
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-Azabicyclo[3.2.1]octane scaffold: synthesis and applications
Mariana
Crespo Monteiro
 ,
João Rafael
Vale
and
Filipa
Siopa
*
,
João Rafael
Vale
and
Filipa
Siopa
*
Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Av. Professor Gama Pinto, 1649-003, Lisbon, Portugal. E-mail: filipasiopa@ff.ulisboa.pt
First published on 12th March 2024
Abstract
2-Azabicyclo[3.2.1]octanes are nitrogen containing heterocycles with significant potential in the field of drug discovery. This core has been applied as key synthetic intermediate in several total synthesis, while their unique structure can make them a challenging scaffold to acquire. This Minireview summarizes the synthetic approaches to access this bicyclic architecture and highlights its presence in the total synthesis of several target molecules.
 Mariana Crespo Monteiro | Mariana Monteiro received her master's degree in Pharmaceutical Sciences from the Faculty of Pharmacy of the University of Lisbon in 2022. She is currently pursuing her Ph.D. in the Research Institute for Medicines of the University of Lisbon (iMed.ULisboa) under the supervision of Dr. Filipa Siopa. Her Ph.D. project mainly focuses on the valorization of biomass derived compounds through photochemical transformations. She is also working on palladium-catalyzed reactions of aziridines. |
 João Rafael Vale | João R. Vale received his PhD in Pharmaceutical and Therapeutical Chemistry in 2023 from the Faculty of Pharmacy of the University of Lisbon and Tampere University, Finland. His research interests currently lie on photochemistry and photocatalysis, with a strong component in synthetic synthetic organic chemistry. |
 Filipa Siopa | Filipa Siopa obtained a Chemistry PhD (2011) from the Faculty of Science and Technology, New University of Lisbon. After a postdoctoral at Faculty of Pharmacy of the University of Lisbon with an experience with Professor Giovanni Poli at Sorbonne Université (Paris), she obtained a researcher position at Faculty of Pharmacy of the University of Lisbon. Her research focuses on the development of new synthetic methodologies, flow chemistry, biomass valorization, synthesis of bioactive molecules and total synthesis. |
1. Introduction
Nitrogen-containing heterocycles can be found in a variety of products of natural and synthetic origin.1 Due to their known bioactive properties, N-based compounds are particularly interesting from a pharmaceutical point of view.1c,2 Among these, the 2-azabicyclo[3.2.1]octane system, consisting of a six-membered nitrogen heterocycle fused to a cyclopentane ring (Fig. 1), has gained significant interest in the past decades due to its synthetic and pharmacological potential.3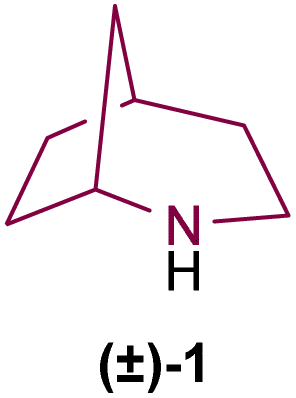 | ||
| Fig. 1 General structure of 2-azabicyclo[3.2.1]octanes. | ||
Interest in this core by the scientific community has spiked since 2014 after Massiot's group reported a series of structurally related cytisine-like alkaloids from the roots and stems of Ormosia hosier, a plant employed in traditional Chinese herbal medicine. These compounds, of the hosieine family (Fig. 2), exhibited high affinity for α4β2 neuronal nicotinic acetylcholine receptor. For instance, (−)-hosieine A, the most potent molecule in this class (IC50 = 0.96 nM, Ki = 0.32 nM), exhibits an activity five times stronger than nicotine.4 The remarkable high activity of (−)-hosieine A and its complex skeleton have already inspired its asymmetric total synthesis, as well as its enantiomer, which are discussed in this manuscript.
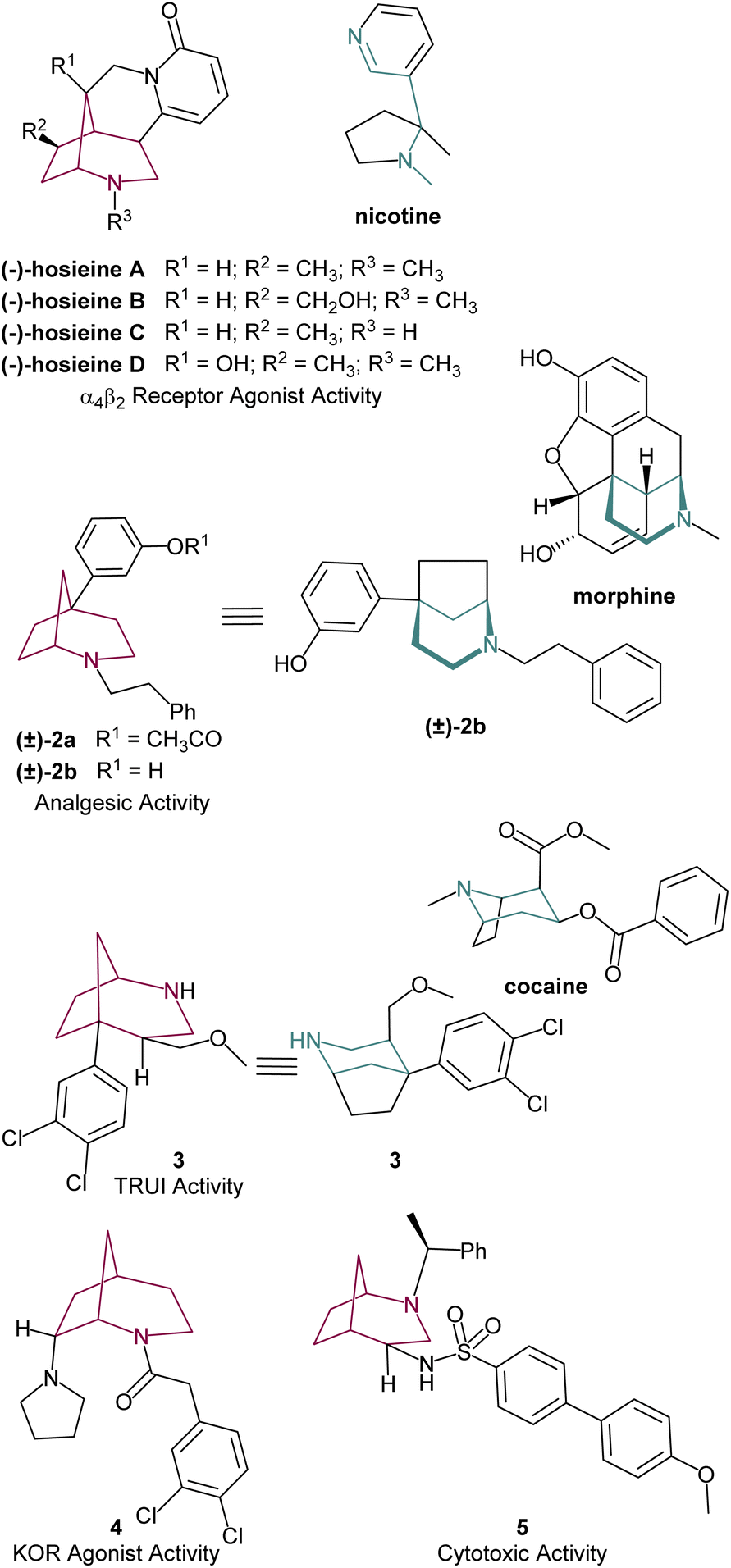 | ||
| Fig. 2 Selected examples of 2-azabicyclo[3.2.1]octane scaffolds with biological activity. | ||
Other synthetic compounds containing the 2-azabicyclo[3.2.1]octane scaffold have also shown diverse biological activities. Ong and Anderson were pioneers in showcasing its medicinal potential as efficient analgesic agents. The most promising compounds 2a and 2b showed a combined agonist-antagonist effect with efficiency close to morphine (Fig. 2).5 In a similar line, Andreotti's group explored the effect of aryl substitution in the bridgehead carbon of the azabicyclo[3.2.1]octane. Compound 3, mimicking the 3D conformation of cocaine, proved to be a potent in vitro triple re-uptake inhibitor (TRUI).3b The introduction of a second nitrogen group attached to the bicyclic structure grants this class of molecules relevant biological activity. Agonist activity in κ-opioid receptors (KOR) was observed for compound 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and cytotoxic activity on different tumor cell lines, such as glioblastoma (GBM), medulloblastoma (MB) and hepatocellular carcinoma (HCC) cell lines (Fig. 2) were reported for 5.7 Overall, the medicinal and biological effect of azabicyclo[3.2.1]octane derivatives appears to derive from its structural similarity with bioactive alkaloids such as nicotine, cocaine and morphine, while its bicyclic backbone provides additional rigidity to the molecular structure, which is an important feature in medicinal chemistry.8
6 and cytotoxic activity on different tumor cell lines, such as glioblastoma (GBM), medulloblastoma (MB) and hepatocellular carcinoma (HCC) cell lines (Fig. 2) were reported for 5.7 Overall, the medicinal and biological effect of azabicyclo[3.2.1]octane derivatives appears to derive from its structural similarity with bioactive alkaloids such as nicotine, cocaine and morphine, while its bicyclic backbone provides additional rigidity to the molecular structure, which is an important feature in medicinal chemistry.8
2-Azabicyclo[3.2.1]octane system is chiral and, unless constructed from enantiomerically pure precursors, it is often present as a mixture of two enantiomers. In this review, we represent it in a consistent 3D configuration for the sake of clarity and consistency, adding (±) notation whenever a racemic mixture is present.
In this Minireview we discuss the reported methodologies for the synthesis of 2-azabicyclo[3.2.1]octanes, namely intramolecular cyclization and rearrangements, such as Beckmann rearrangement. Moreover, their application in total synthesis is also covered in this manuscript. We also include the synthesis of 2-azabicyclo[3.2.1]octan-3-ones, since they are common precursors to the 2-azabicyclo[3.2.1]octane core, typically accessed via reduction with LiAlH4.
2. Synthesis of 2-azabicyclo[3.2.1]octanes by intramolecular cyclization
The assembly of the 2-azabicyclo[3.2.1]octanes is often achieved via nucleophilic attack and concomitant intramolecular cyclization. This strategy typically uses cyclopentanes3b,9 and piperidine derivatives6,9c as starting materials, however other scaffolds have also been successfully applied.10One of the first examples in the literature comes from Gassman and co-workers that, in 1968, reported the preparation of 8-methoxy-2-methyl-2-azabicyclo[3.2.1]octane 8 (Scheme 1). Treatment of amine 6 with aqueous sodium hypochlorite resulted in 7. Reflux of a methanolic solution of 7 in the presence of silver nitrate resulted in product 8 in 55–65% yield.11
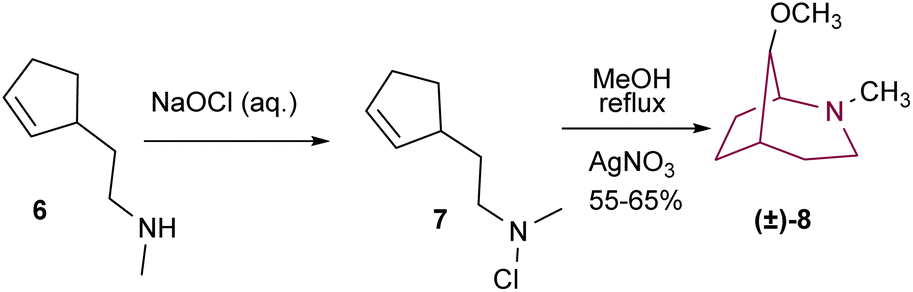 | ||
| Scheme 1 Synthesis of 2-azabicyclo[3.2.1]octane 8. | ||
In 1977, Takeda and co-workers9c were the first to prepare 5-aryl substituted 2-azabicyclo[3.2.1]octane scaffolds (Scheme 2), inspired by the conventional synthesis of phenylmorphan.12 The azabicyclo-core was accessed through intramolecular cyclization using cyclopentanone 10 or cyclopentenone 13 (Scheme 2). The synthetic strategy commences with the α-alkylation of cyclopentanone 9 to afford 10. From there, two synthetic routes emerge: cyclopentanone 10 may be submitted to α-bromination, which is followed by ring closing. Demethylation of the resulting tetraalkyl ammonium is achieved under high temperatures to yield ketone 11 (Scheme 2). In a different approach, 10 may be turned to the corresponding carbamate and then oxidized to the enone 13. Carbamate deprotection leads to intramolecular 1,4-addition of the secondary amine to give bicyclic amino ketone 14 (Scheme 2). Compounds 11 and 14 underwent Wolff–Kishner reduction to yield the desired azabicycloalkane 12. In a convergent manner, ketone 14 could also be obtained via acid catalysed cyclization of 16 (Scheme 2).9c A year later, a similar procedure was described by Ong and co-workers, in which optimization of the reaction conditions applied to 10, 11 and 12 led to improvement of yields and the extension of the reaction scope.5
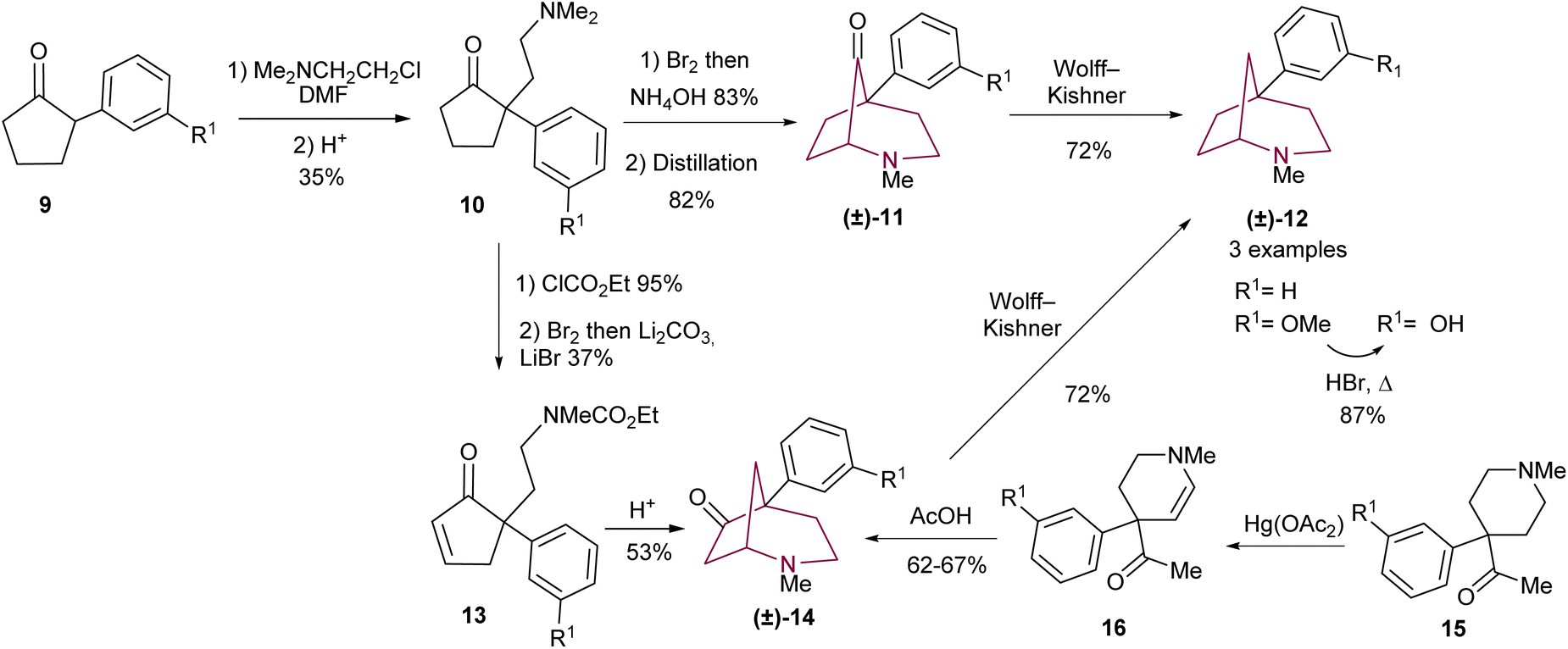 | ||
| Scheme 2 Synthesis of 5-aryl-2-azabicyclo[3.2.1]octanes 12 by intramolecular cyclization. | ||
Fleming and coworkers10a reported the synthesis of 2-azabicyclo[3.2.1]octane 20 from urethane 19 (Scheme 3). Compound 17 was reacted with thionyl chloride and then sodium azide, resulting in the formation of azide 18 through C–C bond cleavage and ring opening. Compound 18 was then heated under reflux in methanol, resulting in 19. Finally, 19 was heated at 200 °C to produce lactam 20, in 87% yield.
