
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA SmI2-mediated reductive cyclisation reaction using the trifluoroacetamide group as the radical precursor†
Kota
Yoshioka
,
Hiroki
Iwasaki
 *,
Mako
Hanaki
,
Saho
Ito
,
Yuzuha
Iwamoto
,
Rio
Ichihara
and
Hisanori
Nambu
*,
Mako
Hanaki
,
Saho
Ito
,
Yuzuha
Iwamoto
,
Rio
Ichihara
and
Hisanori
Nambu
Kyoto Pharmaceutical University, 1 Misasagi-Shichono-cho, Yamashina-ku, Kyoto, 607-8412, Japan. E-mail: iwasaki@mb.kyoto-phu.ac.jp
First published on 16th February 2024
Abstract
A samarium(II)-mediated reductive cyclisation reaction with the aminoketyl radical from the trifluoroacetamide group for synthesising 2-trifluoromethylindolines was developed. This reaction is the first example of using an acyclic amide group, which is considered difficult to react with SmI2, in a reductive cyclisation. Additionally, the conversion of the obtained product into 2-trifluoromethylindole was achieved.
Introduction
Since the simple preparation of SmI2 was reported by Kagan et al. in 1980,1 SmI2 has been recognised as the most versatile single-electron reducing agent.2 One reason for this is that SmI2 is a highly chemoselective reagent, and its functional group selectivity can be fine-tuned by using appropriate ligands and additives.3 Reactions using this useful reagent can be classified into two major categories. One is the reduction of functional groups and the other is the reductive carbon–carbon bond formation reactions. Functional group reductions have been reported for sulfones and sulfoxides, alkyl and aryl halides, epoxides, phosphine oxides, carbonyls, and conjugated double bonds.4 However, reductive carbon–carbon bond formation reactions are often used as important key reactions in the total synthesis of natural products, leveraging the excellent stereochemical control owing to the high oxophilicity of samarium and the ability to coordinate.5 Particularly, cyclisation reactions by single-electron transfer (SET) reduction of carbonyl compounds with SmI2 are superior in that they allow the synthesis of decorative cyclic structures by combining carbonyl moieties with unsaturated functional groups such as alkynes, alkenes, and allenes through radical umpolung.6 Based on this background, many intramolecular cyclisation reactions of ketyl radicals, which can be easily prepared from SmI2 and aldehydes or ketones, have been reported.7 We have also reported the synthesis of spirocyclic compounds by intramolecular cyclisation using ketyl radicals prepared from ketones.8Functional groups such as lactones and acyclic esters, as well as nitrogen-containing compounds such as lactams and cyclic imides, are considered to have difficulty reacting with SmI2. However, in recent years, by adjusting the reactivity of SmI2 using a coordinating additive, cyclisation reactions with those functional groups have been reported (Scheme 1A).9 These studies expanded the adaptive limits of reductive cyclisation with SmI2 and provided useful organic chemical insights. However, there are only a few reports on the reduction reactions of acyclic amides with SmI2 to amines or alcohols (Scheme 1B),10 and to the best of our knowledge, there are no reports on reductive cyclisation reactions.
 | ||
| Scheme 1 SmI2-mediated reductive cyclisation reaction (A) and reduction of amide (B). | ||
Indoles and indolines are basic nitrogen-containing heterocycles that are widely present in various natural products and biologically active compounds.11 Therefore, the development of efficient synthetic methods for indoles and indolines has attracted significant attention from organic chemists. We have reported a reaction for the synthesis of indole derivatives by intramolecular cyclisation using aryl radicals generated from aryl halides with SmI2.12 Incidentally, it is well known that the introduction of fluorine compounds such as trifluoromethyl groups into organic compounds has positive effects on bioactive molecules, such as membrane permeability, lipophilicity, and metabolic oxidation prevention.13 Therefore, the synthesis of 2-trifluoromethylindolines and 2-trifluoromethylindoles has recently attracted much attention, and various approaches have been reported.14
We focused on the functionalised 2-CF3-indole synthesis reported by Nenajdenko et al. (Scheme 2A).14c They synthesised 2-CF3-indoles with various substituents by adding nucleophiles to an indoline intermediate with a cyclic hemiaminal moiety stabilised by a CF3 group using a one-pot reaction. We hypothesised that if aminoketyl radicals generated from acyclic trifluoromethylacetamide groups are trapped by intramolecular alkynes, 2-CF3-indoline with an exo-olefin moiety at the 3-position could be synthesised, and this product could be used as a substrate for the synthesis of functionalised 2-CF3-indoles (Scheme 2B). Herein, we report a new method for synthesising 2-CF3-indoline using SmI2. This reaction is a reductive cyclisation using a chain amide as the radical precursor, which has never been reported before, and is realised by using the captodative effect15 between an electron-withdrawing trifluoromethyl group and an electron-donating amino group. The product was considered useful as a building block for the synthesis of functionalised 2-CF3-indoles by reaction with various nucleophiles.
 | ||
| Scheme 2 2-CF3-indole synthesis via a hemiaminal intermediate and 2-CF3-indoline synthesis via an aminoketyl radical intermediate. | ||
We selected N-(2-ethynylphenyl)-2,2,2-trifluoroacetamide 1a as the model substrate for the synthesis of 2-CF3-indoline derivatives with a hemiaminal structure.16 First, to optimise the reaction conditions for the reductive cyclisation reaction between aminoketyl radicals and alkynes, equivalent amounts of SmI2, additives, and reaction temperatures were examined (Table 1). In our previous studies, we found that cyclisation reactions using SmI2 are promoted when the proton source traps the organic samarium species produced by the single-electron reduction of the radical which was generated after cyclisation.12 Therefore, the reaction of 1a was performed at 0 °C using 1.5 equivalents of SmI2, HMPA as an additive to increase the reduction potential of SmI2,3 and i-PrOH as a proton source. The desired indoline derivative 2a, which has a hemiaminal structure, was obtained in 65% yield without any byproducts (entry 1). To the best of our knowledge, this reaction is the first example of a reductive cyclisation using SmI2 with an acyclic amide group as the radical precursor. Because 11% of the starting material was recovered, we increased the amount of SmI2 and found that the best yield of the desired product (82%) was obtained when 2.5 equivalents of SmI2 were used (entries 2–4). To verify the effect of the proton source, we performed the reaction using 2.0 equivalents of SmI2 and 20 equivalents of i-PrOH and found that the product yield was slightly improved compared to that using 2.0 equivalents of i-PrOH (entry 5). However, at 2.5 equivalents of SmI2, the yield of indoline 2a was lower, resulting in the 3-methyl-2-trifluoromethylindole in 12% yield (entry 6). The absence of a proton source also resulted in a significant decrease in yield (entry 7). These results suggest that the proton source promotes cyclisation by contributing to the protonation of the organic samarium species. Additionally, the reaction temperature was also studied and increasing the reaction temperature to room temperature led to a decrease in the yield (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | SmI2 (eq.) | Additive (eq.) | Temp. (°C) | Yielda (%) 2a |
| a Isolated yield. b Starting material was recovered in 11% yield. c 3-Methyl-2-trifluoromethylindole was obtained by elimination of the hydroxy group, caused by the pushing of the lone pair on the nitrogen atom, and subsequent reduction in 6% yield. d 3-Methyl-2-trifluoromethylindole was obtained in 12% yield. HMPA = hexamethylphosphoramide. | ||||
| 1b | 1.5 | HMPA (5.4), i-PrOH (2.0) | 0 | 65 |
| 2 | 2.0 | HMPA (7.2), i-PrOH (2.0) | 0 | 73 |
| 3 | 2.5 | HMPA (9.0), i-PrOH (2.0) | 0 | 82 |
| 4c | 3.0 | HMPA (10.8), i-PrOH (2.0) | 0 | 61 |
| 5 | 2.0 | HMPA (7.2), i-PrOH (20) | 0 | 80 |
| 6d | 2.5 | HMPA (9.0), i-PrOH (20) | 0 | 61 |
| 7 | 2.5 | HMPA (9.0) | 0 | 47 |
| 8 | 2.0 | HMPA (7.2), i-PrOH (2.0) | rt | 68 |

Our interest shifted to the substrate generality of this reaction. First, we investigated the effects of the substituent positions on the benzene ring. The results obtained using substrates 1b–1e with methyl groups at 3–6 positions of the benzene ring are summarised in Table 2. The desired products 2b–2e were obtained in moderate to good yields. Substrate 1b, with a methyl group next to the alkyne group, yielded the least cyclised product 2b, probably due to the steric repulsion of the methyl group against the exo-olefin caused by cyclisation (entry 1). However, the substrate 1e with a methyl group next to the trifluoroacetamide group gave the desired product 2e in the best yield (entry 4), probably because the steric repulsion between the methyl and trifluoroacetamide groups allowed the aminoketyl radical to easily approach the alkyne, acting as a radical acceptor.
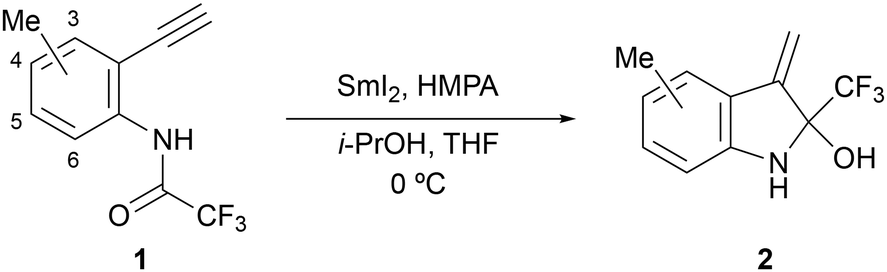
As part of our investigation of the scope and limitations of the substrates, we performed reactions using substrates 1f–1k with electron-donating or electron-withdrawing groups at the para-position of the trifluoroacetamide group, which may affect the stability of the aminoketyl radicals through a captodative effect (Table 3). When the reaction of substrate 1f with a methoxy group as the electron-donating group was performed, 2f was not obtained, unexpectedly (entry 1). 5-Methoxy-3-methyl-2-(trifluoromethyl)-1H-indole 3 and the dimer 4 of 2f were obtained in 4% and 30% yields, respectively. The reason for obtaining the indole derivative is probably because the strong electron-donating nature of the methoxy group increased the electron density on the nitrogen atom, making the hemiaminal structure unstable. Similarly, the increased nucleophilicity of the nitrogen atom is believed to have led to the formation of the dimer of 2f. Subsequently, we examined the use of substrates 1g–1k with halogen, cyano, ester, or nitro groups as electron-withdrawing groups, and found that the yield of the cyclised product decreased as the electron-withdrawing property became stronger (entries 2–6). These results indicate that the presence of an electron-withdrawing group on the benzene ring weakens the electron-donating ability of the nitrogen atom to the aminoketyl radical, resulting in lower yields.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Yieldb (%) | |
| R | |||
a All reactions were performed in THF using SmI2 (2.5 equiv.), i-PrOH (2.0 equiv.), and HMPA (9.0 equiv.) at 0 °C.
b Isolated yield.
c 5-Methoxy-3-methyl-2-(trifluoromethyl)-1H-indole 3 and the dimer 4 of 2f were obtained in 4% and 30% yields, respectively.
d Starting material was recovered in 29% yield.
e Starting material was recovered in 28% yield.
f Starting material was recovered in 57% yield. N.D. = not detected.

|
|||
| 1c | 1f | OMe | 2f: N.D. |
| 2 | 1g | Br | 2g: 62 |
| 3 | 1h | Cl | 2h: 56 |
| 4d | 1i | CN | 2i: 29 |
| 5e | 1j | CO2Me | 2j: 18 |
| 6f | 1k | NO2 | 2k: N.D. |

Using substrates 5a–5e with an electron-donating methyl group on the nitrogen atom, we investigated the effects of steric hindrance adjacent to the nitrogen atom and the captodative effects on the reductive cyclisation reaction (Table 4). With substrate 5a, the desired product 6a was obtained in 69% yield, although there was a slight decrease in the yield compared to the case without a methyl group on the nitrogen atom (entry 1 vs.Table 1, entry 3). However, the methyl group on the nitrogen atom successfully suppressed the dimerisation of substrate 5c with the methoxy group, as expected, and the target product 6c was obtained in 75% yield (entry 3 vs.Table 3, entry 1). Moreover, for substrates 5d and 5e with halogen atoms, the yields were slightly lower than those for the substrate without a methyl group on the nitrogen atom, with the same tendency as shown in entry 1 (entries 4 and 5 vs.Table 3 entries 2 and 3).

Having achieved the cyclisation reaction with terminal alkyne, we further investigated the cyclisation reaction with internal alkyne (Scheme 3). The reaction of substrates having phenyl and trimethylsilyl groups on the alkyne moiety gave the desired products in 87% and 58% yields, respectively.
 | ||
| Scheme 3 Cyclisation reaction with internal alkyne. | ||
As shown in Scheme 4, the reductive cyclisation of 1 proceeded through the aminoketyl radical intermediate A.9 The 5-exo type cyclisation of A occurred to give the exo-vinyl radical intermediate B.9c After further single-electron reduction by another equivalent of SmI2 to an anionic species C, it would be protonated by a proton source to give indoline derivative 2. Radical intermediate B may also abstract hydrogen radicals from the additives and solvents, such as HMPA and THF, thereby forming indoline derivative 2.17
 | ||
| Scheme 4 Plausible reaction mechanism for SmI2-mediated formation of the hemiaminal indoline 2. | ||
As aforementioned (Scheme 2A), the conversion of 2-CF3-indolines into 2-CF3-indoles using nucleophiles in a one-pot reaction was reported by Nenajdenko et al.14c To confirm the utility of 2-CF3-indolines as building blocks in the synthesis of functionalised 2-CF3-indoles, an indole derivative was synthesised using isolated compound 2a and a nucleophile (Scheme 5). When piperidine, an N-nucleophile, was used, the desired indole derivative 9 was obtained in good yield.
 | ||
| Scheme 5 Conversion into 2-CF3-indole by the reaction of 2a with piperidine. | ||
Conclusions
We developed the SmI2-mediated reductive cyclisation reaction using trifluoroacetamide groups as radical precursors and alkynes as radical acceptors to synthesize 2-trifluoromethylindoline derivatives. This reaction is the first example of using an acyclic amide group, which is considered difficult to react with SmI2, in a reductive cyclisation reaction. This reaction provides a finding that significantly contributes to the development of reductive reactions using SmI2 with amide groups. Furthermore, we demonstrated the conversion into 2-CF3-indoles by the reaction of 2a having a hemiaminal structure with piperidine. Therefore, the 2-CF3-indoline product obtained from the present reaction would be useful as a building block for synthesising a variety of indole derivatives. Further studies including reactions using other amide groups as radical precursors and the derivatisation of the products, indolines with exo-olefin and hemiaminal structures, are ongoing in our laboratory.Author contributions
H. I. was responsible for directing the project and editing the draft. K. Y., M. H., S. I., Y. I. and R. I. completed the investigation and data curation. K. Y., H. I. and H. N. completed the original draft. All authors participated in the discussion of the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.References
- (a) J. L. Namy, P. Girard and H. B. Kagan, New J. Chem., 1977, 1, 5–7 CAS; (b) P. GirErd, J.-L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef.
- (a) J. M. Concéllon and H. Rodríguez-Solla, Chem. Soc. Rev., 2004, 33, 599–609 RSC; (b) M. Szostak and D. J. Procter, Angew. Chem., Int. Ed., 2012, 51, 9238–9256 CrossRef CAS PubMed.
- M. Szostak, M. Spain and D. J. Procter, Chem. Soc. Rev., 2013, 42, 9155–9183 RSC.
- S. E. Dibrell, Y. Tao and S. E. Reisman, Acc. Chem. Res., 2021, 54, 1360–1373 CrossRef CAS PubMed.
- (a) M. M. Heravi and A. Nazari, RSC Adv., 2022, 12, 9944–9994 RSC; (b) K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48, 7140–7165 CrossRef CAS PubMed.
- (a) M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed; (b) G. A. Molander and E. P. Cormier, J. Org. Chem., 2005, 70, 2622–2626 CrossRef CAS PubMed; (c) A. Hölemann and H.-U. Reissig, Org. Lett., 2003, 5, 1463–1466 CrossRef PubMed.
- (a) O. Schmalz, R. Brun, J. W. Bats and H.-G. Schmalz, Tetrahedron Lett., 2002, 43, 1009–1013 CrossRef; (b) S. Gross and H.-U. Reissig, Synlett, 2002, 2027–2030 CAS; (c) M. Berndt and H.-U. Reissig, Synlett, 2001, 1290–1292 CrossRef CAS.
- (a) H. Iwasaki, N. Tsutsui, T. Eguchi, H. Ohno, M. Yamashita and T. Tanaka, Tetrahedron Lett., 2011, 52, 1770–1772 CrossRef CAS; (b) H. Ohno, M. Okumura, S. Maeda, H. Iwasaki, R. Wakayama and T. Tanaka, J. Org. Chem., 2003, 68, 7722–7732 CrossRef CAS PubMed.
- (a) C. Morrill, Á. Péter, I. Amalina, E. Pye, G. E. M. Crisenza, N. Kaltsoyannis and D. J. Procter, J. Am. Chem. Soc., 2022, 144, 13946–13952 CrossRef CAS PubMed; (b) H.-M. Huang, J. J. W. McDouall and D. J. Procter, Angew. Chem., Int. Ed., 2018, 57, 4995–4999 CrossRef CAS PubMed; (c) H.-M. Huang and D. J. Procter, Angew. Chem., Int. Ed., 2017, 56, 14262–14266 CrossRef CAS PubMed; (d) S. Shi and M. Szostak, Org. Lett., 2015, 17, 5144–5147 CrossRef CAS PubMed; (e) D. Parmar, L. A. Duffy, D. V. Sadasivam, H. Matsubara, P. A. Bradley and R. A. Flowers II, J. Am. Chem. Soc., 2009, 131, 15467–15473 CrossRef CAS PubMed.
