
Trivalent dialkylaminopyridine-catalyzed site-selective mono-O-acylation of partially-protected pyranosides†
Kalyan
Dey
and
Narayanaswamy
Jayaraman
 *
*
Indian Institute of Science, Bangalore 560012, India. E-mail: jayaraman@iisc.ac.in; Tel: +91-80-2293-2578
First published on 24th May 2024
Abstract
This work demonstrates trivalent tris-(3-N-methyl-N-pyridyl propyl)amine (1) catalyzing the site-selective mono-O-acylation of glycopyranosides. Different acid anhydrides were used for the acylation of monosaccharides, mediated by catalyst 1, at a loading of 1.5 mol%; the extent of site-selectivity and the yields of mono-O-acylation products were assessed. The reactions were performed between 2 and 10 h, depending on the nature of the acid anhydride, where the bulkier pivalic anhydride required a longer duration for acylation. The glycopyranosides are maintained as diols and triols, and from a set of experiments, the site-selectivity of acylations was observed to follow the intrinsic reactivities and stereochemistry of hydroxy functionalities. The trivalent catalyst 1 mediates the reactions with excellent site-selectivities for mono-O-acylation product formation in the studied glycopyranosides, in comparison to the monovalent N,N-dimethylamino pyridine (DMAP) catalyst. This study illustrates the benefits of the multivalency of catalytic moieties in catalysis.
Introduction
The roles of hydroxy functionality-rich carbohydrates in biological functions are diverse.1–3 As a result, an understanding of the structures and functions of carbohydrates has the greatest potential to uncover many intricate biological processes.4 The presence of multiple hydroxy functionalities increases the repertoire of complex oligosaccharides and polysaccharides exponentially. Enzymes exquisitely streamline the assembly of oligosaccharides and polysaccharides, whereas the corresponding synthetic equivalents are also developed impressively.5 The chemical synthesis of complex oligosaccharides encounters the critical roles of protecting groups that mask the reactivities of hydroxy functionalities not involved in the assembly. Protecting group methods enable the chemical synthesis of oligosaccharides and polysaccharides in the most achievable homogeneous manner.6,7 The pKa of primary and secondary hydroxy functionalities are within 16–18, implying that reactivity differences are minimal among these functionalities. As a result, development of methods for the site-selective modification of carbohydrates,8 including one-pot synthetic methods,9 is important. An attractive approach in this direction is exploiting the intramolecular hydrogen bonding of hydroxy functionalities, which increases nucleophilicity. Early demonstrations of this concept are (i) DMAP-catalyzed acylation reactions using Ac2O for the regioselective formation of the C3-OAc surrogate of octyl-β-D-glucopyranoside, as reported by Yoshida and co-workers.10a (ii) Carboxylic or sulfonic acid-linked DMAP-catalyzed site selective acylation at the C6-OH group in octyl glucopyranosides and galactopyranosides.10b The effect of a counter ion in DMAP-catalyzed acylations was studied by Albert and Kattnig.11 Acylation is favored at hydroxy functionalities either at C3, C4 or C6, depending on the nature of the counter anion through the formation of non-covalently linked cyclic transition state between acyl and hydroxy functionalities. The ‘directed protecting group-induced regioselective functionalization’ of partially protected carbohydrates was reported by Moitessier and co-workers, wherein the protecting group at C6 forms intramolecular hydrogen bonding preferentially at the C3 hydroxy functionality and facilitates regioselective acylation.12 A good regioselectivity was observed for the pivaloylation of 4,6-O-benzylidene-protected glycoside derivatives.13 Several other acylating agents, such as benzoyl imidazole,14 BzCN,15 and 1-(benzoyloxy)benzotriazole (BzOBt),16 were developed in the regioselective acylation of 4,6-O-protected pyranoside derivatives. The selectivity was largely dependent on the reaction conditions, acidity and nucleophilicity of reactive functionalities. In the case of BzCN, benzoylation occurs at the axial hydroxy functionality in diols and triols in the presence of DMAP (10 mol%, −78 °C).17 The formation of monodentate hydrogen bonding with cyanide moiety and acylation at the acidic axial hydroxy functionality under kinetic control appeared to favor the regioselectivity, termed as the ‘cyanide effect’.Several structurally-modified DMAP derivatives have been reported to achieve the site-selective functionalization and enantioselective acylation of racemic alcohol.18–26 Kawabata and co-workers27 developed a 4-pyrrolidinopyridine (PPY)-based C2-symmetric chiral catalyst based on L-tryptophan. A good site-selectivity at the hydroxyl functionality at C4 was observed for the installation of isobutyrate in octyl-β-D-glucopyranoside. Kirsch and co-workers generated a library of DMAP-based oligopeptides for the site-selective mono-O-benzoylation in favor of functionalization at C2 in benzylidene-protected α-D-glucopyranoside and at C3 in benzylidene-protected α-D-galactopyranoside derivatives.28 Besides DMAP-derived catalysts, N-methyl imidazole (NMI) has been developed as an acyl transfer reagent. NMI-appended peptide-based catalysts have been developed for the regioselective functionalization of monosaccharides, as reported by Miller and co-workers.29 Schreiner and co-workers recently reported site-selective acylation employing oligopeptide and photo-switchable azo-peptide NMI-catalyst.30 NMI-based chiral oxazolidines catalyze the acylation of the hydroxyl functionality at C3 and the intrinsically less reactive functionality at the C2 carbon in carbohydrate derivatives.31 Chiral benzotetramisole (BTM) catalyst was employed by Tang and co-workers32a to accomplish the regioselective acylation of the trans-1,2-diol moiety in pyranoside derivatives. The hydroxyl functionality at C2 or C3 carbon underwent the reaction selectively by choice of the enantiomeric catalyst. Regioselectivity was observed in this case due to the lone pair of electrons-cation interaction between the acylated catalyst intermediate and either an ether or hydroxyl oxygen of the substrate. In a subsequent study, S-adamantyl group-directed acylation using the same catalyst was also reported.32b
Chiral NHC catalysis33 and chiral NHC-boronic acid dual catalysis34 using aldehyde as an acyl source were applied successfully for the site-selective acylation of carbohydrates. Miller and co-workers developed the site-selective functionalization of methyl 4,6-O-benzylidene-protected pyranosides employing a chiral Ph-Box ligand with Cu(II).35 Site-selectivity was dependent on the chirality of the ligands. Using similar Cu(II)–Ph–Box complexes, Dong and co-workers reported the site-selective acylation of pyranoside-derived triols and furanoside-derived diols.36 Chiral phosphoric acid (CPA) catalyzed the site-selective acetalization of pyranoside-derived 1,2-diols.37a Recently, a polystyrene-supported reusable CPA was used for this purpose and variably protected saccharide derivatives were accomplished in an one-pot reaction.37b Methods were also developed for the site-selective functionalization of unprotected or partially protected pyranosides using salts of tin-,38,39 iron-,40 silicon-41 and copper-42 silver-based43 reagents. Although organotin reagents are considered largely for the regioselective functionalization of unprotected carbohydrates, a major shortcoming is the inherent toxicity of tin reagents. Organoboron reagents, developed by Taylor and Shimada groups, are non-toxic and used for site-selective acylation,44 sulfamoylation,45a sulfation,45b sulfonylation,46,47 carbamoylation,48 and glycosylation49,50 for unprotected carbohydrates having the cis-1,2-diol moiety. The above developments illustrate the necessity to uncover methods that facilitate the site-selective derivatization of multi-functional carbohydrate moieties. An important functionalization in this direction is the protection of the hydroxyl functionality as esters, which are used routinely to derive, for example, partially-protected carbohydrate derivatives. The ester protecting groups are installed by treating the alcohol functionality with carboxylic acid anhydrides and acid chlorides in the presence of DMAP (cat.).51 Several polymer-bound (dialkylamino)pyridine catalysts have been reported to act as nucleophilic catalysts in organic transformations.52 Portnoy and co-workers developed a series of p-aminopyridine53a and imidazole-based53b nucleophilic organocatalysts, functionalized further with either oligoether or long alkyl moieties for the site-selective acylation and phosphorylation of amphiphilic diol and complex natural products.53c,d The choice of base and catalyst were emphasized in order to achieve orthogonal site-selectivities.
Multivalency in catalysis has gained importance in contemporary developments. The covalent attachment of multiple catalytic moieties onto a branched structure leads to efficiencies in catalysis that are over and above the statistical increases in the moieties.54 Elegant examples are in C–C bond formations enabled by multivalent organometallic catalysts.55 Multivalent organocatalysis by a variety of dendritic structures have been reported in a number of reactions.56,57 Good to excellent selectivities were achieved, and such possibilities are not observed with monomeric organocatalysts alone.
The multivalent presentations of DMAP organocatalysts are previously reported, which demonstrated the catalytic properties of Fréchet-type benzyl ether and aliphatic ester dendritic polymers containing 4-(dialkylamino)pyridines in the interior environment.58 Multivalent DMAP organocatalyst plays an important role for the selective labelling of biopolymer under mild and benign condition. Hamachi and co-workers devised an efficient method of the site-selective chemical acylation of different proteins enabled by multivalent affinity-guided DMAP (AGD) catalysts.59 The combined effects of catalyst multivalency and affinity guidance imposed by the DMAP-attached ligand were the key factors for achieving site-selective protein acylation with acyl donors possessing the desired probe. Chelation and coordination assistance in the catalytic sites and substrates lead to increases in the catalytic activities in the studied instances. Such an opportunity is studied herein in the case of site-selectivities in multiple hydroxy functionalities of monosaccharides with the aid of a trivalent DMAP catalyst.
Selectivity herein pertains to the site-selectivity among the secondary hydroxy functionalities. The coordination of a trivalent catalyst with the hydroxyl functionalities might improve the reactivity at a particular site. A series of acylations was performed using the trivalent catalyst, involving several monosaccharides that possess diols and triols and several carboxylic acid anhydrides, with variable steric and electronic features, as acylation agents. The details of the studies are described herein.
Results and discussion
Synthesis of the catalyst
The site-selective functionalization of carbohydrates was undertaken with the aid of a trivalent (dialkylamino)pyridine (DAAP) catalyst. The synthesis of the new DAAP-based nucleophilic organocatalyst 1 was accomplished by the N-alkylation of tris-halide 260 with 4-N-methylamino pyridine 3 in the presence of NaH in THF solution. The reaction occurred in a facile manner to afford the trivalent DAAP catalyst 1, after purification by column chromatography (neutral Al2O3, CHCl3/MeOH, linear gradient), in 88% yield (Scheme 1).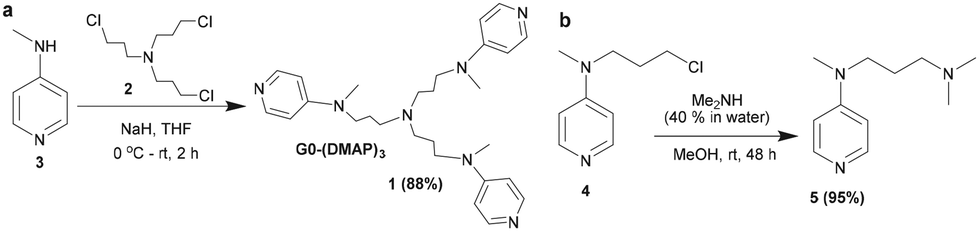 | ||
| Scheme 1 Synthesis of (a) trivalent DAAP catalyst 1 and (b) monovalent (5) DAAP catalysts. | ||
The corresponding monovalent catalyst 5 was secured through the N-alkylation of the corresponding halide 461 with dimethyl amine (40% in water) in MeOH. The newly synthesized DAAP catalysts 1 and 5 were characterized by NMR spectroscopy and mass spectrometry; the details are given in the ESI.†
Study of the site-selective acylation of pyranoside-derived 1,2-diols
The efficacies of catalysts 1 and 5 as acylation transfer agents in monosaccharides having secondary 1,2-diol functionalities were studied initially. Reactivity-based differences are not obvious to equatorial secondary hydroxy functionalities, and in practice, scarcely attainable. For DMAP-catalyzed acylations, acyl halides or anhydrides are commonly employed. Trivalent catalyst 1 might facilitate the reaction of N-acyl-pyridinium intermediate with the hydroxy functionality than that with the monomeric DMAP catalyst. The effect of steric factors on the DMAP-catalyzed acylation was demonstrated by Zipse and co-workers.62 The acylation of methyl 4,6-O-benzylidene-α-D-glucopyranoside (6), presenting a 1,2-trans-diol, was conducted using Ac2O (1.1 molar equiv.), DAAP catalysts 1 (1.5 mol%), 5 (4.5 mol%) and DMAP (4.5 mol%) in MeCN, at 0 °C to room temperature for 3 h, and the formation of the acylation products was ascertained by 1H NMR spectroscopy of the crude reaction mixtures. Mono-O-acylations and di-O-acylation were assessed, along with the overall yield of the reaction and site-selectivity towards the formation of the 2-O-acyl product (Table 1).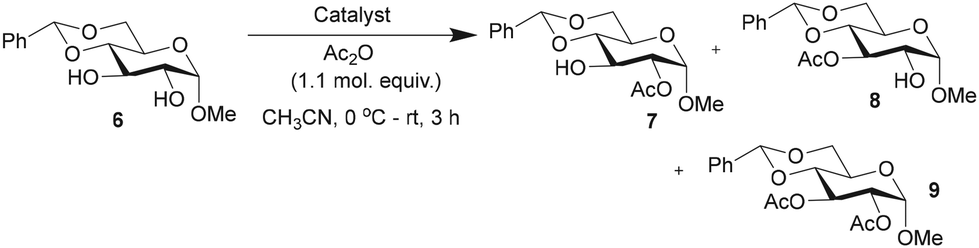
Reactions were conducted in MeCN as the substrates and catalysts are more soluble freely in MeCN than in CHCl3, CH2Cl2 and THF solutions. After the desired duration, the reaction mixture was quenched with MeOH, solvents evaporated under reduced pressure and analyzed by 1H NMR spectroscopy.
The acylation occurred at varying proportions, depending on the catalyst. Among the catalysts, trivalent DAAP 1 led to an efficient conversion to 2-O-acyl derivative 7 than monovalent 5 and DMAP catalysts. Further, an increase to 5 mol% of trivalent catalyst 1 led only to increased di-O-acylation product 9 (ESI†). As a result, the catalytic activities and site-selectivities of acylations mediated by multivalent catalysts were assessed using ∼7 times reduced mol%. The monovalent catalyst 5 and DMAP were maintained at 4.5 mol%, whereas the trivalent catalyst 1 was maintained at one-third (1.5 mol%) throughout the present study. When DMAP derivative 3 was used as the catalyst (4.5 mol%), the conversion and site-selectivity were less, presumably due to acylation at the amine site of 3. In light of the highest site-selectivity afforded by the trivalent catalyst 1, further acylations were performed with this catalyst and in comparison, with DMAP. All the acylation experiments were conducted under auxiliary base-free condition.63 Bases, such as Et3N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 64 and diisopropylethyl amine,65 alone mediate the site-selective acylation of carbohydrate substrates.
64 and diisopropylethyl amine,65 alone mediate the site-selective acylation of carbohydrate substrates.
Glucopyranoside 6 was subjected to acylation with few anhydrides of variable electronic and steric nature, namely, isobutyric anhydride ((iPrCO)2O), pivalic anhydride ((tBuCO)2O) and benzoic anhydride ((PhCO)2O), under the optimized catalyst loading (1.5 mol%), and the observations are given in (Table 2).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Reagent | Time (h) | Catalyst | Products (%) | Conversion (%) | Yieldb (%) | ||
| a Products ratio determined by comparing the anomeric proton integrations in the 1H NMR spectrum. b Yield of the isolated major product for 1-catalyzed acylation reactions. | ||||||||
| (i-PrCO)2O | 2 | 10 | 11 | 12 | ||||
| 1 | 1 | 75 | 10 | 15 | 90 | 61 | ||
| 2 | DMAP | 47 | 30 | 23 | 89 | |||
| (t-BuCO)2O | 16 | 13 | 14 | |||||
| 3 | 1 | 77 | 23 | 70 | 48 | |||
| 4 | DMAP | 64 | 36 | 45 | ||||
| Bz2O | 10 | 15 | 16 | 17 | ||||
| 5 | 1 | 54 | 12 | 34 | 90 | 42 | ||
| 6 | DMAP | 42 | 31 | 26 | 89 | |||
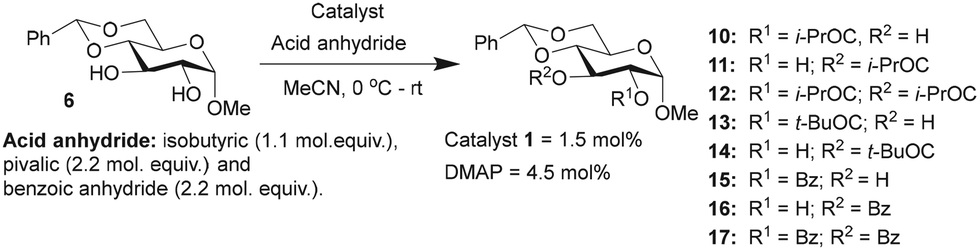
The acylation of glucopyranoside 6 with isobutyric anhydride (1.1 molar equiv.) in the presence of catalyst 1 led to excellent site-selectivity for the 2-O-isobutyroyl product. The promising results of site-selective acetylation catalyzed by 1 warranted experiments using a sterically-hindered acylating agent, pivalic anhydride ((tBuCO)2O). DMAP catalysis is sensitive to steric factors associated with reacting alcohols and carboxylic acid anhydrides. The same was observed in the pivaloylation of glucopyranoside 6 catalyzed by DMAP, which afforded an overall yield of 45% for the mono-O-acylated products, 2-pivaloyl (13) and 3-pivaloyl (14) products, in the ratio of 1.7:1. In these reactions, excess molar equiv. of pivalic anhydride (2.2 molar equiv.) was used, with increased duration of reaction to 16 h. Site-selectivity in benzoylation was conducted subsequently to secure the corresponding benzoates. The reaction was conducted using 2.2 mol. equiv. of benzoic anhydride (PhCO)2O, catalyzed by either 1 or DMAP catalyst for 10 h. In this reaction, a considerable amount of 2,3-di-O-benzoyl product 17 also formed, presumably due to excess benzoic anhydride and longer reaction duration.
In all the cases, the overall conversion refers to the percentages of all the acylated products; the rest of the percentages were unreacted starting material, which were recovered during the purification of the crude reaction mixtures.
Acylation was then undertaken with methyl 4,6-O-benzylidene-α-D-galactopyranoside 18 possessing an equatorial 1,2-trans diol moiety. In this instance, the reactivity at C2 and C3 hydroxy functionalities are hardly distinguishable due to the ‘axial oxy group effect’.17b The acylation of derivative 18 using Ac2O (1.1 mol. equiv.) and catalyst 1 (1.5 mol%) afforded an overall yield of 99% in 2 h of reaction duration (Table 3). A ratio of 1:1 of products 19 and 20 was observed in this case. Similar results were observed in the isobutyration and benzoylation of derivative 18; the site-selectivity was comparable in both cases. The facile site-selectivity for trivalent catalyst 1 might arise due to steric and electronic effects, favoring acylation at the C3 hydroxyl functionality.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Reagent | Time (h) | Catalyst | Products (%) | Conversion (%) | Yieldb (%) | ||
| a Products ratio determined by comparing the anomeric proton integrations in the 1H NMR spectrum. b Yield of the isolated major product for 1-catalyzed acylation reactions. | ||||||||
| Ac2O | 2 | 19 | 20 | 21 | ||||
| 1 | 1 | 18 | 61 | 20 | 99 | 51 | ||
| 2 | DMAP | 49 | 51 | — | 25 | |||
| (i-PrCO)2O | 2 | 22 | 23 | |||||
| 3 | 1 | 21 | 79 | 93 | 64 (22/23 = 30:70) |
|||
| 4 | DMAP | 34 | 66 | 71 | ||||
| (t-BuCO)2O | 16 | 24 | 25 | |||||
| 5 | 1 | 16 | 84 | 95 | 63 (24/25 = 21:79) |
|||
| 6 | DMAP | 38 | 62 | 27 | ||||
| Bz2O | 10 | 26 | 27 | 28 | ||||
| 7 | 1 | 17 | 58 | 25 | 87 | 52 (26/27 = 23:77) |
||
| 8 | DMAP | 32 | 57 | 11 | 68 | |||
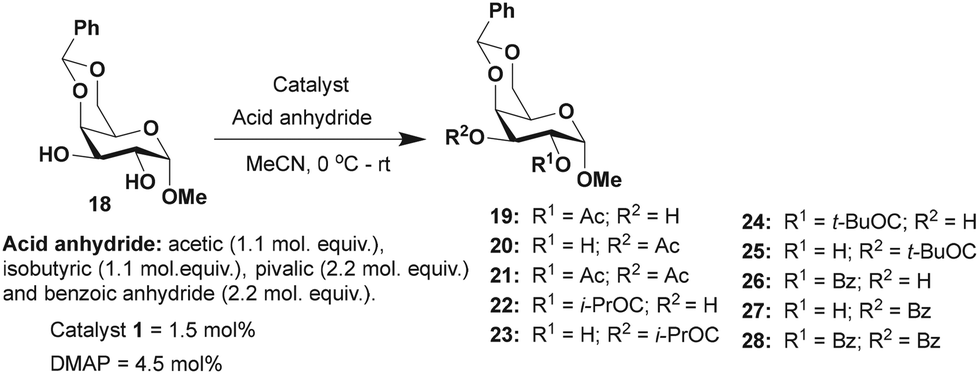
Methyl 4,6-O-benzylidene-α-D-mannopyranoside 29, possessing 1,2-cis-diol, was subjected to acylation subsequently. The results of the acylation of mannopyranoside 29 are summarized in Table 4. Slightly better yield (74%) was observed with catalyst 1 as compared to the DMAP catalyst in the case of O-acetylation occurring at the hydroxyl functionality at the C3 carbon. The lack of a considerable distinction in the site-selective O-acylation of mannopyranoside 29 is likely due to the higher reactivity of the C3 hydroxyl functionality of the 1,2-cis-diol. In pyranoside 1,2-diols, the site-selectivity appears to arise from the intrinsic reactivity difference. The hydrogen bonding at 1,2-cis-diol and the ‘axial oxy group effect’ might favor a site-selectivity towards the hydroxyl functionality at C3.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Reagent | Time (h) | Catalyst | Products (%) | Conversion (%) | Yieldb (%) | ||
| a Products ratio determined by comparing the anomeric proton integrations in the 1H NMR spectrum. b Yield of the isolated major product for 1-catalyzed acylation reactions. | ||||||||
| Ac2O | 3 | 30 | 31 | 32 | ||||
| 1 | 1 | 19 | 58 | 23 | 74 | 36 | ||
| 2 | DMAP | 16 | 75 | 9 | 60 | |||
| (i-PrCO)2O | 2 | 33 | 34 | 35 | ||||
| 3 | 1 | 5 | 81 | 14 | 95 | 68 | ||
| 4 | DMAP | 7 | 82 | 11 | 91 | |||
| (t-BuCO)2O | 16 | 36 | ||||||
| 5 | 1 | 34 | 34 | 26 | ||||
| 6 | DMAP | 28 | 28 | |||||
| Bz2O | 10 | 37 | 38 | 39 | ||||
| 7 | 1 | 11 | 83 | 6 | 78 | 56 | ||
| 8 | DMAP | 19 | 75 | 7 | 82 | |||
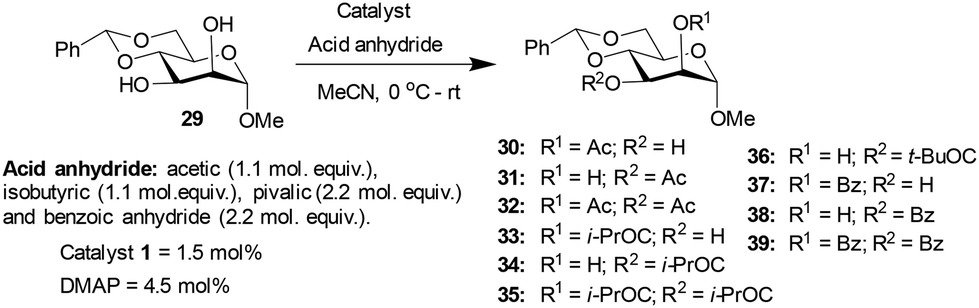
Methyl 4,6-O-benzylidene-α-D-glucopyranoside 6 afforded 2-O-acyl functionalized products, whereas methyl 4,6-O-benzylidene-α-D-galactopyranoside 18 and methyl 4,6-O-benzylidene-α-D-mannopyranoside 29 produced 3-O-acylation products. The synergistic effect of the hydrogen bonding, the disposition of the diols and steric factor associated with the acyl transfer process might play a role in the observed site-selectivities. Other than methyl 4,6-O-benzylidene-α-D-mannopyranoside 29 with 1,2-cis-diol disposition at C2 and C3 hydroxyl functionalities, the remaining pyranoside derivatives possess 1,2-trans-diol dispositions and afforded good yields and site-selectivities using trivalent catalyst 1.
Acylation of 6-O-protected monosaccharides
The functionalization of the C6 primary hydroxyl functionality of pyranosides is facile in the presence of the secondary hydroxy functionalities. Advancements in the site-selective protection of 6-O-protected glycopyranosides are: (i) boron-mediated regioselective acylation of 6-O-protected carbohydrates;44a (ii) DMAP catalyst in the presence of sym-collidine by Kawabata and Muramatsu,66 and (ii) Oriyama's chiral catalyst and TMEDA, as reported by Vasella and co-workers67 (Scheme 2).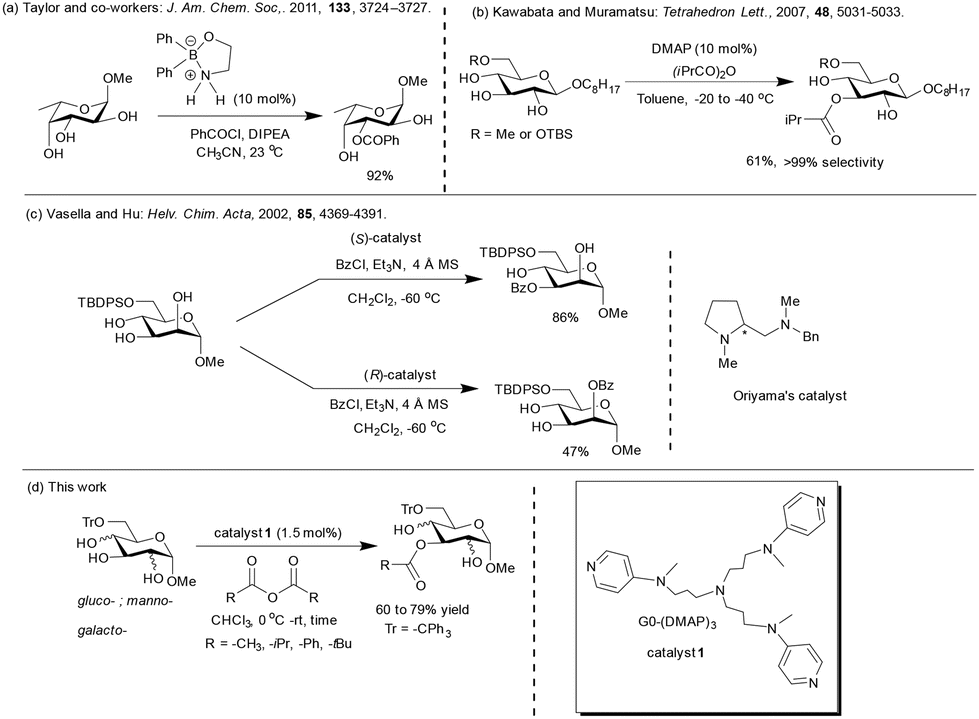 | ||
| Scheme 2 Site-selective acylation of pyranoside-derived triols. | ||
Site-selective acylation employing hydrogen bond acceptor-type protecting groups at the C6 hydroxyl functionality were also studied previously by Moitessier and co-workers.12b,c
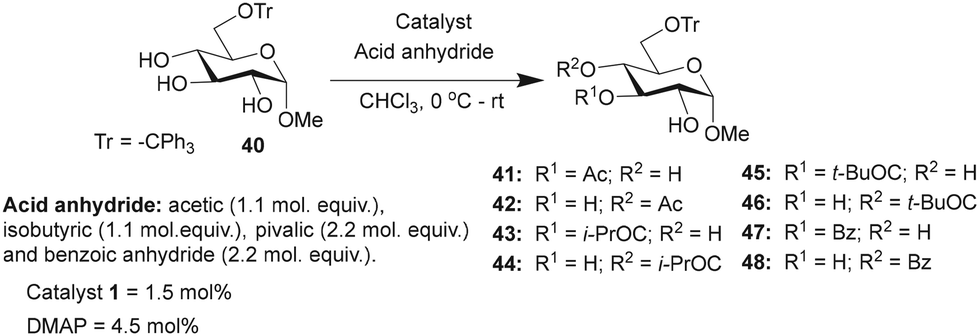
Studies were undertaken to assess the site selectivity in 6-O-protected carbohydrate triols in the presence of trivalent 1 and DMAP catalysts. The reactions were conducted using methyl 6-O-trityl-α-D-glycopyranosides in CHCl3 solution. The acylation of methyl 6-O-trityl-α-D-glucopyranoside (40) was conducted using catalyst 1 (1.5 mol%), Ac2O (1.1 mol. equiv.) at 0 °C to room temperature for 45 min, followed by HPLC analysis. A new intense peak at a retention time of 7.6 min (56%) appeared, along with few other minor peaks. Unreacted starting material 40 appeared at a retention time of 27 min (Fig. 1).
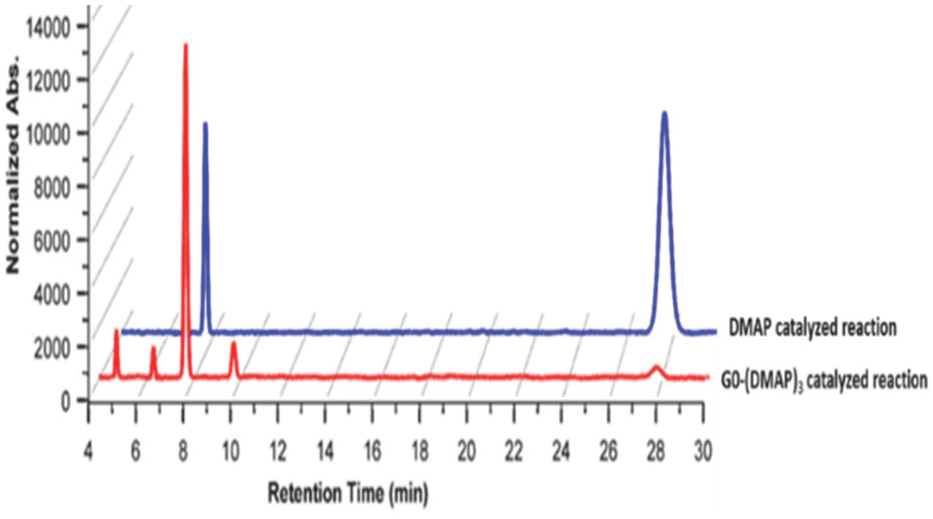 | ||
| Fig. 1 HPLC chromatograms for the acetylation of glucopyranoside 40. Chromatography conditions: Shimadzu Shim-pack (UC-X-Sil) 5 μm column, EtOAc–hexane = 60:40, flow rate 1 mL min−1, PDA detector (254 nm), retention times: 7.6 min (major isomer 41), 27.7 min (starting material 40) and the remaining peaks correspond to the minor isomers. | ||
The crude reaction mixture was subjected to column chromatography purification (SiO2) (pet. Ether/EtOAc linear gradient) to secure a major product in 62% yield, which was identified as the 3-O-acylated product. The peak at 9.70 min corresponds to the 4-O-acetylated product 42. The remaining peaks at 4.70 min and 6.29 min are related to multiple acetylated products, as evident from the mass spectrum of the crude reaction mixture. However, the identities of these minor products were not studied due to very less conversion (Fig. 1).
The observation reiterated the role of trivalent catalyst 1 in the elevated yields of the site-selective acylation product. Acylation glucopyranoside 40 was investigated further with varied acid anhydrides. Acetylation and isobutyration were conducted using acid anhydride (1.1 mol. equiv.), whereas benzoylation and pivaloylation were conducted using acid anhydride (2.2 mol. equiv.). In these instances too, catalyst 1 led to excellent overall yields of the mono-O-acylated products, with good site-selectivity in favor of the 3-O-acylated product. Except acetylation, other acylations afforded only 3-O-acyl and 4-O-acyl products (Table 5). In the case of bulky pivalic anhydride, the mono-3-O-pivaloate product 45 was formed in a good yield (84%) mediated by catalyst 1 than that mediated by the DMAP catalyst.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reagent | Time | Catalyst | Products (%) | Conversion (%) | Yieldb (%) | |
| a Product ratios determined by HPLC analysis. b Yield of the isolated major product for 1-catalyzed acylation reactions. | |||||||
| Ac2O | 45 min | 41 | 42 | ||||
| 1 | 1 | 74 | 9 | 94 | 62 | ||
| 2 | DMAP | 19 | — | 19 | |||
| (i-PrCO)2O | 30 min | 43 | 44 | ||||
| 3 | 1 | 89 | 6 | 95 | 70 | ||
| 4 | DMAP | 35 | 3 | 38 | |||
| (t-BuCO)2O | 16 h | 45 | 46 | ||||
| 5 | 1 | 84 | 14 | 98 | 72 | ||
| 6 | DMAP | 8 | 1 | 9 | |||
| Bz2O | 8 h | 47 | 48 | ||||
| 7 | 1 | 73 | 23 | 96 | 61 | ||
| 8 | DMAP | 12 | 3 | 16 | |||
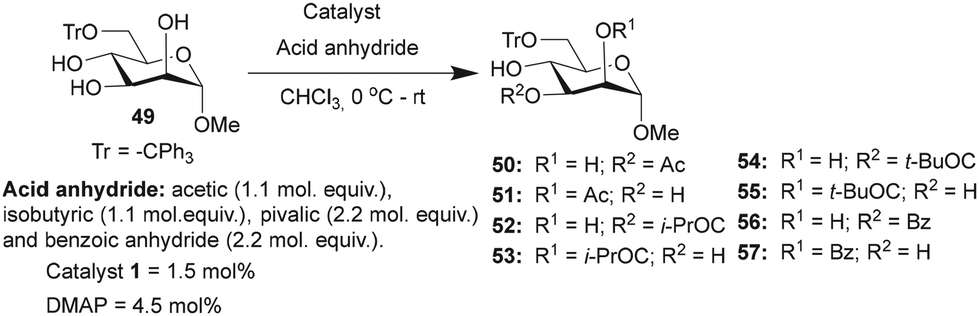
Site-selective acylations were also performed on methyl 6-O-trityl-α-D-mannopyranoside 49 under similar reaction conditions. In all the cases, the regio-isomers of the mono-O-acyl product were observed by HPLC analyses. The acetylation of mannopyranoside 49, mediated by catalyst 1, led to the preferential formation of mono 3-O-acetylated derivative. Mono O-acetylated products were isolated as an inseparable mixture of 2/3-O-acetylated products after purification (Table 6). Isobutyration also provided good yield of the mono-O-isobutyrated products, and the major 3-O-isobutyrated product 52 was isolated as a pure isomer. A good conversion of the pivaloylation also occurred, mediated by catalyst 1, in comparison to DMAP catalysis. However, site-selectivity for acylation at the C3-hydroxyl functionality reduced due to the formation of a considerable amount of the C2-O-pivaloate 55, for example, in the case of mono-O-benzoate 56, at C3-hydroxyl functionality, in the presence of catalyst 1 (Table 6).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reagent | Time | Catalyst | Products (%) | Conversion (%) | Yieldb (%) | |
| a Product ratios determined by HPLC analysis. b Yield of the isolated major product for 1-catalyzed acylation reactions. | |||||||
| Ac2O | 45 min | 50 | 51 | ||||
| 1 | 1 | 59 | 11 | 91 | 52 (50/51 = 70:30) |
||
| 2 | DMAP | 21 | 4 | 26 | |||
| (i-PrCO)2O | 30 min | 52 | 53 | ||||
| 3 | 1 | 65 | 23 | 97 | 58 | ||
| 4 | DMAP | 24 | 4 | 32 | |||
| (t-BuCO)2O | 16 h | 54 | 55 | ||||
| 5 | 1 | 55 | 33 | 91 | 42 | ||
| 6 | DMAP | 11 | 1 | 13 | |||
| Bz2O | 8 h | 56 | 57 | ||||
| 7 | 1 | 63 | 24 | 86 | 47 | ||
| 8 | DMAP | 5 | 6 | 11 | |||
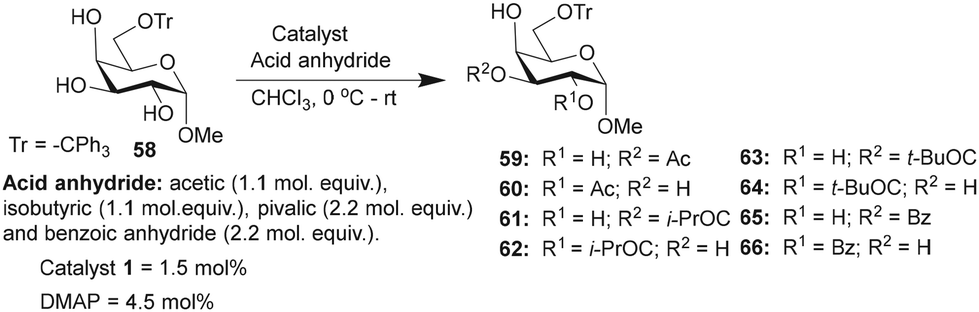
Galactopyranoside, namely, methyl 6-O-trityl-α-D-galactopyranoside-derived triol 58, was subjected to O-acylation, wherein only two regioisomeric products were formed, as evident from the HPLC analysis of the crude reaction mixture. Isolation and NMR characterization suggested the formation of the C3-O-acylated product as the major regio-isomer (Table 7). In this instance too, catalyst 1 afforded the mono-O-acylation products in higher yields and site-selectivity, as compared to the DMAP catalyst. Isobutyration reaction provided excellent overall conversion of the mono-O-isobutyrate products, where C3-O-isobutyrate 61 was isolated as the major product. Pivaloylation also furnished good conversion with C3/C2-O-pivaloate (63 and 64) as an inseparable mixture after purification. Finally, the benzoylation reaction provided the mono-O-benzoyl derivatives 65 and 66, catalyzed by catalyst 1, in higher yields than that by the DMAP catalyst (Table 7).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reagent | Time | Catalyst | Products (%) | Conversion (%) | Yieldb (%) | |
| a Product ratios determined by HPLC analysis. b Yield of the isolated major product for 1-catalyzed acylation reactions. | |||||||
| Ac2O | 45 min | 59 | 60 | ||||
| 1 | 1 | 40 | 27 | 71 | 20 | ||
| 2 | DMAP | 24 | 7 | 31 | |||
| (i-PrCO)2O | 30 min | 61 | 62 | ||||
| 3 | 1 | 61 | 20 | 81 | 42 | ||
| 4 | DMAP | 27 | 10 | 38 | |||
| (t-BuCO)2O | 16 h | 63 | 64 | ||||
| 5 | 1 | 57 | 15 | 72 | 45 (63/64 = 77:23) |
||
| 6 | DMAP | 5 | 1 | 6 | |||
| Bz2O | 8 h | 65 | 66 | ||||
| 7 | 1 | 61 | 25 | 86 | 44 | ||
| 8 | DMAP | 24 | 8 | 32 | |||
Although DMAP-catalyzed acylations of triols appear to provide an edge on the site-selectivity as compared to the trivalent catalyst 1, however, striking differences are observed in the overall conversion and the yield of the major mono-O-acylated product. In all the cases, trivalent catalyst 1 mediated the acylation of pyranoside triol derivatives, with excellent overall conversion and yield of the major regioisomer.
The effect of base on the rate and site-selectivity of acylation reaction is studied previously for monosaccharides11 and acyclic amphiphilic diols.68 Besides being trivalent, catalyst 1 is also constituted with a tertiary amine functionality. In order to assess the role of tertiary amine, the acetylation of 6 was conducted with 4.5 mol% DMAP, 1.1 mol. equiv. Ac2O, along with 1.1 mol. equiv. of Et3N. Interestingly, the reactions of DMAP, with and without Et3N, led to similar conversions and site-selectivities, as evident from the HPLC analyses of the reaction mixtures (Fig. S25†). Further, several unidentified products were observed in the reaction with Et3N. The reaction with Et3N suggests a minimal role, if any, on the enhanced site-selectivities and reaction rates by the trivalent catalyst 1.
Ratios of the major and minor regioisomers were investigated at different time intervals for trivalent 1-catalyzed acylation experiments for glucopyranoside diol 6 and triol 40. At different time intervals, an aliquot of the acylation reaction mixture was analyzed by HPLC after quenching with methanol. No significant change in the regioisomeric ratios was observed at different time intervals for a particular acylation reaction (ESI†).
In order to verify the efficiency of the reaction, the turn-over-frequency (TOF) was calculated for the major regioisomers in different acylations of substrates 6 and 40 catalyzed by trivalent catalyst 1. The results of the experiments are tabulated in the ESI.† The TOF ranged between 7.8 after 4 h for pivaloylation of 6 and 184 after 15 min for the isobutylation of substrate 40 (ESI S7†). All the acylation experiments were conducted in the absence of any auxiliary base. We surmise that the tertiary amine site in the catalyst acts as the protonation site as a result.52b
The above demonstrations of the site-selective acylation of diols and triols mediated by the trivalent catalyst 1 illustrate that a trimeric catalyst gains benefit from both the overall conversion and site-selectivity. The site-selectivity is inferred due to the internal hydrogen bonding among the hydroxy functionalities and the associated increases in the nucleophilicity. The internal hydrogen bonding scheme, coupled with the acyl transfer efficiency of the trivalent catalyst 1, contributes to the impressive site-selectivity of mono-O-acylation in the majority of instances. Increasing the number of catalytic moieties within a molecular scaffold increases the reactivities over and above the statistical increase of the catalytic moiety. Such trends of multivalent catalysis were observed in many occasions earlier in organometallic and organocatalysis reactions.56,57,69 Higher reaction efficiencies by multivalent catalysts are accounted for by the stabilization of the active catalytic moiety, in addition to kinetic advantages. The trivalent catalyst 1 studied herein is an organocatalyst based on DMAP acyl transfer agent. The trivalent nature of catalyst 1 presumably leads to a statistical increase in the acyl transfer and, as a result, the increased reactivity. It is likely that the individual DAAP moiety in 1 might aid in the deprotonation of the pyridinium species and an efficient re-generation of catalytically active DAAP moiety, as advanced by Hamachi and co-workers on multivalent affinity-guided DMAP (AGD) catalysts.59a These combined phenomena are inferred as a plausibility for the observed significantly increased reactivity and site-selectivity of reactions catalyzed by trivalent catalyst 1.
The α-anomeric methoxy moiety would participate in the formation of an internal hydrogen bonding with the hydroxyl functionality at C2 in glucopyranoside diol derivative 6, which subsequently led to 2-O-acyl products predominantly. A similar plausible mechanism of internal hydrogen bonding in the substrate and an enhanced reactivity of the trivalent catalyst would account for the site-selective formation of 3-O-acylation product, in the case of galactopyranoside diol substrate 18.
Catalyst development is critical to achieve both site-selectivity and conversion efficiencies. Reaction conditions, such as low temperatures and presence of a base, play an important role in achieving good selectivities and conversions. Peptide-based monovalent DMAP catalysts provide good selectivities for benzoylations, although with limited conversion efficiency.28a The selectivity and conversion efficiency of the present method are comparable in case of the Cu-bis(oxazoline)-catalyzed site-selective acetylation of glycoside 6 and benzoylation for galactopyranoside 18 using Cu-(R,R)-PhBox35 catalysts. A further case in point is the isobutyration, an acylation encountered with the sterically hindered isobutyryl moiety. Trivalent catalyst 1 mediates this reaction with substrate 6 with good selectivity and yield of C2-isobutyroyl ester. In the case of pyranoside-derived triols, it is noteworthy that the trivalent catalyst 1 affords C3 O-acylated product in much higher yields than either DMAP or substrates possessing hydrogen bond acceptor type protecting group at the C6 hydroxyl functionality in the triol.12b,c Several acylations on 6-O-trityl-protected glucopyranoside triol substrate 40 yielded C3 O-acylated products in higher percentages than that known currently.12b,c Interestingly, the benzoylation of pyranoside derivatives 40, 49 and 61 at C3, employing the catalyst 1, was different from that observed by Evtushenko, using benzoic anhydride, 2,4,6-collidine and copper(II) trifluoroacetate as promoters.42 Under this condition, benzoylation at C2 site was observed. MoO2(acac)2 as a catalyst promoted benzoylation at C3, as reported in an earlier study from the same group,70 which was similar with the observation of the present studies. Without an amplification of the intrinsic reactivity differences, acylations occur equally at the available hydroxyl functionalities in diols and triols. We undertook to exploit the enhanced reactivity of multivalent catalysts. The beneficial effects of the valencies of catalytic sites in a catalyst pertaining to site-selectivity and conversion efficiency are observed in the present study with varied acylations, ranging from acetyl, isobutyryl, pivaloyl and benzoyl esters and with diols and triols derived from D-glucopyranoside, galactopyranoside and mannopyranoside.
Conclusions
This work uncovers a catalytic, site-selective method for the mono-O-acylation of several pyranoside-derived diols and triols, employing a trivalent DAAP catalyst 1. Varied acid anhydrides were used for the acylation; the acylation and the site-selectivity are largely dependent on the intrinsic reactivity of the carbohydrate hydroxyl functionalities. 4,6-O-Benzylidene-protected glucopyranoside diol furnished 2-O-acylated product, whereas the corresponding galactopyranoside diol enabled the formation of the 3-O-acylated derivative in good yield and site-selectivity, mediated by trivalent 1 catalyst. Experiments using monovalent DMAP led to considerably lesser conversions of the desired mono-O-acyl derivatives, with diminished site-selectivity in most instances. Among the glycopyranoside derivatives, 6-O-trityl-protected glycopyranoside triols furnished the 3-O-acylated derivatives in moderate to good yields and good site-selectivities using catalyst 1.Experimental
General methods
Unless otherwise noted, all reagents were purchased from commercial sources and used as received. Solvents were dried and distilled according to literature procedures. TLC analyses were performed on commercial Merck plates coated with silica gel or neutral alumina 60 F254 (type E, 0.2 mm), and visualizations were conducted by UV radiation or using I2 as a staining agent. Silica gel (100–200 and 230–400 mesh) and neutral alumina were used for column chromatography purifications. Mass spectral characterizations were performed on ESI-QTOF operated in the positive ion mode on samples in either MeCN/water or MeOH/water solution. 1H and 13C NMR spectral analyses were performed on a spectrometer operating at 400 and 100 MHz, respectively. Processing of the FID data was performed on Bruker TopSpinTM and Mnova software with default settings. Chemical shifts are reported with respect to tetramethylsilane (TMS) for 1H NMR and the central line (77.0 ppm) of CDCl3 for 13C NMR spectra. Coupling constants (J) are reported in Hertz. Standard abbreviations s, d, t, dd, td, br. s, m, quint. and app. refer to singlet, doublet, triplet, doublet of doublet, triplet of doublet, broad singlet, multiplet, quintet and apparent, respectively. HPLC analyses were performed using an analytical silica column attached with a photodiode array detector and using pet.ether/EtOAc binary gradient elution. The first 0 to ∼3.5 min in the traces cover the void volume of each HPLC analysis, and the HPLC traces are given without the void volume.57 (0.28 g, 1.12 mmol) in THF (25 mL) was introduced to the mixture in a 0 °C ice bath via injection. The solution was stirred further for 24 h at 70 °C, filtered and the solution evaporated under reduced pressure. The resulting crude product was purified by column chromatography (neutral Al2O3) (eluant: MeOH/CH2Cl2 1:99) to secure 1 (0.46 g, 88%) as a colorless gum. 1H NMR (400 MHz, CDCl3) δ 8.16 (app. d, J = 6.4 Hz, 6 H), 6.44 (app. d, J = 6.8 Hz, 6 H), 3.33 (t, J = 7.2 Hz, 6 H), 2.91 (s, 9 H), 2.42 (t, J = 7.2 Hz, 6 H), 1.68 (quint., J = 7.2 Hz, 6 H). 13C NMR (100 MHz, CDCl3) δ 153.4, 149.8, 106.6, 51.4, 49.5, 37.5, 24.4. HRMS (ESI) m/z: [M + H]+ calcd for C27H40N7, 462.3345; found 462.3345.
:1) to secure intermediate 4 (0.14 g, 82%) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 8.18 (app. d, J = 6.4 Hz, 2 H), 6.48 (app. d, J = 6.4 Hz, 2 H), 3.55 (t, J = 6.2 Hz, 2 H), 3.50 (t, J = 7 Hz, 2 H), 2.96 (s, 3 H), 2.02 (qn, J = 6.8 Hz, 2 H). 13C NMR (100 MHz, CDCl3): δ 153.2, 149.9, 106.6, 48.5, 42.3, 37.8, 29.6. HRMS (ESI) m/z: [M + H]+ calcd for C9H14N2Cl, 185.0849; found 185.0846.
A solution of 4 (0.11 g, 0.59 mmol) in MeOH (20 mL) and Me2NH (40% in water) (0.25 mL, 1.47 mmol) was stirred for 48 h at room temperature. Solvents were removed under reduced pressure, the resulting residue diluted with CHCl3 (25 mL), washed with saturated aq. NaHCO3, brine, dried, evaporated under reduced pressure and the resulting crude product was purified (alumina, CH2Cl2/MeOH 99:1) to secure 5 (0.11 g, 95%) as a colorless liquid. 1H NMR (400 MHz, D2O) δ 7.88 (d, J = 4 Hz, 2 H), 6.40 (d, J = 8.0 Hz, 2 H), 3.16 (t, J = 7.2 Hz, 2 H), 2.75 (s, 3 H), 2.15 (t, J = 8 Hz, 2 H), 2.02 (s, 6 H), 1.53 (quint, J = 8 Hz, 2 H). 13C NMR (100 MHz, D2O): δ 154.0, 148.2, 106.7, 55.8, 48.8, 43.8, 36.7, 23.6. HRMS (ESI) m/z: [M + H]+ calcd for C11H20N3, 194.1657, found 194.1658.
30a was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 1:1) as a colorless gum (15.2 mg, 44%). 1H NMR (400 MHz, CDCl3): δ 7.51–7.49 (m, 2 H), 7.38–7.37 (m, 3 H), 5.55 (s, 1 H), 4.95 (d, J = 3.6 Hz, 1 H), 4.80 (dd, J = 9.6, 3.6 Hz, 1 H), 4.30 (dd, J = 10, 4.4 Hz, 1 H), 4.18 (t, J = 9.6 Hz, 1 H), 3.85 (td, J = 10, 4.4 Hz, 1 H), 3.76 (t, J = 10.2 Hz, 1 H), 3.57 (t, J = 9.2 Hz, 1 H), 3.41 (s, 3 H), 2.16 (s, 3 H). 13C NMR (100 MHz, CDCl3): δ 170.8, 137.0, 129.5, 128.5, 126.4, 102.2, 97.7, 81.5, 73.7, 69.0, 68.8, 62.1, 55.6, 21.1. HRMS (ESI) m/z: [M + Na]+ calcd for C16H20O7Na, 347.1107, found 347.1107.
30a was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 4:1), as a colorless gum (22.8 mg, 61%). 1H NMR (400 MHz, CDCl3): δ 7.50–7.48 (m, 2 H), 7.38–7.37 (m, 3 H), 5.54 (s, 1 H), 4.94 (d, J = 3.6 Hz, 1 H), 4.77 (dd, J = 9.6, 4 Hz, 1 H), 4.29 (dd, J = 10, 4.4 Hz, 1 H), 4.18 (t, J = 9.6 Hz, 1 H), 3.84 (td, J = 10, 4.8 Hz, 1 H), 3.75 (t, J = 10.2 Hz, 1 H), 3.55 (t, J = 9.2 Hz, 1 H), 3.39 (s, 3 H), 2.66 (sept, J = 6.8 Hz, 1 H), 1.19 (dd, J = 6.8, 2.4 Hz, 6 H). 13C NMR (100 MHz, CDCl3): δ 177.0, 137.1, 129.4, 128.5, 126.4, 102.1, 97.7, 81.5, 73.4, 69.0, 68.9, 62.1, 55.6, 33.9, 19.1, 18.9. HRMS (ESI) m/z: [M + Na]+ calcd for C18H24O7Na, 375.1420, found 375.1420.
:1), as a colorless gum (18.7 mg, 48%). 1H NMR (400 MHz, CDCl3): δ 7.50–7.48 (m, 2 H), 7.37–7.36 (m, 3 H), 5.54 (s, 1 H), 4.93 (d, J = 3.6 Hz, 1 H), 4.73 (dd, J = 9.6, 3.6 Hz, 1 H), 4.29 (dd, J = 10, 4.4 Hz, 1 H), 4.18 (t, J = 9.6 Hz, 1 H), 3.84 (td, J = 10, 4.8 Hz, 1 H), 3.75 (t, J = 10.2 Hz, 1 H), 3.55 (t, J = 9.4 Hz, 1 H), 3.38 (s, 3 H), 1.24 (s, 9 H). 13C NMR (100 MHz, CDCl3): δ 178.3, 137.1, 129.4, 128.4, 126.4, 102.1, 97.8, 81.5, 73.5, 69.0, 68.9, 62.1, 55.7, 39.0, 27.1. HRMS (ESI) m/z: [M + Na]+ calcd for C19H26O7Na, 389.1576, found 389.1574.
28b was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 3:1), as a colorless gum (17.2 mg, 42%). 1H NMR (400 MHz, CDCl3): δ 8.11–8.09 (m, 2 H), 7.60–7.57 (m, 1 H), 7.53–7.51 (m, 2 H), 7.48–7.44 (m, 2 H), 7.39–7.38 (m, 3 H), 5.58 (s, 1 H), 5.07 (d, J = 6.4 Hz, 1 H), 5.04 (dd, J = 9.6, 4 Hz, 1 H), 4.38–4.31 (m, 2 H), 3.92 (td, J = 10, 4.8 Hz, 1 H), 3.80 (t, J = 10.4 Hz, 1 H), 3.64 (t, J = 9.6 Hz, 1 H), 3.40 (s, 3 H). 13C NMR (100 MHz, CDCl3): δ 166.4, 1371., 133.5, 130.1, 129.6, 129.4, 128.6, 128.5, 126.5, 102.2, 97.9, 81.6, 74.2, 69.1, 69.0, 62.2, 55.6. HRMS (ESI) m/z: [M + Na]+ calcd for C21H22O7Na, 409.1263, found 409.1264.
:2) as a colorless gum (11.7 mg, 51%).1H NMR (400 MHz, CDCl3): δ 7.51–7.49 (m, 2 H), 7.38–7.36 (m, 3 H), 5.51 (s, 1 H), 5.12 (dd, J = 10.4, 2.8 Hz, 1 H), 4.95 (d, J = 3.2 Hz, 1 H), 4.38 (d, J = 2.8 Hz, 1 H), 4.28 (d, J = 12.8 Hz, 1 H), 4.18–4.16 (m, 2 H), 4.07 (d, J = 12.4 Hz, 1 H), 3.73 (s, 3 H), 2.14 (s, 3 H). 13C NMR (100 MHz, CDCl3): δ 171.5, 137.7, 129.1, 128.3, 126.3, 100.9, 100.4, 74.4, 71.8, 69.3, 66.8, 62.6, 55.8, 21.3. HRMS (ESI) m/z: [M + Na]+ calcd for C16H20O7Na, 347.1107, found 347.1106.
:1) as a colorless gum (15.9 mg, 64%) and as an inseparable 2/3-O-isobutyrate regioisomers (30:70). 1H NMR (400 MHz, CDCl3): δ 7.50–7.48 (m, 2.6 H), 7.36–7.35 (m, 3.9 H), 5.57 (s, 0.44 H), 5.52 (s, 1 H), 5.14 (dd, J = 10, 2.8 Hz, 0.43 H), 5.09 (dd, J = 10.4, 3.2 Hz, 1 H), 4.98 (d, J = 3.2 Hz, 0.45 H), 4.95 (d, J = 3.2 Hz, 1 H), 4.38 (d, J = 3.2 Hz, 1 H), 4.32–4.27 (m, 1.95 H), 4.18 (dd, J = 10, 3.2 Hz, 1 H), 4.13–4.05 (m, 2 H), 3.73 (s, 1.42 H), 3.47 (s, 3 H), 3.42 (s, 0.69 H), 2.66 (sept, J = 7 Hz, 1.44 H), 1.19 (d, J = 6.8 Hz, 7.88 H). 13C NMR (100 MHz, CDCl3): δ 177.6, 177.5, 137.8, 137.5, 129.4, 129.0, 128.4, 128.3, 126.4, 126.1, 101.5, 100.7, 100.4, 98.4, 77.3, 76.2, 74.3, 71.6, 71.2, 69.4, 67.4, 66.9, 62.7, 62.6, 55.9, 55.8, 34.1, 34.0, 19.3, 19.1, 19.0, 18.9. HRMS (ESI) m/z: [M + Na]+ calcd for C18H24O7Na, 375.1420, found 375.1422.
28b was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 2:1) as a colorless gum (16.3 mg, 63%) and as an inseparable 2/3-O-pivaloate regioisomers (21:79). 1H NMR (400 MHz, CDCl3): δ 7.51–7.48 (m, 2.5 H), 7.37–7.34 (m, 3.6 H), 5.57 (s, 0.3 H), 5.52 (s, 1 H), 5.06 (dd, J = 10.4, 3.2 Hz, 1 H), 4.97 (d, J = 3.6 Hz, 0.3 H), 4.93 (d, J = 3.6 Hz, 1 H), 4.38 (d, J = 3.2 Hz, 1 H), 4.30–4.27 (m, 1.5 H), 4.18 (dd, J = 10.4, 3.2 Hz, 1 H), 4.10–4.05 (m, 1.4 H), 3.80 (s, 0.14 H), 3.73 (s, 1 H), 3.47 (s, 3 H), 3.41 (s, 0.63 H), 1.24 (s, 10.9 H). 13C NMR (100 MHz, CDCl3) δ 178.9, 178.8, 137.8, 137.5, 130.0, 129.4, 128.9, 128.4, 128.2, 126.4, 125.9, 101.4, 100.5, 100.4, 98.5, 76.2, 74.2, 71.6, 71.2, 69.3, 67.4, 66.9, 62.6, 62.5, 55.9, 55.8, 39.1, 39.0, 27.2, 27.1. HRMS (ESI) m/z: [M + Na]+ calcd for C19H26O7Na, 389.1576, found 389.1574.
71 was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 3:2) as a colorless gum (14.2 mg, 52%) and as an inseparable 2/3-O-benzoate regioisomers (77:23). 1H NMR (400 MHz, CDCl3): δ 8.11–8.09 (m, 2.7 H), 7.63–7.54 (m, 1.5 H), 7.52–7.48 (m, 2.6 H), 7.46–7.42 (m, 2.6 H), 7.39–7.35 (m, 3.2 H), 5.60 (s, 0.34 H), 5.54 (s, 1 H), 5.39 (dd, J = 10, 3.6 Hz, 1.4 H), 5.13 (d, J = 3.2 Hz, 0.3 H), 5.01 (d, J = 3.6 Hz, 1 H), 4.50 (d, J = 3.2 Hz, 1 H), 4.36–4.30 (m, 3 H), 4.14–4.09 (m, 1.5 H), 3.80 (s, 1.4 H), 3.50 (s, 3 H), 3.44 (s, 0.8 H). 13C NMR (100 MHz, CDCl3) δ 166.9, 166.8, 137.8, 137.5, 133.8, 133.4, 130.3, 130.0, 129.9, 129.4, 129.0, 128.8, 128.6, 128.5, 128.4, 128.2, 126.4, 126.2, 101.6, 100.7, 100.5, 98.5, 76.3, 74.6, 72.3, 72.0, 69.4, 69.2, 67.5, 67.1, 62.8, 62.6, 55.9, 55.8. HRMS (ESI) m/z: [M + Na]+ calcd For C21H22O7Na, 409.1263, found 409.1263.
:1) as a colorless gum (10.3 mg, 36%). 1H NMR (400 MHz, CDCl3) δ 7.46–7.44 (m, 2 H), 7.36–7.26 (m, 3 H), 5.55 (s, 1 H), 5.34 (dd, J = 10.4, 3.2 Hz, 1 H), 4.75 (s, 1 H), 4.29 (dd, J = 9.6, 4.4 Hz, 1 H), 4.15 (brs, 1 H), 4.08 (t, J = 9.6 Hz, 1 H), 3.93 (td, J = 10, 4.4 Hz, 1 H), 3.86 (t, J = 10 Hz, 1 H), 3.41 (s, 3 H), 2.13 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 169.9, 137.3, 129.9, 129.2, 128.4, 126.3, 102.0, 101.4, 76.2, 70.8, 69.9, 68.9, 63.8, 55.2, 21.2. HRMS (ESI) m/z: [M + Na]+ calcd for C16H20O7Na, 347.1107, found 347.1105.
30a was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 3:1) as a colorless gum (21.2 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 7.45–7.43 (m, 2 H), 7.35–7.34 (m, 3 H), 5.56 (s, 1 H), 5.34 (dd, J = 10, 2.8 Hz, 1 H), 4.76 (s, 1 H), 4.29 (dd, J = 9.6, 4 Hz, 1 H), 4.14 (br, 1 H), 4.09 (t, J = 9.6 Hz, 1 H), 3.95–3.83 (m, 2 H), 3.41 (s, 3 H), 2.62 (sept, J = 7.2 Hz, 1 H), 1.18 (dd, J = 6.8, 4 Hz, 6 H). 13C NMR (100 MHz, CDCl3) δ 176.1, 137.4, 129.0, 128.3, 126.1, 101.7, 101.5, 76.4, 70.4, 70.1, 68.9, 63.7, 55.2, 34.2, 19.3, 19.0. HRMS (ESI) m/z: [M + Na]+ calcd for C18H24O7Na, 375.1420, found 375.1423.
13 was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 3:1) as a colorless gum (8.4 mg, 26%). 1H NMR (400 MHz, CDCl3) δ 7.43–7.42 (m, 2 H), 7.35–7.33 (m, 3 H), 5.57 (s, 1 H), 5.33 (dd, J = 10.4, 3.2 Hz, 1 H), 4.75 (s, 1 H), 4.30 (dd, J = 9.6, 4 Hz, 1 H), 4.13–4.07 (m, 2 H), 3.94–3.85 (m, 2 H), 3.40 (s, 3 H), 1.23 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 177.3, 137.4, 128.9, 128.3, 126.0, 101.6, 101.5, 76.4, 70.5, 70.1, 68.9, 63.7, 55.2, 39.2, 27.3. HRMS (ESI) m/z: [M + Na]+ calcd for C19H26O7Na, 389.1576, found 389.1576.
72 was secured after column chromatography (SiO2) (eluant: n-hexane/EtOAc 3:1) as a colorless solid (19.1 mg, 56%).1H NMR (400 MHz, CDCl3) δ 8.10–8.06 (m, 2 H), 7.63–7.55 (m, 1 H), 7.49–7.42 (m, 4 H), 7.32–7.31 (m, 3 H), 5.60 (s, 1 H), 5.56 (dd, J = 10, 3.2 Hz, 1 H), 4.80 (s, 1 H), 4.34–4.25 (m, 3 H), 4.00 (td, J = 10, 4.8 Hz, 1 H), 3.91 (t, J = 10.4 Hz, 1 H), 3.44 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 165.6, 137.4, 133.4, 130.3, 129.9, 129.1, 128.6, 128.3, 126.3, 101.9, 101.6, 76.3, 71.6, 69.9, 69.0, 63.9, 55.3. HRMS (ESI) m/z: [M + Na]+ calcd for C21H22O7Na, 409.1263, found 409.1262.
73 was secured after column chromatography (SiO2) (eluant: n-hexane–EtOAc 3:2), as a colorless gum (16.9 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 7.46–7.44 (m, 6 H), 7.33–7.29 (m, 6 H), 7.26–7.23 (m, 3 H), 5.05 (t, J = 9.4 Hz, 1 H), 4.78 (d, J = 4 Hz, 1 H), 3.71–3.66 (m, 1 H), 3.61 (t, J = 9.2 Hz, 2 H), 3.44 (s, 3 H), 3.41–3.39 (m, 2 H), 2.15 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 172.4, 143.8, 128.8, 128.1, 127.3, 99.3, 87.4, 76.5, 71.0, 70.7, 70.3, 64.2, 55.4, 21.2. HRMS (ESI) m/z: [M + Na]+ calcd For C28H30O7Na, 501.1889, found 501.1888.
:n-hexane–EtOAc 7:3), as a colorless gum (20.3 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 7.47–7.45 (m, 6 H), 7.33–7.29 (m, 6 H), 7.24–7.22 (m, 3 H), 5.03 (t, J = 9.4 Hz, 1 H), 4.79 (d, J = 4 Hz, 1 H), 3.73–3.68 (m, 1 H), 3.59–3.54 (m, 2 H), 3.46 (s, 3 H), 3.43–3.35 (m, 2 H), 2.63 (sept, J = 7 Hz, 1 H), 1.19 (dd, J = 6.8, 5.2 Hz, 6 H). 13C NMR (100 MHz, CDCl3) δ 178.7, 143.8, 128.8, 128.0, 127.3, 99.3, 87.3, 76.4, 71.0, 70.6, 70.5, 64.2, 55.4, 34.3, 19.1, 19.0. HRMS (ESI) m/z: [M + Na]+ calcd For C30H34O7Na, 529.2202, found 529.2200.
:n-hexane–EtOAc 4:1) as a colorless gum (25.7 mg, 72%). 1H NMR (400 MHz, CDCl3) δ 7.48–7.46 (m, 6 H), 7.33–7.29 (m, 6 H), 7.26–7.22 (m, 3 H), 5.02 (t, J = 9.4 Hz, 1 H), 4.80 (d, J = 3.6 Hz, 1 H), 3.76–3.71 (m, 1 H), 3.62 (br, 1 H), 3.54–3.52 (m, 1 H), 3.48 (s, 3 H), 3.45–3.44 (m, 1 H), 3.36 (dd, J = 10, 5.6 Hz, 1 H), 1.23 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 180.2, 143.9, 128.8, 128.0, 127.2, 99.2, 87.2, 76.6, 71.0, 70.6, 70.5, 64.1, 55.3, 39.1, 27.2. HRMS (ESI) m/z: [M + Na]+ calcd for C31H36O7Na, 543.2359, found 543.2357.
:n-hexane–EtOAc 3:2) as a colorless gum (18.8 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 8.10–8.06 (m, 2 H), 7.63–7.54 (m, 1 H), 7.49–7.47 (m, 6 H), 7.45–7.41 (m, 2 H), 7.33–7.29 (m, 6 H), 7.25–7.23 (m, 3 H), 5.32 (t, J = 9.2 Hz, 1 H), 4.85 (d, J = 3.6 Hz, 1 H), 3.82–3.75 (m, 3 H), 3.48 (s, 3 H), 3.43 (td, J = 12, 4 Hz, 2 H). 13C NMR (100 MHz, CDCl3) δ 167.9, 143.8, 133.4, 130.2, 130.0, 128.7, 128.4, 128.0, 127.2, 99.3, 87.2, 76.8, 71.0, 70.7, 70.4, 64.1, 55.4. HRMS (ESI) m/z: [M + Na]+ calcd for C33H32O7Na, 563.2046, found 563.2046.
74 was secured after column chromatography (SiO2) (eluant:n-hexane–EtOAc 3:2) as a colorless foam (17.1 mg, 52%) and as an inseparable regioisomeric mixture of 2-OAc/3-OAc (70:30). 1H NMR (400 MHz, CDCl3) δ 7.31–7.3 (m, 7 H), 7.28 (br, 10 H), 7.27–7.26 (m, 2 H), 5.08 (d, J = 4 Hz, 1 H), 5.06 (d, J = 3.2 Hz, 0.4 H), 4.71 (s, 1 H), 4.68 (s, 0.5 H), 4.02 (br, 1 H), 3.97–3.96 (m, 2 H), 3.93–3.91 (m, 1.4 H), 3.87–3.80 (m, 1 H), 3.64–3.57 (m, 1.4 H), 3.38 (s, 3 H), 3.36 (s, 1 H). 13C NMR (100 MHz, CDCl3) δ 171.1, 170.9, 143.8, 143.7, 128.8, 128.7, 128.1, 128.0, 127.3, 127.1, 100.6, 100.5, 87.5, 87.2, 74.3, 71.9, 70.8, 70.7, 70.0, 69.4, 69.3, 67.8, 64.7, 62.8, 55.13, 55.10, 21.3, 21.2. HRMS (ESI) m/z: [M + Na]+ calcd for C28H30O7Na, 501.1889, found 501.1886.
:n-hexane–EtOAc 7:3) as a colorless gum (20.1 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 7.47–7.45 (m, 6 H), 7.32–7.28 (m, 7 H), 7.26–7.22 (m, 2 H), 5.06 (dd, J = 10, 3.4 Hz, 1 H), 4.72 (d, J = 1.2 Hz, 1 H), 3.98 (br, 1 H), 3.91–3.87 (m, 1 H), 3.76–3.71 (m, 1 H), 3.45 (t, J = 4.6 Hz, 2 H), 3.41 (s, 3 H), 2.64 (sept, J = 6.8 Hz, 1 H), 1.19 (d, J = 8 Hz, 6 H). 13C NMR (100 MHz, CDCl3) δ 177.3, 143.8, 128.7, 128.0, 127.3, 100.5, 87.4, 74.1, 71.0, 69.4, 67.9, 64.6, 55.1, 34.1, 19.15, 19.12. HRMS (ESI) m/z: [M + Na]+ calcd for C30H34O7Na, 529.2202, found 529.2206.
:n-hexane–EtOAc 4:1) as a colorless gum (15.0 mg, 42%). 1H NMR (400 MHz, CDCl3) δ 7.48–7.46 (m, 6 H), 7.32–7.28 (m, 7 H), 7.24–7.22 (m, 2 H), 5.03 (dd, J = 9.6, 3.2 Hz, 1 H), 4.73 (d, J = 1.6 Hz, 1 H), 3.98–3.96 (m, 1 H), 3.89–3.82 (m, 1 H), 3.76–3.71 (m, 1 H), 3.49–3.44 (m, 1 H), 3.42 (s, 3 H), 3.40 (s, 1 H), 1.23 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ 178.7, 143.8, 128.8, 128.0, 127.2, 100.5, 87.3, 74.2, 71.1, 69.4, 55.1, 39.1, 27.3. HRMS (ESI) m/z: [M + K] + calcd For C31H36O7K, 559.2098, found 559.2095.
70 was secured after column chromatography (SiO2) (eluant:n-hexane–EtOAc 4:1) as a colorless gum (17.4 mg, 47%). 1H NMR (400 MHz, CDCl3) δ 8.12–8.07 (m, 4 H), 7.63–7.55 (m, 2 H), 7.49–7.42 (m, 8 H), 7.32–7.29 (m, 4 H), 7.24–7.22 (m, 2 H), 5.33 (dd, J = 9.6, 3.2 Hz, 1 H), 4.77 (d, J = 1.2 Hz, 1 H), 4.15–4.07 (m, 2 H), 3.84–3.79 (m, 1 H), 3.49 (d, J = 4.8 Hz, 2 H), 3.43 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 166.6, 143.7, 133.8, 130.3, 130.0, 128.7, 128.6, 128.1, 127.3, 100.6, 87.5, 75.0, 70.8, 69.4, 68.0, 64.7, 55.2. HRMS (ESI) m/z: [M + K]+ calcd for C33H32O7K, 579.1785, found 579.1786.
:2) as a colorless gum (6.5 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 7.45–7.43 (m, 6 H), 7.32–7.28 (m, 6 H), 7.24–7.22 (m, 3 H), 5.03 (dd, J = 10, 2.8 Hz, 1 H), 4.86 (d, J = 4 Hz, 1 H), 4.09 (app. d, J = 2 Hz, 1 H), 4.05 (dd, J = 10, 3.6 Hz, 1 H), 3.86 (t, J = 5.2 Hz, 1 H), 3.44 (s, 3 H), 3.42–3.34 (m, 2 H), 2.16 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 171.0, 143.5, 128.5, 127.9, 127.2, 99.6, 87.2, 73.3, 69.1, 68.7, 67.3, 63.5, 55.4, 21.2. HRMS (ESI) m/z: [M + Na]+ calcd for C28H30O7Na, 501.1889, found 501.1890.
:n-hexane–EtOAc 3:2) as a colorless gum (14.6 mg, 42%). 1H NMR (400 MHz, CDCl3) δ 7.46–7.44 (m, 6 H), 7.32–7.28 (m, 6 H), 7.24–7.22 (m, 3 H), 5.02 (dd, J = 10.4, 2.8 Hz, 1 H), 4.86 (d, J = 4 Hz, 1 H), 4.05 (app. d, J = 2 Hz, 2 H), 3.87 (t, J = 5.4 Hz, 1 H), 3.45 (s, 3 H), 3.38 (d, J = 5.2 Hz, 2 H), 2.66 (sept, J = 7 Hz, 1 H), 1.20 (dd, J = 6.8, 5.2 Hz, 6 H). 13C NMR (100 MHz, CDCl3) δ 177.3, 143.7, 128.7, 128.0, 127.3, 99.8, 87.3, 73.1, 69.2, 68.9, 67.5, 63.6, 55.5, 34.1, 19.2, 19.1. HRMS (ESI) m/z: [M + Na]+ calcd for C30H34O7Na, 529.2202, found 529.2206.
:n-hexane/EtOAc 3:2) as a colorless gum and as an inseparable mixture of 3-O-pivaloyl/2-O-pivaloyl (73:27) regioisomers (15.6 mg, 45%). 1H NMR (400 MHz, CDCl3) δ 7.47–7.44 (m, 8 H), 7.32–7.28 (m, 9 H), 7.24–7.22 (m, 2 H), 5.00 (dd, J = 10.4, 3.2 Hz, 1 H), 4.94 (dd, J = 10, 3.6 Hz, 0.37 H), 4.90 (d, J = 3.6 Hz, 0.36 H), 4.85 (d, J = 4 Hz, 1 H), 4.04–3.94 (m, 3 H), 3.89–3.85 (m, 1.52 H), 3.46 (s, 3 H), 3.42–3.38 (m, 1.27 H), 3.37 (s, 0.9 H), 3.36–3.33 (m, 0.6 H), 1.24 (s, 9 H), 1.23 (s, 2.50 H). 13C NMR (100 MHz, CDCl3) δ 179.1, 178.5, 143.8, 143.7, 128.7, 128.1, 128.0, 127.3, 127.2, 99.7, 97.5, 87.2, 73.2, 71.7, 70.3, 69.1, 69.0, 68.6, 68.5, 67.6, 63.5, 55.6, 55.5, 39.1, 39.0, 27.3, 27.1. HRMS (ESI) m/z: [M + Na]+ calcd for C31H36O7Na, 543.2356, found 543.2358.
70 was secured after column chromatography (SiO2) (eluant:n-hexane–EtOAc 3:2) as a colorless gum (16.3 mg, 44%). 1H NMR (400 MHz, CDCl3) δ 8.11–8.09 (m, 3 H), 7.63–7.54 (m, 2 H), 7.49–7.41 (m, 9 H), 7.32–7.29 (m, 4 H), 7.23–7.22 (m, 2 H), 5.26 (dd, J = 10, 2.8 Hz, 1 H), 4.92 (d, J = 4 Hz, 1 H), 4.24–4.20 (m, 2 H), 3.94 (dd, J = 10.8, 5.6 Hz, 1 H), 3.48 (s, 3 H), 3.41 (dd, J = 5.2, 2 Hz, 2 H). 13C NMR (100 MHz, CDCl3) δ 166.6, 143.6, 133.4, 130.3, 130.0, 128.7, 128.5, 128.0, 127.3, 99.8, 87.3, 74.1, 69.2, 68.9, 67.5, 63.5, 55.6. HRMS (ESI) m/z: [M + Na]+ calcd for C33H32O7Na, 563.2046, found 563.2048.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research is supported by a generous funding by Science and Engineering Research Board, DST, New Delhi. K. D. acknowledges Council of Industrial and Scientific Research, New Delhi, for a research fellowship.References
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- Y. van Kooyk and G. A. Rabinovich, Nat. Immunol., 2008, 9, 593–601 CrossRef CAS PubMed.
- S. Liedtke, H. Geyer, M. Wuhrer, R. Geyer, G. Frank, R. Gerardy-Schahn, U. Zahringer and M. Schachner, Glycobiology, 2001, 11, 373–384 CrossRef CAS PubMed.
- (a) P. H. Seeberger and D. B. Werz, Nat. Rev. Drug Discovery, 2005, 4, 751–763 CrossRef CAS PubMed; (b) S. S. Shivatare, V. S. Shivatare and C.-H. Wong, Chem. Rev., 2022, 122, 15603–15671 CrossRef CAS PubMed; (c) M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS PubMed; (d) L. Krasnova and C.-H. Wong, J. Am. Chem. Soc., 2019, 141, 3735–3754 CrossRef CAS PubMed; (e) C.-W. Cheng, Y. Zhou, W.-H. Pan, S. Dey, C.-Y. Wu, W.-L. Hsu and C.-H. Wong, Nat. Commun., 2018, 9, 5202 CrossRef CAS PubMed; (f) O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed.
- (a) Y. Maki, R. Okamoto, M. Izumi, T. Murase and Y. Kajihara, J. Am. Chem. Soc., 2016, 138, 3461–3468 CrossRef CAS PubMed; (b) X. Qian, K. Sujino, A. Otter, M. M. Palcic and O. Hindsgaul, J. Am. Chem. Soc., 1999, 121, 12063–12072 CrossRef CAS; (c) K. Moremen, A. Ramiah, M. Stuart, J. Steel, L. Meng, F. Forouhar, H. A. Moniz, G. Gahlay, Z. Gao, D. Chapla, S. Wang, J.-H. Yang, P. K. Prabhakar, R. Johnson, M. D. Rosa, C. Geisler, A. V. Nairn, J. Seetharaman, S.-C. Wu, L. Tong, H. J. Gilbert, J. LaBaer and D. J. Jarvis, Nat. Chem. Biol., 2018, 14, 156–162 CrossRef CAS PubMed; (d) C. E. Council, K. J. Kilpin, J. S. Gusthart, S. A. Allman, B. Linclau and S. S. Lee, Org. Biomol. Chem., 2020, 18, 3423–3451 RSC.
- (a) P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, Wiley, Hoboken, NJ, 4th edn, 2007 Search PubMed; (b) P. G. Goekjian and S. Vidal, Regioselective Protection at the Secondary Positions of Carbohydrates with Acyclic Protecting Groups, in Protecting Groups – Strategies and Applications in Carbohydrate Chemistry, ed. S. Vidal, Wiley-VCH, Weinheim, 2019, ch. 4, pp. 109–144 Search PubMed.
- (a) B. Dhakal and D. Crich, J. Am. Chem. Soc., 2018, 140, 15008–15015 CrossRef CAS PubMed; (b) A. A. Hettikankanamalage, R. Lassfolk, F. S. Ekholm, R. Leino and D. Crich, Chem. Rev., 2020, 120, 7104–7151 CrossRef CAS PubMed.
- (a) V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed; (b) K. Yamatsugu and M. Kanai, Chem. Rev., 2023, 123, 6793–6838 CrossRef CAS PubMed; (c) M. Jäger and A. J. Minnaard, Chem. Commun., 2016, 52, 656–664 RSC; (d) M. D. Witte and A. J. Minnaard, ACS Catal., 2022, 12, 12195–12205 CrossRef CAS PubMed; (e) K. Dey and N. Jayaraman, Chem. Commun., 2022, 58, 2224–2227 RSC; (f) K. Dey and N. Jayaraman, Carbohydr. Res., 2022, 520, 108610 CrossRef CAS PubMed.
- (a) A. Francais, D. Urban and J.-M. Beau, Angew. Chem., Int. Ed., 2007, 46, 8662–8665 CrossRef CAS PubMed; (b) S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed; (c) Z. Zhang, I. R. Ollmann, X.-S. Ye, R. Wischnat, T. Baasov and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef CAS.
- (a) T. Kurahashi, T. Mizutani and J.-i. Yoshida, J. Chem. Soc., Perkin Trans. 1, 1999, 465–473 RSC; (b) T. Kurahashi, T. Mizutani and J.-i. Yoshida, Tetrahedron, 2002, 58, 8669–8677 CrossRef CAS.
- E. Kattnig and M. Albert, Org. Lett., 2004, 6, 945–948 CrossRef CAS PubMed.
- (a) J. Lawandi, S. Rocheleau and N. Moitessier, Tetrahedron, 2016, 72, 6283–6319 CrossRef CAS; (b) N. Moitessier, P. Englebienne and Y. Chapleur, Tetrahedron, 2005, 61, 6839–6853 CrossRef CAS; (c) N. Moitessier and Y. Chapleur, Tetrahedron Lett., 2003, 44, 1731–1735 CrossRef CAS; (d) S. Rocheleau, J. Pottel, I. Huskic and N. Moitessier, Eur. J. Org. Chem., 2017, 646–656 CrossRef CAS; (e) J. Lawandi, S. Rocheleau and N. Moitessier, Tetrahedron, 2011, 67, 8411–8420 CrossRef CAS.
- L. Jiang and T.-H. Chan, J. Org. Chem., 1998, 63, 6035–6038 CrossRef CAS PubMed.
- F. A. Carey and K. O. Hodgson, Carbohydr. Res., 1970, 12, 463–465 CrossRef CAS.
- S. A. Abbas and A. H. Haines, Carbohydr. Res., 1975, 39, 358–363 CrossRef CAS.
- S. Kim, H. Chang and W. J. Kim, J. Org. Chem., 1985, 50, 1751–1752 CrossRef CAS.
- (a) P. Peng, M. Linseis, R. F. Winter and R. R. Schmidt, J. Am. Chem. Soc., 2016, 138, 6002–6009 CrossRef CAS PubMed; (b) T. Li, T. Li, M. Linseis, F. Wang, R. F. Winter, R. R. Schmidt and P. Peng, ACS Catal., 2020, 10, 11406–11416 CrossRef CAS.
- G. Ma, J. Deng and M. P. Sibi, Angew. Chem., Int. Ed., 2014, 53, 11818–11821 CrossRef CAS PubMed.
- (a) M. R. Heinrich, H. S. Klisa, H. Mayr, W. Steglich and H. Zipse, Angew. Chem., Int. Ed., 2003, 42, 4826–4828 CrossRef CAS PubMed; (b) R. Tandon, T. Unzner, T. A. Nigst, N. De Rycke, P. Mayer, B. Wendt, O. R. P. David and H. Zipse, Chem. – Eur. J., 2013, 19, 6435–6442 CrossRef CAS PubMed.
- M. R. Crittall, H. S. Rzepa and D. R. Carbery, Org. Lett., 2011, 13, 1250–1253 CrossRef CAS PubMed.
- S. Singh, G. Das, O. V. Singh and H. Han, Org. Lett., 2007, 9, 401–404 CrossRef CAS PubMed.
- N. D. Rycke, G. Berionni, F. Couty, H. Mayr, R. Goumont and O. R. P. David, Org. Lett., 2011, 13, 530–533 CrossRef PubMed.
- (a) J. C. Ruble, H. A. Latham and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 1492–1493 CrossRef CAS; (b) J. C. Ruble, J. Tweddell and G. C. Fu, J. Org. Chem., 1998, 63, 2794–2795 CrossRef CAS.
- (a) T. Kawabata, M. Nagato, K. Takasu and K. Fuji, J. Am. Chem. Soc., 1997, 119, 3169–3170 CrossRef CAS; (b) H. Mandai, K. Fujii, H. Yasuhara, K. Abe, K. Mitsudo, T. Korenaga and S. Suga, Nat. Commun., 2016, 7, 11297 CrossRef PubMed.
- (a) A. C. Spivey, T. Fekner and S. E. Spey, J. Org. Chem., 2000, 65, 3154–3159 CrossRef CAS PubMed; (b) E. Larionov, M. Mahesh, A. C. Spivey, Y. Wei and H. Zipse, J. Am. Chem. Soc., 2012, 134, 9390–9399 CrossRef CAS PubMed.
- R. P. Wurz, Chem. Rev., 2007, 107, 5570–5595 CrossRef CAS PubMed.
- (a) T. Kawabata, W. Muramatsu, T. Nishio, T. Shibata and H. Schedel, J. Am. Chem. Soc., 2007, 129, 12890–12895 CrossRef CAS PubMed; (b) Y. Ueda, W. Muramatsu, K. Mishiro, T. Furuta and T. Kawabata, J. Org. Chem., 2009, 74, 8802–8805 CrossRef CAS PubMed; (c) M. Yanagi, Y. Ueda, R. Ninomiya, A. Imayoshi, T. Furuta, K. Mishiro and T. Kawabata, Org. Lett., 2019, 21, 5006–5009 CrossRef CAS PubMed.
- (a) F. Huber and S. F. Kirsch, Chem. – Eur. J., 2016, 22, 5914–5918 CrossRef CAS PubMed; (b) M. L. Tong, F. Huber, E. S. T. Kaptouom, T. Cellnik and S. F. Kirsch, Chem. Commun., 2017, 53, 3086–3089 RSC.
- (a) K. S. Griswold and S. J. Miller, Tetrahedron, 2003, 59, 8869–8875 CrossRef CAS; (b) M. Sánchez-Roselló, A. L. A. Puchlopek, A. J. Morgan and S. J. Miller, J. Org. Chem., 2008, 73, 1774–1782 CrossRef PubMed.
- (a) A. Seitz, R. C. Wende, E. Roesner, D. Niedek, C. Topp, A. C. Colgan, E. M. McGarrigle and P. R. Schreiner, J. Org. Chem., 2021, 86, 3907–3922 CrossRef CAS PubMed; (b) A. Seitz, R. C. Wende and P. R. Schreiner, Chem. – Eur. J., 2023, 29, e202203002 CrossRef CAS PubMed; (c) D. Niedek, F. R. Erb, C. Topp, A. Seitz, R. C. Wende, A. K. Eckhardt, J. Kind, D. Herold, C. M. Thiele and P. R. Schreiner, J. Org. Chem., 2020, 85, 1835–1846 CrossRef CAS PubMed.
- X. Sun, H. Lee, S. Lee and K. L. Tan, Nat. Chem., 2013, 5, 790–795 CrossRef CAS PubMed.
- (a) G. Xiao, G. A. Cintron-Rosado, D. A. Glazier, B. Xi, C. Liu, P. Liu and W. Tang, J. Am. Chem. Soc., 2017, 139, 4346–4349 CrossRef CAS PubMed; (b) S. A. Blaszczyk, G. Xiao, P. Wen, H. Hao, J. Wu, B. Wang, F. Carattino, Z. Li, D. A. Glazier, B. J. McCarty, P. Liu and W. Tang, Angew. Chem., Int. Ed., 2019, 58, 9542–9546 CrossRef CAS PubMed.
- D. L. Cramer, S. Bera and A. Studer, Chem. – Eur. J., 2016, 22, 7403–7407 CrossRef CAS PubMed.
- W.-X. Lv, H. Chen, X. Zhang, C. C. Ho, Y. Liu, S. Wu, H. Wang, Z. Jin and Y. R. Chi, Chem, 2022, 8, 1518–1534 CAS.
- C. L. Allen and S. J. Miller, Org. Lett., 2013, 15, 6178–6181 CrossRef CAS PubMed.
- I.-H. Chen, K. G. M. Kou, D. N. Le, C. M. Rathbun and V. M. Dong, Chem. – Eur. J., 2014, 20, 5013–5018 CrossRef CAS PubMed.
- (a) E. Mensah, N. Camasso, W. Kaplan and P. Nagorny, Angew. Chem., Int. Ed., 2013, 52, 12932–12936 CrossRef CAS PubMed; (b) S. Wang, O. Zhelavskyi, J. Lee, A. J. Arguelles, Y. Y. Khomutnyk, E. Mensah, H. Guo, R. Hourani, P. M. Zimmerman and P. Nagorny, J. Am. Chem. Soc., 2021, 143, 18592–18604 CrossRef CAS PubMed.
- (a) Z. Zhang and C.-H. Wong, Tetrahedron, 2002, 58, 6513–6519 CrossRef CAS; (b) Y. Zhou, K. S. Liao, T. Y. Chen, Y. S. Y. Hsieh and C.-H. Wong, J. Org. Chem., 2023, 88, 7141–7151 CrossRef CAS PubMed.
- (a) Y. Demizu, Y. Kubo, H. Miyoshi, T. Maki, Y. Matsumura, N. Moriyama and O. Onomura, Org. Lett., 2008, 10, 5075–5077 CrossRef CAS PubMed; (b) F. Iwasaki, T. Maki, O. Onomura, W. Nakashima and Y. Matsumura, J. Org. Chem., 2000, 65, 996–1002 CrossRef CAS PubMed; (c) W. Muramatsu, J. Org. Chem., 2012, 77, 8083–8091 CrossRef CAS PubMed.
- J. Lv, T. Luo, Y. Zhang, Z. Pei and H. Dong, ACS Omega, 2018, 3, 17717–17723 CrossRef CAS PubMed.
- Y. X. Zhou, O. Ramstrom and H. Dong, Chem. Commun., 2012, 48, 5370–5372 RSC.
- E. V. Evtushenko, Carbohydr. Res., 2012, 359, 111–119 CrossRef CAS PubMed.
- H. Wang, J. She, L.-H. Zhang and X.-S. Ye, J. Org. Chem., 2004, 69, 5774–5777 CrossRef CAS PubMed.
- (a) D. Lee and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 3724–3727 CrossRef CAS PubMed; (b) N. Shimada, Y. Nakamura, T. Ochiai and K. Makino, Org. Lett., 2019, 21, 3789–3794 CrossRef CAS PubMed.
- (a) D. J. Gorelik, J. A. Turner and M. S. Taylor, Org. Lett., 2022, 24, 5249–5253 CrossRef CAS PubMed; (b) D. Gorelik, Y. C. Lin, A. L. Briceno-Strocchia and M. S. Taylor, J. Org. Chem., 2019, 84, 900–908 CrossRef CAS PubMed.
- D. Lee, C. L. Williamson, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2012, 134, 8260–8267 CrossRef CAS PubMed.
- S. Kuwano, Y. Hosaka and T. Arai, Chem. – Eur. J., 2019, 25, 12920–12923 CrossRef CAS PubMed.
- J. Alsarraf, L. Petitpoisson and A. Pichette, Org. Lett., 2021, 23, 6052–6056 CrossRef CAS PubMed.
- N. Shimada, T. Sugimoto, M. Noguchi, C. Ohira, Y. Kuwashima, N. Takahashi, N. Sato and K. Makino, J. Org. Chem., 2021, 86, 5973–5982 CrossRef CAS PubMed.
- (a) K. A. D'Angelo and M. S. Taylor, J. Am. Chem. Soc., 2016, 138, 11058–11066 CrossRef PubMed; (b) C. Gouliaras, D. Lee, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 13926–13929 CrossRef CAS PubMed.
- (a) B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef; (b) S. Xu, B. Kempf, H. Mayr, W. Steglich and H. Zipse, Chem. – Eur. J., 2005, 11, 4751–4757 CrossRef CAS PubMed.
- (a) A. Deratani, G. D. Darling, D. Horak and J. M. J. Fréchet, Macromolecules, 1987, 20, 767–772 CrossRef CAS; (b) A. Deratani, G. D. Darling and J. M. J. Fréchet, Polymer, 1987, 28, 825–830 CrossRef CAS; (c) E. J. Delaney, L. E. Wood and I. M. Klotz, J. Am. Chem. Soc., 1982, 104, 799–807 CrossRef CAS; (d) F. M. Menger and D. J. McCann, J. Org. Chem., 1985, 50, 3928–3930 CrossRef CAS; (e) G.-J. Wang and W. K. Fife, J. Am. Chem. Soc., 1998, 120, 883–887 CrossRef CAS; (f) H. T. Chen, S. Huh, J. W. Wiench, M. Pruski and V. S. Y. Lin, J. Am. Chem. Soc., 2005, 127, 13305–13311 CrossRef CAS PubMed.
- (a) A. Fallek, M. Weiss-Shtofman, M. Kramer, R. Dobrovetsky and M. Portnoy, Org. Lett., 2020, 22, 3722–3727 CrossRef CAS PubMed; (b) A. Fallek, O. Fleischer, F. Saady and M. Portnoy, Eur. J. Org. Chem., 2022, e202201089 CrossRef CAS; (c) N. Ashush, R. Fallek, A. Fallek, R. Dobrovetsky and M. Portnoy, Org. Lett., 2020, 22, 3749–3754 CrossRef CAS PubMed; (d) R. Fallek, N. Ashush, A. Fallek, O. Fleischer and M. Portnoy, J. Org. Chem., 2022, 87, 9688–9698 CrossRef CAS PubMed.
- (a) P. I. Kitov and D. Bundle, J. Am. Chem. Soc., 2003, 125, 16271–16284 CrossRef CAS PubMed; (b) L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS PubMed; (c) M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- G. Jayamurugan and N. Jayaraman, Adv. Synth. Catal., 2009, 351, 2379–2390 CrossRef CAS.
- A. M. Caminade, A. Ouali, M. Keller and J. P. Majoral, Chem. Soc. Rev., 2012, 41, 4113–4125 RSC.
- B. Rasmussen and J. B. Christensen, Org. Biomol. Chem., 2012, 10, 4821–4835 RSC.
- B. Helms, C. O. Liang, C. J. Hawker and J. M. J. Fréchet, Macromolecules, 2005, 38, 5411–5415 CrossRef CAS.
- (a) H. Wang, Y. Koshi, D. Minato, H. Nonaka, S. Kiyonaka, Y. Mori, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2011, 133, 12220–12228 CrossRef CAS PubMed; (b) T. Hayashi, Y. Sun, T. Tamura, K. Kuwata, Z. Song, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2013, 135, 12252–12258 CrossRef CAS PubMed; (c) Y. Koshi, E. Nakata, M. Miyagawa, S. Tsukiji, T. Ogawa and I. Hamachi, J. Am. Chem. Soc., 2008, 130, 245–251 CrossRef CAS PubMed.
- F. E. Hah and S. Rupprecht, Chem. Ber., 1991, 124, 481–486 CrossRef.
- W. Storck and G. Manecke, J. Mol. Catal., 1985, 30, 145–169 CrossRef CAS.
- B. Fischer, S. Xu and H. Zipse, Chem. – Eur. J., 2006, 12, 5779–5784 CrossRef PubMed.
- A. Sakakura, K. Kawajiri, T. Ohkubo, Y. Kosugi and K. Ishihara, J. Am. Chem. Soc., 2007, 129, 14775–14779 CrossRef CAS PubMed.
- X.-A. Lu, C.-H. Chou, C.-C. Wang and S.-C. Hung, Synlett, 2003, 1364–1366 CAS.
- B. Ren, L. Gan, L. Zhang, N. Yana and H. Dong, Org. Biomol. Chem., 2018, 16, 5591–5597 RSC.
- W. Muramatsu and T. Kawabata, Tetrahedron Lett., 2007, 48, 5031–5033 CrossRef CAS.
- G. Hu and A. Vasella, Helv. Chim. Acta, 2002, 85, 4369–4391 CrossRef CAS.
- R. Fallek, N. Ashush, A. Fallek and M. Portnoy, Synlett, 2021, 32, 1849–1854 CrossRef CAS.
- R. S. Bagul and N. Jayaraman, Inorg. Chim. Acta, 2014, 409, 34–52 CrossRef CAS.
- E. V. Evtushenko, J. Carbohydr. Chem., 2010, 29, 369–378 CrossRef CAS.
- N. Dang, V. R. N. Munasinghe and W. G. Overend, J. Chem. Soc., Perkin Trans. 1, 1983, 257–264 RSC.
- V. Cachatra, A. Almeida, J. Sardinha, S. D. Lucas, A. Gomes, P. D. Vaz, M. H. Florencio, R. Nunes, D. Vila-Vicosa, M. J. Calhorda and A. P. Rauter, Org. Lett., 2015, 17, 5622–5625 CrossRef CAS PubMed.
- C. G. Casinovi, M. Framondino, G. Randazzo and F. Siani, Carbohydr. Res., 1974, 36, 67–73 CrossRef CAS.
- D. A. MacManus and E. N. Vulfson, Carbohydr. Res., 1995, 279, 281–291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC traces of crude reaction mixture, 1H and 13C NMR spectra of all compounds. See DOI: https://doi.org/10.1039/d4ob00599f |
| This journal is © The Royal Society of Chemistry 2024 |