheet character and a decrease in organization of the SATO units. Release of H2S from the nanofibers was studied through triggering with various thiols. The release rate and total amount of H2S released over both short (5 h) and long (7 d) time scales varied with the charge state: negatively charged and zwitterionic thiols (e.g., Ac-Cys-OH and H-Cys-OH) triggered release slowly while a neutral thiol (Ac-Cys-OMe) showed ∼10-fold faster release, and a positively charged thiol (H-Cys-OMe) triggered H2S release nearly 50-fold faster than the negatively charged thiols. CD spectroscopy studies monitoring changes in secondary structure over time during H2S release showed similar trends. This study sheds light on the driving forces behind self-assembling nanostructures and offers insights into tuning H2S release through thiol charge state modulation.
Introduction
Hydrogen sulfide (H2S) functions as an endogenous gasotransmitter, regulating cell signaling and behavior through various pathways.1–5 It also exhibits cytoprotective and anti-inflammatory properties in certain cell types at sub-toxic concentrations.6,7 Efforts to both probe H2S physiology and capitalize on these endogenous pathways for therapeutic benefits often involve administration of exogenous H2S.8 However, direct administration of gaseous H2S poses inherent hazards due to its flammability and toxicity at moderate doses,9 thus necessitating alternative methodologies to investigate its effects at physiological levels.10–17 Exogenous H2S administration utilizing aqueous solutions of sulfide salts such as Na2S or NaHS were employed in foundational studies.18 Unfortunately, the instantaneous conversion of sulfide salts into H2S generates a rapid surge to supraphysiological levels, making them a poor mimic of natural H2S signaling in comprehensive studies of H2S biology; these salts are also notoriously impure.19 Consequently, researchers have developed diverse classes of compounds and materials capable of controlled H2S release, including synthetic small molecules,20–28 peptide/protein H2S donor conjugates (PHDCs),29–35 and polymeric H2S donors,36–45 among others.46,47 Of these classes of H2S donors, PHDCs may represent the most versatile family. Due to their ability to self-assemble into intricate nanostructures, peptides offer significant advantages over other systems, such as enhanced water solubility, enabling PHDCs to safeguard H2S-donating functional groups from water-induced hydrolysis or degradation while regulating release of H2S.48Efforts to synthesize peptide-based H2S donors focus on tuning peptide sequences to create complex supramolecular structures. In 2015, Matson and coworkers reported a nanofiber-forming hexapeptide, namely Ile-Ala-Val-Glu-Glu-Glu (IAVEEE), conjugated to an H2S donating S-aroylthiooxime (SATO) at the N-terminus.49 Since that initial report, we have further explored the creation of unique PHDC nanostructures, including twisted nanoribbons,50 nanocoils,48 nanotoroids,51 and nanocrescents,52 based on H2S donor–peptide conjugates. Despite the increased understanding of H2S biology that these nanostructures have provided, developing PHDCs capable of achieving prolonged and controlled H2S release over several days remains challenging. Such materials might find use in long-releasing coatings and implants. To address this objective, we have turned our attention to stabilizing peptide sequences, particularly those incorporating multiple phenylalanine (Phe, F) residues, which utilize multiple types of intermolecular interactions to maintain stable self-assembled nanostructures.
The intermolecular forces of aromatic–aromatic interactions, along with hydrogen bonding, play a crucial role in the stability of nanostructures.53–55 The FF dipeptide motif, known for its role in the self-assembly of amyloid β peptides, represents the smallest recognition module identified in a landmark 2003 paper by Gazit.56 It has been widely used in self-assembling peptides and polymer–peptide conjugates.56–59 However, these FF stabilizing motifs often exhibit poor water solubility.60 To address this challenge, Besenius and co-workers reported in 2018 the design of C3-symmetric dendritic peptide amphiphiles, in which a hydrophilic carbohydrate moiety was appended to a tripeptide arm, Phe-Phe-Phe (FFF).61 This modification enhanced the water solubility of the peptide, and the additional F residues enabled self-assembly into rod-like particles in aqueous environments. Building upon this design, a subsequent study detailed the synthesis of an azidoglycine appended FFF tetrapeptide (N3-GFFF) attached to a Newkome-type dendron.62 The introduction of oligoethylene glycol (OEG) chains within the dendron enhanced the amphiphilicity of the peptide–dendron conjugates and promoted nanorod formation in water.
Building on this amphiphilic peptide–dendron system and other related peptide-based materials,63–66 we aimed here to explore the potential of integrating an H2S donor functionality into a peptide-based material containing the GFFF motif. The objective was to develop a stable supramolecular nanostructure capable of tunable and slow release of H2S. In particular, we envisioned incorporating a thiol-triggered SATO H2S donor at one end of the GFFF peptide sequence while introducing a gallic acid based tetraethylene glycol (TEG) dendron at the other. This hydrophilic dendron could be connected to the hydrophobic peptide sequence via a previously reported aminohexanoic spacer in order to shield supramolecular peptide interactions like H-bonding.61–67 By leveraging the self-assembly properties of the GFFF motif, we anticipated the formation of well-defined nanostructures that could effectively shield the SATO group from premature degradation. Furthermore, we hypothesized that the release of H2S from this system could be tuned by using specific thiol-containing molecules with varying charge state as triggering moieties.
