
Glycosylation of n-pentenyl glycosides using bromodiethylsulfonium salt as an activator: interception of the glycosyl intermediate by chloride ion transfer†
Supanat
Buntasana
and
Panuwat
Padungros
 *
*
Green Chemistry for Fine Chemical Production and Environmental Remediation Research Unit, Department of Chemistry, Faculty of Science, Chulalongkorn University, Phayathai Road, Pathumwan, Bangkok 10330, Thailand. E-mail: panuwat.p@chula.ac.th
First published on 23rd November 2023
Abstract
Utilization of n-pentenyl glycosides (NPGs) in modern carbohydrate synthesis may be hindered by their sluggish activation, which results from reversible halogenation and cyclization processes. Bromodiethylsulfonium bromopentachloroantimonate (BDSB) has been previously shown to be a powerful brominating agent for the cation–π polyene cyclization of less reactive and electron-poor polyenes. This study demonstrates the activation of NPGs using BDSB as a powerful brominating agent. BDSB effectively activates the terminal olefins of NPGs and the reaction proceeds through 5-exo-tet cyclization, offering a rapid and mild approach for glycosylation with a wide range of glycosyl donors, including n-pentenyl mannoside, n-pentenyl galactoside, and n-pentenyl glucoside. The success of this approach derives from the chloride ion transfer from the nonnucleophilic SbCl5Br anion to the glycosyl intermediate, which disrupts the equilibrium and produces a glycosyl chloride intermediate that is smoothly converted to 22 coupling products, with yields ranging from moderate to excellent (49–100%). The β-selective glycosylation is accomplished when employing NPGs equipped with a neighboring participating group. The practicality of the BDSB-activated glycosylation is demonstrated by a gram-scale synthesis. This study showcases BDSB as a potent activator for NPG glycosylation through the interception of a glycosyl intermediate that diminishes the equilibration during halogenation and 5-exo-tet cyclization.
Introduction
The chemical synthesis of carbohydrates has emerged as one of the most important techniques for creating well-defined carbohydrate structures.1 The precise structures of synthetic carbohydrates may shed light on the interaction between carbohydrates and biological systems.2–5 Several glycosyl donors are testaments to the establishment of chemical glycosylation, including thioglycoside, glycosyl trichloroacetimidate, glycosyl halide, and n-pentenyl glycoside (NPG).6 Thioglycoside and NPG are among the unique glycosyl donors. The aglycon moieties of these donors serve as robust protecting groups at the anomeric position during protecting group manipulation and as leaving groups upon suitable activation. Despite the existence of well-established activation protocols, new glycosylation methods are still needed to overcome the existing limitations.6n-Pentenyl glycosides (NPGs), introduced by Fraser-Reid, are conventionally activated by halogenation at the olefinic end of the n-pentenyl group.7 Bromination/iodination at the terminal olefin of NPG, followed by a 5-exo-tet cyclization, generates a reactive glycosyl intermediate. The ejection of the 2-(halomethyl)tetrahydrofuran leaving group at the rate-determining step (RDS) followed by the addition of a glycosyl acceptor provided the disaccharide product (Fig. 1A, pathway a).8 The initial two activation stages, halogenation and cyclization, are reversible processes.7,9 The glycosylation may proceed slowly, particularly if low reactivity of activators, e.g., N-bromosuccinimide (NBS) or N-iodosuccinimide (NIS), or unreactive glycosyl donors are employed. In addition, the formation of vicinal dibrominated NPG side products was also observed during activation.9–11
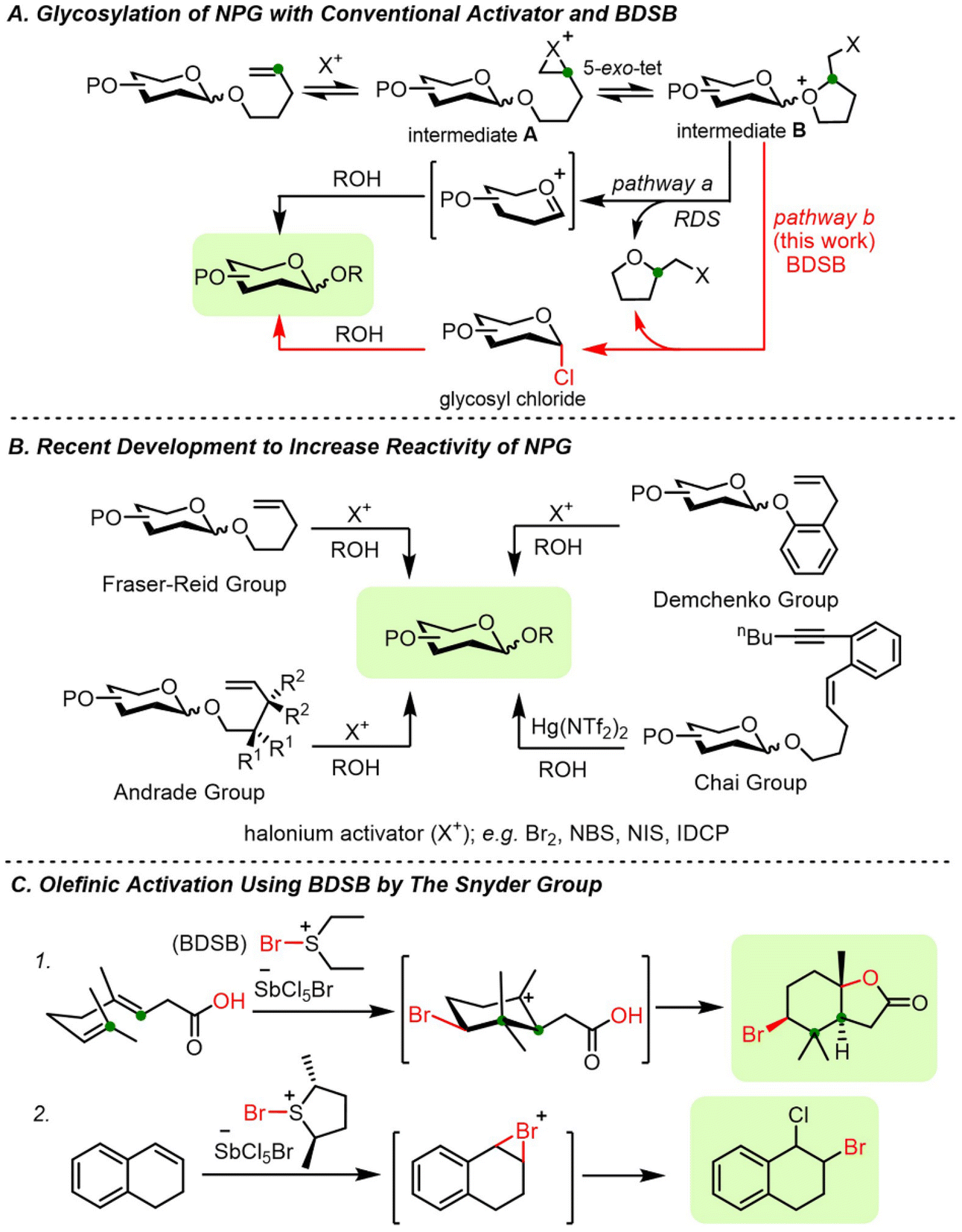 | ||
| Fig. 1 (A) Mechanism of NPG activation, (B) recent development of NPG analogs, and (C) olefinic activation with BDSB. | ||
Other research groups have recognized the sluggish reactivity of certain NPGs and have converted them to more reactive glycosyl donors, including glycosyl trichloroacetimidates,12 glycosyl trifluoroacetimidates,13 glycosyl 1,2-orthoesters,14 and glycosyl phosphates.15 Recently, the development of glycosyl donors equipped with more reactive 4-pentenyl analogs has been pursued, such as gem-dimethyl NPGs (Andrade group),8 2-allyloxy phenyl glycosides (Demchenko group),16 and 4-n-pentenyl-1,5-enynyl glycosides (Chai group),17 as shown in Fig. 1B. Despite the success of 4-pentenyl analogs, the formation of side products resulting from the addition of a glycosyl acceptor to the terminal olefin of 4-pentenyl analogs has been reported.16 In these cases, the improved reactivity of modified 4-pentenyl analogs of the original NPGs was hampered by the increasing molecular weight of the leaving groups at anomeric carbon, which may restrict their application in large-scale synthesis.
Cation–π polyene cyclization of electron-poor polyene was successfully demonstrated with a powerful brominating agent, namely bromodiethylsulfonium bromopentachloroantimonate (BDSB) (Fig. 1C; eqn (1)).18 BDSB is an attractive brominating agent due to its high reactivity, ease of preparation, and handling since it is obtained as an orange crystalline solid after recrystallization.19 BDSB is one of the most powerful brominating agents in its category.20 This high reactivity may be partially attributed to the nonnucleophilic nature of the bromopentachloroantimonate anion (SbCl5Br−), as evidenced by X-ray crystallographic data of BDSB.21 Surprisingly, despite the expected nonnucleophilic nature of the SbCl5Br anion, chloride ion transfer from the SbCl5Br anion was observed during the bromination of 1,2-dihydro naphthalene (Fig. 1C; eqn (2)).22 Similarly, our research group has recently observed the transfer of chloride ions during the synthesis of α-glycosyl chlorides from thioglycosides using BDSB as a mild oxidant.23
Herein, we propose to harness a unique chloride ion transfer for the activation of NPGs (Fig. 1A, pathway b). We envisage that the bromination of the terminal olefin (yielding glycosyl intermediate A) and 5-exo-tet cyclization (yielding glycosyl intermediate B) could be accomplished by the powerful BDSB. Interception of the glycosyl intermediate B by the chloride ion from the SbCl5Br anion facilitated the liberation of the 2-(bromomethyl)tetrahydrofuran leaving group and perturbed equilibrations to move forward. Nucleophilic substitution of glycosyl chloride would yield the desired disaccharide.
Results and discussion
Our investigation began with evaluating the reactivity of BDSB compared to that of well-established halonium activators, such as NBS, NIS, bromodimethylsulfonium bromide (BDMS),24 and NIS with triethylsilyl trifluoromethanesulfonate as a co-activator (NIS/TESOTf). The activations of the n-pentenyl glucosyl donor 1 with haloniums were performed at −78 to 20 °C in CH2Cl2, using trifluoroethanol (2) as the glycosyl acceptor (Scheme 1). Complete consumption of NPG 1 by BDSB was observed at −50 °C, whereas NIS/TESOTf and BDMS required higher temperatures (−20 °C and 20 °C, respectively) for complete consumption. Some NPG 1 was left unreacted when using only NBS (32% conversion) and NIS (18% conversion) as activators (see Section VII in the ESI† for details). The slow consumption of NPG by NBS and NIS activators is consistent with the lower reactivity of these halonium sources. Quantitative 19F NMR analysis revealed that BDSB and NIS/TESOTf facilitated the formation of glucoside 1a in high yields of 86% and 83%, respectively. Other activators, including BDMS, NBS, and NIS, provided the products in lower yields, ranging from 6–10%. Next, the reactivity profiles during the kinetic study between BDSB and NIS/TESOTf were evaluated. At −70 °C, the poor solubility of crystalline BDSB in CH2Cl2 at low temperature may account for the slow conversion (20% conversion) compared to the homogenized mixture obtained when employing NIS/TESOTf (74% conversion). Interestingly, despite a high consumption by NIS/TESOTf at −70 °C, the glucoside product 1a was afforded in only 18% yield along with the hydrolyzed glucosyl donor 1. At −40 °C, NPG 1 was consumed by NIS/TESOTf with 85% conversion; however, only 58% of the glucoside 1a was produced. For the BDSB activation at −40 °C, the NPG was completely consumed (100% conversion) and generated the glucoside 1a in 84% yield. These observations suggest that the NPG was reactive under NIS/TESOTf activation. Still, the reaction between glycosyl intermediates A and B may have reached equilibrium (Fig. 1A), preventing the formation of glucoside 1a.7–9 Lastly, for the BDMS activator, complete conversion of NPG 1 was attained using BDMS at 20 °C; however, the yield of glucoside 1a was low (10%), mostly due to the hydrolysis of NPG 1 and the formation of glucosyl bromide (15). In summary, BDSB was shown to be a potent activator due to its high reactivity (complete consumption of NPG 1 at −50 °C) and provided a good yield (86%) of glucoside 1a.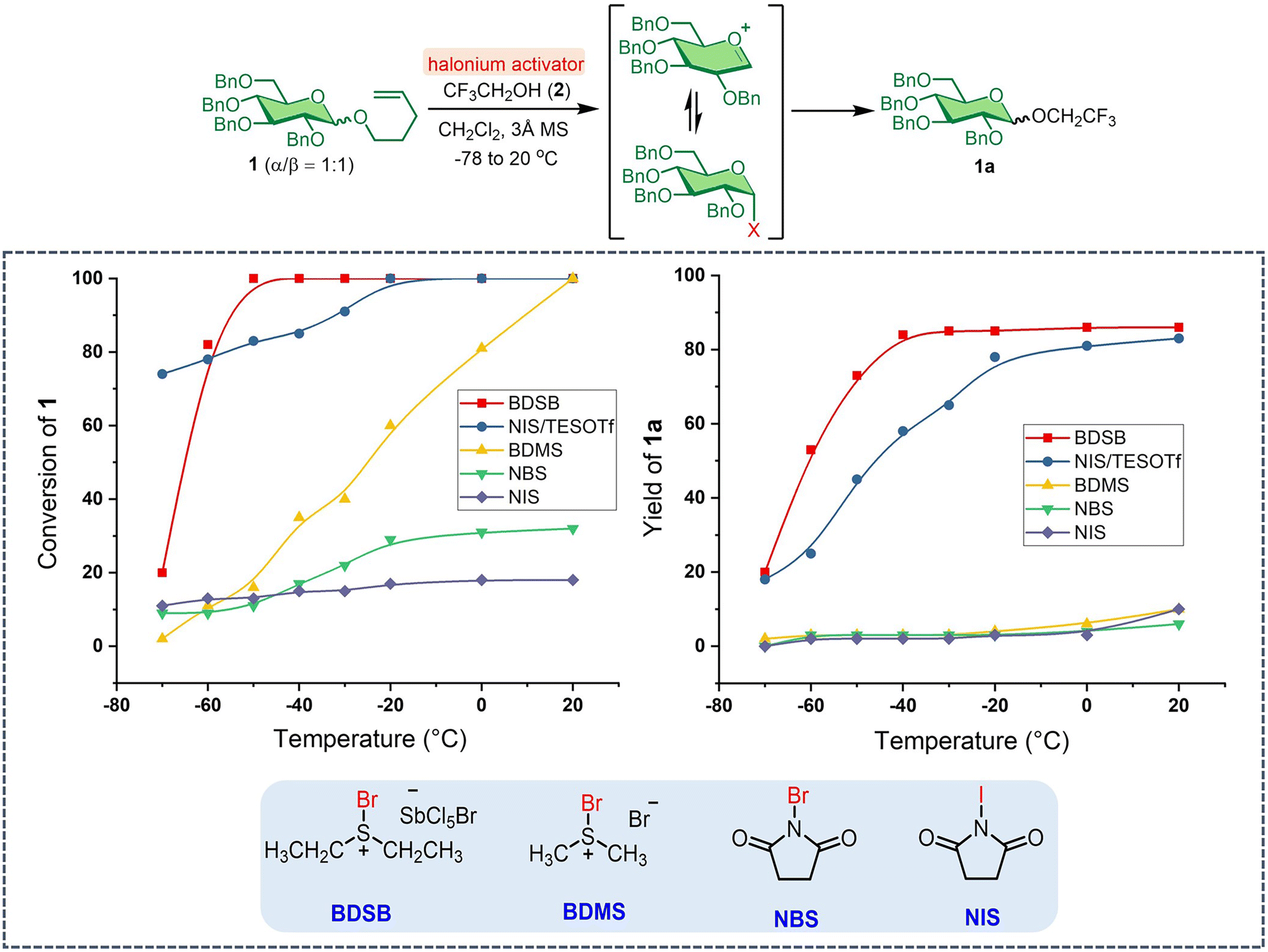 | ||
| Scheme 1 Evaluation of halonium activators towards NPG. | ||
In light of these findings, BDSB was chosen to investigate the glycosylation of NPG using various solvents, temperatures, and glycosyl acceptors (Table 1). We hypothesized that α-mannosyl chloride 4 would serve as an intermediate and subsequently be converted to the mannoside product. The BDSB-activated glycosylation of n-pentenyl mannoside 3 and trifluoroethanol (2) in polar aprotic solvents (CH2Cl2, CH3NO2, and CH3CN) and a non-polar solvent (Et2O) was carried out at low temperatures (−60 °C or −40 °C) (entries 1–4). In solvent screening, mannoside 3a was achieved in the highest yield of 91% with moderate α/β selectivity (α/β = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) when using CH2Cl2 (entry 1). Due to the poor solubility of BDSB in Et2O, mannoside 3a was obtained in a low yield (45%; entry 4). Increasing the temperature from −60 °C to −40 °C and 0 °C also provided similar yields and stereoselectivities of mannoside 3a (entries 5 and 6). Therefore, the optimal conditions involved using CH2Cl2 as the solvent and carrying out the glycosyl coupling at temperatures ranging from 0 °C to room temperature. Changing the glycosyl acceptor to more sterically hindered acceptors, such as benzyl alcohol (5) and glucosyl acceptor 6, drastically decreased the yields of mannosides 3b and 3c to 79% and 35%, respectively (entries 7 and 8). In these cases, sufficient quantities of α-mannosyl chloride 4 were isolated (18% and 59%, respectively; entries 7 and 8). Treatment of mannosyl donor 3 with BDSB alone, without the addition of a glycosyl acceptor, resulted in the α-mannosyl chloride 4 as expected in 75% yield (entry 9). The formation of α-mannosyl chloride 4 supported our hypothesis that the interception of the glycosyl intermediate B by the chloride ion occurred during the course of activation (Fig. 1A).
