
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceΔ-Keto-acid/hydroxy-lactone isomerization in some lichen depsides, depsidones and diphenyl ethers†
Philippe
Uriac
*a,
Solenn
Ferron
a,
Philippe
Jehan
b,
Thierry
Roisnel
 a and
Sophie
Tomasi
a
a and
Sophie
Tomasi
a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes)-UMR 6226, Rennes, France. E-mail: sophie.tomasi@univ-rennes.fr
bUniv Rennes, CNRS, ScanMAT UAR 2025, CRMPO (Centre Régional de Mesures Physiques de l'Ouest), F-35000 Rennes, France. E-mail: philippe.uriac@univ-rennes. fr
First published on 7th February 2024
Abstract
In some compounds in lichens, the carboxylic acid is ortho-substituted by an 2-oxoalkyl chain. This particular structure induces the existence of δ-keto-acid ka or hydroxy-lactone hl isomers, clearly identified by their NMR data and chemical properties, such as dehydration, methylation and behaviour in thermal conditions. Internal hydrogen bonding between the carboxylic acid and substituent in the ortho′ position is proposed as an isomerization modulator: an H-bond acceptor (OCH3) leads to ka isomers, whereas hl isomers are obtained with an H-bond donor (OH).
Introduction
Depsides, depsidones and diphenyl ethers are naturally occurring polyfunctionalized aromatic compounds produced by lichens1,2 or, more rarely, by fungi.3 In some depsides, the C-6 position of the B ring (more commonly referred to C-6′) is substituted by a 2-oxoalkyl chain, as shown in Fig. 1 for seven specialized metabolites selected for this study. This unusual chain was introduced as a linear acyl-CoA precursor involved in the polyenzymatic system of polyketide synthase (PKS).4,5 The presence of a free carboxylic acid on C-1′, in the ortho position to the 2-oxoalkyl chain induces the δ-keto-acid (ka)/hydroxy-lactone (hl) isomerization (Scheme 1). The factors influencing the position of this equilibrium are not well understood although this phenomenon, wrongly termed “tautomerism”, has been reported.6–11 Hence, the occurrence of ka and/or hl isomers should be explored further.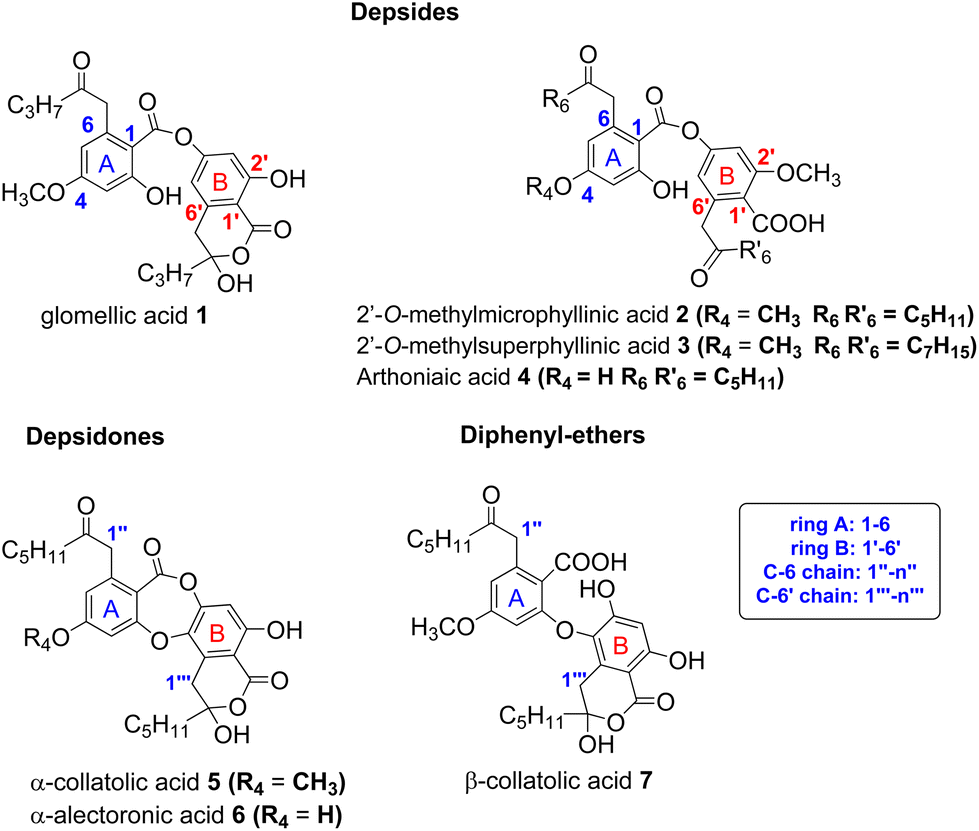 | ||
| Fig. 1 Structure of depsides, depsidones and diphenyl-ethers bearing a 2-oxoalkyl chain considered in this study. Other metabolites are listed in S1.† | ||
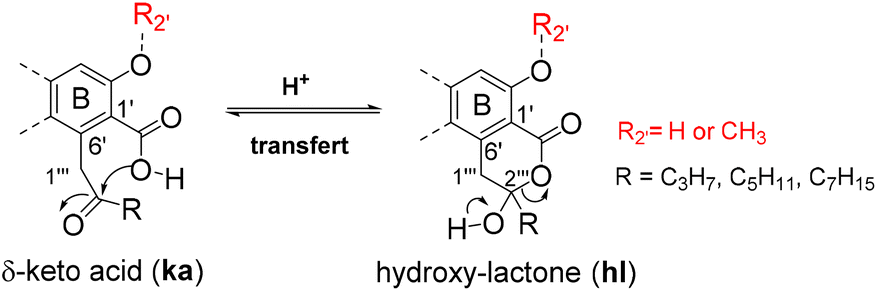 | ||
| Scheme 1 Δ-Keto-acid/hydroxy-lactone (ka/hl) isomerism. | ||
In the present study, new results are presented allowing the establishment of the structure and stability of compounds depicted in Fig. 1. Thus, the first observation of the hydroxy-lactone isomers (hl) of 2′-O-methylmicrophyllinic 2 and of 2′-O-methylsuperphyllinic 3 acids, as well as the obtention of α-collatollic 5 and α-alectoronic 6 acids as δ-keto-acid open form (ka), is reported, and their 1H NMR data analyzed. Then, to validate hl and ka structures, the synthesis of some new derivatives is described. Finally, the hydrogen bonding between the carboxylic acid and vicinal O-R2′ (R2′ = H or CH3) is proposed as the main isomerization modulator.
Results
The structures of compounds 1–4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and 5–77–11 (Fig. 1), isolated after acetone extraction and purification by column chromatography and/or preparative thin-layer chromatography, were confirmed by NMR analyses and matched values stated in the literature.
6 and 5–77–11 (Fig. 1), isolated after acetone extraction and purification by column chromatography and/or preparative thin-layer chromatography, were confirmed by NMR analyses and matched values stated in the literature.
Hydroxylactones isomers 2hl and 3hl
The isomers 2hl and 3hl were observed only in their 1H NMR spectra in a 15/85% mixture with their corresponding keto-acid isomers 2ka and 3ka, whereas 4hl has never been observed. The occurrence of these hl forms could be assessed by the downfield shifting of H-1′′′ and H-3′′′ signals in relation to the transformation of the ketone on C-2′′′ into a hemi-acetal. The C-2′′′ chirality resulted in an AB system for the diastereotopic CH2-1′′′, whereas these protons were enantiotopic in the ka form, leading to a singlet. As an example, the spectrum of 3ka + 3hl mixture is shown on Fig. 2.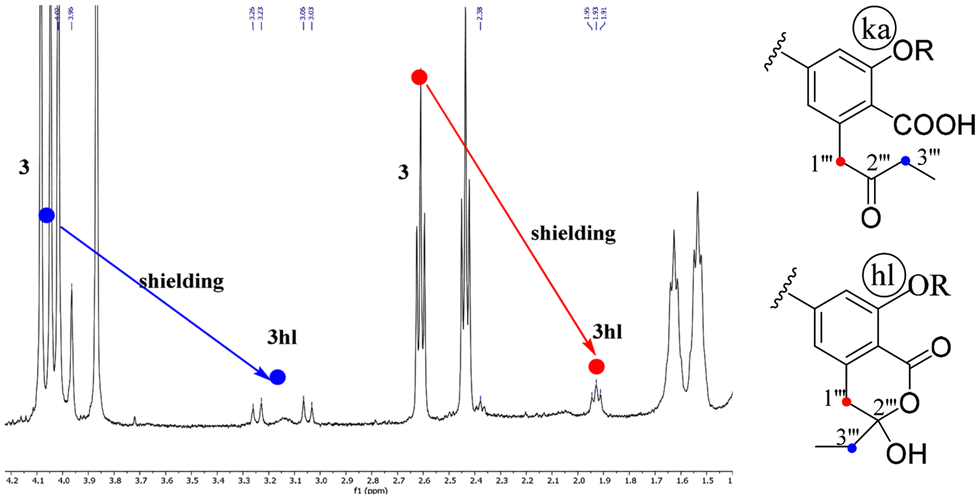 | ||
| Fig. 2 Expansion of the 1H NMR spectrum (2–4 ppm, 500 MHz, CDCl3) of the mixture 3ka + 3hl. | ||
On the spectra of the 3′′′-O-methyl derivatives of 1, 5 and 6 (vide infra), an AB system was observed for CH2-1′′′ and CH2-3′′′.
Keto-acid isomers 5ka and 6ka
If pure depsidones 5hl and 6hl were dissolved in Culberson's solvent G (toluene, EtOAc, HCOOH: 70/40/4),1 they were obtained as δ-keto-acid isomers 5ka and 6ka after evaporation of the solvent under reduced pressure. Likewise, only keto-acid forms could be observed when 5 and 6 were purified from acetone extracts of lichens using the same solvent system (respectively from Tephrolema atra and Ochrolecchia parella). For example, an enlarged version of the 2–4 ppm range of the 1H NMR spectra of 5hl and 5ka is depicted in Fig. 3. The recovery of 5ka and 6ka after G solvent evaporation was observed when (CD3)2CO was used as an NMR solvent. Interestingly, in the case of depside 1, open isomers 1ka have never been obtained. The open structures 5ka and 6ka were clearly attested in 1H NMR by the signal deshielding of 2H-1′′′ and 2H-3′′′ induced by the carbonyl in position 2′′′.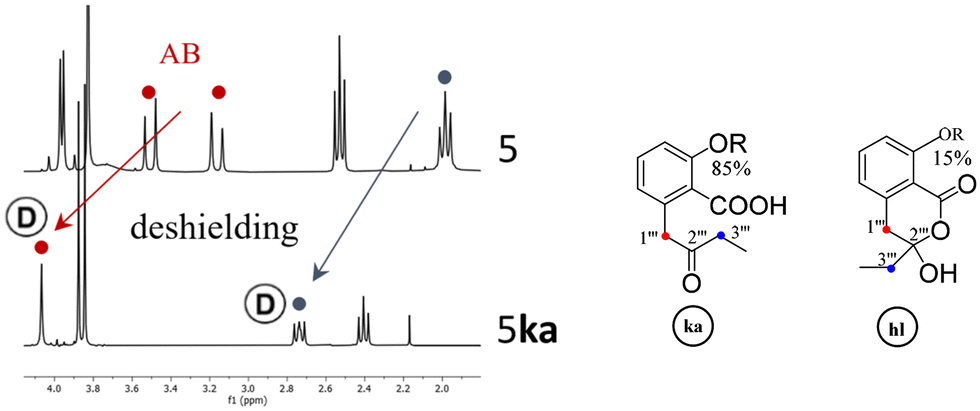 | ||
| Fig. 3 Expansion of the 1H NMR data (300 MHz) of 5 (5hl) and 5ka (CDCl3). Ⓓ: partial H/D exchange. | ||
Finally, the hybridization change of C-1′′′ resulting in significant differences in 1H NMR data allowed the unequivocal attribution of hl or ka isomers, as illustrated for CH2-1′′′ and CH2-3′′′, in Table 1. To explain the coalescence (noted a in Table 1) occurring in the spectra of hl isomers, a “tautomerism equilibrium” has been previously proposed. Nevertheless, this phenomenon is the result of inversion of the hydroxy-lactone ring. This explanation was clearly attested with 67, 78 and 1 (see S4†) by the transformation at low temperature of the coalescent CH2-1′′′ signal into an AB system. The spectra of the ka isomers of 5 and 6 were recorded in d6-acetone, and the disappearance and/or intensity reduction of CH2-1′′′ and CH2-3′′′ signals were observed (Fig. 3). A keto–enol tautomerism, which induced a H/D exchange (noted Ⓓ in Table 1 and Fig. 3) and discussed below, could be proposed (see S4†). On 13C NMR spectra, ring inversion and tautomerism resulted in the disappearance of signals but in some cases C-2′′′ was observed near 206 ppm (ka) or 105 ppm (hl).
Reactivity in relation to the hydroxy-lactone structure (see S6†)
Most reactions were performed with collatolic acids 5 and 7 taken as examples, but new derivatives of α-alectoronic acid 6 and of glomellic acid 1 were also prepared.Dehydration and O-methylation of the hydroxy-lactone ring
First, dehydration and O-methylation of the hydroxy-lactone isomers of 1, 5 and 6 were performed to confirm the stability of the six-membered ring. • In a study dedicated to the lichenochemical investigation of Tephromela atra,7 the presence of the α-deoxycollatolic acid (= α-collatone) was reported. In the present study, such water removal from acids 5 and 7 was achieved by heating these compounds in a CHCl3 or CDCl3/CF3COOH 80/20 mixture at 55 °C, thereby yielding α-collatone 8 and β-collatone 9. The latter was obtained as crystals suitable for X-ray analysis (Fig. 4) clearly disclosing the hydrogen bonding between OH-2′ and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O-1′.
O-1′.
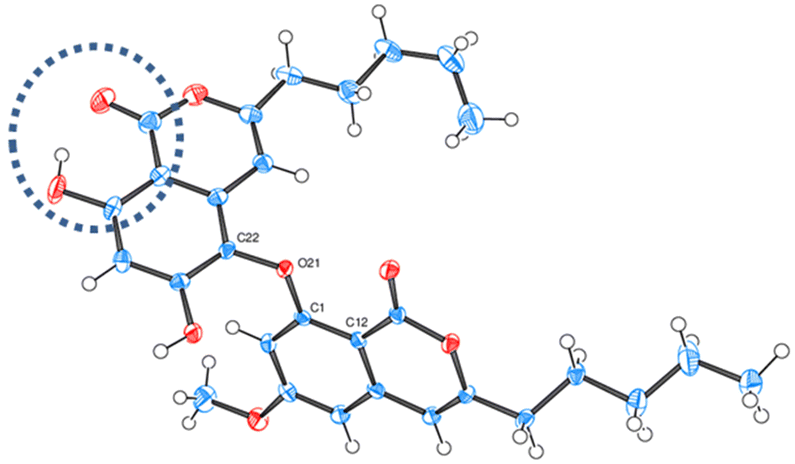 | ||
| Fig. 4 ORTEP diagram of β-collatone 9 (drawn with ellipsoids at 50% probability). H-bonding site is emphasized in the dashed blue circle. | ||
• Using the O-methylation method previously undertaken,9 in the structural determination of α-alecteronic acid 6 (CH3OH, H+ catalytic at room temperature), 2′′′-O-methyl derivatives 10, 11 and 12 from 1, 5 and 7 (Schemes 2 and 3) could be obtained, respectively, in fair yields using CF3COOH as a catalyst. As previously reported, diastereotopic CH2 appeared as an AB system. Interestingly, the same tertiary carbocation intermediate could be postulated in the dehydration and nucleophilic attack of CH3OH.
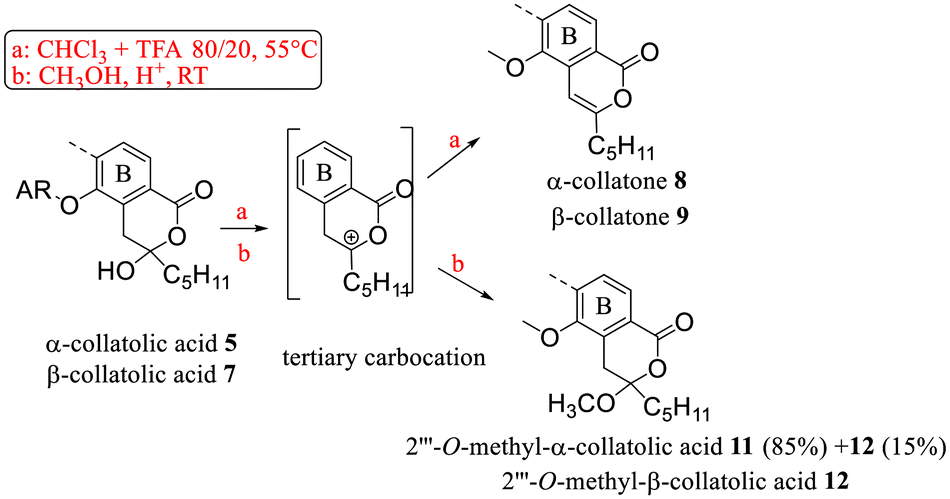 | ||
| Scheme 2 Reactivity of the hydroxy-lactone ring of 5 and 7. | ||
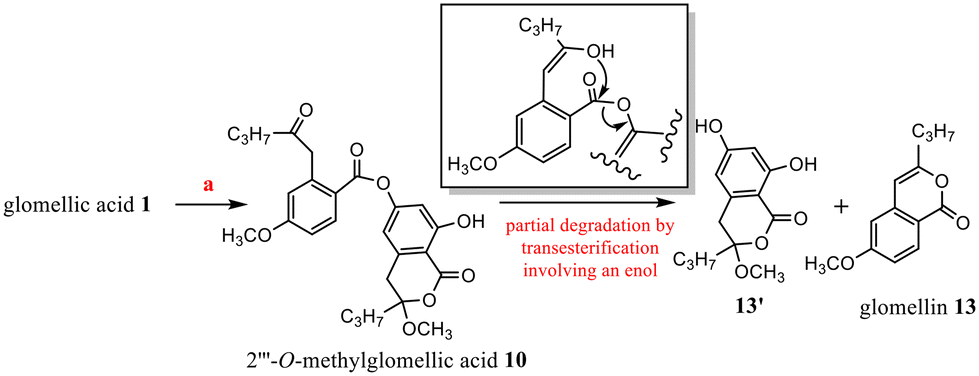 | ||
| Scheme 3 O-Methylation of 1 and degradation of 10. | ||
In these conditions (CH3OH, H+), chemical evolution occurred with 10 and 11. Regarding the specific example of 10, the rapid cleavage of the ester bond led to glomellin613 and to the analogous structure of penidiolactol1213′ (Scheme 3). In the case of 11, analogous transesterification partially led to 12 (Schemes 2 and 5).
The methyl ester of 2 is the main metabolite of Porpidia contraponenda,6 but those of 1, 5 and 7 were never observed under our conditions. Moreover, the synthesis of methyl α-alectoronate 14 has been reported9 if diazomethane is used with 6. The authors suggested that the methylation occurred on the carboxylic acid of 6ka after hydroxy-lactone ring opening. However, another mechanistic sequence in accordance with the hl structure (Scheme 4) can be envisaged in which the driving force would be the OH-lactone deprotonation of 6hl leading to CH3+, which could methylate the alectoronate.
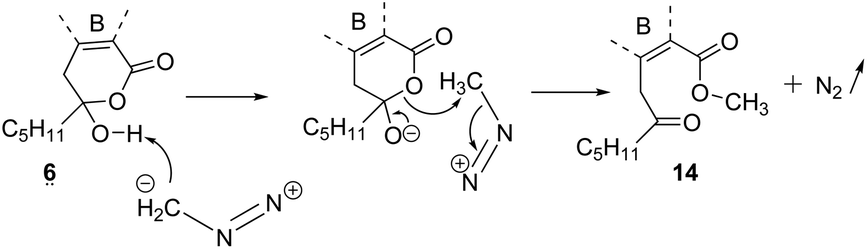 | ||
| Scheme 4 Synthesis of methyl α-alectoronate 14 using CH2N2. Mechanistic proposition. | ||
Thermal isomerization of hydroxy-lactones: examples of α-collatolic acid 5 and physodic acid
The behavior in thermal conditions of α-collatolic acid 5 and physodic acid were compared. The previously observed7 isomerization of α-collatolic acid 5 into corresponding β-isomer 7 was confirmed herein by heating in refluxing toluene or d8-toluene (Scheme 5). With respect to the degradation of 10, involvement of an enol intermediate, assisted by the eventual acidic catalysis of free COOH-1′, can be postulated. In contrast, physodic acid was isomerized and decarboxylated to give physodone,‡15 in same thermal conditions. In this case, acid deprotonation, followed by a electrophilic substitution, has been proposed according to the literature.13 Then, this decarboxylation absence with 5 and 6 is another strong argument in favor of the cyclic structure (Scheme 5 and S5†). Mixtures of α (depsidones) and β isomers (diphenyl-ethers) of 5 and 6 are also signaled in acetonic extracts of lichens (for example, in Xanthoparmelia glabrans14).
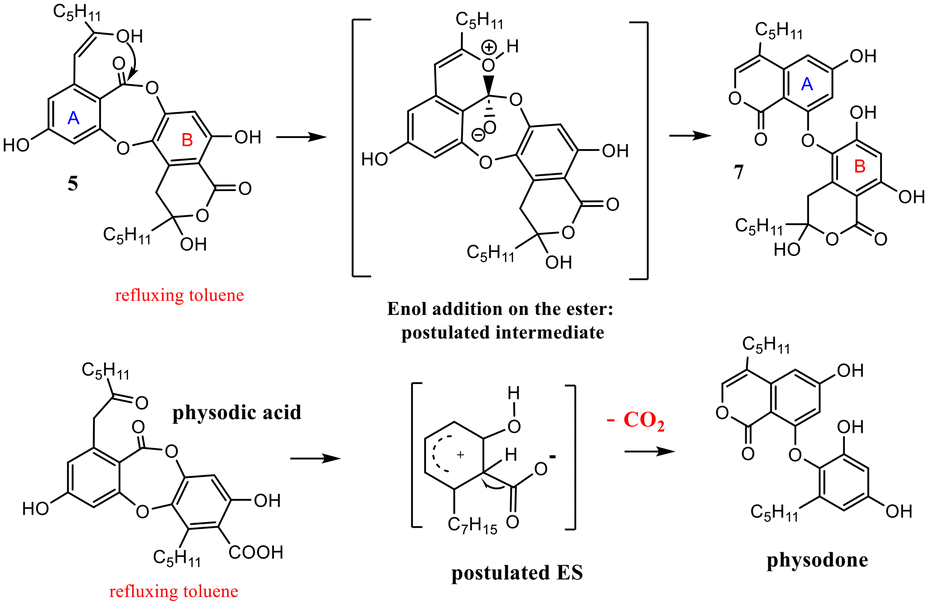 | ||
| Scheme 5 Thermal isomerization of 5 and decarboxylation of physodic acid. | ||
Reactivity in relation of the δ-keto-acid structure
1H NMR spectra, recorded in d6-acetone, revealed a property in accordance with the open ka structure of 2. Thus, disappearance of CH2-1′′′ and CH2-3′′′ signals was observed (see S4†). This phenomenon, also attested in 2H NMR, can be explained by their deuteriation involving an enol. The NMR solvent would provide deuterium atoms and tautomerism would be catalysed by the close carboxylic acid. Thus, H/D exchange never occurred on the C-1′′ and C-3′′ of the A ring bearing an ester on C-1. The less easy enolization of CH2-3′′′ was slower and incomplete. Compounds 5ka and 6ka presented such an H/D exchange.Discussion
The results concerning the preparation and characterization of 2hl, 3hl, 5ka and 6ka, as well as those devoted to the stability of the hl and ka isomers, must be explained. The occurrence of a hydroxy group, acting as an H-bond donor (1, 5–7), is in relation to the hydroxy-lactone ring hl, whereas a methoxy substituent, acting as an H-bond acceptor (2–4), is instead associated with δ-keto-acid ka isomers, as clearly attested by NMR data (Scheme 2). Then, the stabilization energy due to the hydrogen bonding (Scheme 6) between O-R2′ and COOH-1′ could be the main reason accounting for the isomerization control. If R2′ = H (1, 5–7), then the hydrogen bonding between OH-2′ and COOH-1′ promoted the nucleophilic attack of the hydroxy group of the carboxyl on the ketone to give hydroxy-lactone. In contrast, with R2′ = CH3 (2–4), this hydroxy group is bonded with the methoxy group, and δ-keto-acid remains untransformed. Such hydrogen bonding has been widely discussed12,15 and attested through the X-ray structure of 5-methylmellein16 and β-collatone 9 (Fig. 4).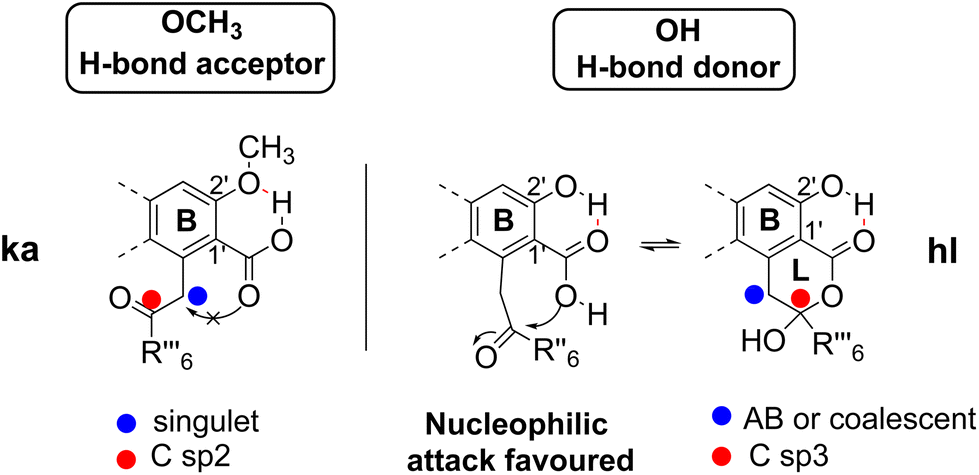 | ||
| Scheme 6 Influence of hydrogen bonding on isomerization. | ||
To explain obtaining 5ka and 6ka during G solvent evaporation, one can propose that, in this solvent, HCOOH would induce breaking of H-bonds, whereas EtOAc would act as an H-bond acceptor. Finally, in depsidones 5 and 6, the ether bridge would induce steric hinderance, disfavouring COOH-1′ attack on the ketone and preventing 5hl and 6hl obtention. On the contrary, the ether absence in depside 1hl could explain why 1ka has never been obtained.
Conclusions
In conclusion, it appears that the structure of specialized metabolites bearing a δ-keto-acid moiety on ring B is the result of isomerization controlled by internal hydrogen bonding. This latter involves the acid function and its ortho OH or OCH3 substituent. If a methoxy is present in C-2′, they should be described as keto-acids (ka). If C-2′ is bearing an OH, they should be termed hydroxy-lactones hl according to their reactivity. Thus, in the latter case, we suggest to delete the frequent mention of tautomerism. To standardize the nomenclature of these compounds according to their reactivity, we propose to name unusual isomers using the prefix “iso” as follows: 5ka = α-isocollatolic acid, 6ka = α-isoalectoronic acid, 2hl = 2′-O-methylisomicrophyllinic acid, and 3hl = 2′-O-methylisosuperphyllinic acid. Finally, this study is another demonstration6 of the interest of the extraction of natural products which allows original structural and/or mechanistic chemical studies.Experimental
Reagents
All reagents were of high quality and purchased from commercial suppliers. They were used as received without further purification or purified/dried according to standard procedures.Preparation of extracts and samples
Lichen thalli (often with minerals) were ground and extracted thrice with warm acetone. After drying, the solvent was evaporated under reduced pressure. Isolation of 1–4 has been previously reported. Depsidone 5 was obtained from Tephromela atra (Huds.) Hafellner, whereas depsidone 6 from Ocholechia parella (L.) Massal using the same procedure described previously.7,8 Both were collected on coastal rocks at Lancieux (France, department 22). To perform the methylation of 2, Porpidia cinereoatra was collected in Néant-sur-Yvel (France, department 56).TLC analysis
Secondary metabolites were identified by thin-layer chromatography (TLC) using standards. Extracts and standards were loaded onto an aluminium sheet (10 × 20 cm) coated by silica gel 60F254 (Merck). Thereafter, plates were developed with solvent C (toluene/acetic acid: 85/15) or solvent G (toluene/ethyl acetate/formic acid: 139/83/8). All TLC plates were observed under UV light at 254 and 365 nm (12 W, 6 W at 365 nm, 6 W at 254 nm; Fischer Bioblock Scientific). TLC examples are given in ESI.†Experimental Rf values in solvent C: 1 (0.30), 5 (0.35), 6 (0.16), 7 (0.22), 8 (0.40), 9 (0.51), 10 (0.51), 13 (0.55), 13′ (0.32), 11 (0.49), and 12 (0.39) (see TLC on ESI†).
Purifications using liquid chromatography
Silica gel 60 (Merck) and mainly solvent system C1 were used for the purification of different extracts. Combined fractions with selected metabolites were washed once with water to eliminate acids, dried over MgSO4 and evaporated under reduced pressure. Preparative TLC was also performed using the same solvent.Tephromela atra: 19.4 g; extract: 1.35 g; 5 after chromatography using solvent C or G: 220 mg recrystallization. In toluol: 160 mg.
Ochrolecchia parella: 8.3 g; extract: 390 mg; 6 after chromatography in solvent C or G: 45 mg.
Porpidia cinereoatra: extract: 320 mg; after chromatography in solvent C. 2: 34 mg and confluentic acid: 160 mg.
High resolution mass spectrometry (HRMS) and tandem mass spectrometry (MS-MS)
The mass spectra of standards were recorded at CRMPO-ScanMat (Rennes, France) on an Orbitrap Thermo Fisher Scientific Q-Exactive instrument with an ESI (electrospray Ionization) source in negative mode by direct introduction at 5–10 μg mL−1. Samples were prepared in methanol at 10 μg mL−1.Synthesis procedures
Reactions were performed on a small amount of extraction products; hence, yields are not given. Only the quantities obtained after working up are indicated.5 (20 mg) gave α-collatone = deoxycollatolic acid 8 (11 mg) with similar data to those previously reported.7 Presence of residues of 5 and of 10 was noted on TLC.
7 (20 mg) gave β-collatone 9 (14 mg).
1 (15 mg) gave 2′′′-O-methylglomellic acid 10 (12 mg). After 1 month, along with 10 (4 mg), hydrolysis compounds glomelline 13 (3 mg) and 13′ (3 mg) were obtained.
7 (30 mg) gave 2′′′-O-methyl-β-collatolic acid 12 (24 mg) and 5 gave 2′′′-O-methyl-α-collatolic acid 11 (14 mg) as a white precipitate after filtration. Isomerization of 11 into 12 was also observed by TLC.
X-ray data
β-Collatone 9 CCDC 2241570.†1H, 2H and 13C NMR spectra
Spectra were recorded at 300/75 MHz or 500/100 MHz (cryoprobe) respectively, using CDCl3 or d6-acetone. Shifts (δ values) are given in parts per million (ppm) and coupling constants (J values) are given in Hertz (Hz). The multiplicity of signals was reported as follows: s (singlet), d (doublet), t (triplet), q (quadruplet), quint (quintet), sext (sextet), m (multiplet), * (broad or coalescent signal), dt (doublet of triplet), and td (triplet of doublet). The references were: CDCl3/7.26 ppm and d6-acetone/2.05 ppm. In some compounds, coalescent signals were missing or badly resolved in 13C NMR.2hl and 3hl in mixture with 2ka and 3ka (15/85%). Only some signals were visible.
2′-O-Methylisomicrophyllinic acid 2hl C30H38O9MW: 542.62 g mol−1. 3-Hydroxy-8-methoxy-3-pentyl-3H-2-benzopyran-1(4H)-on-6-yl and 2-hydroxy-4-methoxy-6-(2-oxoheptyl)benzoate. 1H NMR (300 MHz, CDCl3): 3.22 and 3.03 (AB, J = 16 Hz, 2H), 1.89 (t, 2H). In mixture with 2ka (15/85%).
2′-O-Methylsuperphyllinic acid 3hl C34H46O9MW: 598.72 g mol−1. 3-Heptyl-3-hydroxy-8-methoxy-3-3H-2-benzopyran-1(4H)-on-6-yl and 2-hydroxy-4-methoxy-6-(2-oxononyl)benzoate. 1H NMR (500 MHz, CDCl3): 3.24 and 3.04 (AB, J = 16 Hz, 2H), 1.93 (m, 2H). 13C NMR (125 MHz, CDCl3): 41.1 and 37.8. In mixture with 3ka (15/85%).
In 13C NMR of 5ka and 6ka, some signals of sp2 carbons were not visible, particularly C′′′-1 and C′′′-2.
α-Isocollatolic acid 5ka: [522-52-1] C28H32O9 C29H34O9MW: 526.58 g mol−1. 8-Hydroxy-3-methoxy-11-oxo-1,6-bis(2-oxoheptyl)-11H-dibenzo[b,e][1,4]dioxepin-7-carboxylic acid. 1H NMR (300 MHz, CD3COCD3): 6.87 (d, J = 2.4 Hz, 1H), 6.78 (d, J = 2.4 Hz, 1H), 6.70 (s, 1H), 4.08 (s, 2H), 4.04 (s*, 2H), 3.88 (s, 3H), 2.55 (t, J = 7.5 Hz, 2H), 2.36 (s*, 1.6H), 1.59 (m, 4H), 1.32 (m, 8H), and 0.89 (t, 6H). 13C NMR (100 MHz, CD3COCD3): 163.5, 162.2, 162.1, 141.4, 141.0, 115.8, 114.2, 106.9, 104.2, 55.5, 54.6, 47.2, 42.0, 41.1, 31.5, 31.2, 23.1, 22.3, 22.2, 13.4, and 13.4.
α-Isoalectoronic acid 6ka: [668-80-4] C28H32O9 55 g mol−1. MW: 512.55 g mol−1. 3,8-Dihydroxy-11-oxo-1,6-bis(2-oxoheptyl)-11H-dibenzo[b,e][1,4]dioxepin-7-carboxylic acid. 1H NMR (300 MHz, CD3COCD3): 8.16 (s, 0.4H), 6.75 (d, J = 2.4 Hz, 1H), 6.63 (d, J = 2.4 Hz, 1H), 6.00 (s, 1H), 4.00 (s, 2H), 4.27 (s*, 2H), 3.97 (s, 2H), 2.52 (t, J = 7.5 Hz, 2H), 2.44 (t*, 1.8H), 1.55 (m, 4H), 1.29 (m, 8H), 0.88 (t, J = 7.4 Hz, 3H), and 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CD3COCD3): 162.6, 162.5, 162.1, 141.7, 141.1, 117.0, 112.9, 106.7, 105.8, 54.6, 47.3, 41.9, 41.5, 31.4, 31.2, 23.1, 22.3, 22.3, 13.4, and 13.4.
β-Collatone 9 [520-08-1] 1H NMR (300 MHz, CDCl3): 11.04 (s, 1H), 9.21 (s, 1H), 6.57 (s, 1H), 6.51 (d, J = 2.3 Hz, 1H), 6.37 (d, J = 2.3 Hz, 1H), 6.36 (s, 1H), 6.24 (s, 1H), 3.80 (s, 3H), 2.53 (t, J = 7.5 Hz, 2H), 2.51 (t, J = 7.5 Hz, 2H), 1.70 (m, 4H), 1.37 (m, 4H), 1.33 (m, 4H), 0.93 (t, J = 6.9 Hz, 3H), and 0.91 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): 166.1, 165.9, 162.8, 161.7, 161.1, 159.8, 159.2, 157.8, 142.2, 132.0, 130.2, 104.6, 104.3, 103.5, 103.3, 102.9, 98.6, 98.4, 56.0, 33.7, 33.4, 31.3, 31.2, 26.6, 22.5, 22.5, 14.1, and 14.0. HRMS [M – H]− C29H31O8 calc. 507.20244 found. 507.2024.
2′′′-O-Methylglomellic acid 10 C26H30O9MW: 486.51 g mol−1. (8-Hydroxy-3-methoxy-3-propyl-3H-2-benzopyran-1(4H)-on-6-yl and 2-hydroxy-4-methoxy-6-(2-oxopentyl)benzoate). 1H NMR (300 MHz, CDCl3): 11.21 (s, 1H), 11.19 (s, 1H), 6.62 (d, J = 2.4 Hz, 1H), 6.46 (d, J = 2.4 Hz, 1H), 6.45 (d, J = 2.4 Hz, 1H), 6.3 (d, J = 2.4 Hz, 1H), 4.03 (s, 2H), 3.84 (s, 3H), 3.18 (m, JAB = 16 Hz, 1H), 3.08 (m, JAB = 16 Hz, 1H), 2.39 (m, 2H), 2.02 and 1.98 (2 m, 2H), 1.54 (m, 6H), 1.43 (m, 2H), 1.39 (m, 4H), 1.02 (t, 3H), and 1.02 (t, 3H). 13C NMR (100 MHz, CDCl3): 207.2, 168.8, 168.6, 166.8, 165.2, 163.6, 160.0, 155.4, 139.9, 139.0, 113.6, 112.9, 109.5, 107.6, 106.6, 104.3, 100.2, 51.4, 50.1, 44.5, 37.3, 36.3, 17.2, 17.0, 14.2, and 13.8. HRMS [M – H]− C26H29O9 calc. 485.18171 found. 485.1822.
Compound 13′ C13H16O5MW: 252.27 g mol−1. 3-Pentyl-3,6,8-trihydroxy-3H-2-benzopyrane-1(4H)-one 1H NMR (300 MHz, CDCl3): 11.17 (s, 1H), 6.29 (d, J = 2.4 Hz, 1H), 6.20 (d, J = 2.4 Hz, 1H), 5.55 (s,1H), 3.35 (s, 3H), 3.11 (m, JAB = 16 Hz, 1H), 3.00 (m, JAB = 16 Hz, 1H), 1.44 (m, 6H), 1.44 (t, 3H), and 1.01 (t, 3H). 13C NMR (75 MHz, CDCl3): 168.8, 164.4, 162.5, 140.4, 107.4, 107.1, 101.9, 101.8, 50.1, 37.3, 36.3, 17.0, and 14.2. HRMS [M − H]− C13H15O5 calc. 251.0925 found. 251.0924.
2′′′-O-Methyl-α-collatolic acid 11 C30H36O9MW: 540.62 g mol−1. (2,11-Dimethoxy-5-hydroxy-9-(2-oxoheptyl)-2-pentyl-4H,8H-[1,4]benzodioxepino[2,3-f][2]benzopyran-4(1H),8-dione). 1H NMR (300 MHz, CDCl3): 11.01 (s, 1H), 6.79 (s, 1H), 6.58 and 6.59 (m, 2H), 3.99 and 3.97 (AB, JAB = 17 Hz, 2H), 3.84 (s, 2H), 3.53 and 3.00 (AB, JAB = 17 Hz, 2H), 3.34 (s, 3H), 2.54 (t, J = 7 Hz, 2H), 2.07–1.93 (mAB, 2H), 1.7–1.2 (m, 12H), 0.94, (t, J = 7 Hz, 3H), and 0.89 (t, J = 7 Hz, 3H). 13C NMR (75 MHz, CDCl3): 168.1, 163.4, 161.7, 160.3, 150.5, 141.4, 129.7, 115.0 107.9, 107.5, 104.8, 55.8, 50.0, 48.0, 42.8, 35.0, 31.7, 31.3, 30.4, 23.3, 23.2, 22.5, 22.5, 14.0, and 14.0. HRMS [M – H]− C30H35O9 calc. 539.22866 found. 539.2279.
2′′′-O-Methyl-β-collatolic acid 12 C30H36O9MW: 540.62 g mol−1. (6,8-dihydroxy-3-methoxy-5-(6-methoxy-1-oxo-3-pentyl-1H-2-benzopyran-8-yl)oxy-3,6,8-trihydroxy-3-pentyl-1H-2-benzopyran-1(1H)-one). 1H NMR (500 MHz, CDCl3): 11.12 (s, 1H), 6.58 (s*, 1H), 6.51 (d, J = 2.4 Hz), 6.50 (s, 1H), 6.21 (s, 1H), 3.79 (s, 3H), 3.35 (s, 3H), 3.24 (d, JAB = 16 Hz, 1H), 2.99 (d, JAB = 16 Hz, 1H), 2.51 (t, J = 7.4, 2H), 1.96 (m, 2H), 1.70 (m, 2H), 1.34 (m, 12H), 0.91 (t, J = 7.5, 3H), and 0.88 (t, J = 7.5, 3H). 13C NMR (75 MHz, CDCl3): 168.3, 166.0, 162.6, 162.2, 160.6, 159.4, 157.0, 141.7, 134.3, 131.2, 107.0, 103.8, 103.4, 103.2, 102.8, 99.9, 35.0, 33.3, 31.7, 31.5, 31.2, 29.7, 26.5, 23.1, 22.4, 22.4, and 14.0 HRMS [M − H]− C30H35O9 calc. 539.22866 found. 539.2287.
Data concerning C-1′′′2′′′3′′′ (Fig. 3)
Glomellic acid 1 [52589-14-7] Proposed DCI: 3,8-dihydroxy-3-propyl-3H-2-benzopyran-1(4H)-on-6-yl, and 2-hydroxy-4-methoxy-6-(2-oxopentyl)benzoate. 1H NMR (300 MHz, CDCl3): 4.04 (s, 2H), 3.22*(s, 2H), 2.39 (t, J = 7.5 Hz, 2H), 1.98* (m, 2H). 13C NMR (80 MHz, CD3COCD3): 106.5, 43.5, and 37.2.62′-O-Methylmicrophyllinic acid 2 [79579-62-7] 1H NMR (300 MHz, CDCl3): 4.07 (s, 2H), 4.03 (s, 2H), 2.59 (t, J = 7.5 Hz, 2H), and 2.42 (t, J = 7.5 Hz, 2H).13C NMR (80 MHz, CDCl3): 51.2 and 42.7.6
2′-O-Methylsuperphyllinic acid 3 [108529-20-0] 1H NMR (300 MHz, CDCl3): 4.06 (s, 2H), 4.02 (s, 2H), 2.59 (t, J = 7.5 Hz, 2H), and 2.41 (m, 2H). 13C NMR (100 MHz, CDCl3): 207.3, 49.2, and 42.7.6
Arthoniaic acid 4 [25556-24-5] 1H NMR (300 MHz, CDCl3): 4.17 (s, 2H), 3.85 (s, 2H), 2.51 (t, J = 7.5 Hz, 2H), and 2.48 (t, J = 7.5 Hz, 2H). 13C NMR (80 MHz, CDCl3): 207.4, 47.7, and 42.5.6
Author contributions
Project design by P. Uriac. Synthesis by P. Uriac. Characterization by S. Ferron and P. Jehan. Crystallographic experiments and analyses by T. Roisnel. The manuscript was written by P. Uriac and S. Tomasi with edits from all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Dr Pierre Le Pogam (BIoCIS, Paris Saclay), Dr Elsa Catyan for 2H NMR and Dr Arnaud Bondon (CorInt, ISCR) for the recording of 500 MHz NMR spectra and fruitful discussions. Part of this work has been performed using the PRISM core facility (Biogenouest, Univ Rennes, Univ Angers, INRAE, CNRS,France).References
- S. Huneck and I. Yoshimura, Identification of Lichen Substances, Springer Berlin Heidelberg, Berlin, Heidelberg, 1996 Search PubMed.
- M. J. Calcott, D. F. Ackerley, A. Knight, R. A. Keyzers and J. G. Owen, Chem. Soc. Rev., 2018, 47, 1730–1760 RSC.
- Y. Liang, Y. Zheng, Y. Shen, Q. Li, Y. Lu, S. Ye, X.-N. Li, D. Li, C. Chen, H. Zhu and Y. Zhang, Tetrahedron Lett., 2022, 105, 154046 CrossRef CAS.
- D. Armaleo, X. Sun and C. Culberson, Mycologia, 2011, 103, 741–754 CrossRef CAS PubMed.
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 RSC.
- S. Ferron, P. Jéhan, X. Guillory and P. Uriac, Phytochemistry, 2022, 198, 113139 CrossRef CAS PubMed.
- M. Millot, S. Tomasi, S. Sinbandhit and J. Boustie, Phytochem. Lett., 2008, 1, 139–143 CrossRef CAS.
- M. Millot, S. Tomasi, K. Articus, I. Rouaud, A. Bernard and J. Boustie, J. Nat. Prod., 2007, 70, 316–318 CrossRef CAS PubMed.
- J. Elix, B. Ferguson and M. Sargent, Aust. J. Chem., 1974, 27, 2403 CrossRef CAS.
- S. Huneck, A. Porzel and H. T. Lumbsch, Hergozia, 1997, 39–43 Search PubMed.
- O. E. Krivoshchekova, N. P. Mishchenko, L. S. Stepanenko and O. B. Maksimov, Chem. Nat. Compd., 1983, 19, 11–16 CrossRef.
- A. Saeed, Monatsh. Chem., 2003, 134, 457–463 CrossRef CAS.
- G. E. Dunn, E. G. Janzen and W. Rodewald, Can. J. Chem., 1968, 46, 2905–2909 CrossRef CAS.
- S. Hellou, P. Uriac, F. Le Dévéhat, A. Sauvager, P. Jéhan, A. Zebboudj, J. Boustie and J. Esnault, Herzogia, 2019, 32, 485–502 Search PubMed.
- K. Mustafa, H. G. Kjaergaard, N. B. Perry and R. T. Weavers, Tetrahedron, 2003, 59, 6113–6120 CrossRef CAS.
- K. Krohn, R. Bahramsari, U. Flörke, K. Ludewig, C. Kliche-Spory, A. Michel, H.-J. Aust, S. Draeger, B. Schulz and S. Antus, Phytochemistry, 1997, 45, 313–320 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2241570. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob02026f |
| ‡ Personal communication. See the NMR spectra in S5.† |
| This journal is © The Royal Society of Chemistry 2024 |