
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and isomerization behavior relationships of new push–pull azo-pyrrole photoswitches†
D.
Gallardo-Rosas
a,
J. M.
Guevara-Vela
 b,
T.
Rocha-Rinza
c,
R. A.
Toscano
c,
J. G.
López-Cortés
c and
M. C.
Ortega-Alfaro
*a
b,
T.
Rocha-Rinza
c,
R. A.
Toscano
c,
J. G.
López-Cortés
c and
M. C.
Ortega-Alfaro
*a
aInstituto de Ciencias Nucleares, UNAM, Circuito Exterior, Ciudad Universitaria, Coyoacán C.P. 04510, Ciudad de México, Mexico. E-mail: carmen.ortega@nucleares.unam.mx
bDepartamento de Química Física Aplicada, Universidad Autónoma de Madrid, 28049 Madrid, Spain
cInstituto de Química UNAM, Circuito Exterior, Ciudad Universitaria, Coyoacán C.P. 04510, Cuidad de México, Mexico
First published on 30th April 2024
Abstract
A family of stilbenyl-azopyrroles compounds 2a–d and 3a–d was efficiently obtained via a Mizoroki–Heck C–C-type coupling reaction between 2-(4′-iodophenyl-azo)-N-methyl pyrrole (1a) and different vinyl precursors. The influence of the π-conjugated backbone and the effect of the pyrrole moiety were correlated with their optical properties. Studies via UV-Visible spectrophotometry revealed that the inclusion of EWG or EDG favors a red-shift of the main absorption band in these azo compounds compared with their non-substituted analogues. Furthermore, there is a clear influence between the half-life of the Z isomer formed by irradiation with white light and the push–pull behavior of the molecules. In several cases, the stilbenyl-azopyrroles led to the formation of J-type aggregates in binary MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O solvents, which are of interest for water compatible applications.
:H2O solvents, which are of interest for water compatible applications.
Introduction
The development of strategies to control molecular motion is a central concept in a wide variety of areas, such as molecular machines,1 molecular recognition,2 self-assembly,3 and catalysis.4 Particularly, heteroaryl azo-compounds including nitrogen-based heterocycles such as pyridine,5 pyrrole,6 pyrazole,7 imidazole,8 benzimidazole,9 thiazole,10 benzothiazole,11 and indole12 are emerging as a new class of highly efficient and versatile molecular photoswitches. These compounds are of great interest due to their ability to carry out reversible and selective photoinduced transformations by using a defined wavelength. Additionally, these molecules exhibit nearly quantitative E–Z isomerization with tunable thermal lifetimes.8,9–12In this context, azo compounds containing a push–pull system incorporated into their structure display interesting nonlinear optic and solvatochromic properties, which can be used in the design of opto-electronic materials.12–15 The molecular polarization of push–pull systems has key implications for the optical response at the molecular level. The optimization of π-conjugated linkers, electron-donor, and electron-acceptor groups are relevant design variables to consider in this regard.16 Recently, we demonstrated that push–pull properties are efficiently modulated by using a functionalized biphenyl moiety as a π-linker, in combination with a pyrrole ring on each side of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N azo group. This molecular architecture provokes a red-shift to the visible region of the π/π* band of the azo-group, an increasing in its molar absorption coefficient,13 and a reversible photoisomerization upon irradiation using a white LED. This strategic design has been extended to some examples of push–pull azo-compounds, including a stilbenyl fragment that can be photoisomerized upon two-photon excitations.14,16
N azo group. This molecular architecture provokes a red-shift to the visible region of the π/π* band of the azo-group, an increasing in its molar absorption coefficient,13 and a reversible photoisomerization upon irradiation using a white LED. This strategic design has been extended to some examples of push–pull azo-compounds, including a stilbenyl fragment that can be photoisomerized upon two-photon excitations.14,16
As an ongoing program for the synthesis and photophysical studies of π-extended azo-heteroarene compounds, we report herein a general method for the obtainment of a family of push–pull azopyrroles bearing several stilbenyl motifs directly bonded to the NN azo group. The key synthetic strategy is based on the Mizoroki–Heck coupling reaction catalyzed by a robust palladium source as catalyst, with different vinyl precursors as coupling partners (Scheme 1).
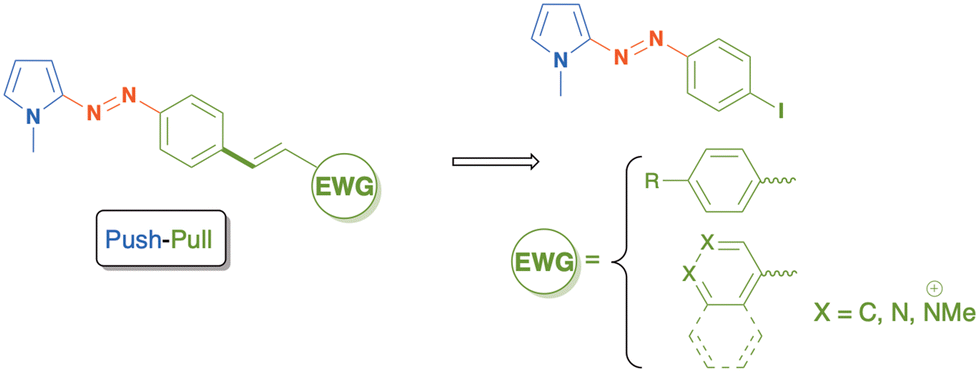 | ||
| Scheme 1 Retrosynthetic approach to obtain push–pull stilbenyl azo-pyrroles. | ||
Results and discussion
The synthesis of this family of π-extended azopyrrole compounds 2a–d began with the preparation of 1, following a well-established azo-coupling reaction between N-methylpyrrole and 4-iodoaniline.14,15 Then, we reacted precursor 1 with diverse p-substituted styrenes in presence of [Pd/(N,N)-pyrrole ligand].This palladium complex demonstrated to be a robust and efficient catalytic precursor to promote the Mizoroki–Heck coupling using demandant substrates as ethylene and other azo-pyrrole compounds (Table 1, entries 5–7).18 Using this methodology, we obtained the new π-extended azo-pyrroles dyes 2a–d as orange or red solids in good to moderated yields. In the case of 2c and 2d, we observed the formation of lateral products. The molecular identities of these compounds agree with the spectroscopical data obtained by NMR, IR and MS analyses. For instance, the 1H NMR spectrum of 2a shows a doublet at 7.76 ppm (J = 9.0 Hz) assigned to 2 hydrogens of a phenyl group adjacent to a CC double bond. Between 7.55 and 7.52 ppm, we observe a multiple signal (6H) assigned to the rest of protons of 1,4-disustituted phenyl rings. Likewise, an AB system (J = 15 Hz) at 7.16 and 7.09 ppm confirm the presence of the double bond with an E geometry. Additionally, three multiplets at 6.88, 6.66 and 6.24 ppm correspond to hydrogens in position 5, 3 and 4 (respectively) on the pyrrole ring. A singlet at 3.9 ppm is assigned to a methyl group. In the 13C NMR spectrum, we can find the corresponding signals for the double bond of the stilbene moiety at 130.6 and 127.7 ppm that confirms the C–C coupling between 1 and 4-trifluoromethyl styrene. Similar spectroscopic data are obtained for this series of azo-compounds 2a–d.
|
|
||||
|---|---|---|---|---|
| Entry | Compound | R | Time (h) | Yieldb (%) |
| a All reactions were performed with 1 mmol of 1a, 1.2 mmol of the corresponding styrene, DMF (5 mL) and 1.2 mmol of NEt3, 0.1% mol of [Pd] at 160 °C. b Isolated yield after SiO2 column chromatography. | ||||
| 1 | 2a | CF3 | 2 | 71 |
| 2 | 2b | CN | 3 | 73 |
| 3 | 2c | Br | 1 | 58 |
| 4 | 2d | OMe | 1 | 57 |
| 5 | 2e | H | 1 | 9015 |
| 6 | 2f | NO2 | 1 | 8215 |
| 7 | 2g | NMe2 | 2 | 7815 |
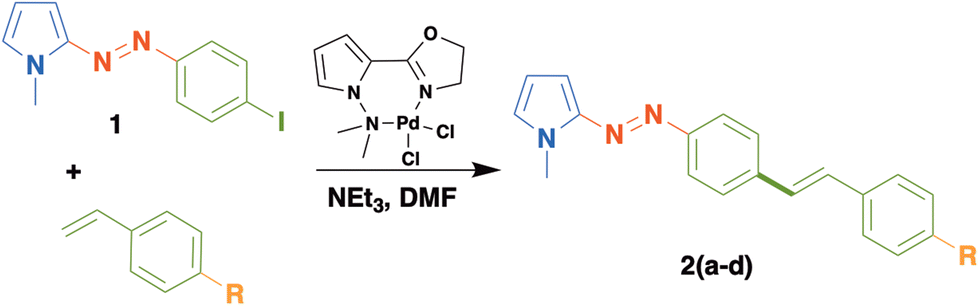
We successfully extended this methodology to other vinyl heteroarenes, thereby obtaining the π-extended azopyrroles 3a–d in good yields (Table 2). In a similar fashion to azo-dyes 2a–d, these compounds were obtained as orange and red solids in yields ranging from 53 to 82%. Again, we observed the formation of some vinyl oligomers as side products derived of lateral oligomerization of vinyl reagents, mainly for the case of the synthesis of 3c. The molecular identities of these compounds were confirmed by conventional spectroscopic techniques. For instance, the 1H NMR spectrum of 3b displays a set of doublets at 8.59 and 7.38 ppm (J = 9 Hz) assigned to the 4-pyridin fragment. At 7.83 and 7.62 ppm, two set of signals are observed for the protons of the phenyl ring adjacent to the azo group. Likewise, an AB system (J = 15 Hz) at 7.34 and 7.07 ppm confirms the double bond formed after the coupling reaction. The protons of the pyrrole ring appear at 6.96, 6.74 and 6.32 ppm. The methyl group bonded to the nitrogen atom of the pyrrole group is identified as a singlet at 3.97 ppm.
|
|
||||
|---|---|---|---|---|
| Entry | Compound | Het | Time (h) | Yieldb (%) |
| a All reactions were performed with 1 mmol of 1a, 1.2 mmol of the corresponding vinyl precursor, DMF (5 mL) and 1.2 mmol of NEt3, 0.1% mol of [Pd] at 160 °C. b Isolated yield after SiO2 column chromatography. | ||||
| 1 | 3a | Fc | 2 | 69 |
| 2 | 3b | 4-Pyridinyl | 1 | 73 |
| 3 | 3c | 3-Pyridinyl | 1 | 53 |
| 4 | 3d | 4-Quinolinyl | 2 | 82 |

Single crystals of 3b allow to confirm its structure by X-ray monocrystal diffraction (Fig. 1). The azo group exhibits an E geometry and the bond distance between the N atoms in the azo group [N(2) and N(3)] is longer than that reported for azobenzene [1.273(3) Å] but shorter than that observed in other azopyrrole dyes.14,15 This behavior reveals that although the pyridine group is a π-deficient group, its electron withdrawing effect is not comparable to that of –CF3 or –NO2 groups. Hence, the molecule is less coplanar displaying a torsion angle of approximately 10° between the plane formed by C12–C13–C6 and C13–C14–C15.
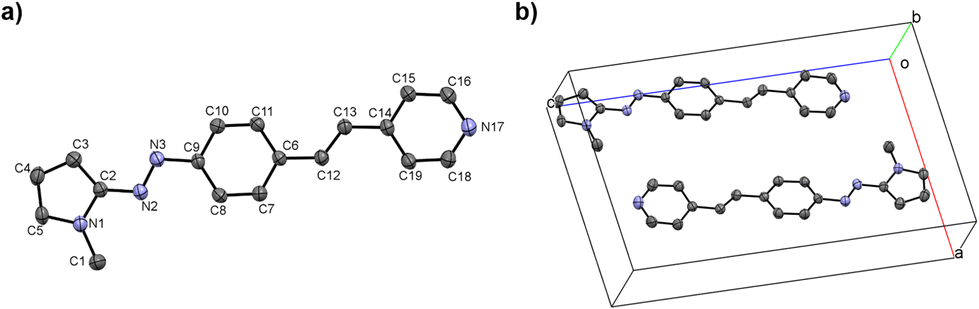 | ||
| Fig. 1 ORTEP representation of 3b. Ellipsoids are shown at 30% of probability. N(1)–C(1) 1.452(4), N(1)–C(2) 1.378(3), N(1)–C(5) 1.350(4), N(2)–N(3) 1.273(3), N(2)–C(2) 1.385(3), N(3)–C(9) 1.419(3), C(2)–C(3) 1.375(4), C(3)–C(4) 1.383(4), C(4)–C(5) 1.365(4), C(6)–C(12) 1.461(3), C(12)–C(13) 1.321(3), C(13)–C(14) 1.467(4), C(14)–C(15) 1.380(3), C(14)–C(19) 1.391(3), C(15)–C(16) 1.381(4), C(16)–N(17) 1.322(4), N(17)–C(18) 1.323(4), C(18)–C(19) 1.367(4), (b) crystal lattice of 3b. | ||
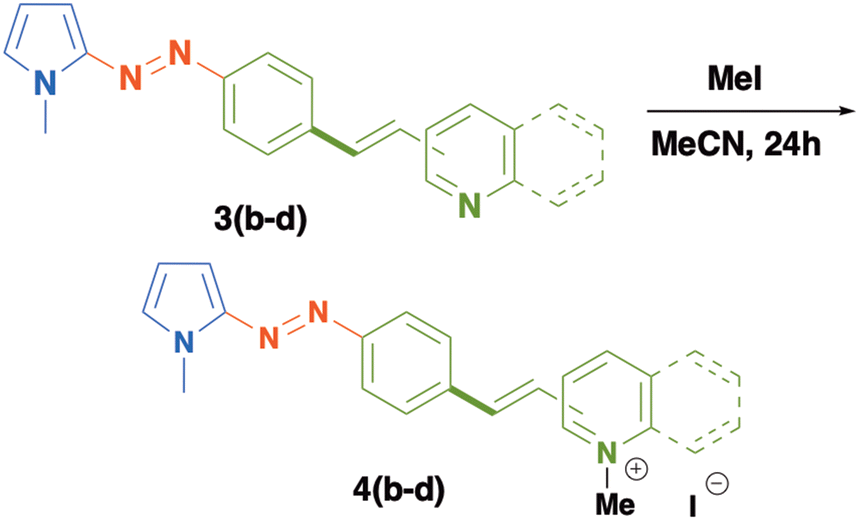
In order to modify the optical properties of 3a–c and to increase the electron acceptor character of the heterocyclic moieties included in these compounds, we carried out an alkylation reaction to obtain a new series of push–pull azo dyes 4b–d as solid salts, in excellent yields (Table 3). The formation of these new azo-compounds was confirmed by conventional spectroscopic techniques. The 1H-NMR spectrum of 4b reveals distinct changes in the chemical shifts of protons of the 4-pyridinum fragment which are shifted to higher frequencies (8.88 and 8.35 ppm) in comparison to those of compound 3b. Likewise, the protons of the double bond are shown as an AB system registered at 8.08 and 6.60 ppm (J = 15 Hz). The protons of 1,4-disubstituted phenyl rings appear as a multiplet at 7.88 ppm and the protons of the pyrrole ring are recorded at 7.38, 6.67 and 6.35 ppm. Two singlets at 4.27 and 3.96 ppm are assigned to the methyl groups included in the pyridinium fragment and in the pyrrole ring, respectively.
UV-Visible studies of stilbenylazopyrrole dyes
The UV/vis spectra of π-extended biphenyl-azopyrroles 2a–d and 3a–d acquired in MeOH are shown in Fig. 2. In general, we observe non-significant changes in the position of λmax by virtue of the substituent. These compounds display a π–π* transition near 420 nm and an n–π* transition around 440 nm as a shoulder on the π–π* transition band. When the phenyl group in azobenzene is replaced by a π-electron rich system like N-methylpyrole, we noted an important bathochromic shift of the main absorption band from 320 nm19 to 385 nm.11,15 We recently informed that this absorption can be red-shifted if a pseudostilbenyl fragment which is p-substituted with an EWG is included in the structure.15 The D–A interaction via a conjugated system and the increase in the size of the π-spacer, result in the formation of a new low-energy molecular orbital and a decrease in the HOMO–LUMO gap.14,15 With purpose of comparison, we included the spectra and optical parameters of compounds 2e–g recently informed by our group.15 | ||
| Fig. 2 UV-Visible spectra of 2a–g (top) and 3a–d (dawn) acquired in MeOH. | ||
We observed that when a moderated electron-acceptor group such as –CF3 or –CN is included in the para-position of the stilbenyl moiety, the main absorption band does not display important changes in comparison to 2e. Only in the case of a strong electron-withdrawing group such as a –NO2 group, a larger intensity and a similar red-shift of both transition bands are observed (Table 4, entries 1, 2 and 6). In contrast, for the electron-releasing group –OMe, we also observe a red-shift, but it is less important than that exhibited by the –NMe2 group15 (Table 4, entries 4 and 7). We also acquired the absorption spectra of compounds 3a–d and 4b–d in methanol (Fig. 2 and 3). Systems 3a–d display a similar behavior as the series of push–pull azodyes 2a–d, with a slight red-shift in comparison to 2e.
 | ||
| Fig. 3 Comparison of UV-Visible spectra of 3b–d (dotted line) and 4b–d (continue line) acquired in MeOH. | ||
| Entry | Comp. | R | MeOH | CHCl3 | λ onset (nm)3 | Op. Eg (eV)3 | Calc. Egap (eV)4 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max (nm) | E exp (eV) | ε (104 cm−1 M−1) | E theo (eV)1 | λ max (nm) | E exp (eV) | ε (104 cm−1 M−1) | E theo (eV)2 | ||||||
| Calculated excitation energies obtained from the computed absorption spectra in 1MeOH and 2CHCl3 at the CAM-B3LYP/Def2-TZVP level of theory using MeOH and CHCl3 as implicit solvent (SMD model). 3Data obtained from experimental UV-visible spectra acquired in MeOH. 4Frontier molecular orbitals in the minimum energy orientation for the E isomers calculated with the PBE/cc-pVDZ level of theory using MeOH as implicit solvent (SMD model). | |||||||||||||
| 1 | 2a | CF3 | 415 | 2.99 | 4.08 | 3.16 | 419 | 2.96 | 3.36 | 3.10 | 497 | 2.50 | 1.92 |
| 2 | 2b | CN | 419 | 2.96 | 3.42 | 3.11 | 423 | 2.94 | 3.30 | 3.07 | 500 | 2.48 | 1.87 |
| 3 | 2c | Br | 415 | 2.99 | 4.08 | 3.14 | 420 | 2.96 | 3.53 | 3.10 | 499 | 2.49 | 1.91 |
| 4 | 2d | OMe | 421 | 2.94 | 3.74 | 3.09 | 424 | 2.93 | 3.25 | 3.06 | 506 | 2.45 | 1.81 |
| 5 | 2e | H | 402 | 3.08 | 3.49 | 3.14 | 415 | 2.99 | 3.07 | 3.11 | 502 | 2.47 | 1.94 |
| 6 | 2f | NO2 | 426 | 2.91 | 5.65 | 3.04 | 431 | 2.88 | 4.01 | 3.03 | 511 | 2.43 | 1.64 |
| 7 | 2g | NMe2 | 460 | 2.70 | 4.99 | 2.90 | 467 | 2.66 | 3.34 | 2.90 | 539 | 2.30 | 1.48 |
With respect to 3a, the inclusion of the ferrocenyl group which behaves like an electron releasing group gave rise to additional absorptions bands between 400 and 600 nm as result of n–π* transitions and an MLCT absorption of the ferrocenyl group.20 However, when we compare the absorption spectra of 3b–d with those of compounds 4b–d, we noticed that the methylation of the nitrogen atom included in these azo-pyrrole dyes induces an important bathochromic effect and increases the intensity of the absorption (ε), in comparison to their neutral counterparts 3b–d and 2e. This observation confirms that the insertion of a stronger electron withdrawing group such as a pyridinium or quinolinium salt helps to generate zwitterionic resonance structures that contributes to decrease the energy of the excited state (Scheme 2).21 This behavior is less important in compound 4c, and hence, the smaller red-shift of its main absorption band. This circumstance occurs because the 3-pyridinium moiety acts as an electron-acceptor group only by inductive effects (Table 5, entry 6).
 | ||
| Scheme 2 Zwitterionic resonance structures of compound 4b. | ||
| Entry | Comp. | Het | MeOH | CHCl3 | λ onset (nm)3 | Op. EGap (eV)3 | Calc. Egap (eV)4 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max (nm) | E exp (eV) | ε (104 cm−1 M−1) | E calc (eV)1 | λ max (nm) | E exp (eV) | ε (104 cm−1 M−1) | E calc (eV)2 | ||||||
| Calculated excitation energies obtained from the computed absorption spectra in 1MeOH and 2CHCl3 at the CAM-B3LYP/Def2-TZVP level of theory using MeOH and CHCl3 as implicit solvent (SMD model). 3Data obtained from experimental UV-visible spectra acquired in MeOH. 4Frontier molecular orbital in the minimum energy orientation for the E isomers calculated with the PBE/cc-pVDZ level of theory using MeOH as implicit solvent (SMD model). | |||||||||||||
| 1 | 3a | Fc | 413 | 3.00 | 3.00 | 3.28 | 415 | 2.99 | 3.12 | 3.28 | 560 | 2.21 | 1.50 |
| 2 | 3b | 4-Py | 416 | 2.98 | 3.61 | 3.17 | 418 | 2.97 | 4.10 | 3.13 | 498 | 2.49 | 1.92 |
| 3 | 3c | 3-Py | 414 | 2.99 | 3.92 | 3.16 | 417 | 2.98 | 2.53 | 3.12 | 499 | 2.49 | 1.94 |
| 4 | 3d | 4-Quin | 418 | 2.97 | 2.53 | 3.16 | 423 | 2.94 | 3.07 | 3.12 | 505 | 2.46 | 1.95 |
| 5 | 4b | 4-Py(N)+Me | 454 | 2.73 | 3.98 | 3.03 | — | — | — | 2.83 | 531 | 2.37 | 1.63 |
| 6 | 4c | 3-Py(N)+Me | 422 | 2.94 | 4.04 | 3.16 | — | — | — | 3.05 | 505 | 2.45 | 1.86 |
| 7 | 4d | 4-Quin(N)+Me | 476 | 2.60 | 3.76 | 2.99 | — | — | — | 2.82 | 564 | 2.20 | 1.56 |
| E = hν = hc/λ. | (1) |
Using the approximation of the first optical absorption edge calculated from UV-vis spectra of these compounds (Fig. S30, S35, S40, S45, S50, S55, S60, S65, S70, S75 and S80†), we estimated the energy difference between the HOMO–LUMO orbitals.15,22 We used the optical absorption spectra of all samples acquired from MeOH solutions, and we compared them with the results obtained from computational studies (Tables 4 and 5) as discussed below.
Then, we calculated the energy values for the frontier orbitals HOMO/LUMO, and from those values, we also calculated the energy gap for all compounds (Tables 4 and 5). The HOMO and LUMO of all E isomers are clearly of π-nature. We observe a good trend in the obtained data, showing that the incorporation of a quinolinium moiety as EWG in 4d favors a better push–pull character (Table 5, entry 7).
We computed the absorption spectrum of the compounds 2a–2g, 3a–3d and 4b–4d in methanol [Fig. 4(a), (c) and (e)]. The error of the computed excitation energies is smaller than 0.3 eV in all cases (except for compound 4d for which the difference between the computed and the experimentally determined excitation energies is 0.39 eV). The experimental trends are overall well reproduced. For example, (i) the general order of the excitation energies throughout the series 2a–2g and 4b–4d (although the calculated energy of 2g is slightly smaller than that 2e) and (ii) the closeness in the excitation energies of the compounds 3a–3d. The calculations also predict in agreement with experiment that (i) the methylation of compounds 3b–3d to generate systems 4b–4d induces a bathochromic shift and (ii) the red-shift for the change 3c to 4c is the smallest one in this regard. We also computed similar results (Fig. 4(b) and (d)) for the excitation energies of the compounds in chloroform, concerning the ordering of the excitation energies and errors with respect to the values experimentally registered. In particular, the theoretical calculations also agree with the experimentally observed modest bathochromism when we change the solvent from methanol to chloroform. These experimental and theoretical results indicate that the electronic structure of the investigated chromophores does not change substantially because of photoexcitation. We exploited wavefunction analyses particularly the Quantum Theory of Atoms in Molecules (QTAIM)23 to inquire further about this hypothesis. The QTAIM divides the 3D-space of an electronic system in atomic basins ΩA,ΩB… based on the topological properties of the electron density, which equals the expectation value of a Dirac observable, viz.,
 | (2) |
 | ||
| Fig. 4 Computed TDDFT absorption spectra of the compounds addressed in this investigation: (a) 2a–2g in methanol, (b) 2a–2g in chloroform, (c) 3a–3d in methanol, (d) 3a–3d in chloroform and (e) 4b–4d in methanol. | ||
The QTAIM atoms are proper open quantum subsystems for which one may compute expectation values of different Dirac observables, for instance, the number of electrons within them, as
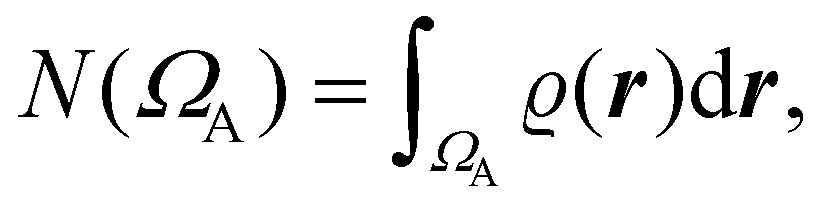 | (3) |
| Q(ΩA) = Z(ΩA) − N(ΩA), | (4) |
| ΔQ(ΩA) = Q*(ΩA) − QS0(ΩA) | (5) |
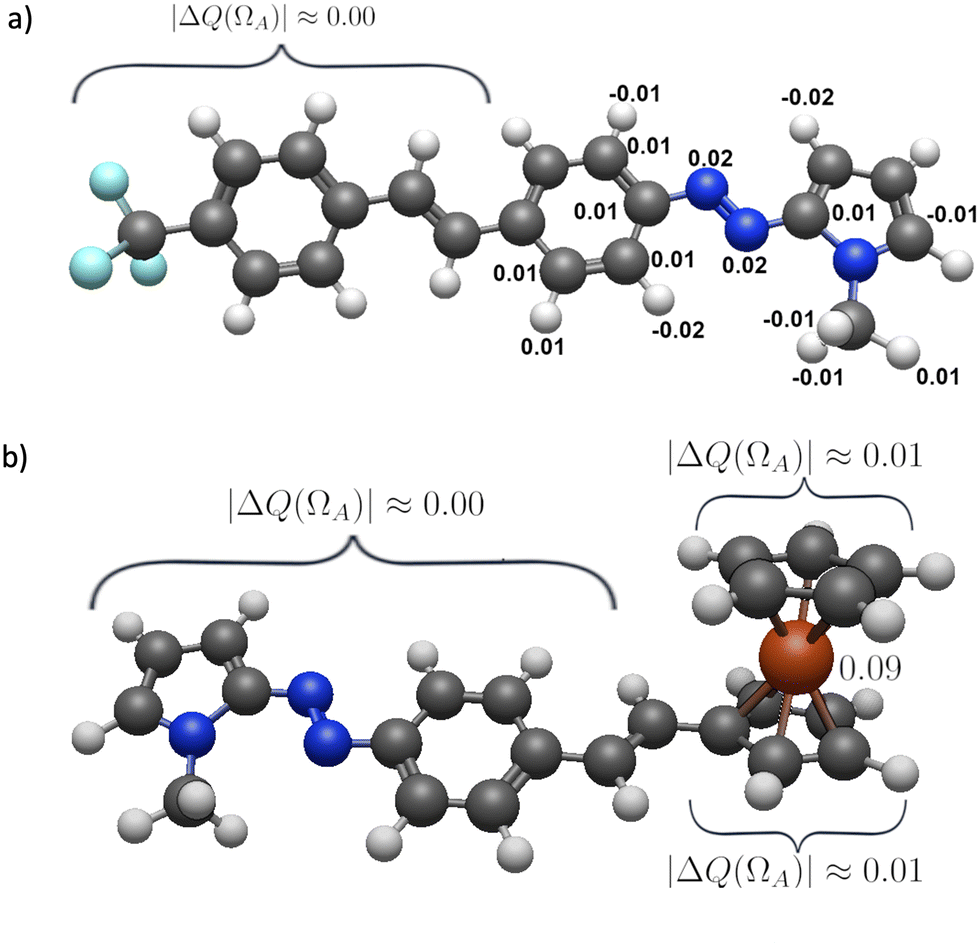 | ||
| Fig. 5 Values of ΔQ(ΩA) (eqn (4)) for the QTAIM basins within compounds (a) 2a and (b) 3a addressed in this investigation. | ||
| Entry | Compound | R | t 1/2 (min) | k (s−1) | PSS (%) | |
|---|---|---|---|---|---|---|
| Z | E | |||||
| a UV-Vis spectra acquired in MeOH at 2 × 10−5 M, E → Z PSS irradiation conducted with a white led at RT. b These data were acquired at 20 °C upon irradiation at 485 nm, using a pulsed laser.15 | ||||||
| 1 | 2a | CF3 | 7.8 | 1.5 × 10−3 | 74 | 26 |
| 2 | 2b | CN | 6.6 | 1.7 × 10−3 | 71 | 29 |
| 3 | 2c | Br | 5.6 | 2 × 10−3 | 70 | 30 |
| 4 | 2d | OMe | 2.7 | 4.2 × 10−3 | 71 | 29 |
| 5 | 2e | H | 4.6 | 2.5 × 10−3 | 72 | 28 |
| 8.6b | 1.3 × 10−3b |
72b | 28b | |||
| 6 | 2f | NO2 | 9.4 | 1.2 × 10−3 | 69 | 31 |
| 12.1b | 9.7 × 10−4b |
83b | 17a | |||
| 7 | 2g | NMe2 | 3.4 | 3.3 × 10−3 | 42 | 58 |
| 4.4b | 2.7 × 10−3b |
67b | 33b | |||
| 8 | 3a | Fc | 2 | 5.5 × 10−3 | 24 | 76 |
| 9 | 3b | 4-Py | 8.7 | 1.3 × 10−3 | 72 | 28 |
| 10 | 3c | 3-Py | 8.7 | 1.3 × 10−3 | 71 | 29 |
| 11 | 3d | 4-Quin | 5.8 | 1.9 × 10−3 | 72 | 28 |
| 12 | 4b | 4-Py(N)Me+ | 5.4 | 2.1 × 10−3 | 66 | 34 |
| 13 | 4c | 3-Py(N)Me+ | 7.8 | 1.5 × 10−3 | 73 | 27 |
| 14 | 4d | 4-Quin(N)Me+ | 0.8 | 1.2 × 10−3 | 45 | 55 |
The emission profile of the white LED exhibits two bands: a narrowband at 452 nm and a broad band in the range of 490–670 nm, which conform with the n–π* transition observed in all the azo-compounds studied allowing its use as an alternative of the common narrowband lasers (Fig. S81†).
The UV-vis spectra of 2a and the corresponding PSS after irradiation, along with several intermediate spectra are showed in Fig. 6 (top). The exponential evolution for the cis → trans isomerization, obtained from the UV-vis data is presented in Fig. 6 (dawn). The corresponding UV-Vis measurements for the series 2b–d, 3a–d and 4b–d are included in the ESI.† From the PSS spectra of Z-2a and the deconvolution of the cis/trans contributions, the absorption maxima for the cis isomer were identified at 312 and 346 nm. These absorption bands correspond to π–π* and n–π* transitions, respectively and they are blue-shifted due to the loss of conjugation in the azo compound. Likewise, we observe that the n–π* transition occurs with a higher molar absorptivity coefficient than that observed for the E-isomer. We also notice that azo-stilbenylpyrroles containing electron donor groups 2d, 2g and 3a exhibit shorter half-life times and larger rate constants in comparison to the other members of this series.
 | ||
| Fig. 6 (Top) UV-vis absorption spectra for 2a in the photo-stationary state after white led irradiation, and spectral evolution during the cis → trans thermal return. (Dawn) Absorption changes during the cis → trans thermal return at three different wavelengths. | ||
On the other hand, the azo-compounds 3b–d, which include π-deficient heteroaryls, show longer half-life times than those of 2a and 2b. The behavior of these compounds changes when the electron withdrawing character in these compounds is increased after nitrogen alkylation (4b–d). Likewise, we observe that the incorporation of a 4-quinoline moiety in 3d and 4d also modifies the kinetic parameter of the photoisomerization process. This circumstance can be attributed to the extended electronic delocalization of this fragment and the high stability of the hydrazo tautomer that enhances the rotation across the single N–N bond (Scheme 3). In general, we observe that the inclusion of a NO2 group as part of the push–pull stilbenyl-azopyrrole 2g increases the thermal stability of the cis-isomer, a condition which leads to the best photoisomerization yield.
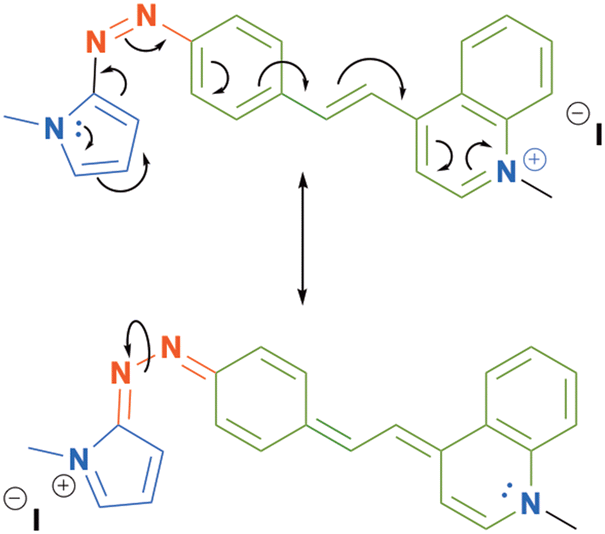 | ||
| Scheme 3 Resonance structures of the azo-compound 4d. | ||
To probe the photostability of all these compounds upon several irradiation-relaxation cycles, we prepared methanolic solutions of the series E-2(a–d), E-3(a–d) and E-4(b–d), which were exposed at least to ten irradiation-relaxation cycles using white led irradiation. These azo-compounds do not show any appreciable photobleaching (Fig. 7 and S29–S79 in the ESI†).
 | ||
| Fig. 7 Photoisomerization fatigue cycles for compounds 2a (top) and 4b (dawn). Samples were irradiated ten times for 2 minutes (5.0 mW). | ||
As the synthesized stilbenyl azopyrroles showed limited solubility in water, we can force their arrangement forming aggregates in partially aqueous medium and we can examine the system via UV/visible absorption spectroscopy.26 To study this possible aggregation process, we used MeOH:H2O as a binary solvent at the same sample concentration.
The results obtained for 3b and 4b are shown in Fig. 8. For instance, 3b exhibits a main absorption band at λmax = 416 nm, but when the proportion of water increases, the absorption band adopts an asymmetrical shape with a slight inflexion around 456 nm, showing a clear isosbestic point, suggesting the formation of J-aggregates.24b A similar behavior is observed for 3c and 3d. This behavior agreed with the molecular arrangement observed in the solid state of this compound analyzed by X-ray diffraction (Fig. 1).
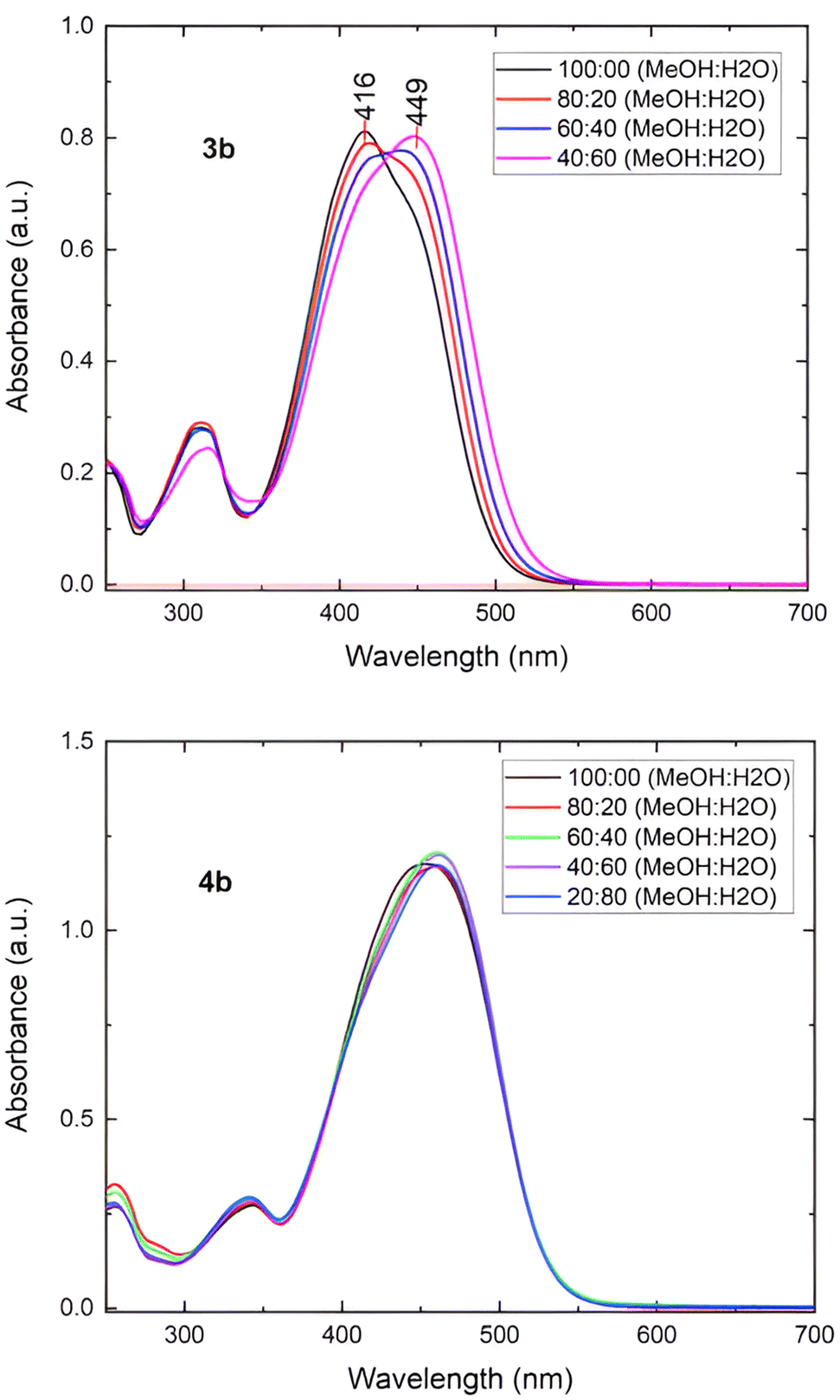 | ||
| Fig. 8 (Top) UV-visible spectra of compound 3b in different ratios MeOH/H2O. (Dawn) UV-visible spectra of compound 4b in different ratios MeOH/H2O. | ||
Likewise, to gain further insights about this behavior, we calculated the association energies of a head-to tail 3b-dimer, as a reduced model of a J-aggregate, in methanol and water (Table 7 and Fig. 9). The calculated values reveal that this compound exhibits a significant tendency to aggregate in an antiparallel form in polar solvents.
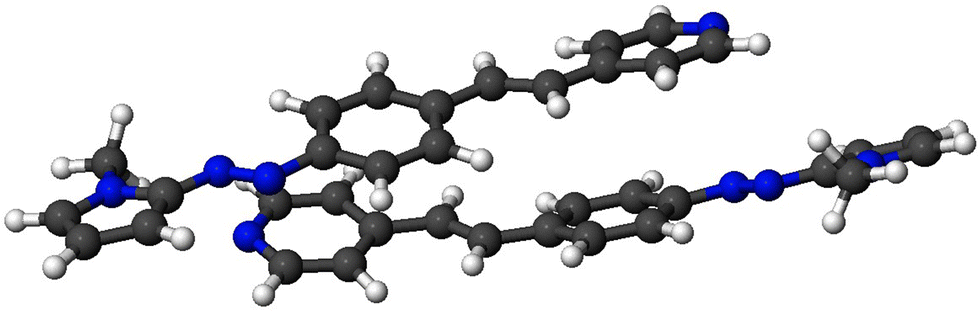 | ||
| Fig. 9 Optimization of the calculated head-to-tail dimer of 3b in MeOH. | ||
| Entry | Solvent | E as (kcal mol−1) |
|---|---|---|
| a Association energies (Eas) computed at the CAM-B3LYP/Def2-TZVP level of theory. | ||
| 1 | MeOH | 12.5 |
| 2 | H2O | 7.9 |
When we explored the aggregation behavior of 4b–d in the same conditions, we only detected a slight bathochromic effect as result of the increase in the binary solvent polarity, characteristic of these push–pull azo-pyrrole compounds.14
Finally, we studied the photoisomerization of 3b, using MeOH/water (80:20) as a binary solvent. After illumination with the white led for 1 minute, we observed a very fast thermal relaxation from the PSS to the trans-isomer, to the extent that we could not record kinetic parameters (Fig. 10). This behavior is consistent with the formation of J-aggregates and it also indicates that the thermal relaxation can be water-assisted. A similar behavior was observed in their closely related biphenyl-azopyrroles, which displayed smaller photoisomerization rate constant and thermal half-lives in these conditions.14 As we stated before, some azo-heteroaryl compounds have been used as photopharmaceutical agents.7f,9b In this kind of applications, the use of a physiological medium mainly composed by water can promote the formation of aggregates, modifying the photoisomerization kinetics of these compounds as we have demonstrated in previous reports.14
 | ||
| Fig. 10 (Top) UV-Vis spectra for 3b (2.9 × 10−5 M, MeOH/H2O 80:20) after irradiation (PSS) and cis–trans return. The spectra were acquired every minute. (Dawn) ln([C]∞ − [C]) vs. time for 3b (2.9 × 10−5 M) after white light irradiation in different ratios MeOH/H2O (the loss of linearity is observed). | ||
Experimental
General considerations
All reagents and solvents were obtained from commercial suppliers and used without further purification. Column chromatography was performed using 70–230 mesh silica gel.All compounds were characterized by IR spectra, recorded on a Perkin-Elmer Spectrum 100 FT-IR equipped with ATR accessory, and all data are expressed in wave numbers (cm−1). Melting points were obtained on a Stuart Melting Point Apparatus SMP10 and are uncorrected. NMR spectra were measured with a Bruker Avance III at 300 MHz for 1H and 75 MHz for 13C using CDCl3, and DMSO-d6, as solvents. Chemical shifts are in ppm (δ), relative to TMS. The MS-FAB spectra were obtained on a JMS-SX102A spectrometer using nitrobenzyl alcohol and polyethylene glycol matrices. MS-DART spectra were obtained on an AccuTOF JMS-T100LC spectrometer. The values of the signals are expressed in mass/charge units (m/z).
UV-Vis absorption spectra were recorded at 298 K on a Thermo-Scientific Evolution 60S UV-Vis spectrophotometer, using spectrophotometric grade solvents purchased from Sigma-Aldrich Co. and 1 cm quartz cell. The solvatochromic study was performed using 10−5 M to 10−4 M solutions of azopyrrole dyes in CHCl3 and MeOH as solvents at room temperature.
The aggregation study was carried out using 5 × 10−5 M solutions of azopyrrole dyes at different MeOH:H2O ratios, from 100:0 to 20:80 at room temperature. ASTM type 1 ultra-pure water (Millipore-Q system, 18.2 MΩ cm) was used as solvent for the aggregation studies.
29
Computational studies
We used the ORCA program30 to compute the structures of molecules at the PBEh-3c level of theory.31 The HOMO–LUMO gaps were computed using the PBE/cc-pVDZ approximation as this functional has been successfully exploited for this purpose previously in our group.14 Subsequently, TDDFT calculations were carried out using the range-separated CAM-B3LYP functional32 and the Def2-TZVP basis set.33 The SMD approach34 was employed to consider solvent effects due to methanol and chloroform. Finally, we performed QTAIM wave function analyses with the aid of the AIMAll software package35 over DFT densities obtained using the GAUSSIAN software36 following the procedure detailed in ref. 37. Finally, we used the Avogadro program for visualization.38Synthetic procedures
Conclusions
We have successfully synthetized a new family of push–pull stilbenyl azopyrrole dyes 2a–g and 3a–d using a Heck–Mizoroki cross-coupling reaction in moderated to good yields. The exploited [Pd/(N,N)-pyrrole ligand] complex was an efficient and robust catalyst for this cross-coupling reaction using compound 1 in combination with diverse vinyl arenes. The alkylation reaction over the nitrogen of pyridine or quinoline moieties in azodyes 3b–d induced an important red-shift to the visible region for the main absorption band.The theoretical studies showed that the stilbenyl-azopyrroles including an efficient EWG as part of the push–pull system (3dvs. 4d) have relatively low optical gaps energies and an effective electronic transfer, as result of the coplanarity across the π-system in the ground state. Likewise, stilbenyl-azopyrrole compounds can be photoisomerized upon irradiation into the first absorption band using a white led. We observe that the incorporation of a 4-quinoline moiety in 3d and 4d modifies the kinetic parameter of the isomerization process, which can be attributed to the extended electronic delocalization of this fragment and the high stability of the hydrazo tautomer that enhances the rotation across the single N–N bond. This behavior can be exploited for tunning the photoisomerization of the azo-compound. We have also demonstrated that these compounds can be auto-ensembled forming J-aggregates in binary-solvents as MeOH/water, and this behavior is consistent with that observed in the solid state. Indeed, we are confident that these findings will be useful to broaden the uses of these push–pull azo-switches in water compatible applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge UNAM for PAPIIT IN216123 and CONAHCYT for the Master. Sc. grant extended to D. G.-R. (887548). The authors are thankful to DGTIC-UNAM for computer time (project LANCAD-DGTIC-UNAM-250). We thank the technical assistance provided by Cesar I. Sandoval Chávez, Martin Cruz Villafañe and M. C. García-González.References
- (a) R. Costil, M. Holzheimer, S. Crespi, N. A. Simeth and B. L. Feringa, Chem. Rev., 2021, 121, 13213–13237 CrossRef CAS PubMed; (b) D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310–433 CrossRef CAS PubMed.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- (a) S. Chen, R. Costil, F. K.-C. Leung and B. L. Feringa, Angew. Chem., Int. Ed., 2021, 60, 11604–11627 CrossRef CAS PubMed; (b) Z. Yu and S. Hecht, Chem. Commun., 2016, 52, 6639–6653 RSC; (c) J. Vapaavuori, C. G. Bazuin and A. Priimagi, J. Mater. Chem. C, 2018, 6, 2168–2188 RSC; (d) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed.
- (a) A. Telleria, P. W. N. M. van Leeuwen and Z. Freixa, Dalton Trans., 2017, 46, 3569–3578 RSC; (b) T. Imahori, R. Yamaguchi and S. Kurihara, Chem. – Eur. J., 2012, 18, 10802–10807 CrossRef CAS PubMed; (c) R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed.
- Selected references: (a) G. Campillo-Alvarado, R. J. Liu, D. W. Davies and Y. Diao, Cryst. Growth Des., 2021, 21, 3143–3147 CrossRef CAS; (b) J. Long, D. Kumar, C. Deo, P. Retailleau, G. V. Dubacheva, G. Royal, J. Xie and N. Bogliotti, Chem. – Eur. J., 2021, 27, 9563–9570 CrossRef CAS PubMed; (c) E. M. Bolitho, H. E. Bridgewater, R. J. Needham, J. P. C. Coverdale, P. D. Quinn, C. Sanchez-Cano and P. J. Sadler, Inorg. Chem. Front., 2021, 8, 3675–3685 RSC; (d) B. Peng, H. Li, Y. Li, Z. Lv, M. Wu and C. Zhao, Chem. Eng. J., 2020, 395, 125079 CrossRef CAS; (e) S. Maity, K. Naskar, T. Bhowmik, A. Bera, T. Weyhermüller, C. Sinha and P. Ghosh, Dalton Trans., 2020, 49, 8438–8442 RSC.
- Selected references: (a) D. M. Adrion, D. S. Kaliakin, P. Neal and S. A. Lopez, J. Phys. Chem. A, 2021, 125, 6474–6485 CrossRef CAS PubMed; (b) M. X. He, Y.-Z. Wu, Y. Yao, Z.-Y. Mo, Y.-M. Pan and H.-T. Tanga, Adv. Synth. Catal., 2021, 363, 2752–2756 CrossRef CAS; (c) T.-T. Yin, Z.-X. Zhao and H.-X. Zhang, New J. Chem., 2017, 41, 1659–1669 RSC; (d) J. Chen and Z. Yin, Dyes Pigm., 2014, 102, 94–99 CrossRef CAS; (e) J. Calbo, C. E. Weston, A. J. White, H. S. Rzepa, J. Contreras-Garcia and M. J. Fuchter, J. Am. Chem. Soc., 2017, 139, 1261–1274 CrossRef CAS PubMed; (f) C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. White and M. J. Fuchter, J. Am. Chem. Soc., 2014, 136, 11878–11881 CrossRef CAS PubMed.
- Selected references: (a) Z.-Y. Zhang, Y. He, Y. Zhou, C. Yu, L. Han and T. Li, Chem. – Eur. J., 2019, 25, 13402–13410 CrossRef CAS PubMed; (b) J. Calbo, A. R. Thawani, R. S. L. Gibson, A. J. P. White and M. J. Fuchter, Beilstein J. Org. Chem., 2019, 15, 2753–2764 CrossRef CAS PubMed; (c) S. Vela, C. Krüger and C. Corminboeuf, Phys. Chem. Chem. Phys., 2019, 21, 20782–20790 RSC; (d) S. Devi, M. Saraswat, S. Grewal and S. Venkataramani, J. Org. Chem., 2018, 83, 4307–4322 CrossRef CAS PubMed; (e) L. Stricker, M. Böckmann, T. M. Kirse, N. L. Doltsinis and B. J. Ravoo, Chem. – Eur. J., 2018, 24, 8639–8647 CrossRef CAS PubMed; (f) C. E. Weston, A. Kramer, F. Colin, Ö. Yildiz, M. G. J. Baud, F.-J. Meyer-Almes and M. J. Fuchter, ACS Infect. Dis., 2017, 3, 152–161 CrossRef CAS PubMed.
- (a) C. Schütt, G. Heitmann, T. Wendler, B. Krahwinkel and R. Herges, J. Org. Chem., 2016, 81, 1206–1215 CrossRef PubMed; (b) G. Heitmann, C. Schütt and R. Herges, Eur. J. Org. Chem., 2016, 3817–3823 CrossRef CAS; (c) T. Wendler, C. Schütt, C. Näther and R. Herges, J. Org. Chem., 2012, 77, 3284–3287 CrossRef CAS PubMed; (d) J. Otsuki, K. Suwa, K. K. Sarker and C. Sinha, J. Phys. Chem. A, 2007, 111, 1403–1409 CrossRef CAS PubMed; (e) J. Otsuki, K. Suwa, K. Narutaki, C. Sinha, I. Yoshikawa and K. Araki, J. Phys. Chem. A, 2005, 109, 8064–8069 CrossRef CAS PubMed.
- (a) S. A. M. Steinmüller, D. Prischich, M. Odaybat, G. Galli, M. J. Fuchter and M. Decker, Chem. Sci., 2024, 15, 5360–5367 RSC; (b) S. A. M. Steinmuller, J. Fender, M. H. Deventer, A. Tutov, K. Lorenz, C. P. Stove, J. N. Hislop and M. Decker, Angew. Chem., Int. Ed., 2023, 62, e202306176 CrossRef PubMed.
- (a) C. Boga, S. Cino, G. Micheletti, D. Padovan, L. Prati, A. Mazzanti and N. Zanna, Org. Biomol. Chem., 2016, 14, 7061–7068 RSC; (b) M. M. M. Raposo, M. C. R. Castro, M. Belsley and A. M. C. Fonseca, Dyes Pigm., 2011, 91, 454–465 CrossRef CAS.
- (a) J. Garcia-Amorós, B. Maerz, M. Reig, A. Cuadrado, L. Blancafort, E. Samoylova and D. Velasco, Chem. – Eur. J., 2019, 25, 7726–7732 CrossRef PubMed; (b) U. Daswani, U. Singh, P. Sharma and A. Kumar, J. Phys. Chem. C, 2018, 122, 14390–14401 CrossRef CAS; (c) J. Garcia-Amorós, M. C. R. Castro, P. Coelho, M. M. M. Raposo and D. Velasco, Chem. Commun., 2013, 49, 11427–11429 RSC; (d) M. C. R. Castro, P. Schellenberg, M. Belsley, A. M. C. Fonseca, S. S. M. Fernandes and M. M. M. Raposo, Dyes Pigm., 2012, 95, 392–399 CrossRef CAS; (e) H. Faustino, C. R. Brannigan, L. V. Reis, P. F. Santos and P. Almeida, Dyes Pigm., 2009, 83, 88–94 CrossRef CAS.
- (a) N. A. Simeth, A. Bellisario, S. Crespi, M. Fagonnni and B. König, J. Org. Chem., 2019, 84, 6565–6575 CrossRef CAS PubMed; (b) S. Crespi, N. A. Simeth, A. Bellisario, M. Fagnoni and B. König, J. Phys. Chem. A, 2019, 123, 1814–1823 CrossRef CAS PubMed; (c) N. A. Simeth, S. Crespi, M. Fagnoni and B. König, J. Am. Chem. Soc., 2018, 140, 2940–2946 CrossRef CAS PubMed.
- (a) J. Geng, Y. Dai, H. F. Qian, N. Wang and W. Huang, Dyes Pigm., 2015, 117, 133–140 CrossRef CAS; (b) A. C. Razus, L. Birzan, M. Cristea, V. Tecuceanu and C. Enache, Dyes Pigm., 2012, 92, 1166–1176 CrossRef CAS; (c) M. M. M. Raposo, A. M. F. P. Ferreira, M. Amaro, M. Belsley and J. C. V. P. Moura, Dyes Pigm., 2009, 83, 59–65 CrossRef CAS.
- J. A. Balam-Villarreal, B. J. López-Mayorga, R. A. Toscano, M. P. Carreón-Castro, V. Basiuk, F. Cortés-Guzmán, J. G. López-Cortés and M. C. Ortega-Alfaro, Org. Biomol. Chem., 2020, 18, 1657–1670 RSC.
- L. Muñoz Rugeles, D. Gallardo-Rosas, J. Durán-Hernández, R. López-Arteaga, R. A. Toscano, N. Esturau-Escofet, J. G. López Cortés, J. Peon Peralta and M. C. Ortega Alfaro, ChemPhotoChem, 2020, 4, 144–154 CrossRef.
- (a) M. C. R. Castro, M. Belsey and M. M. M. Raposo, Dyes Pigm., 2016, 128, 89–95 CrossRef CAS; (b) F. Bures, RSC Adv., 2014, 4, 58826–58861 RSC; (c) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- E. Villatoro, L. Muñoz-Rugeles, J. Durán-Hernández, B. Salcido, N. Esturau-Escofet, J. G. López-Cortés, M. C. Ortega-Alfaro and J. Peón, Chem. Commun., 2021, 57, 3123–3126 RSC.
- F. Hochberger-Roa, S. Cortés-Mendoza, D. Gallardo-Rosas, R. A. Toscano, M. C. Ortega-Alfaro and J. G. López-Cortés, Adv. Synth. Catal., 2019, 361, 4055–4064 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- (a) N. V. Pilipchuk, G. O. Kachkovsky, L. Slominskii Yu and O. D. Kachkovsky, Dyes Pigm., 2006, 71, 1–9 CrossRef CAS; (b) B. López-Mayorga, C. I. Sandoval-Chávez, P. Carreón-Castro, V. M. Ugalde-Saldívar, F. Cortés-Guzmán, J. G. López-Cortés and M. C. Ortega-Alfaro, New J. Chem., 2018, 42, 6106–6113 RSC.
- H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482–2506 CrossRef CAS PubMed.
- (a) J. C. S. Costa, R. J. S. Taveira, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS; (b) T. Michinobu, C. Boudon, J. P. Gisselbrecht, P. Seiler, B. Frank, N. N. P. Moonen, M. Gross and F. Diederich, Chem. – Eur. J., 2006, 12, 1889–1905 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- (a) P. W. Bohn, Annu. Rev. Phys. Chem., 1993, 44, 37–60 CrossRef CAS; (b) K. S. Kumar and A. Patnaik, Chem. – Eur. J., 2011, 17, 5327–5343 CrossRef CAS PubMed.
- (a) J. Jin, L. S. Li, Y. J. Zhang, Y. Q. Tian, S. Jiang, Y. Zhao, Y. Bai and T. J. Li, Langmuir, 1998, 14, 5231–5236 CrossRef CAS; (b) Z. Chen, C. Zhong, Z. Zhang, Z. Li, L. Niu, Y. Bin and F. Zhang, J. Phys. Chem. B, 2008, 112, 7387–7394 CrossRef CAS PubMed.
- M. Kasha, Radiat. Res., 1963, 20, 55–71 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Canalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- Crystal data of 3b: C18H16N4, M = 288.35, monoclinic, a = 11.0543(16), b = 7.6061(11), c = 18.348(3) Å, V = 1529.3(4) Å3, T = 298 K, space group P21, Z = 4, μ(Mo-Kα) = 0.077 mm−1, 8576 reflections measured, 3932 unique (Rint = 0.0604) (which were used in all calculations. The final R and wR(F2) were 0.1570 and 0.1398 respectively (all data), CCDC 2189449.†.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, J. Chem. Phys., 2015, 143, 054107 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. J. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019 (https://aim.tkgristmill.com) Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- E. I. Sánchez-Flores, R. Chávez-Calvillo, T. A. Keith, G. Cuevas, T. Rocha-Rinza and F. Cortés-Guzmán, J. Comput. Chem., 2014, 35, 820–828, DOI:10.1002/jcc.23559.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- (a) J. Pschierer, N. Peschek and H. Plenio, Green Chem., 2010, 12, 636–642 RSC; (b) A. Gordillo, E. de Jesus and C. López-Mardomingo, Chem. Commun., 2007, 39, 4056–4058 RSC; (c) T. Li, F. Guo, Z. Zha and Z. Wang, Chem. – Asian J., 2013, 8, 534–537 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of all compounds. CCDC 2189449 (3b). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00417e |
| This journal is © The Royal Society of Chemistry 2024 |