:1) when using CH2Cl2 (entry 1). Due to the poor solubility of BDSB in Et2O, mannoside 3a was obtained in a low yield (45%; entry 4). Increasing the temperature from −60 °C to −40 °C and 0 °C also provided similar yields and stereoselectivities of mannoside 3a (entries 5 and 6). Therefore, the optimal conditions involved using CH2Cl2 as the solvent and carrying out the glycosyl coupling at temperatures ranging from 0 °C to room temperature. Changing the glycosyl acceptor to more sterically hindered acceptors, such as benzyl alcohol (5) and glucosyl acceptor 6, drastically decreased the yields of mannosides 3b and 3c to 79% and 35%, respectively (entries 7 and 8). In these cases, sufficient quantities of α-mannosyl chloride 4 were isolated (18% and 59%, respectively; entries 7 and 8). Treatment of mannosyl donor 3 with BDSB alone, without the addition of a glycosyl acceptor, resulted in the α-mannosyl chloride 4 as expected in 75% yield (entry 9). The formation of α-mannosyl chloride 4 supported our hypothesis that the interception of the glycosyl intermediate B by the chloride ion occurred during the course of activation (Fig. 1A).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Acceptor | Solvent | Temperature | Yieldb | α/βc | |
| a Standard conditions: n-pentenyl mannoside 3 (0.04–0.22 mmol), BDSB (1.2 equiv.), glycosyl acceptor (5.0 equiv. for 2 and 5 and 2.0 equiv. for 6), 3 Å MS, [c] = 0.1 M. b Isolated yields. c Stereoselectivities (α/β) were determined by 1H NMR integration. | ||||||
| Solvent | 1 | 2 | CH2Cl2 | −60 °C to rt | 3a; 91% | 2:1 |
| 2 | 2 | CH3NO2 | −60 °C to rt | 3a; 76% | 2:1 |
|
| 3 | 2 | CH3CN | −40 °C to rt | 3a; 84% | 1:1 |
|
| 4 | 2 | Et2O | −60 °C to rt | 3a; 45% | 3:1 |
|
| Temperature | 5 | 2 | CH2Cl2 | −40 °C to rt | 3a; 93% | 2:1 |
| 6 | 2 | CH 2 Cl 2 | 0 °C to rt | 3a ; 91% |
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
|
| Acceptor | 7 | 5 | CH2Cl2 | 0 °C to rt | 3b; 79% |
3b; 1:1 |
| 4; 18% |
4; 1:0 |
|||||
| 8 | 6 | CH2Cl2 | 0 °C to rt | 3c; 35% |
3c; 2:1 |
|
| 4; 59% |
4; 1:0 |
|||||
| 9 | None | CH2Cl2 | 0 °C to rt | 4; 75% | 1:0 |
|
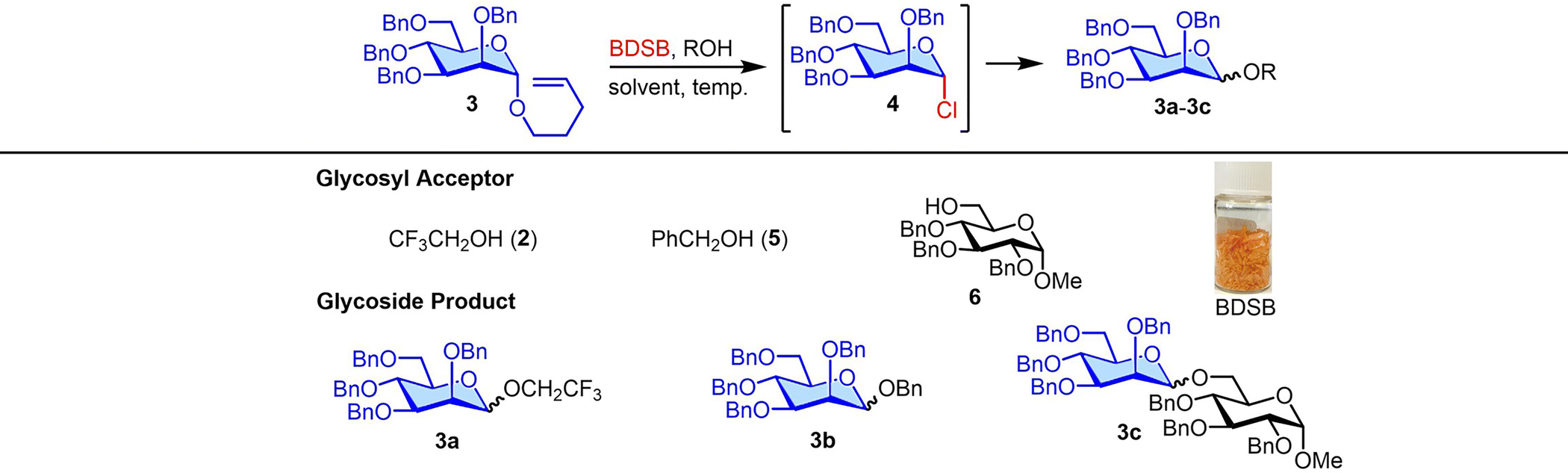
To improve glycosylation efficiency, AgOTf was added to the reaction mixture to activate the remaining glycosyl chloride. The substrate diversity of glycosyl donors and acceptors was next explored (Scheme 2). First, NPG was treated with BDSB at 0 °C in CH2Cl2. After observing the complete consumption of NPG (approximately 15 minutes), a glycosyl acceptor and AgOTf were added to the reaction mixture. The glycosylation between the n-pentenyl mannosyl donor 3 and glucosyl acceptor 6 with the aid of AgOTf significantly improved the yield of the disaccharide 3ca from 35% (Table 1, entry 8) to 81%. Next, the glycosylation of n-pentenyl mannoside 3 with various alcoholic nucleophiles, i.e., trifluoroethanol (2), benzyl alcohol (5), isosorbide 2-acetate (14), and 1-adamantanol, gave the coupling products 3aa, 3ba, 3da, and 3ea in high yields (66–91%) with moderate α/β stereoselectivities. When a glycosyl acceptor was used as a coupling partner for n-pentenyl mannoside 3 and β-pinene was used as an acid scavenger,25 the desired disaccharide 3g was afforded in 61% yield. Due to the modest reactivity of the n-pentenyl per-O-benzoylated mannosyl donor 7, a moderate yield (49%) of disaccharide 7a was obtained. Both the n-pentenyl galactosyl donor 8 and n-pentenyl glucosyl donor 1 produced the coupling products 8a, 8b, and 1a in satisfactory to exceptional yields (71%, 77%, and 100%, respectively). The n-pentenyl glucosyl donor 9 containing a free hydroxyl at the C2 position yielded disaccharide 9a in a moderate yield (64%) with high β-selectivity.26 This accomplishment demonstrated chemoselectivity in the oxidation of BDSB with an olefin moiety over the alcohol of C2 hydroxyl.27 The effect of β-pinene in BDSB-activated glycosylation was also probed. Glycosyl coupling of the mannosyl donor 3 and isosorbide 2-acetate (14) with β-pinene resulted in α-mannoside 3d in 76% yield, comparable to that when there was no addition of β-pinene (71%). Intriguingly, when β-pinene was omitted during the coupling of the glucosyl donor 1, the 1,6-anhydro glucoside 10 was isolated in 84% yield as the main product, along with the desired disaccharide 1ba in 5% yield. A high yield (81%) of disaccharide 1b was restored when β-pinene was added to the glycosylation. We propose that conformational flexibility for the cyclization of the glucosyl donor 1 and a highly acidic environment, in the absence of an acid scavenger, could accelerate the formation of the 1,6-anhydro glucoside 10 side product. The enhancement of coupling yields by β-pinene has also been observed by other research groups.28 It is worth noting that for the synthesis of disaccharide 3fb, β-pinene was not an effective acid scavenger to suppress the decomposition of the acidic labile PMB group at the anomeric position. As a result, Et3N was employed as a base and disaccharide 3fb was successfully produced in 70% yield and with high α-selectivity.
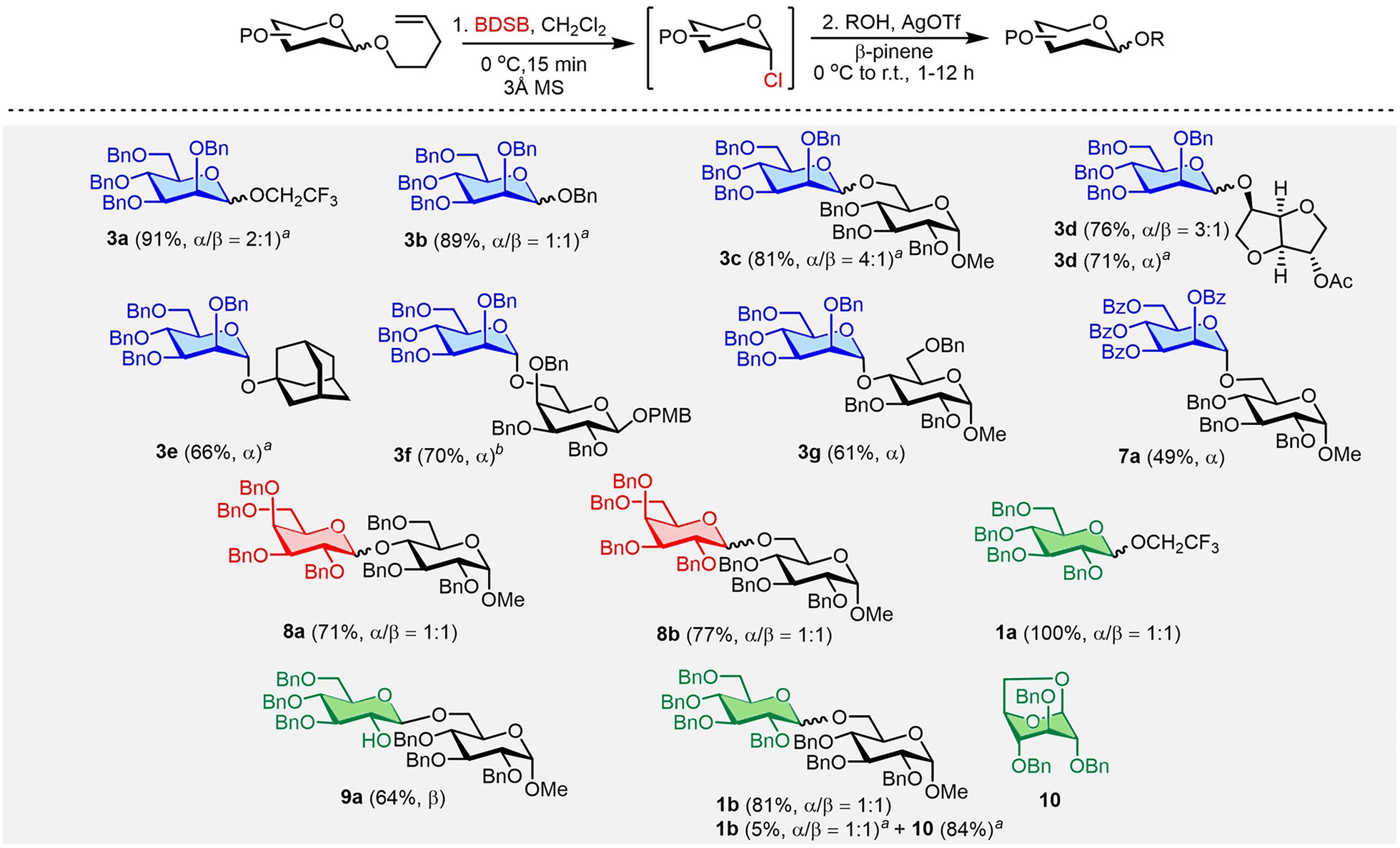 | ||
| Scheme 2 The substrate scope of NPGs and glycosyl acceptors. Standard conditions: n-pentenyl glycosides (0.05–0.27 mmol), BDSB (1.1 equiv.), 3 Å MS, CH2Cl2, [c] = 0.1 M, 0 °C, 15 min then β-pinene (5.0 equiv.), a glycosyl acceptor (0.7–5.0 equiv.), AgOTf (2.0 equiv.) then slowly warming to r.t. (27–30 °C). Isolated yields after column chromatography and stereoselectivities (α/β) were determined by 1H NMR integration. aGlycosylation without using β-pinene. bEt3N (0.08 mmol, 1.0 equiv.) was used as a base instead of β-pinene. | ||
The 1,2-trans stereoselective glycosylation was then investigated using the NPG containing 2-O-acyl as the neighboring participating group (Scheme 3). n-Pentenyl 2-O-pivaloyl glucoside 11 was an excellent glycosyl donor, yielding the β-glucosides 11a–11e with high yields (76–98%) and β-selectivities. β-Selective glycosylation with n-pentenyl 2-O-acetyl glucoside 12 was also achieved and provided disaccharide 12a in 80% yield, albeit with a lower selectivity (α/β = 1:3) compared to the disaccharide 11d. This was possibly due to the less steric hindrance of the 2-O-acetyl group in shielding the α-face of the glycosyl intermediate. Finally, disaccharides 13a–13c were obtained in high yields (75–85%) and with good β-selectivities from the couplings of n-pentenyl 2-O-pivaloyl galactoside 13 with various glycosyl acceptors under the established conditions. Demchenko and co-workers recently reported the successful glycosylation of glycosyl chlorides promoted by substoichiometric amounts of Ag2O (0.5 equiv.) and TfOH (0.25–0.5 equiv.) compared to the conventional practice that required an excess amount of a silver(I) salt.29 The synthesis of disaccharide 13a under Demchenko's conditions was examined (see Section IV in the ESI† for details). The use of Ag2O (0.5 equiv.) and TfOH (0.5 equiv.) as activators resulted in the desired disaccharide 13aa in only 57% yield. Additionally, the α-galactosyl chloride 16a intermediate was afforded in 17% yield. Increasing the amount of Ag2O to 1.5 equivalents consumed all the α-galactosyl chloride 16a and provided disaccharide 13ab in a slightly higher yield (68%). We hypothesized that the need for an excess amount of Ag2O in this work resulted from the coordination between Ag2O or AgOH (postulated as an intermediate in Demchenko's work)29 and diethyl sulfide (liberated from BDSB during the activation, see Scheme 5). The Ag2O–SEt2 complex could deactivate the Ag2O activator, or the AgOH–SEt2 complex could perturb the regeneration of Ag2O from AgOH. Consequently, an excess amount of Ag2O was unavoidable in our work.
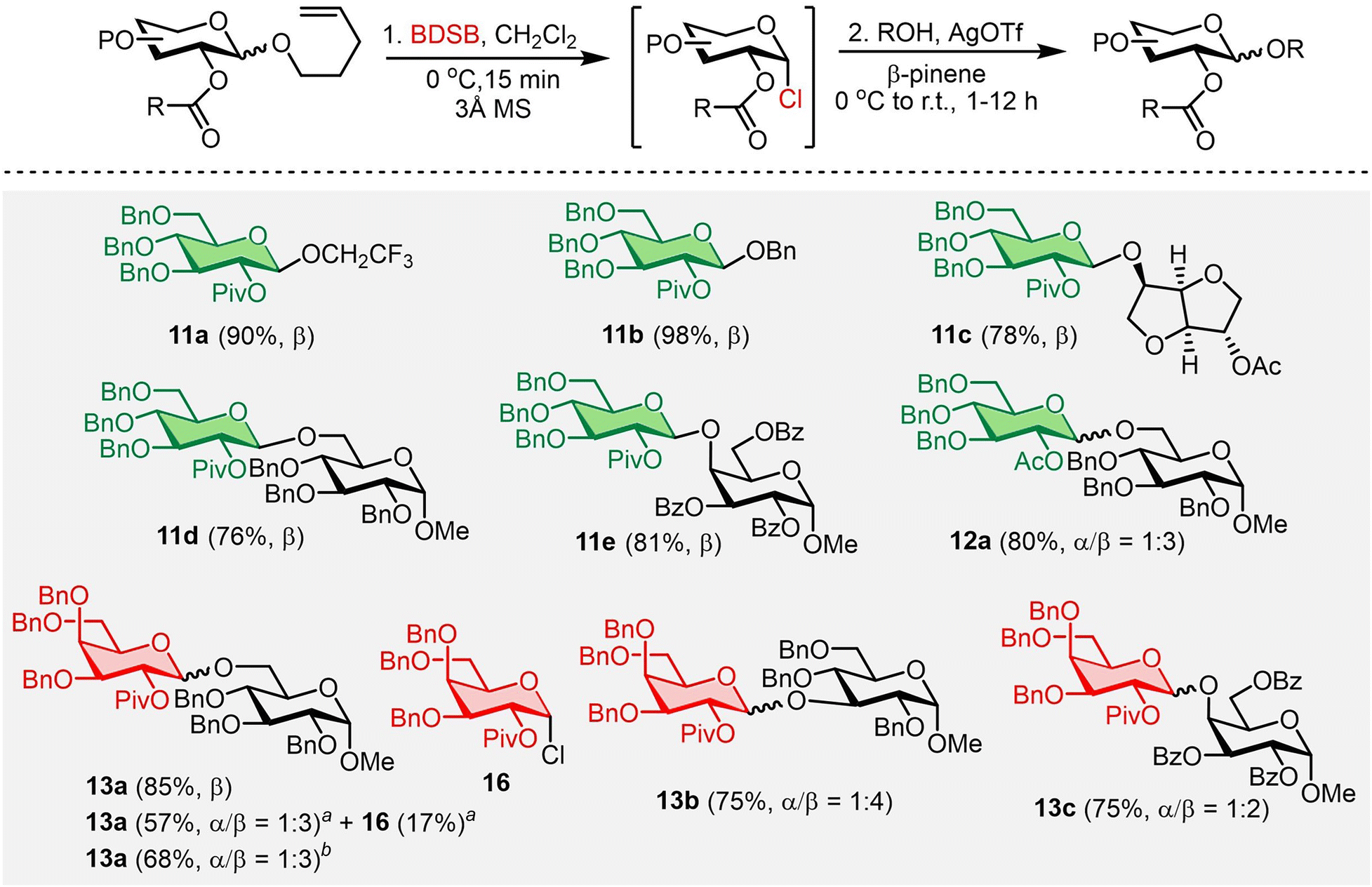 | ||
| Scheme 3 The substrate scope of NPGs containing 2-O-acyl and glycosyl acceptors. Standard conditions: n-pentenyl glycosides (0.05–0.27 mmol), BDSB (1.1 equiv.), 3 Å MS, CH2Cl2, [c] = 0.1 M, 0 °C, 15 min then β-pinene (5.0 equiv.), a glycosyl acceptor (0.7–5.0 equiv.), AgOTf (2.0 equiv.) then slowly warming to r.t. (27–30 °C). Isolated yields after column chromatography and stereoselectivities (α/β) were determined by 1H NMR integration. aAg2O (0.5 equiv.) and TfOH (0.5 equiv.) were used as activators instead of AgOTf. bAg2O (1.5 equiv.) and TfOH (0.5 equiv.) were used as activators instead of AgOTf. | ||
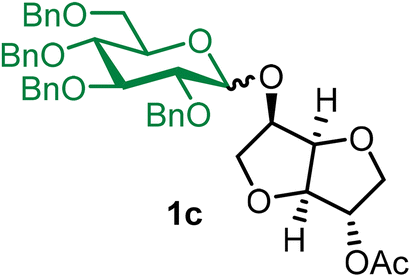
The robustness and scalability of the BDSB-activated glycosylation of NPG were demonstrated through a gram-scale synthesis (Scheme 4A). A glycosyl coupling between n-pentenyl glucoside 1 (1.02 g, 1.7 mmol) and isosorbide 2-acetate (14) smoothly produced glucoside 1c in 78% yield with α/β = 3:1.30 It is important to mention that the activation of NPGs by BDSB can be performed in the presence of a glycosyl acceptor and AgOTf. As a result, the two-step activation process was simplified to a one-step activation (Scheme 4B). The n-pentenyl galactosides 8 and 13 were smoothly coupled with the glucosyl acceptor 6 by the addition of the glucosyl acceptor, BDSB, and AgOTf to a solution of a galactosyl donor at 0 °C. Disaccharides 8b and 13a were acquired in comparable yields (80% and 78%, respectively) when compared with the standard, two-step protocol. This finding strengthens the practicality of the BDSB activation. Next, the comparison between the reactivity of BDSB and NIS/TESOTf activation was made (Scheme 4C). According to our experience, the glycosidic linkage of disaccharide 8a was sensitive and susceptible to hydrolysis during column chromatography or prolonged storage; thus, it was selected as a model for this study. Glycosylations of n-pentenyl galactosides 8 with the glucosyl acceptor 17 at 0 °C and −50 °C activated by both activators were carried out. At 0 °C, BDSB activation produced the disaccharide 8a in a slightly higher yield (71%) than NIS/TESOTf activation (65%). When the glycosylations were performed at −50 °C, BDSB activation provided disaccharide 8a in 69% yield. In contrast, the NIS/TESOTf activation exhibited lower activity, yielding disaccharide 8a in 55% yield and unreacted n-pentenyl galactosides 8 in 9% yield. The results suggest that BDSB is more reactive than NIS/TESOTf, particularly at low temperatures, which may be advantageous for the synthesis of carbohydrates bearing sensitive glycosidic linkages or protecting groups.
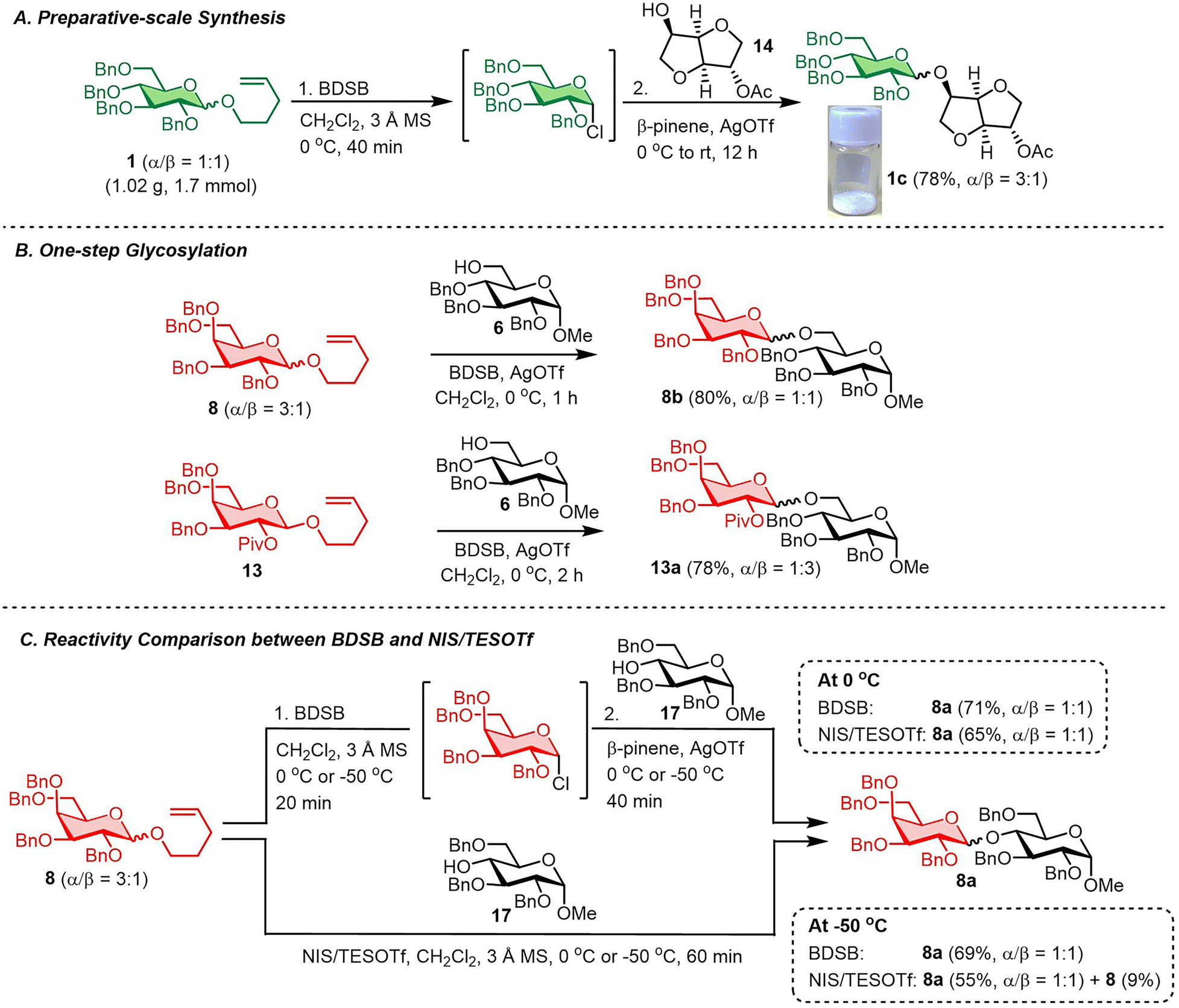 | ||
| Scheme 4 (A) Preparative-scale synthesis, (B) one-step glycosylation and (C) reactivity comparison between BDSB and NIS/TESOTf. | ||
Based on these findings, a possible mechanism is proposed (Scheme 5). The glycosyl intermediate A is generated by brominating the olefinic group on NPG with BDSB. The intramolecular cyclization via 5-exo-tet of the anomeric oxygen yields the glycosyl intermediate B. These reversible processes are disrupted by chloride ion transfer from the SbCl5Br anion,23,31 which could endure an SN1-like pathway through an oxocarbenium intermediate or direct displacement of 2-(bromomethyl)tetrahydrofuran (SN2-like pathway), generating glycosyl chloride.32 Subsequently, nucleophilic substitution by the glycosyl acceptor forms the desired coupling product.
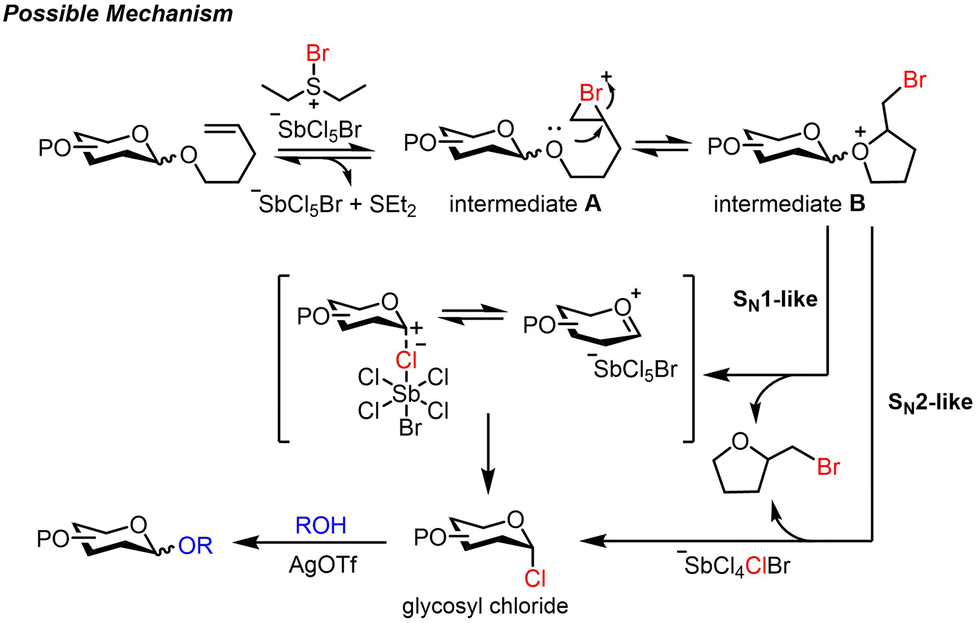 | ||
| Scheme 5 Possible glycosylation mechanism. | ||
Conclusions
In conclusion, the equilibrium that occurs during halonium activation of NPG has impeded the use of NPG in modern carbohydrate synthesis due to the sluggish activation or generation of side products.7–9 This report harnesses the chloride ion transfer during bromination with BDSB and generates the glycosyl chloride intermediate to disrupt such reversible processes. Without the need for an external chloride ion source, chloride ion transfer from the SbCl5Br anion efficiently generates glycosyl chloride, which is efficiently converted to a coupling product. The developed procedure has shown versatility for a wide range of substrates, including mannosyl, galactosyl, and glucosyl donors. Twenty-two substrates were synthesized in moderate to high yields. The β-selective glycosylation was achieved by equipping NPG with a neighboring participating group. A gram-scale synthesis of glycoside was successfully demonstrated to verify the robustness and practicality of this procedure. We believe that BDSB-activated glycosylation of NPG described here will contribute to the future advancement of carbohydrate synthesis.33 Finally, the utilization of chloride ion transfer from a nonnucleophilic anion (SbCl5Br−) during the electrophilic activation described here might be applicable to other synthesis strategies, not limited to carbohydrate synthesis, involving oxidation and trapping of reactive intermediates.Experimental section
All chemicals were purchased from Acros, Merck, Sigma-Aldrich, and TCI. Solvents were purchased from RCI Lab Scan. Reaction monitoring by TLC was performed on silica gel 60 F254 0.2 mm pre-coated aluminum plates purchased from Merck. Chemical spots on TLC were observed by visualization under 254 nm UV light or by staining with a p-anisaldehyde staining solution or iodine (I2). Silica gel 60 (70–230 mesh) from Merck was used for purification by column chromatography. Deuterated solvents for NMR experiments were purchased from Cambridge Isotope Laboratories. Chemical structure characterization was conducted by using a nuclear magnetic resonance (NMR) spectrometer on a Bruker Avance 400 NMR spectrometer operating at 400 MHz for 1H NMR and 101 MHz for 13C{1H} NMR or a JEOL JNM-ECZ500R/S1 spectrometer operating at 500 MHz for 1H NMR, 126 MHz for 13C{1H} NMR, and 471 MHz for 19F NMR. The exact masses of all products were determined by high-resolution mass spectrometry (HRMS) on a Bruker Daltonics micrOTOF-QII-ESI-QqTOF mass spectrometer. Optical rotations were measured on a Jasco P-1010 polarimeter with a cylindrical glass cell (CG3-100) – 3.5 mm × 100 mm path length.General procedure for BDSB-activated glycosylation of NPGs
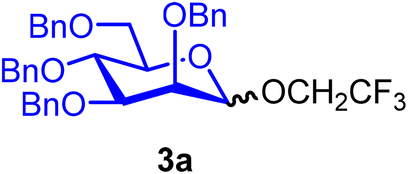
2,2,2-Trifluoroethyl 2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranoside (3a).
The glycosylation without using β-pinene was carried out as described in general procedure A, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (78 mg, 0.13 mmol), BDSB (84 mg, 0.15 mmol), 2,2,2-trifluoroethanol (2) (46 μL, 0.64 mmol), silver trifluoromethanesulfonate (66 mg, 0.26 mmol), and dichloromethane (1.3 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.50). The crude mixture was purified by column chromatography using gradient elution with 10–30% ethyl acetate in hexanes as the eluent. 2,2,2-Trifluoroethyl 2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranoside (3a) was obtained as a yellow syrup (72 mg, 91%, α/β = 2
 :1).
:1).
3aα: [α]24.0D +138.1 (c = 2.4, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.39–7.23 (m, 18H), 7.16–7.14 (m, 2H), 4.93 (d, J = 1.9 Hz, 1H, H-1α), 4.86 (d, J = 10.7 Hz, 1H), 4.76 (d, J = 12.3 Hz, 1H), 4.69 (d, J = 12.3 Hz, 1H), 4.67–4.62 (m, 3H), 4.53 (d, J = 12.1 Hz, 1H), 4.51 (d, J = 11.4 Hz, 1H), 3.99 (t, J = 9.3 Hz, 1H), 3.96–3.86 (m, 2H), 3.86–3.80 (m, 2H), 3.78–3.68 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.4, 138.3, 138.3, 138.1, 128.5, 128.4, 128.1, 127.9, 127.9, 127.8, 127.7, 127.7, 98.7 (C-1α), 79.8, 75.2, 74.6, 74.5, 73.5, 73.1, 72.7, 72.5, 69.1, 64.0 (q, J = 34.9 Hz). 19F NMR (471 MHz, CDCl3) δF −73.96 (t, J = 9.3 Hz). HRMS (ESI-QTOF): m/z calcd for C36H37F3O6Na [M + Na+] 645.24399, found 645.24543. The spectroscopic data of 3aα matched those reported in the literature.34
3aβ: [α]24.0D −13.5 (c = 1.6, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.47–7.42 (m, 2H), 7.36–7.24 (m, 16H), 7.18–7.16 (m, 2H), 4.95 (d, J = 12.4 Hz, 1H), 4.90 (d, J = 10.7 Hz, 1H), 4.84 (d, J = 12.3 Hz, 1H), 4.64–4.43 (m, 6H), 4.26–4.22 (m, 1H), 3.97–3.96 (m, 1H), 3.93–3.85 (m, 2H), 3.79–3.77 (m, 1H), 3.75–3.72 (m, 1H), 3.51–3.49 (m, 1H), 3.47–3.43 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.4, 128.5, 128.5, 128.5, 128.3, 128.2, 127.9, 127.8, 127.7, 127.7, 101.4 (C-1β), 82.0, 76.2, 75.3, 74.6, 74.1, 73.6, 73.1, 72.4, 69.5, 66.0 (q, J = 37.8 Hz). 19F NMR (471 MHz, CDCl3) δF −74.17 (t, J = 11.0 Hz). HRMS (ESI-QTOF): m/z calcd for C36H37F3O6Na [M + Na+] 645.24399, found 645.24536.
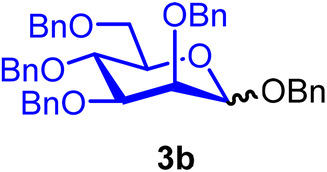
Benzyl 2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranoside (3b).
The glycosylation without using β-pinene was carried out as described in general procedure A, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (140 mg, 0.23 mmol), BDSB (152 mg, 0.28 mmol), benzyl alcohol (5) (35 μL, 0.35 mmol), silver trifluoromethanesulfonate (117 mg, 0.46 mmol), and dichloromethane (2.3 mL). The reaction was monitored by TLC (10% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.20). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Benzyl 2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranoside (3b) was obtained as a colorless syrup (129 mg, 89%, α/β = 1
 :1).
:1).
3bα: [α]24.0D +24.6 (c = 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.38–7.23 (m, 22H), 7.16–7.15 (m, 3H), 4.97 (d, J = 1.8 Hz, 1H, H-1α), 4.88 (d, J = 10.7 Hz, 1H), 4.73–4.68 (m, 4H), 4.61–4.57 (m, 2H), 4.56 (d, J = 12.1 Hz, 1H), 4.51 (d, J = 10.7 Hz, 1H), 4.45 (d, J = 11.9 Hz, 1H), 4.00 (d, J = 9.4 Hz, 1H), 3.95 (dd, J = 9.4, 3.1 Hz, 1H), 3.85–3.77 (m, 3H), 3.75–3.71 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.6, 138.5, 138.5, 138.4, 137.4, 128.5, 128.4, 128.1, 128.0, 127.9, 127.9, 127.8, 127.7, 127.6, 127.6, 97.3 (C-1), 80.3, 75.3, 75.1, 74.7, 73.5, 72.7, 72.3, 72.1, 69.0. HRMS (ESI-QTOF): m/z calcd for C41H42O6Na [M + Na+] 653.28791, found 653.29050. The spectroscopic data of 3bα matched those reported in the literature.35
3bβ: [α]24.0D −19.7 (c = 1.3, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.45 (dd, J = 6.9, 2.6 Hz, 1H), 7.41–7.21 (m, 22H), 7.19–7.17 (m, 2H), 5.00 (d, J = 1.6 Hz, 1H), 4.90–4.84 (m, 2H), 4.73–4.70 (m, 1H), 4.68–4.58 (m, 4H), 4.58–4.48 (m, 2H), 4.44–4.40 (m, 1H), 3.95–3.71 (m, 5H), 3.49–3.45 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.8, 138.3, 137.4, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.9, 127.8, 127.7, 127.7, 127.6, 127.6, 100.4 (C-1β), 82.5, 76.0, 75.2, 75.0, 74.0, 73.9, 73.6, 71.5, 70.9, 69.8. HRMS (ESI-QTOF): m/z calcd for C41H42O6Na [M + Na+] 653.28791, found 653.28921. The spectroscopic data of 3bβ matched those reported in the literature.36
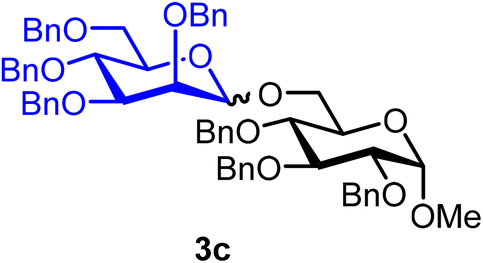
Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranosyl)-α-D-glucopyranoside (3c).
The glycosylation without using β-pinene was carried out as described in general procedure A, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (67 mg, 0.11 mmol), BDSB (66 mg, 0.21 mmol), glycosyl acceptor 6 (28 mg, 0.12 mmol), silver trifluoromethanesulfonate (56 mg, 0.22 mmol), and dichloromethane (1.1 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.50). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranosyl)-α-D-glucopyranoside (3c) was obtained as a colorless syrup (75 mg, 81%, α/β = 4
 :1).
:1).
3cαβ: 1H NMR (500 MHz, CDCl3) δH 7.46–7.11 (m, 35H), 5.01–4.74 (m, 5.8H), 4.74–4.41 (m, 9.8H), 4.16 (d, J = 10.8 Hz, 0.2H), 4.11 (s, 0.2H), 3.98 (dt, J = 11.4, 9.4 Hz, 2H), 3.87–3.77 (m, 3H), 3.73–3.64 (m, 2.8H), 3.62–3.58 (m, 2H), 3.51 (dd, J = 9.6 Hz, 3.2 Hz, 0.2H, H-3′β), 3.48–3.34 (m, 2H), 3.32 (s, C-1β-OCH3, 0.6H), 3.30 (s, C-1α-OCH3, 2.4H). 13C{1H} NMR (126 MHz, CDCl3, α) δC 138.8, 138.5, 138.3, 128.6, 128.5, 128.5, 128.4, 128.4, 128.3, 128.3, 128.1, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7, 127.7, 127.6, 127.6, 127.5, 101.6 (C-1′β), 98.3 (C-1′α), 97.9 (C-1α), 97.9 (C-1α), 82.2, 80.1, 79.7, 75.9, 75.1, 75.0, 74.9, 74.7, 73.4, 72.6, 72.1, 72.0, 69.9, 69.2, 65.9, 55.2 (OCH3α), 55.2 (OCH3β). HRMS (ESI-QTOF): m/z calcd for C62H66O11Na [M + Na+] 1009.45028, found 1009.44908. The spectroscopic data of 3cαβ matched those reported in the literature.37
Isosorbide 2-acetyl 2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranoside (3d).
The glycosylation without using β-pinene was carried out as described in general procedure A, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (115 mg, 0.19 mmol), BDSB (114 mg, 0.21 mmol), isosorbide 2-acetate (14) (28 mg, 0.15 mmol), silver trifluoromethanesulfonate (97 mg, 0.38 mmol), and dichloromethane (1.9 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.30). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Isosorbide 2-acetyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3d) was obtained as a colorless syrup (77 mg, 71%).
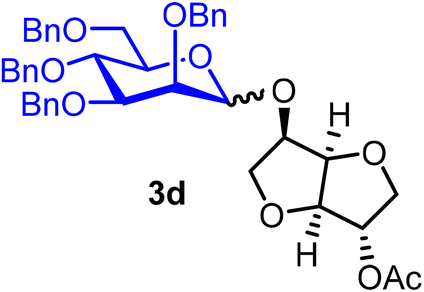
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (167 mg, 0.27 mmol), BDSB (166 mg, 0.30 mmol), isosorbide 2-acetate (14) (47 mg, 0.25 mmol), silver trifluoromethanesulfonate (139 mg, 0.54 mmol), β-pinene (170 μL, 1.1 mmol) and dichloromethane (2.7 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.30). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Isosorbide 2-acetyl 2,3,4,6-tetra-O-benzyl-α/β-D-mannopyranoside (3d) was obtained as a colorless syrup (145 mg, 76%, α/β = 3:1).
3dα: [α]24.0D +65.5 (c = 4.15, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.42–7.23 (m, 18H), 7.16–7.13 (m, 2H), 5.20–5.09 (m, 2H), 4.91–4.85 (m, 1H), 4.79–4.46 (m, 8H), 4.43 (d, J = 4.9 Hz, 1H), 4.36–4.30 (m, 1H), 3.97 (t, J = 9.4, 1H), 3.91–3.77 (m, 5H), 3.77–3.65 (m, 4H), 2.09 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δC 170.2, 138.5, 138.3, 138.3, 138.2, 128.5, 128.4, 128.4, 128.2, 128.1, 127.9, 127.7, 127.7, 97.4 (C-1α), 86.1, 81.0, 79.8, 78.3, 75.7, 75.4, 74.9, 74.3, 73.6, 73.5, 72.4, 72.4, 72.3, 71.6, 69.3, 21.1. HRMS (ESI-QTOF): m/z calcd for C42H46O10Na [M + Na+] 733.29887, found 733.29980.
1-Adamantanyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3e).
The glycosylation without using β-pinene was carried out as described in general procedure A, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (100 mg, 0.16 mmol), BDSB (99 mg, 0.18 mmol), 1-adamantanol (23 mg, 0.15 mmol), silver trifluoromethanesulfonate (82 mg, 0.32 mmol), and dichloromethane (1.6 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.70). The crude mixture was purified by column chromatography using gradient elution with 5–10% ethyl acetate in hexanes as the eluent. 1-Adamantanyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3e) was obtained as a colorless syrup (73 mg, 66%).
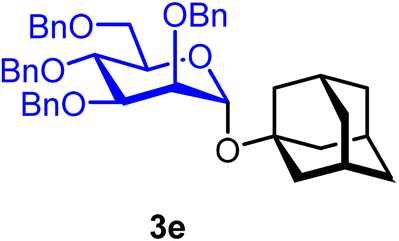
3eα: [α]24.0D +10.9 (c = 3.7, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.40–7.15 (m, 20H), 5.25 (d, J = 2.0 Hz, 1H, H-1α), 4.88 (d, J = 10.6 Hz, 1H), 4.75 (d, J = 12.5 Hz, 1H), 4.70 (d, J = 12.5 Hz, 1H), 4.68 (d, J = 12.2 Hz, 1H), 4.62 (d, J = 11.8 Hz, 2H), 4.52 (d, J = 12.0 Hz, 1H), 4.49 (d, J = 10.6 Hz, 1H), 3.99–3.95 (m, 3H), 3.83–3.78 (m, 1H), 3.72 (d, J = 10.6 Hz, 1H), 3.60–3.58 (m, 1H), 2.13–2.04 (m, 3H), 1.74–1.66 (m, 6H), 1.60–1.57 (m, 6H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.8, 138.7, 138.6, 128.4, 128.3, 128.2, 128.1, 127.8, 127.7, 127.7, 127.6, 127.6, 127.4, 91.0 (C-1α), 80.3, 76.0, 75.4, 75.3, 73.4, 72.5, 72.2, 71.4, 69.6, 42.4, 36.3, 30.6. HRMS (ESI-QTOF): m/z calcd for C44H50O6Na [M + Na+] 697.35051, found 697.35086. The spectroscopic data of 3eα matched those reported in the literature.38
p-Methoxybenzyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-β-D-galactopyranoside (3f).
The glycosylation without using β-pinene was carried out as described in general procedure A, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (49 mg, 0.08 mmol), BDSB (49 mg, 0.09 mmol), a glycosyl acceptor (43 mg, 0.07 mmol), silver trifluoromethanesulfonate (41 mg, 0.16 mmol), triethylamine (11 μL, 0.08 mmol), and dichloromethane (1.0 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde 2 times, Rf = 0.30). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. p-Methoxybenzyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-β-D-galactopyranoside (3f) was obtained as a white solid (56 mg, 70%, α).
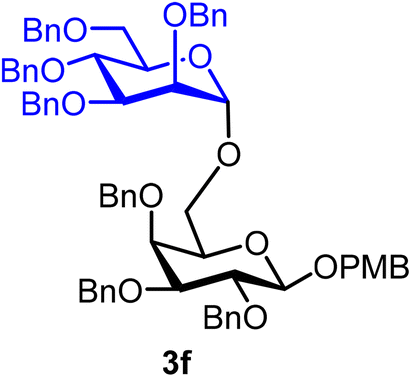
3fα: 1H NMR (500 MHz, CDCl3) δH 7.39–7.12 (m, 37H), 6.78 (d, J = 8.6 Hz, 2H), 4.90 (d, J = 11.2 Hz, 2H), 4.87 (d, J = 10.8 Hz, 1H), 4.80 (d, J = 11.7 Hz, 1H), 4.78–4.64 (m, 8H), 4.61 (d, J = 1.7 Hz, 2H), 4.55–4.47 (m, 5H), 4.37 (d, J = 7.7 Hz, 1H), 4.01 (t, J = 9.2 Hz, 1H), 3.87–3.83 (m, 1H), 3.82 (dd, J = 3.2, 1.4 Hz, 1H), 3.81–3.79 (m, 1H), 3.77 (s, 3H), 3.76–3.72 (m, 1H), 3.72–3.69 (m, 1H), 3.63 (dd, J = 3.1, 1.9 Hz, 1H), 3.51–3.44 (m, 2H), 3.42 (t, J = 6.4 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.8, 138.6, 138.5, 138.4, 129.7, 128.5, 128.5, 128.5, 128.4, 128.3, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.8, 127.8, 127.7, 127.7, 127.6, 102.5 (C-1β), 97.9 (C-1′α), 83.9, 80.0, 79.7, 75.3, 74.9, 74.8, 74.4, 73.5, 73.4, 72.8, 72.6, 72.2, 71.7, 70.6, 68.9, 55.3. HRMS (ESI-QTOF): m/z calcd for C69H72O12Na [M + Na+] 1115.49215, found 1115.48947.
Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-α-D-glucopyranoside (3g).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside (3) (96 mg, 0.16 mmol), BDSB (96 mg, 0.17 mmol), a glycosyl acceptor (59 mg, 0.13 mmol), silver trifluoromethanesulfonate (82 mg, 0.32 mmol), β-pinene (124 μL, 0.79 mmol), and dichloromethane (1.6 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.35). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-α-D-glucopyranoside (3g) was obtained as a yellow syrup (77 mg, 61%, α).
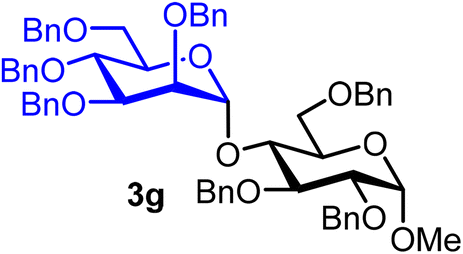
3gα: [α]24.3D +6.8 (c = 1.9, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.38–7.12 (m, 35H), 5.29 (d, J = 2.2 Hz, 1H, H-1′α), 5.08 (d, J = 11.6 Hz, 1H), 4.83 (d, J = 10.9 Hz, 1H), 4.67 (d, J = 12.2 Hz, 1H), 4.61–4.50 (m, 7H), 4.47 (s, 1H), 4.44–4.41 (m, 2H), 4.30 (d, J = 12.1 Hz, 1H), 4.21 (d, J = 12.2 Hz, 1H), 3.97 (t, J = 9.4 Hz, 1H), 3.87–3.85 (m, 1H), 3.85–3.80 (m, 1H), 3.78 (ddd, J = 9.7, 4.9, 1.7 Hz, 1H), 3.74–3.69 (m, 5H), 3.66 (dd, J = 10.7, 4.8 Hz, 1H), 3.56 (dd, J = 10.8, 1.9 Hz, 1H), 3.54–3.52 (m, 1H), 3.39 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.9, 138.7, 138.7, 138.6, 138.5, 138.4, 138.4, 138.3, 138.0, 138.0, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.3, 128.3, 128.2, 128.1, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7, 127.5, 127.3, 126.8, 100.6 (C-1′α), 97.8 (C-1α), 81.7, 80.0, 79.8, 79.6, 77.9, 76.3, 75.5, 75.1, 74.9, 74.7, 73.9, 73.6, 73.5, 73.4, 73.3, 73.0, 72.7, 72.5, 72.4, 72.2, 55.4. HRMS (ESI-QTOF): m/z calcd for C62H66O11Na [M + Na+] 1009.45028, found 1009.44714. The spectroscopic data of 3gα matched those reported in the literature.39
Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-α-D-glucopyranoside (7a).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2,3,4,6-tetra-O-benzoyl-α-D-mannopyranoside (7) (68 mg, 0.10 mmol), BDSB (62 mg, 0.11 mmol), glycosyl acceptor 6 (46 mg, 0.10 mmol), silver trifluoromethanesulfonate (51 mg, 0.20 mmol), β-pinene (79 μL, 0.51 mmol), and dichloromethane (1.0 mL). The reaction was monitored by TLC (25% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.50). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl)-α-D-glucopyranoside (7a) was obtained as a colorless syrup (50 mg, 49%, only α).
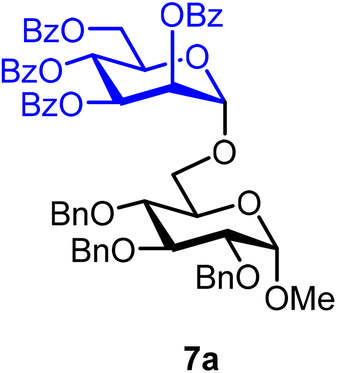
7aα: [α]23.9D −0.1 (c = 2.5, CHCl3); 1H NMR (500 MHz, CDCl3) δH 8.09–8.06 (m, 2H), 8.05–8.02 (m, 2H), 7.91–7.88 (m, 2H), 7.84–7.81 (m, 2H), 7.61–7.27 (m, 27H), 6.06 (t, J = 10.1 Hz, 1H), 5.87 (dd, J = 10.1, 3.3 Hz, 1H), 5.72 (d, J = 1.8 Hz, 1H), 5.15 (d, J = 1.8 Hz, 1H), 5.01 (dd, J = 11.1, 2.8 Hz, 2H), 4.87–4.77 (m, 2H), 4.69 (dd, J = 11.7, 4.4 Hz, 2H), 4.65–4.59 (m, 2H), 4.39 (d, J = 10.2 Hz, 1H), 4.33 (dd, J = 12.1, 4.4 Hz, 1H), 4.03 (t, J = 9.3 Hz, 1H), 3.94 (dd, J = 11.1, 5.2 Hz, 1H), 3.90–3.77 (m, 2H), 3.59–3.52 (m, 2H), 3.45 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δC 166.2, 165.5, 165.4, 165.3, 138.8, 138.3, 138.2, 133.6, 133.3, 133.2, 130.0, 129.9, 129.9, 129.8, 129.4, 129.2, 129.0, 128.7, 128.6, 128.6, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.7, 98.0 (C-1′α), 97.8 (C-1α), 82.2, 80.3, 77.8, 75.8, 75.1, 73.6, 70.3, 70.0, 69.9, 69.0, 66.9, 66.7, 62.8, 55.3. HRMS (ESI-QTOF): m/z calcd for C62H58O15Na [M + Na+] 1065.36734, found 1065.36316. The spectroscopic data of 7aα matched those reported in the literature.40
Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8a).
The glycosylation using β-pinene was carried out as described in general procedure B. At 0 °C, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranoside (8) (35 mg, 0.06 mmol, α/β = 3
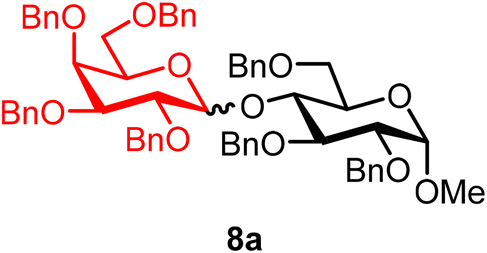 :1), BDSB (35 mg, 0.06 mmol), a glycosyl acceptor (36 mg, 0.08 mmol), silver trifluoromethanesulfonate (30 mg, 0.12 mmol), β-pinene (46 μL, 0.29 mmol), and dichloromethane (0.6 mL). The reaction was monitored by TLC (25% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8a) was obtained as a colorless syrup (41 mg, 71%, α/β = 1:1).
:1), BDSB (35 mg, 0.06 mmol), a glycosyl acceptor (36 mg, 0.08 mmol), silver trifluoromethanesulfonate (30 mg, 0.12 mmol), β-pinene (46 μL, 0.29 mmol), and dichloromethane (0.6 mL). The reaction was monitored by TLC (25% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8a) was obtained as a colorless syrup (41 mg, 71%, α/β = 1:1).
8aαβ: 1H NMR (500 MHz, CDCl3) δH 7.52–7.17 (m, 35H), 5.76 (d, J = 3.8 Hz, 0.5H, α), 5.03 (d, J = 11.6 Hz, 0.5H, β), 4.97 (d, J = 11.5 Hz, 0.5H, α), 4.96 (d, J = 11.4 Hz, 0.5H, β), 4.85–4.73 (m, 2.5H), 4.73–4.64 (m, 3.5H), 4.63–4.51 (m, 4.5H), 4.42 (d, J = 12.3 Hz, 0.5H, α), 4.36 (d, J = 11.8 Hz, 0.5H, β), 4.32 (d, J = 11.9 Hz, 0.5H, β), 4.31–4.23 (m, 2H), 4.06 (dd, J = 9.6, 8.6 Hz, 0.5H, α), 4.03–3.92 (m, 1.5H), 3.91–3.78 (m, 3.5H), 3.74 (dd, J = 9.7, 7.7 Hz, 0.5H, β), 3.70 (dd, J = 10.7, 4.6 Hz, 0.5H, α), 3.64 (dd, J = 10.6, 2.4 Hz, 0.5H, α), 3.60 (m, 0.5H, β), 3.55–3.52 (m, 1H), 3.50–3.40 (m, 2H), 3.37 (s, C-1α-OCH3, 1.5H), 3.36 (s, C-1β-OCH3, 1.5H), 3.35–3.29 (m, 1.5H). 13C{1H} NMR (126 MHz, CDCl3) δC 128.5, 128.4, 128.4, 128.3, 128.3, 128.2, 128.2, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.6, 127.6, 127.5, 126.8, 103.8 (C-1′β), 98.5 (C-1α), 97.8 (C-1′α), 97.5 (C-1α), 83.6, 83.2, 82.1, 80.3, 75.4, 74.6, 74.0, 73.8, 73.5, 72.8, 72.4, 70.4, 68.9, 68.5, 54.5. HRMS (ESI-QTOF): m/z calcd for C62H66O11Na [M + Na+] 1009.45028, found 1009.44701. The spectroscopic data of 8aαβ matched those reported in the literature.41
Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8b).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranoside (8) (71 mg, 0.12 mmol, α/β = 1
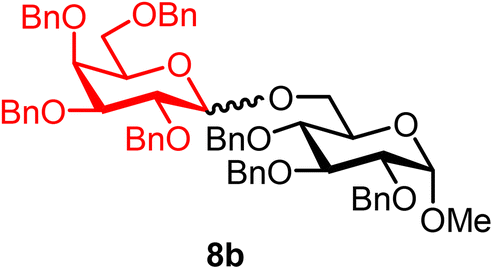 :1), BDSB (71 mg, 0.13 mmol), glycosyl acceptor 6 (42 mg, 0.09 mmol), silver trifluoromethanesulfonate (61 mg, 0.24 mmol), β-pinene (95 μL, 0.60 mmol), and dichloromethane (1.2 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8b) was obtained as a colorless syrup (77 mg, 77%, α/β = 1:1).
:1), BDSB (71 mg, 0.13 mmol), glycosyl acceptor 6 (42 mg, 0.09 mmol), silver trifluoromethanesulfonate (61 mg, 0.24 mmol), β-pinene (95 μL, 0.60 mmol), and dichloromethane (1.2 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8b) was obtained as a colorless syrup (77 mg, 77%, α/β = 1:1).
8bαβ: 1H NMR (500 MHz, CDCl3) δH 7.39–7.13 (m, 35H), 4.98 (d, J = 3.6 Hz, 0.5H), 4.97–4.89 (m, 2.5H), 4.85 (d, J = 10.9, 0.5H, α), 4.80 (d, J = 11.2 Hz, 0.5H, α), 4.78–4.64 (m, 6H), 4.60–4.54 (m, 2.5H), 4.52 (d, J = 3.6 Hz, 0.5H, α), 4.50 (d, J = 11.1 Hz, 0.5H, β), 4.45–4.39 (m, 1.5H), 4.35 (d, J = 11.8 Hz, 0.5H, α), 4.30 (d, J = 7.7 Hz, 0.5H, β), 4.14 (dd, J = 10.9, 2.0 Hz, 0.5H, β), 4.02 (dd, J = 9.3, 3.7 Hz, 0.5H, α), 3.99–3.93 (m, 1.5H), 3.92–3.88 (m, 1.5H), 3.87–3.71 (m, 2.5H), 3.63–3.54 (m, 2H), 3.53–3.44 (m, 3H), 3.41 (dd, J = 9.6, 3.6 Hz, 0.5H, α), 3.29 (s, 3H, C-1α-OCH3, 1.5H), 3.29 (s, 3H, C-1β-OCH3, 1.5H). 13C{1H} NMR (126 MHz, CDCl3) δC 139.0, 138.8, 138.8, 138.6, 138.5, 138.3, 138.0, 128.6, 128.5, 128.4, 128.4, 128.3, 128.3, 128.2, 128.1, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7, 127.6, 127.6, 127.5, 104.3 (C-1′β), 98.0 (C-1′α), 98.0 (C-1α), 98.0 (C-1α), 82.4, 82.2, 82.1, 80.2, 79.9, 79.4, 78.3, 78.2, 78.1, 75.8, 75.8, 75.1, 75.0, 74.9, 73.6, 73.6, 73.5, 73.4, 73.0, 72.9, 72.7, 69.5, 55.3, 55.2. HRMS (ESI-QTOF): m/z calcd for C62H66O11Na [M + Na+] 1009.45028, found 1009.45272. The spectroscopic data of 8bαβ matched those reported in the literature.41
2,2,2-Trifluoroethyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1a).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1) (72 mg, 0.12 mmol, α/β = 1
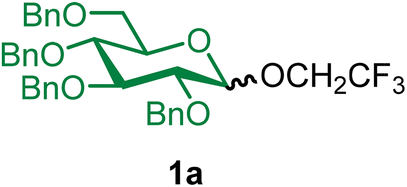 :1), BDSB (72 mg, 0.13 mmol), 2,2,2-trifluoroethanol (2) (42 μL, 0.59 mmol), silver trifluoromethanesulfonate (61 mg, 0.24 mmol), β-pinene (92 μL, 0.59 mmol), and dichloromethane (1.2 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.50). The crude mixture was purified by column chromatography using gradient elution with 0–5% ethyl acetate in hexanes as the eluent. 2,2,2-Trifluoroethyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1a) was obtained as a white solid (74 mg, 100%, α/β = 1:1).
:1), BDSB (72 mg, 0.13 mmol), 2,2,2-trifluoroethanol (2) (42 μL, 0.59 mmol), silver trifluoromethanesulfonate (61 mg, 0.24 mmol), β-pinene (92 μL, 0.59 mmol), and dichloromethane (1.2 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.50). The crude mixture was purified by column chromatography using gradient elution with 0–5% ethyl acetate in hexanes as the eluent. 2,2,2-Trifluoroethyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1a) was obtained as a white solid (74 mg, 100%, α/β = 1:1).
1aαβ: 1H NMR (500 MHz, CDCl3) δH 7.37–7.23 (m, 18H), 7.13 (td, J = 7.5, 2.4 Hz, 2H), 4.98 (d, J = 10.9 Hz, 0.5H, α), 4.93 (d, J = 10.7 Hz, 0.5H, β), 4.92 (d, J = 10.9 Hz, 0.5H, β), 4.85–4.75 (m, 3H), 4.68 (d, J = 10.6 Hz, 0.5H, β), 4.64–4.56 (m, 1.5H), 4.55–4.43 (m, 2.5H), 4.21 (dq, J = 12.3, 8.8 Hz, 0.5H, β), 4.01–3.93 (m, 1H), 3.88 (q, J = 8.7 Hz, 1H, α), 3.78–3.56 (m, 4.5H), 3.49 (t, J = 8.1 Hz, 0.5H, β), 3.45 (m, 0.5H, β). 13C{1H} NMR (126 MHz, CDCl3) δC 138.8, 138.6, 138.1, 138.1, 138.0, 137.8, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 128.0, 127.9, 127.8, 127.8, 127.8, 124.9 (q, J = 278.3 Hz, β), 122.7, (q, J = 278.2 Hz, α), 103.7 (C-1β), 97.9 (C-1α), 84.4, 81.8, 81.7, 79.7, 75.9, 75.9, 75.3, 75.2, 75.1, 75.1, 73.6, 73.5, 71.0, 68.7, 68.2, 66.3, 66.0, 64.9 (β, q, J = 34.5 Hz), 64.6 (α, q, J = 38.0 Hz). 19F NMR (471 MHz, CDCl3) δF −73.4 (t, J = 10.9 Hz, α), −74.0 (t, J = 9.2 Hz, β). HRMS (ESI-QTOF): m/z calcd for C36H37F3O6Na [M + Na+] 645.24399, found 645.24105. The spectroscopic data of 1aαβ matched those reported in the literature.42
Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (1b).
The glycosylation without using β-pinene was carried out as described in general procedure A, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1) (87 mg, 0.14 mmol, α/β = 1
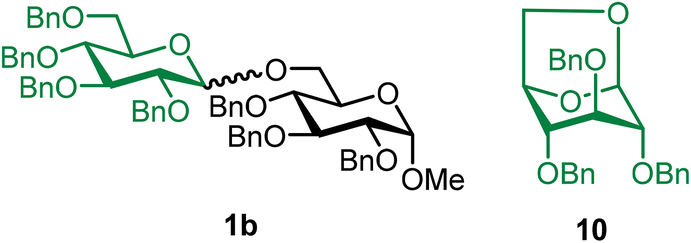 :1), BDSB (86 mg, 0.16 mmol), glycosyl acceptor 6 (57 mg, 0.12 mmol), silver trifluoromethanesulfonate (71 mg, 0.28 mmol), and dichloromethane (1.4 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (1b) was obtained as a white solid (7 mg, 5%, α/β = 1: 1) and 1,6-anhydro-2,3,4-tri-O-benzyl-β-D-glucopyranose (10) was afforded as a side product (101 mg, 84%).
:1), BDSB (86 mg, 0.16 mmol), glycosyl acceptor 6 (57 mg, 0.12 mmol), silver trifluoromethanesulfonate (71 mg, 0.28 mmol), and dichloromethane (1.4 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (1b) was obtained as a white solid (7 mg, 5%, α/β = 1: 1) and 1,6-anhydro-2,3,4-tri-O-benzyl-β-D-glucopyranose (10) was afforded as a side product (101 mg, 84%).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1) (79 mg, 0.13 mmol), BDSB (78 mg, 0.14 mmol), glycosyl acceptor 6 (50 mg, 0.11 mmol), silver trifluoromethanesulfonate (66 mg, 0.26 mmol), β-pinene (100 μL, 0.65 mmol), and dichloromethane (1.3 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (1b) was obtained as a white solid (86 mg, 81%, α/β = 1:1).
1bαβ: 1H NMR (500 MHz, CDCl3) δH 7.36–7.11 (m, 35H), 4.99–4.94 (m, 2H), 4.91 (d, J = 11.1, 1H), 4.84–4.75 (m, 4H), 4.74–4.69 (m, 1H), 4.67–4.62 (m, 3H), 4.60–4.58 (m, 1H), 4.57–4.51 (m, 3H), 4.44 (d, J = 11.0 Hz, 0.5H), 4.41 (d, J = 12.1 Hz, 0.5H), 4.35 (d, J = 7.8 Hz, 0.5H), 4.18 (dd, J = 10.9, 2.0 Hz, 0.5H), 4.02–3.93 (m, 1H), 3.85–3.75 (m, 2H), 3.74–3.58 (m, 4H), 3.57–3.40 (m, 4H), 3.35 (s, C-1α-OCH3, 1.5H), 3.32 (s, C-1β-OCH3, 1.5H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.9, 138.6, 138.6, 138.5, 138.5, 138.4, 138.3, 138.3, 138.2, 138.2, 138.1, 128.6, 128.5, 128.5, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7, 127.7, 127.6, 103.9 (C-1′β), 98.2 (C-1′α), 98.1 (C-1α), 97.4 (C-1α), 84.9, 82.3, 82.2, 82.1, 81.8, 80.2, 80.1, 79.8, 78.1, 78.0, 77.9, 77.7, 75.9, 75.8, 75.6, 75.1, 75.1, 75.0, 73.5, 72.5, 70.4, 70.3, 69.9, 69.1, 68.7, 68.5, 66.1. HRMS (ESI-QTOF): m/z calcd for C62H66O11Na [M + Na+] 1009.45028, found 1009.44712. The spectroscopic data of 1bαβ matched those reported in the literature.43
10: 1H NMR (500 MHz, CDCl3) δH 7.42–7.18 (m, 15H), 5.46 (s, 1H), 4.64–4.51 (m, 5H), 4.47–4.37 (m, 2H), 3.91 (d, J = 7.2 Hz, 1H), 3.68 (dd, J = 7.2, 6.9 Hz, 1H), 3.59 (s, 1H), 3.35–3.34 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δC 138.0, 138.0, 128.6, 128.6, 128.5, 128.5, 128.4, 128.1, 128.0, 127.9, 100.7 (C-1β), 76.7, 76.1, 76.0, 74.5, 72.1, 71.9, 71.3, 65.5. The spectroscopic data of 10 matched those reported in the literature.44
Methyl 2,3,4-tri-O-benzyl-6-O-(3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-α-D-glucopyranoside (9a).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 3,4,6-tri-O-benzyl-β-D-glucopyranoside (9) (85 mg, 0.16 mmol), BDSB (85 mg, 0.16 mmol), glycosyl acceptor 6 (53 mg, 0.11 mmol), silver trifluoromethanesulfonate (82 mg, 0.32 mmol), β-pinene (110 μL, 0.69 mmol), and dichloromethane (1.6 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-α-D-glucopyranoside (9a) was obtained as a yellow syrup (65 mg, 64%, only β).
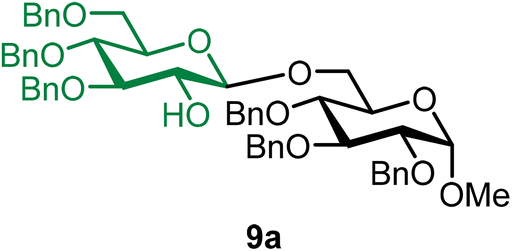
9aβ: [α]23.7D +9.4 (c = 1.5, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.39–7.20 (m, 28H), 7.15 (dd, J = 7.4, 2.2 Hz, 2H), 4.97 (d, J = 10.9 Hz, 1H), 4.89 (t, J = 11.3 Hz, 2H), 4.84–4.75 (m, 4H), 4.67–4.47 (m, 6H), 4.24–4.20 (m, 1H), 4.14 (dd, J = 11.0, 2.3 Hz, 1H), 3.99 (t, J = 9.3 Hz, 1H), 3.83–3.80 (m, 1H), 3.75–3.63 (m, 3H), 3.58–3.44 (m, 6H), 3.37 (s, 3H), 2.48 (s, 1H). 13C{1H} NMR (101 MHz, CDCl3) δC 138.8, 138.3, 138.1, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 128.0, 127.9, 127.9, 127.9, 127.8, 127.7, 127.7, 127.6, 127.6, 127.3, 103.5 (C-1′β), 98.1 (C-1α), 84.5, 82.0, 79.8, 78.1, 77.5, 75.7, 75.4, 75.1, 75.0, 74.5, 73.5, 73.4, 69.9, 69.0, 68.8, 55.3. HRMS (ESI-QTOF): m/z calcd for C55H60O11Na [M + Na+] 919.40333, found 919.39968. The spectroscopic data of 9aβ matched those reported in the literature.45
2,2,2-Trifluoroethyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11a).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11) (67 mg, 0.11 mmol), BDSB (68 mg, 0.12 mmol), 2,2,2-trifluoroethanol (2) (40 μL, 0.36 mmol), silver trifluoromethanesulfonate (58 mg, 0.22 mmol), β-pinene (87 μL, 0.56 mmol), and dichloromethane (1.2 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.50). The crude mixture was purified by column chromatography using gradient elution with 5–15% ethyl acetate in hexanes as the eluent. 2,2,2-Trifluoroethyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11a) was obtained as a colorless syrup (62 mg, 90%, only β).
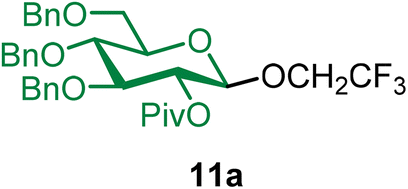
11aβ: [α]24.0D −1.7 (c = 3.1, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.33–7.23 (m, 13H), 7.13 (dd, J = 6.8, 2.3 Hz, 2H), 5.09 (dd, J = 9.5, 7.7 Hz, 1H, H-2), 4.75 (dd, J = 10.9, 6.6 Hz, 2H), 4.69 (d, J = 11.1 Hz, 1H), 4.60 (d, J = 12.2 Hz, 1H), 4.55–4.49 (m, 3H), 4.11–4.00 (m, 1H), 3.99–3.89 (m, 1H), 3.77–3.66 (m, 4H), 3.54–3.49 (m, 1H), 1.18 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δC 177.0, 138.0, 137.9, 137.9, 128.5, 128.5, 128.0, 128.0, 127.9, 127.8, 127.5, 102.3 (C-1β), 84.8, 77.5, 75.5, 75.2, 75.1, 73.6, 72.5, 68.6, 64.9 (q, J = 33.9 Hz), 39.7, 27.1. 19F NMR (471 MHz, CDCl3) δF −73.88 (t, J = 9.4 Hz). HRMS (ESI-QTOF): m/z calcd for C34H39O7F3Na [M + Na+] 639.25456, found 639.25381.
Benzyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11b).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11) (59 mg, 0.10 mmol), BDSB (60 mg, 0.11 mmol), benzyl alcohol (5) (30 μL, 0.30 mmol), silver trifluoromethanesulfonate (51 mg, 0.20 mmol), β-pinene (77 μL, 0.49 mmol), and dichloromethane (1.0 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.50). The crude mixture was purified by column chromatography using gradient elution with 5–15% ethyl acetate in hexanes as the eluent. Benzyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11b) was obtained as a colorless syrup (60 mg, 98%, only β).
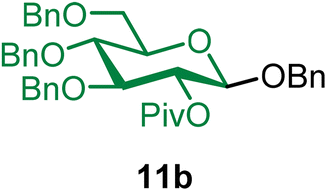
11bβ: [α]23.3D −5.0 (c = 3.0, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.38–7.21 (m, 18H), 7.18–7.10 (m, 2H), 5.15 (dd, J = 9.1, 7.9 Hz, 1H, H-2), 4.88 (d, J = 12.0 Hz, 1H), 4.74 (t, J = 11.0 Hz, 2H), 4.69–4.61 (m, 2H), 4.61–4.51 (m, 3H), 4.45 (d, J = 7.9 Hz, 1H, H-1β), 3.79–3.74 (m, 1H), 3.74–3.65 (m, 3H), 3.54–3.45 (m, 1H), 1.13 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δC 176.9, 138.2, 138.0, 137.2, 128.5, 128.5, 128.4, 128.0, 127.9, 127.9, 127.8, 127.7, 127.5, 99.9 (C-1β), 83.4, 77.9, 75.3, 75.0, 73.6, 73.1, 71.1, 69.3, 39.8, 27.2. HRMS (ESI-QTOF): m/z calcd for C39H44O7Na [M + Na+] 647.29847, found 647.29904. The spectroscopic data of 11bβ matched those reported in the literature.8
Isosorbide 2-acetyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11c).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11) (79 mg, 0.13 mmol), BDSB (79 mg, 0.14 mmol), isosorbide 2-acetate (14) (16 mg, 0.10 mmol), silver trifluoromethanesulfonate (67 mg, 0.26 mmol), β-pinene (100 μL, 0.65 mmol) and dichloromethane (1.3 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.20). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Isosorbide 2-acetyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11c) was obtained as a colorless syrup (45 mg, 78%, only β).
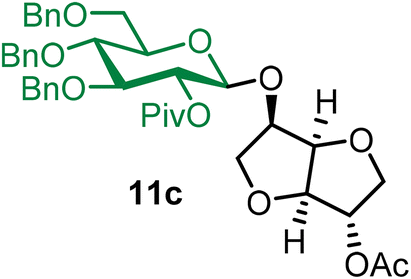
11cβ: [α]23.4D +7.4 (c = 2.3, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.34–7.20 (m, 13H), 7.15 (dd, J = 7.2, 2.3 Hz, 2H), 5.15 (dd, J = 9.2, 8.0 Hz, 1H, H-2), 5.12 (d, J = 4.0 Hz, 1H), 4.78–4.70 (m, 2H), 4.69 (s, 1H), 4.63 (d, J = 4.4 Hz, 1H), 4.60–4.49 (m, 4H), 4.44 (d, J = 4.1 Hz, 1H), 4.32–4.25 (m, 1H), 4.02 (d, J = 3.8 Hz, 1H), 3.96 (d, J = 4.3 Hz, 1H), 3.87–3.82 (m, 1H), 3.74–3.61 (m, 4H), 3.59–3.55 (m, 1H), 3.54–3.50 (m, 1H), 2.04 (s, 3H), 1.17 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δC 176.9, 170.2, 138.2, 138.1, 137.9, 128.5, 128.5, 128.1, 128.0, 127.8, 127.5, 100.6 (C-1β), 85.6, 83.2, 81.5, 79.2, 78.3, 77.7, 75.6, 75.1, 73.8, 73.5, 72.3, 69.6, 69.0, 38.9, 27.2, 21.0. HRMS (ESI-QTOF): m/z calcd for C40H48O11Na [M + Na+] 727.30943, found 727.30970.
Methyl 2,3,4-tri-O-benzyl-6-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-α-D-glucopyranoside (11d).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11) (59 mg, 0.10 mmol), BDSB (59 mg, 0.11 mmol), glycosyl acceptor 6 (35 mg, 0.076 mmol), silver trifluoromethanesulfonate (51 mg, 0.20 mmol), β-pinene (76 μL, 0.49 mmol), and dichloromethane (1.0 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.60). The crude mixture was purified by column chromatography using gradient elution with 5–15% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-α-D-glucopyranoside (11d) was obtained as a colorless syrup (56 mg, 76%, only β).
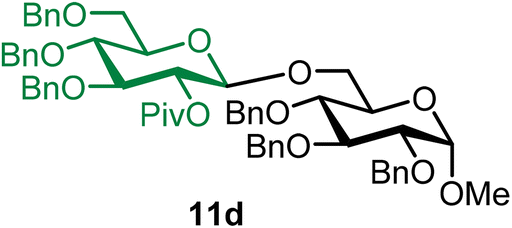
11dβ: [α]24.3D +6.2 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.36–7.20 (m, 28H), 7.16–7.09 (m, 2H), 5.09 (dd, J = 8.8, 7.8 Hz, 1H, H-2′), 4.96 (d, J = 11.0 Hz, 1H), 4.86–4.69 (m, 5H), 4.65 (d, J = 11.6 Hz, 1H), 4.64 (d, J = 12.1 Hz, 1H), 4.60–4.45 (m, 5H), 4.40 (d, J = 7.9 Hz, 1H, H-1′β), 4.02 (dd, J = 10.6, 1.9 Hz, 1H), 3.97 (t, J = 9.2 Hz, 1H), 3.82–3.77 (m, 1H), 3.72–3.62 (m, 4H), 3.57–3.52 (m, 1H), 3.50–3.44 (m, 3H), 3.34 (s, 3H), 1.14 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3) δC 176.6, 138.9, 138.3, 138.2, 138.0, 128.4, 128.4, 128.3, 128.1, 127.9, 127.8, 127.8, 127.8, 127.7, 127.6, 127.6, 127.5, 127.5, 127.3, 101.1 (C-1′β), 97.7 (C-1α), 83.3, 82.0, 80.0, 78.1, 77.8, 75.6, 75.4, 74.9, 74.7, 73.5, 73.2, 72.9, 69.9, 68.9, 68.1, 55.2, 38.8, 27.2. HRMS (ESI-QTOF): m/z calcd for C60H68O12Na [M + Na+] 1003.46085, found 1003.45811. The spectroscopic data of 11dβ matched those reported in the literature.8
Methyl 2,3,6-tri-O-benzoyl-4-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-α-D-galactopyranoside (11e).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (11) (31 mg, 0.05 mmol), BDSB (31 mg, 0.057 mmol), a glycosyl acceptor (27 mg, 0.053 mmol), silver trifluoromethanesulfonate (26 mg, 0.10 mmol), β-pinene (36 μL, 0.25 mmol), and dichloromethane (0.5 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.35). The crude mixture was purified by column chromatography using gradient elution with 5–15% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzoyl-4-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-glucopyranosyl)-α-D-galactopyranoside (11e) was obtained as a colorless syrup (42 mg, 81%, only β).
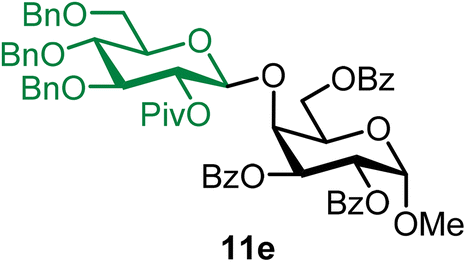
11eβ: [α]24.0D +18.1 (c = 2.1, CHCl3); 1H NMR (500 MHz, CDCl3) δH 8.12–8.09 (m, 2H), 8.08–8.05 (m, 2H), 7.96–7.92 (m, 2H), 7.62–7.20 (m, 22H), 7.10–7.04 (m, 2H), 5.89 (dd, J = 10.7, 3.1 Hz, 1H), 5.39 (dd, J = 10.7, 3.6 Hz, 1H), 5.24 (d, J = 3.6 Hz, 1H), 5.19 (dd, J = 9.5, 7.9 Hz, 1H, H-2′), 4.77 (d, J = 11.2 Hz, 1H), 4.72 (d, J = 7.9 Hz, 1H), 4.69–4.64 (m, 3H), 4.54–4.45 (m, 4H), 4.40 (d, J = 12.1 Hz, 2H), 4.33–4.29 (m, 1H), 3.72 (t, J = 9.4 Hz, 1H), 3.62–3.52 (m, 2H), 3.36 (s, 3H), 3.27 (d, J = 9.8 Hz, 1H), 1.21 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δC 176.8, 166.3, 165.7, 165.6, 138.1, 133.7, 133.3, 133.1, 129.9, 129.8, 128.7, 128.5, 128.5, 128.2, 128.0, 127.8, 127.7, 127.6, 127.2, 100.4 (C-1′β), 97.1 (C-1α), 83.5, 78.0, 75.3, 75.1, 75.0, 73.6, 72.9, 72.9, 70.3, 69.9, 68.9, 68.2, 64.8, 55.4, 38.9, 27.3. HRMS (ESI-QTOF): m/z calcd for C60H62O15Na [M + Na+] 1045.39864, found 1045.39853.
Methyl 2,3,4-tri-O-benzyl-6-O-(2-O-acetyl-3,4,6-tri-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (12a).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-acetyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (12) (78 mg, 0.14 mmol), BDSB (84 mg, 0.15 mmol), glycosyl acceptor 6 (50 mg, 0.110 mmol), silver trifluoromethanesulfonate (72 mg, 0.28 mmol), β-pinene (110 μL, 0.69 mmol), and dichloromethane (1.4 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.40). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2-O-acetyl-3,4,6-tri-O-benzyl-α/β-D-glucopyranosyl)-α-D-glucopyranoside (12a) was obtained as a colorless syrup (81 mg, 80%, α/β = 1
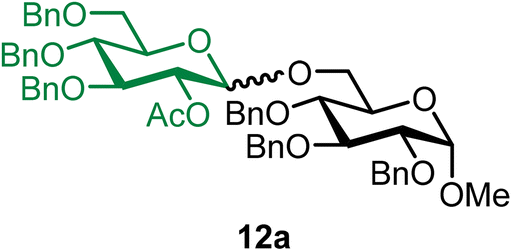 :3).
:3).
12aβ: [α]22.9D +9.5 (c = 1.6, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.37–7.21 (m, 28H), 7.19–7.16 (m, 2H), 5.05 (t, J = 8.4 Hz, 1H, H-2′), 4.96 (d, J = 10.8 Hz, 1H), 4.85–4.73 (m, 5H), 4.66–4.61 (m, 2H), 4.59–4.48 (m, 5H), 4.39 (d, J = 7.9 Hz, 1H, H-1′β), 4.09 (dd, J = 10.7, 1.8 Hz, 1H), 3.96 (t, J = 9.3 Hz, 1H), 3.78–3.59 (m, 6H), 3.55–3.40 (m, 3H), 3.34 (s, 3H), 1.86 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δC 169.3, 138.9, 138.3, 138.2, 138.2, 137.9, 128.6, 128.6, 128.5, 128.5, 128.5, 128.3, 128.1, 128.1, 128.0, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.8, 127.7, 127.6, 101.0 (C-1′β), 98.1 (C-1α), 83.1, 82.1, 79.9, 78.1, 77.8, 75.8, 75.4, 75.2, 75.1, 75.0, 73.5, 73.0, 69.8, 68.9, 68.0, 55.2, 21.0. HRMS (ESI-QTOF): m/z calcd for C57H62O12Na [M + Na+] 961.41390, found 961.41479. The spectroscopic data of 12aβ matched those reported in the literature.43
Methyl 2,3,4-tri-O-benzyl-6-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (13a).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-galactopyranoside (13) (43 mg, 0.07 mmol), BDSB (43 mg, 0.079 mmol), glycosyl acceptor 6 (25 mg, 0.0534 mmol), silver trifluoromethanesulfonate (36 mg, 0.14 mmol), β-pinene (56 μL, 0.35 mmol), and dichloromethane (0.7 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.45). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-α-D-glucopyranoside (13a) was obtained as a colorless syrup (45 mg, 85%, only β).
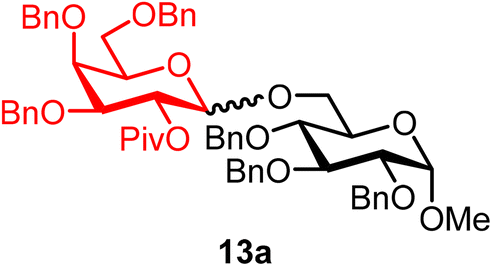
13aβ: [α]23.7D +3.0 (c = 2.2, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.41–7.20 (m, 30H), 5.45 (dd, J = 10.1, 7.9 Hz, 1H, H-2′), 4.99–4.86 (m, 2H), 4.85–4.72 (m, 3H), 4.66–4.58 (m, 2H), 4.59–4.47 (m, 3H), 4.44–4.30 (m, 4H), 4.04–3.90 (m, 3H), 3.78 (m, 1H), 3.65–3.44 (m, 7H), 3.33 (s, 3H), 1.15 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δC 176.8, 139.0, 138.6, 138.4, 138.3, 137.9, 128.5, 128.5, 128.4, 128.3, 128.2, 128.2, 128.0, 127.9, 127.7, 127.6, 127.3, 101.5 (C-1′β), 97.7 (C-1α), 82.2, 81.0, 80.0, 78.2, 75.7, 74.8, 74.5, 73.8, 73.6, 73.3, 72.7, 72.3, 70.8, 69.8, 68.6, 67.5, 57.9, 38.8, 27.3. HRMS (ESI-QTOF): m/z calcd for C60H68O12Na [M + Na+] 1003.46085, found 1003.45913.
Methyl 2,4,6-tri-O-benzyl-3-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (13b).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-galactopyranoside (13) (36 mg, 0.06 mmol), BDSB (36 mg, 0.066 mmol), a glycosyl acceptor (20 mg, 0.043 mmol), silver trifluoromethanesulfonate (31 mg, 0.12 mmol), β-pinene (28 μL, 0.18 mmol), and dichloromethane (0.7 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.30). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,4,6-tri-O-benzyl-3-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (13b) was obtained as a colorless syrup (32 mg, 75%, α/β = 1
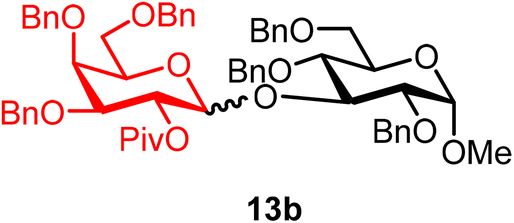 :4).
:4).
13bαβ: 1H NMR (500 MHz, CDCl3) δH 7.42–7.11 (m, 28H), 7.16–6.95 (m, 2H), 5.53 (dd, J = 10.3, 8.0 Hz, 1H, H-2′), 5.08 (d, J = 8.0 Hz, 1H), 5.01 (dd, J = 10.5, 5.1 Hz, 2H), 4.71 (m, 3H), 4.67–4.27 (m, 10H), 4.08–4.04 (m, 1H), 3.77–3.43 (m, 8H), 3.26 (s, C-1β-OCH3, 2.4H), 3.23 (s, C-1α-OCH3, 0.6H), 1.22 (s, C-2β-OPiv, 7.2H), 1.20 (s, C-2α-OPiv, 1.8H). 13C{1H} NMR (126 MHz, CDCl3) δC 177.3, 138.9, 138.2, 138.1, 138.0, 137.9, 129.2, 128.5, 128.5, 128.5, 128.4, 128.3, 128.1, 128.1, 128.0, 127.9, 127.8, 127.7, 127.4, 127.3, 100.4 (C-1′β), 97.5 (C-1α), 81.5, 81.1, 76.5, 75.5, 75.4, 74.9, 73.7, 73.6, 73.2, 73.1, 73.1, 72.0, 69.6, 68.4, 67.8, 55.0, 39.0, 27.4 (β), 27.0 (α). HRMS (ESI-QTOF): m/z calcd for C60H68O12Na [M + Na+] 1003.46085, found 1003.46091.
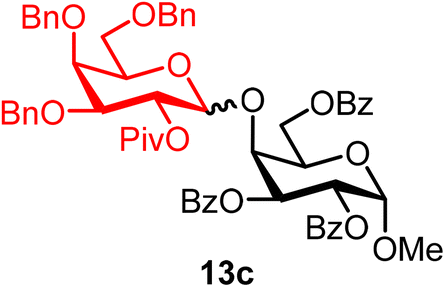
Methyl 2,3,6-tri-O-benzoyl-4-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-α/β-D-galactopyranosyl)-α-D-galactopyranoside (13c).
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-galactopyranoside (13) (101 mg, 0.17 mmol), BDSB (101 mg, 0.18 mmol), a glycosyl acceptor (74 mg, 0.15 mmol), silver trifluoromethanesulfonate (86 mg, 0.34 mmol), β-pinene (131 μL, 0.84 mmol), and dichloromethane (1.7 mL). The reaction was monitored by TLC (20% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.30). The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzoyl-4-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-α/β-D-galactopyranosyl)-α-D-galactopyranoside (13c) was obtained as a colorless syrup for both the α-isomer (42 mg) and the β-isomer (70 mg) (112 mg, 75%, α/β = 1
 :2).
:2).
13cα: [α]23.5D +20.8 (c = 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δH 8.09–8.01 (m, 2H), 7.99–7.92 (m, 2H), 7.92–7.84 (m, 2H), 7.60–7.13 (m, 22H), 7.01 (dd, J = 7.2, 2.4 Hz, 2H), 5.81–5.70 (m, 2H), 5.39 (dd, J = 10.5, 3.7 Hz, 1H, H-2′), 5.31 (d, J = 3.7 Hz, 1H, H-1′α), 5.20 (d, J = 3.4 Hz, 1H), 4.85 (d, J = 11.2 Hz, 1H), 4.80–4.76 (m, 2H), 4.65–4.59 (m, 1H), 4.51 (d, J = 2.8 Hz, 1H), 4.45–4.32 (m, 3H), 4.21 (t, J = 7.0 Hz, 1H), 4.18–4.11 (m, 1H), 3.95–3.82 (m, 3H), 3.42 (s, 3H), 3.18 (t, J = 8.4 Hz, 1H), 2.99 (dd, J = 8.7, 5.8 Hz, 1H), 1.28 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δC 178.3, 166.2, 166.1, 166.0, 138.6, 138.3, 138.1, 133.3, 129.9, 129.8, 129.4, 129.4, 128.6, 128.5, 128.2, 128.1, 127.7, 127.5, 127.5, 97.5 (C-1′α), 96.8 (C-1α), 76.3, 74.8, 74.5, 73.0, 72.8, 71.1, 70.3, 69.7, 69.2, 68.4, 67.9, 55.5, 39.0, 27.4. HRMS (ESI-QTOF): m/z calcd for C60H62O15Na [M + Na+] 1045.39864, found 1045.39675.
13cβ: [α]24.6D +28.4 (c = 2.4, CHCl3); 1H NMR (500 MHz, CDCl3) δH 8.15–8.10 (m, 2H), 8.08–8.05 (m, 2H), 7.97–7.93 (m, 2H), 7.64–7.12 (m, 24H), 5.87 (dd, J = 10.8, 3.0 Hz, 1H), 5.52 (dd, J = 10.2, 7.9 Hz, 1H, H-2′), 5.42 (dd, J = 10.8, 3.7 Hz, 1H), 5.23 (d, J = 3.6 Hz, 1H), 4.88 (d, J = 11.8 Hz, 1H), 4.73 (d, J = 7.9 Hz, 1H, H-1′β), 4.67 (dd, J = 11.9, 4.4 Hz, 1H), 4.62 (d, J = 12.0 Hz, 1H), 4.53–4.48 (m, 3H), 4.47 (t, J = 6.0 Hz, 1H), 4.30 (m, 1H), 4.24 (s, 2H), 3.86 (d, J = 2.6 Hz, 1H), 3.57–3.50 (m, 1H), 3.39 (dd, J = 10.3, 2.8 Hz, 1H), 3.36 (m, 1H), 3.34 (s, 3H), 3.29 (m, 1H), 1.22 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δC 177.0, 166.3, 165.7, 165.6, 138.6, 137.9, 137.8, 133.6, 133.3, 133.0, 130.3, 130.0, 129.9, 129.8, 129.8, 129.5, 129.5, 128.6, 128.5, 128.5, 128.4, 128.3, 128.2, 128.0, 127.9, 127.9, 127.7, 127.6, 127.5, 127.2, 100.7 (C-1′β), 97.1 (C-1α), 81.2, 74.4, 73.7, 73.5, 72.4, 72.3, 72.2, 71.1, 70.5, 69.8, 68.3, 68.1, 64.4, 55.4, 38.9, 27.3. HRMS (ESI-QTOF): m/z calcd for C60H62O15Na [M + Na+] 1045.39864, found 1045.40139.
The glycosylation using β-pinene was carried out as described in general procedure B, using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1) (1.02 g, 1.7 mmol), BDSB (1.01 g, 1.8 mmol), isosorbide 2-acetate (14) (287 mg, 1.5 mmol), silver trifluoromethanesulfonate (775 mg, 3.0 mmol), β-pinene (710 μL, 4.5 mmol), and dichloromethane (15 mL). The reaction was monitored by TLC (30% ethyl acetate in hexanes; p-anisaldehyde, Rf = 0.25). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Isosorbide 2-acetyl 2,3,4,6-tetra-O-benzyl-α/β-D-glucopyranoside (1c) was obtained as a white solid (652 mg) and a colorless syrup (188 mg) for the α-isomer and the β-isomer, respectively (840 mg, 78%, α/β = 3
 :1).
:1).
1cα: [α]23.4D +81.5 (c = 0.4, CHCl3); 1H NMR (500 MHz, CDCl3) δH 7.43–7.21 (m, 18H), 7.13–7.08 (m, 2H), 5.19 (d, J = 15.2 Hz, 1H), 5.16 (d, J = 3.6 Hz, 1H), 5.01 (d, J = 10.8 Hz, 1H), 4.85–4.77 (m, 2H), 4.75 (t, J = 4.8 Hz, 1H), 4.69 (d, J = 11.8 Hz, 1H), 4.60 (d, J = 12.2 Hz, 1H), 4.49 (d, J = 5.5 Hz, 2H), 4.47–4.41 (m, 2H), 4.29 (m, 1H), 4.16 (dd, J = 10.8, 3.8 Hz, 1H), 4.00 (t, J = 9.2 Hz, 1H), 3.93–3.83 (m, 2H), 3.80–3.75 (m, 1H), 3.74–3.67 (m, 2H), 3.67–3.58 (m, 3H), 2.09 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δC 170.2, 138.9, 138.3, 138.1, 137.9, 128.5, 128.5, 128.5, 128.1, 128.1, 128.0, 128.0, 127.9, 127.8, 127.7, 95.7 (C-1α), 86.3, 81.6, 80.3, 79.7, 78.5, 75.8, 75.3, 75.0, 73.8, 73.6, 72.5, 71.0, 70.7, 68.5, 21.1. HRMS (ESI-QTOF): m/z calcd for C42H46O10Na [M + Na+] 733.29887, found 733.29723.
Glycosylation using n-pentenyl galactosyl 8 as a donor. n-Pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranoside (8) (63 mg, 0.10 mmol, α/β = 3
:1), BDSB (63 mg, 0.11 mmol), glycosyl acceptor 6 (37 mg, 0.08 mmol), silver trifluoromethanesulfonate (53 mg, 0.21 mmol), and dichloromethane (1.0 mL) were used. The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8b) was obtained as a colorless syrup (63 mg, 80%, α/β = 1:1).
Glycosylation using n-pentenyl galactosyl 13 as a donor. n-Pentenyl 2-O-pivaloyl-3,4,6-tri-O-benzyl-β-D-galactopyranoside (13) (87 mg, 0.14 mmol), BDSB (86 mg, 0.16 mmol), glycosyl acceptor 6 (59 mg, 0.13 mmol), silver trifluoromethanesulfonate (73 mg, 0.29 mmol), and dichloromethane (1.4 mL) were used. The crude mixture was purified by column chromatography using gradient elution with 5–20% ethyl acetate in hexanes as the eluent. Methyl 2,3,4-tri-O-benzyl-6-O-(2-O-pivaloyl-3,4,6-tri-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (13a) was obtained as a colorless syrup (98 mg, 78%, α/β = 1
:3).
Glycosylation using NIS/TESOTf at 0 °C or −50 °C. In a typical procedure, n-pentenyl glycosides (0.06–0.11 mmol), a glycosyl acceptor (1.2 equivalents), freshly activated 3 Å molecular sieves, and dichloromethane, [c] = 0.1 M, were added to a round bottom flask and stirred under an argon atmosphere for 30 minutes at room temperature. Then the flask was placed in a cooling bath at 0 °C or −50 °C (ice bath or isopropanol/dry ice), followed by the addition of NIS (1.1 equivalents) and TESOTf (0.5 equivalents). The reaction was monitored using thin-layer chromatography until the glycosyl donor was completely consumed. The reaction mixture was quenched by adding saturated aqueous Na2S2O3 (1.5 mL) and saturated aqueous NaHCO3 (1.5 mL), extracted with dichloromethane (3 × 10 mL) and washed with brine (10 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated to dryness using a rotary evaporator. The crude product was purified by column chromatography on silica gel using gradient elution with ethyl acetate and hexanes.
Glycosylation was performed at 0 °C using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranoside (8) (36 mg, 0.06 mmol, α/β = 3:1), NIS (15 mg, 0.065 mmol), glycosyl acceptor 17 (36 mg, 0.08 mmol), TESOTf (7 μL, 0.03 mmol), and dichloromethane (0.6 mL). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8a) was obtained as a colorless syrup (38 mg, 65%, α/β = 1:1).
Glycosylation was performed at −50 °C using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranoside (8) (66 mg, 0.11 mmol, α/β = 3:1), NIS (27 mg, 0.12 mmol), glycosyl acceptor 17 (61 mg, 0.13 mmol), TESOTf (17 μL, 0.05 mmol), and dichloromethane (1.1 mL). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8a) was obtained as a colorless syrup (59 mg, 55%, α/β = 1:1) and the glycosyl donor 8 (9%) was recovered.
Glycosylation using BDSB at −50 °C. The glycosylation using β-pinene was carried out as described in general procedure B, except that it was performed at −50 °C instead of 0 °C using n-pentenyl 2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranoside (8) (53 mg, 0.09 mmol, α/β = 3
:1), BDSB (53 mg, 0.10 mmol), glycosyl acceptor 17 (49 mg, 0.10 mmol), silver trifluoromethanesulfonate (45 mg, 0.17 mmol), β-pinene (68 μL, 0.44 mmol), and dichloromethane (0.9 mL). The crude mixture was purified by column chromatography using gradient elution with 5–30% ethyl acetate in hexanes as the eluent. Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-benzyl-α/β-D-galactopyranosyl)-α-D-glucopyranoside (8a) was obtained as a colorless syrup (59 mg, 69%, α/β = 1:1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported financially by the Thailand Science Research and Innovation Fund Chulalongkorn University (CU_FRB65_bcg (14)_082_23_12 for P. P.). P. P. and S. B. are thankful for the 90th Anniversary of Chulalongkorn University Scholarship (GCUGR1125661023D). S. B. is thankful for the Science Achievement Scholarship of Thailand.References
- T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS.
- Y. Wu, D.-C. Xiong, S.-C. Chen, Y.-S. Wang and X.-S. Ye, Nat. Commun., 2017, 8, 14851 CrossRef CAS PubMed.
- T. T. H. Dinh, P. Tummamunkong, P. Padungros, P. Ponpakdee, L. Boonprakong, W. Saisorn, A. Leelahavanichkul, P. Kueanjinda and P. Ritprajak, Front. Cell. Infect. Microbiol., 2021, 10, 566661 CrossRef PubMed.
- (a) K. Wangpaiboon, P. Padungros, S. Nakapong, T. Charoenwongpaiboon, M. Rejzek, R. A. Field and R. Pichyangkura, Sci. Rep., 2018, 8, 8340 CrossRef PubMed; (b) T. N. Y. Nguyen, P. Padungros, P. Wongsrisupphakul, N. Sa-Ard-Iam, R. Mahanonda, O. Matangkasombut, M. K. Choo and P. Ritprajak, Sci. Rep., 2018, 8, 17123 CrossRef PubMed.
- S. Arayatham, S. Buntasana, P. Padungros and T. Tharasanit, Theriogenology, 2022, 181, 16–23 CrossRef CAS.
- A. V. Demchenko, General Aspects of the Glycosidic Bond Formation, in Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, ed. A. V. Demchenko, Wiley-VCH, Weinheim, 2008, ch. 1 Search PubMed.
- B. Fraser-Reid, P. Konradsson, D. R. Mootoo and U. Udodong, J. Chem. Soc., Chem. Commun., 1988, 823–825 RSC.
- M. Fortin, J. Kaplan, K. Pham, S. Kirk and R. B. Andrade, Org. Lett., 2009, 11, 3594–3597 CrossRef CAS.
- B. Fraser-Reid, J. R. Merritt, A. L. Handlon and C. W. Andrews, Pure Appl. Chem., 1993, 65, 779–786 CrossRef CAS.
- B. Fraser-Reid, Z. Wu, U. E. Udodong and H. Ottosson, J. Org. Chem., 1990, 55, 6068–6070 CrossRef CAS.
- R. Rodebaugh, J. S. Debenham, B. Fraser-Reid and J. P. Snyder, J. Org. Chem., 1999, 64, 1758–1761 CrossRef CAS.
- J. Lu and B. Fraser-Reid, Chem. Commun., 2005, 862–864 RSC.
- A. N. Zakharova, S. I. Awan, F. Nami, C. H. Gotfredsen, R. Madsen and M. H. Clausen, Molecules, 2018, 23, 327 CrossRef.
- S. A. Thadke and S. Hotha, Org. Biomol. Chem., 2014, 12, 9914–9920 RSC.
- P. Pale and G. M. Whitesides, J. Org. Chem., 1991, 56, 4547–4549 CrossRef CAS.
- H. D. Premathilake and A. V. Demchenko, Beilstein J. Org. Chem., 2012, 8, 597–605 CrossRef CAS PubMed.
- Y. Zu, C. Cai, J. Sheng, L. Cheng, Y. Feng, S. Zhang, Q. Zhang and Y. Chai, Org. Lett., 2019, 21, 8270–8274 CrossRef CAS PubMed.
- D. S. Treitler and S. A. Snyder, Encyclopedia of Reagents for Organic Synthesis (EROS) 2015, pp. 1–3, DOI:10.1002/047084289X.rn01740.
- S. A. Snyder and D. S. Treitler, Org. Synth., 2011, 88, 54–69 CrossRef CAS.
- K. D. Ashtekar, N. S. Marzijarani, A. Jaganathan, D. Holmes, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2014, 136, 13355–13362 CrossRef CAS.
- S. A. Snyder and D. S. Treitler, Angew. Chem., Int. Ed., 2009, 48, 7899–7903 CrossRef CAS.
- A. P. Brucks, D. S. Treitler, S.-A. Liu and S. A. Snyder, Synthesis, 2013, 45, 1886–1898 CrossRef CAS.
- T. Chooppawa, P. Janprasert and P. Padungros, Synlett, 2022, 33, 1391–1398 CrossRef CAS.
- L. H. Choudhury, T. Parvin and A. T. Khan, Tetrahedron, 2009, 65, 9513–9526 CrossRef CAS.
- X. Gu, L. Chen, X. Wang, X. Liu, Q. You, W. Xi, L. Gao, G. Chen, Y.-L. Chen, B. Xiong and J. Shen, J. Org. Chem., 2014, 79, 1100–1110 CrossRef CAS.
- P. Padungros, L. Alberch and A. Wei, J. Org. Chem., 2014, 79, 2611–2624 CrossRef CAS.
- N. Venkatasubramanian and V. Thiagarajan, Can. J. Chem., 1969, 47, 694–697 CrossRef CAS.
- G. Chen, Q. Yin, J. Yin, X. Gu, X. Liu, Q. You, Y.-L. Chen, B. Xiong and J. Shen, Org. Biomol. Chem., 2014, 12, 9781–9785 RSC.
- S. A. Geringer, Y. Singh, D. J. Hoard and A. V. Demchenko, Chem. – Eur. J., 2020, 26, 8053–8063 CrossRef CAS.
- D. J. Claffey, M. F. Casey and P. A. Finan, Carbohydr. Res., 2004, 339, 2433–2440 CrossRef CAS PubMed.
- F. T. Schevenels, M. Shen and S. A. Snyder, Org. Lett., 2017, 19, 2–5 CrossRef CAS.
- P. R. Andreana and D. Crich, ACS Cent. Sci., 2021, 7, 1454–1462 CrossRef CAS.
- (a) T. Ratthachag, S. Buntasana, T. Vilaivan and P. Padungros, Org. Biomol. Chem., 2021, 19, 822–836 RSC; (b) P. Padungros, L. Alberch and A. Wei, Org. Lett., 2012, 14, 3380–3383 CrossRef CAS PubMed; (c) P. Padungros, R.-H. Fan, M. D. Casselman, G. Cheng, H. R. Khatri and A. Wei, J. Org. Chem., 2014, 79, 4878–4891 CrossRef CAS.
- S. Chatterjee, S. Moon, F. Hentschel, K. Gilmore and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 11942–11953 CrossRef CAS PubMed.
- S. R. Koppolu, R. Niddana and R. Balamurugan, Org. Biomol. Chem., 2015, 13, 5094–5097 RSC.
- M. S. Tambie and N. K. Jalsa, J. Carbohydr. Chem., 2015, 34, 545–559 CrossRef CAS.
- J. Dinkelaar, A. R. de Jong, R. van Meer, M. Somers, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Org. Chem., 2009, 74, 4982–4991 CrossRef CAS.
- T. A. Boebel and D. Y. Gin, Angew. Chem., Int. Ed., 2003, 42, 5874–5877 CrossRef CAS.
- P. Wen and D. Crich, Org. Lett., 2017, 19, 2402–2405 CrossRef CAS PubMed.
- P. Wen, C. J. Simmons, Z.-x. Ma, S. A. Blaszczyk, P. G. Balzer, W. Ye, X. Duan, H.-Y. Wang, D. Yin, C. M. Stevens and W. Tang, Org. Lett., 2020, 22, 1495–1498 CrossRef CAS PubMed.
- M.-H. Zhuo, D. J. Wilbur, E. E. Kwan and C. S. Bennett, J. Am. Chem. Soc., 2019, 141, 16743–16754 CrossRef CAS PubMed.
- J. L. Xue, S. Cecioni, L. He, S. Vidal and J.-P. Praly, Carbohydr. Res., 2009, 344, 1646–1653 CrossRef CAS PubMed.
- H. He and X. Zhu, Org. Lett., 2014, 16, 3102–3105 CrossRef CAS.
- D. Hager, C. Paulitz, J. Tiebes, P. Mayer and D. Trauner, J. Org. Chem., 2013, 78, 10784–10801 CrossRef CAS.
- S. Izumi, Y. Kobayashi and Y. Takemoto, Org. Lett., 2019, 21, 665–670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experiment procedures, characterization data and NMR spectra of all compounds. See DOI: https://doi.org/10.1039/d3ob01618h |
| This journal is © The Royal Society of Chemistry 2024 |