Results and discussion
PHDC design and assembly characterization
We designed a PHDC termed SATO-GFFF-TEGdendron, simplified here to PHDC, that consisted of three units. It included a hydrophobic SATO unit, a type of thiol-triggered H2S donor,68 attached to the N-terminus of the GFFF sequence, and a hydrophilic TEG dendron to the C-terminus. The PHDC (Fig. 1) was synthesized utilizing solid-phase peptide synthesis techniques, starting from the Fmoc-protected tetrapeptide Fmoc-GFFF-OH. Next, the peptide was cleaved from the resin and conjugated to a TEG functionalized gallic acid dendron via a hexamethylenediamine linker through a PyBOP-mediated amidation. After Fmoc group removal, a SATO group was added to the peptide N-terminus by a PyBOP-mediated reaction of the N-terminal amine with the carboxylic acid group of a SATO unit, affording the final PHDC product. The complete synthetic pathway is shown in the ESI (Scheme S1†).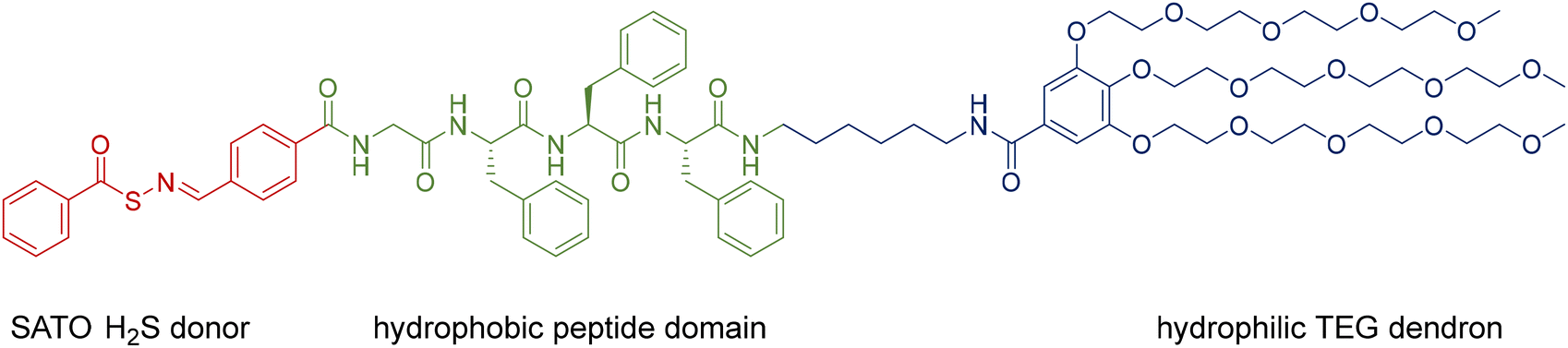 | ||
| Fig. 1 Chemical structure of the H2S-releasing amphiphilic SATO-GFFF-TEG dendron PHDC (referred to simply as the PHDC), including the SATO H2S donor (red); the hydrophobic peptide domain GFFF (green); and the hydrophilic TEG dendron segment (blue). | ||
To investigate the supramolecular self-assembly of the PHDC, a solution of 50 μM PHDC was prepared in phosphate buffer (PB, 10 mM, pH 7.4) and allowed to stand at rt without stirring, and the structural changes during self-assembly were monitored using circular dichroism (CD) spectroscopy at rt. CD spectroscopy is a sensitive technique that can measure the differential absorption of left- and right-handed circularly polarized light, providing insights into the secondary structures of self-assembling chiral systems. In β-sheet forming systems, a positive band near 195 nm and a negative band near 220 nm are a common signature.69 The CD spectra exhibited distinct intensity changes over time in both the peptide region (190–260 nm) and the SATO region (300–360 nm), indicating the involvement of both the GFFF segment and the SATO domain in the formation of ordered nanostructures (Fig. 2A). Over the course of 30 h, the signal near 195 nm appeared noisy and somewhat distorted, likely due to the salt concentration from the buffer, but the increasing intensity of the negative band at 215 nm indicated an increasing β-sheet signature. Additionally, the positive signal near 335 nm progressively increased, suggesting interactions among the SATO groups during self-assembly. Drawing from the signal intensity at 215 nm specifically, the results revealed that the self-assembled nanostructure reached an equilibrium state after approximately 20 h (Fig. 2B) under these stagnant (unstirred) conditions at rt.
 | ||
| Fig. 2 (A) CD kinetics study of self-assembled PHDCs at rt in phosphate buffer (PB, 10 mM, pH 7.4) at 50 μM without stirring. The plots include the initial spectrum immediately after dissolution and spectra at 1 h, 3 h, 10 h, 20 h, and 30 h. Raw spectra were smoothed by the FFT filtering method in Origin to remove high-frequency noise. The full CD spectroscopic investigation of the self-assembly process is available in ESI (Fig. S22†). (B) CD signals at 215 nm as a function of time. The data were fitted via Origin with the Boltzmann function (black line). (C) Conventional TEM images in aqueous solution of the PHDC after 3 d, with schematic illustration in the top right corner. Solution concentration: 50 μM in PB (10 mM, pH 7.4). | ||
Conventional transmission electron microscopy (TEM) was employed to visualize the nanostructures after 3 d. The TEM images revealed spherical structures with an average diameter of (10 ± 2) nm. Considering the extended length of a SATO group (∼1.2 nm) and the extended length of the entire PHDC (∼6.3 nm) (as estimated using ChemDraw, Fig. S24†), the observed spherical structure can be attributed to a core–shell arrangement, with the SATO functional groups aggregating in the core, the GFFF units aligning in the middle layer via aromatic–aromatic interactions and hydrogen bonding, and the TEG dendron units distributing near the surface due to hydrophilic and solvation interactions. The weak CD signal in the peptide region, indicative of limited structural ordering, is unusual for peptides that contain the GFFF sequence,70 which typically drives the formation of β-sheets that dominate the CD spectrum. The lack of a strong β-sheet signal suggested to us the potential for further organization and transformation into alternative supramolecular nanostructures given the propensity for the GFFF sequence to form β-sheets. We speculate that the micellar structures in Fig. 2C are in a kinetically trapped state driven by the spontaneous aggregation of SATO functional groups, which promotes the formation of a spherical morphology without substantial ordering of the peptide units.
