
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid diazotransfer for selective lysine labelling†
Susannah H.
Calvert
 ab,
Tomasz
Pawlak
a,
Gary
Hessman
a and
Joanna F.
McGouran
*ab
ab,
Tomasz
Pawlak
a,
Gary
Hessman
a and
Joanna F.
McGouran
*ab
aSchool of Chemistry, Trinity Biomedical Science Institute, Trinity College Dublin, D02 R590, Ireland. E-mail: jmcgoura@tcd.ie
bSSPC, The SFI Research Centre for Pharmaceuticals, Ireland
First published on 16th September 2024
Abstract
Azide functionalization of protein and peptide lysine residues allows selective bioorthogonal labeling to introduce new, site selective functionaltiy into proteins. Optimised diazotransfer reactions under mild conditions allow aqueous diazotransfer to occur in just 20 min at pH 8.5 on amino acid, peptide and protein targets. In addition, conditons can be modified to selectively label a single lysine residue in both protein targets investigated. Finally, we demonstrate selective modification of proteins containing a single azidolysine using copper(I)-catalyzed triazole formation.
Introduction
Protein modification is common in nature, significantly enhancing the diversity of protein structures and functions by up to two orders of magnitude.1 However, our ability to replicate these natural modifications synthetically is largely constrained by the available chemistry. For these reasons, a broad range of protein modification methodologies have been investigated to achieve precise control over these macromolecules.2,3 Cysteines have been of particular focus for modification, due to the reactivity of the side chain thiol. Cysteine also has a low abundance of 1.7%.4 Methods to target cysteine residues often utilise electrophiles. However, amines and alcohols such as those in lysine, serine, and threonine, can also react with electrophiles targeting cysteine residues which can lead to a loss of specificity.5,6 Other amino acids can also be selectively targeted, including tyrosine,7–9 tryptophan,10–12 and lysine.13–18Lysine is an attractive residue to target for modification due to its relative abundance, around 6%, and the presence of a nucleophilic amine.19 Ligandable lysines have been targeted in a number of methods20–24 including, but not limited to, NHS- and STP-esters,15,25 sulfonyl fluorides,26 and isothiocyanates.27 Furthermore, lysines are often the targets of post-translational modifications (PTMs), including acetylation, methylation and ubiquitylation.14 These PTMs regulate the activity, interaction and stability of the protein. However, understanding the biological significance or physiological roles of specific modification sites can be difficult due to their dynamic and sub-stoichiometric characteristics.28
Introduction of an azide into a protein or peptide is an attractive, as yet underutilised, modification due to its unique biorthogonal reactivity. Additionally, as a result of the small size of this group it leads to a negligible disruption to the overall structure and function of the protein. The installation of one or more azides provides a handle to carry out a variety of reactions including Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC),29,30 Staudinger ligation,31,32 or strain promoted cycloaddition.33,34 Incorporation of azides with a diazo transfer reagent, imidazole-1-sulfonyl azide, has been shown using protein substrates for bioconjugation applications.13,35–37 However, the reaction times in these cases have been lengthy taking between 6–48 h, which is considerably longer than the analogous cysteine modification reactions.38 In addition, this reaction often requires a high pH environment, compromising protein stability and function.39
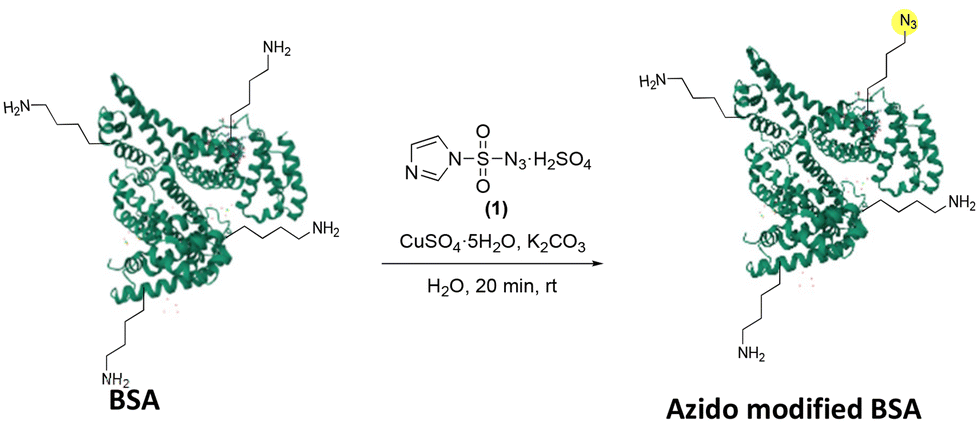
Herein, we present work towards rapid formation of azidolysine residues using the mild diazo transfer reagent imidazole-1-sulfonyl azide hydrogen sulfate 1 at a reduced pH of 8.5. The reaction is demonstrated on two model proteins, both showing distinct lysine reactivity patters. In addition, we use this reaction and subsequent modification to selectively Ubiquitinate a model protein for the synthesis of a diubiquitin activity-based probe.