- (a) S. R. Huq, S. Shi, R. Diao and M. Szostak, J. Org. Chem., 2017, 82, 6528–6540 CrossRef CAS PubMed; (b) M. Szostak, M. Spain, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2014, 136, 2268–2271 CrossRef CAS PubMed.
- (a) F. Shirai, T. Tsumura, Y. Yashiroda, H. Yuki, H. Niwa and S. Sato, J. Med. Chem., 2019, 62, 3407–3427 CrossRef CAS PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491–502 CrossRef CAS.
- H. Iwasaki, K. Suzuki, M. Yamane, S. Yoshida, N. Kojima, M. Ozeki and M. Yamashita, Org. Biomol. Chem., 2014, 12, 6812–6815 RSC.
- (a) H. Mei, A. M. Remete, Y. Zou, H. Moriwaki, S. Fustero, L. Kiss, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2020, 31, 2401–2413 CrossRef CAS; (b) D. O'Hagan, J. Fluor. Chem., 2010, 131, 1071–1081 CrossRef.
- (a) K. H. Min, N. Iqbal and E. J. Cho, Org. Lett., 2022, 24, 989–994 CrossRef CAS PubMed; (b) K. Mo, X. Zhou, J. Wu and Y. Zhao, Org. Lett., 2022, 24, 2788–2792 CrossRef CAS PubMed; (c) V. M. Muzalevskiy, Z. A. Sizova and V. G. Nenajdenko, Org. Lett., 2021, 23, 5973–5977 CrossRef CAS PubMed; (d) L. Mei, J. Moutet, S. M. Stull and T. L. Gianetti, J. Org. Chem., 2021, 86, 10640–10653 CrossRef CAS PubMed; (e) W. Ji, H.-H. Wu, W. Li and J. Zhang, Chem. Commun., 2021, 57, 4448–4451 RSC; (f) E. Vitaku, D. T. Smith and J. T. Njardarson, Angew. Chem., Int. Ed., 2016, 55, 2243–2247 CrossRef CAS PubMed. For recent synthesis of fluorine atom containing compounds: (g) Y. Cheng, X. Zhang, G. An, G. Li and Z. Yang, Chin. Chem. Lett., 2023, 34, 107625 CrossRef CAS; (h) L. Wen, B. Li, Z. Zou, N. Zhou, C. Sun, P. Feng and H. Li, Org. Chem. Front., 2024, 11, 142–148 RSC; (i) D. Komatsu, K. Yamada and T. Hanamoto, Org. Biomol. Chem., 2023, 21, 6762–6771 RSC.
- (a) M. Liu, X. Yang, Q. Sun, T. Wang, R. Pei, X. Yang, Y. Zhao, L. Zhao, G. Frenking and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300068 CrossRef CAS PubMed; (b) J. P. Peterson and A. H. Winter, J. Am. Chem. Soc., 2019, 141, 12901–12906 CrossRef CAS PubMed; (c) L. Duque, C. Zapata, B. Rojano, B. Schneider and F. Otálvaro, Org. Lett., 2013, 15, 3542–3545 CrossRef CAS PubMed; (d) H. G. Viehe, R. Merényi and Z. Janousek, Pure Appl. Chem., 1988, 60, 1635–1644 CrossRef CAS.
- We performed the reactions with trichloro- and tribromoacetamide groups. However, dehalogenated compounds were obtained and cyclised products could not be obtained.
- (a) D. Parmar, H. Matsubara, K. Price, M. Spain and D. J. Procter, J. Am. Chem. Soc., 2012, 134, 12751–12757 CrossRef CAS PubMed; (b) G. A. Molander and E. P. Cormier, J. Org. Chem., 2005, 70, 2622–2626 CrossRef CAS PubMed; (c) A. Hölemann and H.-U. Reissig, Chem. – Eur. J., 2004, 10, 5493–5506 CrossRef PubMed; (d) A. Hölemann and H.-U. Reissig, Org. Lett., 2003, 5, 1463–1466 CrossRef PubMed; (e) D. P. Curran, T. L. Fevig, C. P. Jasperse and M. J. Totleben, Synlett, 1992, 943–961 CrossRef CAS; (f) M. Matsukawa, J. Inanaga and M. Yamaguchi, Tetrahedron Lett., 1987, 28, 5877–5878 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob02040a |
| This journal is © The Royal Society of Chemistry 2024 |