To promote the formation of β-sheet structures by the GFFF units, we introduced mild energy input during the self-assembly process and again monitored the changes using CD spectroscopy. The samples were prepared as before, and this time the solution was maintained at 37 °C with continuous gentle stirring using a magnetic stir bar. Under these conditions, the CD spectra obtained over 30 h exhibited a distinctive pattern characteristic of well-defined β-sheet structures. The intensity of both the positive band near 196 nm and a corresponding negative band near 217 nm increased with time and to a much greater magnitude than in the previous experiments, indicating the successful formation of β-sheet structures during self-assembly (Fig. 3A and B grey and red curves). Intriguingly, the signal centered around 335 nm, which reflects the interaction among the SATO functional groups, was barely discernible under these conditions (Fig. 3A). Even though a slight increase in the 335 nm signal was observed within the first 10 h, it exhibited a subsequent decline over time (Fig. 3B blue curve). These results suggest that the SATO functional groups tended to aggregate during the early stages of self-assembly at 25 °C, but the application of stirring and temperature increase to 37 °C lessened their ability to form well-ordered aggregates. In other words, as the self-assembly progressed and the β-sheet structures became more pronounced, the interactions among the SATO groups gradually diminished, thus indicating that the GFFF units dominated the self-assembly process under these conditions. This is also consistent with large signal changes at 196 nm and 217 nm (from 0 to about 250 L mol−1 cm−1), compared to the minor changes at 335 nm (0 to about 5 L mol−1 cm−1). Heating to 37 °C without stirring showed little change in the CD signal over time, indicating the need for stirring to promote these changes in secondary structure. In summary, the very slow self-assembly time scales on the order of hours rather than minutes, as previously reported in our group,66 combined with the temperature-dependent results, are indicative of a kinetically controlled process and competing self-assembly pathways. To elucidate the detailed role of primary and secondary nucleation processes, as well as the role of kinetically trapped intermediates in the supramolecular assembly process, future efforts will focus on concentration-dependent kinetic investigations and seeding experiments to guide the complex self-assembly pathways.
 | ||
| Fig. 3 (A) CD kinetics study of PHDC self-assembled at 37 °C in PB (10 mM, pH 7.4) at 50 μM under constant stirring. The plots include the initial spectrum immediately after dissolution and spectra at 1 h, 3 h, 10 h, 20 h, and 30 h. Raw spectra were smoothed by the FFT filtering method in Origin to remove the high-frequency noise. The full CD spectroscopic investigation of the self-assembly is available in ESI (Fig. S23†). (B) CD signals at 196 nm, 217 nm, and 335 nm as a function of time. (C) Conventional TEM images in aqueous solution of the PHDC under constant stirring after 3 d at a PHDC concentration of 50 μM in PB (10 mM, pH 7.4), with schematic illustration in the top right corner. | ||
Further insights into the resulting supramolecular nanostructures were gained by TEM analysis after both 30 h and 72 h (Fig. S25† and Fig. 3C). The TEM images revealed the presence of nanofibers with varying lengths from 60 nm to 720 nm, exhibiting an average diameter of 12 ± 2 nm after 30 h. Alternatively, at the 72 h mark, these nanofibers had extended to the micrometer scale, exceeding the measurable range of the TEM camera frame. Comparing the diameters of nanofibers and the extended length of an individual PHDC (∼7 nm), we speculate that the nanofibers encompass loosely aggregated SATO functional groups in the core with highly ordered GFFF-dendron units protruding outward (schematic illustration in Fig. 3C). Taken together, these findings underscore the effectiveness of the applied energy input strategy and the subsequent formation of β-sheet structures in modifying the aggregation of SATO functional groups and driving the generation of more intricate and well-organized supramolecular nanostructures.
The distinct outcomes observed in the self-assembly of PHDCs under static and stirred heated conditions can be attributed to activation by energy input. The initial driving force for self-assembly in many peptide amphiphiles in aqueous media is the hydrophobic effect, and the individual peptide units then organize into regular nanostructures.71 The hydrophobic effect is a strong but nondirectional interaction. Here, this leads to an initial self-assembly into spherical micelles as a kinetically trapped state because no conversion into one-dimensional fibers was observed even over longer periods of time at 25 °C. However, the introduction of a stirring force and increase to a physiologically relevant temperature of 37 °C provides enough energy to overcome the kinetic barrier, leading to the formation of ordered β-sheet secondary structures and anisotropic fibers, in contrast to the nondirectional supramolecular nanostructures observed in the spherical assemblies. This phenomenon has previously been described in a series of coarse-grained molecular dynamics simulations on a peptide amphiphile, where raising the temperature above a critical micellization temperature led to a shift from spherical micelles to one-dimensional nanofibers.71
H2S release triggered by thiols with varying charge states
SATOs release H2S in response to a thiol trigger, and we aimed to measure the rate of release from the nanofibers using various thiols. We used the methylene blue colorimetric assay, a well-established and reliable method for H2S detection, provided that appropriate controls are conducted,72 to investigate the kinetics of H2S release. In this experimental setup, the PHDC was initially dissolved in PB (10 mM, pH 7.4) with a small amount of DMSO to maintain solubility during the experiment and was kept within a temperature window of 37–40 °C while applying gentle stirring. After allowing 3 d for self-assembly, Zn(OAc)2 was added, followed by an excess of the thiol trigger. At predetermined time points, aliquots of the release solution were removed, then FeCl3 and N,N-dimethyl-p-phenylenediamine were added to generate the dye for quantification. The amount of released H2S was determined using a calibration curve generated from Na2S (Fig. S27†).We first used the commonly employed thiol-containing molecule, Cys (i.e., H-Cys-OH), to trigger and study the H2S release behavior of the self-assembled PHDC in nanofibers. However, after a 5 h release period, we measured only 2 μM of H2S in the solution, with an initial release rate of 0.7 μM h−1 and less than 1% of the theoretical maximum (Fig. S28†). To account for the potential shielding of the SATO group within the core of nanofibers, we extended the release time to 7 d. At this time, 17% of the theoretical H2S release amount was observed (Fig. S27†).