Results & discussion
Initial studies to assess decreasing the reaction time were carried out using a protected amino acid model system, Fmoc-Lys-OH 2. Conversion of the sidechain amine to the azide, resulting in the formation of protected azidolysine derivative 3 has been reported, however, effort was placed on reducing the reaction time of 5 h, Table 1 entry 1, significantly.40 A range of conditions were screened (Table 1). The reaction progress, with a particular focus on starting material consumption, was monitored by TLC analysis. TLC samples were taken every min up until 10 min, and then followed with every 5 min until reaction completion was observed or the time stated in Table 1. Initially, a fivefold and tenfold increase in both the eq. of the diazo transfer reagent and the catalytic loading of copper were investigated, Table 1, entries 1–3. However, after 2 h, the starting material had not been consumed. Increasing the amount of base in the reaction from 3 eq. to 6 and 10 eq. (Table 1, entries 3–5) still did not yield a reaction. Measuring the pH of these reactions showed a suboptimal pH of under 3, to counter this in the following attempts the pH was adjusted to 8.5 by further increasing the amount of base used in the reaction (Table 1, entries 6–10). Table 1, entry 8 showed the best results with full consumption of the starting material by TLC analysis in 4 min, using ten eq. of the azido transfer reagent, imidazole-1-sulfonyl azide hydrogen sulfate 1 and a stoichiometric quantity of copper(II) sulfate. Further analysis of the reaction mixture by NMR and HRMS showed no starting material and the presence of the azide containing product 3. Final attempts using a catalytic amount of Cu(II), were tried as copper is toxic to cells and can be difficult to remove from proteins.41,42 However, further increases in 1 with lower catalytic loading of Cu(II) did not afford a reaction time improvement (Table 1, entries 9 and 10). Overall, through the modification of reagent eq., the reaction time was decreased significantly from 5 h to 4 min affording a fast and reliable diazo transfer procedure.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 (Eq.) | CuSO4·5H2O (Eq.) | K2CO3 (Eq.) | Time | SM consumed by TLCa | pH |
a TLC conditions DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH:AcOH (4:1:0.01, v/v).
b Not all reagents fully soluble. :MeOH:AcOH (4:1:0.01, v/v).
b Not all reagents fully soluble.
|
||||||
| 1 | 1 | 0.01 | 3 | 5 h | ✓ | 8.5 |
| 2 | 5.5 | 0.05 | 3 | 2 h | ✗ | ND |
| 3 | 10 | 0.05 | 3 | 2 h | ✗ | ND |
| 4 | 10 | 0.05 | 6 | 30 min | ✗ | 2 |
| 5 | 10 | 0.05 | 10 | 30 min | ✗ | 3 |
| 6 | 10 | 0.05 | 20 | 25 min | ✓ | 8.5 |
| 7 | 10 | 1 | 20 | 8 min | ✓ | 8 |
| 8 | 10 | 1 | 20 | 4 min | ✓ | 8 |
| 9 | 20 | 0.05 | 20 | 30 minb | ✗ | 6 |
| 10 | 20 | 0.05 | 40 | 20 minb | ✓ | 8.5 |

Having significantly reduced the reaction time on the amino acid system, these reaction conditions were translated onto a larger system, model peptide 4. This substrate was chosen as it contains a range of reactive residues including tyrosine, serine, tryptophan, and glutamic acid. The conditions optimised in Table 1 entry 8 were utilised for azide installation, however, the reaction was not complete after 10 min, Table 2 entry 1. Further increase of reagents to account for both the lysine side chain amine and the N-terminus afforded complete conversion by TLC after 4 min, Table 2 entries 2 and 3. Analysis of the reaction mixture using infrared spectroscopy showed azide formation. Further analysis using HRMS showed the presence of the di-azide containing product 5 but also remaining starting material 4. Further analysis using NMR spectrometry to quantify the starting material to product ratio was hampered due to the presence of the paramagnetic copper. However, we showed that this system was effective on larger models and tolerant of a wide range of amino acid side chains, and as such we next turned our attention to validate the system using protein targets.

The transport protein bovine serum albumin (BSA) has been widely investigated as a model protein.43–45 It contains 59 lysines and has a free N-terminus and as such was chosen as the initial protein to test this system. Optimised conditions from the prior model systems were undertaken with a reaction time of 20 min. This increased reaction time was chosen to ensure completion of the reaction given the complexity of the system used when compared to the amino acid and peptide models.
Following this, a size exclusion and concentration step were performed to remove the excess reagents from the protein material. Tryptic digest of the protein sample, followed by LC-MS/MS analysis showed that when using our initial conditions, 8% of the lysines were modified (Table 3, entry 1). Furthermore, replacing the trypsin digest with an elastase digest showed very similar results (Table S1,† entries 1 and 2). Following this initial result, in which several, but not all lysine residues were labelled the effects of both increase and decrease of reagents were investigated. This was used to assess whether full coverage and/or selective modification of a single lysine could be achieved. Further increasing the eq. of reagent four-fold to 89 eq. per amine saw 10 lysines modified after 20 min, Table 3 entry 2, and decreasing this number of eq. two-fold to 44, Table 3 entry 3, saw fewer lysines, only 8, being modified. Interestingly, when further decreasing the eq. of diazo transfer reagent 1 used to 28, Table 3 entry 4, the same number modified lysines was observed. However, this can likely be explained by the larger scale of the reaction leading to a 4% increase in protein sequence coverage compared to Table 3 entry 3. However, further decreasing eq. showed fewer modified lysines (Table 3 entries 5 and 6). By decreasing the number of eq. of diazo transfer reagent 1 to less that single equivalent, selective modification of K413 was seen (Table 3 entry 7). It was noted that the number of lysines being modified was affected by reagent concentration, however, the order in which they were modified remained surprisingly consistent with K413 being the most reactive lysine. The differing reactivities observed between these lysine residues is caused by differences in the surface accessibility and pKaH of the side chain amines. A qualitative assessment of the degree of modification was performed by comparing extracted ion chromatograms for the azide modified and unmodified peptides for each modified lysine observed (ESI page 18–35†).
