
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst-free photo-induced aerobic radical synthesis of lactams from N-alkenyl trichloroacetamides in 2-methyltetrahydrofuran as the radical initiator under violet light†
Faïza
Diaba
 * and
Gisela
Trenchs
* and
Gisela
Trenchs
Laboratori de Química Orgànica, Facultat de Farmàcia, Universitat de Barcelona, Av. Joan XXIII 27-31, 08028-Barcelona, Spain. E-mail: faiza.diaba@ub.edu
First published on 12th December 2023
Abstract
The first violet light-mediated synthesis of γ- and δ-lactams from N-alkenyl trichloroacetamides is reported in tetrahydrofuran or 2-methyltetrahydrofuran alone. These catalyst and additive-free reactions are achieved with non-anhydrous solvents and under an air atmosphere where the solvent serves as the radical initiator.
Introduction
Alkyl radicals play a pivotal role as intermediates in the construction of carbon–carbon bonds1–3 usually by addition to alkenes.4 Traditionally, they are generated from alkyl halides through a homolytic cleavage of a C–X (X = I, Br, Cl) bond in the presence of toxic tributyltin hydride (TBTH) and azobisisobutyronitrile (AIBN) as the radical initiator. In this process, tributyltin hydride serves a dual purpose facilitating the generation of Bu3Sn˙ radical species as the radical chain carrier and also operating as a hydrogen donor (Scheme 1a).5,6 Alkyl radicals from alkyl halides were also generated under tin free conditions specifically in the presence of triethylborane and oxygen as the radical initiator (Scheme 1b).7,8 Over the past decade, photoredox catalysis has also become a reliable tool for the formation of these radicals under mild conditions, usually in the presence of a photocatalyst, an amine and under light irradiation (Scheme 1c).9 Finally, alkyl radicals generation could also be carried out under catalyst-free photochemical reactions via electron donor–acceptor (EDA) complexes.10Scheme 1d depicts an example of inter single-electron-transfer (SET) from transient enamines to alkyl bromides ending in the formation of an alkyl radical for the α-alkylation of aldehydes.11 | ||
| Scheme 1 Selected examples for the strategies used to generate alkyl radicals from alkyl halides. | ||
Recently we reported the first air-tolerant photocatalyzed synthesis of γ- and δ-lactams from trichloroacetamides under blue LEDs irradiation and non-anhydrous conditions.12 In this investigation the best results were achieved in the presence of 1 mol% of Ir(ppy)3 as the photocatalyst, N,N-diisopropylethylmine (5 equiv.) in a mixture of THF and acetone as a solvent. The reaction carried out with 19 different substrates, were also undertaken in the absence of amines providing the corresponding lactams with acceptable yields despite the extended reaction time. As a continuation of this work we decided to start an investigation with the goal of exploring the same reaction under photocatalyst and base free conditions.
Herein we present our findings related to the green and sustainable synthesis of γ- and δ-lactams from N-alkenyl trichloroacetamides in the presence of tetrahydrofuran or 2-methyltetrahydrofuran alone as the radical initiators and the solvents under violet light irradiation.
Results and discussion
As mentioned earlier, reaction of N-alkenyl trichloroacetamides under photoredox conditions allowed us recently to access a wide range of lactams under mild conditions. The best results, under blue LEDs irradiation, were achieved in a mixture of THF/acetone (Table 1, entry 1) or in THF (entry 2). In acetone using the same conditions, the yield was lower (entry 3). During that stage of the investigation we decided to check the exact reaction time for the full conversion of 1a to 2a. Thus, we conducted a TLC analysis to track the reaction's progress in THF and found that it had reached completion within just 2 hours of irradiation providing lactam 2a with a good yield (entry 4). The quantity of amine was reduced to 1 equiv. without affecting significantly the course of the reaction (entries 5 and 6). As part of our efforts to use economical green procedures, we decided to run the reaction in the presence of ammonia instead of DIPEA. Thereby, the reactions not only concluded in a shorter time but also provided better yields (entries 7 and 8).13 It is worth noting that when the reactions were carried out in the absence of amines, the cyclization process was extremely slow (entries 9–10). Some control reactions were also undertaken in the absence of the catalyst and despite the sluggishness of the reaction, lactam 2a was isolated with very good yields (entries 11 and 12). Further control experiments established that, in the absence of the catalyst and the amine, light is necessary for reactivity (entries 13 and 14 in contrast with entries 15 and 16). The reactions achieved under photocatalyst and amine-free conditions suggest that another reaction mechanism is operating in the formation of lactam 2a involving the solvent and trichloroacetamide 1a under blue LEDs’ irradiation.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ir(ppy)3 (mol%) | Amine (equiv.) | Solvent | Light | Time (h) | 2a yield (%) | 3a yield (%) |
| a Reactions were performed with 1a (0.2 mmol scale) in solvent (4 mL, 0.05 M) at rt and under air atmosphere, blue LEDs (435–445 nm), violet LEDs (395–405 nm). b See ref. 12. c For the results achieved from other substrates see the ESI.† d Conversion 57%. e Reaction achieved from 2 mmol of 1a in 40 mL of solvent. f Traces. g In toluene, DCM, AcOEt, ethanol, acetone, DMSO, MeCN, water, dioxane or diisopropylether no reaction took place and 1a was recovered. h Conversion 27%. | |||||||
| 1 | 1 | DIPEA (5) | THF/acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
Blue LEDs | 14 | 81b | — |
| 2 | 1 | DIPEA (5) | THF | Blue LEDs | 14 | 86b | — |
| 3 | 1 | DIPEA (5) | Acetone | Blue LEDs | 14 | 60b | — |
| 4 | 1 | DIPEA (5) | THF | Blue LEDs | 2 | 78 | — |
| 5 | 1 | DIPEA (1) | THF/acetone (1:1) |
Blue LEDs | 2 | 70 | — |
| 6 | 1 | DIPEA (1) | THF | Blue LEDs | 2 | 73 | — |
| 7 | 1 | NH3 (11)c | THF/acetone (1:1) |
Blue LEDs | 1 | 82 | — |
| 8 | 1 | NH3 (4) | THF | Blue LEDs | 0.5 | 88 | — |
| 9 | 1 | — | THF/acetone (1:1) |
Blue LEDs | 62 | 29 | 29 |
| 10 | 1 | — | THF | Blue LEDs | 62 | 54 | 12 |
| 11 | — | DIPEA (5) | THF/acetone (1:1) |
Blue LEDs | 120 | 89 | — |
| 12 | — | NH3 (11) | THF/acetone (1:1) |
Blue LEDs | 180 | 96 | — |
| 13 | — | — | THF/acetone (1:1) |
Blue LEDs | 216 | 54 | — |
| 14 | — | — | THF | Blue LEDs | 216 | 44d | 11 |
| 15 | — | — | THF/acetone (1:1) |
— | 160 | — | — |
| 16 | — | DIPEA (5) | THF/acetone (1:1) |
— | 160 | — | — |
| 17 | — | — | THF/acetone (1:1) |
Violet LEDs | 14 | 57 | 25 |
| 18 | — | — | THF | Violet LEDs | 14 | 82 | 2 |
| 19e | — | — | THF | Violet LEDs | 14 | 94 | 2 |
| 20 | 1 | DIPEA (1) | THF | Violet LEDs | 1 | 93 | —f |
| 21 | — | — | Solventg | Violet LEDs | 14 | — | — |
| 22 | — | — | Diethyl etherh | Violet LEDs | 14 | 8 | 8 |
| 23 | — | — | 2-MeTHF | Violet LEDs | 14 | 92 | — |
| 24e | — | — | 2-MeTHF | Violet LEDs | 14 | 90 | — |
| 25 | — | — | 2-MeTHF | Violet LEDs | 4 | 85 | —f |

With these results in hand and knowing that 1a absorbs at λ < 400 nm in almost all solvents used in organic synthesis (Fig. 1), we decided to examine the reaction under different light sources to ascertain if the cyclization process could be accelerated. The first reaction was carried out from 1a (0.2 mmol) with available violet light (395–405 nm) and in a mixture of THF/acetone (1:1) as the solvent.
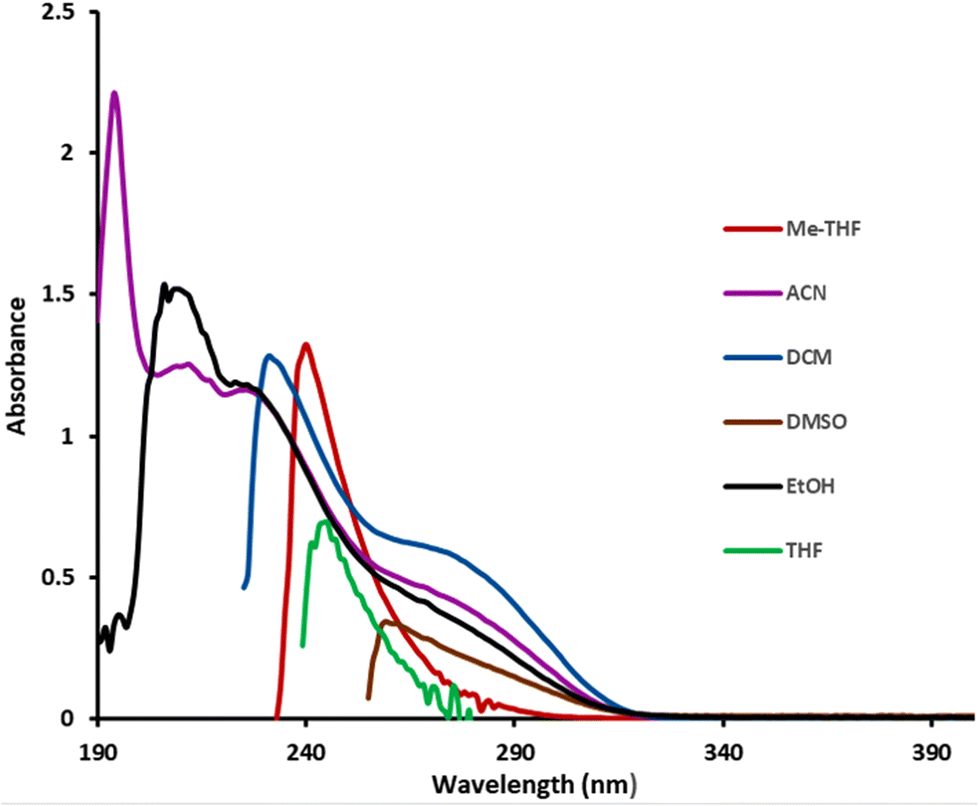 | ||
| Fig. 1 UV absorption spectra of 1a in different solvents. | ||
To our great surprise, the reaction reached completion after a mere 14 hours of irradiation providing the corresponding γ-lactam as a mixture of 2a and 3a in an 82% global yield (entry 17). In THF a similar yield was achieved (entry 18) with almost the exclusive formation of 2a. Scaling up the reaction to 2 mmol had no impact on the reaction outcome (entry 19). In the presence of the photocatalyst (1 mol%) and the amine (1 equiv.) the reaction concluded in only 1 h providing 2a with a very good yield (entry 20). Notably, the same reaction in toluene, DCM, AcOEt, ethanol, acetone, DMSO, MeCN, water, dioxane or diisopropylether did not take place, leaving trichloroacetamide 1a unaltered (entry 21). Only in diethy ether, a slight conversion was observed (entry 22). Next, and in line with our commitment to upholding green chemistry protocols, we decided to carry out the reaction in 2-methyltetrahydrofuran as a sustainable green solvent.14 Thus, when 1a was irradiated with violet light in 2-methyltetrahydrofuran for 14 h, 2a was isolated with an excellent yield (entries 23 and 24). Moreover, when the reaction was monitored by 1H NMR spectroscopy, we discovered that it had concluded within just 4 hours (entry 25). As it was commented in our previous work,12 here also the reactions were carried out under non-anhydrous conditions in an aerobic environment showing a full tolerance to air and moisture.