We hypothesized that the slow triggering behavior of H-Cys-OH might come from the charge state at neutral pH. Although H-Cys-OH exists predominately as a zwitterion at pH 7.4, it has a partial negative charge with a reported pI of 5.07.73 Therefore, we chose three additional molecules, namely N-acetyl Cys (Ac-Cys-OH), N-acetyl Cys methyl ester (Ac-Cys-OMe), and Cys methyl ester (H-Cys-OMe) (Fig. 4A), to further probe how the charge and hydrophobicity of the thiol trigger affected H2S release. The relative net charge, Qnet, of each thiol was estimated by calculation from Henderson–Hasselbalch-derived equations and is listed in Table S3† with charges ranging from −1.01 to +0.37.
 | ||
| Fig. 4 (A) Chemical structures of Cys derivatives. The direction of the black arrow indicates the increasing net charge of all four thiols at pH 7.4. (B) H2S release profiles determined using the methylene blue assay (250 μM PHDC) triggered by different Cys derivatives (2.5 mM) at rt in buffer (10 mM PB, pH 7.4, 800 μM Zn(OAc)2, 10% DMSO). Error bars indicate standard deviations of three separate experiments. (C) Change in CD signals of PHDC solutions monitored at 337 nm as a function of time. The concentration of each component in solution was the same as in the H2S release studies described in panel (B). Data were smoothed by the FTT filtering method and normalized to [0,1] range. | ||
Interestingly, the methylene blue assay results demonstrated that the H2S release rate was significantly influenced by the charge state (Fig. 4B). H2S release triggered by the positively charged thiol (H-Cys-OMe, Qnet = +0.37) displayed an initial rate of 9.1 μM h−1, followed by the neutral thiol (Ac-Cys-OMe, Qnet = −0.02) with an initial rate of 2.5 μM h−1. In contrast, the negatively charged Ac-Cys-OH (Qnet = −1.01) displayed an initial release rate of 0.2 μM h−1, even lower than the 0.7 μM h−1 previously observed with H-Cys-OH (Qnet = −0.14). The neutral thiols H-Cys-OH and Ac-Cys-OMe showed similar net charge Qnet but strongly deviating initial H2S release rates. We hypothesize that the difference is attributed to the different ionic character of these triggers. While Ac-Cys-OMe has a nearly neutral, hydrophobic character that can penetrate into the hydrophobic pocket of the PHDC easily, H-Cys-OH is present as a zwitterion with poor penetration probably due to electrostatic interaction. Total H2S release over 7 d exhibited a similar trend across the various thiol triggers (Fig. S28†).
The differences in H2S release rates were further supported by measuring the signal change using CD spectroscopy under the same release conditions (Fig. 4C). PHDC and Cys derivative concentrations as well as temperature and solvents were adjusted to match the experimental conditions used in the methylene blue colorimetric assay. The peptide region (190–260 nm) was obscured due to the high absorbance of DMSO. Instead, we followed the signal change at 315 nm (in the SATO region) to evaluate whether the self-assembled structure changed during the H2S release process. Upon the reaction of the SATO functional group with the triggering thiol, the CD signal in the SATO region gradually decreased over time. The observed trend was consistent with the H2S release data, with the fastest signal decrease for H-Cys-OMe, followed by Ac-Cys-OMe. Conversely, H-Cys-OH and Ac-Cys-OH exhibited minimal signal decreases within the 5 h timeframe. These results suggest that negatively charged thiols exhibit limited interaction with the PHDC nanofibers, resulting in poor penetration into the core structure and impairing their ability to trigger H2S release. On the other hand, faster H2S release from the positively charged thiol suggests a more favorable interaction, while the moderate release rate from the neutral, hydrophobic thiol suggests moderate penetration capabilities.
To gain insight into the trend, we tested the zeta potential of the PHDC nanofibers in buffer (10 mM PB with 7.7 vt% DMSO) and obtained a result of −17 ± 2 mV, consistent with other PEGylated materials. Due to electrostatic interactions, the negatively charged surface likely repels negatively charged H-Cys-OH or Ac-Cys-OH, resulting in slow penetration, while it attracts positively charged H-Cys-OMe, leading to faster penetration. This difference in penetration ability leads to varying accumulated concentrations of free thiol in the core of nanofibers, further influencing the observed differences in H2S release rates. Future efforts involving simulations will focus on the interactions between the various thiols and the PHDCs to trigger H2S release.
