
Developing a straightforward route toward the synthesis of arylaminomaleimides by palladium-catalyzed arylation of one-pot synthesized aminomaleimides†
Elmira
Taheri
 and
Farnaz
Jafarpour
*
and
Farnaz
Jafarpour
*
School of Chemistry, College of Science, University of Tehran, 14155-6455 Tehran, Iran. E-mail: jafarpour@ut.ac.ir
First published on 1st December 2023
Abstract
3-Aryl-4-aminomaleimides have well-demonstrated applications, such as being used as fluorophores and inhibitors. However, their previous synthesis methods have involved tedious multi-step procedures or methods that need pre-functionalized maleimides and toxic or unstable reagents. Here, a feasible method is developed to synthesize these useful compounds. This includes the one-pot preparation of 3-aminomaleimides, followed by their direct arylation through a palladium-catalyzed Heck reaction with various aryl iodides regioselectively at the β-position of their amine substituents. The results show that this method efficiently exhibits a broad scope.
1. Introduction
Maleimide is one of the valuable core moieties of various natural organic compounds, including crucial bioactive compounds and drug candidates.1–3 For instance, it has been demonstrated that the existence of the maleimide core in related compounds is crucial in delivering antimicrobial effects.4 According to the previous reports, functionalization of the maleimide ring at its carbons at positions 3 and 4 has important beneficial effects on their potential activities.5–8 These compounds have vast applications, such as angiogenesis inhibition, antibacterial activity, and enzyme inhibition, e.g. COX-2 inhibitors for anti-inflammation.9,10 The properties of the maleimide ring can be influenced by aryl and amino functionalities at C-3 and C-4, which can extend the resulting conjugated systems.11 Furthermore, the substituent group directly attached to the nitrogen of the maleimide core can prominently change the characteristics of the aimed compound. In accordance with those mentioned, bisarylmaleimides and especially 3-aryl-4-aminomaleimides with proven activities, e.g.SB-415286 compound (Fig. 1), are well-known as inhibitors of glycogen synthase kinase-3 (GSK-3).12–14 Furthermore, Schmöle et al. demonstrated that indolylmaleimide II, inhibited GSK-3b and subsequently increased β-catenin concentration significantly in human neural progenitor cells (Fig. 1).15 Moreover, Tantray et al. identified a number of maleimide-based compounds and evaluated their GSK-3 inhibition activities where the compounds III and IV showed intriguing outcomes (Fig. 1).16 It should be noted that GSK-3 as a key serine/threonine kinase for the phosphorylation of glycogen synthase has a potential effect on blood glucose levels, Alzheimer's disease, bipolar disorder, etc.17,18 Besides the mentioned merits, maleimides have drawn considerable attention as tunable organic fluorophores with a wide range of applications, including in organic light-emitting diode (OLED) materials, sensors, tissue imaging, and protein labeling compounds.19 While unsubstituted maleimides because of low energy transition of n–π* are considered quenchers of fluorescence through a non-emission pathway for the decay of the excited state,20,21 maleimide derivatives comprising 3-aminomaleimides and 3-aryl-4-aminomaleimides have shown fascinating fluorescence activities.22–26 They can present aggregation-induced emission (AIE) characteristics, i.e., non-self-quenching due to aggregation, for developing solid-state emission materials.11,19 This feature also provides the possibility of developing mechanochromic luminescent sensors, memory devices, and security systems based on tailored maleimide compounds that respond due to a change of molecular arrangement or planarity.27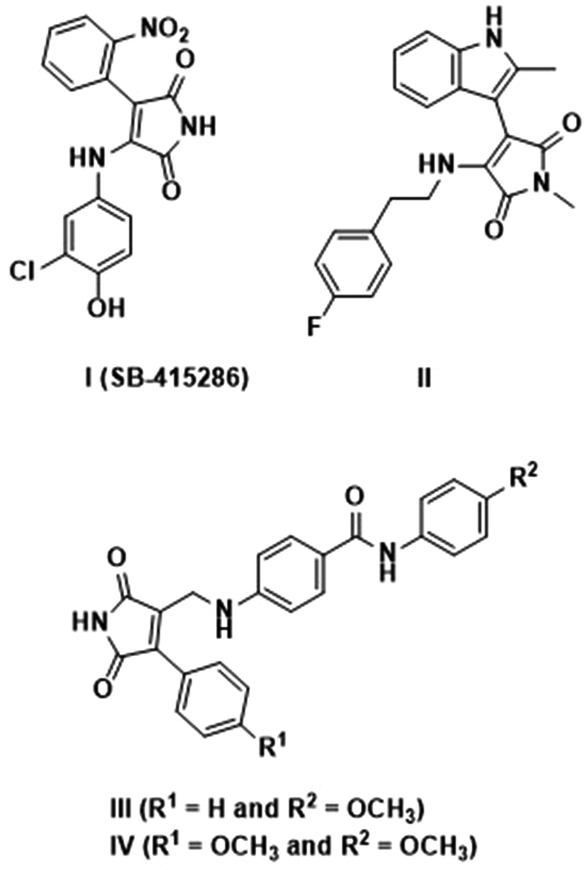 | ||
| Fig. 1 Maleimide-based compounds with GSK-3 inhibition activities. | ||
Besides all the above-mentioned desired features of maleimide derivatives, they can be converted into various prominent heterocyclic compounds, such as succinimides, pyrrolidines, γ-lactams, and lactims, which makes the synthesis of functionalized maleimides and the methods toward them more interesting and beneficial.28,29
The construction of new C–C bonds toward extending the structure of compounds to tailored desired ones is gaining significant attention.30 One of the noticeable strategies for addressing this goal is the direct C–H functionalization of organic moieties as an atomic economical method.31 Accordingly, the Heck cross-coupling reaction catalyzed by palladium is a well-known coupling reaction for direct functionalization of olefin's C–H with vinyl and aryl halides.32,33 Despite the electron deficiency of maleimides, they suffer from hydrolysis and ring-opening of the maleimide core under basic conditions.34 However, our group previously reported the challenging diarylation of maleimides by the Heck coupling and also the cross-dehydrogenative reaction.10,31 In addition, monoarylation of maleimides by the Heck reaction using potassium acetate as a weak base has been reported.35 It is worth mentioning that according to the literature, electron-donating group-bearing olefins, such as enamines, have been reported to show selectivity mostly for arylation through the Heck cross-coupling reaction with aryl halides at the α-position of the amine substituent instead of the β-position.36,37
Generally, there are two ubiquitous strategies for the synthesis of 3,4-disubstituted maleimides, which include the synthesis of the aimed compound either through a sequential procedure toward the formation of the maleimide core or by functionalization of a suitable pre-functionalized maleimide core.6,10 A literature survey for the synthetic methods of aminomaleimides shows that they mainly include a two-step method involving the Michael addition of amines to the maleimide core, followed by oxidation of the obtained 3-aminosuccinimides.23 The second method is the coupling of amines through dehydrohalogenation or dehydration with 3-halo- or 3-hydroxymaleimides, respectively.38–40 Amination of 1,4-dihydro-1,4-diarsininetetracarboxylic acid dianhydride with excess amounts of arylamines is another procedure to obtain 3-aminomaleimides.26 Furthermore, the desired amino maleimides can also be synthesized by a simple one-pot synthetic method via the reaction of acetylenedicarboxylate and alkyl or arylamines.24,27
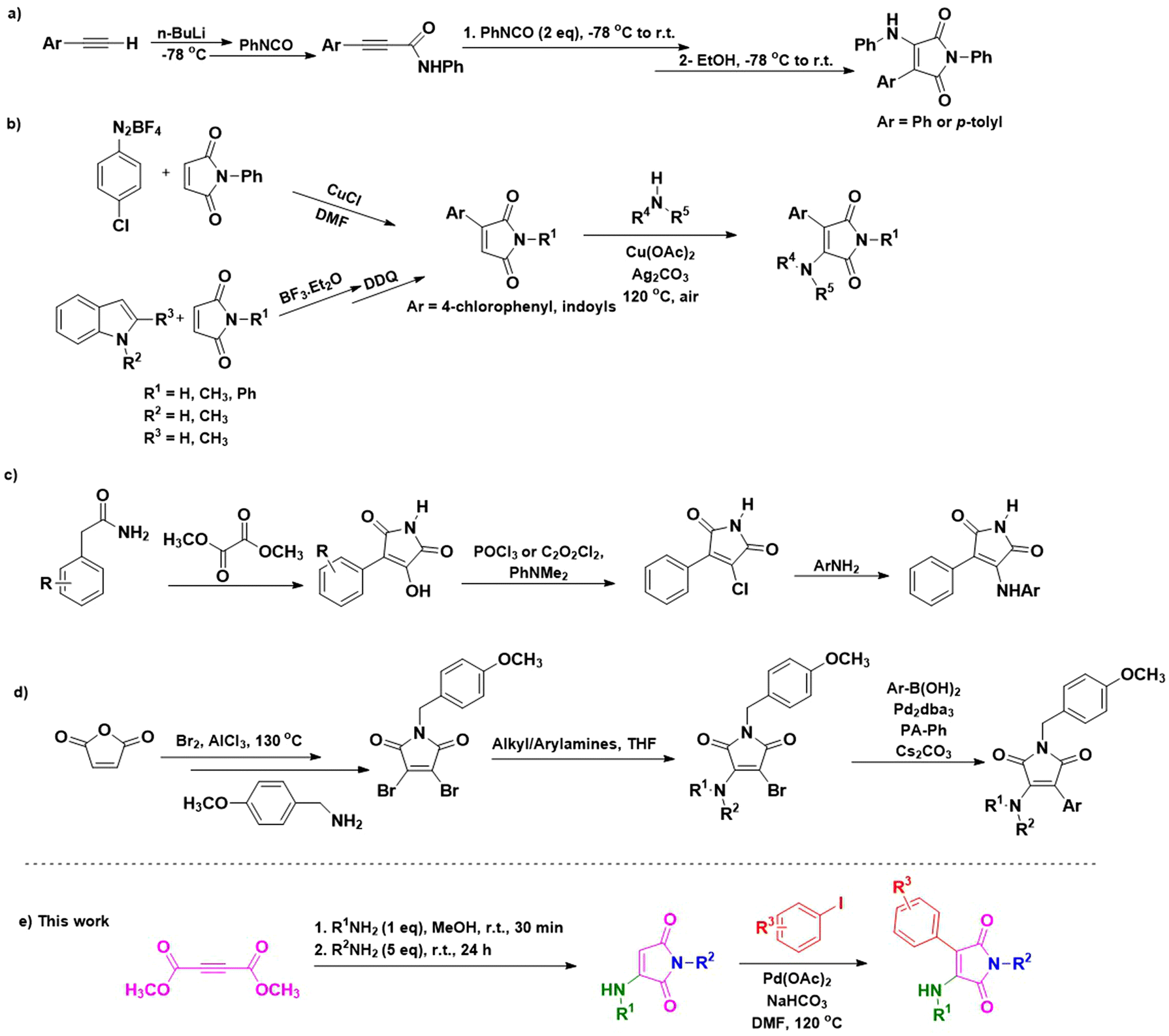
Various studies in recent years have stated the production of 3-aryl-4-aminomaleimides through different methods. Most of them, besides the preparation of maleimide core moieties under tedious and complicated conditions or through multi-step methods, need pre-functionalized maleimides for step-by-step alkylation/arylation and amination using appropriate reagents and/or catalysts. These include Adams and coworkers’ report based on the cyclization of aryl acetylides with phenyl isocyanate at −78 °C using n-BuLi (Scheme 1a).41 The reported method by An et al. proceeds through the arylation of maleimides via arenediazonium tetrafluoroborate intermediates,42 and suffers from the general concerns with diazonium salts, including their instability and tendency to decompose.43,44 Moreover, the procedure involves the synthesis of 3-indolylmaleimides in two steps in the presence of BF3·Et2O, followed by the oxidation of the resulting 3-indolylsuccinimide intermediates using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), which is a costly and hazardous compound.45 Then, they subjected the resulting 3-arylmaleimides to Cu/Ag catalyzed oxidative amination (Scheme 1b).42 Smith et al. reported a three-step method based on the condensation of arylacetamide with dimethyl oxalate under basic conditions, followed by chlorination of 3-aryl-4-hydroxymaleimide intermediates using the toxic phosphorus oxychloride or oxalyl chloride. Finally, the obtained 3-aryl-4-chloromaleimides were subjected to amination using anilines (Scheme 1c).46 On the other hand, N-(p-methoxybenzyl)-3,4-dibromomaleimide, synthesized via a multi-step procedure, has been utilized by Awuah and her colleague for a two-step functionalization toward 3-aryl-4-aminomaleimides. Their method includes the amination of pre-dibrominated N-(p-methoxybenzyl)-3,4-dibromomaleimide using different alkyl/arylamines in the presence of Na2CO3. Next, the obtained 3-amino-4-bromomaleimides were subjected to Suzuki coupling with arylboronic acids in the presence of Cs2CO3 as the base and 1,3,5,7-tetramethyl-2,4,8-trioxa-6-phenyl-6-phosphaadamantane (PA-Ph) as the ligand (Scheme 1d).38 The latter method not only needs the synthesis of the 3,4-dibromomaleimide core via a tedious three-step method from maleic anhydride and the bromination of the corresponding N-(p-methoxy)benzylmaleimide but also needs a two-step functionalization of the obtained pre-dibrominated maleimide toward the aimed final products. Furthermore, because of using the high-cost PA-Ph and Cs2CO3, it might not be an economical procedure.47,48
 | ||
| Scheme 1 Synthetic methods of 3-aryl-4-aminomaleimides. | ||
By considering the above-mentioned merits and potential applications of 3-aryl-4-aminomaleimides and the necessity of developing a straightforward, economical, and environmentally friendly method for the synthesis of these prominent compounds, we provide an efficient procedure for synthesizing this class of maleimides. In the present study, the one-pot synthesized 3-aminomaleimides from dimethyl acetylenedicarboxylate (DMAD) and amines were subjected to direct arylation. In other words, we carried out arylation of 3-aminomaleimides under ligand-free conditions through palladium-catalyzed Heck cross-coupling with aryl iodides in a regioselective manner at the β-position of amine substituents toward synthesizing the aimed 3-aryl-4-aminomaleimides (Scheme 1e).
2. Results and discussion
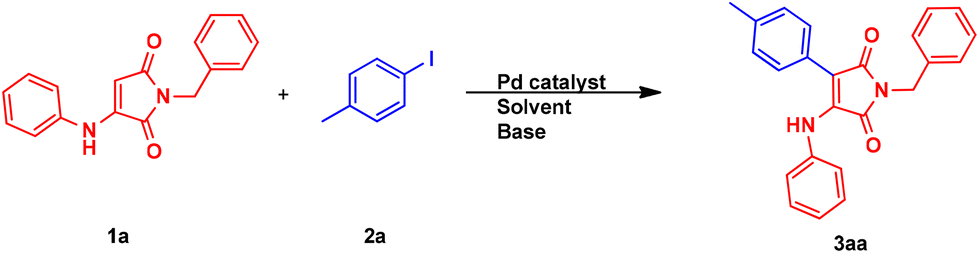
In order to find the optimized reaction conditions for the synthesis of the desired 3-aryl-4-aminomaleimide 3aa in a palladium-catalyzed Heck cross-coupling reaction, the reaction between the synthesized 1-benzyl-3-(phenylamino)-1H-pyrrole-2,5-dione 1a and 4-iodotoluene 2a under various reaction conditions was investigated. This includes investigating the type of base, solvent, and palladium catalyst source used, along with the effect of using the ligand in that reaction. Furthermore, the temperature, the amount of base, and the 2a reagent were also screened. In the first step, through the use of the Pd(OAc)2 catalyst in dimethylformamide (DMF) solvent at 120 °C, various bases were investigated, where NaHCO3 showed the best result (95% yield) (Table 1, entries 1–15). With this result in hand, the presence of the ligand, including 1,10-phenanthroline (Phen) and triphenylphosphine (PPh3), was also studied. The obtained results for Phen and PPh3, respectively, present much and moderately lower yields under ligand-free conditions (entries 16 and 17). Next, the model reaction was performed using PdCl2, Pd(acac)2, Pd(dba)2, and Pd(ACN)2Cl2 instead of Pd(OAc)2 as the catalyst and except Pd(acac)2, all others showed somewhat lower yields. However, in the absence of Pd(OAc)2, the product was not formed (entries 15 and 18–22). Solvent screening showed DMF to be the best choice. Using dimethylsulfoxide (DMSO) and toluene both resulted in a significantly lower yield. When using chlorobenzene (PhCl) or 1,4-dioxane, only a trace amount of the product was detected (entries 15 and 23–27). Various amounts of NaHCO3 base were also tested. Increasing the amount to 0.3 mmol did not improve the yield, while decreasing it to 0.1 mmol resulted in a lower yield of 75% (entries 15 and 28–29). Furthermore, the activity of the catalyst was investigated at different temperatures, 140 °C and 100 °C, and both of them led to lower yields (entries 15 and 30–31). It is worth mentioning that increasing the temperature might result in decomposition of the reagent and/or the product. In the end, the effect of molar ratios of reactants 1a and 2a was investigated. Decreasing the amount of 2a to 0.1 mmol led to a lower yield of the desired product (entries 15 and 32). Hence, the best result was obtained by the reaction of 1a (0.1 mmol) with 2a (0.2 mmol) in the presence of Pd(OAc)2 (10 mol%) and NaHCO3 (0.2 mmol) in DMF (1 mL) at 120 °C for 24 h (entry 15).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Yield (%) |
| Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Pd catalyst (10 mol%), base (0.2 mmol), and solvent (1 mL) at 120 °C for 24 h.a Ligand (20 mol%).b 0.1 mmol of NaHCO3.c 0.3 mmol of NaHCO3.d 140 °C.e 100 °C.f 0.1 mmol of 2a. | |||||
| 1 | Pd(OAc)2 | — | Cs2CO3 | DMF | 0 |
| 2 | Pd(OAc)2 | — | K2CO3 | DMF | 0 |
| 3 | Pd(OAc)2 | — | Na2HPO4 | DMF | 5 |
| 4 | Pd(OAc)2 | — | KOAc | DMF | 90 |
| 5 | Pd(OAc)2 | — | NaOAc | DMF | 83 |
| 6 | Pd(OAc)2 | — | K2SO4 | DMF | 0 |
| 7 | Pd(OAc)2 | — | Na2CO3 | DMF | 87 |
| 8 | Pd(OAc)2 | — | NaH2PO4 | DMF | 6 |
| 9 | Pd(OAc)2 | — | KHCO3 | DMF | 0 |
| 10 | Pd(OAc)2 | — | Et3N | DMF | 23 |
| 11 | Pd(OAc)2 | — | (NH)4OAc | DMF | 14 |
| 12 | Pd(OAc)2 | — | CaCO3 | DMF | 0 |
| 13 | Pd(OAc)2 | — | DABCO | DMF | 50 |
| 14 | Pd(OAc)2 | — | K2HPO4 | DMF | 91 |
| 15 | Pd(OAc) 2 | — | NaHCO 3 | DMF | 95 |
| 16a | Pd(OAc)2 | Phen | NaHCO3 | DMF | 25 |
| 17a | Pd(OAc)2 | PPh3 | NaHCO3 | DMF | 72 |
| 18 | PdCl2 | — | NaHCO3 | DMF | 90 |
| 19 | Pd(acac)2 | — | NaHCO3 | DMF | 77 |
| 20 | Pd(dba)2 | — | NaHCO3 | DMF | 86 |
| 21 | Pd(ACN)2Cl2 | — | NaHCO3 | DMF | 92 |
| 22 | — | — | NaHCO3 | DMF | 0 |
| 23 | Pd(OAc)2 | — | NaHCO3 | DMSO | 15 |
| 24 | Pd(OAc)2 | — | NaHCO3 | DMA | 93 |
| 25 | Pd(OAc)2 | — | NaHCO3 | PhCl | Trace |
| 26 | Pd(OAc)2 | — | NaHCO3 | Toluene | 35 |
| 27 | Pd(OAc)2 | — | NaHCO3 | Dioxane | Trace |
| 28b | Pd(OAc)2 | — | NaHCO3 | DMF | 75 |
| 29c | Pd(OAc)2 | — | NaHCO3 | DMF | 95 |
| 30d | Pd(OAc)2 | — | NaHCO3 | DMF | 79 |
| 31e | Pd(OAc)2 | — | NaHCO3 | DMF | 83 |
| 32f | Pd(OAc)2 | — | NaHCO3 | DMF | 81 |
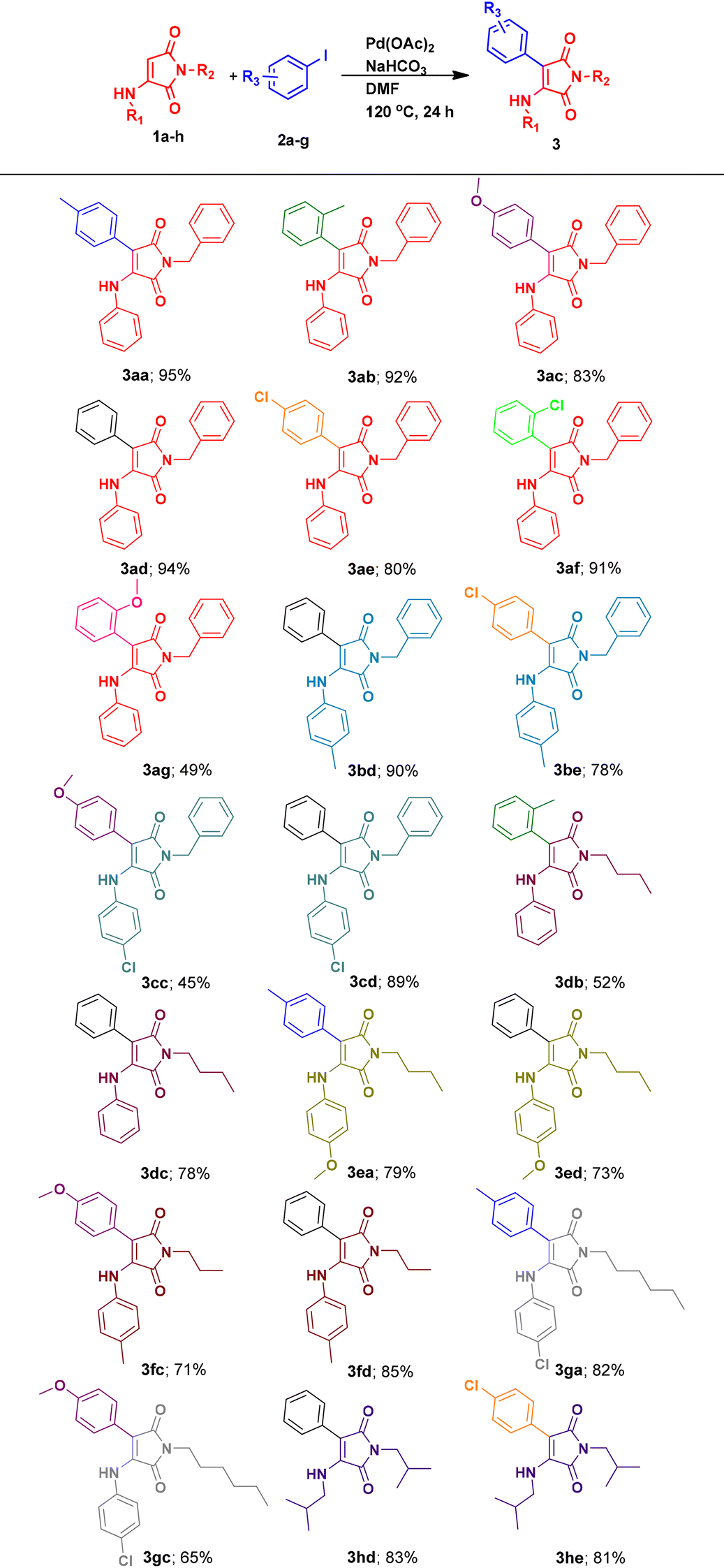
After obtaining the optimized reaction conditions, the scope of the proposed procedure for synthesizing various 3-aryl-4-aminomaleimides was also screened. Hence, various 3-aminomaleimides were synthesized using different arylamines and alkylamines (1a–h; see the ESI†). Then, these 3-aminomaleimides 1a–h were subjected to Heck cross-coupling with diverse aryl iodides having substituents at different positions with varied steric and electronic properties. The results are presented in Table 2. At first, employing 3-aminomaleimide 1a with various o-methyl, p-methoxy, o-methoxy, p-chloro and o-chloro-substituted iodobenzenes as well as the unsubstituted iodobenzene resulted in high to excellent yields of the arylated maleimides (3ab–3ag; Table 2). These results showed that 3-aminomaleimide 1a due to its special structure with two carbonyl groups, despite the presence of the 3-aniline moiety, can efficiently undergo the Heck reaction with various iodoarenes. In other words, it can be arylated regioselectively at the β-position. According to the Heck reaction in the literature,36,49,50 it possibly includes the palladium acetate-maleimide core's double bond complex formation, which then proceeds through the oxidative addition of aryl iodides to the palladium complex, followed by the migratory insertion of the maleimide core's double bond (as the olefin) from its β-position to the formed arylpalladium iodide and the β-hydride elimination from the maleimide moiety to result in the final product. It should be noted that the slight decrease in the yields observed in comparison with the 3aa yield can be attributed to the greater electron deficiency or steric repulsion of the substituted iodoarenes used. Moreover, for 2-methoxyiodobenzene, the formed arylpalladium iodide species during the catalytic heck coupling can be subjected to methoxy C–H activation via a five-membered palladium heterocyclic intermediate and therefore result in byproducts.51–53 Hence, this could be another reason for the lower yield of 3ag. In the next step, by synthesizing and employing 3-substituted N-benzylmaleimides with 4-methylaniline 1b and 4-chloroaniline moieties 1c, average to high yields of the desired products 3bd and 3cd were obtained. It seems that due to the electron-donating characteristics of CH3 and Cl substituents of aniline moieties through induction and resonance, lower reactivity and yields were observed. Notably, when replacing N-benzylmaleimide with N-butylmaleimide 1d, lower yields for both 2-iodotoluene and iodobenzene (3db and 3dc; Table 2) were obtained. These results can be attributed to higher inductive electron-donation of the butyl moiety in 1d with respect to the benzyl group in 1a. As anticipated, the developed 3-substituted N-butylmaleimide with the 4-methoxyaniline moiety 1e due to its p-methoxy group exhibited lower reactivity toward iodobenzene in contrast to the reactivity exhibited by 1d (3ed; Table 2). Using 4-iodotoluene with 1e showed almost a similar result (3ea; Table 2). Also, N-propylmaleimide 1f and N-hexylmaleimide 1g produced satisfactory yields under the optimized conditions (3fc and 3gc; Table 2). Finally, the tolerance of 3-aminomaleimides was further examined by utilizing maleimide 3h as a model reagent of N-alkyl functionalized 3-alkylaminomaleimides in the execution of Heck reactions with iodobenzene and p-chloroiodobenzene and gratefully, the corresponding products 3hd and 3he were obtained in high yields (Table 2).
| Reaction conditions: 1a–h (0.1 mmol), 2a–g (0.2 mmol), Pd(OAc)2 (10 mol%), NaHCO3 (0.2 mmol), and DMF (1 mL) at 120 °C for 24 h. |
|---|
|
3. Conclusion
In summary, we have proposed and developed a feasible, straightforward, relatively environmentally friendly, and economical method for synthesizing diverse 3-aryl-4-aminomaleimides. This class of compounds can have vast potential applications, including as enzyme inhibitors and organic fluorophores. In this method, 3-aminomaleimides are first prepared from dimethyl acetylenedicarboxylate and amines in one pot, and then they undergo a palladium-catalyzed direct arylation, that is a Heck coupling reaction with aryl iodide derivatives toward the desired products. The results showed that due to the unique properties of 3-aminomaleimides with two electron-withdrawing carbonyl groups, they can be efficiently subjected to the Heck arylation at the β-position with iodoarenes. Additionally, the analysis of reagent scope indicates that this approach is a new reliable route with a broad scope for producing different derivatives of 3-aryl-4-aminomaleimides with moderate to high yields.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support from the University of Tehran.References
- J. Yang, J. Liu, R. Jackstell and M. Beller, Chem. Commun., 2018, 54, 10710–10713 RSC.
- K. R. Bettadapur, M. S. Sherikar, V. Lanke and K. R. Prabhu, Asian J. Org. Chem., 2018, 7, 1338–1342 CrossRef CAS.
- M. S. Sherikar and K. R. Prabhu, Org. Lett., 2019, 21, 4525–4530 CrossRef CAS.
- N. S. Patil, G. B. Deshmukh, S. V. Patil, A. D. Bholay and N. D. Gaikwad, Eur. J. Med. Chem., 2014, 83, 490–497 CrossRef CAS.
- B. E. Uno, K. K. Deibler, C. Villa, A. Raghuraman and K. A. Scheidt, Adv. Synth. Catal., 2018, 360, 1719–1725 CrossRef CAS.
- L. Bouissane, J. P. r. Sestelo and L. A. Sarandeses, Org. Lett., 2009, 11, 1285–1288 CrossRef CAS PubMed.
- K. Canto, R. da Silva Ribeiro, A. F. P. Biajoli and C. R. D. Correia, Eur. J. Org. Chem., 2013, 8004–8013 CrossRef CAS.
- D. Basavaiah, B. Devendar, K. Aravindu and A. Veerendhar, Chem. – Eur. J., 2010, 16, 2031–2035 CrossRef CAS PubMed.
- M. Saroha, G. Bartwal and J. M. Khurana, Tetrahedron, 2019, 75, 130486 CrossRef.
- F. Jafarpour, M. Shamsianpour, S. Issazadeh, M. Dorrani and H. Hazrati, Tetrahedron, 2017, 73, 1668–1672 CrossRef CAS.
- H. Imoto, K. Nohmi, K. Kizaki, S. Watase, K. Matsukawa, S. Yamamoto, M. Mitsuishic and K. Naka, RSC Adv., 2015, 5, 94344–94350 RSC.
- M. Boominathan, V. Sathish, M. Nagaraj, N. Bhuvanesh, S. Muthusubramanian and S. Rajagopal, RSC Adv., 2013, 3, 22246 RSC.
- P. Sivaprakasam, A. Xie and R. J. Doerksen, Bioorg. Med. Chem., 2006, 14, 8210–8218 CrossRef CAS.
- M. Akhtar and P. V. Bharatam, Chem. Biol. Drug Des., 2012, 79, 560–571 CrossRef CAS.
- A. C. Schmöle, A. Brennfuhrer, G. Karapetyan, R. Jaster, A. Pews-Davtyan, R. Hubner, S. Ortinau, M. Beller, A. Rolfs and M. J. Frech, Bioorg. Med. Chem., 2010, 18, 6785–6795 CrossRef.
- M. A. Tantray, I. Khan, H. Hamid, M. S. Alam, A. Dhulap and A. Kalam, New J. Chem., 2016, 40, 6109–6119 RSC.
- J. Fang, D. Huang, W. Zhao, H. Ge, H. B. Luo and J. Xu, J. Chem. Inf. Model, 2011, 51, 1431–1438 CrossRef CAS.
- T. A. Engler, S. Malhotra, T. P. Burkholder, J. R. Henry, D. Mendel, W. J. Porter, K. Furness, C. Diefenbacher, A. Marquart, J. K. Reel, Y. Li, J. Clayton, B. Cunningham, J. McLean, C. O'Toole J, J. Brozinick, E. Hawkins, E. Misener, D. Briere, R. A. Brier, J. R. Wagner, R. M. Campbell, B. D. Anderson, R. Vaughn, D. B. Bennett, T. I. Meier and J. A. Cook, Bioorg. Med. Chem. Lett., 2005, 15, 899–903 CrossRef CAS.
- Q. Zhu, Z. Ye, W. Yang, X. Cai and B. Z. Tang, J. Org. Chem., 2017, 82, 1096–1104 CrossRef CAS.
- Y. Xie, J. T. Husband, M. Torrent-Sucarrat, H. Yang, W. Liu and R. K. O'Reilly, Chem. Commun., 2018, 54, 3339–3342 RSC.
- A. B. Mabire, M. P. Robin, W. D. Quan, H. Willcock, V. G. Stavros and R. K. O'Reilly, Chem. Commun., 2015, 51, 9733–9736 RSC.
- M. Staniforth, W. D. Quan, T. N. V. Karsili, L. A. Baker, R. K. O'Reilly and V. G. Stavros, J. Phys. Chem. A, 2017, 121, 6357–6365 CrossRef CAS.
- Y. Guo, L. Yao, L. Luo, H.-X. Wang, Z. Yang, Z. Wang, S.-L. Ai, Y. Zhang, Q.-C. Zou and H.-L. Zhang, Org. Chem. Front., 2021, 8, 239–248 RSC.
- K. Kizaki, H. Imoto, T. Kato and K. Naka, Tetrahedron, 2015, 71, 643–647 CrossRef CAS.
- H. Imoto, K. Kizaki, S. Watase, K. Matsukawa and K. Naka, Chemistry, 2015, 21, 12105–12111 CrossRef CAS PubMed.
- T. Kato and K. Naka, Chem. Lett., 2012, 41, 1445–1447 CrossRef CAS.
- H. Imoto, K. Kizaki and K. Naka, Chem. – Asian J., 2015, 10, 1698–1702 CrossRef CAS.
- Z.-H. Yang, H.-R. Tan, Y.-L. An, Y.-W. Zhao, H.-P. Lin and S.-Y. Zhao, Adv. Synth. Catal., 2018, 360, 173–179 CrossRef CAS.
- S. Dana, A. Mandal, H. Sahoo and M. Baidya, Org. Lett., 2017, 19, 1902–1905 CrossRef CAS.
- A. S. Baghel, Y. Jaiswal and A. Kumar, Org. Lett., 2020, 22, 1908–1913 CrossRef CAS PubMed.
- F. Jafarpour and M. Shamsianpour, RSC Adv., 2016, 6, 103567–103570 RSC.
- Y. J. Du, X. X. Sheng, J. H. Li, J. M. Chen, S. Yang and M. Chen, Chem. Sci., 2023, 14, 3580–3586 RSC.
- L. Lv, D. Zhu and C. J. Li, Nat. Commun., 2019, 10, 715 CrossRef PubMed.
- A. I. Roshchin and E. V. Polunin, Mendeleev Commun., 2008, 18, 332–333 CrossRef CAS.
- L. H. Lim and J. Zhou, Org. Chem. Front., 2015, 2, 775–777 RSC.
- J. Mo, L. Xu and J. Xiao, J. Am. Chem. Soc., 2005, 127, 751–760 CrossRef CAS.
- J. Ruan, O. Saidi, J. A. Iggo and J. Xiao, J. Am. Chem. Soc., 2008, 130, 10510–10511 CrossRef CAS PubMed.
- E. Awuah and A. Capretta, J. Org. Chem., 2011, 76, 3122–3130 CrossRef CAS PubMed.
- M. Tanaka, S. Sagawa, J. Hoshi, F. Shimoma, I. Matsuda, K. Sakoda, T. Sasase, M. Shindo and T. Inaba, Bioorg. Med. Chem. Lett., 2004, 14, 5171–5174 CrossRef CAS PubMed.
- B. Budke, J. H. Kalin, M. Pawlowski, A. S. Zelivianskaia, M. Wu, A. P. Kozikowski and P. P. Connell, J. Med. Chem., 2013, 56, 254–263 CrossRef CAS PubMed.
- H. Adams, M. Baker, H. Hodson and M. J. Morris, Tetrahedron Lett., 2017, 58, 1695–1698 CrossRef CAS.
- Y.-L. An, H.-H. Zhang, Z.-H. Yang, L. Lin and S.-Y. Zhao, Eur. J. Org. Chem., 2016, 5405–5414 CrossRef CAS.
- A. A. Bondarev, E. V. Naumov, A. Z. Kassanova, E. A. Krasnokutskaya, K. S. Stankevich and V. D. Filimonov, Org. Process Res. Dev., 2019, 23, 2405–2415 CrossRef CAS.
- J. D. Firth and I. J. S. Fairlamb, Org. Lett., 2020, 22, 7057–7059 CrossRef CAS PubMed.
- M. A. Alsharif, Q. A. Raja, N. A. Majeed, R. S. Jassas, A. A. Alsimaree, A. Sadiq, N. Naeem, E. U. Mughal, R. I. Alsantali, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 29826–29858 RSC.
- D. G. Smith, M. Buffet, A. E. Fenwick, D. Haigh, R. J. Ife, M. Saunders, B. P. Slingsby, R. Stacey and R. W. Ward, Bioorg. Med. Chem. Lett., 2001, 11, 635–639 CrossRef CAS PubMed.
- A. Gómez-Herrera, F. Nahra, J. Wu, F. Izquierdo, M. Brill, C. S. J. Cazin and S. P. Nolan, ChemistrySelect, 2019, 4, 5–9 CrossRef.
- G. Dhangar, J. L. Serrano, C. Schulzke, K. C. Gunturu and A. R. Kapdi, ACS Omega, 2017, 2, 3144–3156 CrossRef CAS PubMed.
- Q. Yao, E. P. Kinney and Z. Yang, J. Org. Chem., 2003, 68, 7528–7531 CrossRef CAS PubMed.
- P. Surawatanawong and M. B. Hall, Organometallics, 2008, 27, 6222–6232 CrossRef CAS.
- F. Jafarpour, S. Rahiminejadan and H. Hazrati, J. Org. Chem., 2010, 75, 3109–3112 CrossRef CAS PubMed.
- G. Dyker, Angew. Chem., Int. Ed. Engl., 1992, 31, 1023–1025 CrossRef.
- G. Dyker, J. Org. Chem., 1993, 58, 6426–6428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01765f |
| This journal is © The Royal Society of Chemistry 2024 |