Beside the simplicity of setting up the reaction which consists in mixing trichloroacetamide 1a and the non-anhydrous solvent followed by irradiation with violet light, the reactions achieved in THF or in 2-MeTHF, were simply concentrated by the end of the process to give lactam 2a almost pure as a white solid (Fig. 2). As it is shown in the latter, the spectrum of the crude displays only the signals belonging to lactam 2a alone. Thereafter, a simple crystallization in the scarce quantity of ether or a rapid filtration using a short chromatography column provided analytically pure 2a as a white solid.
 | ||
| Fig. 2 Spectra of 1a (A), the reaction after 4 or 14 h with the reaction flask after concentration (B) and pure 2a (C). | ||
With the optimized conditions in hand, we then evaluated the scope of the reaction to assess its potential applications and limitations (Fig. 3). This involved mainly testing different substrates to access diverse structural lactam motifs. From trichloroacetamides 1b–1g, as evidenced in our previous research, the substituent on the nitrogen has no influence on the course of the reaction since γ-lactams 2b–2g were isolated with good yields. The reaction from 1h and 1i with a substituted allyl chain furnished lactams 2h and 2i with excellent to acceptable yields. From N-propargyl trichloroacetamide 1j, 2j was isolated with a modest yield probably due to side reactions on the lactam since a full conversion was observed. Operating from 1k with a butenyl chain, the corresponding δ-lactams 2k was isolated with a very good yield. From its analogue 1l, a diastereoselective cyclization was observed furnishing 2l with a modest yield. From N-pentenyl trichloroacetamide 1m, although with a low yield, only the challenging 8-endo cyclization took place to give lactam 2m. The methodology was successfully applied to achieve the bicyclic indol (2n and 2o), isoquinoline (2q) and the challenging morphan (2r and 3s) scaffolds respectively.
 | ||
| Fig. 3 Substrate scope in the synthesis of β-, γ- and δ-lactams from N-alkenyl-trichloacetamides 1b–1s. Unless otherwise stated all reactions were performed with trichloroacetamides 1b–1s (0.2–0.4 mmol scale) in THF (A) or 2-MeTHF (B) (4–8 mL, 0.05 M) at rt and under violet LEDs (395–405 nm) irradiation for 14 h. | ||
Finally, from tricloroacetamide 1p, unlike what was previously reported under reductive conditions (in the presence of TBTH and AIBN in refluxing benzene),15 β-lactam 4p was isolated as the main compound. The latter results from a 4-exotrig cyclization followed by chlorine atom transfer from 1p and then elimination. This unprecedented behaviour is probably due to conformational preferences since the reaction is performed at room temperature. It is worth mentioning that the reaction from N-allyl-2,2,2-trichloroacetamide or allyl 2,2,2-trichloroacetate did not proceed to give the corresponding γ-lactam or γ-lactone showing the importance of having a substituent on the nitrogen in the trichloroacarbamoyl alkenyl derivative for the cyclization process.16
Mechanistic studies
In this work, we have reported the first photo-induced intramolecular cyclization for the synthesis of β-, γ- and δ-lactams from N-alkenyl-trichloacetamides in THF or 2-MeTHF alone as solvents under violet LEDs irradiation. It should be highlighted that the presence of all three chloro atoms in trichloroacetamides 1 is crucial for the reaction to take place, as replacing one or two of these atoms with hydrogens resulted in no reaction even after 2 days of irradiation.To gain a deeper understanding of the reaction mechanism, a series of control experiments was performed to capture the possible intermediates. First, to unmistakably establish that radicals are involved in the cyclization process, (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) was used as a radical inhibitor. In the presence of TEMPO, the reaction did not take place and only few amounts of the radical-TEMPO adduct 6a identified by 1H NMR was isolated (Fig. 6).17 Additionally, when the reaction was carried out from 1a in THF-D8, in contrast to the reaction in THF the deuterated analog of 2a was not detected and trichloroatemide 1a was recovered. This result can be explained by the strong isotope effect concerning the rate of a hydrogen abstraction from THF by carbon-centred radicals which is found to be approximately 8 times faster than a deuterium abstraction from THF-D8.18 To further establish the dependence of the reaction on light exposure, an on–off experiment was performed. Hence, the reaction flask was irradiated alternately with 1 hour of exposure to violet light and 1 h of darkness.
The conversion ratio, analysed by 1H NMR spectroscopy, indicates that no product formation was observed in the dark phase, and thus establishes the light-dependent nature of the reaction eliminating the possibility of a light-induced chain mechanism (Fig. 4).
 | ||
| Fig. 4 Light on/off intermittent experiment for the synthesis of 2a followed by 1H NMR. | ||
Additionally, cyclic voltammetry studies in acetonitrile, the more common solvent used for these measurements or THF the solvent of our reaction, detailed in Fig. 5a showed an irreversible behaviour for 1a in a tetra-n-butylammonium hexafluorophosphate solution. This is probably due to the high reactivity of the dichloromethylcarbamoyl radical species I (see Fig. 6) which adds rapidly to the alkene to generate primary radical II even more reactive that the first one.
 | ||
| Fig. 5 (a) Cyclovoltameric study of 1a in THF and in acetonitrile. (b) Cyclovoltammetric study of trichloroacetamide 1a and its analogues α,α-dichloro and α-chloro acetamides in acetonitrile. | ||
 | ||
| Fig. 6 Proposed mechanism for the formation of lactams 2 and 3. | ||
Moreover, the higher potentials necessary for the single electron transfer in 1a analogues where one of both chlorine atoms were replaced by hydrogens could explain the inertia of these substrates when submitted to the cyclization optimized conditions (Fig. 5b).
In this work we have reported the first catalyst- and amine-free photocyclization19 of trichloroacetamides demonstrating that formation of lactams 2 and 3 involves radical species. Given that the reaction effectively occurs solely in THF or 2-MeTHF under violet light irradiation, we concluded that the mechanism involved herein may suggest oxygen promoted generation of radicals or peroxides from the solvent to initiate the radical process in the synthesis of lactams.
Indeed, in the literature oxidation of tetrahydrofuran and 2-methyltetrahydrofuran with oxygen, in the presence of UV light was reported to give the corresponding hydroperoxides.20 In another investigation, direct oxidative hydroperoxidation of α-ethereal C–H bond of aliphatic ethers e.g. THF was achieved via direct C–H bond insertion using singlet O2. The reaction was carried out in the presence of a photosensitizer (typically meso-TPP or rose bengal) and Lewis acid (γ-Al2O3) under irradiation using a 100 W Hg lamp or a blue-LEDs array at room temperature under a 1 atm oxygen pressure.21 Additionally, aerobic generation of THF radical A (Fig. 6) was established in the functionalisation of ethereal-based saturated heterocycles at 80 °C.22 On the basis of the above-mentioned experiments and our own control experiments we decided to investigate the behaviour of THF and 2-MeTHF alone under violet light. Thus, when both solvents were irradiated separately overnight, after concentration, the residue contained mainly peroxides B which NMR data were identical to those reported previously.21
In view of these results, we propose the mechanism shown in Fig. 6 for the formation of lactams 2 and 3. First, as it was reported in the literature (see ref. 22), oxygen under violet light irradiation interact with THF or 2-MeTHF to generate radical species A that acts as radical initiators23 generating (carbamoyl)dichloromethyl radical I which evolves into radical II through cyclization.
Indeed, analysis of the reaction mixture before concentration by 1H NMR spectroscopy unequivocally revealed the presence of the signals corresponding to chloro derivative C (R′ = H).24 Finally, intermediate II abstracts a hydrogen from the solvent to form lactam 2 or a chlorine atom from trichloroacetamide 1 to produce 3 through an atom transfer process. The probability of the latter to take place increases with the stability of radical II. Indeed, with trichloroacetamide 1s, where a tertiary radical is formed, only 3s was isolated (Fig. 3). 3s evolves to 4s through elimination.
Conclusions
In summary, we have reported herein an unprecedented catalyst- and amine-free one-step radical cyclization for the effective synthesis of γ- and δ-lactams from N-alkenyl trichloroacetamides. The reactions are achieved under mild conditions in THF Or 2-MeTHF alone as solvents under air atmosphere and violet light irradiation. In this investigation we have reported a sustainable green strategy to access a wide range of lactams including indoles, isoquinoline and morphan scafolds. We have also demonstrated that the solvent (THF or 2-MeTHF) acts as the radical initiator in the presence of oxygen allowing the radical cyclization to take place using the simplest reaction setup. Further work is underway to explore the potentiel of this reaction for the synthesis of advanced intermediates towards the synthesis of natural compounds.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support for this research was provided by projects Bosch i Gimpera Foundation (no. 310959 and 311976, PI: F. Diaba),References
- Radicals in organic synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed.
- S. Z. Zard, Radical Reactions in Organic Synthesis, Oxford University Press, 2003 Search PubMed.
- K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC and the references therein.
- B. Giese, J. A. González-Gómez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69–70 CrossRef.
- (a) W. P. Neumann, Synthesis, 1987, 665–683 CrossRef CAS; (b) A. G. Davies, J. Chem. Res., 2006, 141–148 CrossRef CAS.
- A. Padwa, H. Nimmesgern and G. S. K. Wong, J. Org. Chem., 1985, 50, 5620–5627 CrossRef CAS.
- (a) Y. Yamamoto, Triethylborane, in Encycl. Reagents Org. Synth, 2007, DOI:10.1002/047084289X.rt219; (b) V. Darmency and P. Renaud, Top. Curr. Chem., 2006, 263, 71–106 CrossRef CAS.
- M. Ikeda, H. Teranishi, N. Iwamura and H. Ishibachi, Heterocycles, 1997, 45, 863–866 CrossRef CAS.
- For alkyl radicals generation from alkyl halides using photoredox catalysis, see inter alia: (a) X. Zhuang, X. Shi, R. Zhu, B. Sun, W. Su and C. Jin, Org. Chem. Front., 2021, 8, 736–742 RSC; (b) Q. S. Zhao, G. Q. Xu, J. T. Xu, Z. Y. Wang and P. F. Xu, Chem. Commun., 2020, 56, 2206–2209 RSC; (c) X. Ju, Y. Liang, P. Jia, W. Li and W. Yu, Org. Biomol. Chem., 2012, 10, 498–501 RSC.
- For reviews see: (a) Y. Q. Yuan, S. Majumder, M. H. Yang and S. R. Guo, Tetrahedron Lett., 2020, 61, 1–15 Search PubMed; (b) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- G. Trenchs and F. Diaba, Org. Biomol. Chem., 2022, 20, 3118–3123 RSC.
- For additional results achieved from other substrates in the presence of ammonia see the ESI.†.
- (a) C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed; (b) F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- S. Jansana, F. Diaba and J. Bonjoch, Org. Lett., 2019, 21, 5757–5761 CrossRef CAS PubMed.
- E. W. Della and A. M. Knill, Aust. J. Chem., 1995, 48, 2047–2051 CrossRef CAS.
- For the characterization of 6a see ref. 12.
- L. Zhang, J. Cradlebaugh, G. Litwinienko, B. E. Smart, K. U. Ingold and J. W. R. Dolbier, Org. Biomol. Chem., 2004, 2, 689–694 RSC.
- For reviews, see: (a) W. Liu and C. J. Li, Synlett, 2017, 28, 2714–2754 CrossRef CAS; (b) M. L. Deb, B. S. Saikia, P. J. Borpatra and P. K. Baruah, Asian J. Org. Chem., 2022, 11, e202100706 CrossRef CAS.
- G. I. Nikishin, V. G. Glukhovtsev, M. A. Peikova and A. V. Ignatenko, Russ. Chem. Bull., 1971, 20, 2202–2204 CrossRef.
- A. Sagadevan, K. C. Hwang and M. D. Su, Nat. Commun., 2017, 8, 1–8 CrossRef CAS PubMed.
- N. Ahmed, R. J. Spears, T. D. Sheppard and V. Chudasama, Chem. Sci., 2022, 13, 8626–8633 RSC.
- M. Newcomb, R. M. Sanchez and J. Kaplan, J. Am. Chem. Soc., 1987, 109, 1195–1199 CrossRef CAS.
- S. Lee, P. S. J. Kaib and B. List, J. Am. Chem. Soc., 2017, 139, 2156–2159 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterization of new compounds with 1H and 13C NMR copies. See DOI: https://doi.org/10.1039/d3ob01804k |
| This journal is © The Royal Society of Chemistry 2024 |