Conclusions
In summary, this study presents the synthesis and self-assembly of a PHDC incorporating a GFFF peptide flanked by a TEG dendron and an H2S donor. Under aqueous conditions, this PHDC exhibited distinct self-assembled nanostructures, spheres and fibers, in the presence and absence of stirring with gentle heat, respectively. CD spectroscopy and TEM images revealed that the spherical structures formed in the absence of stirring and at ambient temperature due to the aggregation of SATO functional groups via the hydrophobic effect, but nanofibers formed when β-sheet formation dominated the molecular packing with gentle stirring at physiological temperature of 37 °C. The methylene blue colorimetric assay revealed that Cys derivatives with various net charge states ranging from −1.01 to +0.37 triggered the release of H2S from the nanofibers, resulting in different rates and total amounts released. The CD signal in the SATO region showed similar trends, decreasing more quickly in response to the thiols that triggered the fastest H2S release. This PHDC has potential applications as a slowly releasing H2S donor in response to glutathione, an abundant cellular thiol with an overall negative charge. Stable H2S release over several days could be valuable in studying slow and complex biological processes where H2S plays a critical role, such as in wound healing, angiogenesis, and tumor progression. In sum, these results provide a valuable understanding on the influence of activation barriers involved in self-assembling peptide-based nanostructures and highlight the impact of the net charge on the interactions between small molecules and the resulting supramolecular nanostructures. Additionally, the data highlight how tuning the charge state of trigger molecules offers a route to regulate H2S release in a wide variety of systems.Author contributions
Z. L.: investigation, formal analysis, methodology, writing – original draft. M. T.: investigation, formal analysis, methodology, writing – original draft. C. M. B.: investigation, methodology, writing – review & editing. P. B.: conceptualization, funding acquisition, project administration, supervision, writing – review & editing. J. B. M.: conceptualization, funding acquisition, project administration, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health (R01GM123508), the Humboldt Foundation and the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (ERC CoG SUPRAVACC 819856). Christian M. Berač was supported through the DFG Graduate School “Materials Science in Mainz” (GSC 266) and the Max Planck Graduate Center with the Johannes Gutenberg-University Mainz (MPGC). We also acknowledge the financial support by the Deutsche Forschungsgemeinschaft (DFG) through the Research Training Group RTG 2516 (grant no. 405552959) [OSS, PB]. We thank Sarah Swilley for careful reading of the manuscript and Prof. Emily Mevers in the Chemistry department at Virginia Tech for the help with LC-MS. We also thank Patrick Ahlers for the synthesis of TEG dendron HMDAGAEG4. This work used shared facilities at the Nanoscale Characterization and Fabrication Laboratory, which is funded and managed by Virginia Tech's Institute for Critical Technology and Applied Science. Additional support is provided by the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), supported by NSF (ECCS 1542100 and ECCS 2025151).References
- D. J. Lefer, A new gaseous signaling molecule emerges: Cardioprotective role of hydrogen sulfide, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17907–17908 CrossRef CAS PubMed.
- R. Wang, Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- H. Kimura, Production and Physiological Effects of Hydrogen Sulfide, Antioxid. Redox Signal., 2014, 20, 783–793 CrossRef CAS PubMed.
- C. Szabo, Gasotransmitters in cancer: from pathophysiology to experimental therapy, Nat. Rev. Drug Discovery, 2016, 15, 185–203 CrossRef CAS PubMed.
- G. Cirino, C. Szabo and A. Papapetropoulos, Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs, Physiol. Rev., 2023, 103, 31–276 CrossRef CAS PubMed.
- J. W. Calvert, S. Jha, S. Gundewar, J. W. Elrod, A. Ramachandran, C. B. Pattillo, C. G. Kevil and D. J. Lefer, Hydrogen Sulfide Mediates Cardioprotection Through Nrf2 Signaling, Circ. Res., 2009, 105, 365–374 CrossRef CAS PubMed.
- A. L. King, D. J. Polhemus, S. Bhushan, H. Otsuka, K. Kondo, C. K. Nicholson, J. M. Bradley, K. N. Islam, J. W. Calvert, Y. X. Tao, T. R. Dugas, E. E. Kelley, J. W. Elrod, P. L. Huang, R. Wang and D. J. Lefer, Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3182–3187 CrossRef CAS.
- M. D. Hartle and M. D. Pluth, A practical guide to working with H2S at the interface of chemistry and biology, Chem. Soc. Rev., 2016, 45, 6108–6117 RSC.
- S. L. M. Rubright, L. L. Pearce and J. Peterson, Environmental toxicology of hydrogen sulfide, Nitric Oxide, 2017, 71, 1–13 CrossRef PubMed.
- C. M. Levinn, M. M. Cerda and M. D. Pluth, Activatable Small-Molecule Hydrogen Sulfide Donors, Antioxid. Redox Signal., 2020, 32, 96–109 CrossRef CAS.
- Y. Zhao, T. D. Biggs and M. Xian, Hydrogen sulfide (H2S) releasing agents: chemistry and biological applications, Chem. Commun., 2014, 50, 11788–11805 RSC.
- S. Xu, A. Hamsath, D. L. Neill, Y. Wang, C. T. Yang and M. Xian, Strategies for the Design of Donors and Precursors of Reactive Sulfur Species, Chemistry, 2019, 25, 4005–4016 CrossRef CAS PubMed.
- K. Kaur, R. J. Carrazzone and J. B. Matson, The Benefits of Macromolecular/Supramolecular Approaches in Hydrogen Sulfide Delivery: A Review of Polymeric and Self-Assembled Hydrogen Sulfide Donors, Antioxid. Redox Signal., 2020, 32, 79–95 CrossRef CAS PubMed.
- C. R. Powell, K. M. Dillon and J. B. Matson, A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef CAS PubMed.
- K. M. Dillon, R. J. Carrazzone, J. B. Matson and K. Kashfi, The evolving landscape for cellular nitric oxide and hydrogen sulfide delivery systems: A new era of customized medications, Biochem. Pharmacol., 2020, 176, 113931 CrossRef CAS PubMed.