|
|
||||
|---|---|---|---|---|
| Entry | Equiv. of 1 per amine | Protein coverage (%) | Unmodified lysines observed | Modified lysines observed |
| a Reaction performed on 500 μg scale. | ||||
| 1 | 22 | 20.26 | K51, K204, K239, K431 | K211, K221, K224, K232, K413, K524 |
| 2 | 89 | 25.54 | K12, K20, K51, K388, K533 | K204, K211, K221, K224, K232, K239, K377, K413, K431, K524 |
| 3 | 44 | 23.89 | K51, K204, K388, K533 | K211, K221, K224, K232, K239, K413, K431, K524 |
| 4a | 28 | 24.8 | K20, K51, K204, K533, K537, K544, K556 | K40, K211, K221, K224, K232, K239, K413, K524 |
| 5 | 8 | 24.05 | K51, K204, K239, K375, K431, K533 | K211, K221, K224, K232, K413 |
| 6 | 4 | 22.57 | K51, K204, K239, K224, K232, K388, K524, K533 | K211, K221, K413 |
| 7 | 0.8 | 19.11 | K51, K204, K221, K224, K232, K239, K388, K524, K533 | K413 |
| 8 | 0 | 18.62 | K51, K204, K232, K273, K388, K413, K533 | — |
Having optimised the diazo transfer reaction on a protein system, and observed surprising levels of site selectivity, the reaction was then explored to functionalise the protein ubiquitin (Ub). The addition of monomeric ubiquitin or poly-ubiquitin chains are common post-translational modifications and many biochemical processes in eukaryotic cells are controlled using ubiquitin signalling.46 Ub is a 76 amino acid protein, which has seven lysines (K6, K27, K29, K33, K48 and K63), all of which can undergo ubiquitination, with the best characterised modifications occurring at K48 and K63.47 In addition, the biological function of the specific poly-Ub chain is dictated by the lysine residue of Ub utilised in Ub–Ub conjugation.48
Previous work in our group has focused on the generation of ubiquitin activity-based probes with alkene warheads that are activated upon irradiation with UV light.49 Diubiquitin activity-based probes have also been shown to be highly effective.50 In addition, the synthesis of several diubquitin probes utilised CuAAC chemistry with the resulting triazole shown to be uncleavable, making it a robust probe.51,52 To investigate making a diubiquitin probe using this selective diazo transfer methodology a HA-tagged Ub1–75 construct containing a C-terminal alkene 6 was generated (Fig. 1(a)) and diazo transfer conditions were applied. Even with a large excesses of imidazole-1-sulfonyl azide hydrogen sulfate 1 (125 eq. per amine), LC-MS/MS showed only a single azide substitution at K48 Fig. 1(c) when compared to the initial construct (Fig. 1(b)).
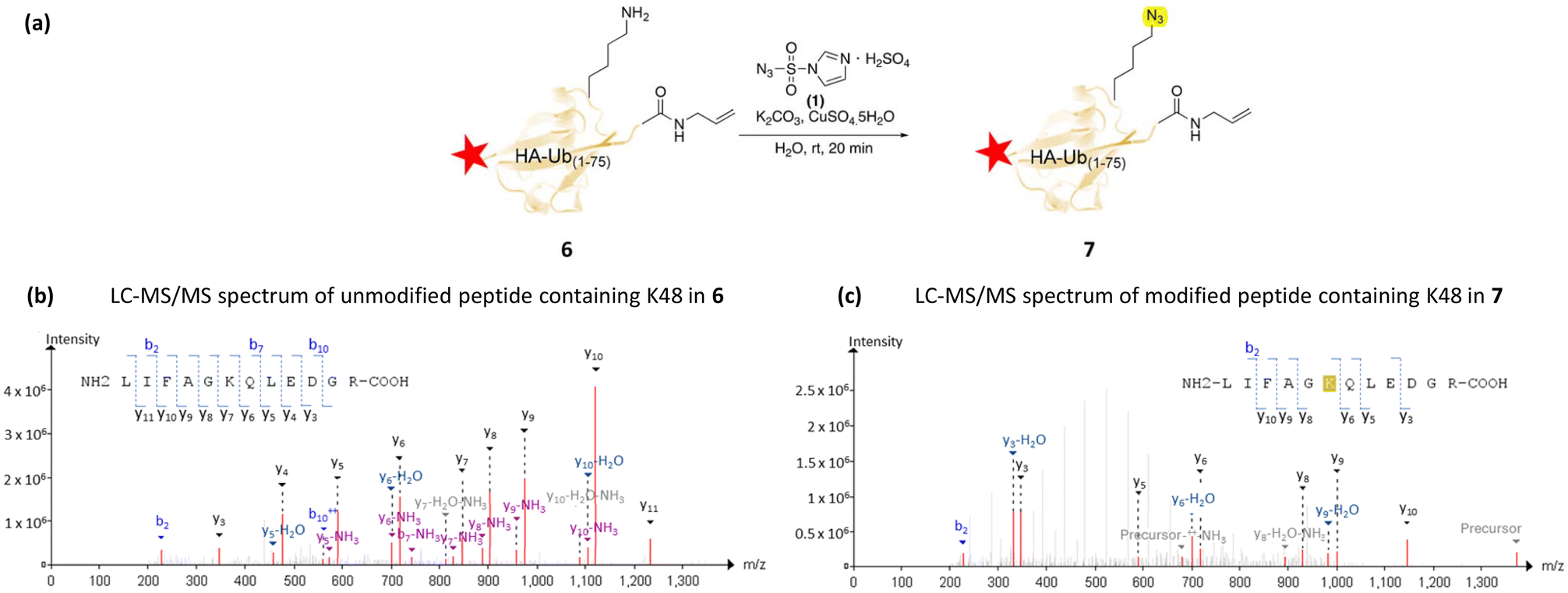 | ||
| Fig. 1 (a) Azide modification of K48 of a HA tagged radical Ubiquitin-based Activity-Based Probe 6, HA tag depicted by red star (b) LC-MS/MS identification of Ub75 C-terminal peptide of probe 6 full peptide m/z = 1346.5498. (c) LC-MS/MS identification of Ub75 C-terminal peptide following azido transfer. Mass of product 7 containing azido-modified K48, mass increased by 26 Da, corresponding to a single substitution of an NH2 to N3, m/z = 1372.5498 Precursor peptides containing an azide and 2+ ions have also been noted to increase in m/z. | ||
CuAAC chemistry has previously been shown to be an efficient method to generate Ub dimers51,53,54 and as such the azido-K48 derivative 7 was chosen as a suitable substrate for this reaction. A HA-tagged Ub1–75 with a C-terminal alkyne 8 was generated. The two Ub derivatives 7 and 8 were incubated in the presence of a CuAAC cocktail of copper(II) sulfate, tris-hydroxypropyltriazolylmethylamine (THPTA) and sodium ascorbate for 3 h, Fig. 2(a). The reaction was analysed by gel electrophoresis and visualised by anti-HA western blot, Fig. 2(b). Lanes 1–2 contain HA-tagged Ub1–75 Propene and HA-tagged Ub1–75 Propagylamine alone (2 μg protein), Lanes 3–4 contain both proteins (2.6 μg protein) in the presence and absence of CuAAC coupling reagents. Following the CuAAC reaction a new band, at around 22 kDa, corresponding to the expected molecular weight of the diubiquitin adduct 9 is observed in Fig. 2(b) lane 3, blue arrow. A control reaction was also undertaken, where no CuAAC cocktail is present, and this new additional band was not observed, Fig. 2(b) lane 4. This newly synthesised di-ubiquitin-alkene probe 9 can be explored for its potency and selectivity as an activity-based probe. Images of the full gel lanes corresponding silver stain are available in the ESI (Fig. S1†).
 | ||
| Fig. 2 Modified diubiquitin formation. (a) Schematic of modified diubiquitin formation. (b) Anti-HA western blot of modified ubiquitin dimerization. K48 azido modified HA-Ub1–75-propene (0.6 μg) and HA-Ub1–75-propargylamine (2 μg) were incubated in the presence of CuAAC cocktail containing copper(II) sulfate, tris-hydroxypropyltriazolylmethylamine (THPTA) and sodium ascorbate for 3 hours (lane 3). Controls of K48 azido modified HA-Ub1–75-propene (2 μg) alone (lane 1), HA-Ub1–75-propargylamine (2 μg) alone (lane 2) and the reaction in the absence of the CUAAC cocktail (lane 4). | ||
Conclusions
In summary, we have presented a simple and efficient way to rapidly install azides into amino acids, peptides, and proteins. Both protein samples tested showed that selective modification of a single lysine could be achieved. In addition, we used an azide installed in this manner in a CuAAC click reaction to synthesise a ubiquitin dimer, showing the utility of the method in mimicking lysine post translational modifications. The mild modification conditions and small steric footprint of the azide modification highlight the potential for retaining the biological activity of proteins following modification, however we anticipate that this will vary from protein to protein.Author contributions
S.H.C: conceived and carried out experiments on amino acid and peptide, formal analysis, funding acquisition, writing. T.P: conceived and carried out experiments on proteins, formal analysis, funding acquisition. J.F.M.: conceived experiments, supervision, resources, funding acquisition. All authors were involved in the writing and editing of the manuscript.Data availability
Characterisation data for all compounds is available in the ESI,† as are further supporting experimental data referenced in the manuscript. Chemical and biochemical experimental methods are also included as are NMR spectra. Mass spec data will be made uploaded to a public repository and will be freely accessible upon publication of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Irish Research Council, GOIPG/2019/3950 (S.H.C) and GOIPG/2021/648 (T.P), and the SSPC, 12/RC/2275_P2 (J.F.M).References
- C. Walsh, Posttranslational modification of proteins: expanding nature's inventory, Roberts and Co. Publishers, Englewood, Colo, 2006 Search PubMed.