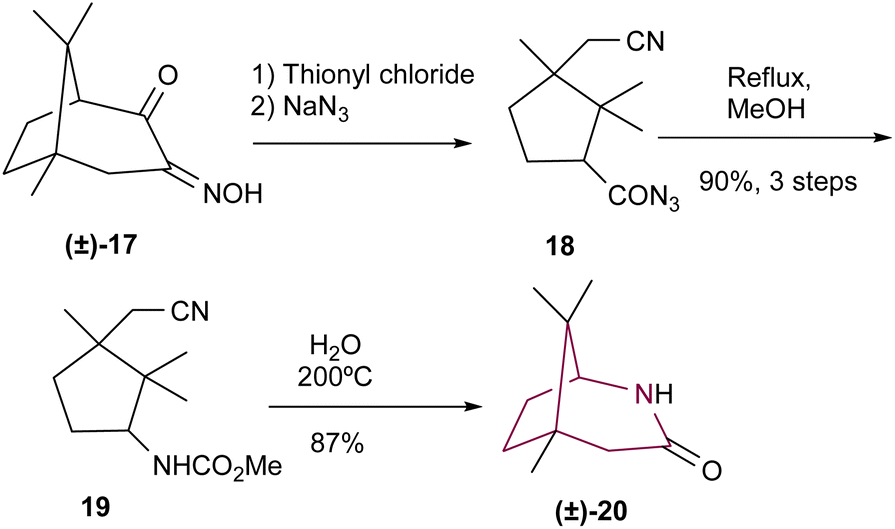 | ||
| Scheme 3 Synthesis of 2-azabicyclo[3.2.1]octane (±)-20via intramolecular amide formation. | ||
Fourrey and co-workers10b reported in 1999 the preparation of 2-azabicyclo[3.2.1]octane core through Tsuji–Trost reaction followed by intramolecular cycloaddition. Palladium catalyzed intramolecular cyclization of hydrazine derivative 22 delivered the compound 23 in high yield. Bicyclic 23 was transformed into cyclopentane 24, a key intermediate in the synthesis of homocarbocyclic nucleosides (Scheme 4).
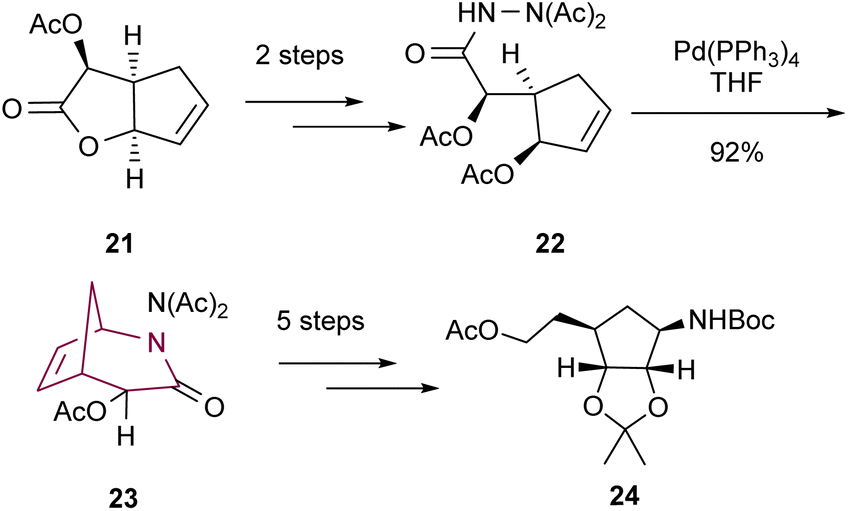 | ||
| Scheme 4 Synthesis of 2-azabicyclo[3.2.1]octan-3-one 23 by palladium catalyzed intramolecular cyclization. | ||
In the same year, the 2-azabicyclo[3.2.1]octan-3-one core was shown once more to be an important precursor to other carbocyclic nucleosides by Lundt's group.10c Bicyclic system 27 was accessed from 26via epoxide ring opening with ammonia followed by subsequent nucleophilic attack of the amine to the ester (Scheme 5). Amide cleavage and other operations led to the desired highly functionalized cyclopentane 28.
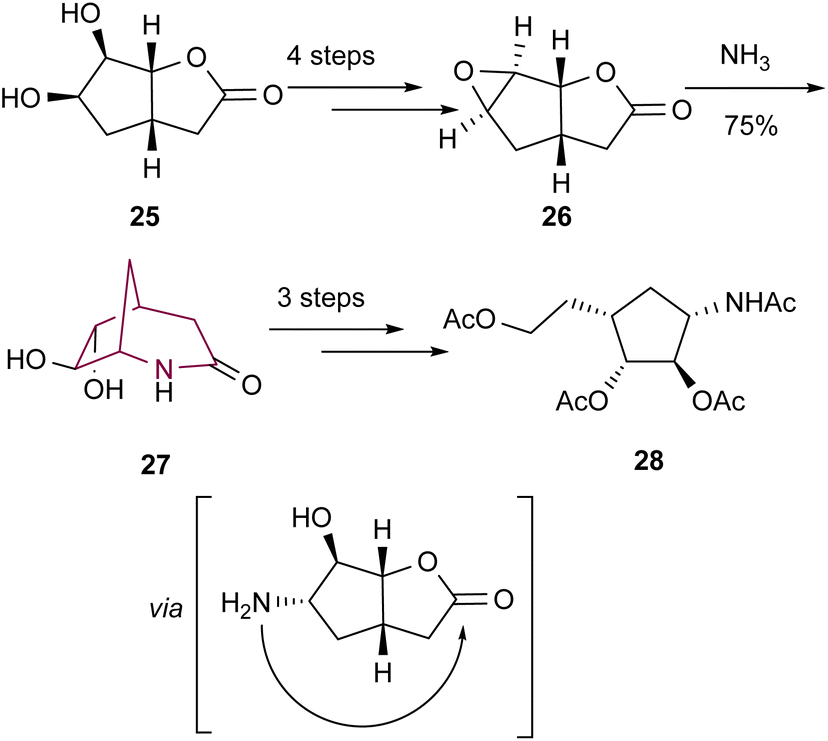 | ||
| Scheme 5 Synthesis of bicyclic system 27 by intramolecular cyclization. | ||
In a similar manner, 2-azabicyclo[3.2.1]octane core 32a–c, was prepared via intramolecular epoxide ring opening reaction of 31a–c with trifluoroacetamide (Scheme 6). Trifluoroacetamide removal led to bicyclic compound 33a–c, the precursor for the Aza-Cope-Mannich rearrangement, the key transformation in the total synthesis of Strychnos alkaloids: (±)-dehydrotubifoline 35, (±)-akuammicine 3613 and (−)-strychnine 41.14 The synthetic strategy to obtain (±)-dehydrotubifoline 35 and (±)-akuammicine 36 is very similar and commences from compound 29. While the synthesis of (−)-strychnine started with an enantioselective hydrolysis of cis-3,5 diacetoxycyclopentene to access compound 30. Epoxide 31a–c was synthesized by applying several standard manipulations, that include hydroxyl protection, Pd-catalyzed carbonylation, epoxide formation and Wittig methylenation towards compound 29 and 30. The desired azabicyclo[3.2.1]octane 33a–c, was accessed via epoxide ring opening with trifluoracetamide, followed by trifluoroacetamide removal. The Aza-Cope-Mannich rearrangement of 33a–c proceeds via the formation of an iminium intermediate A, that undergoes a [3,3] rearrangement to intermediate B. Cyclization leads exclusively to the formation of tricyclic compound 34a–c already containing three stereogenic centers present in the final molecules. Hydrolysis of 34a delivered the desired (±)-dehydrotubifoline 35. (±)-Akuammicine 36 was obtained after acylation, that furnished the correspondent β-keto ester, and acid catalyzed hydrolysis. (−)-Strychnine 41 was furnished after carbomethoxylation of 34c, that afforded the correspondent β-keto ester derivative mostly in the enol form, followed by tert-butyl and triazone removal. Selective reduction of vinylogous carbamate double bond provided a mixture of α-ester 38 and β-ester 39, that under basic conditions, efficiently converted to the desired β-ester 39. Finally, ester reduction with DIBALH led to Wieland-Gumlich aldehyde 40 that, upon treatment with malonic acid under Perkin condensation conditions, originated the desired (−)-strychnine 41.
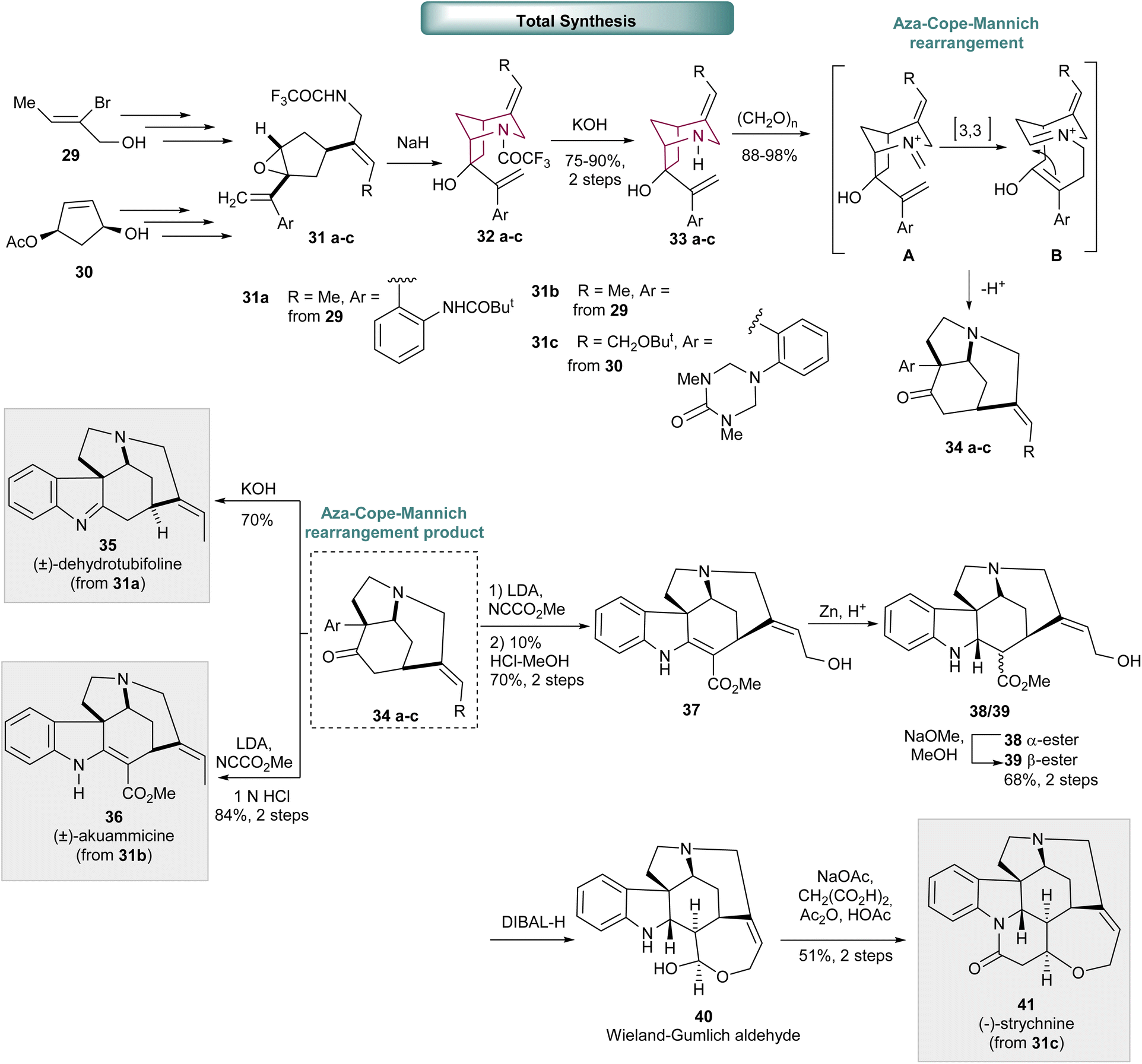 | ||
| Scheme 6 Synthesis of bicyclic system 32a–c by intramolecular epoxide ring opening reaction. Aza-Cope-Mannich rearrangement of 33a–c to 34a–c, a key intermediate in the total synthesis of (±)-dehydrotubifoline 35, (±)-akuammicine 36 and (−)-strychnine 41. | ||
In 2018, Wood and co-workers reported the total synthesis of (+)-hosieine A, an alkaloid containing the 2-azabicyclo[3.2.1]octane core, in just 7 steps with an overall yield of 16% (Scheme 7).15 The synthesis of (+)-hosieine A commences with N-alkylation of pyridinone 42 to provide (±)-43 by modification of Lee and Knochel's organozinc allylation reaction, nucleophilic alkynylation and sequential homologation, O-acylation and desilylation. The key step, a catalytic asymmetric Rautenstrauch reaction, was applied to (±)-43 and provided intermediate 45 with good enantiocontrol (90:10 er). Sequential Michael-cyclization, reductive amination and lactamization delivered azabicyclo[3.2.1]octan-3-one core (+)-48 in moderate yield (42% from 45). Amide reduction with BH3/THF furnished (+)-hosieine A 49.