- C. Szabo and A. Papapetropoulos, International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H(2)S Levels: H2S Donors and H2S Biosynthesis Inhibitors, Pharmacol. Rev., 2017, 69, 497–564 CrossRef CAS PubMed.
- E. Magli, E. Perissutti, V. Santagada, G. Caliendo, A. Corvino, G. Esposito, G. Esposito, F. Fiorino, M. Migliaccio, A. Scognamiglio, B. Severino, R. Sparaco and F. Frecentese, H2S Donors and Their Use in Medicinal Chemistry, Biomolecules, 2021, 11, 1899 CrossRef CAS PubMed.
- S. Jha, J. W. Calvert, M. R. Duranski, A. Ramachandran and D. J. Lefer, Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling, Am. J. Physiol.: Heart Circ. Physiol., 2008, 295, 801–806 CrossRef PubMed.
- W. H. Smith, J. Birnbaum and C. A. Wolden, Production and purification of anhydrous sodium sulfide, J. Sulphur Chem., 2021, 42, 426–442 CrossRef CAS.
- L. Li, G. Rossoni, A. Sparatore, L. C. Lee, P. Del Soldato and P. K. Moore, Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative, Free Radicals Biol. Med., 2007, 42, 706–719 CrossRef CAS PubMed.
- Z. W. Lee, J. B. Zhou, C. S. Chen, Y. J. Zhao, C. H. Tan, L. Li, P. K. Moore and L. W. Deng, The Slow-Releasing Hydrogen Sulfide Donor, GYY4137, Exhibits Novel Anti-Cancer Effects In Vitro and In Vivo, PLoS One, 2011, 6, e21077 CrossRef CAS PubMed.
- Y. Zhao, H. Wang and M. Xian, Cysteine-Activated Hydrogen Sulfide (H2S) Donors, J. Am. Chem. Soc., 2011, 133, 15–17 CrossRef CAS PubMed.
- C. M. Park, Y. Zhao, Z. H. Zhu, A. Pacheco, B. Peng, N. O. Devarie-Baez, P. Bagdon, H. Zhang and M. Xian, Synthesis and evaluation of phosphorodithioate-based hydrogen sulfide donors, Mol. Biosyst., 2013, 9, 2430–2434 RSC.
- B. Szczesny, K. Modis, K. Yanagi, C. Coletta, S. Le Trionnaire, A. Perry, M. E. Wood, M. Whiteman and C. Szabo, AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro, Nitric Oxide, 2014, 41, 120–130 CrossRef CAS PubMed.
- A. K. Steiger, S. Pardue, C. G. Kevil and M. D. Pluth, Self-Immolative Thiocarbamates Provide Access to Triggered H2S Donors and Analyte Replacement Fluorescent Probes, J. Am. Chem. Soc., 2016, 138, 7256–7259 CrossRef CAS PubMed.
- C. M. Levinn, M. M. Cerda and M. D. Pluth, Development and Application of Carbonyl Sulfide-Based Donors for H2S Delivery, Acc. Chem. Res., 2019, 52, 2723–2731 CrossRef CAS PubMed.
- K. Kaur, P. Enders, Y. M. Zhu, A. F. Bratton, C. R. Powell, K. Kashfi and J. B. Matson, Amino acid-based H2S donors: N-thiocarboxyanhydrides that release H2S with innocuous byproducts, Chem. Commun., 2021, 57, 5522–5525 RSC.
- Q. W. Hu, S. I. Suarez, R. A. Hankins and J. C. Lukesh, Intramolecular Thiol- and Selenol-Assisted Delivery of Hydrogen Sulfide, Angew. Chem., Int. Ed., 2022, 61, e202210754 CrossRef CAS PubMed.
- A. Longchamp, K. Kaur, D. Macabrey, C. Dubuis, J.-M. Corpataux, S. Déglise, J. B. Matson and F. Allagnat, Hydrogen sulfide-releasing peptide hydrogel limits the development of intimal hyperplasia in human vein segments, Acta Biomater., 2019, 97, 374–384 CrossRef CAS PubMed.
- K. Kaur, Y. Wang and J. B. Matson, Linker-Regulated H2S Release from Aromatic Peptide Amphiphile Hydrogels, Biomacromolecules, 2020, 21, 1171–1178 CrossRef CAS PubMed.
- Y. Wang, K. M. Dillon, Z. Li, E. W. Winckler and J. B. Matson, Alleviating Cellular Oxidative Stress through Treatment with Superoxide-Triggered Persulfide Prodrugs, Angew. Chem., Int. Ed., 2020, 59, 16698–16704 CrossRef CAS PubMed.
- R. Ali, R. Hameed, D. Chauhan, S. Sen, M. Wahajuddin, A. Nazir and S. Verma, Multiple Actions of H2S-Releasing Peptides in Human beta-Amyloid Expressing C. elegans, ACS Chem. Neurosci., 2022, 13, 3378–3388 CrossRef CAS PubMed.
- H. Chen, X. Y. Guan, Q. Q. Liu, L. C. Yang, J. Guo, F. Gao, Y. H. Qi, X. T. Wu, F. Zhang and X. M. Tian, Co-assembled Nanocarriers of De Novo Thiol-Activated Hydrogen Sulfide Donors with an RGDFF Pentapeptide for Targeted Therapy of Non-Small-Cell Lung Cancer, ACS Appl. Mater. Interfaces, 2022, 14, 53475–53490 CrossRef CAS PubMed.
- Y. Zhu, W. R. Archer, K. F. Morales, M. D. Schulz, Y. Wang and J. B. Matson, Enzyme-Triggered Chemodynamic Therapy via a Peptide-H2S Donor Conjugate with Complexed Fe2+, Angew. Chem., Int. Ed., 2023, 62, e202302303 CrossRef CAS PubMed.
