
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of the monomeric counterpart of Marinomycin A and B†
Frederic
Ballaschk
a,
Kathrin
Bensberg
 a,
Benedikt
Crone
bc,
Stefan F.
Kirsch
*a and
Helge
Menz
bd
a,
Benedikt
Crone
bc,
Stefan F.
Kirsch
*a and
Helge
Menz
bd
aOrganic Chemistry, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: sfkirsch@uni-wuppertal.de
bDepartment Chemie, Technische Universität München, Lichtenbergstr. 4, 85747 Garching, Germany
cBASF SE, Carl-Bosch-Str. 38, 67056 Ludwigshafen am Rhein, Germany
dPharmpur GmbH, Messerschmittring 33, 86343 Königsbrunn, Germany
First published on 7th June 2024
Abstract
The synthesis of polyketide natural products has been a captivating pursuit in organic chemistry, with a particular focus on selectively introducing 1,3-polyol units. Among these natural products, Marinomycins A–D have garnered substantial interest due to their exceptional structural features and potent cytotoxicity. In this paper, we present a novel approach for synthesising the monomeric counterparts of Marinomycin A and B. Our method employs a previously established iterative cycle in conjunction with a standardised polyketide building block. Through this strategy, we showcase a promising pathway towards total and partial syntheses of these intriguing natural products.
Introduction
The Marinomycins, a family of four compounds referred to as Marinomycin A–D (1–4, Fig. 1), were initially discovered in 2006 by Fenical et al. from a marine actinomycete, Marinospora, on the coast of La Jolla, California.1 These compounds have shown antibiotic properties against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecium (VREF), with MIC values ranging from 0.1 to 0.6 μM, as well as potent anti-cancer activity against NCI's 60 cancer cell line panel, with LC50 values ranging from 0.2 to 2.7 μM, displaying especially high selectivity against six of the eight melanoma cell lines.1 Marinomycin A, B, and C share a similar structure, differing only in the conformation of the double bond adjacent to the aromatic cores (Δ8–9), while Marinomycin D possesses one extra carbon in its backbone. Notably, the macrocycles Marinomycin A (1) and B (2) feature a unique structural motif with a C2 symmetry. Fenical et al. demonstrated that under ambient light irradiation, a solution of pure Marinomycin A in methanol undergoes complete isomerisation at double bond Δ8–9, forming a mixture of Marinomycins A–C in less than an hour. Harsh UV irradiation, as shown by Evans, Mackenzie, and Goss in 2019, vastly accelerates the isomerisation process of Marinomycins A–D to below 60 seconds, while encapsulation can extend its half-life.2 The process of isomerisation as well as its complex macrocyclic polyketide structure make the synthesis of Marinomycin natural products a rather challenging task.1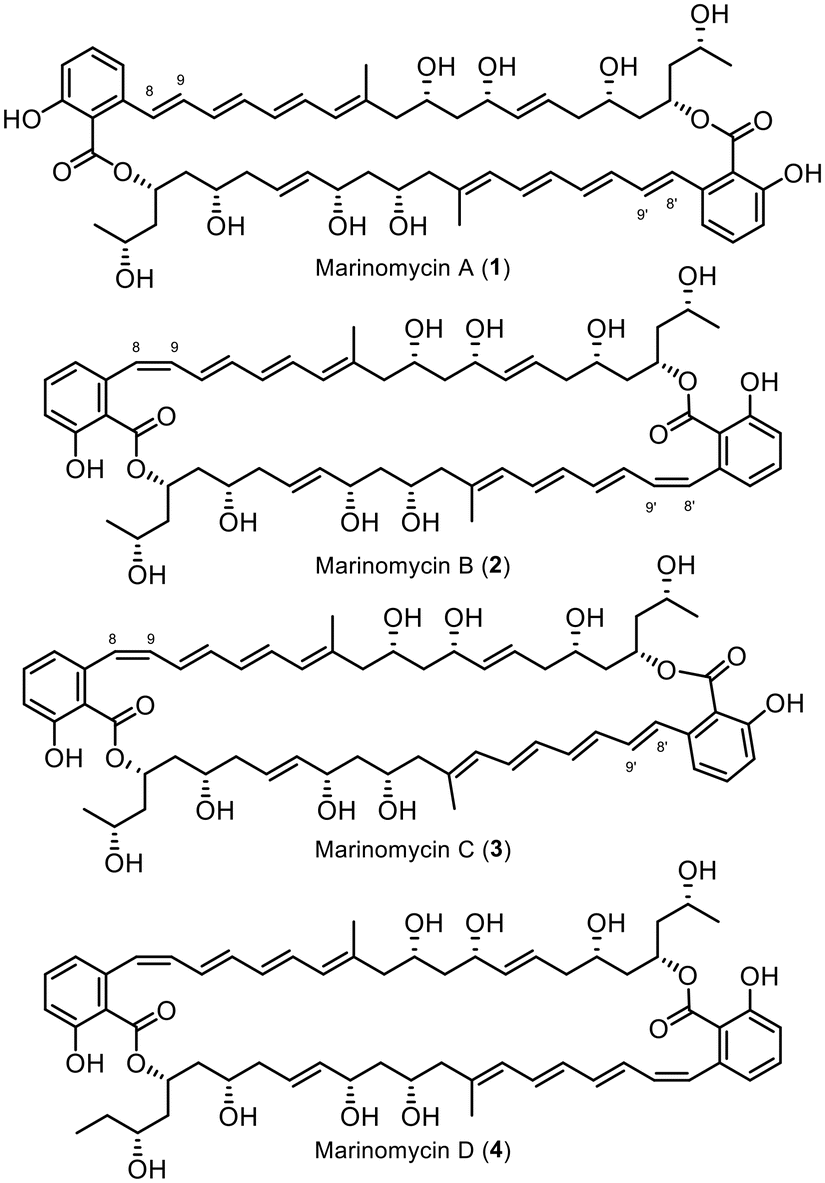 | ||
| Fig. 1 Structures of Marinomycins A–D (1–4). | ||
To date, only three groups have successfully synthesised Marinomycin A, including Nicolaou et al. in 2006,3,4 Evans et al. in 2012,5 and Hatakeyama et al. in 2014.6
While Nicolaou et al. and Evans et al. employed consecutive construction strategies to synthesise the target molecule, Hatakeyama et al. chose a highly convergent direct dimerisation strategy of the monomeric compound. In addition to the completed total syntheses of Marinomycin A, Cossy et al. presented two synthetic approaches for the monomeric counterpart in 20077 and 2009.8 Rajesh et al. further demonstrated a synthetic route for the C13–C28 fragment.9
Results and discussion
Our approach to synthesise Marinomycin A and B revolves around utilising key monomer 5 that could be cyclised through a direct dimerisation via Sonogashira cross coupling (Scheme 1).10,11 The triple bond in the resulting dimer (C-8–9 in 1 and 2) would allow for selective E- or Z-reduction, thereby enabling access to both natural products.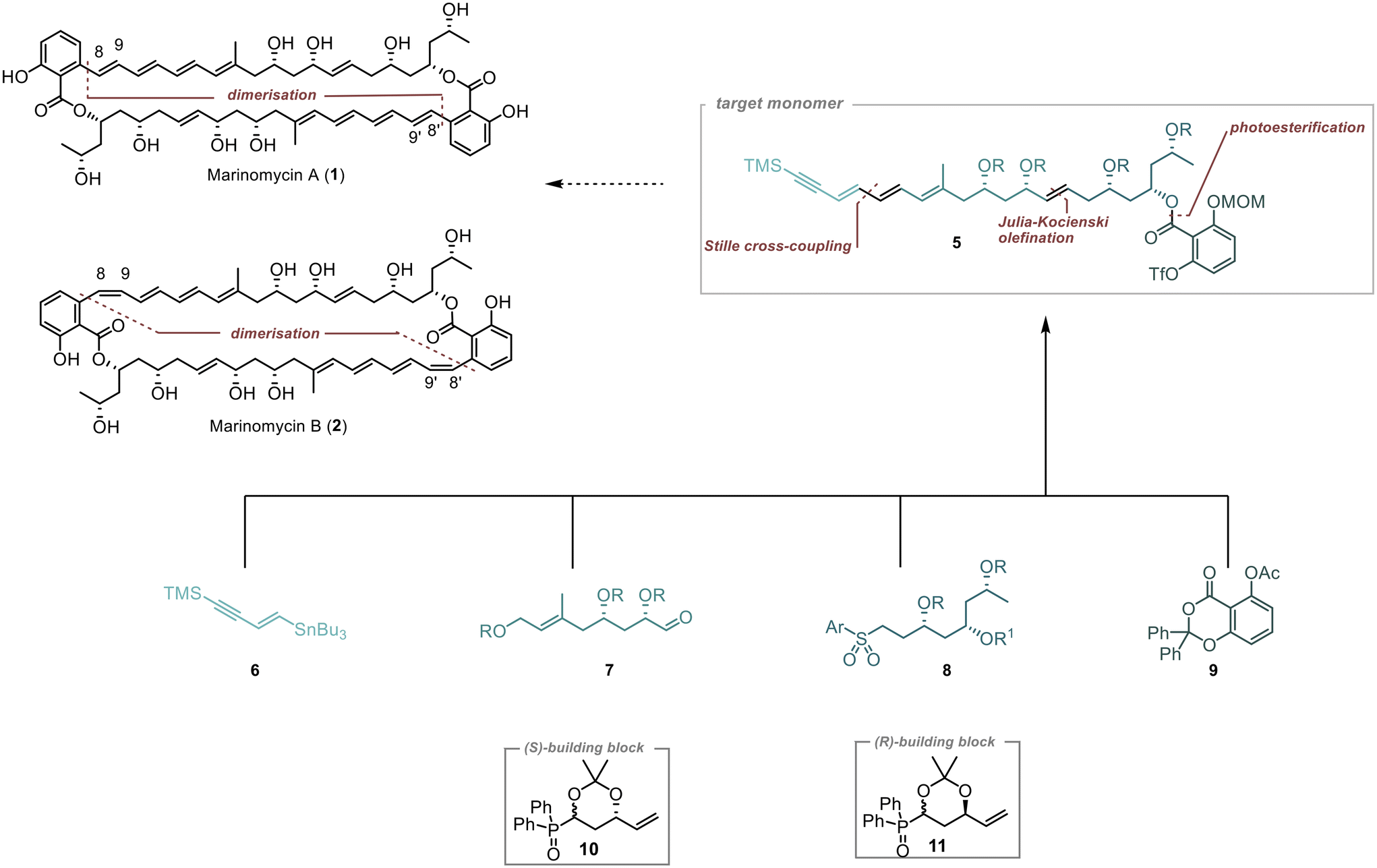 | ||
| Scheme 1 Planned approach for the synthesis of 1 and 2, R = TBS, R1 = PMB, Ar = phenyltetrazole. | ||
To form Monomer 5, we utilised four building blocks: 6–9. Fragment 6, which contains the terminal alkyne and a double bond with a tributylstannyl moiety, allows for its connection to 7via Stille cross-coupling.12 The polyol fragments 7 and 8 should be linked together using Julia–Kocienski olefination.13–15 The introduction of the densely substituted aromatic core was planned through a photochemical esterification method following the protocol established by de Brabander et al.16–18
1,3-Polyols were introduced on the basis of our previously described strategies.19,20 we planned to employ the chiral building blocks 10 and 11 in an iterative fashion for the construction of the polyol-containing fragments 7 and 8.21,22 This strategic approach facilitates the controlled introduction of two stereogenic centres, resulting in an efficient and streamlined synthesis.
Our synthetic journey began with the assembly of aromatic core 9 from 15 (Scheme 2). Initially, we synthesised the benzodioxinone scaffold 16 through a reaction involving benzophenone, thionyl chloride, and DMAP. The obtained product was subsequently transformed into the final fragment 9 using acetic anhydride, DMAP, and pyridine, with an overall yield of 27%.
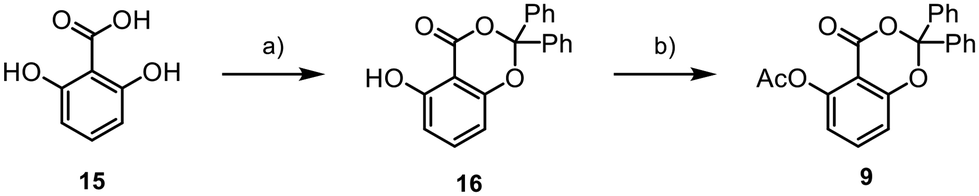 | ||
Scheme 2 Synthesis of 9; (a) Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1.3 equiv.), SOCl2 (1.3 equiv.), DMAP (5 mol%), 0 °C to rt, 18 h, (DME), 30%; (b) Ac2O (1.8 equiv.), pyridine (5 equiv.), DMAP (10 mol%), 0 °C to rt, 2 h, (DCM), 89%. O (1.3 equiv.), SOCl2 (1.3 equiv.), DMAP (5 mol%), 0 °C to rt, 18 h, (DME), 30%; (b) Ac2O (1.8 equiv.), pyridine (5 equiv.), DMAP (10 mol%), 0 °C to rt, 2 h, (DCM), 89%. | ||
Fragment 6 was obtained through synthesis of vinylstannane 17 starting from propargyl alcohol 12, which underwent a palladium-catalysed hydrostannylation (Scheme 3).23,24 Through MnO2-mediated oxidation Enal 18 was obtained in a high yield of 97%,25,26 followed by conversion into the corresponding alkyne 19via Colvin rearrangement.27 Lastly, terminal alkyne 19 was TMS-protected, which gave the product 6 in 31% overall yield.
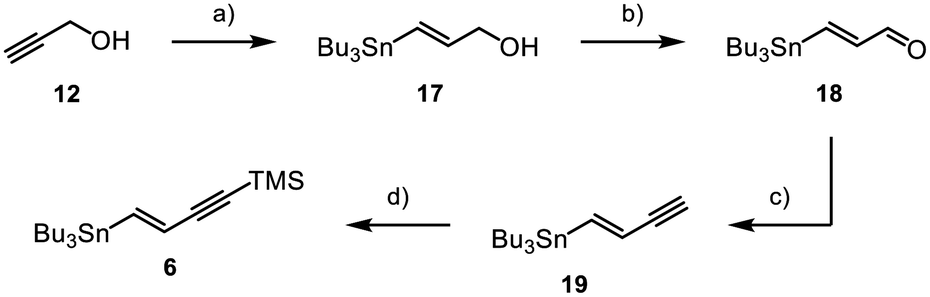 | ||
| Scheme 3 Synthesis of 6; (a) Pd2(dba)3 (0.25 mol%), PCy3 (2 mol%), Bu3SnH (1.15 equiv.), 0 °C, 3 h, (DCM), 56%; (b) MnO2 (20 equiv.), rt, 18 h, (acetone), 97%; (c) (i) nBuLi (1.35 equiv.), TMSCHN2 (1.5 equiv.), −78 °C, 30 min, (ii) aldehyde 18 (1 equiv.), −78 °C to 0 °C, 90 min, (THF), 59%; (d) (i) nBuLi (1.2 equiv.), −78 °C, 1 h, (ii) TMSCl (1.2 equiv.), −78 °C, 1 h, (iii) −78 °C to rt, 16 h, (THF), 97%. | ||
With the successful preparation of the first two fragments 6 and 9, we were now poised to tackle the synthesis of the polyol fragments 7 and 8.
The synthesis of fragment 7 started with the TBS-protection of (Z)-but-2-ene-1,4-diol (13) and subsequent ozonolysis of the double bond. Reaction with the commercially available Wittig reagent 21 then afforded aldehyde 22 in 94% yield with excellent selectivity of E/Z > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 4).28,29
:1 (Scheme 4).28,29
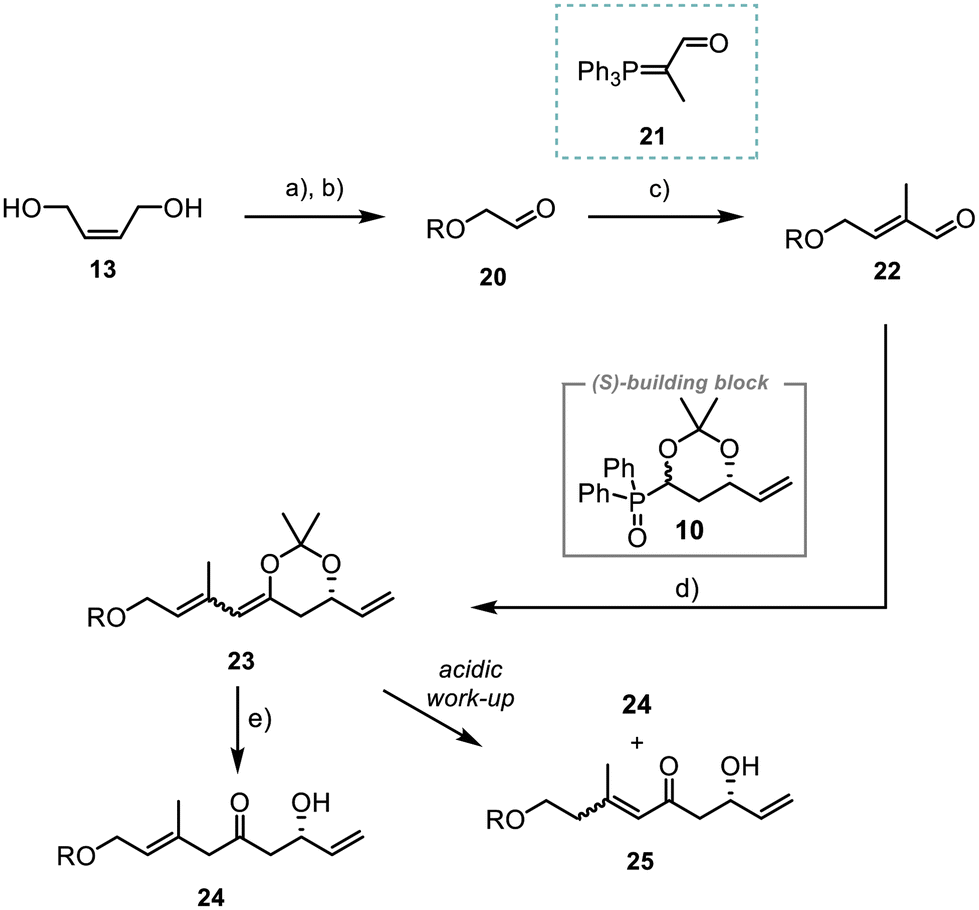 | ||
| Scheme 4 Approach for the introduction of the first two stereogenic centres; R = TBS; (a) imidazole (4.8 equiv.), TBSCl (2.5 equiv.), rt, 16 h, (DCM), quant.; (b) (i) O3, −78 °C, 20 min, (ii) PPh3 (1.17 equiv.), rt, 1.5 h, (DCM), 81%; (c) 21 (1.2 equiv.), rt, 60 h, (benzene), 94%; (d) (i) DIPA (1.15 equiv.), nBuLi (1.15 equiv.), −78 °C, 15 min, (ii) 10 (1 equiv.), −78 °C, 60 min, (iii) aldehyde 22 (1.3 equiv.), −78 °C to rt, 90 min, (iv) KOtBu (1.2 equiv.), rt, 60 min, (THF), 83%, (e) CeCl3 (2 equiv.), oxalic acid (25 mol%), H2O (2 equiv.), −78 °C, 16 h, (THF), 63%. | ||
Next, the unsaturated aldehyde 22 was reacted with the polyketide building block 10, using a Horner–Wittig reaction previously established by us.21,22 The acetonide-protected alcohol 23 was obtained in 83% yield (dr 1:1). However, attempts to remove the acetonide during an acidic work-up gave a mixture of the desired compound 24 and the conjugated system 25. Alternative tests utilising various Lewis acids or sterically demanding proton acids for the acetonide deprotection (ESI, Table 1 entries 1–3†)30–32 led to the formation of complex mixtures. Best results were obtained with a variation of the methodology by Bai et al.33 Further optimisation showed that the use of anhydrous CeCl3 and addition of water improved the yield and 24 was obtained in 63% (ESI, Table 1, entry 7†).
Consequently, we performed a syn-selective Narasaka–Prasad reduction with β-hydroxyketone 24 to yield 1,3-diol 26 with excellent yield and diastereoselectivity (>20:1) (Scheme 5).34–37 TBS-protection of the diol gave substrate 27 under standard conditions. However, all our attempts to convert terminal alkene 27 into aldehyde 7 were unsuccessful.
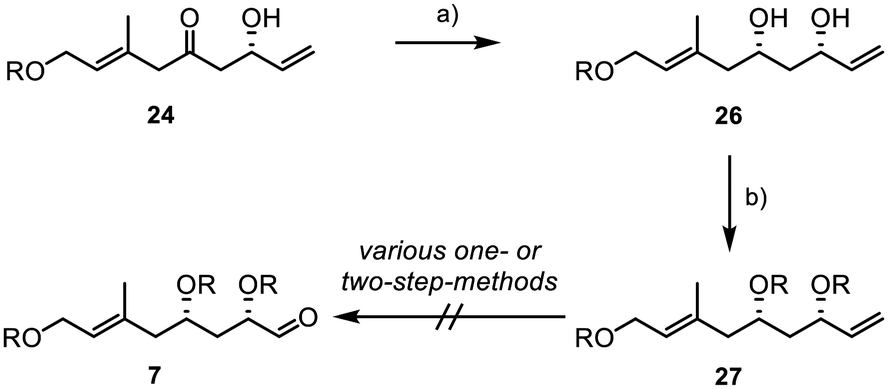 | ||
| Scheme 5 Unsuccessful attempts to introduce the aldehyde function of fragment 7; R = TBS; (a) Et2BOMe (1.2 equiv.), NaBH4 (1.1 equiv.), −78 °C, 2 h, (THF), 99%, dr > 20:1; (b) imidazole (4.8 equiv.), TBSCl (2.5 equiv.), 0 °C to rt, 2 h, (DMF), 90%. | ||
Despite testing a broad range of reaction conditions, the internal double bond was more reactive, and the direct conversion of the terminal double bond was never achieved. Also, several two-step sequences, involving the introduction of a 1,2-diol followed by its conversion into the aldehyde through glycol cleavage, did not yield the desired product. As a result, we decided to replace the TBS groups with an acetonide (Scheme 6).
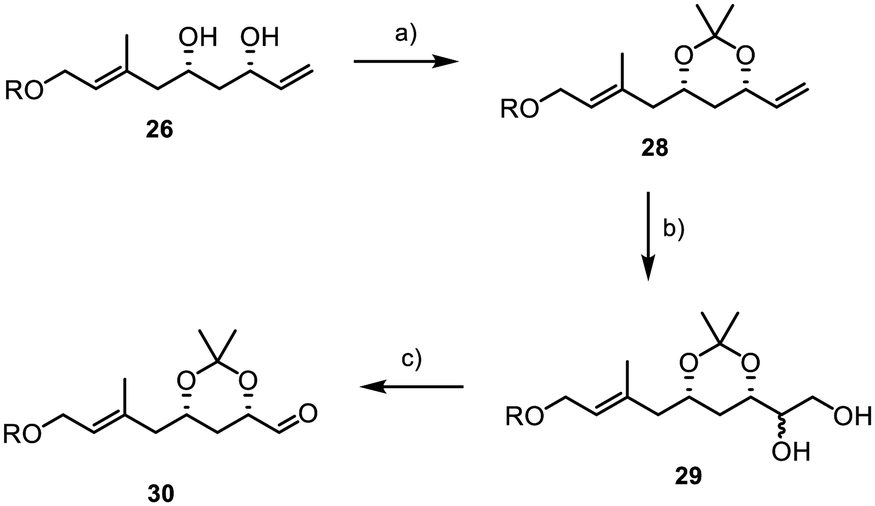 | ||
| Scheme 6 Introduction of the aldehyde moiety; R = TBS; (a) 2,2-dimethoxypropane (25 equiv.), PPTS (10 mol%), 330 mbar, 45 °C, 2 h, 98%; (b) (i) B2pin2 (2 equiv.), Cs2CO3 (30 mol%), 70 °C, 16 h, (ii) NaOH (6 equiv.), H2O2 (23.4 equiv.), 0 °C to rt, 4 h, (THF/MeOH, 3:1), dr 1:1; (c) NaIO4 (2.2 equiv.), rt, 10 min, (THF/water, 1:1), 85% over two steps. | ||
We were delighted to find that under dihydroxylation conditions by Morken et al., previously unsuccessful with TBS protected diol 27, acetonide 28 was successfully and selectively converted to the 1,2-diol 29.38 Treating 28 with B2pin2 and catalytic amounts of caesium carbonate led to the formation of diol 29 with a diastereomeric ratio of 1:1. Subsequently, the diol was reacted with sodium periodate in a mixture of THF and water, affording the desired aldehyde 30 in 85% yield over two steps. However, it was later found that retaining the acetonide protecting group was not feasible, as subsequent results showed when subjecting 30 and 8 to a Julia–Kocienski olefination, where the acetonide function presented significant challenges in terms of selectivity and yield. Disappointingly, we were also unable to directly convert acetonide 30 into the desired TBS-protected substrate 7 due to the formation of various by-products resulting from the presence of the free hydroxyl groups and the aldehyde during deprotection.39 In consequence, this necessitated a complex and multi-step deprotection–reprotection sequence involving the secondary alcohols to obtain 7, as outlined in Scheme 7. First, the aldehyde function was reduced to primary alcohol 31, followed by the installation of a PMB group to yield 32 (Scheme 7). Subsequently, reprotection with TBS chloride was carried out to form 33. Finally, PMB deprotection and oxidation with IBX were performed, resulting in the successful acquisition of the desired aldehyde 7.
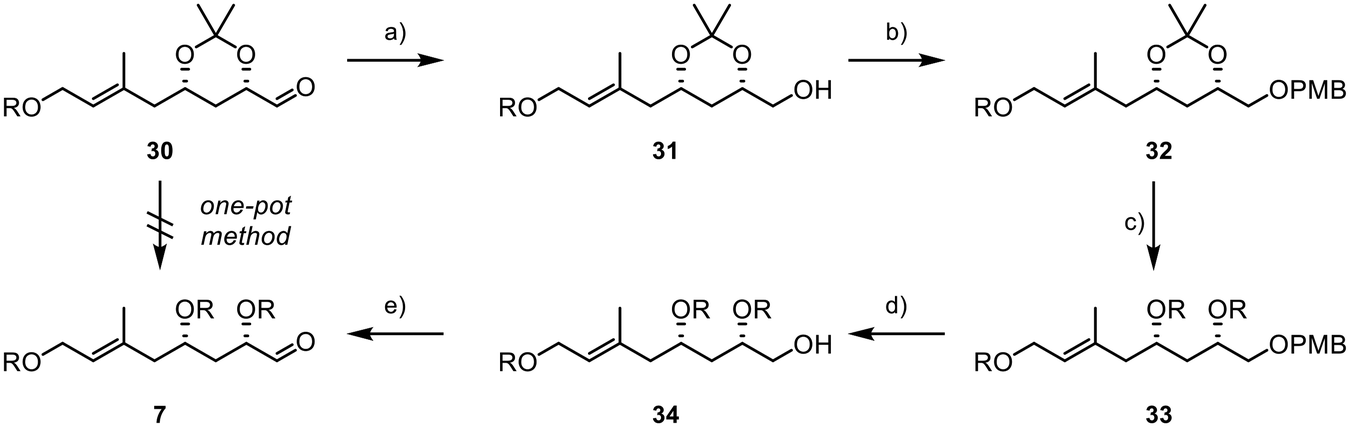 | ||
| Scheme 7 Final steps in the synthesis of fragment 7; R = TBS; (a) DIBAL-H (1.3 equiv.), −78 °C, 1.5 h, (DCM), 92%; (b) NaH (1.2 equiv.), PMBCl (1.2 equiv.), TBAI (20 mol%), 0 °C to rt, 18 h, (DMF), 98%; (c) (i) PPTS (25 mol%), rt, 4 h, (MeOH),44 (ii) TBSCl (6 equiv.), imidazole (10 equiv.), rt, 16 h, (DMF), 86% (2 steps); (d) DDQ (1.5 equiv.), 0 °C, 3 h, (DCM/pH7-buffer, 1:1), 64%; (e) IBX (1.5 equiv.), rt, 16 h, (DMSO), 79%. | ||
The synthesis of fragment 8 commenced with (R)-methyl lactate 14 as a commercially available chiral starting material having the needed pre-existing stereogenic centre (Scheme 8). Installation of a TBS group at the free hydroxyl function and subsequent reduction of the ester to the aldehyde yielded the desired compound 36 in 87% yield over two steps.40 The Horner–Wittig reaction between 36 and 11 proceeded smoothly, having 98% yield. However, direct deprotection with aqueous work-up was impractical due to elimination reactions. Previous efforts in our group indicated that such systems tended to undergo elimination when directly deprotected with hydrochloric acid, as typically done for acetonide deprotection. Fortunately, we successfully deprotected the acetonide moiety under the same conditions we used to produce 24, yielding β-hydroxyketone 38 in 75% yield. This method demonstrated good selectivity and prevented the formation of unwanted elimination products. Selective anti-reduction was then accomplished using samarium(II) iodide and acetaldehyde under Evans–Tishchenko conditions.41 The reaction provided excellent results with 96% yield and a dr of >20:1 for 39. Deprotection lead to the formation of 40 in 99% yield. Alternative one-step methods, like the anti-reduction under Evans–Saksena conditions,42,43 lead to the formation of 40 in good yields albeit with poor diastereoselectivities (see ESI†).
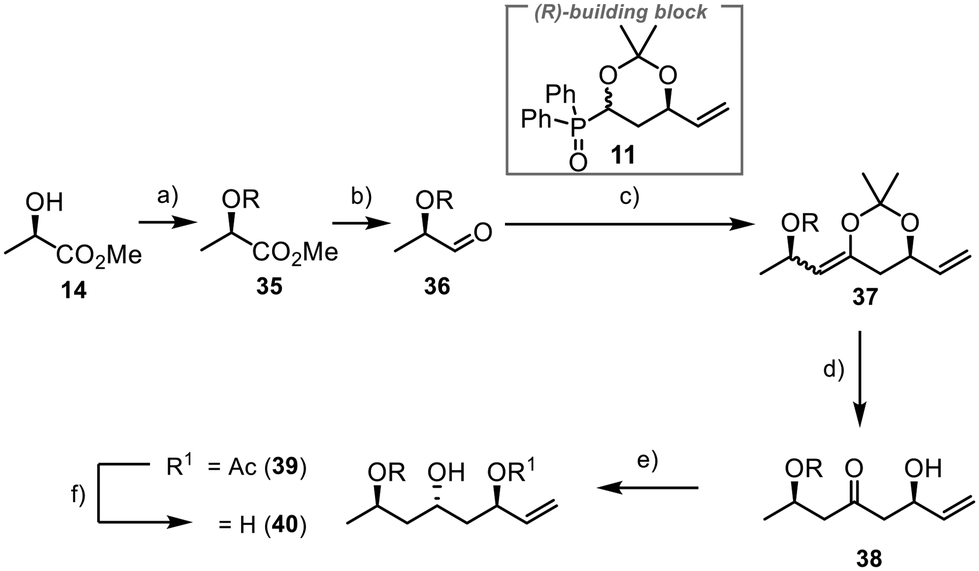 | ||
| Scheme 8 Introduction of the three stereogenic centres of fragment 8; R = TBS; (a) TBSCl (1.2 equiv.), imidazole (1.5 equiv.), rt, 30–60 min, (DMF), quant.; (b) DIBAL-H (1.2 equiv.), −78 °C, 1.5 h, (DCM), 87%; (c) (i) DIPA (2 equiv.), nBuLi (2 equiv.), −78 °C, 15 min, (ii) 11 (1 equiv.), −78 °C, 60 min, (iii) 35 (3 equiv.), −78 °C to rt, 90 min, (iv) KOtBu (1.05 equiv.), rt, 60 min, (THF), 98%; (d) CeCl3 (2 equiv.), oxalic acid (25 mol%), H2O (2 equiv.), −78 °C, 16 h, (THF), 75%; (e) SmI2 (0.75 equiv.), CH3CHO (3.5 equiv.), −50 °C to −20 °C, 16 h, (THF), 96%, dr > 20:1; (f) K2CO3 (2 equiv.), 0 °C to rt, 60 min, (MeOH/H2O, 3:1), 99%. | ||
To introduce an orthogonal protecting group at C25 that allows for the late connection with the aryl unit through ester formation, we used 1-(dimethoxymethyl)-4-methoxybenzene to protect the 1,3-diol 40 (Scheme 9). Reductive opening of acetal 41 with DIBAL-H resulted in excellent selectivity, yielding the desired product 42.45,46 Olefin 43 was obtained by TBS protection of the alcohol moiety, followed by the conversion of the double bond to the primary alcohol 44 in 97% yield using hydroboration and oxidative work-up.47 The aryl sulfide was incorporated through a Mitsunobu reaction.48 The final step involved the oxidation of the sulfide 45 to the sulfone 8, which was achieved in high yield (90%), completing the synthesis of the final fragment.
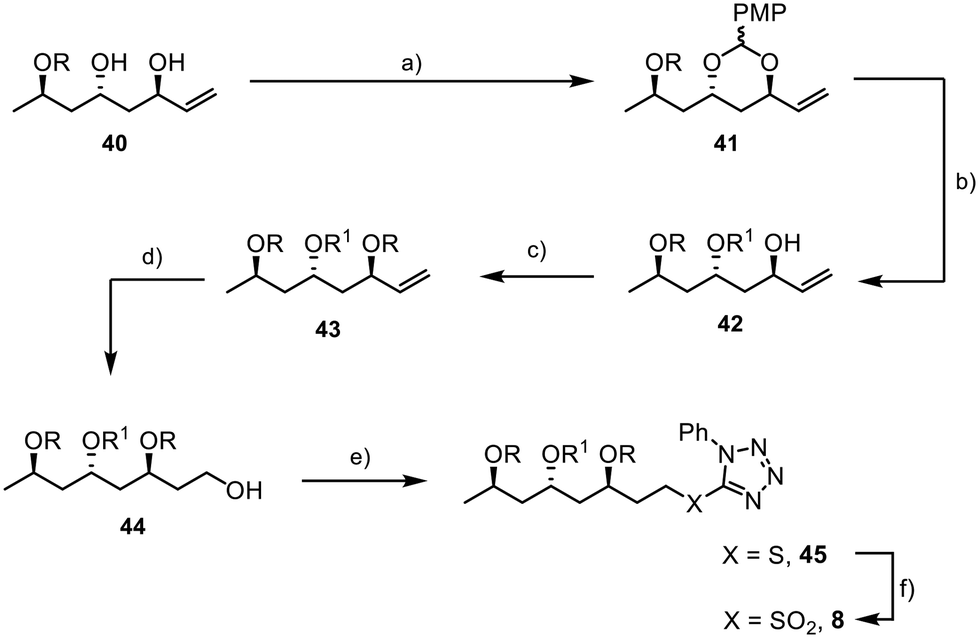 | ||
| Scheme 9 Final steps for the synthesis of fragment 8; R = TBS; R1 = PMB; PT = phenyltetrazole; PMP = p-methoxyphenyl; (a) 1-(dimethoxymethyl)-4-methoxybenzene (1.5 equiv.), PPTS (7 mol%), rt, 16 h, (DCM), 89%; (b) DIBAL-H (4.45 equiv.), 0 °C, 10 min, (DCM), 85%; (c) TBSCl (1.5 equiv.), imidazole (3 equiv.), rt, 16 h, (DMF), 95%; (d) 9-BBN (3 equiv.), rt, 15 min, then H2O2 (12 equiv.), NaOH (3 equiv.), rt, 4 h, (THF), 97%; (e) HS-PT (2 equiv.), DIAD (1.8 equiv.), PPh3 (1.5 equiv.), 0 °C, 4 h, (THF), 94%; (f) (NH4)6Mo7O24·4H2O (20 mol%), H2O2 (10 equiv.), rt, 3 h, (EtOH), 90%. | ||
The assembly of the fragments started with a Julia–Kocienski reaction between 7 and 8. The internal double bond of 46 was formed with an excellent E/Z selectivity of >20:1 and good yield of 85% (Scheme 10). The PMB group was then efficiently removed using DDQ in a buffered dichloromethane/water suspension, providing the desired product 47 in 90% yield (E/Z > 20:1).
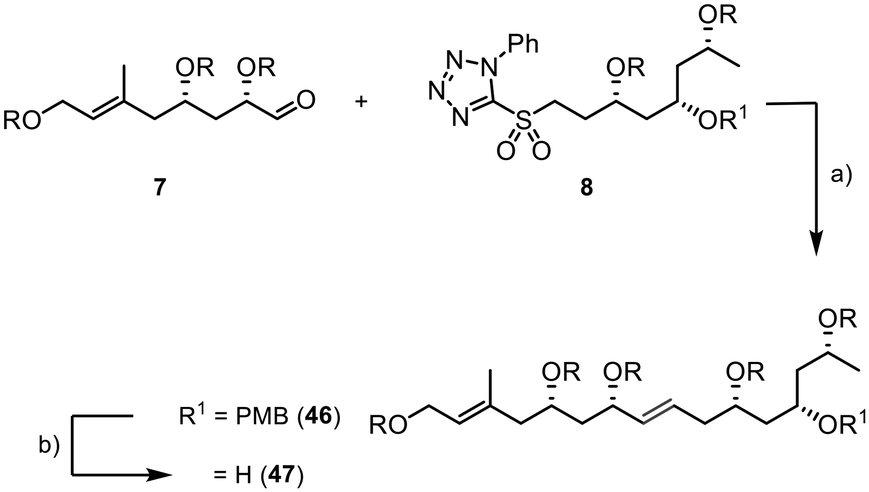 | ||
| Scheme 10 Initial fragment assembly via Julia–Kocienski olefination; R = TBS; (a) (i) sulfone 8 (1.15 equiv.), KHMDS (1.2 equiv.), −78 °C, 30 min, (ii) aldehyde 7 (1 equiv.), −78 °C, 2.5 h, (DME), 85%, E/Z > 20:1; (b) DDQ (1.5 equiv.), 0 °C, 2 h, (DCM/pH7-buffer, 1:1), 90%. | ||
The planned photoesterification represented a critical stage in the synthesis of the ortho-disubstituted benzoate 49 (Scheme 11). De Brabander et al. initially introduced this reaction, utilising light at a wavelength of 310 nm to generate the quinoketene 48 as the reactive intermediate.16 Subsequently, the quinoketene was expected to be captured by the secondary alcohol 47 to yield the desired ester 49. Following reaction optimisation, we achieved 49 with a moderate yield of 62%. Subsequently, we successfully protected the free phenol group as a triflate, in a high yield of 91%. However, unexpected instability issues arose with the preinstalled acetate group. Consequently, we opted for the deprotection and introduction of a MOM group, providing improved stability to the system 50 for subsequent reactions. This two-step transformation was achieved in a high yield of 95%.
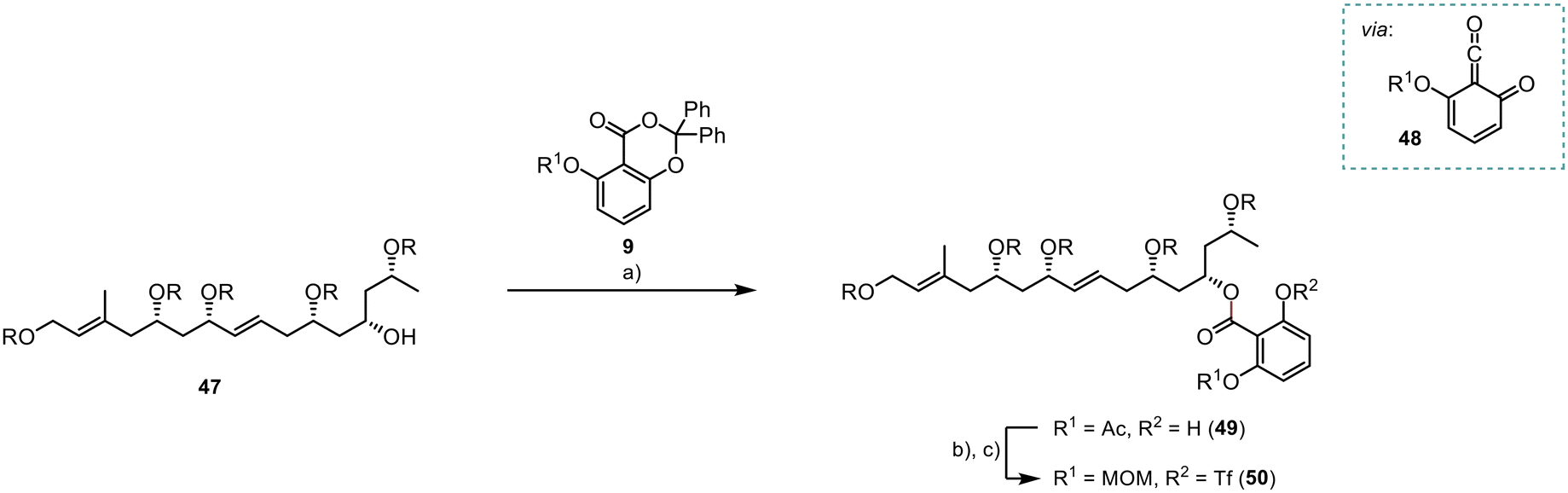 | ||
| Scheme 11 Photochemical introduction of the aromatic core; R = TBS; (a) 9 (2.5 equiv.), 310 nm, rt, 5 h, (DCM), 62%; (b) Tf2O (2 equiv.), pyridine (5 equiv.), 0 °C to rt, 2.5 h, (DCM), 91%; (c) (i) K2CO3 (0.5 equiv.), rt, 1 h, (MeOH/THF, 1:1), (ii) MOMCl (2 equiv.), DIPEA (5 equiv.), 0 °C to rt, 16 h, (DCM), 95% (two steps). | ||
Selective deprotection of the primary TBS group in the presence of the other secondary TBS groups proved to be a significant hurdle in the final steps of the synthesis of 51 (Scheme 12). However, after several unsuccessful attempts, we found suitable conditions for achieving the desired selectivity, adapting a protocol developed by Menche et al.,49 where they overcame this obstacle by utilising an excess of sodium periodate in an aqueous THF solution. Adapting the protocol to substrate 50 yielded the desired free hydroxyl group in 41% yield. Although the yield was modest, the remaining starting material was recoverable during chromatography, resulting in a yield of 87% (brsm). Subsequent oxidation with MnO2 and Colvin rearrangement led to the formation of the terminal alkyne 51 in 67% yield over the two steps.25,27 To generate the vinyl iodide 52, a modified procedure from Lipshutz et al. was employed,50 utilising an in situ generated Schwartz reagent and NIS as iodine source.51 The desired iodide 52 was obtained in 47% yield. Due to the high light sensitivity and limited stability of this unsaturated compound, immediate utilisation was required for its subsequent Stille cross-coupling with stannane 6 under slightly modified conditions from Evans et al.5 The synthesis of the monomeric counterpart 5 was accomplished under the specified conditions, resulting in a modest yield of 36% for the desired product. Nevertheless, we successfully attained our objective of synthesising this crucial intermediate for the preparation of Marinomycin A and B.
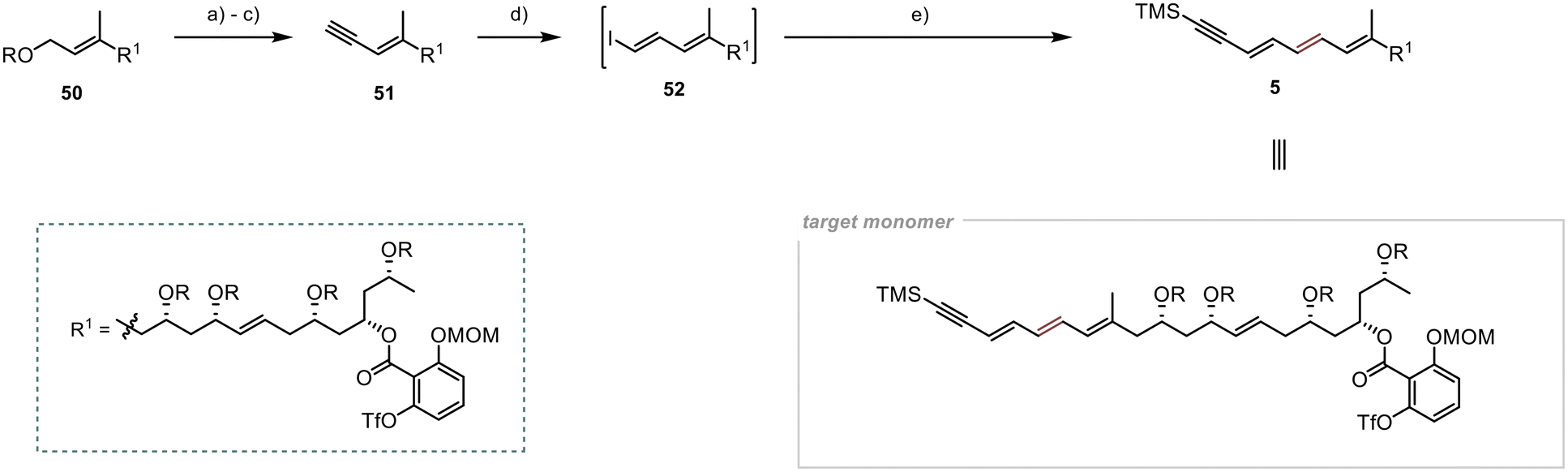 | ||
| Scheme 12 Final steps in the synthesis of the monomeric counterpart 5 of Marinomycin A and B; R = TBS; (a) NaIO4 (6 equiv.), rt, 5 h, (THF/H2O, 4:1), 41% (87% brsm); (b) MnO2 (50 equiv.), rt, 16 h, (DCM), quant.; (c) (i) nBuLi (1.5 equiv.), TMSCHN2 (1.8 equiv.), −78 °C, 1 h, (ii) aldehyde (1 equiv.), −78 °C to rt, 4.5 h, (THF), 67%; (d) (i) Cp2ZrCl2 (4 equiv.), LiHBEt3 (4 equiv.), rt, 60 min, (ii) alkyne 51 (1 equiv.), rt, 60 min, (iii) NIS (5 equiv.), rt, 15 min, (THF), 47%; (e) 6 (1.1 equiv.), Pd2(dba)3 (10 mol%), Ph3As (80 mol%), LiCl (15 equiv.), 40 °C, 2 h, (DMF), 36%. | ||
Conclusions
Our research outlines the synthesis of a novel monomeric precursor to Marinomycins A and B, accomplished via a convergent four-fragment approach. Our strategy integrated a polyketide building block previously established in literature, resulting in the final product attained through 24 steps for the longest linear sequence, with an overall yield of 0.6%. Looking ahead we will investigate the dimerisation of this monomer to synthesise Marinomycin A and B. Furthermore, we aim to enhance the synthesis by refining a more streamlined and efficient route for the fragments, while also improving the reliability of the final steps.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Research support from TUM and BUW is gratefully acknowledged.References
- H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Am. Chem. Soc., 2006, 128, 1622 CrossRef CAS PubMed.
- C. S. Bailey, J. S. Zarins-Tutt, M. Agbo, H. Gao, A. Diego-Taboada, M. Gan, R. B. Hamed, E. R. Abraham, G. Mackenzie, P. A. Evans and R. J. M. Goss, Chem. Sci., 2019, 10, 7549 RSC.
- K. C. Nicolaou, A. L. Nold, R. R. Milburn and C. S. Schindler, Angew. Chem., Int. Ed., 2006, 45, 6527 CrossRef CAS PubMed.
- K. C. Nicolaou, A. L. Nold, R. R. Milburn, C. S. Schindler, K. P. Cole and J. Yamaguchi, J. Am. Chem. Soc., 2007, 129, 1760 CrossRef CAS PubMed.
- P. A. Evans, M.-H. Huang, M. J. Lawler and S. Maroto, Nat. Chem., 2012, 4, 680 CrossRef CAS PubMed.
- T. Nishimaru, M. Kondo, K. Takeshita, K. Takahashi, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2014, 53, 8459 CrossRef CAS PubMed.
- D. Amans, V. Bellosta and J. Cossy, Org. Lett., 2007, 9, 1453 CrossRef CAS PubMed.
- D. Amans, L. Bareille, V. Bellosta and J. Cossy, J. Org. Chem., 2009, 74, 7665 CrossRef CAS PubMed.
- A. Rajesh, G. Sharma and K. Damera, Synthesis, 2015, 47, 845 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- D. Wang and S. Gao, Org. Chem. Front., 2014, 1, 556 RSC.
- D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636 CrossRef CAS.
- J. B. Baudin, G. Hareau, S. A. Julia and O. Ruel, Tetrahedron Lett., 1991, 32, 1175 CrossRef CAS.
- P. R. Blakemore, W. J. Cole, P. J. Kocieński and A. Morley, Synlett, 1998, 26 CrossRef CAS.
- F. Mittendorf, I.-E. Celik and S. F. Kirsch, J. Org. Chem., 2022, 87, 14899 CrossRef CAS PubMed.
- O. Soltani and J. K. de Brabander, Angew. Chem., Int. Ed., 2005, 44, 1696 CrossRef CAS PubMed.
- O. Soltani and J. K. de Brabander, Org. Lett., 2005, 7, 2791 CrossRef CAS PubMed.
- S. Bolshakov and J. L. Leighton, Org. Lett., 2005, 7, 3809 CrossRef CAS PubMed.
- H. Menz and S. F. Kirsch, Org. Lett., 2009, 11, 5634 CrossRef CAS PubMed.
- S. Kirsch, P. Klahn and H. Menz, Synthesis, 2011, 3592 CrossRef CAS.
- A. Bredenkamp, M. Wegener, S. Hummel, A. P. Häring and S. F. Kirsch, Chem. Commun., 2016, 52, 1875 RSC.
- A. Bredenkamp, Z.-B. Zhu and S. F. Kirsch, Eur. J. Org. Chem., 2016, 252 CrossRef CAS.
- L. Ferrié, J. Fenneteau and B. Figadère, Org. Lett., 2018, 20, 3192 CrossRef PubMed.
- A. Darwish, A. Lang, T. Kim and J. M. Chong, Org. Lett., 2008, 10, 861 CrossRef CAS PubMed.
- C. T. Brain, A. Chen, A. Nelson, N. Tanikkul and E. J. Thomas, Tetrahedron Lett., 2001, 42, 1247 CrossRef CAS.
- C. T. Brain, A. Chen, A. Nelson, N. Tanikkul and E. J. Thomas, Tetrahedron, 2010, 66, 6613 CrossRef CAS.
- E. W. Colvin and B. J. Hamill, J. Chem. Soc., Chem. Commun., 1973, 151 RSC.
- J. A. Lafontaine, D. P. Provencal, C. Gardelli and J. W. Leahy, J. Org. Chem., 2003, 68, 4215 CrossRef CAS PubMed.
- K.-V. Tran and D. Bickar, J. Org. Chem., 2006, 71, 6640 CrossRef CAS PubMed.
- J. Zhu and D. Ma, Angew. Chem., Int. Ed., 2003, 42, 5348 CrossRef CAS PubMed.
- T. J. Tewson and M. J. Welch, J. Org. Chem., 1978, 43, 1090 CrossRef CAS.
- K. S. Kim, Y. H. Song, B. H. Lee and C. S. Hahn, J. Org. Chem., 1986, 51, 404 CrossRef CAS.
- X. Xiao and D. Bai, Synlett, 2001, 535 CrossRef CAS.
- K.-M. Chen, K. G. Gunderson, G. E. Hardtmann, K. Prasad, O. Repic and M. J. Shapiro, Chem. Lett., 1987, 16, 1923 CrossRef.
- K.-M. Chen, G. E. Hardtmann, K. Prasad, O. Repič and M. J. Shapiro, Tetrahedron Lett., 1987, 28, 155 CrossRef CAS.
- K. Narasaka and F.-C. Pai, Tetrahedron, 1984, 40, 2233 CrossRef CAS.
- K. Narasaka and H. C. Pai, Chem. Lett., 1980, 9, 1415 CrossRef.
- T. P. Blaisdell, T. C. Caya, L. Zhang, A. Sanz-Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9264 CrossRef CAS PubMed.
- F. Ballaschk, Y. Özkaya and S. F. Kirsch, Eur. J. Org. Chem., 2020, 6078 CrossRef CAS.
- S. M. Gibson, R. M. Lanigan, L. Benhamou, A. E. Aliev and T. D. Sheppard, Org. Biomol. Chem., 2015, 13, 9050 RSC.
- D. A. Evans and A. H. Hoveyda, J. Am. Chem. Soc., 1990, 112, 6447 CrossRef CAS.
- D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560 CrossRef CAS.
- A. K. Saksena and P. Mangiaracina, Tetrahedron Lett., 1983, 24, 273 CrossRef CAS.
- R. van Rijsbergen, M. J. O. Anteunis and A. de Bruyn, J. Carbohydr. Chem., 1983, 2, 395 CrossRef CAS.
- J. Janssens, M. D. P. Risseeuw, J. van der Eycken and S. van Calenbergh, Eur. J. Org. Chem., 2018, 6405 CrossRef CAS.
- H. Kumar, A. S. Reddy and B. S. Reddy, Tetrahedron Lett., 2014, 55, 1519 CrossRef CAS.
- H. C. Brown and J. Chen, J. Org. Chem., 1981, 46, 3978 CrossRef CAS.
- O. Mitsunobu and M. Yamada, Bull. Chem. Soc. Jpn., 1967, 40, 2380 CrossRef CAS.
- J. Li and D. Menche, Synthesis, 2009, 1904 Search PubMed.
- B. H. Lipshutz, R. Keil and E. L. EIIsworth, Tetrahedron Lett., 1990, 31, 7257 CrossRef CAS.
- P. C. Wailes and H. Weigold, J. Organomet. Chem., 1970, 24, 405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00742e |
| This journal is © The Royal Society of Chemistry 2024 |