
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of moiramide B derivatives†
Oliver
Andler
 and
Uli
Kazmaier
*
and
Uli
Kazmaier
*
Organic Chemistry, Saarland University, P.O. Box 151150, 66041 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
First published on 6th June 2024
Abstract
Moiramide B is a peptide–polyketide hybrid with a bacterial origin and interesting antibiotic activity. Besides its structurally conserved peptide part, it contains a highly variable fatty acid side chain. We modified this part of the molecule by introducing a terminal alkyne, and we then subjected it to click reactions and Sonogashira couplings. This provided a library of moiramide B derivatives with high and selective in vivo activities against S. aureus.
Introduction
There is absolutely no question that the development of antimicrobial resistance is a global threat to humanity and that the development of new antibiotics with new modes of action is urgently needed to counter this danger. Broad-spectrum antibiotics are essential for the treatment of infections with often unknown pathogens, while narrow-spectrum antibiotics are predestined for the targeted treatment of a known pathogen, as collateral damage to the human microbiome can be avoided more easily.In 1987, Komura et al. described the isolation of andrimid from Enterobacter sp.1 A few years later, Andersen et al. isolated andrimid together with moiramides A–C (Fig. 1) from Pseudomonas fluorescens, with andrimid and moiramide B showing potent in vivo antibiotic activity.2 The biosyntheses of these compounds occur in a nonribosomal peptide synthetase–polyketide synthase hybrid, generating the unsaturated fatty acid (green), the β-phenylalanine (red) and the unusual valinyl-succinimide unit (blue) from valine, glycine and malonyl-CoA.3
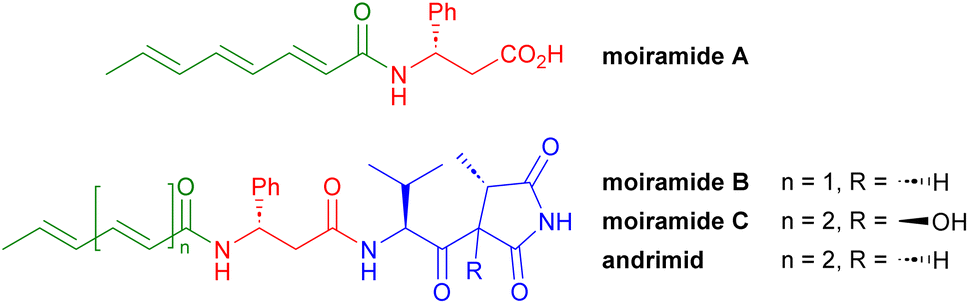 | ||
| Fig. 1 Structures of moiramides A–C and andrimid. | ||
As an inhibitor of the carboxyltransferase component of acetyl-CoA carboxylase (ACC), moiramide B affects fatty acid biosynthesis in bacteria and thus their cell wall assembly.4 The crystal structure of moiramide B bound to carboxyltransferase revealed that the succinimide head group tightly binds to the oxyanion hole of the enzyme in its enol or enolate form, while the valine-derived sidechain fills a hydrophobic pocket. The β-phenylalanine fragment increases the binding affinity by forming additional hydrogen bonds with the target. In contrast, the fatty acid unit does not bind strongly to the target but plays an important role in the in vivo activity, as it is likely responsible for the transport of the compound into the bacterial cell.5
It is therefore not surprising that this class of substances came to the attention of synthetic chemists. Two years after its isolation, Komura et al. accomplished the first total synthesis of andrimid. The succinimide unit was constructed by enolate alkylation of a valine-derived β-ketoamide, followed by ring closure.6 In an alternative approach, Rao et al. acylated racemic 3-methylsuccinimide with the imidazolide of protected valine and separated the obtained diastereomers.7 The first total synthesis of moiramide was reported by Davies et al. in 1996. The stereogenic center in the pyrrolidinedione fragment was constructed via an auxiliary-based enolate alkylation. After cyclization to the succinimide, the valinyl unit was introduced by acylation with the Boc-protected N-carboxyanhydride of L-valine (Boc-Val-NCA).8 Very recently, we reported an alternative approach generating the succinimide subunit via a Pd-catalysed allylic alkylation.9
Since the first total syntheses of andrimid and moiramide B, several derivatives for structure–activity relationship (SAR) studies have been prepared by both chemical synthesis4a,6,10 and combinatorial biosynthesis.11 SAR studies showed that the succinimide fragment allows little variation, whereas the valine side chain can be replaced by other nonpolar groups. Substitution of the β-phenylalanine side chain with other aromatic residues is also possible.
In particular, the fatty acid unit is variable as long as apolar residues are employed. Therefore, this part of the molecule should be best suited for further derivatisations. Recently, moiramide B-based bivalent inhibitors of both the biotin carboxylase and the carboxyltransferase subunit of ACC have been reported, where the fatty acid moiety was replaced by a lipophilic linker.12
Results and discussion
For several years, our group has been involved in the total synthesis of natural products,13 preferentially with anticancer14 and antibiotic activities,15 and we thus became interested in moiramides as well. As previous SAR studies on moiramide B and andrimid derivatives indicated, the fatty acid side chain is structurally highly variable. Therefore, we aimed to develop a synthesis that allows late-stage modifications of this subunit. For the construction of succinimide building block 1, we used the established protocols by Davies et al.8 The conversion of 1 into natural product 4 was accomplished by slightly modified literature procedures, that is, Boc deprotection to hydrochloride 2 and the PyBOP/DMAP-mediated coupling with acid 3 (Scheme 1). The first moiramide derivative, 5, could be obtained in quantitative yield by catalytic hydrogenation of the fatty acid side chain.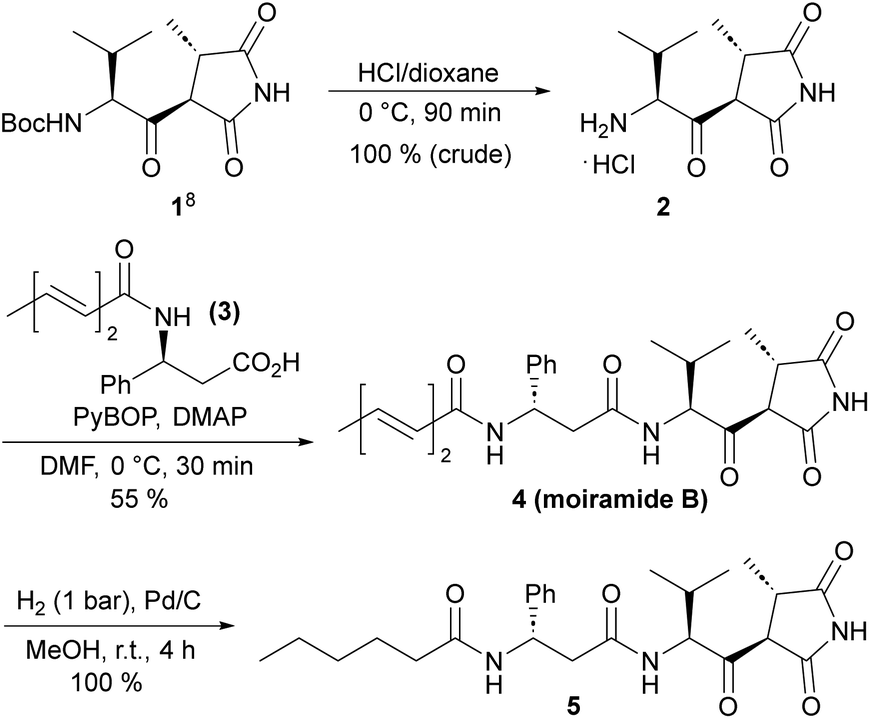 | ||
| Scheme 1 Synthesis of moiramide B and its tetrahydro derivative 5. | ||
Next, we focused on the preparation of a modified fatty acid building block bearing a terminal alkyne (Scheme 2). Starting from nonadiynoic acid 6, we prepared pentafluorophenyl ester 7, which could be smoothly and selectively isomerized to dienoic acid ester 8 using triphenylphosphine as a catalyst.16 Remarkably, the terminal alkyne remained unaffected under these conditions because the isomerization is limited to electron-deficient alkynes.16a The reaction of active ester 8 with (S)-β-phenylalanine methyl ester proceeded slowly but cleanly, yielding amide 9. After saponification of the methyl ester, reaction of 10 with amine 2 provided moiramide derivative 11.
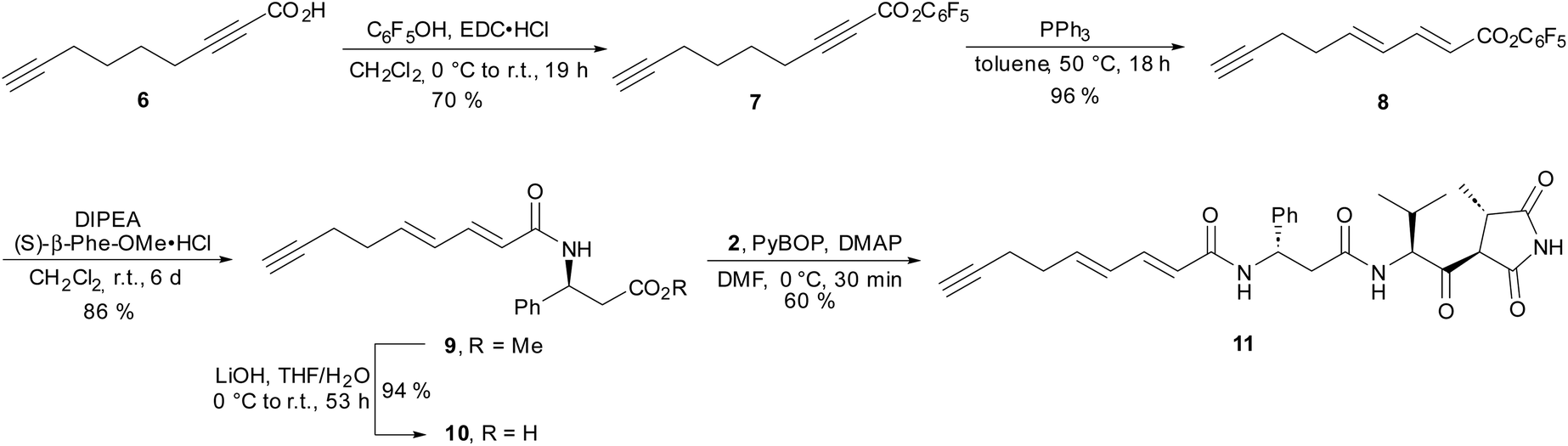 | ||
| Scheme 2 Synthesis of moiramide derivative 11 with a modified fatty acid bearing a terminal alkyne. | ||
Starting from compound 11, we prepared several triazole derivatives 12a–f using copper-catalysed azide–alkyne cycloadditions (CuAAC) using nonpolar (12a–c) as well as polar (12d) azides and linkers bearing terminal carboxylic acid or amino groups (12e–g) (Scheme 3).
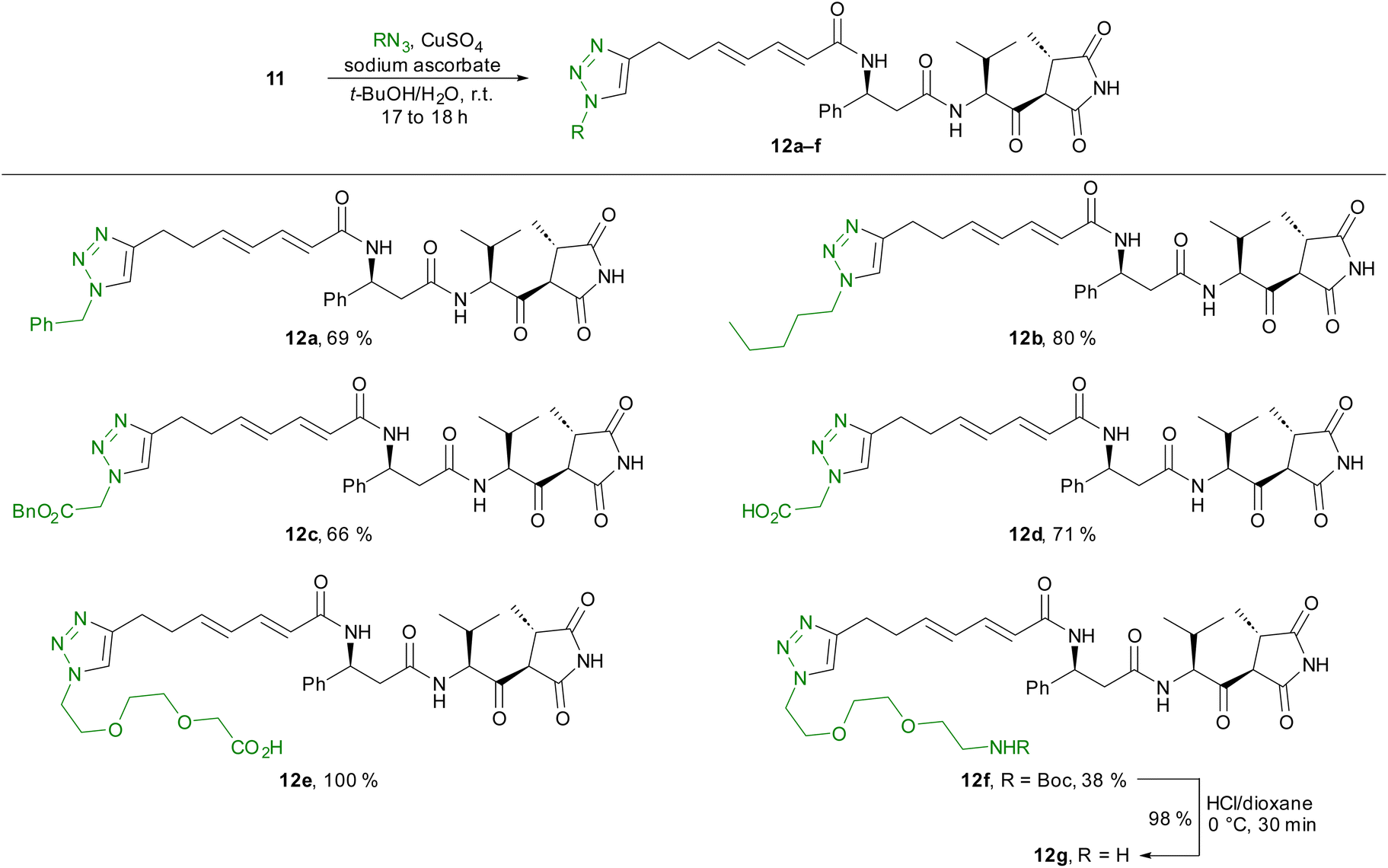 | ||
| Scheme 3 Synthesis of triazole derivatives 12a–gvia CuAAC. | ||
As an additional modification, we subjected alkyne 11 also to Sonogashira couplings using various aryl iodides. If we used the classical conditions [Pd(OAc)2/PPh3/CuI or PdCl2(PPh3)2/CuI as the catalyst and Et2NH or NEt3 as the solvent],17 we observed no conversion, at least at room temperature. After increasing the reaction temperature to 50 °C, full consumption of the starting material was observed but we could isolate only trace amounts of the desired cross coupling products. Unfortunately, switching to a copper-free protocol (XPhos Pd G3 as the catalyst, Cs2CO3 as base and MeCN as the solvent)18 did not increase the yield or the purity of the Sonogashira coupling products either. We assumed that the problems stemmed from the β-ketoamide moiety, which may be coordinating to the palladium or copper and/or leading to side reactions under the basic conditions.
To circumvent these difficulties, we then performed the cross-coupling reaction at an earlier stage without the β-ketoamide in place. While the conversion of carboxylic acid 10 was slow due to the formation of an insoluble ammonium carboxylate, the Sonogashira coupling of methyl ester 9 with several aryl iodides succeeded (Scheme 4). Unsubstituted iodobenzene (13a), as well as electron-poor (13b–c) and electron-rich (13d–f) para-substituted aryl iodides, could be employed in this reaction. A free aniline or phenol was also tolerated, but the yields decreased in these examples because of competing alkyne homodimerisation.
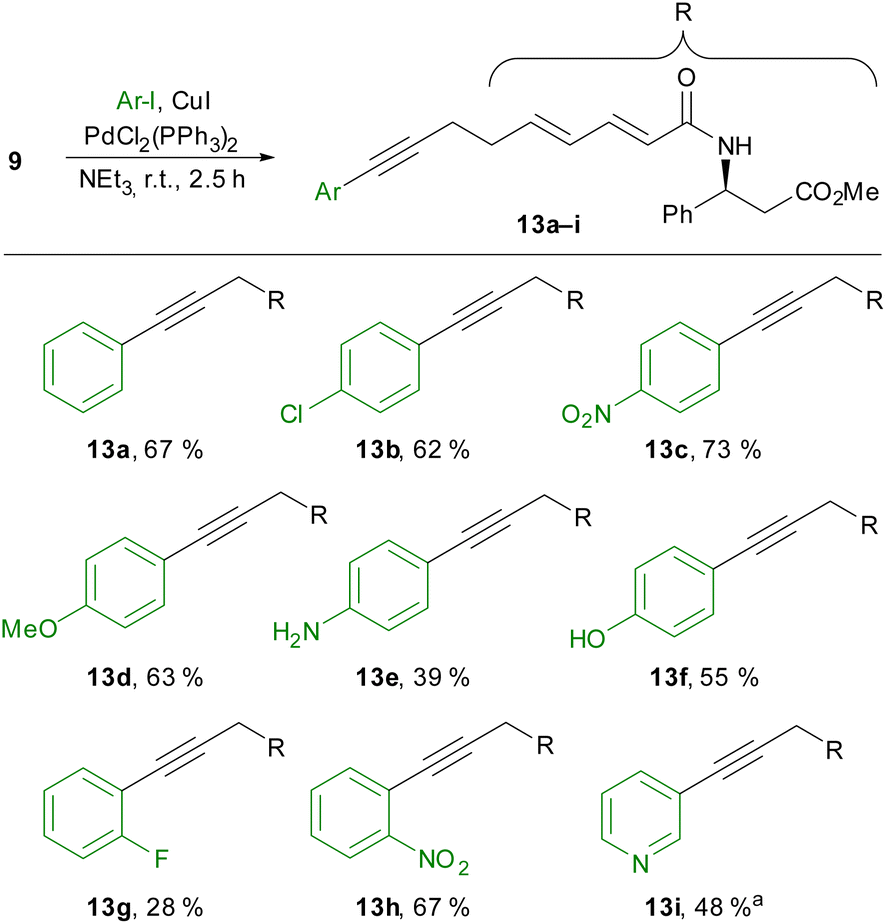 | ||
Scheme 4 Sonogashira coupling of alkyne 9 with various aryl iodides. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was performed for 2 h at 60 °C. The reaction was performed for 2 h at 60 °C. | ||
In principle, ortho-substituted aryl iodides could also be used (13g,h), although a rather low yield was obtained in the case of 1-fluoro-2-iodobenzene (13g). With 3-iodopyridine, we could also employ a heteroaryl iodide if the reaction temperature was increased to 60 °C (13i).
We completed the synthesis of moiramide derivatives 14a-I by saponification of the methyl ester, followed by coupling with amine 2 (Scheme 5). In most cases, we obtained the desired coupling products in good yields. Notably, amide coupling in the presence of a free aniline succeeded without difficulty (14e). In contrast, crude product 14f bearing a free phenol was initially impure and required further purification via preparative HPLC. In the case of ortho-fluorophenyl and 3-pyridyl derivatives 14g and 14f, the yields decreased significantly. Nevertheless, these products could also be isolated in sufficient quantity for biological testing.
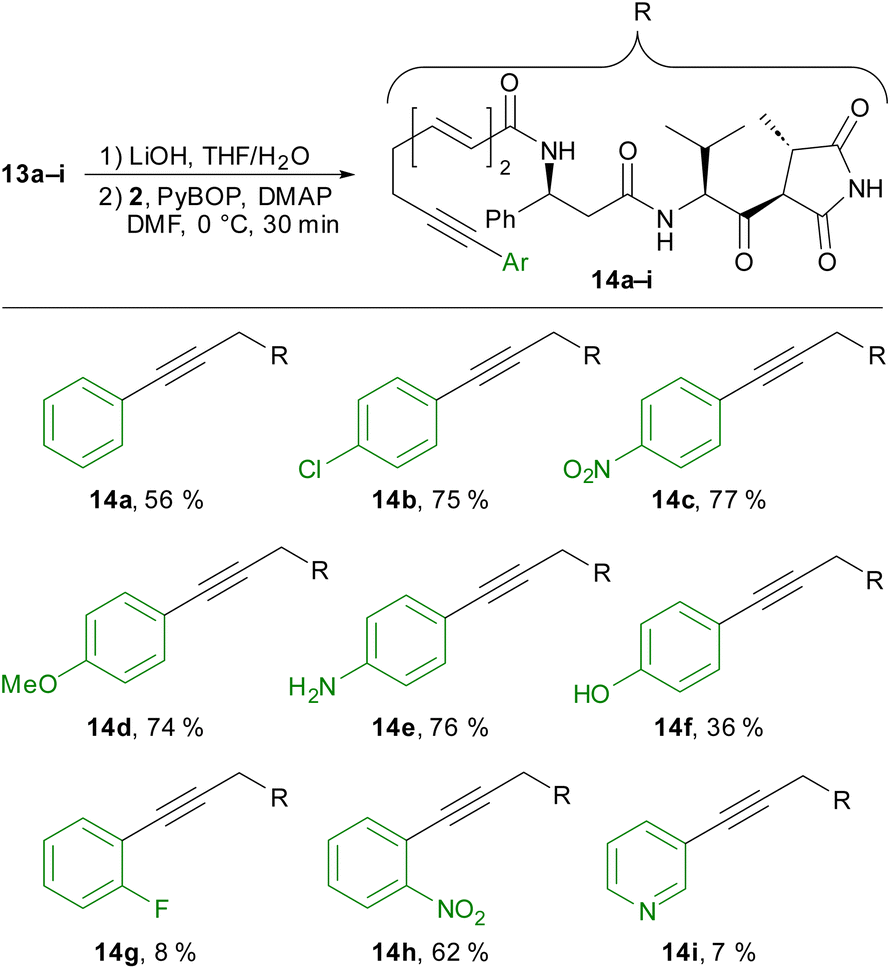 | ||
| Scheme 5 Synthesis of arylated moiramide derivatives 14a–i. | ||
Next, we measured the antibiotic activities of all compounds against B. subtilis and S. aureus as Gram-positive bacteria and E. coli acrB as Gram-negative bacteria (Table 1). Although moiramide B (4) was highly active against all bacterial strains tested, the MIC (minimum inhibitory concentration) values increased by a factor of 4–16 for the saturated derivative 5. To our delight, alkynyl derivative 11 also displayed antibiotic activity against all three strains, although with increased MIC values compared with the natural product. Introducing a triazole moiety was poorly tolerated independent of the substituent on the triazole. None of the triazole derivatives showed any significant activity.
| compound | S. aureus Newman | B. subtilis [DSM-10] | E. coli acrB [JW0451-2] | HepG2 |
|---|---|---|---|---|
| 4 (moiramide B) | 1–2 | 1–2 | 4 | >37 |
| 5 | 4–8 | 16 | 32 | >37 |
| 11 | 4 | 4–8 | 16–32 | >37 |
| 12a | >64 | >64 | >64 | >37 |
| 12b | >64 | >64 | >64 | >37 |
| 12c | 64 | 64 | >64 | >37 |
| 12d | >64 | >64 | >64 | >37 |
| 12e | >64 | >64 | >64 | >37 |
| 12f | >64 | >64 | >64 | >37 |
| 12g | >64 | >64 | >64 | >37 |
| 14a | 1 | 2–4 | >64 | >37 |
| 14b | 2 | 4 | >64 | >37 |
| 14c | 2 | 8–16 | >64 | >37 |
| 14d | 2 | 16 | >64 | >37 |
| 14e | 1 | 8–16 | 64 | >37 |
| 14f | 1 | 4 | 32–64 | >37 |
| 14g | 2 | 8 | 64 | >37 |
| 14h | 2 | 8–16 | 32–64 | >37 |
| 14i | 8 | >64 | >64 | >37 |
Better results were obtained with the aryl-substituted derivatives. For these compounds, the activities against Gram-positive bacteria were similar to those of the natural products and significantly higher than for the unsubstituted alkyne 11.
The phenyl, para-amino and para-hydroxy derivatives, 14a, 14e and 14f, respectively, were notable with MIC values of 1 μg mL−1 against S. aureus. All derivatives 14a–i showed potent activity against S. aureus, and a little weaker activity against B. subtilis. Interestingly, little or almost no activities was observed against E. coli, in contrast to moiramid B. Also no activity was observed against a wide range of Gram-negative and -positive bacteria as well as fungi and yeasts such as Candida albicans (for details see ESI†). Additionally, none of the compounds tested showed cytotoxicity against HepG2 cells.
Conclusions
For the synthesis of moiramide derivatives, we prepared a central building block 9, with a modified moiramide fatty acid side chain via triphenylphosphine-catalysed regioselective isomerisation. Starting from this building block, we prepared various moiramide derivatives in only a few steps. Triazole derivatives could be prepared from moiramide analogue 11via CuAAC, but showed almost no antibiotic activity. In contrast, various aryl-substituted derivatives prepared via Sonogashira coupling were highly active and selective against S. aureus. This late-stage modification should allow the synthesis of moiramide B libraries to further optimize antibacterial activities and physicochemical properties.Data availability
Copies of 1H and 13C NMR spectra, GC chromatograms and experimental details.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Saarland University and the DFG (Bruker Neo 500 – 447298507) is gratefully acknowledged. Thanks to Jennifer Herrmann, Alexandra Amann and Sari Rasheed from Helmholtz Institute for Pharmaceutical Research Saarland (HIPS) for measuring MIC and IC50 values.References
- A. Fredenhagen, S. Y. Tamura, P. T. M. Kenny, H. Komura, Y. Naya and K. Nakanishi, J. Am. Chem. Soc., 1987, 109, 4409–4411 CrossRef CAS.
- (a) J. Needham, M. T. Kelly, M. Ishige and R. J. Andersen, J. Org. Chem., 1994, 59, 2058–2063 CrossRef CAS; (b) M. P. Singh, M. J. Mroczenski-Wildey, D. A. Steinberg, R. J. Andersen, W. M. Maiese and M. Greenstein, J. Antibiot., 1997, 50, 270–273 CrossRef CAS.
- (a) M. Jin, M. A. Fischbach and J. Clardy, J. Am. Chem. Soc., 2006, 128, 10660–10661 CrossRef CAS PubMed; (b) N. A. Magarvey, P. D. Fortin, P. M. Thomas, N. L. Kelleher and C. T. Walsh, ACS Chem. Biol., 2008, 3, 542–554 CrossRef CAS PubMed; (c) X. Liu, P. D. Fortin and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13321–13326 CrossRef CAS PubMed; (d) F. Ishikawa, H. Sugimoto and H. Kakeya, ChemBioChem, 2016, 17, 2137–2142 CrossRef CAS PubMed; (e) M. A. Matilla, V. Nogellova, B. Morel, T. Krell and G. P. C. Salmond, Environ. Microbiol., 2016, 18, 3635–3650 CrossRef CAS PubMed; (f) H. L. Messiha, K. A. P. Payne, N. S. Scrutton and D. Leys, ACS Synth. Biol., 2021, 10, 228–235 CrossRef CAS PubMed.
- (a) C. Freiberg, N. A. Brunner, G. Schiffer, T. Lampe, J. Pohlmann, M. Brands, M. Raabe, D. Häbich and K. Ziegelbauer, J. Biol. Chem., 2004, 279, 26066–26073 CrossRef CAS PubMed; (b) C. Freiberg, H. P. Fischer and N. A. Brunner, Antimicrob. Agents Chemother., 2005, 49, 749–759 CrossRef CAS PubMed.
- M. A. Silvers, S. Pakhomova, D. B. Neau, W. C. Silvers, N. Anzalone, C. M. Taylor and G. L. Waldrop, Biochemistry, 2016, 55, 4666–4674 CrossRef CAS PubMed.
- W. McWhorter, A. Fredenhagen, K. Nakanishi and H. Komura, J. Chem. Soc., Chem. Commun., 1989, 299–301 RSC.
- A. V. Rama Rao, A. K. Singh and C. V. N. S. Varaprasad, Tetrahedron Lett., 1991, 32, 4393–4396 CrossRef.
- (a) D. J. Dixon and S. G. Davies, Chem. Commun., 1996, 1797–1798 RSC; (b) S. G. Davies and D. J. Dixon, J. Chem. Soc., Perkin Trans. 1, 1998, 2635–2644 RSC.
- C. Prudel and U. Kazmaier, Synlett, 2024 DOI:10.1055/s-0042-1751578 , in press.
- (a) J. Pohlmann, T. Lampe, M. Shimada, P. G. Nell, J. Pernerstorfer, N. Svenstrup, N. A. Brunner, G. Schiffer and C. Freiberg, Bioorg. Med. Chem. Lett., 2005, 15, 1189–1192 CrossRef CAS PubMed; (b) C. Freiberg, J. Pohlmann, P. G. Nell, R. Endermann, J. Schuhmacher, B. Newton, M. Otteneder, T. Lampe, D. Häbich and K. Ziegelbauer, Antimicrob. Agents Chemother., 2006, 50, 2702–2712 CrossRef PubMed.
- B. S. Evans, Y. Chen, W. W. Metcalf, H. Zhao and N. L. Kelleher, Chem. Biol., 2011, 18, 601–607 CrossRef CAS PubMed.
- (a) M. A. Silvers, G. T. Robertson, C. M. Taylor and G. L. Waldrop, J. Med. Chem., 2014, 57, 8947–8959 CrossRef CAS PubMed; (b) M. T. Cifone, Y. He, R. Basu, N. Wang, S. Davoodi, L. A. Spagnuolo, Y. Si, T. Daryaee, C. E. Stivala, S. G. Walker and P. J. Tonge, J. Med. Chem., 2022, 65, 16510–16525 CrossRef CAS PubMed.
- (a) A. Ullrich, Y. Chai, D. Pistorius, Y. A. Elnakady, J. E. Herrmann, K. J. Weissman, U. Kazmaier and R. Müller, Angew. Chem., 2009, 121, 4486–4489 ( Angew. Chem. Int. Ed. , 2009 , 47 , 4422–4425 ) CrossRef; (b) L. Junk and U. Kazmaier, Angew. Chem., 2018, 130, 11602–11606 ( Angew. Chem. Int. Ed. , 2018 , 57 , 11432–11435 ) CrossRef; (c) L. Junk and U. Kazmaier, J. Org. Chem., 2019, 84, 2489–2500 CrossRef CAS PubMed.
- (a) L. Karmann, K. Schulz, J. Herrmann, R. Müller and U. Kazmaier, Angew. Chem., 2015, 127, 4585–4590 ( Angew. Chem. Int. Ed. , 2015 , 54 , 4502–4507 ) CrossRef; (b) J. Gorges and U. Kazmaier, Org. Lett., 2018, 20, 2033–2036 CrossRef CAS PubMed; (c) O. Andler and U. Kazmaier, Org. Biomol. Chem., 2021, 19, 4866–4870 RSC; (d) M. Tost, O. Andler and U. Kazmaier, Eur. J. Org. Chem., 2021, 6459–6471 CrossRef CAS.
- (a) P. Barbie and U. Kazmaier, Org. Lett., 2016, 18, 204–207 CrossRef CAS PubMed; (b) U. Kazmaier and L. Junk, Mar. Drugs, 2021, 19, 446 CrossRef CAS PubMed; (c) O. Andler and U. Kazmaier, Org. Lett., 2022, 24, 2541–2545 CrossRef CAS PubMed; (d) J. Greve, A. Mogk and U. Kazmaier, Mar. Drugs, 2022, 20, 632 CrossRef CAS PubMed.
- (a) B. M. Trost and U. Kazmaier, J. Am. Chem. Soc., 1992, 114, 7933–7935 CrossRef CAS; (b) U. Kazmaier, Chem. Commun., 1997, 2305–2306 RSC; (c) P. Servatius, T. Stach and U. Kazmaier, Eur. J. Org. Chem., 2019, 3163–3168 CrossRef CAS.
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 50, 4467–4470 CrossRef; (b) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS; (c) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993–5996 ( Angew. Chem. , 2003 , 115 , 6175–6178 ) CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra, GC chromatograms and experimental details. See DOI: https://doi.org/10.1039/d4ob00856a |
| This journal is © The Royal Society of Chemistry 2024 |