- T. L. Foley and M. D. Burkart, Curr. Opin. Chem. Biol., 2007, 11, 12–19 CrossRef CAS.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed.
- A. J. de Graaf, M. Kooijman, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2009, 20, 1281–1295 CrossRef CAS.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- K. Maruyama, T. Ishiyama, Y. Seki, K. Sakai, T. Togo, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2021, 143, 19844–19855 CrossRef CAS.
- P. Wang, Y. Cheng, C. Wu, Y. Zhou, Z. Cheng, H. Li, R. Wang, W. Su and L. Fang, Org. Lett., 2021, 23, 4137–4141 CrossRef CAS PubMed.
- W. Zeng, J. Xue, H. Geng, X. Liu, J. Yang, W. Shen, Y. Yuan, Y. Qiang and Q. Zhu, Biotechnol. Bioeng., 2024, 121, 799–822 CrossRef CAS.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS.
- S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef CAS PubMed.
- Y. Seki, T. Ishiyama, D. Sasaki, J. Abe, Y. Sohma, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2016, 138, 10798–10801 CrossRef CAS PubMed.
- S. Asano, J. T. Patterson, T. Gaj and C. F. Barbas III, Angew. Chem., Int. Ed., 2014, 53, 11783–11786 CrossRef CAS.
- K. C. Tang, J. Cao, L. M. Boatner, L. Li, J. Farhi, K. N. Houk, J. Spangle, K. M. Backus and M. Raj, Angew. Chem., Int. Ed., 2022, 61, e202112107–e202112107 CrossRef CAS PubMed.
- S. M. Hacker, K. M. Backus, M. R. Lazear, S. Forli, B. E. Correia and B. F. Cravatt, Nat. Chem., 2017, 9, 1181–1190 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T.-A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. am Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- L. H. Jones, in Annual Reports in Medicinal Chemistry, ed. R. A. Ward and N. P. Grimster, Academic Press, 2021, vol. 56, pp. 95–134 Search PubMed.
- R. M. Reja, W. Wang, Y. Lyu, F. Haeffner and J. Gao, J. Am. Chem. Soc., 2022, 144, 1152–1157 CrossRef CAS PubMed.
- F. Tekaia, E. Yeramian and B. Dujon, Gene, 2002, 297, 51–60 CrossRef CAS.
- C. Wan, D. Yang, C. Song, M. Liang, Y. An, C. Lian, C. Dai, Y. Ye, F. Yin, R. Wang and Z. Li, Chem. Sci., 2024, 15, 5340–5348 RSC.
- K. Nong, Y.-L. Zhao, S. Yi, X. Zhang, S. Wei and Z.-J. Yao, Bioconjugate Chem., 2024, 35, 286–299 CrossRef CAS PubMed.
- J. Li, J. Chen, Q.-L. Hu, Z. Wang and X.-F. Xiong, Chin. Chem. Lett., 2024, 110126 CrossRef.
- Y. Xie, S. Du, Z. Liu, M. Liu, Z. Xu, X. Wang, J. X. Kee, F. Yi, H. Sun and S. Q. Yao, Angew. Chem., 2022, 134, e202200303 CrossRef.
- K. Shiraiwa, R. Cheng, H. Nonaka, T. Tamura and I. Hamachi, Cell Chem. Biol., 2020, 27, 970–985 CrossRef CAS.
- C. C. Ward, J. I. Kleinman and D. K. Nomura, ACS Chem. Biol., 2017, 12, 1478–1483 CrossRef CAS.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS.
- X. Wang, A. J. Di Pasqua, S. Govind, E. McCracken, C. Hong, L. Mi, Y. Mao, J. Y.-C. Wu, Y. Tomita, J. C. Woodrick, R. L. Fine and F.-L. Chung, J. Med. Chem., 2011, 54, 809–816 CrossRef CAS PubMed.
- J. Chen and Y.-H. Tsai, J. Mol. Biol., 2022, 434, 167424 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- A. A. H. Ahmad Fuaad, F. Azmi, M. Skwarczynski and I. Toth, Molecules, 2013, 18, 13148–13174 CrossRef CAS PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- S. S. van Berkel, M. B. van Eldijk and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 8806–8827 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Org. Lett., 2008, 10, 3097–3099 CrossRef CAS.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648 CrossRef CAS PubMed.
- J. Lohse, L. J. Y. M. Swier, R. C. Oudshoorn, G. Médard, B. Kuster, D. J. Slotboom and M. D. Witte, Bioconjugate Chem., 2017, 28, 913–917 CrossRef CAS PubMed.
- S. F. M. Van Dongen, R. L. M. Teeuwen, M. Nallani, S. S. Van Berkel, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. Van Hest, Bioconjugate Chem., 2009, 20, 20–23 CrossRef CAS PubMed.
- S. Schoffelen, M. B. van Eldijk, B. Rooijakkers, R. Raijmakers, A. J. R. Heck and J. C. M. van Hest, Chem. Sci., 2011, 2, 701–705 RSC.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS.
- A.-S. Yang and B. Honig, J. Mol. Biol., 1993, 231, 459–474 CrossRef CAS.
- J. Pícha, M. Buděšínský, K. Macháčková, M. Collinsová and J. Jiráček, J. Pept. Sci., 2017, 23, 202–214 CrossRef.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS PubMed.
- M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168–1184 CrossRef CAS PubMed.
- Y. U. Moon, R. A. Curtis, C. O. Anderson, H. W. Blanch and J. M. Prausnitz, J. Solution Chem., 2000, 29, 699–718 CrossRef CAS.
- A. Erfani, S. Khosharay, N. H. Flynn, J. D. Ramsey and C. P. Aichele, J. Mol. Liq., 2020, 318, 114067 CrossRef CAS.
- A. K. Gaigalas, J. B. Hubbard, M. McCurley and S. Woo, J. Phys. Chem., 1992, 96, 2355–2359 CrossRef CAS.
- O. Kerscher, R. Felberbaum and M. Hochstrasser, Annu. Rev. Cell Dev. Biol., 2006, 22, 159–180 CrossRef CAS.
- D. K. Stringer and R. C. Piper, Cell Cycle, 2011, 10, 3067–3071 CrossRef CAS.
- W. Li and Y. Ye, Cell. Mol. Life Sci., 2008, 65, 2397–2406 CrossRef CAS PubMed.
- N. C. Taylor, G. Hessman, H. B. Kramer and J. F. McGouran, Chem. Sci., 2020, 11, 2967–2972 RSC.
- N. C. Taylor and J. F. McGouran, Front. Chem., 2019, 7, 914 CrossRef CAS.
- J. F. McGouran, S. R. Gaertner, M. Altun, H. B. Kramer and B. M. Kessler, Chem. Biol., 2013, 20, 1447–1455 CrossRef CAS PubMed.
- D. Flierman, G. J. van der Heden van Noort, R. Ekkebus, P. P. Geurink, T. E. T. Mevissen, M. K. Hospenthal, D. Komander and H. Ovaa, Cell Chem. Biol., 2016, 23, 472–482 CrossRef CAS PubMed.
- S. Eger, M. Scheffner, A. Marx and M. Rubini, J. Am. Chem. Soc., 2010, 132, 16337–16339 CrossRef CAS PubMed.
- N. D. Weikart and H. D. Mootz, ChemBioChem, 2010, 11, 774–777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Including chemical synthesis and characterisation and further biological methods. See DOI: https://doi.org/10.1039/d4ob01094a |
| This journal is © The Royal Society of Chemistry 2024 |