- E. K. Jeong, B. Selvaraj, S. Clovis, Y. J. Son, T. H. Park, A. Veeramanoharan, H.-I. Kim, K.-Y. Yoo, J. W. Lee and C.-M. Park, Synthesis and neuroprotective effects of H2S-donor-peptide hybrids on hippocampal neuronal cells, Free Radicals Biol. Med., 2023, 194, 316–325 CrossRef CAS PubMed.
- U. Hasegawa and A. J. van der Vlies, Design and Synthesis of Polymeric Hydrogen Sulfide Donors, Bioconjugate Chem., 2014, 25, 1290–1300 CrossRef CAS PubMed.
- C. R. Powell, J. C. Foster, B. Okyere, M. H. Theus and J. B. Matson, Therapeutic Delivery of H2S via COS: Small Molecule and Polymeric Donors with Benign Byproducts, J. Am. Chem. Soc., 2016, 138, 13477–13480 CrossRef CAS PubMed.
- J. C. Foster, S. C. Radzinski, X. L. Zou, C. V. Finkielstein and J. B. Matson, H2S-Releasing Polymer Micelles for Studying Selective Cell Toxicity, Mol. Pharmaceutics, 2017, 14, 1300–1306 CrossRef CAS PubMed.
- C. R. Powell, J. C. Foster, S. N. Swilley, K. Kaur, S. J. Scannelli, D. Troya and J. B. Matson, Self-amplified depolymerization of oligo(thiourethanes) for the release of COS/H2S, Polym. Chem., 2019, 10, 2991–2995 RSC.
- Z. H. Li, D. A. Li, L. Wang, C. J. Lu, P. F. Shan, X. L. Zou and Z. Y. Li, Photocontrollable water-soluble polymeric hydrogen sulfide (H2S) donor, Polymer, 2019, 168, 16–20 CrossRef CAS.
- A. V. Do, R. Smith, P. Tobias, D. Carlsen, E. Pham, N. B. Bowden and A. K. Salem, Sustained Release of Hydrogen Sulfide (H2S) from Poly(Lactic Acid) Functionalized 4-Hydroxythiobenzamide Microparticles to Protect Against Oxidative Damage, Ann. Biomed. Eng., 2019, 47, 1691–1700 CrossRef PubMed.
- C. Q. Yang, X. F. Li and Q. Yan, Polythionoester Vesicle: An Efficient Polymeric Platform for Tuning H2S Release, ACS Macro Lett., 2022, 11, 1230–1237 CrossRef CAS PubMed.
- A. J. van der Vlies, M. Ghasemi, B. M. Adair, J. H. Adair, E. D. Gomez and U. Hasegawa, Reactive Oxygen Species-Triggered Hydrogen Sulfide Release and Cancer-Selective Antiproliferative Effect of Anethole Dithiolethione-Containing Polymeric Micelles, Adv. Healthcare Mater., 2023, 12, e2201836 CrossRef PubMed.
- N. V. Dao, F. Ercole, L. M. Kaminskas, T. P. Davis, E. K. Sloan, M. R. Whittaker and J. F. Quinn, Trisulfide-Bearing PEG Brush Polymers Donate Hydrogen Sulfide and Ameliorate Cellular Oxidative Stress, Biomacromolecules, 2020, 21, 5292–5305 CrossRef CAS PubMed.
- J. P. Grace and N. B. Bowden, Synthesis and Hydrogen Sulfide Releasing Properties of Diaminodisulfides and Dialkoxydisulfides, ACS Omega, 2021, 6, 17741–17747 CrossRef CAS PubMed.
- F. E. Chen, R. M. Mandel, J. J. Woods, J. H. Lee, J. Kim, J. H. Hsu, J. J. Fuentes-Rivera, J. J. Wilson and P. J. Milner, Biocompatible metal-organic frameworks for the storage and therapeutic delivery of hydrogen sulfide, Chem. Sci., 2021, 12, 7848–7857 RSC.
- P. K. Allan, P. S. Wheatley, D. Aldous, M. I. Mohideen, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. De Weireld, S. Vaesen and R. E. Morris, Metal-organic frameworks for the storage and delivery of biologically active hydrogen sulfide, Dalton Trans., 2012, 41, 4060–4066 RSC.
- Y. Wang, K. Kaur, S. J. Scannelli, R. Bitton and J. B. Matson, Self-Assembled Nanostructures Regulate H2S Release from Constitutionally Isomeric Peptides, J. Am. Chem. Soc., 2018, 140, 14945–14951 CrossRef CAS PubMed.
- J. M. Carter, Y. Qian, J. C. Foster and J. B. Matson, Peptide-based hydrogen sulphide-releasing gels, Chem. Commun., 2015, 51, 13131–13134 RSC.
- Y. Wang and J. B. Matson, Supramolecular Nanostructures with Tunable Donor Loading for Controlled H2S Release, ACS Appl. Bio Mater., 2019, 2, 5093–5098 CrossRef CAS PubMed.
- Z. Li, S. Y. Joshi, Y. Wang, S. A. Deshmukh and J. B. Matson, Supramolecular Peptide Nanostructures Regulate Catalytic Efficiency and Selectivity, Angew. Chem., Int. Ed., 2023, 62, e202303755 CrossRef CAS PubMed.
- Y. Wang, Z. Li, Y. Shmidov, R. J. Carrazzone, R. Bitton and J. B. Matson, Crescent-Shaped Supramolecular Tetrapeptide Nanostructures, J. Am. Chem. Soc., 2020, 142, 20058–20065 CrossRef CAS PubMed.
- S. Marchesan, A. V. Vargiu and K. E. Styan, The Phe-Phe Motif for Peptide Self-Assembly in Nanomedicine, Molecules, 2015, 20, 19775–19788 CrossRef CAS PubMed.
- E. Mayans and C. Aleman, Revisiting the Self-Assembly of Highly Aromatic Phenylalanine Homopeptides, Molecules, 2020, 25, e6037 CrossRef PubMed.
- S. Bera, B. Xue, P. Rehak, G. Jacoby, W. Ji, L. J. W. Shimon, R. Beck, P. Kral, Y. Cao and E. Gazit, Self-Assembly of Aromatic Amino Acid Enantiomers into Supramolecular Materials of High Rigidity, ACS Nano, 2020, 14, 1694–1706 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Casting metal nanowires within discrete self-assembled peptide nanotubes, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- J. L. Warren, P. A. Dykeman-Bermingham and A. S. Knight, Controlling Amphiphilic Polymer Folding beyond the Primary Structure with Protein-Mimetic Di(Phenylalanine), J. Am. Chem. Soc., 2021, 143, 13228–13234 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Formation of Closed-Cage Nanostructures by Self-Assembly of Aromatic Dipeptides, Nano Lett., 2004, 4, 581–585 CrossRef CAS.
- C. G. Pappas, P. W. Frederix, T. Mutasa, S. Fleming, Y. M. Abul-Haija, S. M. Kelly, A. Gachagan, D. Kalafatovic, J. Trevino, R. V. Ulijn and S. Bai, Alignment of nanostructured tripeptide gels by directional ultrasonication, Chem. Commun., 2015, 51, 8465–8468 RSC.
- R. Sarma, K. Y. Wong, G. C. Lynch and B. M. Pettitt, Peptide Solubility Limits: Backbone and Side-Chain Interactions, J. Phys. Chem. B, 2018, 122, 3528–3539 CrossRef CAS PubMed.
- D. Strassburger, N. Stergiou, M. Urschbach, H. Yurugi, D. Spitzer, D. Schollmeyer, E. Schmitt and P. Besenius, Mannose-Decorated Multicomponent Supramolecular Polymers Trigger Effective Uptake into Antigen-Presenting Cells, ChemBioChem, 2018, 19, 912–916 CrossRef CAS PubMed.
- O. S. Stach, K. Breul, C. M. Berac, M. Urschbach, S. Seiffert and P. Besenius, Bridging Rigidity and Flexibility: Modulation of Supramolecular Hydrogels by Metal Complexation, Macromol. Rapid Commun., 2022, 43, e2100473 CrossRef PubMed.
- V. Lewe, M. Preuss, E. A. Woznica, D. Spitzer, R. Otter and P. Besenius, A clickable NHC-Au(i)-complex for the preparation of stimulus-responsive metallopeptide amphiphiles, Chem. Commun., 2018, 54, 9498–9501 RSC.
- C. M. Berac, L. Zengerling, D. Straβburger, R. Otter, M. Urschbach and P. Besenius, Evaluation of Charge-Regulated Supramolecular Copolymerization to Tune the Time Scale for Oxidative Disassembly of β-Sheet Comonomers, Macromol. Rapid Commun., 2020, 41, e1900476 CrossRef PubMed.
- D. Strassburger, S. Herziger, K. Huth, M. Urschbach, R. Haag and P. Besenius, Supramolecular polymerization of sulfated dendritic peptide amphiphiles into multivalent L-selectin binders, Beilstein J. Org. Chem., 2021, 17, 97–104 CrossRef CAS PubMed.
- M. Thomas, V. Lewe, J. Koelsch, M. Urschbach, J. Erlenbusch, O. S. Stach and P. Besenius, Impact of sample history and solvent effects on pathway control in the supramolecular polymerisation of Au(I)-metallopeptide amphiphiles, Polym. Chem., 2023, 14, 1888–1892 RSC.
- R. Appel, S. Tacke, J. Klingauf and P. Besenius, Tuning the pH-triggered self-assembly of dendritic peptide amphiphiles using fluorinated side chains, Org. Biomol. Chem., 2015, 13, 1030–1039 RSC.
- J. C. Foster, C. R. Powell, S. C. Radzinski and J. B. Matson, S-aroylthiooximes: a facile route to hydrogen sulfide releasing compounds with structure-dependent release kinetics, Org. Lett., 2014, 16, 1558–1561 CrossRef CAS PubMed.
- C. A. Bush, S. K. Sarkar and K. D. Kopple, Circular dichroism of beta turns in peptides and proteins, Biochemistry, 1978, 17, 4951–4954 CrossRef CAS PubMed.
- R. Appel, J. Fuchs, S. M. Tyrrell, P. A. Korevaar, M. C. Stuart, I. K. Voets, M. Schönhoff and P. Besenius, Steric Constraints Induced Frustrated Growth of Supramolecular Nanorods in Water, Chem. – Eur. J., 2015, 21, 19257–19264 CrossRef CAS PubMed.
- Y. S. Velichko, S. I. Stupp and M. O. de la Cruz, Molecular simulation study of peptide amphiphile self-assembly, J. Phys. Chem. B, 2008, 112, 2326–2334 CrossRef CAS PubMed.
- L. M. Siegel, A Direct Microdetermination for Sulfide, Anal. Biochem., 1965, 11, 126–132 CrossRef CAS PubMed.
- G. L. E. Turner, Crc Handbook of Chemistry and Physics - a Ready-Reference Book of Chemical and Physical Data, 70th edn, Weast, Rc, Lide, Dr Ann. Sci., 1991, vol. 48, pp. 496–497 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01148a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |