
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical synthesis of site-selective advanced glycation end products in α-synuclein and its fragments†
Clara
Bosbach
ab,
Luisa Maria
Gatzemeier
ab,
Katja Ilme
Bloch von Blottnitz
a,
Annekatrin
König
b,
Ulf
Diederichsen‡
a,
Claudia
Steinem
 *a and
Tiago Fleming
Outeiro§
*bcd
*a and
Tiago Fleming
Outeiro§
*bcd
aInstitute of Organic and Biomolecular Chemistry, Georg-August-University Göttingen, Tammannstraße 2, 37077 Göttingen, Germany
bDepartment of Experimental Neurodegeneration, Center for Biostructural Imaging of Neurodegeneration, University Medical Center Göttingen, Waldweg 33, 37073 Göttingen, Germany. E-mail: touteir@gwdg.de
cTranslational and Clinical Research Institute, Faculty of Medical Sciences, Newcastle University, Framlington Place, Newcastle Upon Tyne, NE2 4HH, UK
dMax Planck Institute for Multidisciplinary Sciences, Hermann-Rein-Straße 3, 37075 Göttingen, Germany
First published on 8th March 2024
Abstract
Advanced glycation end products (AGEs) arise from the Maillard reaction between dicarbonyls and proteins, nucleic acids, or specific lipids. Notably, AGEs are linked to aging and implicated in various disorders, spanning from cancer to neurodegenerative diseases. While dicarbonyls like methylglyoxal preferentially target arginine residues, lysine-derived AGEs, such as N(6)-(1-carboxymethyl)lysine (CML) and N(6)-(1-carboxyethyl)lysine (CEL), are also abundant. Predicting protein glycation in vivo proves challenging due to the intricate nature of glycation reactions. In vitro, glycation is difficult to control, especially in proteins that harbor multiple glycation-prone amino acids. α-Synuclein (aSyn), pivotal in Parkinson's disease and synucleinopathies, has 15 lysine residues and is known to become glycated at multiple lysine sites. To understand the influence of glycation in specific regions of aSyn on its behavior, a strategy for site-specific glycated protein production is imperative. To fulfill this demand, we devised a synthetic route integrating solid-phase peptide synthesis, orthogonal protection of amino acid side-chain functionalities, and reductive amination strategies. This methodology yielded two disease-related N-terminal peptide fragments, each featuring five and six CML and CEL modifications, alongside a full-length aSyn protein containing a site-selective E46CEL modification. Our synthetic approach facilitates the broad introduction of glycation motifs at specific sites, providing a foundation for generating glycated forms of synucleinopathy-related and other disease-relevant proteins.
Introduction
Post-translational modifications (PTMs)1 are among other crucial factors responsible for the accumulation of misfolded proteins.2 As an example, α-synuclein (aSyn), a 140 amino acid protein strongly associated with Parkinsons disease (PD) and related synucleinopathies, is prone to misfolding and self-association into high molecular weight species, ultimately forming Lewis bodies. PTMs have a direct impact on the aggregation and toxicity of this protein. aSyn undergoes several PTMs such as acetylation and glycation,3 as well as oxidation, phosphorylation, nitration, sumoylation, and ubiquitination.4 Previously, we have shown that acetylation protects aSyn from aggregation and toxicity,5 whereas glycation exacerbates oligomerization and cytotoxicity, which is a generally observed phenomenon in neurodegenerative diseases,6,7 inducing dopaminergic neuronal death.3 Besides misfolding and aggregation of aSyn being a key process in synucleinopathies, glycation can also affect the protein's normal physiological function, which is still not fully understood but is thought to be partially related to synaptic vesicle biology.3,8–12To directly address glycation in aSyn, we here focussed on a synthetic route to produce advanced glycation end products (AGEs).13 AGEs are formed mainly under oxidative stress and can initiate a proinflammatory cascade by binding to the multiligand surface receptor for AGEs (RAGE), leading to impaired cellular defense mechanisms and increased cell death.14 AGEs are predominantly found at the amino side-chains of lysine (Lys) and arginine (Arg), either through direct glycation with reactive dicarbonyls or via the formation of a Schiff base and Amadori product as early-stage glycation intermediates along the Maillard pathway.15,16 To date, almost 20 different and very diverse AGEs have been described.15 Among them, N(6)-(1-carboxymethyl)lysine (CML) and N(6)-(1-carboxyethyl)lysine (CEL) are particularly highly abundant modifications (Fig. 1).17,18 Both are often referred to as glycoxidation products because of the requirement for glycation as well as oxidation in their formation.19
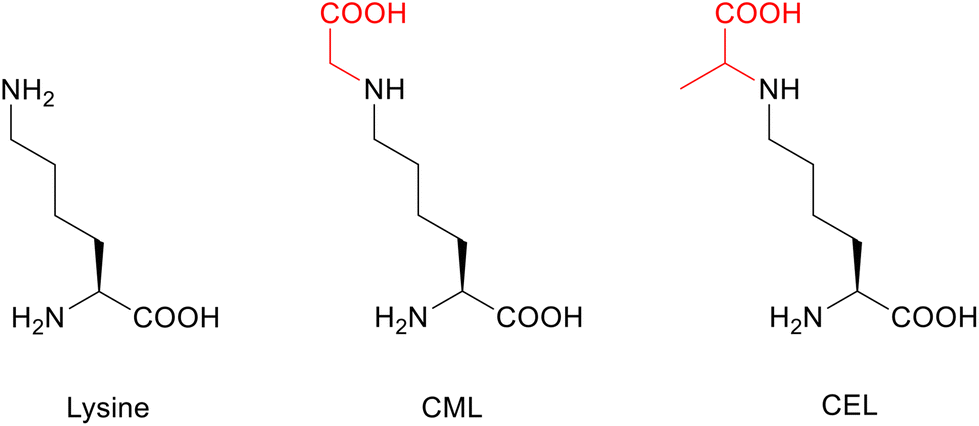 | ||
| Fig. 1 Chemical structures of the amino acid lysine (Lys) and its glycated forms CML and CEL. Glycations are highlighted in red. | ||
The amphiphilic N-terminus (amino acids 1–60, segment 1) (Fig. 2a) of aSyn harbors a large number of lysine residues (11 of the total 15 Lys in aSyn) providing a significant positive charge density strongly contributing to the protein's interaction with membranes that are negatively charged.20 The hydrophobic core region, also known as the non-amyloid component (NAC) region, (amino acids 61–95, segment 2) is prone to aggregation and is distinguished from the C-terminus (amino acids 96–140, segment 3) (Fig. 2a).21
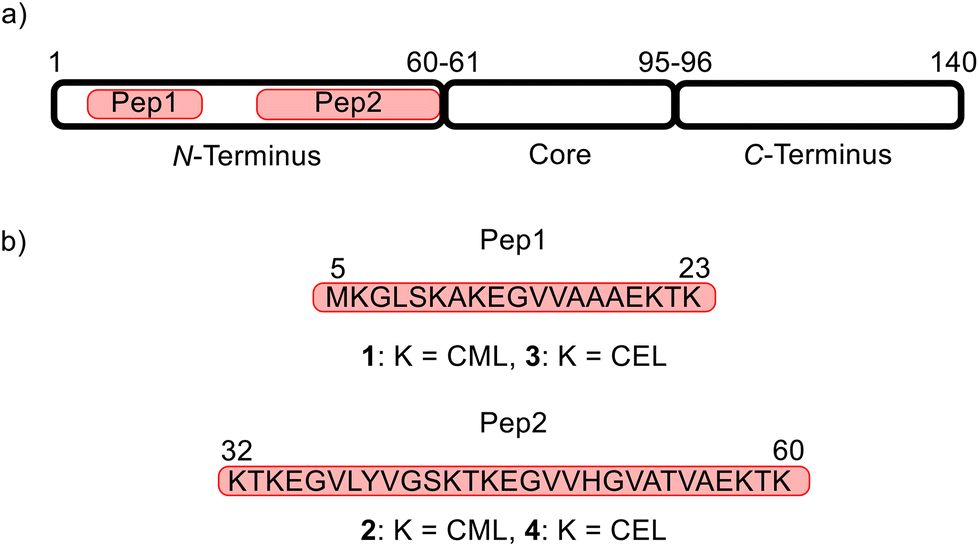 | ||
| Fig. 2 Design of model peptides. (a) Localization of aSyn model peptides in the full-length sequence (highlighted in red). The three functional fragments of aSyn are depicted as black-surrounded blocks. The numbers are the amino acid positions. (b) Amino acid sequence of the target peptides Pep1 (amino acids 5–23) and Pep2 (amino acids 32–60) with carboxymethylated (1 and 2) and carboxyethylated variants (3 and 4). | ||
Glycation targeting one or several lysine residues reduces the molecule's total charge, thereby affecting the binding and folding of aSyn.20,22 Thus, glycation can induce misfolding and accumulation of oligomeric species, which can lead to neuronal dysfunction and death.3,23,24 However, despite the intense study of aSyn glycation, current in vitro glycation methods, involving methylglyoxal (MGO) for CEL and ribose for CML, lack specificity and do not allow selectively targeting multiple defined positions within the protein's sequence.25,26 Recently, Mariño et al. yielded a homogeneously CEL-glycated aSyn with all 15 Lys residues glycated.27 Others made great efforts in incorporating single CML and CEL as designed building blocks for solid-phase peptide synthesis (SPPS) into peptide structures.28,29 However, to the best of our knowledge, the selective glycation of more than two Lys residues in one peptide has not yet been achieved.30
Here, we address these limitations and report on the modification of two N-terminal fragments of aSyn with five and six CML and CEL glycations by combining methods for orthogonal peptide protection and on-resin reductive amination techniques with SPPS. Furthermore, the native chemical ligation (NCL) of chemically synthesized aSyn fragments yielded the full-length protein with a defined E46CEL glycation, modeling the possible effect of the E46K substitution found in familial forms of PD.31 The newly developed approach will facilitate access to multiple defined, site-selective glycations in aSyn and other proteins prone to glycation.
Results and discussion
Design of CML and CEL-glycated peptides and synthetic strategy
The Lys residues of the N-terminus of aSyn (Fig. 2a) are particularly affected by glycation.3 To develop a method that allows the simultaneous glycation of several lysine (Lys, K) residues in one peptide, two aSyn peptide mimics consisting of 19 and 29 amino acids were designed (Fig. 2). These peptides contain all 11 Lys positions in the N-terminus of aSyn and their lengths make them readily accessible by common SPPS procedures. Pep1 harbors five Lys residues with a total length of 19 amino acids, whereas Pep2 contains six Lys residues with a total length of 29 amino acids (Fig. 2b). The goal was to obtain the target structures 1 (carboxymethylated Pep1), and 2 (carboxymethylated Pep2), as well as 3 (carboxyethylated Pep1) and 4 (carboxyethylated Pep2).We started the synthesis with N-terminal fluorenylmethoxy-carbonyl (Fmoc)-protected and side-chain tert-butyloxycarbonyl/tert-butyl (Boc/tBu)-protected CML and CEL building blocks based on the work of Gruber et al. with slight modifications.28 However, using these building blocks during SSPS did not result in the desired peptides 2, 3, and 4. Only 1 could be synthesized in pure form with moderate yield.
We thus pursued a different approach. First, Pep1 and Pep2 were synthesized with orthogonal side-chain protected Lys that are selectively deprotected after SPPS while maintaining all other side-chain protecting groups of the peptide on resin.32 For the orthogonal protection we introduced all Lys as Fmoc-Lys(Alloc)-OH protected building blocks during SPPS (Fig. 3). The Alloc-group was effectively and selectively removed by treatment with a palladium catalyst and phenylsilane.33 Then, the free primary amino groups in the Lys side-chains were glycated by reductive amination with the protected carbonyl components (aldehyde or ketone, respectively). This two-step procedure inspired by Pels et al. follows the natural mechanism of the reductive amination reaction.34 Acidic cleavage from the resin yielded all desired modified peptides with Lys residues replaced by CML or CEL. Of note, the general approach with Alloc- or nosylamide-(Ns) protected Lys is literature known. However, reports on the simultaneous glycation of more than one position are rare.35–37
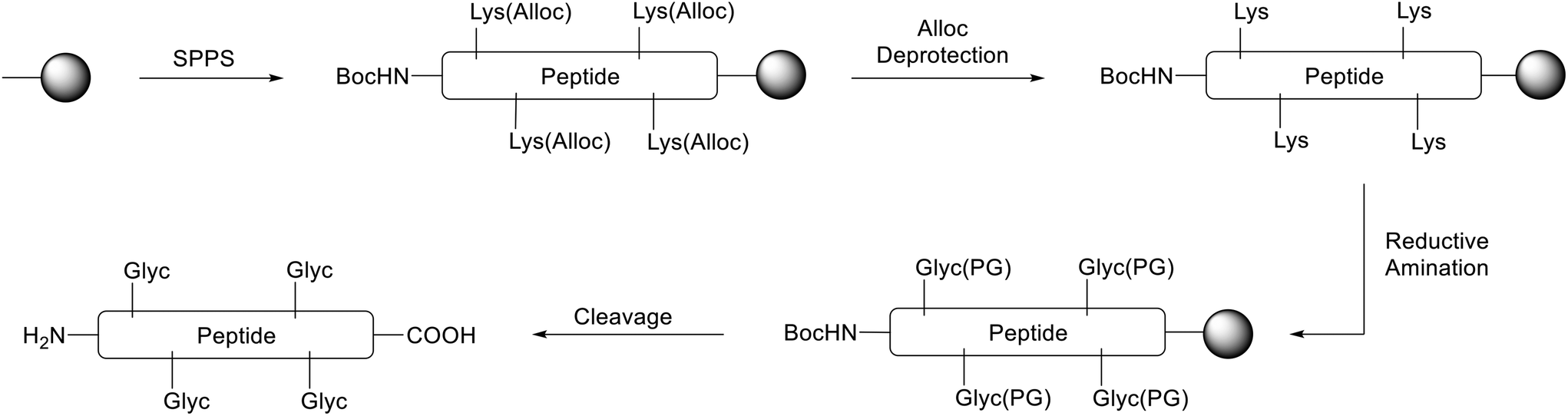 | ||
| Fig. 3 Synthetic strategy to obtain glycated peptides (example with four Lys, schematically highlighted). The solid phase resin is depicted as black dot. First, SPPS with Fmoc-Lys(Alloc)-OH for each Lys in the peptide was used to obtain the protected peptide. After orthogonal Alloc deprotection, reductive amination with the protected carbonyl compound yields the side-chain protected glycated Lys residues (Glyc(PG)). The final cleavage step releases the modified glycated peptide carrying glycated Lys residues (Glyc). | ||
Synthesis of CML-glycated aSyn peptides 1 and 2
First, the tBu-protected aldehyde 6 (Fig. 4), required for reductive amination, was synthesized according to Yao et al. and Yamamoto et al. yielding pure tBu-protected oxoacetate in 19% yield over two steps.38,39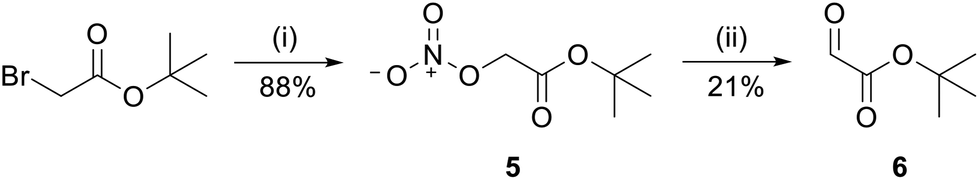 | ||
| Fig. 4 Synthesis of reactive aldehyde 6 for CML peptides. tBu-bromoacetate was treated with AgNO3 in acetonitrile (MeCN) to obtain nitrate 5 which was subsequently oxidized with sodium acetate (NaAc) in dimethyl sulfoxide (DMSO) to target aldehyde 6. Reagents and conditions: (i) 2.00 eq. AgNO3, MeCN, rt, 48 h; (ii) 0.88 eq. NaAc, DMSO, rt, 20 min. | ||
The peptides Pep1 and Pep2 were coupled automatically on a LibertyBlue® Synthesizer with Fmoc-Lys(Alloc)-OH preloaded Wang resin using N-terminal Fmoc-protected and acid labile side-chain protected amino acids and Alloc side-chain protected Lys residues. The final amino acid was introduced with an N-terminal Boc-protection. After Alloc deprotection, the resin was treated with 15 eq. of aldehyde 6 for each Lys to obtain the respective imine intermediate. The latter was finally reduced with NaBH4 yielding the carboxymethylated compound (Fig. 5). After acidic cleavage from the resin and HPLC purification, peptides 1 and 2 were obtained in 8% and 4% yield, respectively. Both peptides were of high purity (>99% for 1 and 95% for 2 as calculated from UHPLC analysis).
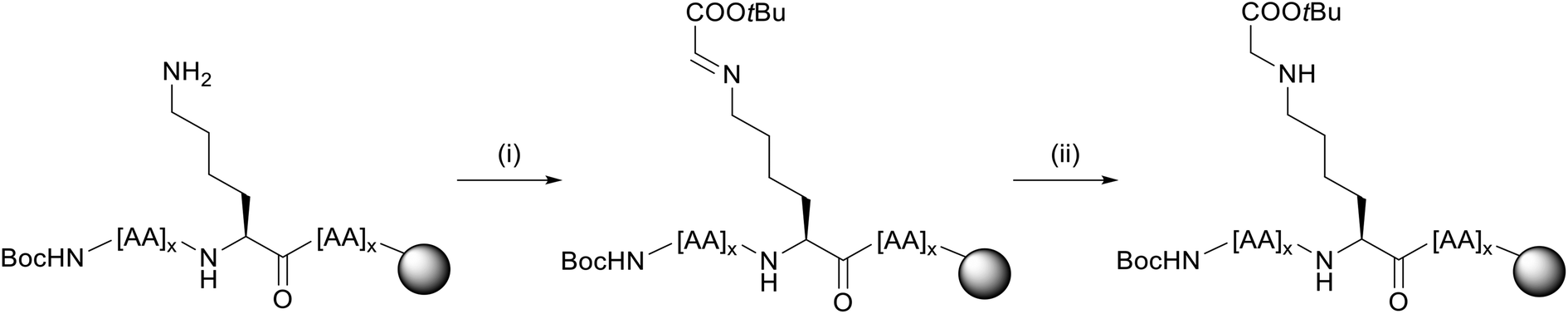 | ||
Fig. 5 Reductive amination procedure for CML peptides 1 and 2 (example for a single Lys). For each Lys in the sequence, 15 eq. of aldehyde 6 and 10 eq. of reducing agent NaBH4 were used. Reagents and conditions: (i) 15 eq. 6, dimethylformamide (DMF), rt, 1 h; (ii) 10 eq. NaBH4, dichlormethane/methanol (DCM/MeOH) 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, rt, 1 h. :1, rt, 1 h. | ||
Synthesis of CEL-glycated aSyn peptides 3 and 4
For peptides 3 and 4, a similar procedure was pursued. In this case, the ketone 7 was required for reductive amination. Ketone 7 was synthesized by protecting pyruvic acid with tert-butanol in the presence of pyridine and mesyl chloride (Fig. 6).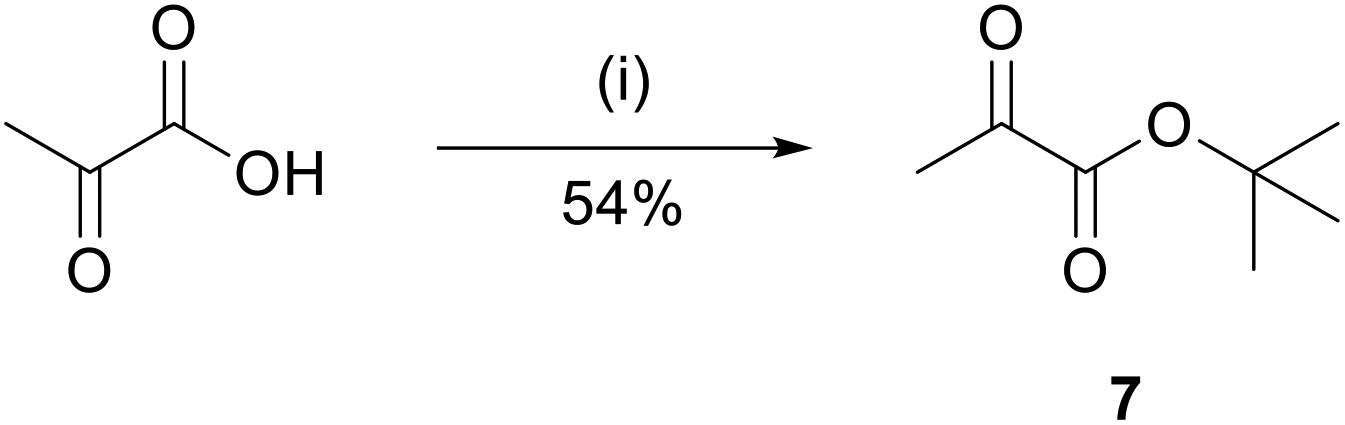 | ||
| Fig. 6 Synthesis of tBu-protected ketone 7. Pyruvic acid was treated with tert-butanol, pyridine and mesyl chloride in tetrahydrofuran (THF). Reagents and conditions: (i) 2.0 eq. tert-butanol, 2.5 eq. pyridine, 1.2 eq. mesyl chloride, THF, 0 °C to rt, over night. | ||
The product was obtained in 54% yield.40 For this procedure, the reductive amination was performed in one step, with 15 eq. of ketone 7 and 15 eq. of reducing agent NaBH3CN per Lys present in the peptide sequence (Fig. 7).41 Upon cleavage and HPLC, the purified peptides were obtained in 14% yield and 95% purity (3) and 3% yield and 95% purity (4).
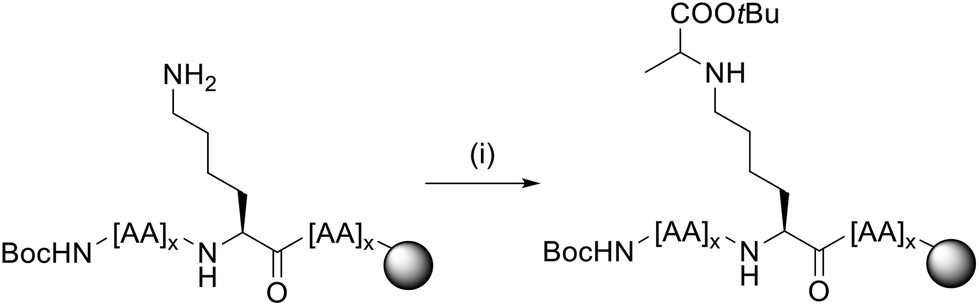 | ||
| Fig. 7 Reductive amination procedure for CEL peptides 3 and 4 (example for a single Lys). For each Lys in the sequence, 15 eq. of ketone 7 and 15 eq. of reducing agent NaBH3CN were used. Reagents and conditions: (i) 15 eq. 7, 15 eq. NaBH3CN, N-methyl-2-pyrrolidone (NMP)/isopropyl alcohol 3:1 + 5% acetic acid, rt, 24 h. | ||
Synthesis of full-length aSyn with E46K and E46CEL
Based on the successfully established carboxymethylation and carboxylethylation strategy of defined aSyn motifs using Alloc side-chain protected Lys, we further exploited the method to modify the full-length aSyn with one CEL glycation at amino acid position 46. In natural aSyn, amino acid 46 is a glutamic acid (Glu, E). This position is of particular interest, as the point mutation E46K is related to an early onset of familiar forms of Parkinson's disease.31 It was found that the exchange of the acidic, negatively charged amino acid Glu with a basic, positively charged Lys induces subtle conformational changes in the protein that alters its aggregation behavior.42 The mutation E46K is associated with an increased ability of the protein to bind to negatively charged vesicles and an increased fibril formation.43,44 Besides these findings, the E46K mutation also raises the question, of whether the introduced Lys is yet another site for glycation in the protein and whether this alters its structure. However, as yet, the influence of the glycation of K46 on the structure and function of aSyn is largely unexplored, as conventional glycation methods do not allow the site-selective glycation of the Lys position.3Recently, we published a new strategy for ligating three aSyn fragments of defined length to obtain the native full-length aSyn sequence, several disease-relevant mutations, and a PTM using native chemical ligation (NCL).45 This general strategy enabled us to synthesize aSyn with an E46K mutation and the corresponding glycated variant.
NCL relies on the reaction between a C-terminal peptide thioester and a peptide fragment carrying an N-terminal cysteine (Cys, C).46 Thioesters can be obtained by on-resin precursor strategies or by in situ thioesterification and were introduced with the hydrazide technique for our peptides.47,48 As aSyn contains no native Cys, we used the ligation–desulfurization method that allows reversible incorporation of a Cys for ligation at an alanine (Ala) position and conversion back to Ala after ligation.49 This approach was used for the sequential assembly of other synuclein family members as well.50 The ligation sites were further chosen to enable good reaction kinetics at non-hindered glycines (Gly) and to obtain fragments of suitable length for SPPS.51 In detail, an N-terminal fragment with 68 (1–68), a central fragment with 38 (69–106) and a C-terminal one with 34 amino acids (107–140) were designed, encompassing the ligation sites at amino acids 68–69 and 106–107 with two Gly-Ala motifs. All peptide fragments were synthesized via microwave-assisted SPPS (ESI†), and then ligated (Fig. 8).
 | ||
| Fig. 8 Schematic ligation pathway of aSyn variants with NCL from the C- to N-terminus. First, Thz-protected hydrazide peptide 10 (green) and C-terminal Cys peptide 11 (red) were ligated and subsequently Thz deprotected to obtain Cys peptide 12. The latter fragment was further ligated either with N-terminal fragment 8 with an E46K mutation or with fragment 9 with E46CEL. The resulting aSyn peptides with two Cys mutations underwent desulfurization to the final aSyn variants 13 (E46K) and 14 (E46CEL). | ||
As the solid phase, either a functionalized 2-chlorotrityl chloride (CTC) resin for hydrazide thioester precursor peptides 8, 9, and 10 (highlighted in blue and green) or an Ala-preloaded CTC resin for the C-terminal fragment 11 (red) was used.45 Amino acids 107 and 69 were introduced as Cys or as the Cys precursor thiazolidine (Thz) to prevent side reactions for the central fragment.52 Peptide 10 was obtained in 23% yield with 96% purity, peptide 11 with 8% yield and 95% purity, respectively. For the N-terminal fragments, either E46 was replaced for SPPS by a Fmoc-Lys(Boc)-OH building block for peptide 8 or by Fmoc-Lys(Alloc)-OH for CEL peptide 9.
The glycated peptide variant CEL 9 was obtained by on-resin Alloc deprotection and reductive amination as described for peptides 3 and 4. The yield of peptide 8 was 2% with 99% purity and 1% with 86% purity for 9, sufficient for NCL. For comparison, the native N-terminal sequence (peptide 15, ESI†) was obtained in around 2% yield. The coupling of a Lys building block instead of a Glu during SPPS did not affect the overall yield.
The first NCL between hydrazide Thz peptide 10 and Cys peptide 11 was performed in ligation buffer with 4-mercaptophenylacetic acid (MPAA) and tris(2-carboxyethyl)phosphine (TCEP) at pH 7. Beforehand, the peptide hydrazide precursor 10 was converted to the respective MPAA thioester in the presence of acetylacetone in situ. Subsequently, after completed ligation, a one-pot Thz deprotection at pH 4 with methoxylamine was performed. Peptide 12 was obtained in 19% yield and 99% purity after HPLC purification and was either ligated with N-terminal fragment 8 for the E46K mutation or fragment 9 for the CEL mutation. After final desulfurization to the native aSyn sequence, both peptides were successfully purified and isolated. aSyn E46K 13 was obtained in 8% overall yield with 80% purity after three steps, aSynCEL 14 in 4% and 89%, respectively. As a reference, wild type (wt) aSyn 16 was also ligated in the same way by using N-terminal fragment 15, with a final 8% overall yield and 78% purity (not shown in Fig. 8, detailed synthesis in ESI†).
Circular dichroism of glycated aSyn protein and fragments
All three aSyn variants (wt 16, E46K 13 and E46CEL 14) as well as the three N-terminal hydrazide fragments (wt 15, E46K 8, and E46CEL 9) were investigated with CD spectroscopy.53 The method allows gathering structural information by distinguishing secondary structure elements like α-helices, β-sheets and turns.54Fig. 9a displays the CD spectra of the native full-length aSyn and the mutated E46K and E46CEL proteins. Fig. 9c shows the respective spectra of the N-terminal variants. Using the BeStSel algorithm, the relative fractions of secondary structure elements were calculated (Fig. 9b and d).
 | ||
| Fig. 9 Spectroscopic CD analysis of native and modified aSyn and its N-terminal fragments. (a) and (c): CD spectra of aSyn variants and N-terminal fragments at 10 μM in water. (b) and (d): Estimation of the secondary structure composition (α-helix, antiparallel β-sheet, β-turns, other structures) from the CD spectra. | ||
The results show that independent of the mutation, more than 50% of the proteins adopt a random coil structure with a characteristic minimum at 198 nm.53 These results are in agreement with those obtained for aSyn and aSyn E46K that were recombinantly expressed. In these studies, CD analysis as well as nuclear magnetic resonance (NMR) and small-angle scattering (SAS) revealed that aSyn appears as an intrinsically disordered protein (IDP).55,56 Also for E46CEL, the same secondary structure elements were observed. This result indicates that neither the E46K nor the E46CEL mutation alters the structure of aSyn in a way that it can explain the observed increase in fibril formation. The same results were found for the N-terminal peptides.
Conclusions
aSyn is a central player in PD and related synucleinopathies. The protein tends to fold incorrectly and forms large molecular structures through self-association. The likelihood of protein aggregation is influenced by alterations in the natural amino acid sequence and PTMs. Therefore, obtaining a pure protein that can be selectively modified chemically is crucial for understanding the impact of PTMs on its aggregation behavior. Chemical peptide synthesis is a powerful tool for the controlled production of various modifications. Our developed synthetic route, utilizing solid-phase peptide synthesis, enables the incorporation of multiple modifications (CML and CEL) into aSyn peptide fragments. This synthetic approach provides a versatile method for introducing glycation motifs at specific sites, serving as the foundation for creating glycated forms of synucleinopathy-related and other disease-relevant proteins.Author contributions
The project was conceptualized by T. F. O., A. K. and C. B. Short N-terminal peptides were synthesised by C. B. and L. M. B. ASyn peptide fragments, ligations and CD measurements were performed by K. I. B. v. B. The manuscript was written, reviewed, and edited by C. B., T. F. O. and C. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. Ulf Diederichsen for supporting the aSyn project at the beginning. C. S. and T. F. O. are supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy, EXC 2067/1-390729940, and by the SFB 1286 (projects B4 and B8).References
- D. Virág, B. Dalmadi-Kiss, K. Vékey, L. Drahos, I. Klebovich, I. Antal and K. Ludányi, Chromatographia, 2020, 83, 1–10 CrossRef.
- R. Gupta, M. Sahu, D. Srivastava, S. Tiwari, R. K. Ambasta and P. Kumar, Ageing Res. Rev., 2021, 68, 101336 CrossRef CAS PubMed.
- H. Vicente Miranda, É. M. Szegő, L. M. A. Oliveira, C. Breda, E. Darendelioglu, R. M. de Oliveira, D. G. Ferreira, M. A. Gomes, R. Rott, M. Oliveira, F. Munari, F. J. Enguita, T. Simões, E. F. Rodrigues, M. Heinrich, I. C. Martins, I. Zamolo, O. Riess, C. Cordeiro, A. Ponces-Freire, H. A. Lashuel, N. C. Santos, L. V. Lopes, W. Xiang, T. M. Jovin, D. Penque, S. Engelender, M. Zweckstetter, J. Klucken, F. Giorgini, A. Quintas and T. F. Outeiro, Brain, 2017, 140, 1399–1419 CrossRef PubMed.
- A. Oueslati, M. Fournier and H. A. Lashuel, Prog. Brain Res., 2010, 183, 115–145 CAS.
- R. M. De Oliveira, H. Vicente Miranda, L. Francelle, R. Pinho, É. M. Szegö, R. Martinho, F. Munari, D. F. Lázaro, S. Moniot and P. Guerreiro, PLoS Biol., 2017, 15, e.2000374 CrossRef PubMed.
- Y.-G. Choi, H.-Y. Shin, J.-I. Kim, E.-K. Choi, R. I. Carp and Y.-S. Kim, Mol. Neurobiol., 2016, 53, 3102–3112 CrossRef CAS.
- N. Ahmed, U. Ahmed, P. J. Thornalley, K. Hager, G. Fleischer and G. Münch, J. Neurochem., 2005, 92, 255–263 CrossRef CAS PubMed.
- L. Tagliafierro and O. Chiba-Falek, Neurogenetics, 2016, 17, 145–157 CrossRef CAS PubMed.
- J. Burré, M. Sharma, T. Tsetsenis, V. Buchman, M. R. Etherton and T. C. Südhof, Science, 2010, 329, 1663–1667 CrossRef PubMed.
- S. Anwar, O. Peters, S. Millership, N. Ninkina, N. Doig, N. Connor-Robson, S. Threlfell, G. Kooner, R. M. Deacon, D. M. Bannerman, J. P. Bolam, S. S. Chandra, S. J. Cragg, R. Wade-Martins and V. L. Buchman, J. Neurosci., 2011, 31, 7264–7274 CrossRef CAS PubMed.
- L. V. Kalia, S. K. Kalia, P. J. McLean, A. M. Lozano and A. E. Lang, Ann. Neurol., 2013, 73, 155–169 CrossRef CAS PubMed.
- E. A. Coon and W. Singer, Continuum, 2020, 26, 72–92 Search PubMed.
- J. Li, D. Liu, L. Sun, Y. Lu and Z. Zhang, J. Neurol. Sci., 2012, 317, 1–5 CrossRef CAS PubMed.
- V. P. Reddy, P. Aryal and E. K. Darkwah, Microorganisms, 2022, 10, 1848 CrossRef CAS.
- A. Twarda-Clapa, A. Olczak, A. M. Białkowska and M. Koziołkiewicz, Cells, 2022, 11, 1312 CrossRef CAS.
- A. Kuzan, Biomed. Rep., 2021, 14, 1–8 Search PubMed.
- S. Reddy, J. Bichler, K. J. Wells-Knecht, S. R. Thorpe and J. W. Baynes, Biochemistry, 1995, 34, 10872–10878 CrossRef CAS.
- D. Deluyker, L. Evens and V. Bito, Amino Acids, 2017, 49, 1535–1541 CrossRef CAS PubMed.
- J. W. Baynes, Diabetes, 1991, 40, 405–412 CrossRef CAS PubMed.
- A. B. Uceda, L. Mariño and M. Adrover, Neural Regener. Res., 2020, 15, 1840–1841 CrossRef.
- S. L. Leong, M. G. Hinds, A. R. Connor, D. P. Smith, E. Illes-Toth, C. L. L. Pham, K. J. Barnham and R. Cappai, PLoS One, 2015, 10, e0116497 CrossRef PubMed.
- P. Semenyuk, K. Barinova and V. Muronetz, Int. J. Biol. Macromol., 2019, 127, 278–285 CrossRef CAS.
- D. Lee, C. W. Park, S. R. Paik and K. Y. Choi, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 421–430 CrossRef CAS PubMed.
- A. Chegão, M. Guarda, B. M. Alexandre, L. Shvachiy, M. Temido-Ferreira, I. Marques-Morgado, B. Fernandes Gomes, R. Matthiesen, L. V. Lopes, P. R. Florindo, R. A. Gomes, P. Gomes-Alves, J. E. Coelho, T. F. Outeiro and H. Vicente Miranda, npj Parkinson's Dis., 2022, 8, 51 CrossRef PubMed.
- I. Ban, H. Sugawa and R. Nagai, Int. J. Mol. Sci., 2022, 23, 1224 CrossRef CAS PubMed.
- G. E. Sroga and D. Vashishth, JBMR Plus, 2021, 5, e10548 CrossRef CAS PubMed.
- L. Mariño, R. Ramis, R. Casasnovas, J. Ortega-Castro, B. Vilanova, J. Frau and M. Adrover, Chem. Sci., 2020, 11, 3332–3344 RSC.
- P. Gruber and T. Hofmann, J. Pept. Res., 2005, 66, 111–124 CrossRef CAS.
- T. M. Woods, M. Kamalov, P. W. R. Harris, G. J. S. Cooper and M. Brimble, Org. Lett., 2012, 14, 5740–5743 CrossRef CAS PubMed.
- J. Ng, H. Kaur, T. Collier, K. Chang, A. E. S. Brooks, J. R. Allison, M. A. Brimble, A. Hickey and N. P. Birch, J. Biol. Chem., 2019, 294, 8806–8818 CrossRef PubMed.
- J. J. Zarranz, J. Alegre, J. C. Gómez-Esteban, E. Lezcano, R. Ros, I. Ampuero, L. Vidal, J. Hoenicka, O. Rodriguez, B. Atarés, V. Llorens, E. G. Tortosa, T. del Ser, D. G. Muñoz and J. G. de Yebenes, Ann. Neurol., 2004, 55, 164–173 CrossRef CAS PubMed.
- O. Dangles, F. Guibe, G. Balavoine, S. Lavielle and A. Marquet, J. Org. Chem., 1987, 52, 4984–4993 CrossRef CAS.
- K. R. Wilson, S. Sedberry, R. Pescatore, D. Vinton, B. Love, S. Ballard, B. C. Wham, S. K. Hutchison and E. J. Williamson, J. Pept. Sci., 2016, 22, 622–627 CrossRef CAS PubMed.
- K. Pels and T. Kodadek, ACS Comb. Sci., 2015, 17, 152–155 CrossRef CAS PubMed.
- M. Kamalov, P. W. R. Harris, G. J. S. Cooper and M. A. Brimble, Synlett, 2014, 1835–1838 CAS.
- U. Greifenhagen, V. D. Nguyen, J. Moschner, A. Giannis, A. Frolov and R. Hoffmann, J. Proteome Res., 2015, 14, 768–777 CrossRef CAS PubMed.
- D. Li and D. L. Elbert, J. Pept. Res., 2002, 60, 300–303 CrossRef CAS.
- Z. Yao, J. Bhaumik, S. Dhanalekshmi, M. Ptaszek, P. A. Rodriguez and J. S. Lindsey, Tetrahedron, 2007, 63, 10657–10670 CrossRef CAS.
- Y. Yamamoto, T. Shirai and N. Miyaura, Chem. Commun., 2012, 48, 2803–2805 RSC.
- J. Zhu, Y. Yuan, S. Wang and Z.-J. Yao, ACS Omega, 2017, 2, 4665–4677 CrossRef CAS PubMed.
- S. F. Loibl, Z. Harpaz and O. Seitz, Angew. Chem., Int. Ed., 2015, 54, 15055–15059 CrossRef CAS.
- R. A. Fredenburg, C. Rospigliosi, R. K. Meray, J. C. Kessler, H. A. Lashuel, D. Eliezer and P. T. Lansbury, Biochemistry, 2007, 46, 7107–7118 CrossRef CAS.
- E. A. Greenbaum, C. L. Graves, A. J. Mishizen-Eberz, M. A. Lupoli, D. R. Lynch, S. W. Englander, P. H. Axelsen and B. I. Giasson, J. Biol. Chem., 2005, 280, 7800–7807 CrossRef CAS PubMed.
- W. Choi, S. Zibaee, R. Jakes, L. C. Serpell, B. Davletov, R. Anthony Crowther and M. Goedert, FEBS Lett., 2004, 576, 363–368 CrossRef CAS PubMed.
- L. M. Gatzemeier, F. Meyer, U. Diederichsen and T. F. Outeiro, Chem. – Eur. J., 2023, 29, e202300649 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS.
- S. Wang, Q. Zhou, Y. Li, B. Wei, X. Liu, J. Zhao, F. Ye, Z. Zhou, B. Ding and P. Wang, J. Am. Chem. Soc., 2022, 144, 1232–1242 CrossRef CAS PubMed.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS PubMed.
- D. Bang and S. B. H. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534–2538 CrossRef CAS.
- D. Corrêa and C. Ramos, Afr. J. Biochem. Res., 2009, 3, 164–173 Search PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- M. Schwalbe, V. Ozenne, S. Bibow, M. Jaremko, L. Jaremko, M. Gajda, M. R. Jensen, J. Biernat, S. Becker and E. Mandelkow, Structure, 2014, 22, 238–249 CrossRef CAS.
- T. R. Alderson and J. L. Markley, Intrinsically Disord. Proteins, 2013, 1, e26255 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of synthesized small molecules and peptides/proteins (NMR spectra, UHPLC chromatograms and HR-MS ESI spectra. See DOI: https://doi.org/10.1039/d4ob00225c |
| ‡ Deceased. |
| § Scientific employee with an honorary contract at Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE), Göttingen, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |