
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D self-assembled polar vs. non-polar NiO nanoparticles nanoengineered from turbostratic Ni3(OH)4(NO3)2 and ordered β-Ni(OH)2 intermediates†
Biljana
Pejova
*a,
Arej
Eid
bc,
Leonardo
Lari
bd,
Ahmad
Althumali
be,
Lidija
Šiller
f,
Adam
Kerrigan
 d,
Ljupcho
Pejov
a and
Vlado K.
Lazarov
*bd
d,
Ljupcho
Pejov
a and
Vlado K.
Lazarov
*bd
aInstitute of Chemistry, Faculty of Natural Sciences and Mathematics, SS. Cyril and Methodius University, POB 162, 1000 Skopje, Macedonia. E-mail: biljana@pmf.ukim.mk
bSchool of Physics Engineering and Technology, University of York, York, UK. E-mail: vlado.lazarov@york.ac.uk
cUniversity of Tabuk, Tabuk, Saudi Arabia
dThe York-JEOL Nanocentre, University of York, York, UK
eDepartment of Physics, Faculty of Science, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia
fSchool of Engineering, Newcastle University, Newcastle upon Tyne NE1 7RU, UK
First published on 20th September 2024
Abstract
A surfactant-free ammonia and carbamide precursor-modulated engineering of self-assembled flower-like 3D NiO nanostructures based on ordered β-Ni(OH)2 and turbostratic Ni3(OH)4(NO3)2 nanoplate-structured intermediates is reported. By employing complementary structural and spectroscopic techniques, fundamental insights into structural and chemical transformations from intermediates to NiO nanoparticles (NPs) are provided. FTIR, Raman and DSC analyses show that the transformation of intermediates to NiO NPs involves subsequent loss of NO3− and OH− species through a double-step phase transformation at 306 and 326 °C corresponding to the loss of free interlayer ions and H2O species, respectively, followed by the loss of chemically bonded OH− and NO3− ions. Transformation to NiO NPs via the ammonia route proceeds as single-phase transition, accompanied with a loss of OH− species at 298 °C. The full transformation to NiO NPs of both intermediates is achieved at 350 °C through annealing in the air atmosphere. Ammonia-derived NPs maintain nanoflower morphology by self-assembling into nanoplates, which is enabled by H2O-mediated adhesion on the NiO NPs’ {100} neutral surfaces. Structural transformations of turbostratic Ni3(OH)4(NO3)2 nanoplates result in the formation of NiO NPs dominantly shaped by inert polar OH-terminated (111) atomic planes, leading to the loss of the initial self-assembled 3D structure. DFT calculations support these observations, confirming that H2O adsorbs dissociatively on polar {111} surfaces, while only physisorption is energetically feasible on {100} surfaces. NiO NPs obtained via two different routes have overall different properties: carbamide-derived NPs are 3 times larger (15.5 vs. 5.4 nm), possess a larger band gap (3.6 vs. 3.2 eV) and are more Ni deficient. The intensity ratio of surface optical (SO) modes to transversal and longitudinal optical modes is ∼40 times higher in the NiO NPs obtained from β-Ni(OH)2 compared to Ni3(OH)4(NO3)2-derived NPs. The SO phonon lifetime is an order of magnitude shorter in NiO obtained from β-Ni(OH)2, reflecting a much smaller NP size. The choice of a precursor defines the size, morphology, crystallographic surface orientations and band gap of the NiO NPs, with Ni deficiency providing pathways for utilizing them as p-type materials, allowing for the precise nanoengineering of polar and neutral surface-dominated NiO NPs, which is of exceptional importance for use in catalysis.
1. Introduction
Transition metal oxide-based nanostructures1–5 have emerged as a crucial class of materials with large compositional and structural diversity and a huge potential for device applications. Compared to their bulk counterparts, these nanostructures are characterized by unique optical, electrochemical, mechanical, thermal and magnetic properties, which are closely related to their increased surface area as their size decreases. Transition metal oxide-based nanostructures have been widely used in various fields, including energy storage, solar cells and heterogeneous catalysis. Besides doping, the properties of transition metal oxide NPs can be tuned by manipulating their size, shape and surface chemistry, which is crucial for new potential applications. In addition, metal–oxide-based nanostructures are widely used as model systems for exploring a large number of phenomena, such as structural changes upon interaction with different gases, i.e. oxidation and reduction, under strictly controlled experimental conditions and variable temperature and gas pressure on an atomic scale, possible with the development of environmental STEM/TEM techniques6–8 and other in situ techniques such as ambient XPS.Among transition metal oxide-based nanostructures, NiO NPs have attracted significant interest due to their excellent chemical stability, durability, low toxicity, and low-cost processing as well as desirable magnetic, electrical, optical and catalytic properties required for many applications in electrochromic devices, solar cells, supercapacitors, smart windows, UV photo-detectors, alkaline batteries, ceramics, and chemical sensors.9–25 NiO NPs have also been used in wastewater dye degradation through photocatalysis,26,27 adsorption of toxic environmental pollutants, dyes and heavy metals, as well as in biomedicines due to their anti-inflammatory and antibacterial properties.28,29 Many studies have successfully tested the anti-cancerous and antiparasitic properties of NiO NPs,30–32 as well as their antioxidant nature.33 Additionally, it has been shown that NiO NPs can facilitate seed germination and increase the seedling growth rate, which implies their potential application in agriculture.30
Nickel(II) oxide is naturally found in its mineralogical form, known as bunsenite. It crystallizes in the cubic system and is characterized by a rock-salt structure with Ni2+ and O2− ions, which occupy the octahedral sites of the crystal lattice with the characteristic fcc-stacking structure,34 and a lattice parameter a of 4.168 Å. With regard to the electronic band structure, considering the experimental and theoretical results, the Ni 3d states mainly participate in the conduction band, while the valence band is mainly formed by O 2p states.35,36 The band gap of NiO is considerably wide, in the range of 3.6–4.3 eV. At room temperature, NiO prepared by both chemical and physical methods is usually non-stoichiometric NiOx, which leads to improved electronic conductivity compared to stoichiometric NiO.34,37 According to published results from both experimental and theoretical studies,34,37,38 non-stoichiometric NiOx is most likely to be nickel-deficient because Ni vacancies are readily formed in the structure under oxygen-rich conditions. In addition, the results from defect calculations have shown that acceptor Ni vacancies have two transition levels in the band gap with low formation energies under oxygen-rich conditions, while the donor oxygen vacancy defect has much higher formation energies and hence is less likely to form.37,38 Therefore, the acceptor levels lead to the p-type semiconductor behavior of the non-stoichiometric NiOx. This property includes NiOx in the group of a few p-type semiconducting oxides in addition to SnO and Cu2O, thus making it very important for numerous emerging technologies. Compared to SnO and Cu2O, which are metastable at room temperature, NiOx has attracted increased scientific interest, particularly in the development of devices based on its unique electrical and magnetic properties.24 However, the development of nickel oxide-based optical and optoelectronic devices is in its initial state. Optoelectronic applications, including solar cells, have opened up a promising field of research based on this p-type material.
In the literature, numerous reports are related to the preparation of nickel oxide NPs using techniques based on the bottom-up, top-down and hybrid approaches.39–49 In addition, NiO NPs have been synthesized by green routes, which include the use of plant extracts, bacteria and fungi as reducing agents.50–55
In this work, we develop synthesis methods for nickel oxide nanostructures using two solution-based routes without the use of surfactants, which provide advantages in terms of composition (stoichiometric vs. nonstoichiometric, etc.), size and morphology control and scalability. We show that the choice of the precursor and hence intermediates plays a crucial role in NiO nanoparticle (NP) assembly in 3D nanostructures, their atomic surface structure, stoichiometry, and physical-optical properties. We also elucidate the structural/chemical pathways from the intermediates to NiO NP formation by utilizing several spectroscopic and structural techniques.
2. Experimental section
2.1. Synthesis of nickel oxide and hydroxide-based NPs
NiO and Ni hydroxide-based NPs were synthesized using two routes based on the colloidal approach. Two reaction systems were designed with the goal of including the lowest possible number of components to obtain a pure phase of the desired product while avoiding the incorporation of impurities. We used Ni(NO3)2·6H2O as Ni2+ ion precursors, while carbamide (NH2CONH2) and ammonia (NH3) played a role as hydroxide ion precursors. The developed routes do not include the use of surfactant and use two simple inorganic and organic precursors. The formation of NiO NPs was a result of the thermal treatment of the obtained hydroxide-based intermediates.In the case of the carbamide-based reaction system, the optimal initial concentrations of relevant components Ni(NO3)2 and NH2CONH2 have been determined to be 0.5 mol L−1 and 0.25 mol L−1, respectively. The chemical synthesis occurred at ∼90 °C. The initial chemical composition of the reaction system when ammonia was used as a hydroxide precursor can be expressed as 0.03 mol dm−3 [Ni(NH3)4]2+, and the synthesis was carried out at ∼50 °C. The aqueous solution of octahedral [Ni(NH3)6]2+ was obtained by dissolving the cation precursor in water and adding a stoichiometry required volume of a concentrated solution of ammonia, i.e. with w(NH3) = 25%.
The described procedures produced two intermediates, which were annealed at ∼350 °C for 4 hours, resulting in a phase transformation to nickel oxide NiO.
2.2. Experimental methods
Structural and chemical properties of the synthesized materials have been investigated by X-Ray Diffraction (XRD), Transmission Electron Microscopy (TEM), Differential Scanning Calorimetry (DSC), Fourier-Transform Infrared (FTIR), Raman and UV-VIS Spectroscopy, while the morphological properties were studied by Scanning Electron Microscopy (SEM).The XRD patterns were collected using the Rigaku Ultima IV X-ray diffractometer using CuKα radiation in the 2θ/θ scanning mode and step-scan of Δ(2θ) = 0.02°.
SEM images and EDX spectra were collected using a JEOL 7800F Prime SEM. TEM-selected area electron diffraction (SAED) patterns and high-resolution TEM (HRTEM) images were obtained using JEOL 2100+ TEM and aberration-corrected JEOL-NeoARM STEM/TEM. The diffraction patterns were simulated using JEMS software, and the TEM-SAD patterns were calibrated using Au nanoparticles as a reference. The atomic interplanar spacings were measured from selected areas of HRTEM images using the fast-Fourier transform (FFT).
The IR measurements were carried out using a PerkinElmer System 2000 FTIR interferometer in the spectral range of 4000–500 cm−1 with a resolution of 2.0 cm−1. All FTIR spectra were recorded in the attenuated total reflection (ATR) mode using a Graseby Specac zinc selenide ATR accessory to avoid any matrix effects.
The Raman spectra were measured using a LabRam300 spectrometer by Horiba Jobin–Yvon, with the green 532 nm Nd:Yag laser line and a backscattering mode.
Optical absorption spectra in the UV-VIS spectral region were measured using a Cary 50 spectrophotometer.
Differential scanning calorimetry (DSC) measurements were carried out with a heat-flux Netzsch DSC 204 F1 Phoenix instrument. The samples were placed into aluminium pans with perforated lids throughout the measurements. DSC analysis was performed in the temperature range from room temperature up to 300 °C in a dynamic N2 atmosphere with a volume flow of 30 mL min−1. In all measurements, the heating rate was kept constant at 10 K min−1.
The X-ray photoemission spectra (XPS) analysis was obtained by applying a Thermo Scientific K-alpha X-ray Photoelectron Spectrometer™ (ThermoScientific, UK). A monochromatic Al Kα X-ray source (1486.7 eV) was used to determine the oxidation states of the elements near the surfaces of all the films. The XPS data were charge-corrected to the adventitious C 1s spectra at a binding energy of 284.8 eV.56,57 The Ni 2p3/2 and O 1s XPS spectra were analyzed using CasaXPS software (version 2.3.25).58 The spectra were fitted with a normal Shirley electron background and a Gaussian (70%)–Lorentzian (30%) peak shape, referred to as GL(30) in CasaXPS software.56,57
We based our analysis of the XPS data on the conceptual model proposed by Gupta and Sen (GS)59 and the other literature data concerning the relevant nickel ion and oxygen species.56–58,60–63 The GS model59 is based on the consideration of both multiplets and the so-called shake-up-related satellite bands.
2.3. Computational details
The VASP code for running DFT calculations was used,64 along with the PAW pseudopotentials supplied by the package. An effective U-value of 5.3 eV was applied to the d-orbitals of Ni obtained from the literature using the same pseudopotentials.65 This value was optimised against multiple parameters, such as magnetic ordering and phase stability. A plane wave cut-off of 450 eV was used along with a k-point spacing of 0.05 2π Å−1.Surface slab models were created for the (100) surface and three terminations of the (111) surface, OH terminated, octopolar reconstructed and faceted. Each slab was separated from its periodic repeat by a vacuum gap of 15 Å. Doing this exposes two surfaces to the vacuum, the top and bottom of the slab, and consideration was taken to ensure that the exposed surfaces were equivalent. The region between these exposed surfaces contained enough layers such that the surface energy of the structure converged to a tolerance of 1 × 10–3 eV Å−2. For the (100) surface, a (4 × 4) supercell of the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m unit cell was used for coverage comparable to the faceted (111) surface, which is a p(4 × 4) reconstruction. For the octopolar and OH-terminated structures, a p(2 × 2) sized supercell was used.
m unit cell was used for coverage comparable to the faceted (111) surface, which is a p(4 × 4) reconstruction. For the octopolar and OH-terminated structures, a p(2 × 2) sized supercell was used.
For the adsorption of H2O molecules and their dissociative components, a molecule was adsorbed onto each exposed surface. Rotational symmetry around the center of the surface slab was maintained to ensure that no dipole was formed in the cell. The molecules were geometrically optimized to a force tolerance of 1 × 10–2 eV Å−1.
3. Results and discussion
3.1. Structure and morphology of synthesized nanostructures
In this section, we present the structural and morphological results of β-Ni(OH)2 and Ni3(OH)4(NO3)2 intermediates following up with NiO after the thermal treatment of the intermediates.Fig. 1 shows clearly that the chemistry of precursor conversion goes through different intermediates. When ammonia is used as a precursor, the diffraction peaks (Fig. 1a) correspond to hexagonal β-Ni(OH)2. When carbamide is used as an anionic precursor, the diffraction peaks (Fig. 1b) can be assigned to hexagonal nickel(II) tetrahydroxide nitrate, Ni3(OH)4(NO3)2, a phase categorized as a hydroxide-rich salt of nickel with a layered structure of close-packed adjacent layers. The significant broadening of the XRD peaks (Fig. 1b), especially at 2θ values higher than 30°, indicates the presence of additional chemical species, e.g. H2O, NO3− within atomic planes, which leads to an increase in the interlayer spacing and ultimately affects layer alignment, resulting in randomly oriented layers, i.e. a turbostratic structure,66 as discussed and shown in the next paragraph by electron microscopy imaging.
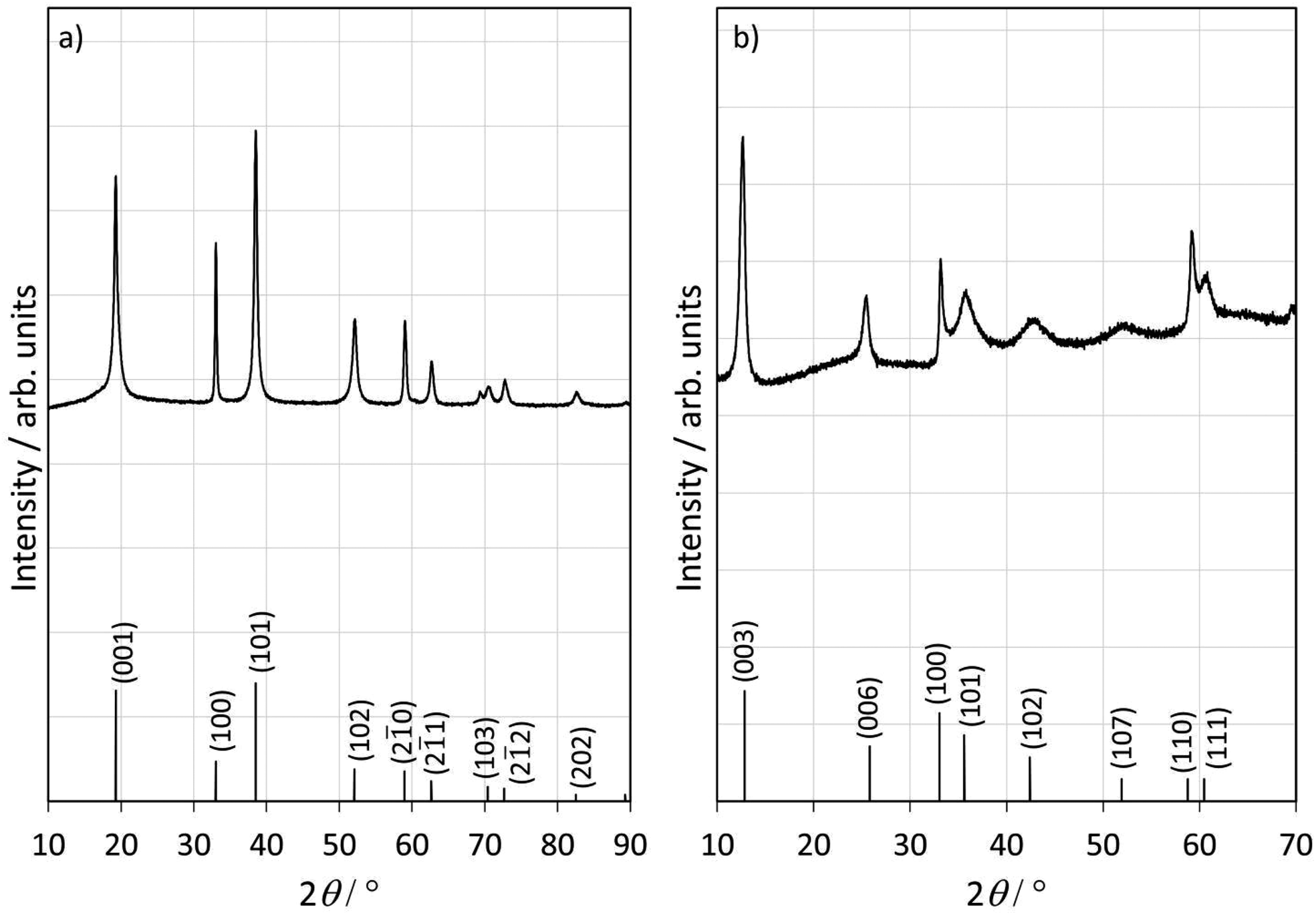 | ||
| Fig. 1 XRD patterns of intermediates in the case of ammonia (a) and carbamide (b) as hydroxide precursors and the corresponding bulk reference XRD of β-Ni(OH)2 and Ni3(OH)4(NO3)2. Reference diffraction peaks are taken from ref. 66 and 67. | ||
The SEM images presented in Fig. 2a and d show that both intermediates have flower-like morphology, consisting of β-Ni(OH)2 and Ni3(OH)4(NO3)2 nanoplates as illustrated by the TEM images in Fig. 2b, c, e and f.
 | ||
| Fig. 2 SEM and TEM images of the intermediates. Flower-like self-assembly of (a) β-Ni(OH)2 and (d) Ni3(OH)4(NO3)2 obtained through SEM, and TEM images of β-Ni(OH)2 (b and c) and Ni3(OH)4(NO3)2 (e and f) at different magnifications, showing their overall morphology and single nanoplates. | ||
The electron diffraction taken from a single flower-like nanostructure, Fig. 3a, shows diffraction rings corresponding to β-Ni(OH)2, which agrees with XRD results,67 while the (001) atomic planes of the β-Ni(OH)2 nanoplates imaged by HRTEM are shown in Fig. 3b and c. The digital diffractograms (i.e. sharpness of diffraction spots in Fig. 3d) from the HRTEM images illustrate the well-ordered atomic structure of this intermediate. Similarly, electron diffraction from a single nano-flower particle, Fig. 4a, of carbamide-derived intermediate confirms the turbostratic structure of Ni3(OH)4(NO3)2, where broadened diffraction rings are due to the variation in atomic interplanar distances, as implied by XRD results.
 | ||
| Fig. 3 Electron diffraction of a single flower-like nanostructure (a) with diffraction rings corresponding to β-Ni(OH)2 (the inset shows that they are from where diffraction was taken). (b and c) HRTEM images of β-Ni(OH)2 nanoplates with (001) planes outlined, and (d) a digital diffractogram from (b), outlining {001} plane ‘reflections’. | ||
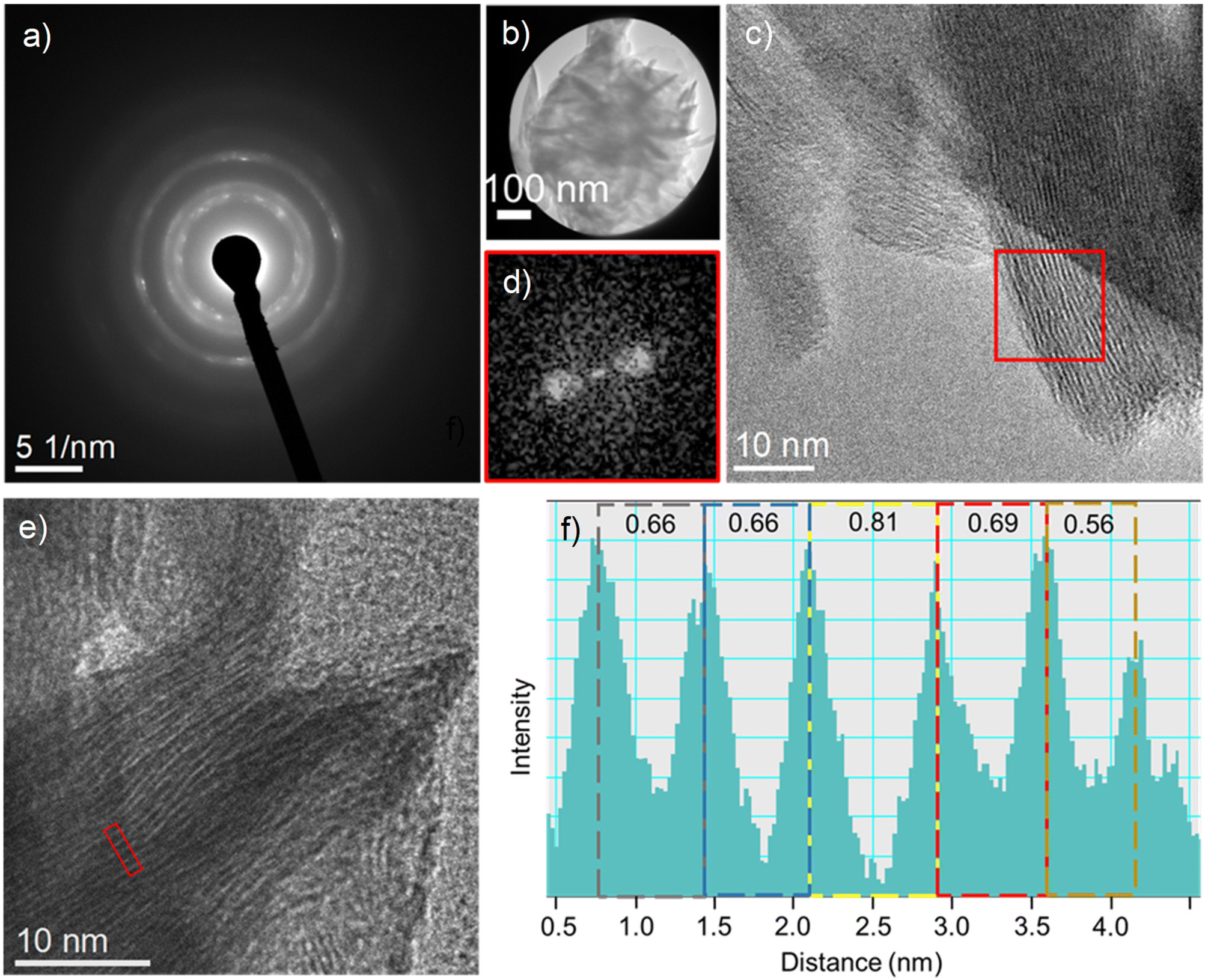 | ||
| Fig. 4 (a) Electron diffraction of a single flower-like nanostructure (b), diffraction rings corresponding to Ni3(OH)4(NO3)2, (c) an HRTEM image of Ni3(OH)4(NO3)2 nanoplates with digital diffractogram from highlighted regions shown in (d) showing the variation in atomic spacing planes highlighted by the real space intensity profile of (003) atomic spacing (f) taken from a region of HRTEM image of the nanoplate in (e). | ||
A direct visualization of Ni3(OH)4(NO3)2 atomic planes obtained by HRTEM is shown in Fig. 4c and e. Fig. 4d shows an FFT (digital diffractogram) from the outlined area in Fig. 4c, where the diffuse spot-shape reflects the variation in atomic plane distances, characteristic of the turbostratic arrangement of atomic planes, due to the presence of free-like NO3− and H2O. The non-homogeneous presence of these species induces a local change in the atomic distances between the layers, as well as their corresponding orientation (Fig. 4c and e), where atomic plane bending and variation of the interplanar distances on the nanoscale are observed, as shown in Fig. 4f. These results clearly show the randomness of the turbostratic nature of the Ni3(OH)4(NO3)2 nanoplates. The presence of ‘free’-like species of NO3−, H2O and their role in the transformation of the intermediates to NiO is confirmed by spectroscopic measurements that are discussed hereinafter.
3.2. Structural and morphological changes in the intermediates of NiO NPs during thermal treatment
Thermal treatment at ∼350 °C of the intermediates results in phase structural changes to NiO NPs, as discussed and shown in detail hereinafter. XRD peak (Fig. 5) analysis of annealed intermediates in the air atmosphere for four hours shows that both intermediates transform to rock salt fcc-NiO. The broadening of the XRD peaks indicates a size reduction in the NiO. The peaks broadening in the case of NiO synthesized from the carbamide precursor are smaller, reflecting the size difference between the NiO nanostructures, as directly shown by the SEM and TEM imaging described hereinafter. We note that the average sizes of NiO NPs obtained by ammonia and carbamide routes (for simplicity, we refer to them as NiO-a and NiO-c, respectively) are different. Their size follows the log–normal distribution, as shown in Fig. SI-1a and b.† The average NiO-a and NiO-c NP sizes are 5.41 ± 2.19 nm and 15.47 ± 5.16 nm, with narrow size dispersion σ/μ = 40% and 33%, respectively (Fig. SI-1a and b†). Therefore, using ammonia as an ion precursor leads to the formation of smaller NiO-a NPs. | ||
| Fig. 5 The left panel represents the XRD patterns of reference (a) cubic NiO71 and (b) non-stoichiometric NiOx,70,71 and NiO obtained using ammonia (c) and carbamide (d) precursors. The right panel shows the (200) reference peaks in comparison to the experimental peaks of NiO obtained by both precursors. | ||
The final product of both intermediates is NiO, and the differences in their peak intensity ratios, e.g. (111) vs. (200) peaks, indicate subtle differences in their structure, which can be associated with a variation of NiO stoichiometry.68,69 Comparison of the intensities of the relative peaks of annealed products with reference XRD files for stoichiometric and non-stoichiometric NiO (Fig. 5) indicates that the NiO products are non-stoichiometric, i.e. Ni deficient in both cases. The analysis of (200) peak positions shows that they shift towards smaller Bragg 2θ angles with respect to the reference peaks, i.e. 43.254° and 43.249°, for carbamide and ammonia precursors, respectively. This is an additional indicator of the non-stoichiometry of the synthesized NiO.70,71
The NiO-a obtained by annealing of nanoplate β-Ni(OH)2 retains the flower-like morphology, as observed by SEM and TEM (Fig. 6a and b). The selected area electron diffraction from the obtained NPs, as expected, shows their fcc-cubic structure (Fig. SI-2†). Even though the nanoplates appear to have smooth surfaces, a closer look by HRTEM shows that they are composed of nanoparticles (Fig. 6c). We also note that the NPs are mainly terminated with a neutral {001} type of surface for additional images outlining the {001} type of NiO-a NP faceting (see Fig. SI-3†).
 | ||
| Fig. 6 (a) SEM image of NiO obtained with thermal treatment of β-Ni(OH)2 flower-like nanostructure, (b) TEM image of annealed NiO nanoflower particle, and (c) HRTEM images of the red marked region in (b) outlining the NiO NPs with (111) atomic planes of NiO from a single particle shown in the inset. | ||
However, NiO-c NPs, obtained by annealing of the Ni3(OH)4(NO3)2 intermediate, are dominantly rod shaped (Fig. 7a and b). Digital diffractogram analysis has shown that NiO-c NP long side is terminated by {111} polar NiO facets. Hence, these nanorods are an example where NiO NPs are dominantly facetted with polar (111) surfaces, as further illustrated by the number of HRTEM images taken from NPs in different zone axes (Fig. SI-3†).
 | ||
| Fig. 7 (a) SEM image of NiO NPs obtained with thermal treatment of Ni3(OH)4(NO3)2, (b) TEM image of NiO NPs, and (c) HRTEM images of selected NiO NPs, outlined in (b) with the red square. The inset in (c) shows the (111) atomic planes of NiO. | ||
Atomic scale imaging by HRTEM of NiO-a and NiO-c NPs shows clearly their atomic ordering (Fig. 6c and 7c), and besides their size difference, as outlined before, very interestingly, the NiO-a and NiO-c exhibit distinctive crystallographic surface facets. For example, the NiO-a NPs are predominantly terminated by a neutral {002} type of surface, while the NiO-c NPs are surface terminated by a polar {111} type of surface facet (Fig. SI-3†). We note that the ability to synthesize NiO NPs terminated with different types of surface facets, especially polar surface-terminated NPs is of exceptional importance to the potential application of the synthesized materials in catalysis.
The above results clearly show that intermediates affect the shape and size of the NPs, and their overall morphology is either retained, i.e. NiO-a preserves the 3D-flower-like nanostructure, or they fully transform to non-self-assembled NiO-c NPs. A common characteristic of Ni3(OH)4(NO3)2 and β-Ni(OH)2 nanoplates is their hexagonal symmetry-layered structure, with the tendency to assemble into flower-like nanostructures.72
Hence, it is rather important to understand how the intermediate transformation leads to different surface faceting in the NiO NPs and, consequently, what is a likely driving force for NP assembling. The surface chemistry of the H2O with NiO NPs is expected to be very sensitive and depends on the atomic structure, i.e. crystallography of the exposed NP surfaces, due to distinct polar vs. neutral surface terminations of NiO-c and NiO-a NPs, respectively. We postulate that water–NiO NP surface interactions could play a significant role in either the occurrence or absence of NP self-assembly, as elaborated in more detail in the next section.
3.3. NiO–H2O interaction: T-dependent FTIR, XPS, and DFT analyses
To gain more in-depth insights into the interactions and forces that maintain the nanoparticle assemblies upon thermal treatment, especially in the presence of OH and H2O species on NP surfaces, we perform temperature-dependent FT-IR and XPS studies of NiO nanostructures obtained by ammonia and carbamide routes, accompanied by DFT modeling of water adsorption on relevant NiO surfaces present in the NPs, that is, the (100) and (111) surfaces.The spectral region of O–H stretching vibrations in the attenuated total reflection (ATR) – FTIR spectra is shown in Fig. 8a and b. Additionally, Fig. SI-4† shows only the first and last spectra from Fig. 8a and b for a clearer observation of the general trend.
 | ||
| Fig. 8 The O–H stretching region in the temperature-dependent ATR FT-IR spectra of (a) NiO-a and (b) NiO-c NPs; the temperature increases from 20 °C to 200 °C from the uppermost curve downwards. The moving-window two-dimensional correlation spectra are constructed from the calculated autocorrelation functions of the spectral responses in the NiO-a (c) and NiO-c NPs (d). | ||
A much wider spectral band in this region, extending significantly to the lower wavenumber side in the spectra of NiO-a NPs, strongly implies the dominance of the O–H stretching vibrations of surface adsorbed non-dissociated water molecules. This band gradually loses intensity upon heating and practically disappears in the spectra recorded at 200 °C (Fig. SI-4a†). The behavior of the spectral region corresponding to the H–O–H bending modes is also consistent with the observations in the higher-wavenumber region, as shown in Fig. SI-5a and c.†
In contrast, the NiO-c NPs show a band that is due to the O–H stretching modes, which is narrower with the centroid shifted towards the higher wavenumbers, corresponding to the position of surface hydroxyl groups.72 Such a spectral appearance is consistent with the physical picture involving predominantly physisorbed water on the {100} surfaces of the NiO-a and dissociatively adsorbed water dominating on the {111} surfaces of the NiO-c NPs.
The presence of H2O and OH groups is further demonstrated by the moving-window two-dimensional correlation spectra constructed based on the calculated autocorrelation functions of the spectral responses; see the ESI† for more details. Fig. 8c and d clearly demonstrate the dominance of processes, leading to loss of the physisorbed water in NiO-a NPs, in line with the previous discussions, while in the case of NiO-c NPs, the dominant surface species are OH groups. In both cases, the physisorbed H2O and chemisorbed OH groups remained on the NiO NP surfaces after the temperature treatment. As implied before, although surface–exposed water molecules could maintain the nanocrystal assemblies through favorable hydrogen-bonding interactions, the presence of predominantly surface hydroxyl groups is expected to lead to unfavorable inter-nanoparticle interactions, as a consequence of the repulsive H⋯H interactions.
Further evidence of surface termination species on NiO NP surfaces is shown by the XPS measurements presented hereinafter.
Fig. 9 shows the XPS spectra of the two O 1s multiplet bands of both sets of NiO NPs, and the complementary Ni 2p3/2 spectra are shown in Fig. SI-6.† The O 1s spectra clearly reflect the presence of surface hydroxyl groups in both products. The spectrum from pure NiO is characterized by an O 1s single peak at ∼529 eV. In the case of OH and H2O surface presence, in addition to the main O 1s peak, additional peaks at ∼530 and ∼531 eV are expected to appear, respectively.56–58,60–63 In the case of NiO-a, a rather intense H2O peak is observed in the O 1s XPS spectrum at 531.8 eV, which is absent in the case of NiO-c (Fig. 9a and b and ESI Table SI-1†). Simultaneously, the XPS spectra of the products from both routes have an O 1s peak corresponding to the surface OH groups appearing at 530.6 and 531 eV in both NiO-a and NiO-c NPS, respectively. As illustrated in Fig. 9, the H2O peak is the most prominent in NiO-a. The appearance of a peak at ∼527 eV in the O 1s region (i.e. on the lower binding energy side of the peak multiplet – denoted by an asterisk in Fig. 9) strongly implies that the studied samples are charge separation active. Assuming the existence of positive trapping sites on the sample surface, some of the electrons can become trapped. The surface thus becomes negatively charged, which shifts the XPS signal to a lower binding energy value than expected and is allowed from the immediate chemical environment in question.
 | ||
| Fig. 9 XPS spectra of NiO synthesized via the ammonia (a) and carbamide (b) routes in the regions of O 1s multiplet bands. | ||
The 3D structure of NiO-a NPs suggests strong H2O retention, which is responsible for retaining nanoparticle self-assembly into nanoplatelets. This is also reflected in the XPS Ni 2p3/2 spectra. In the case of NiO-c, the appearance of this region is simpler and can be straightforwardly explained with the GS model, as described in much more detail in the ESI.† The Ni 2p3/2 XPS region in NiO-a is, however, dominated by the presence of surface adsorbed water; see Fig. SI-6 and Table SI-1† for details.
The above experimental data clearly show that the NiO NPs are preferentially decorated with either OH or H2O. These experimental observations agree with how polar and neutral surfaces, i.e. {111} or {100} of NiO interact with water. The DFT total energy calculations show that H2O can be easily chemisorbed on both (111) and (100) surfaces, using ∼0.5 eV per molecule, with a small variation depending on the atomic site. However, H2O molecules can easily dissociate on the polar (111) surface; the energy gain is −1.74 eV per molecule on the (111) surface, while dissociation on (100) is 0.89 eV per molecule; hence, it is not feasible. This is also in agreement with literature reports based on single-crystal adsorption calorimetry, where it has been shown that H2O adsorption on NiO(111) is dissociative.73
The driving mechanism behind water–NiO polar vs. neutral surface interactions is the huge surface energy difference between polar and neutral surfaces, and the need for significant charge transfer required for polar NiO(111) surfaces to be stabilized. Various surface stabilization mechanisms have been studied and reported in the literature for various polar surfaces, including the (111) rock salt oxide surfaces.74 We note that OH surface stabilization of the (111) surfaces is one of the most dominant atomic reconstruction mechanisms also experimentally observed on the MgO (111) surface.74–76 Finally, when energies of various NiO terminations are evaluated with respect to the chemical potential of the H2O, it has been shown that the presence of water makes the OH surface stabilization mechanism even more favorable.77
3.4. From intermediates to NiO NPs: correlation between structural transitions and vibrational spectroscopy
To further elucidate the chemical changes in the intermediate hydroxide-based phases and their structural transformation under the thermal treatment of NiO NPs, FTIR, UV-VIS and Raman spectroscopic techniques were employed.Fig. 10a shows an FTIR spectrum of the β-Ni(OH)2 intermediate in the 4000–500 cm−1 spectral range, with a note that four IR active vibrational modes for β-Ni(OH)2 are predicted and also observed at 332–354 cm−1, 440–475 cm−1, 510–553 cm−1 and 3630–3650 cm−1.78–81 In the higher wavenumber region, the well-defined band at ∼3630 cm−1 (Fig. 10a) is assigned to the stretching mode from lattice hydroxide (OH) groups. The combination of two lower-frequency modes (Eg + A2u(TO)), however, is most likely a cause for the weak band at ∼1400 cm−1. In the low-wavenumber region, the sharp band at ∼510 cm−1 is assigned to the bending Ni–O–H mode. This spectrum also includes a very weak band at ∼1630 cm−1 due to the H–O–H bending mode, which indicates traces of water in the sample, likely adsorbed on the surface and/or trapped in the structure, originating from the closest environment.
 | ||
| Fig. 10 FTIR spectra of hydroxide phases synthesized from an ammonia (a) and carbamide (b) based reaction system and corresponding NiO phases (c and d). The band at ∼500 cm−1 is assigned to the NiO stretching mode,79,80 while the broad band in the low wavelength part and the weak band at ∼1600 cm−1 imply traces of water in the sample. | ||
The FTIR spectrum of the hydroxide-based phase Ni3(OH)4(NO3)2 intermediate, Fig. 10b, in addition to the bands observed in β-Ni(OH)2 intermediate, shows few additional bands at ∼1495 cm−1, ∼1305 cm−1, ∼994 cm−1 and ∼2189 cm−1. Additionally, compared to the FTIR spectrum presented in Fig. 10a, the band assigned to the bending Ni–O–H mode is blue-shifted at ∼625 cm−1. The three bands at ∼1495 cm−1, ∼1305 cm−1, and ∼994 cm−1 are assigned to the stretching modes of loosely bonded nitrate ions, while the band at ∼2189 cm−1 arises due to the stretching mode of isocyanate ions (NCO−). The presence of isocyanate ions is because they are a coproduct of the process of hydrolysis of carbamide, which during synthesis is likely incorporated as a ‘free’ species between atomic layers. The band at ∼1630 cm−1 assigned to the H–O–H bending mode from free H2O molecules is weak although it is more pronounced in comparison to β-Ni(OH)2. Additionally, the broadening of the band at ∼3630 cm−1 assigned to the stretching mode from lattice OH is likely due to the stretching of the OH mode from H2O involved in hydrogen bonding interactions (ν(HO–H⋯O)).79 Therefore, the intermediate phase synthesized using carbamide as an anion precursor is an hydroxide-rich basic nitrate salt of nickel.
In addition to lattice nitrate ions, the structure of Ni3(OH)4(NO3)2 includes nitrate ions along with isocyanate ions and free water molecules, which are placed between atomic planes. This provides an environment for the development of a turbostratic structure, as indicated by the XRD patterns, and is directly imaged by HRTEM. This intermediate, compared to the alpha-Ni(OH)2, is characterized by a larger interlayer distance due to the excess of H2O between atomic planes.
The FTIR spectra in Fig. 10c and d correspond to NiO-a and NiO-c obtained by annealing intermediate phases by ammonia and carbamide precursors, respectively. The sharp bands due to the stretching and bending vibration modes from lattice OH disappear, and the band is related to the bending Ni–O–H mode. Therefore, FTIR spectroscopy confirms that the intermediate phases transform completely to NiO upon annealing.
To gain further insights into certain structural specificities of the intermediate phases, we performed analyses of the experimental FTIR data based on nonlinear curve-fitting procedures.
The band due to the fundamental vibrational transition of the lattice OH group in the FTIR spectrum of the β-Ni(OH)2 intermediate could be precisely modeled by fitting with three component bands (Fig. 11a), in which each is a linear combination of a Gaussian and Lorentzian function (see the ESI† for details concerning the fitting procedure).
 | ||
| Fig. 11 Reconstruction of the region of the fundamental vibrational transition of the lattice OH group in the FTIR spectrum of the β-Ni(OH)2 (a) and Ni3(OH)4(NO3)2 (b) intermediates. | ||
In the case of Ni3(OH)4(NO3)2, however, successful reconstruction of the experimental band requires fitting with more than 10 component bands, i.e. 14 bands are used to generate the reconstructed band shape in Fig. 11b. The need for only three component bands to reconstruct the experimentally measured OH stretching band in the β-Ni(OH)2 intermediate (spanning a narrow range of less than 100 cm−1) in comparison to 14 component bands for Ni3(OH)4(NO3)2 (spanning a wider range of ∼300 cm−1) indicates the presence of few distinct lattice OH oscillators in the former and a much larger number in the latter case, reflecting the turbostratic structure of Ni3(OH)4(NO3)2. The appearance of the IR absorption spectrum of a given molecular/ionic oscillator embedded in a solid state compound is affected by the local electrostatic effects, causing the site-symmetry splitting and shift, as well as by the effects related to the coupling with other identical oscillators within the unit cell of a crystal, which leads to unit cell group splitting and accidental degeneracy effects. We note that few distinct in-crystal oscillators strongly imply a significantly higher order in the lattice, usually accompanied by a higher symmetry, as in the case of β-Ni(OH)2. The turbostratic effect evidenced in the case of Ni3(OH)4(NO3)2 implies the presence of different intercalated species at variable positions within the crystal lattice. Such higher degrees of disorder cause different local environments around the lattice OH species, with different local perturbations to the OH stretching potentials, resulting in a wide IR band with a well-resolved substructure, as shown in Fig. 11b. These results complement the structural analysis by X-ray diffraction and electron microscopy of both intermediates.
In line with the conclusion drawn from the results obtained by XRD and FTIR spectroscopy, Raman measurements confirm the chemical identity of β-Ni(OH)2 and Ni3(OH)4(NO3)2 intermediates. Raman spectra of intermediates and final products (NiO-a and NiO-c) are shown in Fig. SI-7a–d.† The Raman spectra contain all the expected bands characteristic for both intermediates. Along with the Raman-active modes involving lattice OH stretching and bending vibrations, the spectrum of Ni3(OH)4(NO3)2 intermediate contains additional bands due to NO3− and H2O species. Positions of some of Ni3(OH)4(NO3)2 bands are close to the positions of band characteristics for free NO3− ions and H2O molecules, confirming that the free-like nature of intercalated NO3− ions and H2O molecules in the interlayer spacings is as expected for the turbostratic structure of this intermediate. The Raman spectra of the samples annealed at ∼400 °C for at least 1 h have practically completely lost the bands related to the vibration modes of lattice OH or hydroxide lattice modes (apart from those arising from the surface-adsorbed water albeit with very low intensity), confirming that upon thermal treatment at 400 °C, intermediate hydroxide-based phases, a complete transformation to nickel oxide occurred (Fig. SI-7c and d†). See the ESI† section for a more detailed assignment of the bands in the Raman spectra of the final products (NiO-a and NiO-c).
To gain additional information on the temperature phase transitions of intermediates to NiO NPs and the nature of transitions, we perform DSC calorimetry on both intermediates from RT to 350 °C. The results are presented and discussed in the next section.
3.5. Differential scanning calorimetry measurements: phase transition from intermediate to NiO-NPs
The DSC curves for the intermediate compounds obtained using ammonia and carbamide hydroxide ion precursors are shown in Fig. 12, with clear endothermic peaks that correspond to chemical and structural transformations during thermal treatment associated with the transformation of intermediates to NiO NPs. | ||
| Fig. 12 DSC curves of the β-Ni(OH)2 and Ni3(OH)4(NO3)2 intermediates obtained using ammonia (a) and carbamide (b) as hydroxide ion precursors, respectively. | ||
The initial baseline upward shifts, from 50 to ∼275 °C, for both intermediates can be attributed to the release of physisorbed H2O molecules, with a clear indication that Ni3(OH)4(NO3)2 contains more water molecules, with a significant amount in the interlayers released during the initial structural transformation of the nanoplates. The energy required for such changes is low due to the turbostratic nature of these interlayers.78 In contrast, the H2O presence in β-Ni(OH)2 is surface limited, e.g. no free species between atomic planes. Therefore, this effect is less pronounced. In the higher temperature region, the single endotherm peak (∼298 °C) associated with β-Ni(OH)2 represents the phase transition to NiO. In contrast, the phase transformation associated with Ni3(OH)4(NO3)2 proceeds in two steps. At ∼306 °C, the first broadened peak can be associated with the release of the ‘free’ species within interlayers, e.g. H2O and NO3−, followed by a sharp endotherm peak at ∼326 °C that corresponds to transformation to NiO. As expected after this transition temperature, the full transformation to NiO occurs, as also confirmed by the absence of the OH−, H2O and NO3− bulk-related bands in the FTIR and Raman discussed above. Similarly, the sharp β-Ni(OH)2 endotherm peak, which is accompanied by loss of the OH, corresponds well with the loss of bulk Ni–O–H bending and OH stretching modes observed in the FTIR and Raman spectra. The DSC results show that β-Ni(OH)2 to NiO-a NPs proceed in single-step structural transformation, while Ni3(OH)4(NO3)2 to NiO-c NPs is more complex with two distinctive structural transformations.
The thermal treatment of the products is carried out at 350 °C, which is well beyond the DSC peak maxima for both hydroxide products, to provide a complete transformation to oxide and removal of all impurities consistently and comparably.
3.6. Low dimensionality effects on phonon modes and band gaps in NiO NPs using Raman and UV-VIS spectroscopies
In this final subsection, we explore how quantum confinement affects NP band and phonon modes by utilizing Raman and UV-VIS spectroscopy.We start by presenting Raman Spectroscopy results with a focus on the spectral range where the first order transverse optical (TO) and longitudinal optical (LO) transitions appear, at ∼400 cm−1 in TO and 500 cm−1 in LO modes. The particular range of Raman shift values of the TO and LO modes is affected by several factors, such as lattice imperfections and magnon-phonon interactions.82 The small size of the NiO nanoparticles also induces the appearance of the surface modes (SO). At a relatively smaller NiO NP size (<12 nm), the position of the SO modes could overlap with the TO and LO.83Fig. 13 shows the results of the fitting of the band appearing ∼500 cm−1 with three-component Lorentzian functions, and the peak assignments are in line with experimental and theoretical data in the literature.84,85 Table SI-2† illustrates a summary of the relevant fitting parameters, along with the intensity ratio of the SO with respect to the TO and LO modes. The average lifetime values (τ) for phonons in the LO, TO and SO modes are calculated from the Lorentzian band parameters, using the relation τ = 1/(2πcw).86,87
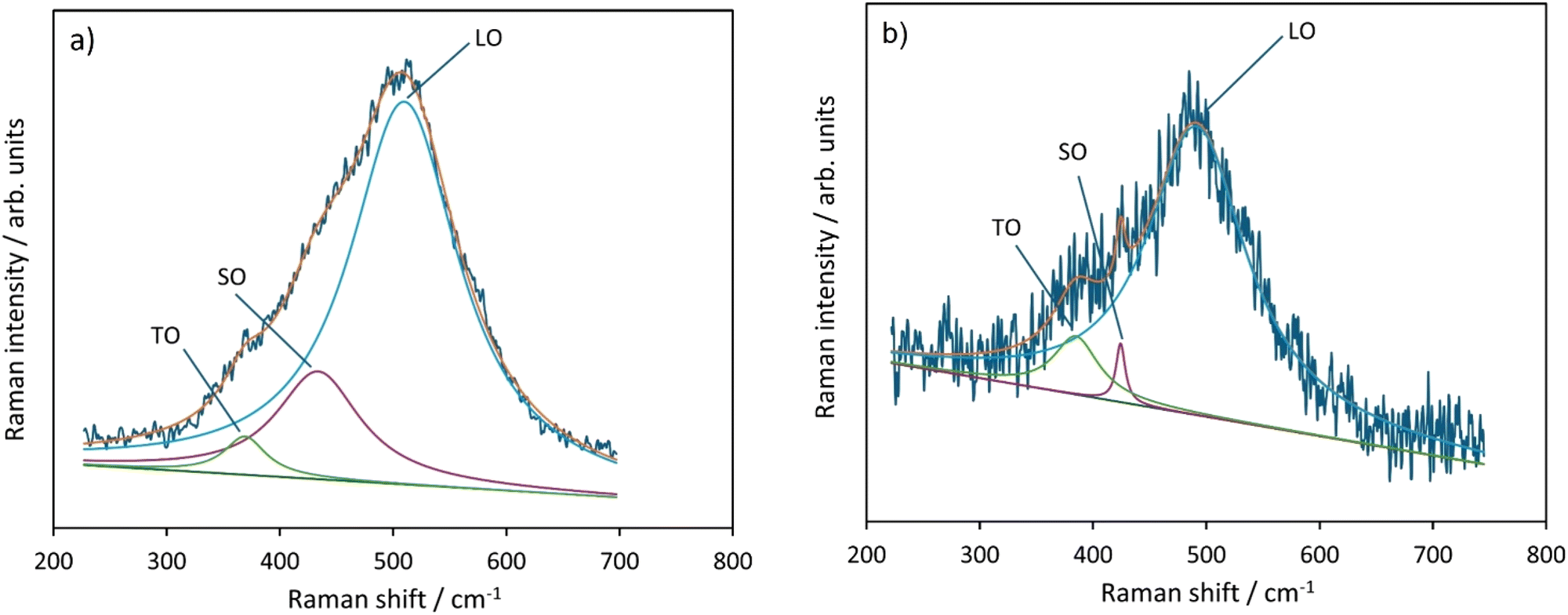 | ||
| Fig. 13 Reconstruction of the region of the fundamental TO, SO and LO phonon bands in the Raman spectrum of the (a) NiO-a phase obtained from β-Ni(OH)2 and (b) NiO-c obtained from Ni3(OH)4(NO3)2. | ||
It could be concluded that the intensity ratio of the SO modes to both the TO and LO modes is much higher (∼40 times higher) in the NiO-a NPs obtained from β-Ni(OH)2 as compared to the NiO-c from Ni3(OH)4(NO3)2. Additionally, while the average lifetimes of the TO and LO modes between the two final products are similar, the average lifetimes of the SO modes are different by an order of magnitude, reflecting the size difference between the two sets of NiO NPs.
Finally, we present the results of the optical absorption spectroscopy data. Fig. 14 shows the semiconductor absorption functions for direct-type band to band transitions constructed from the UV-VIS spectroscopy data for NiO-a and NiO-c.
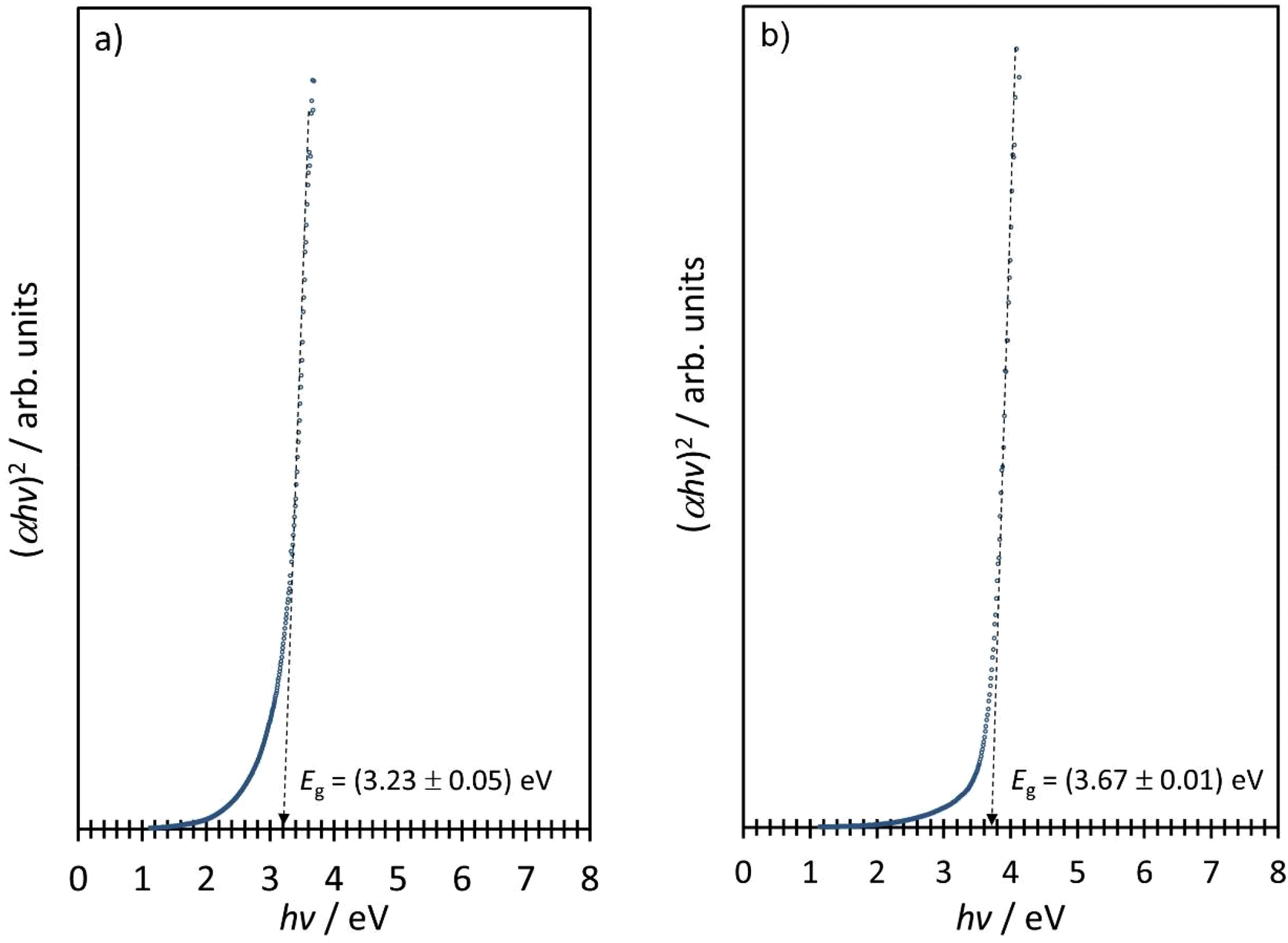 | ||
| Fig. 14 (αhv)2vs. hv dependency of (a) NiO-a and (b) NiO-c NPs obtained through the thermal treatment of the β-Ni(OH)2 and Ni3(OH)4(NO3)2 intermediates, respectively. | ||
Within the framework of the parabolic approximation for the dispersion relation, the band gap energies of the NiO-a and NiO-c were determined as 3.23 eV and 3.67 eV, respectively. Despite the small size of the NPs in both cases, Bohr's excitonic radius for NiO is smaller than 1 nm, and no quantum confinement effects are observed. The difference in the band gap energies of the two sets of NiO NPS can be attributed to the increased density of the surface states of the NiO-a NPs, which ultimately decreases the band gap of NiO-a compared to NiO-c NPs.
3.7. Chemistry of precursor conversion to hydroxide-based intermediate NPs and their conversion to NiO NPs
The previous results allow us to elucidate the details of the chemical pathways to the hydroxide intermediates and their subsequent transformation to NiO NPs upon thermal annealing.The reaction mechanism of hydroxide-based nanoplate formation includes few equilibria, which are mutually and also pH value dependent. When ammonia was used as a hydroxide ion precursor, the following equilibria are of relevance:
| [Ni(NH3)6]2+ (aq) ⇌ Ni2+ (aq) + 6 NH3 (aq), | (1) |
| NH3 (aq) + H2O (l) ⇌ NH4+ (aq) + OH− (aq). | (2) |
The ammonia as a Lewis base forms complex ions (equilibrium (1)) and maintains the equilibrium concentration of metal cations at an optimal level, and it also provides hydroxide ions throughout the equilibrium (2). As discussed before, the results from our measurements performed using various structural and spectroscopic techniques confirm that the formed intermediate is β-Ni(OH)2 phase. Its formation starts when the critical supersaturation has been reached according to the following equilibrium:88
| Ni(OH)2 (s) ⇌ Ni2+ (aq) + 2 OH− (aq) Ksp = 5.48 × 10–16. | (3) |
Therefore, ammonia controls the rate of precursor conversion, which further influences the monomer concentration and the balance between the processes of nucleation and crystalline NP growth. This balance is important for NP size. Upon thermal treatment of the intermediate, Ni(OH)2, it converted to NiO.
In the case of a carbamide-based reaction system, the synthesis occurs at a higher temperature (∼90 °C). The decomposition of carbamide is an endothermic reaction that leads to the formation of ammonia and carbon dioxide:
| NH2CONH2 + H2O ⇌ CO2 + 2 NH3. | (4) |
The already introduced equilibrium (3) is also relevant in the case of a carbamide-based solution, but hydroxide ions, which are generated as a result of the protonation of ammonia, in this case, originate from carbamide as an anion precursor.
According to our results, the choice of the hydroxide precursor results in a difference in the chemical composition of intermediates. When the carbamide is included as a hydroxide ionic precursor, instead of the formation of β-Ni(OH)2, the reaction path progresses through a different intermediate, Ni3(OH)4(NO3)2:
| Ni3(OH)4(NO3)2 (s) ⇌ 3 Ni2+ (aq) + 4 OH− (aq) + 2 NO3− (aq). | (5) |
Such conclusions are supported by analysis of the intermediates by Raman and FTIR Spectroscopy, and by DSC.
The equilibrium concentration of hydroxide ions in the reaction solution is not sufficient to provide deposition of nickel(II) hydroxide but is sufficient to fulfill the conditions for the precipitation of nickel(II) tetrahydroxide nitrate, which is less soluble than β-Ni(OH)2. By designing this route, we provide the conditions for the formation of a turbostratic nanostructure, which leads to a different product built up by NiO NPs with different size, morphological and optical properties.
Upon thermal treatment in the air atmosphere at ∼350 °C for at least 1 h, both intermediates, β-Ni(OH)2 and Ni3(OH)4(NO3)2, convert to NiO NPs. The phase transition is followed by a color change from green to gray, which indicates Ni deficiency in the final products. This deficiency is also confirmed by XRD. As discussed before, the final products from the two routes (taking place through different intermediates) differ in their size, morphology, surface faceting and optical properties.
4. Conclusions
The present paper provides a fundamental understanding of the chemical pathway to engineer NiO nanostructures by utilizing ammonia and carbamide precursors, which direct the synthetic route towards β-Ni(OH)2 and Ni3(OH)4(NO3)2 intermediates that are further transformed to NiO NPs (for simplicity, denoted as NiO-a and NiO-c, respectively). The precursor-engineered intermediates are self-assembled flower-like 3D nanostructures based on nanoplates, which upon thermal annealing at 350 °C, fully transform into two distinctive sets of NiO-a and NiO-c NPs that have different sizes, structures and morphologies, as well as optical and surface properties.Fundamental insight into the structural transformation from intermediates to NiO NPs is gained by employing various complementary structural and spectroscopic techniques. FTIR, Raman and DSC analyses show that the transformation of turbostratic arrangement of Ni3(OH)4(NO3)2 atomic planes, directly confirmed by HRTEM, to NiO NPs goes through subsequent loss of NO3− and OH− species, as a two-step phase transformation at 306 °C and 326 °C. These steps correspond to free interlayer ions and H2O species loss, followed by the loss of chemically bonded OH− and NO3− ions. The β-Ni(OH)2 intermediate transforms to NiO NPs via loss of OH− species, observed as a single-step phase-transition at 298 °C. The temperature-dependent FTIR from intermediates shows that NiO NPs obtained by ammonia and carbamide route are preferentially retaining H2O and OH groups, respectively. This is directly related to the surface terminations of the NiO NPs, where nanorod-shaped NiO-c NPs are terminated predominantly with polar {111} facets, while the NiO-c NPs are faceted by mostly neutral {100} types of surfaces, as shown by HRTEM. Total energy calculations show that H2O can dissociate on polar NiO(111) surfaces, while H2O is physiosorbed on neutral NiO(100)-type surfaces. Such findings are further supported by XPS measurements of the NPs. The NiO-a NPs are H2O terminated, while the surfaces of the NiO-c NPs are OH terminated.
These structural findings provide insight into the driving forces that determine overall NP morphology. For example, the water interaction with the neutral {100} facets of the NiO-a NPs is a likely driving mechanism that allows weak bonding via hydrogen bonds involving water molecules, which leads to the preservation of the nanoflower morphology even after annealing at 350 °C. Indeed, the XPS of NiO-a NPs shows a large amount of water present within the NiO-a 3D self-assembled nanostructures. In contrast, the NiO-c NPs obtained by thermal annealing of the Ni3(OH)4(NO3)2 flower-like 3D nanostructures result in the loss of the 3D self-assembly due to the inert behavior of the dominantly OH – terminated (111) NP facets.
Besides the overall surface structure/morphology differences, the NiO-c NPs are almost 3 times larger (15.5 nm) than NiO-a NPs, and their band gap of 3.6 eV is 0.4 eV larger than the band gap of NiO-a NPs. Finally, we showed that both sets of NiO NPs are Ni deficient: the Ni deficiency is more pronounced in NiO-c NPs obtained from carbamide precursor through the Ni3(OH)4(NO3)2 intermediate.
In summary, the present paper elucidates the possibility of engineering NiO NPs via different precursors, ammonia and carbamide. The size, morphology and band gap energy of the NiO NPs can be modulated by the choice of the precursor. This choice also defines the Ni deficiency in the final product, which provides pathways to utilize their application as p-type materials in energy conservation and transformation technologies and numerous other applications. Moreover, our synthetic approach combined with the post-deposition annealing treatment allows precise nanoengineering of the NiO NP surfaces, a property that is crucial for the application of NiO NPs as catalysts.
Data availability
The datasets generated and analysed during the current study are available from the authors on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the international exchange project by The Royal Society (grant no. IES\R2\222147), and Engineering and Physical Sciences Research Council (grant no. EP/S033394/1).References
- S. Yadav, N. Rani and K. Saini, A review on transition metal oxides-based nanocomposites, their synthesis techniques, different morphologies and potential applications, IOP Conf. Ser.: Mater. Sci. Eng., 2022, 1225, 012004 CrossRef CAS.
- J. Yang, Z. Zeng, J. Kang, S. Betzler, C. Czarnik, X. Zhang, C. Ophus, C. Yu, K. Bustillo, M. Pan, J. Qiu, L. W. Wang and H. Zheng, Formation of two-dimensional transition metal oxide nanosheets with nanoparticles as intermediates, Nat. Mater., 2019, 18, 970–976 CrossRef CAS PubMed.
- G. Guo, L. Ji, X. Shen, B. Wang, H. Li, J. Hu, D. Yang and A. Dong, Self-assembly of transition-metal-oxide nanoparticle supraparticles with designed architectures and their enhanced lithium storage properties, J. Mater. Chem. A, 2016, 4, 16128–16135 RSC.
- L. Tao, Y. Xu, X. Qian, Q. Yue and Y. Kang, Low-temperature molten salt synthesis for ligand-free transition metal oxide nanoparticles, ACS Appl. Energy Mater., 2020, 3, 3984–3990 CrossRef.
- P. Peng, X. M. Lin, Y. Liu, A. S. Filatov, D. Li, V. R. Stamenkovic, D. Yanng, V. B. Prakapenka, A. Lei and E. V. Shevchenko, Binary transition-metal oxide hollow nanoparticles for oxygen evolution reaction, ACS Appl. Mater. Interfaces, 2018, 10, 24715–24724 CrossRef CAS.
- E. Boyes, M. R. Ward, L. Lari and P. L. Gai, ESTEM imaging of single atoms under controlled temperature and gas environment conditions in catalyst reaction studies, Ann. Phys., 2013, 525, 423–429 CrossRef CAS.
- A. P. La Grow, S. Famiani, A. Sergides, L. Lari, D. Loyd, M. Takahashi, S. Maenosono, P. L. Gai, E. D. Boyes and N. T. K. Thanh, Environmental STEM study of the oxidation mechanism for iron and iron carbide nanoparticles, Materials, 2022, 15(4), 1557 CrossRef CAS PubMed.
- L. Lari, S. Steinhauer and V. K. Lazarov, In situ TEM oxidation study of Fe thin-film transformation to single-crystal magnetite nanoparticles, J. Mater. Sci., 2020, 55, 2897–12905 CrossRef.
- Progress in optical science and photonics, ed. V. Kumar, V. Sharma, I. Ayoub and H. C. Swart, Springer Nature Singapore Pte Ltd., Singapore, 2023 Search PubMed.
- M. El-Kemary, N. Nagy and I. El-Mehasseb, Nickel oxide nanoparticles: Synthesis and spectral studies of interactions with glucose, Mater. Sci. Semicond. Process., 2013, 16, 1747–1752 CrossRef CAS.
- Padma, S. Ranju, Yeshas, S. L. Kavya, S. K. Sukrutha, M. R. A. Kumar, A. N. Kumar, M. Kumaraswamy, B. Purushotham and S. B. Boppana, A comparative study of green and chemically synthesized nano nickel oxide for multifunctional applications, Appl. Surf. Sci. Adv., 2022, 12, 100318 CrossRef.
- V. Venkata, S. Varma, C. S. Srikar, J. Ruchitha, P. A. Varma and B. V. S. Praveen, Nickel oxide nanoparticles: a brief review of their synthesis, characterization and applications, Chem. Eng. Technol., 2022, 45, 397–409 CrossRef.
- M. Bonomo, Synthesis and characterization of NiO nanostructures: a review, J. Nanopart. Res., 2018, 20, 222 CrossRef.
- K. M. Dooley, S. Y. Chen and J. R. H. Ross, Stable nickel-containing catalysts for the oxidative coupling of methane, J. Catal., 1994, 145, 402–408 CrossRef CAS.
- E. L. Miller, Electrochemical behavior of reactively sputtered iron-doped nickel oxide, J. Electrochem. Soc., 1997, 144, 3072 CrossRef CAS.
- M. Awais, E. Gibson, J. G. Vos, D. P. Dowling, A. Hagfeldt and D. Dini, Fabrication of efficient NiO photocathodes prepared via RDS with novel routes of substrate processing for p-type dye-sensitized solar cells, ChemElectroChem, 2014, 1, 384–391 CrossRef.
- S. Sheehan, G. Naponiello, F. Odobel, D. P. Dowling, A. D. Carlo and D. Dini, Comparison of the photoelectrochemical properties of RDS NiO thin films for p-type DSCs with different organic and organometallic dye-sensitizers and evidence of a direct correlation between cell efficiency and charge recombination, J. Solid State Electrochem., 2015, 19, 975–986 CrossRef CAS.
- P. A. Sheena, K. P. Priyanka, N. A. Sabu, S. Ganesh and T. Varghese, Effect of electron beam irradiation on the structure and optical properties of nickel oxide nanocubes, Bull. Mater. Sci., 2015, 38, 825–830 CrossRef CAS.
- M. Bonomo, G. Naponiello, I. Venditti, V. Zardetto, A. D. Carlo and D. Dini, Electrochemical and photoelectrochemical properties of screen-printed nickel oxide thin films obtained from precursor pastes with different compositions, J. Electrochem. Soc., 2017, 164, H137–H147 CrossRef CAS.
- H. X. Yang, Q. F. Dong, X. H. Hu, X. P. Ai and S. X. Li, Preparation and characterization of LiNiO2 synthesized from Ni(OH)2 and LiOH·H2O, J. Power Sources, 1999, 79, 256–261 CrossRef CAS.
- G. Wang, X. Lu, T. Zhai, Y. Ling, H. Wang, Y. Tong and Y. Li, Free-standing nickel oxide nanoflake arrays: synthesis and application for highly sensitive non-enzymatic glucose sensors, Nanoscale, 2012, 4, 3123–3127 RSC.
- M. P. Browne, H. Nolan, N. C. Berner, G. S. Duesberg, P. E. Colavita and M. E. G. Lyons, Electrochromic nickel oxide films for smart window applications, Int. J. Electrochem. Sci., 2016, 11, 6636–6647 CrossRef CAS.
- H. Yan, D. Zhang, J. Xu, Y. Lu, Y. Liu, K. Qiu, Y. Zhang and Y. Luo, Solution growth of NiO nanosheets supported on Ni foam as high-performance electrodes for supercapacitors, Nanoscale Res. Lett., 2014, 9, 424 CrossRef PubMed.
- M. Taeno, D. Maestre and A. Cremades, An approach to emerging optical and optoelectronic applications based on NiO micro and nanostructures, Nanophotonics, 2021, 10, 1785–1799 CrossRef CAS.
- M. Dare-Edwards, J. Goodenough, A. Hamnett and N. Nickolson, Photoelectrochemistry of nickel(II) oxide, J. Chem. Soc., Faraday Trans., 1981, 77, 643–661 RSC.
- A. Haider, M. Ijaz, S. Ali, J. Haider, M. Imran, H. Majeed, I. Shahzadi, M. M. Ali, J. A. Khan and M. Ikram, Green synthesized phytochemically (zingiber officinale and allium sativum) reduced nickel oxide nanoparticles confirmed bactericidal and catalytic potential, Nanoscale Res. Lett., 2020, 15, 50 CrossRef CAS.
- Y. Yulizar, I. Abdullah, R. M. Surya, N. Parwati and D. O. B. Apriandanu, Two-phase synthesis of NiCo2O4 nanoparticles using Bryophyllum pinnatum (Lam) Oken leaf extract with superior catalytic reduction of 2,4,6-trinitrophenol, Mater. Lett., 2022, 311, 131465 CrossRef CAS.
- Y. Yulizar, D. O. B. Apriandanu and Z. A. Zahra, SiO2/NiFe2O4 nanocomposites: Synthesis, characterization and their catalytic activity for 4-nitroaniline reduction, Mater. Chem. Phys., 2021, 261, 124–243 CrossRef.
- K. Lingaraju, H. R. Naika, H. Nagabhushana, K. Jayanna, S. Devaraja and G. Nagaraju, Biosynthesis of nickel oxide nanoparticles from Euphorbia heterophylla (L.) and their biological applications, Arabian J. Chem., 2020, 13, 4712–4719 CrossRef CAS.
- H. A. Ariyanta, T. A. Ivandini and Y. Yulizar, Novel NiO nanoparticles via phytosynthesis method: Structural, morphological and optical properties, J. Mol. Struct., 2021, 1227, 129543 CrossRef CAS.
- A. Ezhilarasi, J. J. Vijaya, K. Kaviyarasu, L. J. Kennedy, R. J. Ramalingam and H. A. Al-Lohedan, Green synthesis of NiO nanoparticles using Aegle marmelos leaf extract for the evaluation of in vitro cytotoxicity, antibacterial and photocatalytic properties, J. Photochem. Photobiol., B, 2018, 180, 39–50 CrossRef.
- N. Alrushaid, F. A. Khan, E. A. Al-Suhaimi and A. Elaissari, Nanotechnology in Cancer Diagnosis and Treatment, Pharmaceutics, 2023, 15, 1025 CrossRef CAS PubMed.
- J. Iqbal, B. A. Abbasi, T. Mahmood, S. Hameed, A. Munir and S. Kanwal, Green synthesis and characterizations of nickel oxide nanoparticles using leaf extract of Rhamnus virgata and their potential biological applications, Appl. Organomet. Chem., 2019, 33, e4950 CrossRef.
- M. Napari, T. N. Huq, R. L. Z. Hoye and J. L. MacManus-Driscoll, Nickel oxide thin films grown by chemical deposition techniques: Potential and challenges in next-generation rigid and flexible device applications, InfoMat, 2021, 3, 536–576 CrossRef CAS.
- G. Sawatzky and J. Allen, Magnitude and origin of the band gap energy in NiO, Phys. Rev. Lett., 1984, 53, 2339–2342 CrossRef CAS.
- M. Taguchi, M. Matsunami, Y. Ishida, R. Eguchi, A. Chainani, Y. Takata, M. Yabashi, K. Tamasaku, Y. Nishino, T. Ishikawa, Y. Senba, H. Ohashi and S. Shin, Revising the valence-band and core-level photoemmision spectra of NiO, Phys. Rev. Lett., 2008,(100), 206401 CrossRef CAS PubMed.
- L. Alberts and E. W. Lee, Magnetostriction in antiferromagnetic nickel oxide, Proc. Phys. Soc., 1961, 78(5), 728–733 CrossRef CAS.
- J. Marmeggi and J. Baruchel, Antiferromagnetic domains in nickel oxide by magnetic neutron Laue diffraction, J. Magn. Magn. Mater., 1979, 10(1), 14–24 CrossRef CAS.
- M. Niederberger, M. H. Bartl and G. D. Stucky, Benzyl alcohol and transition metal chlorides as a versatile reaction system for the nonaqueous and low-temperature synthesis of crystalline nano-objects with controlled dimensionality, J. Am. Chem. Soc., 2002, 24, 13642–13643 CrossRef PubMed.
- R. Cinnsealach, G. Boschloo, R. S. Nagaraja and D. Fitzmaurice, Coloured electrochromic windows based on nanostructured TiO2 films modified by adsorbed redox chromophores, Sol. Energy Mater. Sol. Cells, 1999, 57, 107–125 CrossRef CAS.
- A. Surca, B. Orel, B. Pihlar and P. Bukovec, Optical, spectroelectrochemical and structural properties of sol-gel derived Ni-oxide electrochromic film, J. Electroanal. Chem., 1996, 408, 83–100 CrossRef.
- M. Alagiri, S. Ponnusamy and C. Muthamizhchelvan, Synthesis and characterization of NiO nanoparticles by sol-gel method, J. Mater. Sci.: Mater. Electron., 2012, 23, 728–732 CrossRef CAS.
- G. Zhang, Y. Chen, B. Qu, L. Hu, L. Mei, D. Lei, Q. Li, L. Chen, Q. Li and T. Wang, Synthesis of mesoporous NiO nanospheres as anode materials for lithium ion batteries, Electrochim. Acta, 2012, 80, 140–147 CrossRef CAS.
- J. Zhao, H. Liu and Q. Zhang, Preparation of NiO nanoflakes under different calcination temperatures and their supercapacitive and optical properties, Appl. Surf. Sci., 2017, 392, 1097–1106 CrossRef CAS.
- Y. Zheng and M. Zhang, Preparation and electrochemical properties of nickel oxide bymolten-salt synthesis, Mater. Lett., 2007, 61, 3967–3969 CrossRef CAS.
- Z. Zhu, N. Wei, H. Liu and Z. He, Microwave-assisted hydrothermal synthesis of Ni(OH)2 architectures and their in situ thermal convention to NiO, Adv. Powder Technol., 2011, 22, 422–426 CrossRef CAS.
- N. N. M. Zorkipli, N. H. M. Kaus and A. A. Mohamad, Synthesis of NiO nanoparticles through sol-gel method, Procedia Chem., 2016, 19, 626–631 CrossRef CAS.
- C. S. Carney, C. J. Gump and A. W. Weimer, Rapid nickel oxalate thermal decomposition for producing fine porous nickel metal powders. Part 3: mechanism, Mater. Sci. Eng., A, 2006, 431, 26–40 CrossRef.
- Z. Wei, H. Qiao, H. Yang, C. Zhanga and X. Yan, Characterization of NiO nanoparticles by anodic arc plasma method, J. Alloys Compd., 2009, 479, 855–858 CrossRef CAS.
- W. Ahmad and A. Rawat, New Frontiers in the Bio-inspired Green Synthesis of NiO NPs and Their Applications: An Overview, Nat., Environ. Pollut. Technol., 2023, 22(3), 1353–1362 CrossRef CAS.
- W. Ahmad, S. C. Bhatt, M. Verma, V. Kumar and H. Kim, A review on current trends in the green synthesis of nickel oxide nanoparticles, characterizations, and their applications, Environ. Nanotechnol., Monit. Manage., 2022, 18, 100674 CAS.
- J. Iqbal, B. A. Abbasi, R. Ahmad, M. Mahmoodi, A. Munir, S. A. Zahra, A. Shahbaz, M. Shaukat, S. Kanwal, S. Uddin, T. Mahmood and R. Capasso, Phytogenic synthesis of nickel oxide nanoparticles (NiO) using fresh leaves extract of Rhamnus triquetra (wall.) and investigation of its multiple in vitro biological potentials, Biomedicines, 2020, 8, 117 CrossRef CAS PubMed.
- Z. Wang, Y. Bi, K. Li, Z. Song, C. Pan, S. Zhang, X. Lan, N. S. Foulkes and H. Zhao, et al., Nickel oxide nanoparticles induce developmental neurotoxicity in zebrafish by triggering both apoptosis and ferroptosis, Environ. Sci.: Nano, 2023, 10, 640–655 RSC.
- Y. Singh, R. S. Sodhi, P. P. Singh and S. Kaushal, Biosynthesis of NiO nanoparticles using Spirogyra sp. cell-free extract and their potential biological applications, Mater. Adv., 2022, 3, 4991–5000 RSC.
- S. G. Firisa, G. G. Muleta and A. A. Yimer, Synthesis of nickel oxide nanoparticles and copper-doped nickel oxide nanocomposites using Phytolacca dodecandra L'Herit leaf extract and evaluation of its antioxidant and photocatalytic activities, ACS Omega, 2022, 7, 44720–44732 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Paune, L. W. M. Lau, A. Gerson and R. St. C. Smart, X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- M. C. Biesinger, B. P. Paune, A. P. Grosvenor, L. W. M. Lau, A. Gerson and R. St. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- N. Fairley, et al., Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- R. P. Gupta and S. K. Sen, Calculation of multiplet structure of core p-vacancy levels. II, Phys. Rev. B: Solid State, 1975, 12, 15–19 CrossRef CAS.
- N. Weidler, J. Schuch, P. Knaus, S. Hoch, A. Maljusch, B. Schafer and W. Jaegermann, X-ray photoelectron spectroscopic investigation of plasma-enhanced chemical vapor deposited NiOx, NiOx(OH)y, and CoNiOx(OH)y: Influence of the chemical composition on the catalytic activity for the oxygen evolution reaction, J. Phys. Chem. C, 2024, 121, 6455–6463 CrossRef.
- M. A. Peck and M. A. Langell, Comparison of nanoscaled and bulk NiO structural and environmental characteristics by XRD, XAFS, and XPS, Chem. Mater., 2012, 24, 4483–4490 CrossRef CAS.
- A. F. Orliukas, T. Salkus, A. Kezionis, V. Venckute, V. Kazlauskiene, J. Miskinis, G. Laukaitits and J. Dudonis, XPS and impedance spectroscopy of some oxygen vacancy conducting solid electrolyte ceramics, Solid State Ionics, 2011, 118, 36–40 CrossRef.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, Use of oxygen/nickel ratios in the XPS characterisation of oxide phases on nickel metal and nickel alloy surfaces, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 159–166 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- A. Rohrbach, J. Hafner and G. Kresse, Molecular adsorption on the surface of strongly correlated transition-metal oxides: A case study for CO/NiO(100), Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075413 CrossRef.
- JCDPs no. 22-0752.
- Crystallography open database ID: 1011134.
- P. Dubey, N. Kaurav, R. S. Devan, G. S. Okram and Y. K. Kuo, The effect of stoichiometry on the structural, thermal and electronic properties of thermally decomposed nickel oxide, RSC Adv., 2018, 8, 5882–5890 RSC.
- Y. J. Lin, T. H. Su, P. C. Kuo and H. C. Chang, A source of free holes in NiO thin films with different nickel content that are prepared using the sol-gel method, Mater. Chem. Phys., 2022, 276, 125345 CrossRef CAS.
- Crystallography open database ID: 1010093.
- Crystallography open database ID: 1526380.
- J. Jickson, R. Ranjusha, S. S. Anoop, V. Amruthalakshmi, A. Amrutha, V. N. Shantikumar and B. Avinash, Shape tailored Ni3(NO3)2(OH)4 nano-flakes simulating 3-D bouquet-like structures for supercapacitors: exploring the effect of electrolytes on stability and performance, RSC Adv., 2014, 4, 39378–39385 RSC.
- W. Zhao, M. Bajdich, S. Carey, A. Vojvodic, J. K. Norskov and C. T. Campbell, Water dissociative adsorption on NiO(111): Energetics and structure of the hydroxylated surface, ACS Catal., 2016, 6, 7377–7384 CrossRef CAS.
- J. Goniakowski, F. Finocchi and C. Noguera, Polarity of oxide surfaces and nanostructures, Rep. Prog. Phys., 2008, 71, 016501 CrossRef.
- V. K. Lazarov, Z. Cai, K. Yoshida, K. H. L. Zhang, M. Weinert, K. S. Ziemer and P. J. Hasnip, Dynamically stabilized growth of polar oxides: The case of MgO(111), Phys. Rev. Lett., 2011, 107, 056101 CrossRef PubMed.
- V. K. Lazarov, R. Plass, H.-C. Poon, D. K. Saldin, M. Weinert, S. A. Chambers and M. Gajdardziska-Josifovska, Structure of the hydrogen-stabilized MgO(111)-(1 × 1) polar surface: Integrated experimental and theoretical studies, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115434 CrossRef.
- A. Kerrigan, K. Pande, D. Pingstone, S. A. Cavill, M. Gajdardziska-Josifovska, K. P. McKenna, M. Weinert and V. K. Lazarov, Nano-faceted stabilization of polar-oxide thin films: The case of MgO(111) and NiO(111) surfaces, Appl. Surf. Sci., 2022, 596, 153490 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Nickel hydroxide and related materials: A review of their structures, synthesis and properties, Proc. R. Soc. A, 2014, 471, 20140792 CrossRef PubMed and references therein.
- D. S. Hall, D. J. Lockwood, S. Poirier, C. Bock and B. R. MacDougall, Raman and infrared spectroscopy of α and β phases of thin nickel hydroxide films electrochemically formed on nickel, J. Phys. Chem. A, 2012, 116, 6771–6784 CrossRef CAS PubMed.
- X. Liu, J. Fu, S. He and H. Meng, Spectral analysis of the nickel hydroxyl and deuteroxyl nitrate powders, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 774, 012011 CAS.
- C. Pigenet and F. Fievet, Infrared absorption of NiO microcrystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 2785–2792 CrossRef CAS.
- W. Reichardt, W. Wagner and W. Kress, Lattice dynamics of NiO, J. Phys. C: Solid State Phys., 1975, 8, 3955–3962 CrossRef CAS.
- M. M. Lacerda, F. Kargar, E. Aytan, R. Samnakay, B. Debnath, J. X. Li, A. Khitun, R. K. Lake, J. Shi and A. A. Balandin, Variable-temperature inelastic light scattering spectroscopy of nickel oxide: Disentagling phonons and magnons, Appl. Phys. Lett., 2017, 110, 202406 CrossRef.
- D. Wang, S. Xu, L. Wu, Z. Li, P. Zhu and D. Wang, Spin-phonon coupling in NiO nanoparticle, J. Appl. Phys., 2020, 128, 133905 CrossRef CAS.
- A. Sunny and K. Balasubramanian, Raman spectral probe on size-dependent surface optical phonon modes and magnon properties of NiO nanoparticles, J. Phys. Chem. C, 2020, 124, 12636–12644 CrossRef CAS.
- D. Caso, A. Serrano, M. Jaafar, P. Prieto, A. Kamra, C. Gonzales-Ruano and F. G. Aliev, Microwave field-induced changes in Raman modes and magnetic force images of antiferromagnetic NiO films, Condens. Matter, 2024, 9, 7 CrossRef CAS.
- B. Karthikeyan, Raman spectral probed electron-phonon coupling and phonon lifetime properties of Ni-doped CuO nanoparticles, Appl. Phys. A, 2021, 127, 205 CrossRef CAS.
- J. Rumble, CRC Handbook of chemistry and physics, 102nd edn, 2021–2022 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03255a |
| This journal is © The Royal Society of Chemistry 2024 |