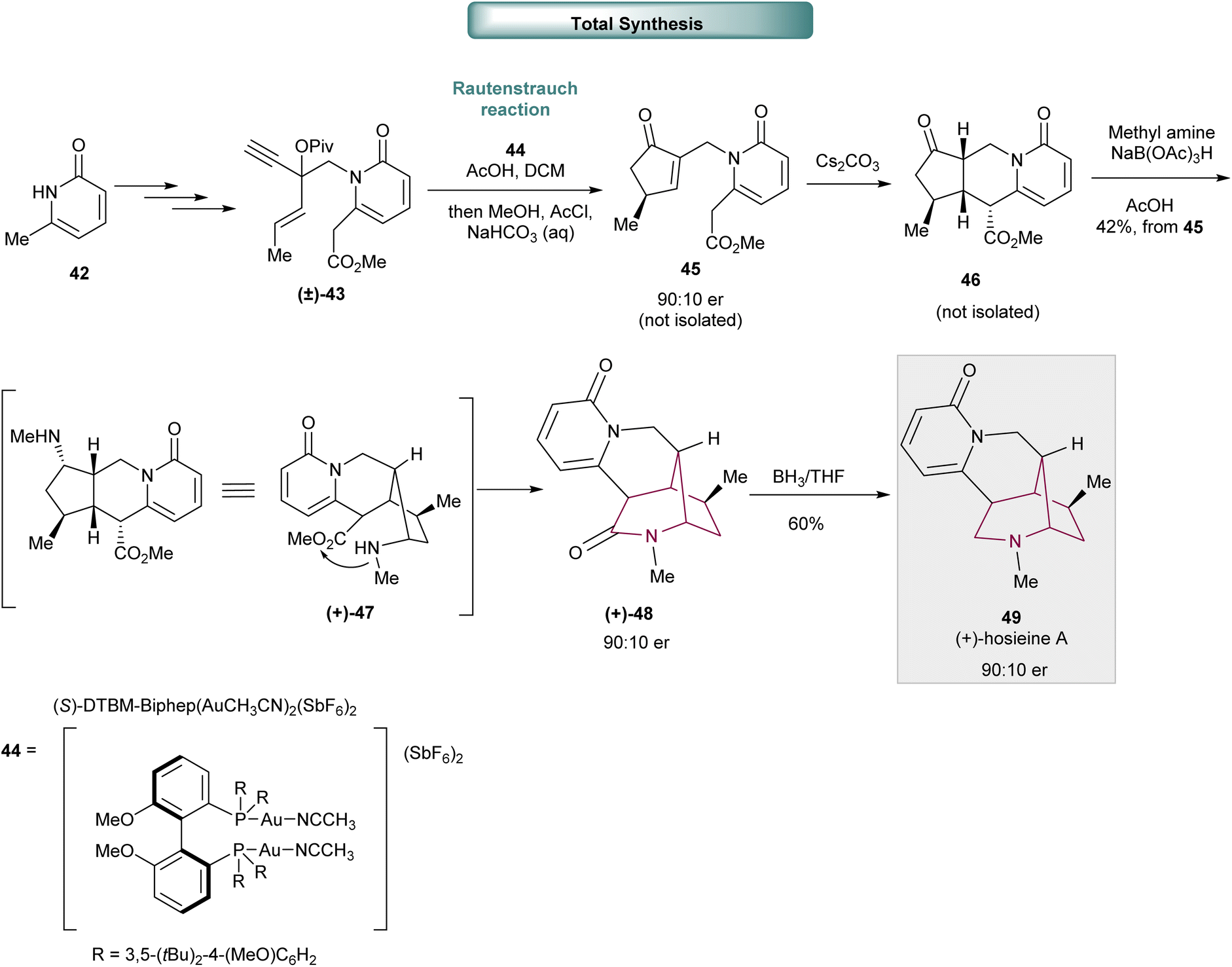 | ||
| Scheme 7 Total synthesis of (+)-hosieine A 49. | ||
In 2010, Profeta and Andreotti3b employed cyclopentanone 50 for the synthesis of the 2-azabicyclo[3.2.1]octane system in a candidate molecule as TRUI (Scheme 8). The synthetic methodology involved the conversion of ketone 50 to 51 through Knoevenagel condensation. After a sequence of reactions involving cuprate 1,4-addition, Boc cleavage with TFA and basic treatment, a mixture of isomers 53 (anti/syn = 5/95) was obtained in 43% yield over three steps. Preparative HPLC separated both isomers which were subjected to amide and ester reduction and alcohol methylation (through amine protection/deprotection sequence), to give the desired cocaine analogue 3.
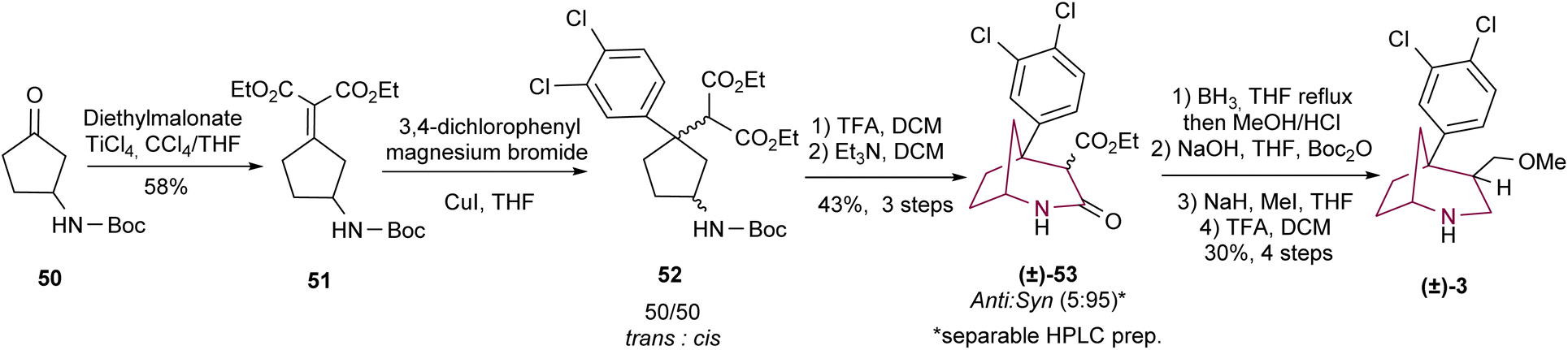 | ||
| Scheme 8 Synthesis of 5-aryl-2-azabicyclo[3.2.1]octane 3 by intramolecular cyclization. | ||
More recently, Wünsch and co-workers9b reported the synthesis of several 2-azabicyclo[3.2.1]octan-7-amines to study their KOR agonist activity. In this procedure, sulfonamide 55 afforded good yields of bicyclic compound 56 as a racemic mixture by intramolecular SN2 reaction upon deprotonation with NaH (Scheme 9). Then, tosylate 56 reacted with NaN3 to originate the corresponding azide, with unexpected retention of configuration, which after reduction yielded the primary amine 57. Alkylation of 57 afforded amines 58a and 58b. Tosyl removal and acylation provided the correspondent bicyclic compounds 59a and 59b.
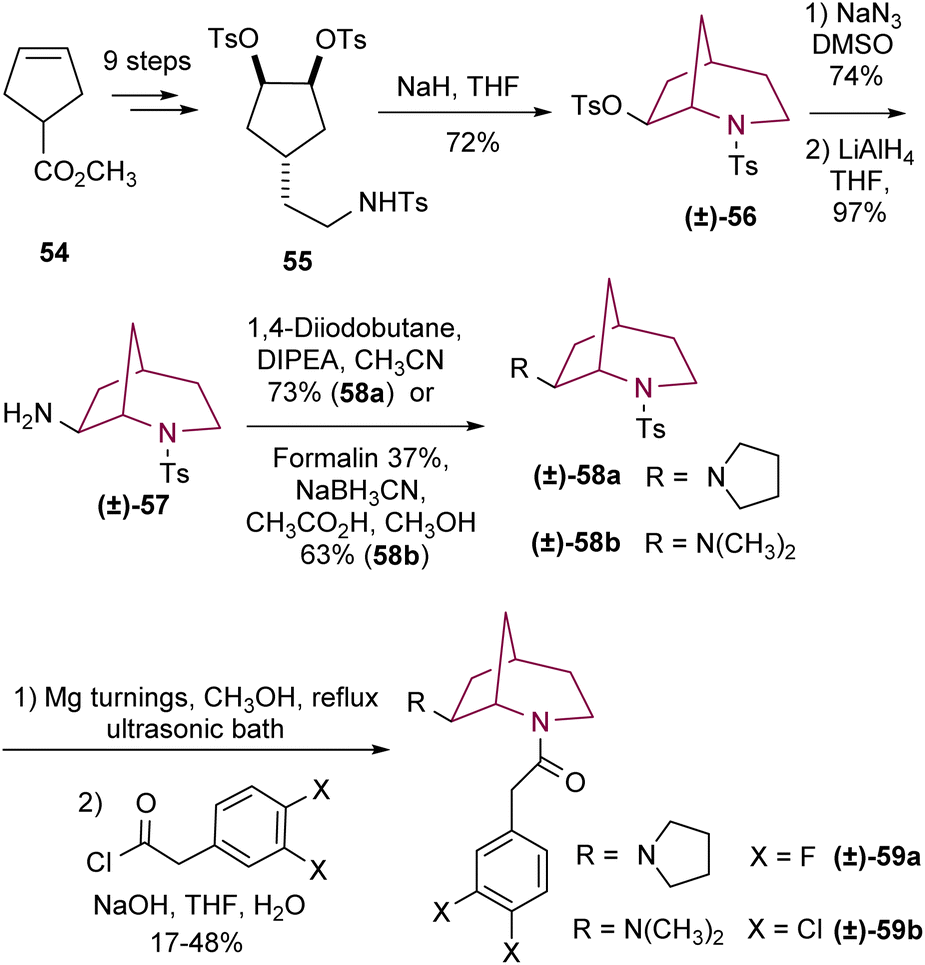 | ||
| Scheme 9 Synthesis of 2-azabicyclo[3.2.1]octane by intramolecular cyclization. | ||
The same group6 has also described the synthesis of 2-azabicyclo[3.2.1]octane core through Dieckmann cyclization of piperidine derivative 60 (Scheme 10). High temperatures were necessary to ensure that both ester substituents on 60 have an axial orientation and to prompt the inversion of configuration of trans-60. Compounds 61 were obtained in good yields, as a mixture of enantiomers (1S,5R) and (1R,5S) in a 3:1 ratio. Deethoxycarbonylation using Krapcho conditions, followed by Boc removal with EtOH and H2O and subsequent acylation resulted in bicyclic amide 62. Compound 62 underwent reductive amination. NaBH(OAc)3 adds to the less hindered side of the iminium intermediate, resulting in 2-azabicyclo[3.2.1]octanes 4 and 63 with an endo-configuration.
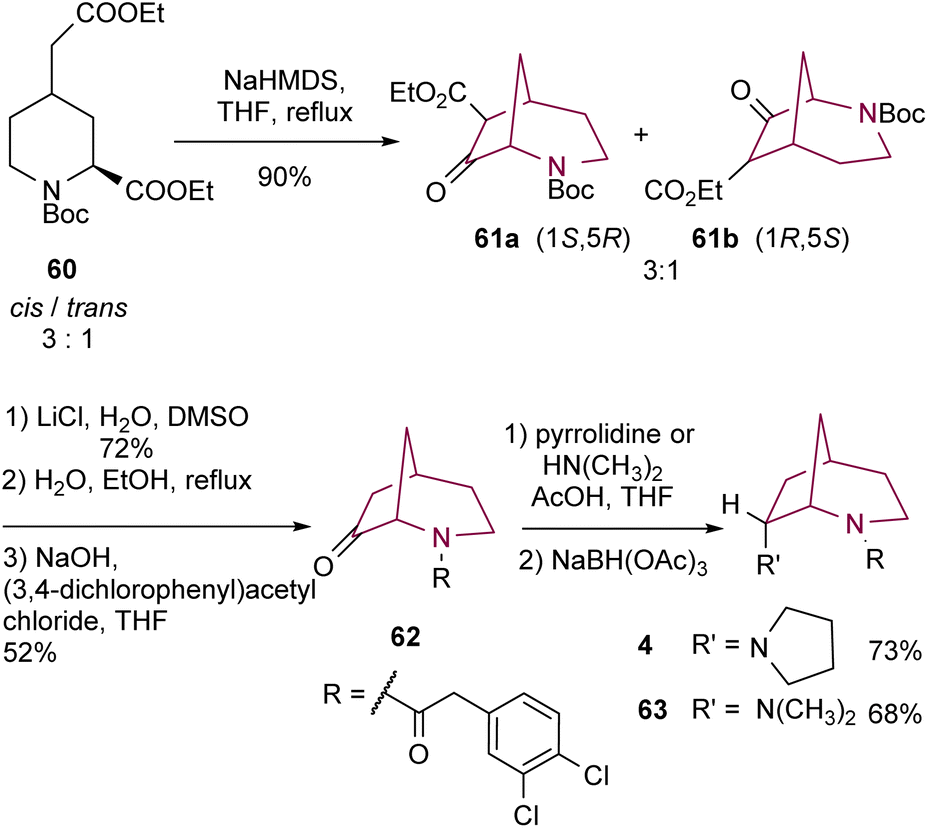 | ||
| Scheme 10 Synthesis of 2-azabicyclo[3.2.1]octanes 4 and 63 by Dieckmann cyclization of a piperidine derivative. | ||
Dondas and co-workers synthesized bicyclic N-hydroxylamine 66a and amine 66b from compounds 64a and 64b, respectively (Scheme 11). Upon treatment with phenylselenyl bromide, aldoxime 64a was converted to bridged nitrone salt 65avia phenylselenyl induced cyclization, in a stereo- and regioselective manner. After reaction with NaBH4, compound 65a resulted in 2-azabicyclo[3.2.1]octane 66a in 69% yield. However, when the same conditions were applied to oxime 64b, only trace amounts of the desired product 66b were obtained. The authors attributed this lack of reaction to the steric inhibition of the gem-dimethyl group. The synthesis of azabicyclo[3.2.1]octane 69 was achieved in 34% yield, by using analogous conditions, starting from compound 67.9a
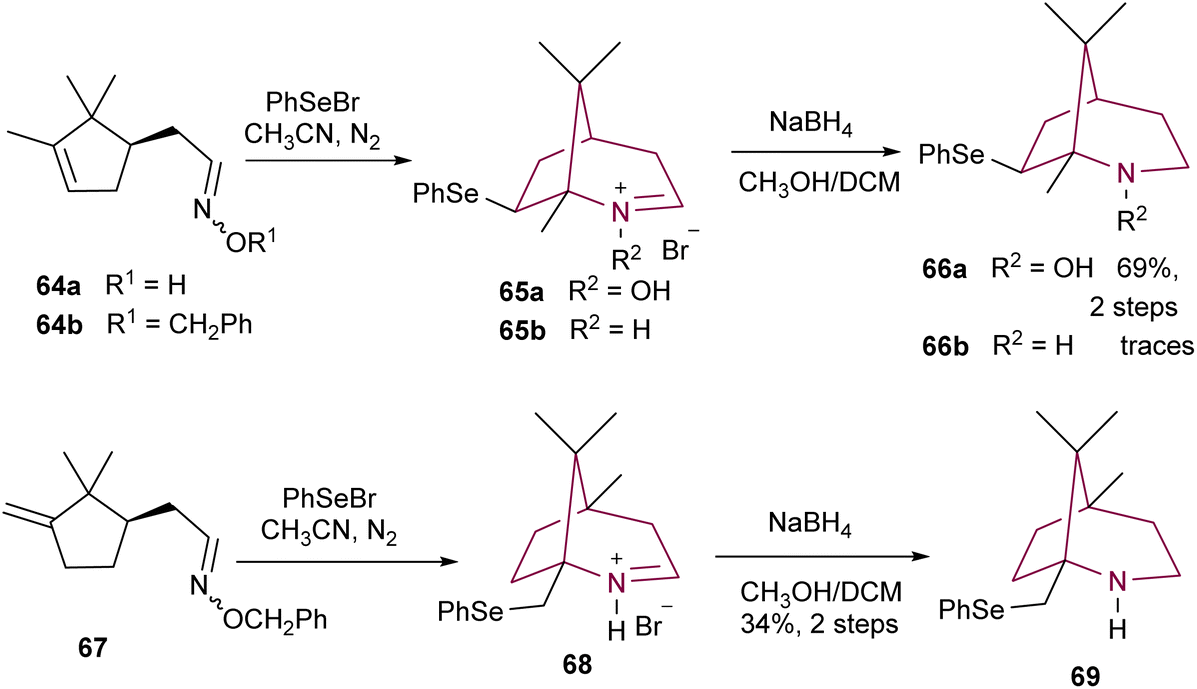 | ||
| Scheme 11 Synthesis of 2-azabicyclo[3.2.1]octanes 66a,b and 69via phenylselenyl induced cyclization. | ||
A unique way to access the azabicyclo[3.2.1]octane core was discovered by Namba and Tanino via construction of a N-acyl-N-tosylhydrazine 72 from an intramolecular palladium(II)-catalyzed cyclization reaction of 70 (Scheme 12). Treatment of 72 with trifluoracetic anhydride followed by epoxide formation provided access to 74. Then, samarium(II) iodide promoted the sequential N–N bond cleavage, epoxide ring opening/cyclization to furnish the azabicyclo[3.2.1]octane core.9d
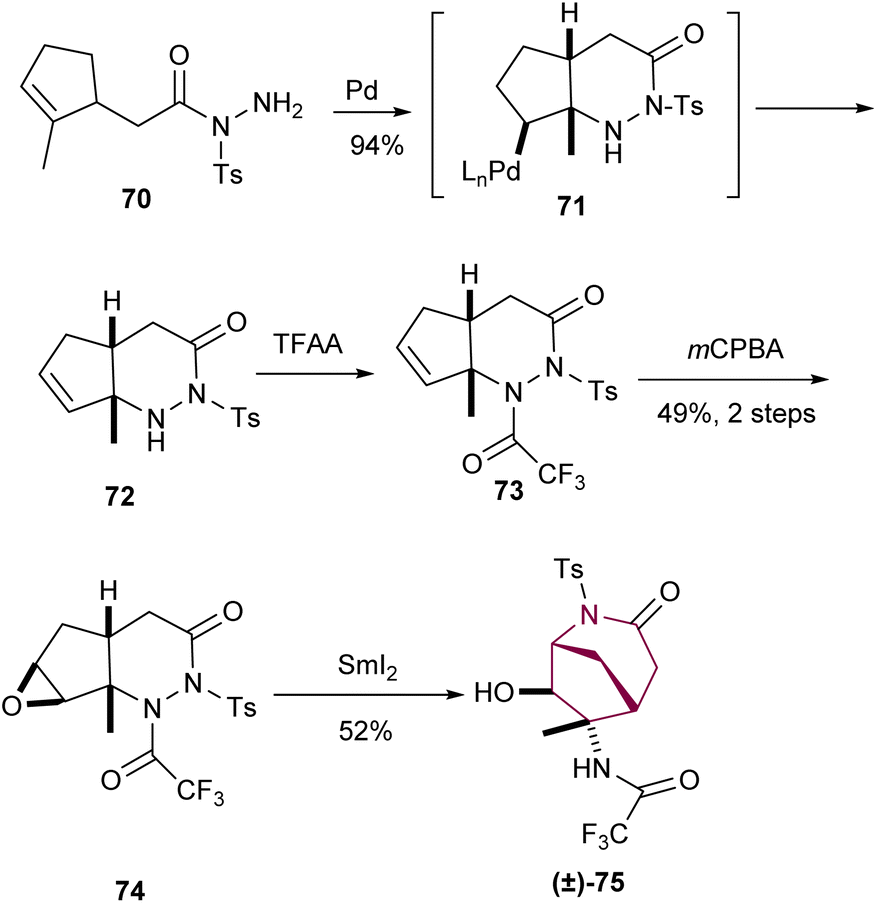 | ||
| Scheme 12 Synthesis of azabicyclo[3.2.1]octane core taking profit of an intramolecular palladium(II)-catalyzed cyclization. | ||
In 2015, Hong and co-workers achieved the first asymmetric synthesis of (−)-hosieine A, an alkaloid containing the 2-azabicyclo[3.2.1]octane core (Scheme 13).3c The key transformation was the elegant nitroso-ene cyclization to form the 2-azabicyclo[3.2.1]octane core. The synthetic approach began with α-methyl aldehyde 76, that after several standard operations such as Horner–Wadsworth–Emmons olefination, 1,4-addition, and ring closing metathesis (RCM) provided hydroxamic acid 77. Hydroxamic acid oxidation delivered 78 that underwent retro-Diels–Alder to afford the unstable acylnitroso intermediate 79, that suffers intramolecular nitroso–ene cyclization. The major product 2-aza-bicyclo[3.2.1]octane 80, was obtained via 6-endo-type reaction, in 9:1 ratio, and the minor 2-azabicyclo[3.3.0]octane 81, obtained via 5-endo-type. N-hydroxy acetylation and treatment with SmI2 resulted in the cleavage of N–O bond, followed by amide methylation to give compound 82. Bromohydrination, using an innovative methodology with NBS and phosphine, afforded the desired bicyclic ring 84, obtained in 84% combined yield (2:1) with a minor product 83. Radical debromination delivered compound 85. Hydroxyl activation and N-alkylation with 2-pyridone furnished 86. 1,6-Conjugate addition and subsequent protonation resulted in 87. Dehydrogenation with activated MnO2 and amide reduction in the presence of borane finally originated (−)-hosieine A 49.
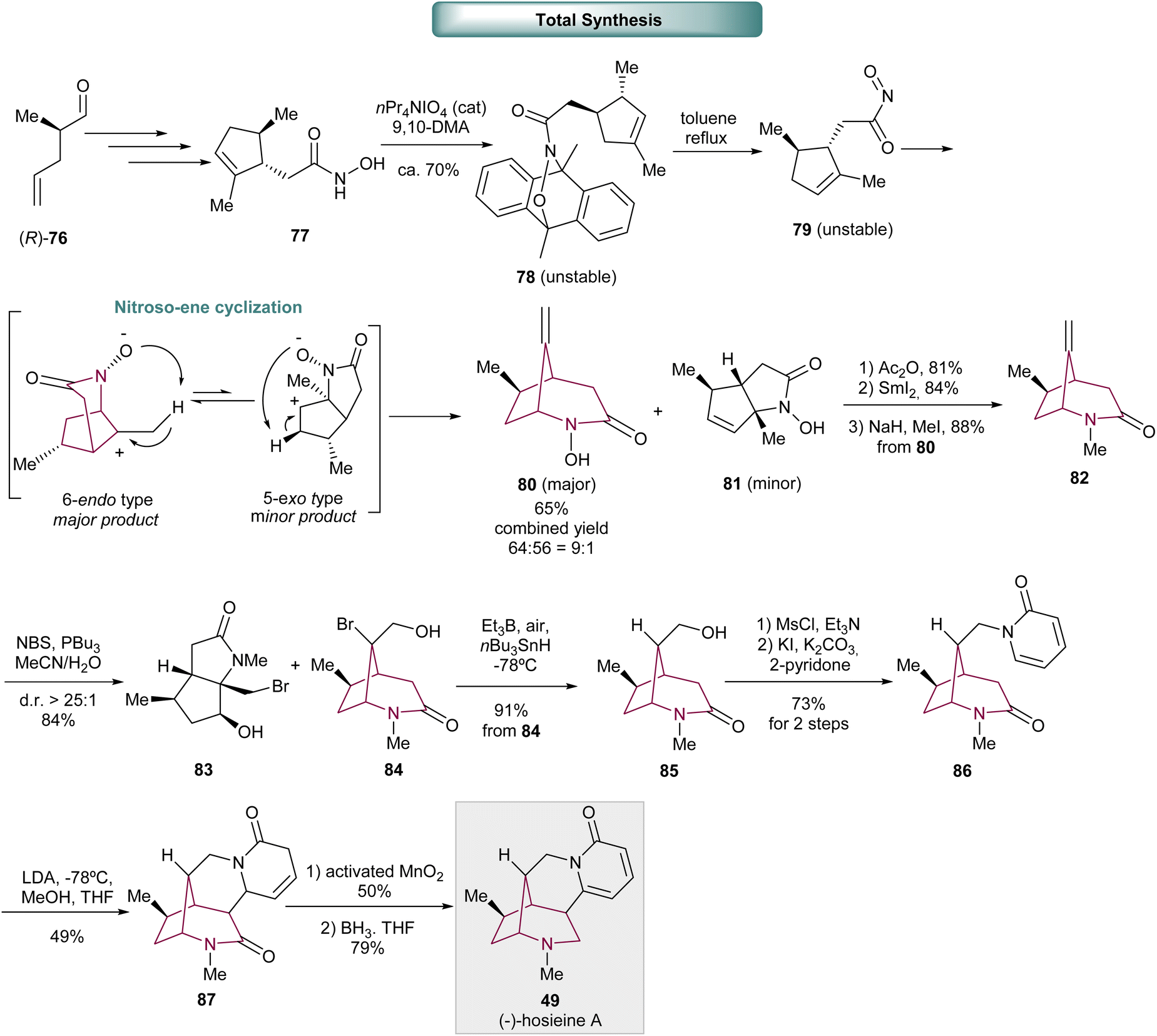 | ||
| Scheme 13 Total synthesis of (−)-hosieine A 49. | ||
3. Synthesis of 2-azabicyclo[3.2.1]octanes by rearrangements
The bicyclic core of the 2-azabicyclo[3.2.1]octane scaffold may also be accessed through different types of rearrangement reactions, typically through ring-enlargement reactions. Such reactions include Beckmann rearrangements,16 norbornadiene cascade rearrangements,17 and rearrangement of bicyclo[2.2.1]heptanes.183.1. Beckmann rearrangement
In 1960, Hall16a reported the first example of a Beckmann rearrangement in norcamphor derived oximes to 2-azabicyclo[3.2.1.]octan-3-one. Treatment of norcamphor derived oximes with benzenesulfonyl chloride or sulfuric acid/NH4OH, provided 2-azabicyclo[3.2.1.]octan-3-one 89a in 38% yield after migration of the bridgehead C–C bond (Scheme 14). Since then, a series of research groups have published their findings in such rearrangement in similar cores, with yields of 89a ranging from 35 to 90%. These results have been already summarized in an excellent review work by Krow in 1981 and will not be covered here extensively.19 Nevertheless, several of the old papers before 1981 report unspecified yields and uncertainty on the nature of some molecules, as well as unreproducible results.20Scheme 14 presents the main limitations associated with this transformation, which for norcamphor is the formation of a regiosiomer 89′a, via migration of the other C–C bond (depicted in green). The related fenchone oxime 88b also undergoes Beckmann rearrangement with additional side-reactions involving formation of unsaturated nitriles such as 90via cleavage of the bridgehead C–C bond (depicted in green, Scheme 14) forming a stable tertiary carbocation 91. Interestingly, camphor fails to give the 2-azabicyclo[3.2.1]octanone regioisomer in more than 12% yield,21 giving as major product the 3-aza isomer in 42% yield.19 Photo-Beckmann conditions have been explored in attempts to improve the yield and selectivity of the process but without success.22 For more complex substrates, the Beckmann rearrangement has also been observed to fail.21a Despite the various challenges associated with this transformation, the Beckmann rearrangement remains a useful protocol for the synthesis of 2-azabicyclo[3.2.1]octane core since the obtained amides can be cleanly reduced with LiAlH4.23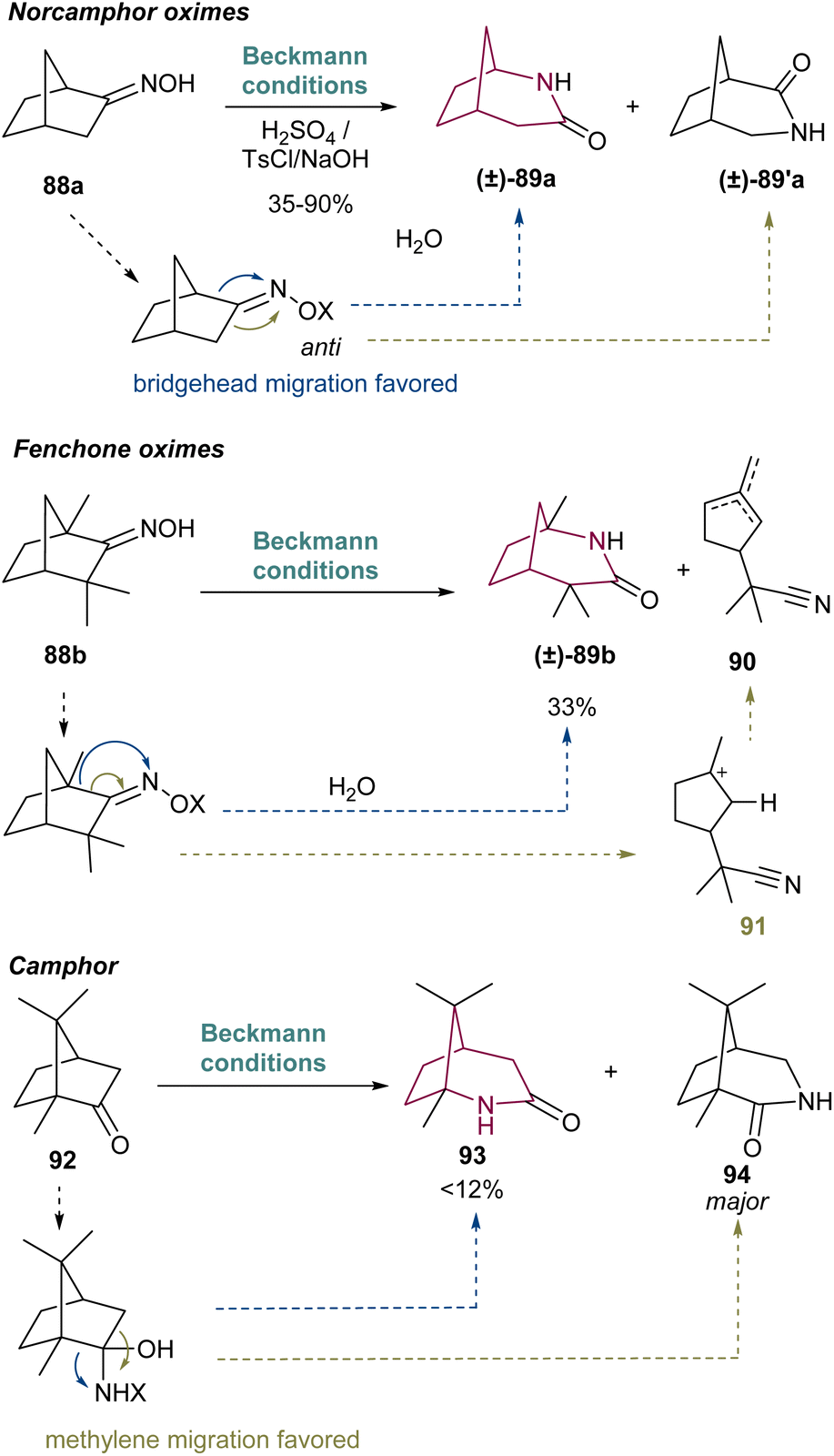 | ||
| Scheme 14 Synthesis of 2-azabicyclo[3.2.1.]octan-3-ones 89a,b. | ||
In 2008, Gin and co-workers applied the Beckmann rearrangement towards the synthesis of chlorocyclopentane cores of the proposed original and revised structures of palau'amine 95a and 95b, respectively (Scheme 15).24 For that, ketone 97 and 103 were prepared in several steps from dihydroquinone 96. The corresponding oxime was formed, and thionyl-chloride mediated Beckmann rearrangement in a regioselective manner to 2-azabicyclo[3.2.1.]octan-3-one 98 (Scheme 15A), and 104, the last obtained after Boc protection (Scheme 15B). Regarding the proposed original structure of palau'amine (Scheme 15A), 98 suffers cyclopentene ozonolysis with subsequent reductive work up and diol acetylation to furnish 99. Lactam Boc protection gave 100. Lastly, acetal saponification, imide hydrolysis and subsequent acidic treatment resulted in lactone 102 in 96% yield, over the 2 steps. Compounds 101 and 102 represent the cyclopentane cores of the originally proposed structure of palau'amine, 95a. Concerning the revised chlorocyclopentane cores of palau'amine 95b (Scheme 15B), from compound 104 two synthetic routes emerge. Lemieux–Johnson oxidation of 104 and subjection to silica gel and Et3N provided di-aldehyde 105 with C12 epimerization. The bridged chlorocyclopentanes 105 and 100 are stereochemically distinct at C12 and C17. Cyclopentene ring of 104 also undergoes ozonolysis followed by reduction with NaBH4 and treatment with TsOH. This led to intramolecular alcoholysis of the imide and lactone formation. Hydroxy protection delivers 106 in 91% yield, over the 2 steps. Benzyl ether removal led to the formation of 107. Finally, lactone hydrolysis, methyl ester formation, and C12 oxidation/epimerization yielded aldehyde 108, that is the C12,C17 diastereomer of chlorocyclopentane 102 (Scheme 15A).
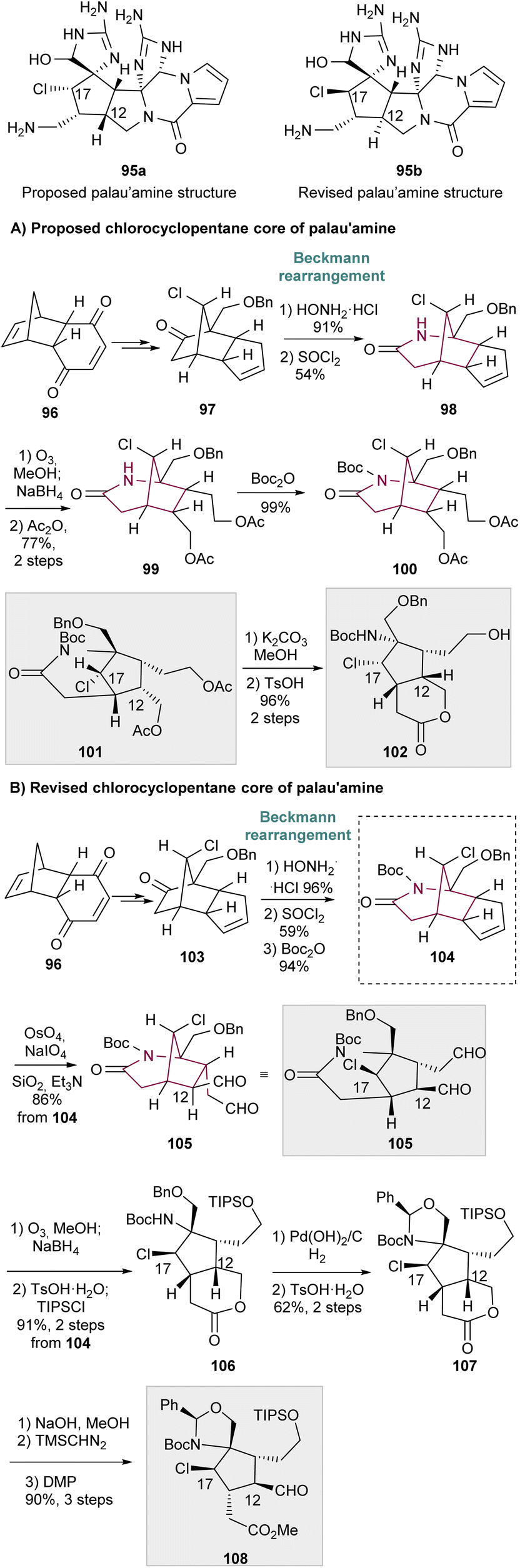 | ||
| Scheme 15 (A) Synthesis of chlorocyclopentane cores of the proposed original structure of palau'amine. (B) Synthesis of chlorocyclopentane cores of the revised structures of palau'amine. | ||
A way to access the bicyclic core from camphor is to initiate rearrangement with camphor hydrazone 109, as demonstrated by Tezuka and co-workers (Scheme 16). Treatment of 109 with m-chloroperbenzoic acid resulted in compounds 110 in high yield, as a 1:1 mixture of endo (110a) and exo (110b) isomers. Upon reflux in ethanol in the presence of acid or base, lactam 112 was formed in 40% yield from 110a. This reaction proceeds via formation of the corresponding alcohol intermediate 111a (Scheme 16).16c The thermal decomposition reaction of α-azohydroperoxide 113 was also reported by the same group. When 113 was refluxed in benzene, a mixture of products were obtained, among them lactam 114 in 22% yield (Scheme 16B).16b
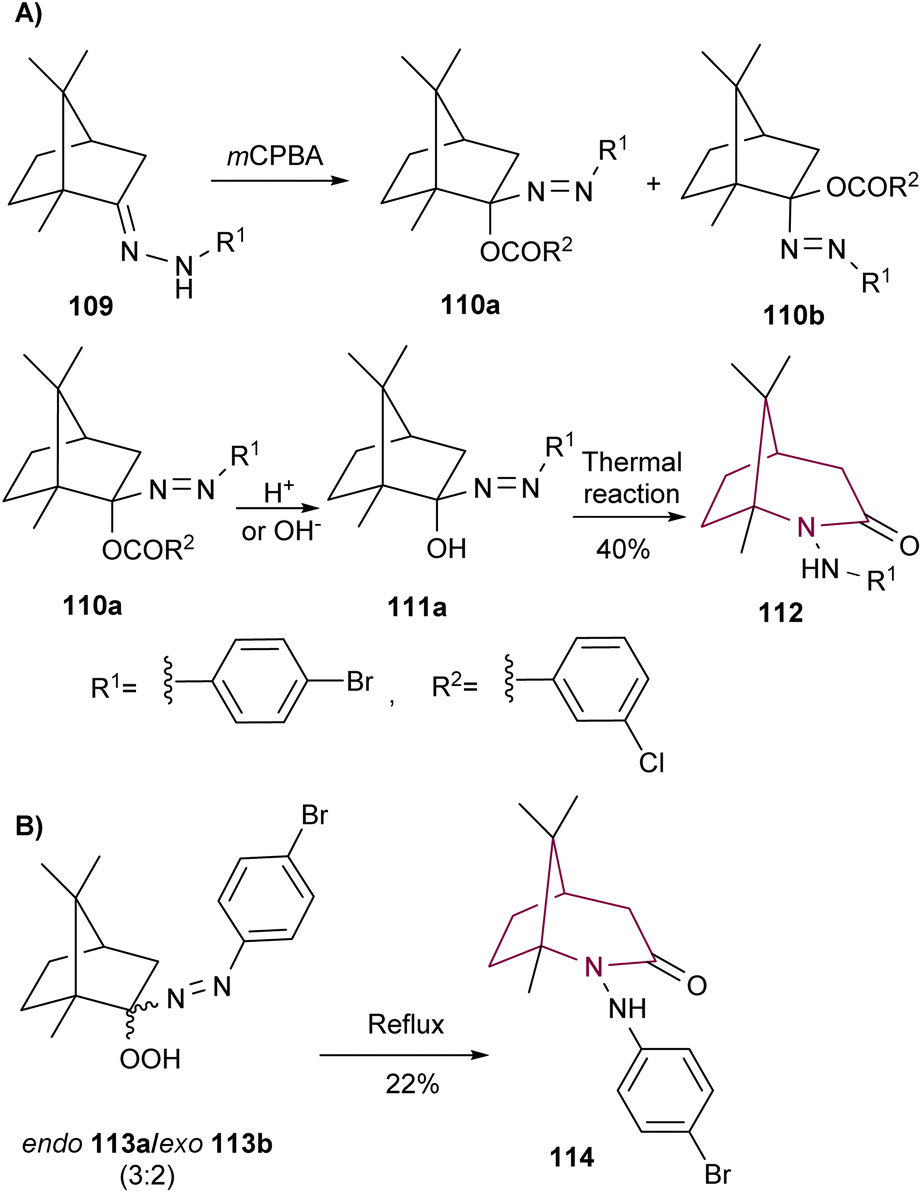 | ||
| Scheme 16 Synthesis of 2-azabicyclo[3.2.l]octane 112 and 114. | ||
3.2. Schmidt reaction
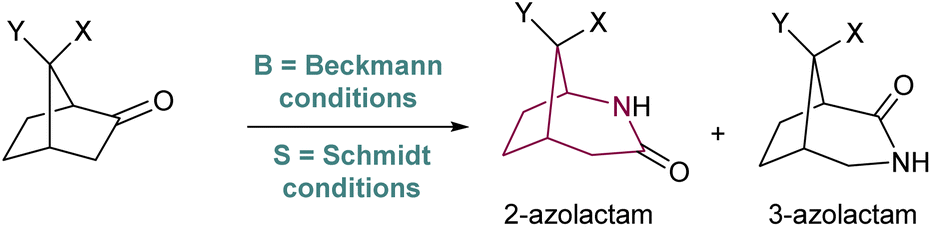
The synthesis of azabicyclo[3.2.1]octane can also be achieved by Schmidt reaction of ketones with hydroxyalkyl azides (Scheme 17). However, the preferred product from such rearrangement in most norcamphor derivatives is the 3-azolactam regioisomer, in low yields,25 which is in contrast with the Beckmann rearrangement. This difference in regioselectivity was studied in detail by Krow in 1996,26 who subjected various norcamphor derivatives to Beckmann and Schmidt conditions (Table 1). For most cases, Beckmann conditions delivered almost exclusively the 2-azolactam isomer, while Schmidt conditions gave a mixture of isomers, in which 3-azolactam was predominant (except for entry 5). Krow postulates that the difference in regioselectivity arises from the poor selectivity in the formation of the iminodiazonium intermediate in Schmidt conditions, whilst the oxime-O-sulfonic acid intermediate in Beckmann conditions forms with syn selectivity (Scheme 17).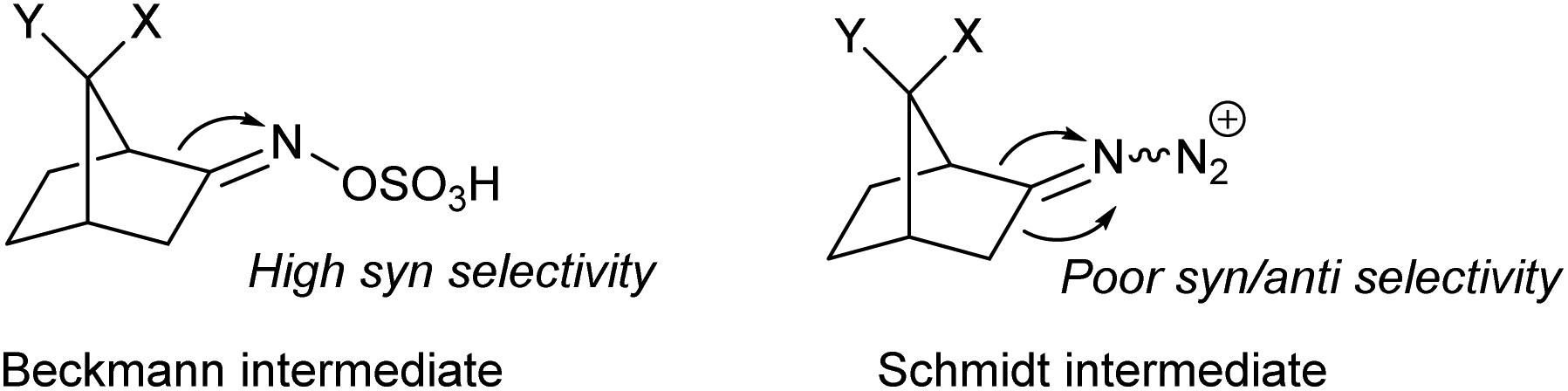 | ||
| Scheme 17 Beckmann and Schmidt intermediates for the norcamphor system. | ||
|
|
||||
|---|---|---|---|---|
| Entry | X | Y | Lactams yield | Ratio of lactams (2-aza:3-aza) |
| Beckmann conditions (B) = hydroxylamine-O-sulfonic acid in acetic acid; Schmidt conditions (S) = sulfuric acid and sodium azide in chloroform. | ||||
| 1 | H | H | B = 37% | B = (90:10) |
| S = 35% | S = (0:100) |
|||
| 2 | OMe | H | B = 27% | B = (100:0) |
| S = 48% | S = (27:73) |
|||
| 3 | Cl | H | B = 49% | B = (100:0) |
| S = 22% | S = (15:85) |
|||
| 4 | Br | H | B = 53% | B = (95:5) |
| S = 34% | S = (19:81) |
|||
| 5 | OTs | H | B = 27% | B = (100:0) |
| S = 52% | S = (65:35) |
|||
| 6 | H | COOMe | B = 29% | B = (100:0) |
| S = 31% | S = (34:66) |
|||
| 7 | H | Cl | B = 23% | B = (100:0) |
| S = 29% | S = (17:83) |
|||
| 8 | H | Br | B = 57% | B = (100:0) |
| S = 41% | S = (0:100) |
|||
| 9 | H | OTs | B = 38% | B = (100:0) |
| S = 40% | S = (64:36) |
|||
| 10 | H (exo-5-Br) | COOMe | B = 64% | B = (100:0) |
| S = 52% | S = (48:52) |
|||
In 2016, Aubé and co-workers18f implemented a new version of Schmidt reaction using triflic acid, in the presence of HFIP (Scheme 18). The reaction proceeds in two stages, first the ketone reacts with hydroxyalkyl azide and then is trapped intramolecularly by the hydroxyl to form the correspondent iminium ether. Finally, the iminium ether is hydrolyzed to deliver the amide. This new methodology was applied towards the synthesis of azabicyclo[3.2.1]octanes 116 and 117. While HFIP and the intramolecular hydroxyl group significantly improve the reaction yield, the poor selectivity in the C–C migration step continues to limit the applicability of the Schmidt rearrangement in the synthesis of azabicyclo[3.2.1]octanes as the resulting regioisomers are usually inseparable.
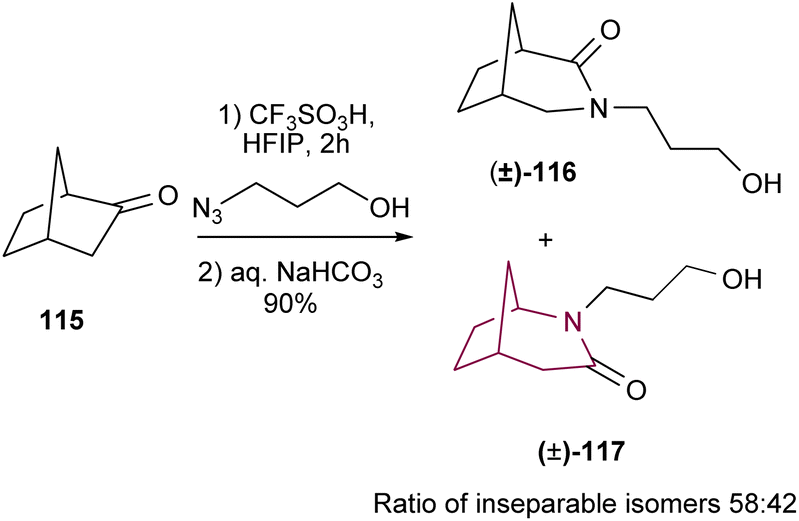 | ||
| Scheme 18 Synthesis of azabicyclo[3.2.1]octane core via Schmidt reaction. | ||
3.3. Norbornadiene rearrangement
A more robust synthetic method for the obtention of 2-azabicyclo[3.2.1]octane is through the rearrangement of norbornadiene 118. Reaction with tosyl azide proceeds through (2 + 3)-cycloaddition to form a transient triazoline 119, which is labile, and ring opens to the diazonium betaine 120. Subsequent loss of nitrogen leads to formation of the fused-ring aziridine 121. Compound 121 then slowly undergoes electrocyclic reaction opening the aziridine ring and providing the bicyclo[3.1.0]hexene imine 122. Then, through an aza-Cope rearrangement, N-tosyl-2-azabicyclo[3.2.1]octa-3,6-diene 123 is formed (Scheme 19), which is the main precursor for the 2-azabicyclo[3.2.1]octane family.17a,c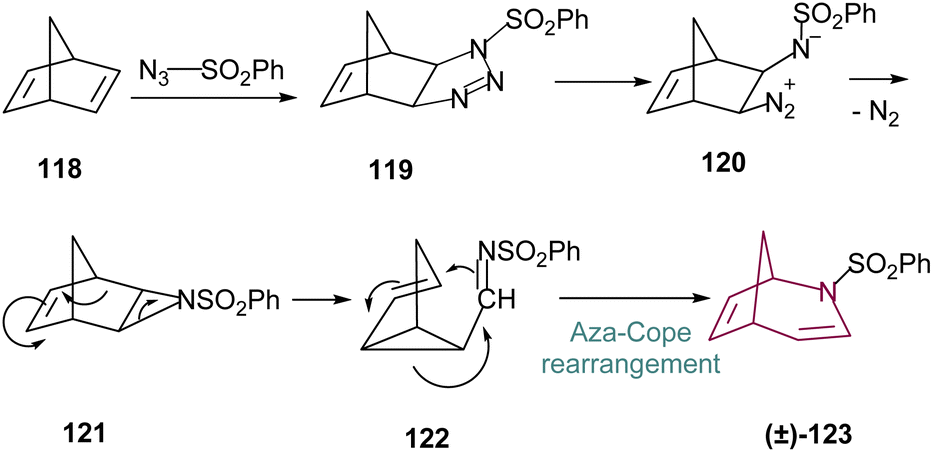 | ||
| Scheme 19 Proposed mechanistic pathway for the synthesis of N-tosyl-2-azabicyclo[3.2.1]octa-3,6-diene 123. | ||
Following the synthesis of 2-azabicyclo[3.2.1]octadiene 123 from norbornadiene 118, Young and co-workers17b obtained the desired 2-azabicyclo[3.2.1]octane 125 through a two-step sequence reduction, using LiAlH4 and hydrogenation. Direct hydrogenation of 2-azabicyclo[3.2.1]octadiene 126 with palladium catalyst results in 2-azabicyclo[3.2.1]octane 127 (Scheme 20).
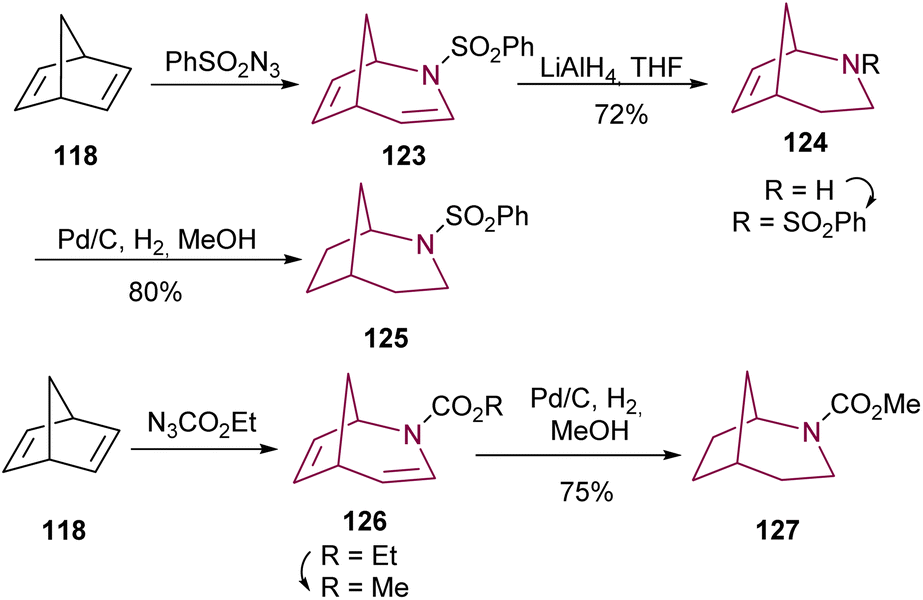 | ||
| Scheme 20 Reduction of 2-azabicyclo[3.2.1]octadiene 123 and 126 to deliver 2-azabicyclo[3.2.1]octane 125 and 127, respectively. | ||
Another approach to prepare 2-azabicyclo[3.2.1]octane core from norbornadiene was reported by Bergmeier and co-workers.17d In this case, a dihydroxylation of the C6–C7 olefin of 128 was performed using either AD-mix α or β, achieving excellent yields in both cases. The resulting diol was protected to yield acetonide 129. This was followed by insertion of a hydroxyl group at C4, yielding 4-exo-hydroxy-6,7-exo-isopropylidenedioxy-N-tosyl-2-azabicyclo[3.2.1]octane 130a in 65% yield, together with the minor 4-endo isomer 130b (Scheme 21).
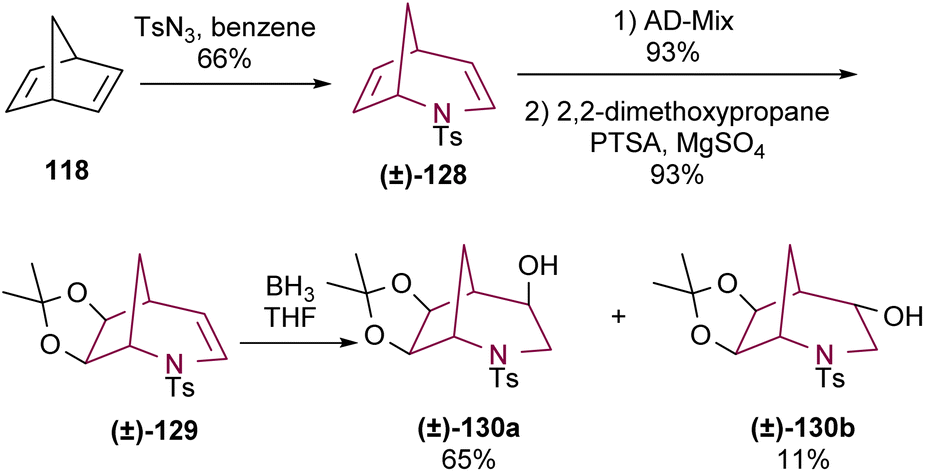 | ||
| Scheme 21 Selective functionalization of 2-azabicyclo[3.2.1]octadiene 128. | ||
The same research group reported another example of the selective functionalization of the double bonds of compound 128.17e Selective reduction of the C6–C7 double bond of 128 resulted in bicyclic compound 131. 4-Arylated and alkenylated products 132 were obtained with good yields via Suzuki coupling. Finally, hydrogenation of the C3–C4 olefin afforded saturated azabicyclo 133 (Scheme 22).
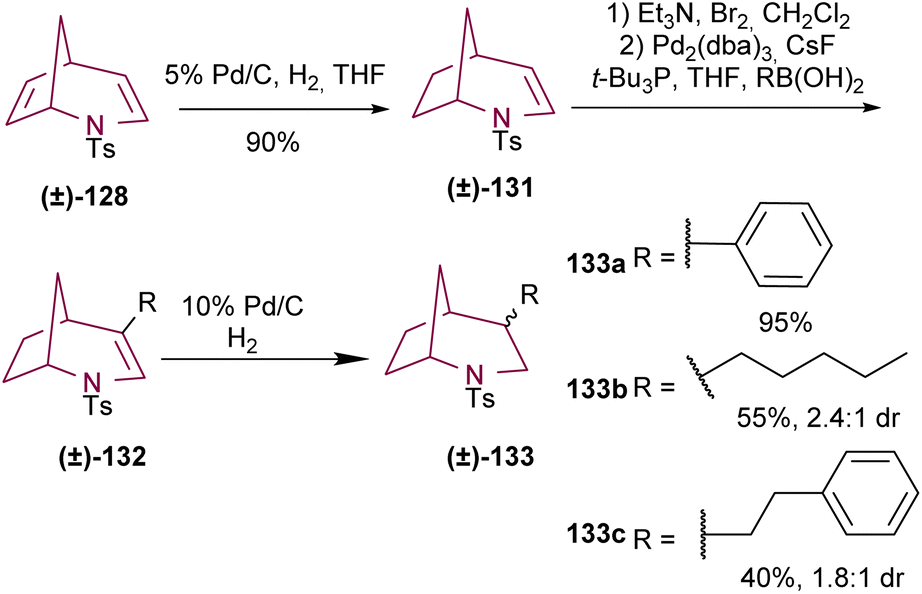 | ||
| Scheme 22 Selective functionalization of 2-azabicyclo[3.2.1]octadiene 128. | ||
2-Azabicyclo[3.2.1]octane can also be prepared taking profit of traditional Cope rearrangement, which allows an equilibrium between compounds 134 and 135, which are formed via epoxidation and rearrangement of norbornadiene. The mixture, when treated with excess methylamine, undergoes imine formation and 1,6-addition of methylamine, yielding 136 and 137 in equilibrium (Scheme 23). Only 137 reacts under hydrogenation conditions, resulting in 2-azabicyclo[3.2.1]octane 138 in 86% yield.18b
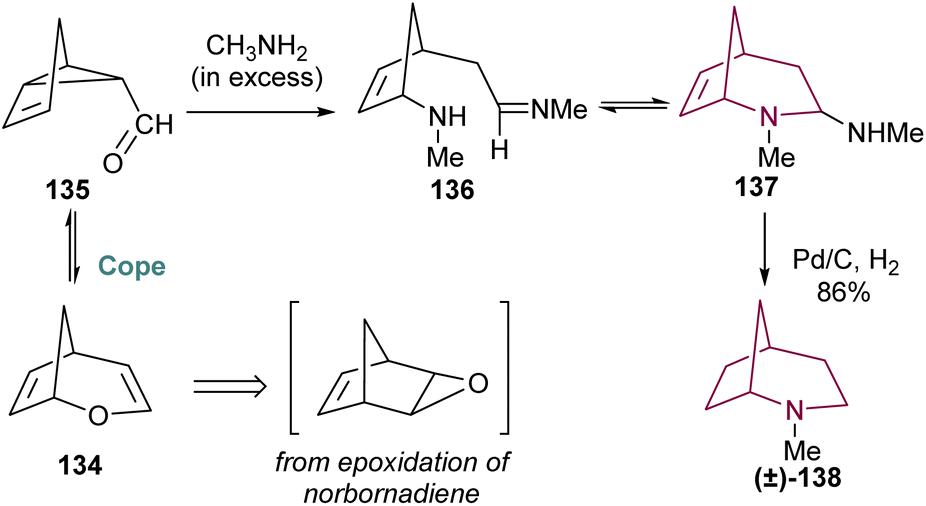 | ||
| Scheme 23 Synthesis of 2-azabicyclo[3.2.l]octane 138 from 134. | ||
3.4. Photolysis
In addition to the Schmidt rearrangement, the azide group has also provided access to the azabicyclo[3.2.1]octane via photolysis. Lwowski and co-workers studied the irradiation of azidonorbornane 139 with UV light (310–410 nm or 254 nm) (Scheme 24). In alcoholic solvents, this reaction resulted in the formation of hemiaminal ethers 140 and 141 in 52–54% and 23–24% yields, respectively. Reduction of 140a with LiAIH4 resulted in 2-azabicyclo[3.2.l]octane 1a in 33% yield, while reduction of 140b gave 1b in a much better 91% yield.18a A few years later, Sheridan and colleagues confirmed the reaction mechanism by methanol trapping experiments, showing that products 140 and 141 were formed through nucleophilic addition of alcohol to the highly reactive bridgehead imine intermediates.18e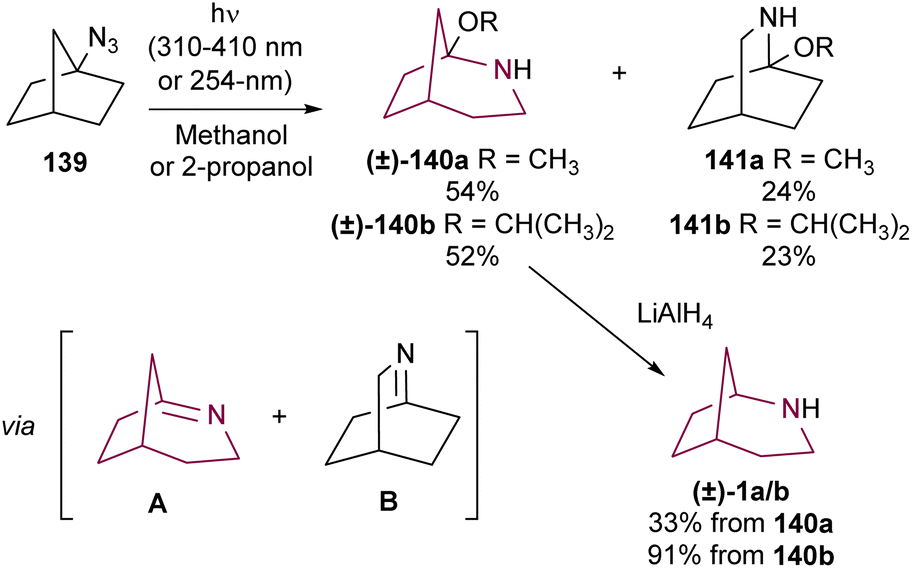 | ||
| Scheme 24 Synthesis of 2-azabicyclo[3.2.1]octanes 140a,bvia photolysis of azidonorbornane 139. | ||
While some research groups have observed the formation of the 2-azabicyclo[3.2.1]octane system via rearrangement of oxaziridines of camphor27 and norcamphor,28 the low yields and poor selectivity have limited their applicability. Remarkably, Yamada and co-workers have tamed their reactivity through photochemical rearrangement of alkanenitronate anions like bicyclic nitro compounds 142a and 142b. After deprotonation and irradiation, isomerization of an oxaziridine intermediate gives access to the azabicyclo[3.2.1]octane hydroxamic acids 143a and 143b in good yields and selectivity. Then, reduction allowed the preparation of the corresponding azabicyclo[3.2.1]octane amides 144a and 144b (Scheme 25).18c,d
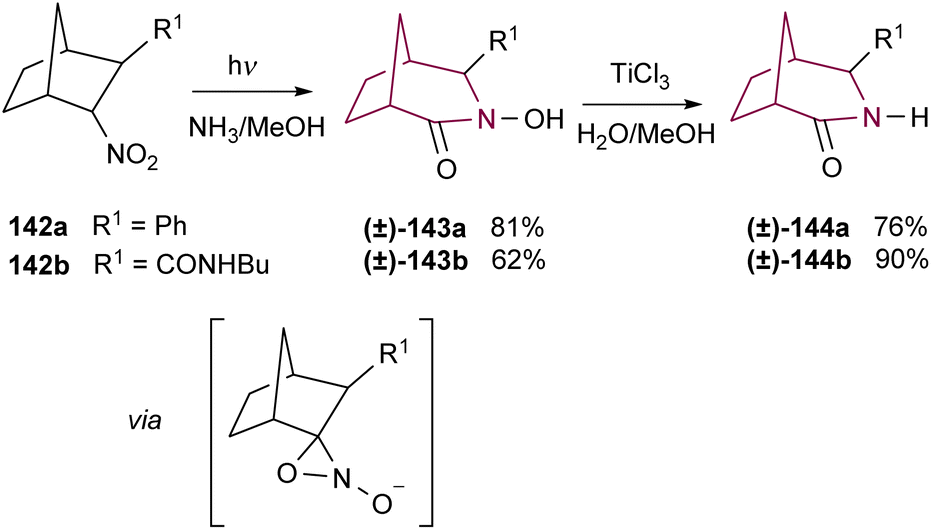 | ||
| Scheme 25 Synthesis of azabicyclo[3.2.1]octane core via rearrangement of alkanenitronate anions. | ||
3.5. Mitsunobu promoted rearrangement
Wojaczyńska and co-workers reported the synthesis of 2-azabicyclo[3.2.1]octane system via rearrangement of 2-azabicyclo[2.2.1]heptane derivatives under Mitsunobu conditions18g or sulfonyl chloride/base (Scheme 26).29 The reaction mechanism proceeds through activation of the primary alcohol and intramolecular nitrogen nucleophilic attack forming an aziridinium intermediate, which was then regioselectively opened by nucleophilic attack at the more substituted carbon, releasing ring strain in the [2.2.1]heptane system (Scheme 26A).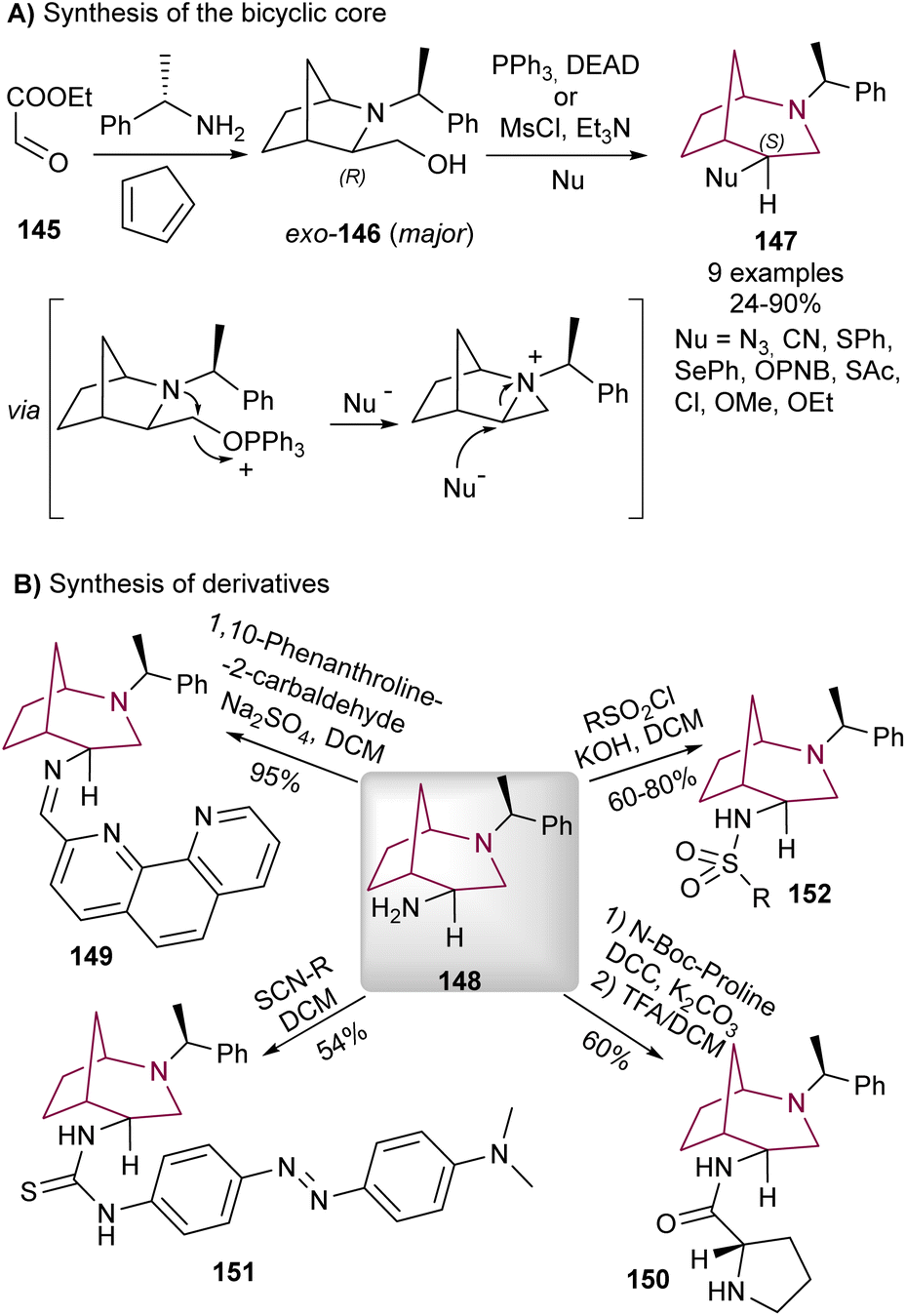 | ||
| Scheme 26 Synthesis of 2-azabicyclo[3.2.1]octane 147 through Mitsunobu reaction (A), and synthesis of derivatives (B). | ||
The correspondent rearranged products were obtained with inversion of configuration expected for SN2 aziridine ring-opening reaction. The minor endo-146 also undergoes the same aziridinium formation/regioselective ring opening isomer. The product derived from azide addition proved to be especially useful since after reduction to the amine 148, it provided the bicyclic core a synthetic handle to install further functionalities (Scheme 26B).
The same authors installed a 1,10-phenanthroline moiety via imine formation (149),30 and a proline via amide bond (150).29 These molecules were evaluated as catalysts for asymmetric aldol reactions, as the chiral and rigid core of the 2-azabicyclo[3.2.1]octane system could induce stereoselectivity. While a pioneer study in the utilization of the 2-azabicyclo[3.2.1]octane system in asymmetric organocatalysis, the des and ees obtained were not outstanding, and no other studies were performed in this topic. The free amine additionally proved useful for the synthesis of chiral thioureas (151)31 and sulfonamides (152), the latter displaying promising antiviral32 and antiproliferative activity,18g and also cytotoxicity to cancer cell lines.7
4. Summary and outlook
The 2-azabicyclo[3.2.1]octane system represents an important scaffold that is present in natural and synthetic products. The major interest in this structure lies in its prospective application as a class of bioactive molecules. Different biological activities have already been demonstrated by several research groups. On the other hand, it has also been shown that this core is often present as an important intermediate in the synthesis of other biologically active molecules. Therefore, efficient access to this scaffold is likely to bring new advancements within drug discovery. Current methods for its synthesis are still lacklustre, both in yield and in regioselectivity in most rearrangements. One major hindrance to the application of the 2-azabicyclo[3.2.1]octane in modern organic chemistry is the lack of direct asymmetric synthetic methods. Being a chiral highly rigid bicyclic system with a functional handle in the form of amine, its nearly absent use in asymmetric organocatalysis is surprising, which can be attributed by its synthetic challenge. In this Minireview, we have summarized several synthetic approaches for the obtention of the 2-azabicyclo[3.2.1]octane scaffold, highlighting their advantages and challenging limitations. Efforts are expected to be devoted to the development of more and efficient approaches to synthesize this bicyclic system, particularly in an asymmetric fashion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Fundação para a Ciência e Tecnologia (FCT) for financial support (2022.08559.PTDC, UIDB/04138/2020, UIDP/04138/2020 and 2023.03748.BD). The project leading to this application has received funding from the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement No. 951996.References
- (a) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247 RSC; (b) J. Liu, J. Jiang, L. Zheng and Z. Q. Liu, Adv. Synth. Catal., 2020, 362, 4876 CrossRef CAS; (c) C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2021, 122, 8181 CrossRef PubMed.
- (a) U. Ghani, Eur. J. Med. Chem., 2015, 103, 133 CrossRef CAS PubMed; (b) G. Li and H. X. Lou, Med. Res. Rev., 2018, 38, 1255 CrossRef PubMed; (c) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770 CrossRef CAS PubMed; (d) A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, The International Natural Product Sciences Taskforce and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200 CrossRef CAS PubMed.
- (a) N. Emelda and S. C. Bergmeier, Tetrahedron Lett., 2008, 49, 5363 CrossRef CAS PubMed; (b) R. Profeta, J. Klein, S. Spada, F. Ferroni, A. Paio and D. Andreotti, Tetrahedron Lett., 2010, 51, 5521 CrossRef CAS; (c) J. Ouyang, R. Yan, X. Mi and R. Hong, Angew. Chem., Int. Ed., 2015, 54, 10940 CrossRef CAS PubMed.
- I. Pouny, M. Batut, L. Vendier, B. David, S. Yi, F. Sautel, P. B. Arimondo and G. Massiot, Phytochemistry, 2014, 107, 97 CrossRef CAS PubMed.
- (a) H. H. Ong and V. B. Anderson, J. Med. Chem., 1978, 21, 758 CrossRef CAS PubMed; (b) H. H. Ong, V. B. Anderson, J. C. Wilker, T. C. Spaulding and L. R. Meyerson, J. Med. Chem., 1980, 23, 726 CrossRef CAS PubMed.
- H. Jonas, D. Aiello, B. Frehland, K. Lehmkuhl, D. Schepmann, J. Köhler, P. Diana and B. Wünsch, Org. Biomol. Chem., 2021, 19, 8384 RSC.
- M. Samadaei, M. Pinter, D. Senfter, S. Madlener, N. Rohr-Udilova, D. Iwan, K. Kamińska, E. Wojaczyńska, J. Wojaczyński and A. Kochel, Molecules, 2020, 25, 2355 CrossRef CAS PubMed.
- A. D. G. Lawson, M. Maccoss and J. P. Heer, J. Med. Chem., 2018, 61, 4383 CrossRef PubMed.
- (a) H. A. Dondas and N. Yaktubay, Heterocycl. Commun., 2003, 9, 337 CAS; (b) D. Ilari, S. Maskri, D. Schepmann, J. Köhler, C. G. Daniliuc, O. Koch and B. Wünsch, Org. Biomol. Chem., 2021, 19, 4082 RSC; (c) K. Noguchi, M. Takeda and S. Nurimoto, Chem. Pharm. Bull., 1977, 25, 890 CrossRef CAS PubMed; (d) K. Namba, Y. Shobo, K. Fujimoto, I. Shoji, M. Yoshida and K. Tanino, Eur. J. Org. Chem., 2014, 5196 CrossRef CAS.
- (a) E. H. Billett, I. Fleming and S. W. Hanson, J. Chem. Soc., Perkin Trans. 1, 1973, 1661 RSC; (b) C. M. Gonzalez-Alvarez, L. Quintero, F. Santiesteban and J. L. Fourrey, Eur. J. Org. Chem., 1999, 3085 CrossRef CAS; (c) S. K. Johansen, H. T. Kornø and I. Lundt, Synthesis, 1999, 171 CrossRef CAS.
- P. G. Gassman, F. Hoyda and J. Dygos, J. Am. Chem. Soc., 1968, 90, 2716 CrossRef.
- (a) E. L. May and J. G. Murphy, J. Org. Chem., 1955, 20, 1197 CrossRef CAS; (b) M. E. Rogers and E. L. May, J. Med. Chem., 1974, 17, 1328 CrossRef CAS PubMed.
- S. R. Angle, J. M. Fevig, S. D. Knight, R. W. Marquis and L. E. Overman, J. Am. Chem. Soc., 1993, 115, 3966 CrossRef CAS.
- (a) S. D. Knight, L. E. Overman and G. Pairaudeau, J. Am. Chem. Soc., 1993, 115, 9293 CrossRef CAS; (b) S. D. Knight, L. E. Overman and G. Pairaudeau, J. Am. Chem. Soc., 1995, 117, 5776 CrossRef CAS.
- Y.-W. Huang, K. Kong and J. L. Wood, Angew. Chem., Int. Ed., 2018, 57, 7664 CrossRef CAS PubMed.
- (a) H. K. Hall, J. Am. Chem. Soc., 1960, 82, 1209 CrossRef CAS; (b) T. Tezuka and T. Otsuka, Chem. Lett., 1989, 18, 1051 CrossRef; (c) T. Tezuka, T. Otsuka and H. Kasuga, Tetrahedron Lett., 1990, 31, 7633 CrossRef CAS.
- (a) A. C. Oehlschlager and L. H. Zalkow, Chem. Commun., 1965, 70 RSC; (b) P. Barraclough, S. Bilgic and D. W. Young, Tetrahedron, 1978, 35, 91 CrossRef; (c) E. Huda, H. Martin, B. Mayer, K. Somnitz, A. Steigel, H. Haddad, G. Distefano and A. Modelli, Chem. Ber., 1991, 124, 2879 CrossRef CAS; (d) D. D. Reed and S. C. Bergmeier, J. Org. Chem., 2007, 72, 1024 CrossRef CAS PubMed; (e) I. S. Armstrong and S. C. Bergmeier, Synthesis, 2017, 2733 CAS.
- (a) J. O. Reed and W. Lwowski, J. Org. Chem., 1971, 36, 2864 CrossRef; (b) P. Barraclough and D. W. Young, J. Chem. Soc., Perkin Trans. 1, 1975, 2354 RSC; (c) K. Yamada, T. Kanekiyo, S. Tanaka, K. Naruchi and M. Yamamoto, J. Am. Chem. Soc., 1981, 103, 7003 CrossRef CAS; (d) K. Yamada, S. Tanaka, K. Naruchi and M. Yamamoto, J. Org. Chem., 1982, 47, 5283 CrossRef CAS; (e) R. S. Sheridan and G. A. Ganzer, J. Am. Chem. Soc., 1983, 105, 6158 CrossRef CAS; (f) H. F. Motiwala, M. Charaschanya, V. W. Day and J. Aubé, J. Org. Chem., 2016, 81, 1593 CrossRef CAS PubMed; (g) D. Iwan, K. Kamińska, E. Wojaczyńska, M. Psurski, J. Wietrzyk and M. Daszkiewicz, Materials, 2020, 13, 5010 CrossRef CAS PubMed.
- G. R. Krow, Tetrahedron, 1981, 37, 1283 CrossRef CAS.
- P. A. Hunt and C. J. Moody, Tetrahedron Lett., 1989, 30, 7233 CrossRef CAS.
- (a) P. Deslongchamps, A. Bélanger, D. J. F. Berney, H. J. Borschberg, R. Brousseau, A. Doutheau, R. Durand, H. Katayama, R. Lapalme, D. M. Leturc, C.-C. Liao, F. N. Maclachlan, J. P. Maffrand, F. Marazza, R. Martino, C. Moreau, L. Ruest, L. Saint-Laurent, R. Saintonge and P. Soucy, Can. J. Chem., 1990, 68, 153 CrossRef CAS; (b) Y. Zhang, T. J. Zhang, X. Y. Li, J. W. Liang, S. Tu, H. L. Xu, W. H. Xue, X. H. Qian, Z. H. Zhang, X. Zhang and F. H. Meng, Eur. J. Med. Chem., 2021, 210, 112988 CrossRef CAS PubMed.
- (a) B. L. Fox and H. M. Rosenberg, J. Chem. Soc. D, 1969, 46, 1115 RSC; (b) H. Suginome, K. Furukawa and K. Orito, J. Chem. Soc., Chem. Commun., 1987, 1004 RSC; (c) H. Suginome, K. Furukawa and K. Orito, J. Chem. Soc., Perkin Trans. 1, 1991, 917 RSC.
- B. L. Fox and J. E. Reboulet, J. Org. Chem., 1968, 33, 3639 CrossRef CAS.
- M. S. Bultman, J. Ma and D. Y. Gin, Angew. Chem., Int. Ed., 2008, 47, 6821 CrossRef CAS PubMed.
- (a) J. Aubé, G. L. Milligan and C. J. Mossman, J. Org. Chem., 1992, 57, 1635 CrossRef; (b) P. Desai, K. Schildknegt, K. A. Agrios, C. J. Mossman, G. L. Milligan and J. Aubé, J. Am. Chem. Soc., 2000, 122, 7226 CrossRef CAS.
- G. R. Krow, O. H. Cheung, Z. Hu and Y. B. Lee, J. Org. Chem., 1996, 61, 5574 CrossRef CAS.
- P. C. B. Page, C. Limousin and V. L. Murrell, J. Org. Chem., 2002, 67, 7787 CrossRef CAS PubMed.
- J. Aubé, M. Hammond, E. Gherardini and F. Takusagawa, J. Org. Chem., 1991, 56, 499 CrossRef.
- D. Iwan, K. Kamińska and E. Wojaczyńska, Molecules, 2021, 26, 5166 CrossRef CAS PubMed.
- E. Wojaczyńska, J. Skarżewski, Ł. Sidorowicz, R. Wieczorek and J. Wojaczyński, New J. Chem., 2016, 40, 9795 RSC.
- K. Kamińska, D. Iwan, A. Iglesias-Reguant, W. Spałek, M. Daszkiewicz, A. Sobolewska, R. Zaleśny, E. Wojaczyńska and S. Bartkiewicz, J. Mol. Liq., 2022, 363, 119869 CrossRef.
- D. Iwan, K. Kamińska, M. Denel-Bobrowska, A. B. Olejniczak and E. Wojaczyńska, Biomed. Pharmacother., 2022, 153, 113473 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |