
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic applications of covalent organic frameworks: synthesis, characterization, and utility
Jared G.
Doremus
,
Bertha
Lotsi
,
Aadarsh
Sharma
and
Psaras L.
McGrier
 *
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: mcgrier.1@osu.edu
First published on 21st October 2024
Abstract
Photocatalysis has emerged as an energy efficient and safe method to perform organic transformations, and many semiconductors have been studied for use as photocatalysts. Covalent organic frameworks (COFs) are an established class of crystalline, porous materials constructed from organic units that are easily tunable. COFs importantly display semiconductor properties and respectable photoelectric behaviour, making them a strong prospect as photocatalysts. In this review, we summarize the design, synthetic methods, and characterization techniques for COFs. Strategies to boost photocatalytic performance are also discussed. Then the applications of COFs as photocatalysts in a variety of reactions are detailed. Finally, a summary, challenges, and future opportunities for the development of COFs as efficient photocatalysts are entailed.
1 Introduction
1.1 Principles of photocatalysis
Traditional synthetic methods can be costly, require high temperatures and harsh conditions, and need extensive workups. Thus, there has been a push to perform conventional reactions under more ambient and less energy-intensive means. One method to do this is photocatalysis and the use of light to drive chemical reactions. Total solar irradiation (TSI) provides an average annual solar radiation of 1361 W m−2.1 Although direct solar radiation at the Earth's surface is less due to absorption of sunlight by atmospheric gases and aerosols, this has the potential to power the world. Solar irradiation mainly has wavelengths around 300–2500 nm which includes ultraviolet (UV), visible, and near infrared (NIR) light. Markedly, solar irradiation is not evenly made up of UV, visible, and NIR light, and the actual composition is 7–8% UV, 42–44% visible, and 48–50% NIR light (Fig. 1).1,2 This illustrates that there is a clear demand for photocatalysts with high visible and NIR light absorption.3 Semiconductors are the most extensively studied materials for use as photocatalysts, especially metal-containing semiconductors. The most commonly used are TiO2, ZnO, WO3, CdS, BiVO4, and Fe2O3; however, organic dyes are proposed as non-metal alternatives.4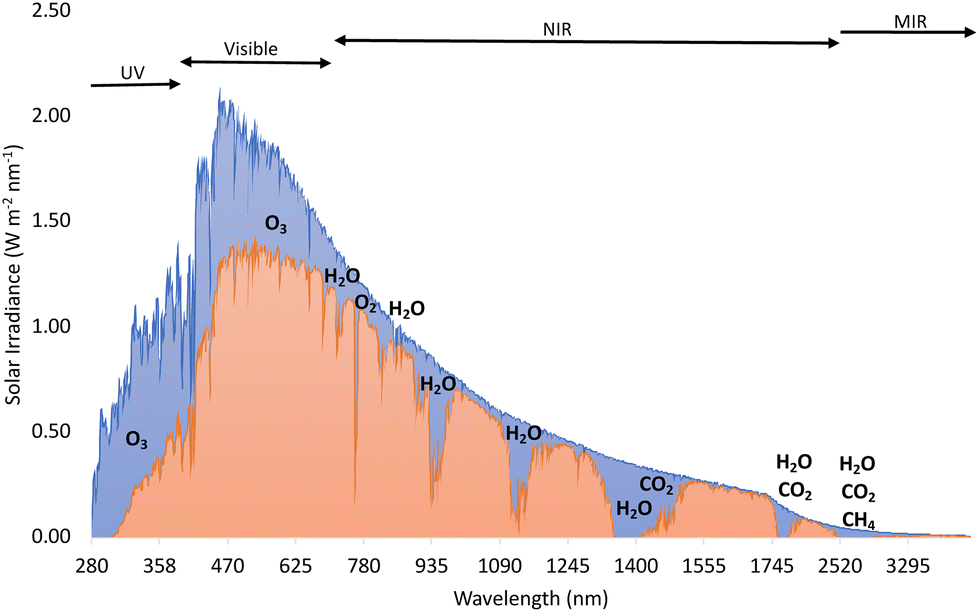 | ||
| Fig. 1 Total solar irradiance (area in blue) and direct solar irradiance (area in orange) with gases shown that cause the difference between them.2 | ||
Since there are discrepancies in the literature, it is important to define photocatalysis and photocatalysts. IUPAC's definition of photocatalysis is the “change in the rate of a chemical reaction or its initiation under the action of ultraviolet, visible or infrared radiation in the presence of a substance—the photocatalyst—that absorbs light and is involved in the chemical transformation of the reaction partners,”5 and its definition of a photocatalyst is a “catalyst able to produce, upon absorption of light, chemical transformations of the reaction partners…”.5 Thus in a conventional definition of catalysis the thermodynamics is not changed but rather the kinetics through the creation of a new route. Therefore, if a material or substance uses light to drive a downhill thermodynamic process, it is a photocatalyst. On the other hand, when a material or substance uses light to facilitate uphill thermodynamic reactions the reaction should be called photosynthesis instead, and the material or substance is not a photocatalyst. The discrepancy arises from the fact that if the photon is considered as a reactant, then the material or substance could be classified as a photocatalyst. Herein, we will use the broader definition of a photocatalyst that can drive either uphill or downhill thermodynamic processes.
Photocatalysis can broadly be depicted in four steps. (1) Light is absorbed. (2) Electron–hole pairs, or excitons, are generated, and the electron–hole pairs are separated by charge. (3) The separated electrons and holes move from the bulk material to the surface. (4) The charges on the surface participate in redox chemical reactions (Fig. 2). However, there are competing events that can occur that limit the photocatalytic process. Foremost, like any substance in an excited state, the photocatalyst can relax and return to the ground state after being excited by light. Due to coulombic interactions, recombination of the separated electrons and holes can occur either in the bulk material or at the surface. Recombination disperses the initial absorbed light energy by emitting heat (non-radiative) or light (radiative). Trapping of the charges in the bulk material can also occur and the trapping states have little to no reactivity, and the charges eventually hop trap-to-trap until they recombine. Notably, materials displaying high crystallinity, and thus few defects, can suppress recombination events as the charges can separate easier and have higher mobility.6–8 Defect sites can trap electrons and holes and cause recombination. Smaller particle sizes and thickness can also limit recombination, as the charges have shorter distances and require less time to travel to the surface.6–8
 | ||
| Fig. 2 Photocatalytic process in semiconductors. The (i) light absorption, (ii) generation and separation of electron–hole pairs, (iii) migration of electrons and holes to the surface of the semiconductor, (iv) redox reactions at the surface of the semiconductor, (v) relaxation of the excited electron and recombination with the hole, (vi) recombination of electron–hole pairs in the bulk or on the surface of the semiconductor, (vii) trapping of electrons and holes in the bulk semiconductor. | ||
In semiconductors, there is an energy gap between the valence band and conduction band, called the band gap, and this is crucial in photocatalytic applications (Fig. 3). When a photon has the energy equal to or greater than the band gap of the semiconductor, it will absorb the photon and become excited, which promotes an electron from the valence band to the conduction band and leaves a hole in the valence band, thus creating the electron–hole pair. Again, these electrons and holes participate in oxidation and reduction half reactions, and the potentials are derived from the valence band maximum and conduction band minimum, respectively. The conduction band potential of the photocatalyst must be lower, or more negative, than the potential of the substrate, and likewise the valence band potential must be higher, or more positive, than the substrate's potential. For example, in the reduction of N2, at pH = 0 versus NHE, the conduction band of the photocatalyst needs to be below 0.55 V and the valence band of the photocatalyst needs to be above 1.33 V. In saying this, it is ideal for a photocatalyst to have low valence band and high conduction band potentials to increase the thermodynamic driving force. However, this leads to a large band gap and thus narrow light absorption. Therefore, there is a give-and-take relationship in creating optimal semiconductor photocatalysts.
 | ||
| Fig. 3 Energy level and band diagram for semiconductors. | ||
Photocatalytic activity has been defined in multiple ways in the literature. Experimentally, yields are typically measured by the moles of product per unit mass of photocatalyst per time, such as μmol g−1 h−1, though other units can be seen as well, including μmol g−1 or μM h−1. This way of defining photocatalytic activity makes it hard to compare various systems because of the difference in the power density and wavelength of light used, the use of any sacrificial reagents, the amount or moles of catalyst used, the overall reaction time, the pH of the solution, and other factors that affect photoactivity. A better method to determine photocatalytic activity is the quantum yield (QY).9 This is the ratio of the number of electrons consumed in the reaction to the number of absorbed photons in a certain wavelength range. The drawback of this method is the lack of accuracy in determining the actual number of absorbed photons, especially in heterogeneous systems, as photons can scatter, reflect, and not be absorbed, and because of monochromaticity and other experimental design reasons. A way around this is by using only incident light, and this is termed the apparent quantum efficiency (AQE) or apparent quantum yield (AQY). AQY is the number of product molecules multiplied by the number of electrons consumed over the total number of incident photons. However, photon scattering still affects this measurement but to a lesser degree, and this method assumes light absorbed is the same in each experiment.9 Notably, coloured reactants, intermediates, and products can skew these values.
1.2 Photophysical properties of conjugated polymers
Conjugated polymers have widely been used as semiconducting materials due to their photophysical properties.10–12 π-Conjugated polymers have even been applied as photocatalysts as far back as 1985 by Yanagida et al. using poly(p-phenylene) for photocatalytic water splitting.13 A significant reason conjugated polymers have been used as photocatalysts is their heterogeneous nature that allows for easy separation of them from the reaction mixture. Conjugated polymers also have limited photobleaching and avoid bimolecular encounters that homogeneous catalysts experience in solution. Moreover, conjugated polymers that are two-dimensional or three-dimensional display anisotropy, allowing for more tunable properties. For instance, the band gap of common semiconductors increases with decreasing layer thickness.14 Comparatively to the aforementioned inorganic semiconductor photocatalysts, organic polymer photocatalysts have more tunable characteristics as the molecular backbone can be modified by varying the monomers, changing the ratio of monomers, or by adding functional groups. This affords conjugated polymers the capacity to alter their band gaps along with their photophysical properties and photocatalytic activity. Conjugated polymers with high molecular weights typically display higher photostability than homogeneous systems.15,16 Even simple two-dimensional organic systems such as graphene, graphene oxide, and g-C3N4 are more diverse and tunable than traditional inorganic semiconductors. π-Conjugated polymers are commonly used in photocatalysis because they have wide-ranging light absorption so they can more readily absorb visible light and induce a π–π* transition to generate excitons.13 Furthermore, these polymers are large π-conjugated systems with low electric resistivities, so charge carriers have high mobilities and protracted lifetimes, which leads to better photocatalytic performance.1.3 Brief introduction to covalent organic frameworks
Covalent organic frameworks (COFs) are a class of crystalline, porous polymers.17,18 COFs are constructed from organic building blocks linked through covalent bonds in dynamic and reversible processes through reticular chemistry. COFs feature low density, and good chemical, mechanical and thermal stability which make them a good platform for a myriad of applications.19–21 Their bottom-up approach also allows for superior tunability and creation of designable structures, topology, morphology, pore shape and size, functionality, and more. Importantly for photocatalysis, COFs exhibit semiconductor behaviour and alterable band structure. COFs share the same advantages in photocatalysis as conjugated polymers as mentioned previously. The tunability of COFs allows for the incorporation of efficient light-absorbing units and certain moieties to increase their photocatalytic performance. A COF's crystallinity helps to create high charge carrier mobility and efficient charge separation of the excitons. Their high porosity enables good exposure to active sites, high diffusion rates, and convective mass transfer. The stability of COFs also provides resistance to photocorrosion, resulting in long photocatalytic lifetimes. Finally, their well-defined structures make it easier to study and understand the structure–property relationship and catalytic mechanism. This will be further discussed in the following sections.1.4 Summary of review
COFs have been catapulted to the forefront as materials for photocatalytic applications in recent years, yet only a handful of reviews have been reported.22–29 Since many of these reviews only discuss hydrogen evolution, water splitting, carbon dioxide reduction, or pollution degradation, we aim to present a comprehensive overview of COFs for photocatalysis with an emphasis on the most recent work that has been reported in the field. We begin by exploring the design, linkages, morphologies, and synthesis of COFs. This is followed by the characterization methods of COFs, especially ones pertaining to photocatalytic applications. A brief elucidation about techniques for photocatalytic product testing and mechanistic studies is conducted. The advantages of COFs towards photocatalysis and strategies to improve performance are examined. Lastly, the application of COFs as photocatalysts in a wide array of reactions is discussed along with the current weaknesses, future opportunities, and proposed areas for improvement.2 COF design and synthesis methods
2.1 Engineering functional design of monomers
COFs are formed through the combination of symmetrical monomers linked by covalent bonds. COFs can be tailored and engineered for a variety of applications by varying the organic monomers and the linkages of the materials. Any conceivable COF can be synthesized using a variety of methods, and they can also be post-synthetically modified to alter their properties.While covalent organic frameworks can be made from a plethora of exotic linkers, there are common archetypes. Typical organic linkers are based on benzene, biphenyl, triphenylamine, triphenylbenzene, triphenyltriazine, tetraphenylmethane, phenylethynylbenzene, pyrene, thiophene, porphyrin, and more (Fig. 4). There is also the ability to design the organic monomer to host a metal and incorporate it into the COF whether pre- or post-synthesis.30 Common metal-binding ligands are based on bipyridine, phenanthroline, pyrazole, catecholate, porphyrin, phthalocyanine, N-heterocyclic carbene (NHC), salen, dehydrobenzoannulene, and triphenylphosphine (Fig. 5). Notably, there is a trade-off between commercial availability and synthetic cost versus complexity when designing and obtaining the building blocks.
 | ||
| Fig. 4 Typical COF organic building motifs. | ||
 | ||
| Fig. 5 Common COF units for metalation. | ||
The first reported COFs by Yaghi, including COF-1 and COF-5, were based on a boroxine and boronate ester linkage, respectively.31 Other boron-based linkages include borosilicate,32 borazine,33 and spiroborate34 (Fig. 6). These linkages are highly reversible in nature and result in highly crystalline COFs but suffer from instability in humid or protic conditions. Therefore, they must be utilized under anhydrous conditions, which limits their use in practical applications.
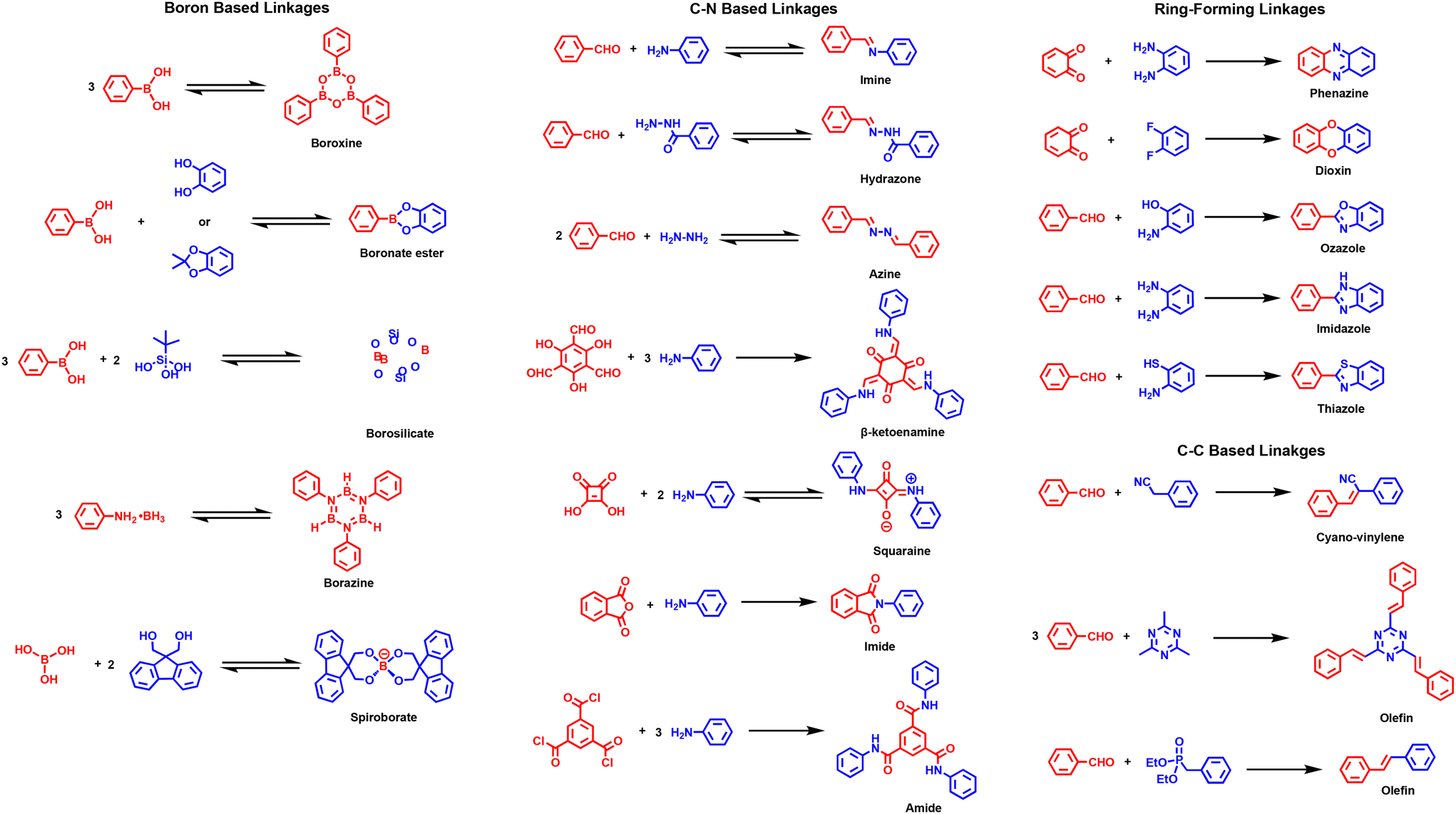 | ||
| Fig. 6 Various formation of COF linkages. | ||
The most studied linkages in COFs are based on C–N bonds. These include, but are not limited to, imine, hydrazone, azine, β-ketoenamine, squaraine, amide, and imide linkages (Fig. 6). The first C–N-based COF was COF-300 and based on an imine linkage.35 Although imine bonds are much more stable than their boron-based counterparts, they still suffer from limited stability in highly acidic conditions and in the presence of strong nucleophiles. Imines can also be converted to other linkages such as amide,36 quinoline,37 benzoxazole,38 benzothiazole,39 and others.40 The chemical stabilities of boron- and imine-linkages can be improved by incorporating hydrogen bonding interactions41–43 and alkyl or alkoxy chains.44,45
The first hydrazone-based COFs, COF-42 and COF-43, were reported in 2011.46 Hydrazones have similar properties to imines but are slightly more stable due to additional hydrogen-bonding interactions. Hydrazones are less susceptible to hydrolysis in acidic conditions but are still fragile to nucleophiles. However, azine-linked COFs, first reported in 2013,47 have superior stability in aqueous solution, acidic or basic conditions, in the presence of nucleophiles and a plethora of polar solvents. The β-ketoenamine linkage has also been used due to its superior chemical stability.48 By adding hydroxyl groups adjacent to the aldehydes, enol–keto tautomerism is enabled where the crystallinity of the COF is dictated by the initial reversible Schiff-base imine formation followed by the irreversible tautomerism to form the highly stable β-ketoenamine linkage. However, this linkage has limited utility, as 1,3,5-triformylphloroglucinol is the typical monomer of choice due to the synthetic difficulty of installing alcohol substituents in the ortho position of aldehydes on most building blocks.
A squaraine-linked COF was synthesized in 2013.49 The zigzag conformation inhibits the layered COF from sideslip and exhibits high stability in most solvents. Its zwitterionic nature also allows for extended π-conjugation through resonance. Similarly, imide-linked COFs have outstanding stabilities due to the overall irreversibility of the final bond formation. Although the reaction as a whole is considered irreversible, the first step of the nucleophilic attack of the alcohol is reversible, which allows the formation of a crystalline polymer. The first imide-linked COF was reported in 2014, and it required high reaction temperatures of 200–250 °C for 5–7 days for defect healing.50 The first directly synthesized amide-linked COF was documented in 2017.51 Since the reaction between acyl chlorides and amines is irreversible, amide-linked COFs are very difficult to synthesize directly and result in lower crystallinities. In the first documented amide COF work, the synthesis involved stirring at 0 °C followed by heating at 250 °C under vacuum to allow for slow reaction time followed by high heat for error correcting. More common routes for amide-linked COFs involve post-synthetic modification, which will be discussed more in Section 2.2.9. The amide linkage produces the COF with exceptional stability in a variety of aqueous and organic solvents and in the presence of many chemical reagents.
In addition to C–N-based COF linkages, C–C-based linkages are also of interest due to their robustness, lack of bond polarization, and ability to delocalize π-electrons permitting COF-based systems with extended conjugation. The two main types of C–C bond linkages consist of cyano-olefin and olefin (Fig. 6). Due to the complete irreversibility of the olefin bond, COFs linked through C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds suffer from low crystallinities. The first cyano-vinylene COF was synthesized in 2016 though Knoevenagel condensation of an aldehyde and a benzyl cyanide.52 Adjusting the base and temperature, the Knoevenagel condensation can be fairly reversible in the initial steps before the CC bond is formed resulting in a crystalline material. The presence of the cyano substituent reduces the overall stability of the olefin bond due to its electron-withdrawing nature, and it can also react with harsh nucleophiles such as Grignard and lithium reagents. To remedy this, olefin-linked COFs can be synthesized by an aldol condensation to avoid these issues.53,54 The initial reversibility of the aldol condensation permits error correcting before the irreversible olefin bond is formed creating a crystalline material. Although the olefin-linkage is highly stable, the design is limited for this type of reaction and typically relies on monomers such as 2,5-dimethylpyrazine, 2,4,6-trimethyltriazine, and 2,4,6-trimethylpyridine. There are also rare reports of making a heteroatom-free olefin-linked COF using the Horner–Wadsworth–Emmons reaction.55,56
C bonds suffer from low crystallinities. The first cyano-vinylene COF was synthesized in 2016 though Knoevenagel condensation of an aldehyde and a benzyl cyanide.52 Adjusting the base and temperature, the Knoevenagel condensation can be fairly reversible in the initial steps before the CC bond is formed resulting in a crystalline material. The presence of the cyano substituent reduces the overall stability of the olefin bond due to its electron-withdrawing nature, and it can also react with harsh nucleophiles such as Grignard and lithium reagents. To remedy this, olefin-linked COFs can be synthesized by an aldol condensation to avoid these issues.53,54 The initial reversibility of the aldol condensation permits error correcting before the irreversible olefin bond is formed creating a crystalline material. Although the olefin-linkage is highly stable, the design is limited for this type of reaction and typically relies on monomers such as 2,5-dimethylpyrazine, 2,4,6-trimethyltriazine, and 2,4,6-trimethylpyridine. There are also rare reports of making a heteroatom-free olefin-linked COF using the Horner–Wadsworth–Emmons reaction.55,56
Ring-forming reactions have also been explored to create COF linkages due to ring systems exhibiting exceptional stability and their capacity to extend conjugation. The first example of a ring-forming linkage was a phenazine-linked COF reported in 2013.57 In 2018, dioxin-linked COFs were synthesized through a nucleophilic aromatic substitution reaction.58 It was proposed that the nucleophilic attack is reversible, and the ring closure is irreversible, yielding crystalline materials that were highly stable. Also, benzoxazole-linked COFs were reported in 2018 displaying excellent stability in aqueous, basic, acidic, and other media.59 The reaction first occurs in a reversible imine bond formation, allowing for error correction, followed by the irreversible oxazole ring cyclization and oxidation/dehydrogenation. In 2019, an imidazole-linked COF was synthesized which exhibited great stability.60 Thiazole-linked COFs were directly synthesized in 2020 using elemental sulfur and DMSO.61 A highly crystalline material was made by undergoing a reversible imine condensation followed by an electrophilic attack with sulfur which undergoes irreversible cyclization and oxidative aromatization to give the highly stable COF. Notably, some of these mentioned ring-linked COFs, and others, are commonly made through post-synthetic modification of the imine linkage, which will be discussed further in Section 2.2.9.
1D COFs provide a unique platform for photocatalysis64,65 and other applications.66–68 In 2D and 3D COFs the active sites and/or functional moieties typically reside within their pores and channels, which may inhibit access to them or require longer reaction times to allow for diffusion. Comparatively, 1D COFs are extended in a linear fashion by using a C2 monomer to essentially cap the COF and prevent extension in a second direction (Fig. 7). The one-dimensionality can also facilitate easier exfoliation of layers to reduce aggregation-based quenching while also allowing for exterior functionalization at the lateral positions. Currently, there is a lack of development of 1D COFs for photocatalysis.
 | ||
| Fig. 7 Synthesis of 1D Tpy-COF.65 | ||
2D COFs are the most studied and prevalent in the literature due to the reliance of utilizing rigid, planar molecules with π-backbones that induce π–π stacking, and the diversity of monomers. The building blocks can have a variety of symmetries to afford isotropic topologies and anisotropic topologies. Some typical 2D COF topologies are hexagonal, tetragonal, rhombic, kagome, and triangular (Fig. 8). Hexagonal COFs can be made from C2 + C2 + C2, C2 + C3, and C3 + C3. Tetragonal COFs can be made from C2 + C4 and C4 + C4. Rhombic COFs can be made from C2 + C2. Kagome COFs can be made from C2 + C2 or C2 + C4. Triangular COFs can be made from C2 + C6 or C2 + C3. These topologies all result from a two-component 1 + 1 strategy (e.g. one unique aldehyde monomer combined with one unique amine monomer), but a multicomponent 1 + 2 or 1 + 3 strategy (e.g. one unique aldehyde monomer combined with two or three different amine monomers) can result in dozens of asymmetric topologies. Notably, changing the monomers and topologies will affect the pore size and column π-density. Additionally, 2D COFs provide a good platform for making thin films whether through interfacial synthesis, exfoliation methods, or fabrication on a substrate. These methods will be discussed in Section 2.2.7. Concerning photocatalysis, 2D COFs are what is typically used, but again can suffer from aggregation-based quenching due to the strong π–π stacking from the layers.
 | ||
| Fig. 8 Reported periodic nets of 2D COFs. | ||
3D COFs are less prevalent than their 2D counterparts but are important and just as, if not more, catalytically active. To synthesize a 3D COF, there needs to be at least one monomer that has Td, D3h, or D2h symmetry (tetrahedral, triangular prism, or cubic symmetry, respectively) that extends it out into three dimensions. There are not many examples of Td monomers in the literature and these monomers are archetypally based on tetraphenylmethane, tetraphenylsilane, or adamantane. The synthetic difficulty of making 3D building units or building units with high connectivity (≥5) has limited the construction of many unique 3D COFs. The first 3D COFs were reported in 2007 by Yaghi.69Td monomers have been used to synthesize 3D COFs with ctn,69 bor,69 dia,35 pts,70 rra,71 lon,72 ljh,73 and qtz74 topologies. D3h monomers have been used to make 3D COFs with tbo,75 stp,76 ceq,77 acs,77 hea,78 and nia79 topologies. D2h monomers have been used to synthesize COFs with ffc,80 fjh,81 scu,82 bcu,83 pcb,84 the,85 flu,86 pto,87 mhq-z,87 and crb88 topologies. Using the linkage design, spiroborate-linked COFs have also taken nbo89 and rra71 topologies and a silicate-linked COF making a srs90 topology. The use of Cu metal complexes as building units has unlocked other “woven” topologies.91 The soc and spn topologies were also achieved by using a TiIV complex as a building unit.92,93 pcu,94 she,95 sql-c,96 tty97 topologies have also been reported through the use of complex building units. Although new topologies of 3D COFs are reported every year, the number of 3D COF topologies that exist are still limited in comparison with the topologies of other 3D materials and the >3500 topologies that are theoretically plausible.98 Due to multi-folding and interpenetration, it is difficult to try and pre-design a 3D COF since the parameters that control the folding mechanism are not fully understood.99 In the same vein, 3D COFs typically result in much lower porosity than theoretically determined due to folding, interpenetration, and the inability to remove unreacted monomer from the pores. Overall, 3D COFs have excellent potential in many applications due to their capacity to achieve high porosities and many topologies. Specifically, for photocatalysis, the three-dimensional nature can also affect the photophysical properties and reduce aggregation-based quenching due to the absence of 2D layers.
2.2 Synthetic methods
The synthesis of COFs is dependent on controlling the rate at which the covalent bonds are formed. If a reaction proceeds too quickly an amorphous material will be produced. Allowing the covalent linkages to error correct enables the construction of a crystalline material. In essence, COF synthesis requires a self-healing period to correct any defects during the nucleation and growth processes. Thus, there are many synthetic methods that have been used and developed in order to prepare highly crystalline COFs.
 | ||
| Fig. 10 Synthesis scheme of COF thin films. (a) Schematic representation of the interfacial crystallization process used to synthesize the Tp-Bpy thin film. The bottom colorless layer corresponds to aldehyde in dichloromethane solution, the blue layer contains only water as the spacer solution, and the top yellow layer is the Bpy amine-PTSA aqueous solution. (b and c) SEM and AFM images (with corresponding height profile), respectively, of the Tp-Bpy thin film synthesized as illustrated in (a). (d) Chemdraw structures of all the COFs used for synthesizing the thin films via interfacial crystallization process.123 Reproduced with permission from ref. 123. Copyright 2017, American Chemical Society. | ||
The first single crystal of a COF was reported by the Yaghi and Wuest groups in 2013.132,133 Wuest achieved this through the reversible self-addition polymerization of tetranitroso monomers to form a 3D COF. Zhao and co-workers were able to make micron-sized single-crystalline COFs by studying the mechanism of the growth and nucleation through intermediate tracing.134 Yaghi and co-workers developed a modulation method to grow large single-crystals.72 Wei et al. performed crystallization of COFs using supercritical CO2 to vastly reduce times to form single-crystalline COFs, and these showed heightened photoconductive properties than the polycrystalline counterpart.135 Wang et al. were the first to synthesize a 3D COF with a high (6-fold) connectivity.136 This lab also was the first to construct a non-interpenetrated 3D COF single crystal.137 Loh and co-workers were the first to synthesize single crystals of a 1D COF.138 Zheng et al. were able to grow 2D COF thin films with single-crystalline domains.127 They also explored the use of amino-acid derivatives to grow gram-scale single-crystalline imine COFs.139 The authors believe that the amino acid based compounds can self-assemble into micelles and regulate the polymerization and crystallization processes. Dichtel developed a method to grow single-crystalline 2D COFs by slow monomer addition and seeded growth.140 The Jiang group also performed post-synthetic modification on single crystals of an imine-linked COF, USTB-5, where the imine was reduced or oxidized to yield amine and amide linkages, respectively.141 Whereas COF single-crystal growth takes weeks to months, Wang et al. created a method to grow COF single crystals in only one or two days using trifluoroacetic acid (TFA) as a catalyst and trifluoroethylamine as a modulator.142 Although a sizable amount of work has been performed to grow single-crystalline COFs, there still is a need to create better, quicker, translatable, and scalable methods that can be widely applied beyond imine- and boronate ester-linked COFs.
3 COF characterization
Characterization techniques are essential for evaluating the structural, compositional, and functional properties of COFs. The choice of techniques depends on the specific properties that researchers aim to investigate. The following are some general techniques and the corresponding broad evaluations for insights into such materials.3.1 Basic COF characterization methods
Understanding the intricate world of covalent organic frameworks (COFs) hinges on a battery of investigative techniques. These methods act like a key, unlocking the secrets of a COF's structure and properties, which ultimately determine its potential applications. Powder X-ray Diffraction (PXRD) serves as a foundational tool, offering a glimpse into the COF's internal organization and order. Fourier-Transform Infrared Spectroscopy (FTIR) acts as a chemical detective, scrutinizing the building blocks and ensuring the COF is free from unwanted remnants. Visualizing the COF's physical form comes into play with scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM provides a surface-level perspective, revealing the COF's external features and size. TEM delves deeper, showcasing the intricate network of pores and the overall framework organization at an incredible nanoscale. For applications like gas storage or catalysis, N2 sorption analysis is a crucial technique. It assesses the COF's capacity to encapsulate gas molecules, providing vital details about the available surface area, pore size distribution, and total pore volume. Solid-state nuclear magnetic resonance (SS-NMR) spectroscopy, specifically 13C cross-polarization magic angle spinning (13C CPMAS-NMR), acts as an advanced probe, examining the COF's internal chemical environment. By analysing the magnetic fields surrounding specific atoms, researchers can confirm the successful formation of the desired linkages and identify any functionality flaws or impurities within the COF. This suite of characterization methods empowers researchers to unlock the potential of COFs and pave the way for their advancement in various technological fields.3.2 COF characterization for photocatalytic application
The photoelectric potential of COFs can be evaluated using characterization techniques as well. The following are some general techniques and the corresponding photoelectrical evaluations for insights into such materials.4 Product detection and mechanistic studies in photocatalytic experiments
After conducting various photocatalytic reactions, it is crucial to use a range of analytical instruments and techniques to analyse the reaction mixture. Here we present some of the most generally used methods for analyses of product and intermediate formation in photocatalytic reactions and generic tests for mechanistic studies.4.1 Nuclear magnetic resonance (NMR)
NMR has been widely utilized as an analytical tool for most photocatalytic reactions due to its ability to provide extensive information about a compound's molecular structure, including the chemical environment of specific nuclei and the relative arrangement of atoms.165–167 Within a molecule, NMR can identify functional groups, allowing for the confirmation of product structure.168,169 This analytical technique can also detect by-products and impurities in samples.170,171 NMR is useful for quantitative analysis, enabling the calculation of the relative amounts of different constituents in mixtures.172,173 NMR provides benefits for investigating molecular dynamics, allowing for real-time monitoring of chemical reactions and improving the comprehension of reaction kinetics and mechanisms.174,175 It can also be employed for analysing isotopic labelling of compounds, providing more detailed insights into the mechanistic pathways of various reactions.174,175 LED-NMR has been used to study photocatalytic processes under light irradiation.176–179 Specifically, in heterogeneous photocatalytic reactions, operando NMR can be utilized to monitor proton transfer.180 As a non-destructive technique, NMR allows the same sample to be reused for other analyses.4.2 Gas chromatography (in situ and ex situ)
Gas chromatography (GC) is a high-resolution method to separate complex mixtures, enabling the identification and quantification of various constituents.181,182 GC efficiently separates and detects sample components which can enable the determination of by-products, intermediates, and impurities. This unique capability makes it valuable in photocatalytic reactions.183,184In situ GC is employed for real-time observation and monitoring of reaction mechanisms and kinetics during the reaction process, allowing for the tracking of dynamic changes. In situ GC is also invaluable for analysing gaseous products.185–187Ex situ GC is useful after a reaction is complete, providing thorough details on the composition of the final reaction mixture, including the quantification and identification of products, by-products, intermediates, and impurities, which helps optimize reaction conditions to improve yields.188 The versatility of GC to be compatible with different detectors, such as a Mass Spectrometer (MS),189 Flame Ionization Detector (FID),190 and Electron Capture Detector (ECD),190,191 gives it an advantage in analysing various types of sample products. Both in situ and ex situ GC are indispensable for their in-depth characterization of reaction mixtures, especially since small sample amounts are required.4.3 Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) is a crucial analytical tool extensively used in the characterization of polymers, catalysts, and solid samples overall.192 One of its advantages over general transmission IR techniques is its ability to amplify weak signals for improved detection of solid surfaces. Due to its surface sensitivity, DRIFTS can identify surface functional groups and detect species adsorbed on the surfaces of solid materials.193,194 It is employed in dynamic studies such as phase transitions, adsorption, and desorption.192–194In situ and operando DRIFTS enables the observation and characterization of the material or catalyst during the reaction process.195–197 By analysing the multiple output IR spectra, DRIFTS facilitates real-time monitoring of chemical reactions, aiding in developing reaction kinetics and mechanisms. In situ DRIFTs has been applied successfully to monitor photocatalytic transformations.198–202 Additionally, DRIFTS is user-friendly as it allows for the analysis of materials in their solid state, avoiding the use of solvents and possible contamination and requiring only a small sample size for analysis. As a non-destructive technique, it allows the same sample to be used in subsequent analyses by other methods.4.4 Scavenger tests
Scavenger tests (STs) are vital, particularly in photocatalytic studies, as they provide an in-depth understanding of photocatalytic processes.203 These tests can verify reactive oxygen species (ROS) such as superoxide radicals (˙O2−), singlet oxygen (1O2), hydroxyl radicals (˙OH), hydrogen peroxide (H2O2), and others. The scavenging of photogenerated electrons and holes can also be done to verify their use in photocatalytic reactions. By investigating the reactive species involved, scavenger tests help elucidate the mechanisms in photocatalytic processes.204 By determining the reactive species and mechanism, STs can lead to improved reaction conditions, resulting in higher reaction efficiency. They can also serve as benchmarks for comparing the performance and capabilities of various photocatalysts based on their generation and utilization of reactive species. Additionally, these tests can help explain the degradation pathways of reactions and identify the roles played by all involved species.203Since STs are useful for detecting and identifying reactive species and intermediates formed during a reaction, a mechanistic pathway can be determined by adding scavengers to inhibit various reactive species and observing the impact on the reaction. This is done by adding a specific scavenger to a photocatalytic reaction where the scavenger selectively reacts with the targeted reactive species and efficiently scavenges them if present. Various scavengers are used to inhibit specific reactive species in photocatalytic reactions. For hydroxyl radicals, common scavengers include alcohols (however, the accuracy is questioned),203,205–207 carboxylic acids,208,209 or dimethylsulfoxide.210p-Benzoquinone and its derivatives or superoxide dismutase are primarily used for scavenging superoxide radicals203,211 and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) is a good trap for detection.212,213 Singlet oxygen is typically scavenged by sodium azide or 1,4-diazabicyclo[2.2.2]octane (DABCO),203,214,215 and 2,2,6,6-tetra-methyl-piperidine (TEMP) is a good trap for detection.216 Ethylenediaminetetraacetic acid (EDTA), oxalic acid, alcohols, thiols, and amines serve as scavengers for holes.217 Silver nitrate, copper chloride, iodates, and persulfates are used to scavenge electrons.217 For instance, in a photocatalytic reaction, if the addition of p-benzoquinone significantly decreases the product, this indicates that superoxide radicals play a vital role in the reaction. Similarly, if the addition of silver nitrate reduces product formation, it indicates the involvement of photogenerated electrons. Overall, scavenger tests are instrumental in testing hypotheses related to photocatalytic reactions.
4.5 Product, by-product, and intermediate detection tests
Photocatalytic reactions can produce various products, by-products, and intermediates depending on the reaction conditions, the selectivity of the catalyst, and how long the reaction is conducted. Here we detail the products, by-products, and intermediates of common photocatalytic reactions and tests to identify them. For example, in photocatalytic water oxidation, the O2 product can react with protons and electrons to form hydrogen peroxide.218,219 Similarly in water splitting reactions, water can react with photogenerated holes to form hydrogen peroxide.220,221 In photocatalytic CO2 reduction, the products and intermediates can include carbon monoxide, formate, formaldehyde, alcohols, and hydrocarbons depending on the desired product.222–224 Additionally, in aqueous conditions the reaction competes with HER.224 Photocatalytic hydrogen peroxide production may generate water and oxygen from the decomposition of hydrogen peroxide, especially under light irradiation or with a suitable catalyst.225–227 Photocatalytic aryl boronic acid oxidation generates boric acid, (BOH)3, as the by-product, and likewise oxidation of boronic esters yields B(OH)(OR)2 as the by-product.228–230 During photocatalytic sulfide oxidation reactions, sulfoxides and sulfones can be made.231–233 To afford the sulfone, the sulfoxide is first made and is the intermediate oxidation state; however, sulfoxides are important products so oxidation to the sulfone is not always wanted. Common by-products and intermediates produced from photocatalytic benzyl alcohol oxidation include H2O2, which can form during advanced oxidation processes, and benzoic acid from overoxidation which can undergo esterification with benzyl alcohol to form benzyl benzoate.234–236 In photocatalytic oxidative amine coupling reactions, typical intermediates are the imine and benzaldehyde derivatives, and the by-products include ammonia and H2O2, which can form during advanced oxidation processes.237–239 In the photocatalytic degradation of organic pollutants, various intermediates and by-products are generated, including small organic acids such as acetic acid, oxalic acid, and formic acid, which result from the partial oxidation of larger organic molecules.240 Additionally, aromatic compounds, including benzene derivatives and phenols, can be produced from the incomplete degradation of more complex aromatic compounds.240With this in mind, it is important to verify these compounds via qualitative or quantitative tests to elucidate the reaction mechanism. As discussed in Sections 4.1–4.4, various analytical techniques can be utilized to detect products, by-products, and intermediates resulting from photocatalytic reactions. Another technique to characterize products and by-products in the post-reaction mixture is UV-Vis spectroscopy. Nosaka and Nosaka detail the many methods including UV-Vis, IR, EPR, chemiluminescence reactions, and fluorescence products to detect reactive oxygen species.241 Additionally, detecting hydrogen peroxide through a coloration method can be done using iodide,242 Ti4+,243 or N,N-dimethyl-p-phenylenediamine244 or by fluorescence methods.241 This can also be done through titration or calibrated UV-Vis to obtain quantitative data, but colorimetric test strips can also be used for qualitative detection.245 To illustrate, UV-Vis spectroscopy is one of the employed analytical techniques to confirm the formation of H2O2 in a photocatalytic reaction, as H2O2 can oxidize iodide ions (I−) to form iodine (I2), which subsequently generates tri-iodide (I3−) in the presence of excess iodide. I3− exhibits two characteristic peaks at approximately 300 and 350 nm in UV-Vis spectra.241 Ammonia can be detected using a coloration method using Nessler's reagent or the indophenol blue method, or by ion chromatography.246–248 Notably, Nessler's reagent can be used in any conditions but is very toxic and should be avoided if possible. The iodophenol blue method can only be used in neutral or basic conditions, and ion chromatography can only be done in acidic or neutral conditions. These methods all have their advantages and drawbacks, including varying accuracy and interference of other substances, but can easily be used in qualitative detection.246–248 Safin et al. detailed a plethora of tests for analysing different functional groups and compounds including aldehydes, carboxylic acids, amines, esters, and sugars.249 These simple coloration tests can easily be applied for qualitative detection of compounds post reaction. Boric acid can be detected using Azomethine H as a colorimetric reagent.250 Undoubtedly, there are other quantitative and qualitative tests for photocatalytic reactions not described here.
5 Gearing COFs for photocatalysis – advantages of COFs and enrichment
5.1 Extending conjugation and UV-visible light absorption
In order for COFs to be eligible for photocatalytic application, they have to be able to absorb light especially in the ultraviolet (UV), visible light, and/or near IR spectrum. This is essential, as the harvesting of light is what initiates the formation of excitons, or electron–hole pairs, due to the excess energy gained. 2D COFs have an advantage with light absorption as the layered sheets stack and afford conjugation though π–π stacking. However, 1D and 3D COFs do not have this capability. Thus, in order to achieve a COF that has good light-absorbing properties, the electronic properties of the monomers and the chemical linkage that is used are imperative. For example, Jiang and co-workers synthesized a COF using triphenylene hexamine and tert-butylpyrene tetraone to form a phenazine-linked COF that exhibited extended π-delocalization.57 This COF also showed utility for high on–off ratio photoswitches and photovoltaic cells. Bromley and Bredas conducted theoretical studies showing the importance of π-conjugation of COFs and the ability of monomer design to realize conjugation.251,252 De Smet and co-workers even showed that the degree of methylation of monomer units affected the absorption and the band gap of the COF.253 Van Der Voort and cohort explicitly detailed that extending the π-conjugation, through monomer and linkage, led to more efficient photocatalytic hydrogen peroxide production.254 Zhang et al. illustrated that adding an alkynyl unit in the monomer led to broader visible-light absorption, narrower optical bandgaps, and much higher photocatalytic amine coupling performance than its counterpart without the alkyne.2555.2 Band gap engineering
Beyond the absorption of light, band gap engineering of the COF is just as crucial. The band gap, or the HOMO–LUMO gap, is the energy difference between the valence band and the conduction band of the semiconductor. The band gap is imperative because as the COF is excited using light, an electron in the highest occupied molecular orbital (HOMO) is promoted into the lowest unoccupied molecular orbital (LUMO), which formally creates the hole in the HOMO and a charge is made and separated; this is called the photoexcitation state. Thus, the HOMO and LUMO energy levels of the COF are important, and are derived from the monomer units. In terms of photocatalysis, the HOMO energy level is essential, as the HOMO of the photocatalyst (i.e. the COF) must be lower than that of the substrate, so the oxidation reaction can occur at the photocatalyst with the photogenerated holes. Also, the LUMO of the photocatalyst must be higher than the energy of the substrate, so the reduction reaction occurs at the substrate with the photogenerated electrons. Furthermore, to create the photoexcitation state a photon with equal or greater energy than that of the COF's band gap is needed, so a small band gap is ideal.For example, Baeg and co-workers performed band gap engineering on imine-linked COFs with various amounts of alcohol substituents that could allow for ketoenamine tautomerization and push–pull effects.256 This work demonstrated that push–pull effects had a direct effect on the band gap of the COF, illustrating that a lower band gap could yield better photocatalytic efficiency. Chen and cohort showed similar results using the same method for photocatalytic hydrogen evolution from water.257 Qiu et al. synthesized various olefin-linked COFs with an increasing amount of alkynyl units in the aldehyde monomer.258 They showed that by increasing the amount of alkynyl units, the band gap was reduced and showed superior photodegradation of bacteria and other contaminants. More commonly, band gap engineering is often achieved by using donor–acceptor systems.
5.3 Donor–acceptor systems
One challenge of photocatalysis is the fast recombination of the photogenerated electrons and holes. Donor–acceptor (D–A) systems are a unique way to address this problem. Donor sites localize holes and acceptor sites localize electrons to cause efficient charge separation. One key parameter for D–A systems to create this charge separation is that the HOMO must be centered on the donor and the LUMO must be centered on the acceptor, or a mix of the linkage and the acceptor, otherwise the holes and electrons will not be split across the material. Furthermore, the D–A system is an effective method to tune band gaps. In fact, the utilization of donor units and acceptor units to perform band gap engineering in polymers is not new.10–12 One strategy to create a D–A system is to add electron-donating and withdrawing substituents, respectively. To increase the electron-withdrawing ability of the monomers, substituents such as nitriles, carbonyls, and nitro groups can be added as acceptors. To increase the electron-donating ability of the monomers, substituents such as amines, thiols, and alkoxy groups can be added as donors. Another strategy to create D–A systems is to construct the material using monomers based on electron-donating and accepting units (Fig. 11). Some donor monomer archetypes include pyrenes, thiophenes, fluorenes, carbazoles, phenothiazines, and triphenylamines. Some acceptor archetypes include triazines, diimides, diketopyrrolopyrroles, quinones, thiadiazoles, and oxadiazoles. The tunability and bottom-up design approach for COFs make them a perfect platform to incorporate these archetypes to make D–A systems. | ||
| Fig. 11 Donor and acceptor moieties. | ||
For example, Liu and cohort synthesized two multicomponent D–A COFs using 1,3,5-tris-(4-formyl-phenyl)triazine (TFPT) and benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene-2,5,8-tri-carbaldehyde (BTT) monomers with either 1,4-phenyldiacetonitrile (PDAN) or 1,4-phenylenediamine (PDA) to create an olefin and imine-linked version, respectively.259 They showed the D–A system had a small band gap and good charge separation which enabled high photocatalytic hydrogen evolution. Dong and co-workers synthesized an imine-linked D–A COF with a narrow band gap using benzo[c][1,2,5]thiadiazole and pyrene-based monomers for photocatalytic hydrogen evolution from water.260 Thomas et al. exemplified a triphenylamine-linked triazine-based COF for photocatalytic cleavage of CC bonds.261 Jiang and co-workers engineered the linkages of D–A COFs using triphenylene donor and benzothiadiazole acceptor units for efficient photocatalytic hydrogen peroxide production from water and air.262 The authors state that the D–A system allowed for fast photoinduced electron transfer, charge separation, localized oxidation and reduction active sites, and segregated columnar π-arrays for yielding and carrying of photogenerated charges. Thomas et al. explored constitutional isomerism in imine-linked D–A COFs based on triphenylamine and triazine units for photocatalytic hydrogen evolution.263 Notably they showed that the bond polarization in D–A systems is important for the optical properties, band gap, and thus photocatalytic activity.
5.4 Photosensitizers
In photocatalysis and other photobased applications, light absorption is necessary for exciton formation and photocatalytic performance. Some materials do not harvest light efficiently, or have low absorption coefficients, or have competing processes such as fluorescence. Photosensitizers are a solution to a lack of light harvesting as they are compounds that have intense light absorption which can then transform light to energy and transfer this electronic excitation energy to the photocatalyst or a substrate in an antenna effect.264 Common photosensitizers include organic dyes (i.e. methylene blue, Nile Blue, and acridine orange), fluoresceins, rhodamines, azulenes, porphyrins, chlorins, phthalocyanines, ruthenium bipyridine complexes, iridium phenylpyridine complexes, and rhenium bipyridine carbonyl complexes (Fig. 12). Even though COFs have a high degree of π-conjugation and wide-ranging UV-visible light absorption, they can suffer from poor absorption coefficients so photosensitizers can be added to increase the light absorption efficacy and overall efficiency of the system. | ||
| Fig. 12 Common photosensitizers. | ||
For instance, Yuan et al. synthesized a boroxine-linked COF and doped it with phthalocyanine-based photosensitizers for effective photodynamic therapy.265 Liao and Hu constructed a vinylene-linked COF to host a p-type thiophene-based photosensitizer, g2T-T, to perform photocatalytic hydrogen evolution.266 Lu and co-workers synthesized Pd-doped TpPa-1 COF which was sensitized by Eosin Y dye, which showed 10 times higher photocatalytic hydrogen evolution than that of Pd/C.267 Wu et al. made a porphyrin-bipyridine-based COF that could host Ru(bpy)3Cl2 photosensitizer for photocoupled electroreduction of carbon dioxide.268 These represent just a handful of examples of COFs utilizing photosensitizers.
5.5 Sacrificial reagents
Sacrificial reagents are useful in photocatalytic reactions in order to scavenge holes or electrons to prevent recombination and thus achieve high catalytic efficacy.217 Most commonly used are electron donors, or hole scavengers. A hole scavenger is oxidized irreversibly with the photogenerated holes to destroy them, and importantly should form a by-product that does not interfere with the reaction. This means that electron donors are fully expended during the course of the reaction, and stoichiometric or continuous addition is required to perform the photocatalytic reaction efficiently. Some common electron donors include potassium iodide, triethylamine (TEA), triethanolamine (TEOA), 1,3-dimethyl-2-phenylbenzimidazoline (BIH), 1-benzyl-1,4-dihydronicotinamide (BNAH), ascorbic acid, oxalate, ethylenediaminetetraacetic acid (EDTA), and thiols (Fig. 13). Electron acceptors, or electron scavengers, are used to destroy photogenerated electrons. Electron scavengers are reduced irreversibly and need to be added in stoichiometric amounts or continuously added just like hole scavengers. However, most photocatalytic reactions use the photogenerated electrons to promote the reaction, so electron scavengers are mainly used to study the mechanism and for proof of electron and hole separation. Common electron acceptors include silver nitrate, copper chloride, potassium chromate, sodium persulfate, sodium iodate, potassium iodate, and oxygen. | ||
| Fig. 13 Common sacrificial electron donors. | ||
5.6 Doping
Doping a material with elements, compounds, or nanoparticles to enhance its optical properties and electronic behaviour is an effective approach for enhancing photocatalytic activity in polymers.269 For example, Pang and cohort synthesized COFs and embedded them with CuSe270 or Ag2Se271 nanoparticles for phototherapy. Su et al. constructed crystalline CTFs and doped them with elemental sulfur for photocatalytic hydrogen evolution using water.272 They demonstrated that the sulfur-doped CTFs showed both greater photocatalytic performance and stability. Bi et al. synthesized a β-ketoenamine COF, TpMa, and combined it with CdS for photocatalytic hydrogen peroxide production.273 The material showed 2.5 times higher H2O2 production than the pristine COF and 4 times higher than CdS alone. Zhang and co-workers synthesized TpPa-2-COF and doped it with α-Fe2O3 for photocatalytic hydrogen evolution.274 The material showed 53 times higher production than the pristine COF, and the authors showed that this was due to the efficient charge separation from transfer of photogenerated electrons from the conduction band of the α-Fe2O3 to the valence band of the COF. Zhang et al. showed that TpTa-COF could encapsulate Ti(OnBu)4 and be converted to Fe–TiO2 nanoparticles (Fig. 14).275 This Fe–TiO2@COF material was highly efficient at photocatalytic degradation of methylene blue dye. Islam and co-workers synthesized TP-TAPM COF and doped it with TiO2 for highly efficient photoreduction of CO2 to CH3OH and N2 to NH3.276 These provide just a small assortment of examples to show the utility of doping in COFs for photoenhancement. | ||
| Fig. 14 Schematic presentation of the synthesis of TpTa-COF (a) and Fe–TiO2@COF (b).275 Reproduced with permission from ref. 275. Copyright 2019, The Royal Society of Chemistry. | ||
5.7 Polymeric composites and hybrid materials
COFs provide a unique platform for constructing composites or hybrid materials due to their porous nature. By combining two or more materials, the chemical, optical, and electrical properties can be enhanced. Some examples of COF composites include COF@COF, polymer@COF, carbon allotrope nanostructure (CAN)@COF, and metal organic framework (MOF)@COF. For instance, Li et al. constructed a triazine-based imine-linked COF (TATF-COF) and combined it with a perylene diimide urea polymer (PUP).277 The PUP@COF material showed 3.5 times higher photocatalytic hydrogen production than that of the pure TATF-COF due to charge-transfer channels and prolonged exciton lifetimes. Pan and co-workers fabricated a melamine sponge COF hybrid, and the MS@TpTt material exhibited excellent photodegradation of tetracycline antibiotics.278 Lu and co-workers produced a Co-porphyrin based COF, COF-366-Co, and covalently anchored it on graphene oxide (GO).279 The GO-COF-366-Co composite was used for photoreduction of carbon dioxide and showed about 4 times higher activity than the pristine COF. This was corroborated by photoelectrical experiments illustrating the higher charge separation and transfer of the composite. Wang and cohort synthesized a TpBD COF and fabricated it on carbon nanotubes for efficient photodegradation of mordant black 17 dye.280 Zhang et al. synthesized NH2-MIL-68 and coated it with TPA-COF, and the NH2-MIL-68@TPA-COF showed high photocatalytic degradation of rhodamine B dye.281 Kim and Sun constructed NH2-MIL-125(Ti), or Ti-MOF, encapsulated Pt nanoparticles in it, forming Ti-MOF@Pt, and then drop-cast DMLZU1 COF onto it giving Ti-MOF@Pt@DMLZU1 (Fig. 15).282 This hybrid material was used for photohydrogenation of olefins and showed quantitative conversion in near perfect selectivity in 40 min using only 1 atm of H2 for styrene. Zang et al. performed similar experiments using a Pd@Ti-MOF@TpTt hybrid for photocatalytic hydrogenation of nitroarenes.283 Interestingly, Yan et al. constructed a MOF@COF@GO composite, UiO-66-NH2/TAPT-TP-COF/GO, for photocatalytic hydrogen evolution.284 These examples provide a small demonstration of the ability to construct composite/hybrid materials using COFs to boost photocatalytic performances. | ||
| Fig. 15 Two-step method to prepare Ti-MOF@DM-LZU1.282 Reproduced with permission from ref. 282. Copyright 2020, American Chemical Society. | ||
6 Photocatalytic applications
6.1 Hydrogen evolution
High population growth significantly increases energy demand, making energy shortages one of the most pressing global challenges.285 This issue requires a multifaceted approach for ensuring a sustainable energy supply while minimizing the environmental impact. Hydrogen production under visible-light irradiation, often called photocatalytic hydrogen evolution, is an area of significant interest in renewable energy research.286–288 COFs are promising materials for photocatalytic hydrogen production due to their porosity, structural designs, visible light harvesting, and chemical stability.24 COFs synthesized from porphyrins, triazines, and pyrene building blocks with extended π-conjugated planar structures have been extensively studied for photocatalytic hydrogen evolution.24 Indeed, one of the most fascinating properties of COFs is their structural tunability,289,290 enabling unique designs. Numerous studies have concentrated on COF-based photocatalysts for H2 evolution, demonstrating that altering their building blocks,291,292 functional groups293,294 and linkages295–299 can enhance photocatalytic performance. The mechanism of photocatalytic H2 evolution using COFs has also been well developed.24In 2014, the Lotsch group introduced the first COF photocatalyst, TFPT-COF, for hydrogen evolution.300 The crystalline hydrazone-based TFPT-COF demonstrated a photocatalytic hydrogen evolution rate of 1970 μmol g−1 h−1 under visible light irradiation (300 W Xe lamp) with 2.2 wt% Pt as the cocatalyst and 10 vol% TEOA as the electron donor, and a smaller 230 μmol g−1 h−1 using sodium ascorbate as the electron donor. Under identical standard conditions, TFPT-COF/Pt demonstrated superior performance compared with the results obtained with other highly effective photocatalytic systems, specifically a Pt-modified amorphous melon, g-C3N4, synthesized as described in the literature,301 and crystalline poly(triazineimide). Notably, the TFPT-COF loses crystallinity after photocatalysis but can be reconverted into the crystalline material by resubjecting it to the initial synthesis conditions.
In 2022, the Li group examined a series of COFs synthesized using 2,4,6-tris(4-aminophenyl)-1,3,5-triazine with 1,3,5-triformylbenzene, 2,4,6-triformylphloroglucinol, 2,4,6-triformylphenol, and 2,4,6-triformylresorcinol, respectively. These were denoted as COF-OH-n (n = 0–3), representing the number of –OH groups.257 COF-OH-0 and COF-OH-3 were synthesized according to the procedure from the literature.302,303 The varying degrees of proton tautomerism in COF-OH-n, due to the different numbers of β-ketoenamine linkages in their structures, regulated the visible light absorption ability, band gap, and band edge positions. Photocatalytic hydrogen evolution experiments were conducted using visible light (300 W Xe lamp), ascorbic acid as the sacrificial agent, and 1 wt% Pt as the cocatalyst. Among the COF-OH-n series, COF-OH-3 exhibited the highest hydrogen evolution rate of 9.89 mmol g−1 h−1. This superior performance was attributed to the irreversible proton tautomerism in COF-OH-3, which resulted in the most favourable photoelectronic properties. The apparent quantum yield of COF-OH-3 was 0.15% at 420 nm.
Recently, metalated or metal-associated COFs have been employed as photocatalysts for H2 evolution.304,305 Metal ions can influence the charge carrier dynamics within the resulting COF frameworks. Recently, a series of planar porphyrin-based MPor-DETH-COFs (M = H2, Co, Ni, and Zn) were designed and synthesized through the condensation reaction of metalated porphyrinic aldehydes with 2,5-diethoxyterephthalohydrazide (DETH).305 The visible-light-driven hydrogen production by these COF photocatalysts was examined under visible light using a 300 W Xe lamp in the presence of Pt and TEOA, with Pt acting as the cocatalyst and TEOA as the sacrificial reagent. Among the MPor-DETH-COFs, ZnPor-DETH-COF exhibited the highest hydrogen production rate of 413 μmol g−1 h−1. H2Por-DETH-COF, CoPor-DETH-COF, and NiPor-DETH-COF had rates of 80, 25, and 211 μmol g−1 h−1, respectively. These findings indicated an increasing driving force from charge carrier dynamics, as supported by transient emission decays and photocurrent tests. ZnPorDETH-COF had an apparent quantum yield of 0.32% at 450 nm. Notably, when the incident wavelength was above 500 nm no hydrogen was produced.
COFs with donor–acceptor (D–A) moieties have been widely utilized for photocatalytic H2 evolution.306 Recently, the Dong group designed and synthesized a novel imine-linked COF, DABT-Py-COF, by condensing a newly constructed donor–acceptor–donor (D–A–D) type monomer, 4,4′,4′′,4′′′-(benzo[c][1,2,5]thiadiazole-4,7-diylbis(9,9-dimethyl-9,10-dihydroacridine-10,2,7-triyl))tetrabenzaldehyde, with 1,3,6,8-tetrakis(4-aminophenyl)pyrene.307 By optimizing the reaction conditions using an aqueous medium containing ascorbic acid as the sacrificial electron donor, DABT-Py-COF as the photocatalyst, and Pt as the cocatalyst under visible light irradiation (300 W Xe lamp), they achieved a hydrogen evolution rate of 5458 μmol g−1 h−1. Without Pt cocatalyst, a rate of 299 μmol g−1 h−1 was achieved. The remarkable performance of DABT-Py-COF was attributed to its high crystallinity, permanent porosity, broad light absorption range, and exceptional chemical stability. Notably, after five cycles, the DABT-Py-COF had a decrease in crystallinity.
Thomas and co-workers have recently explored COFs and COF films with uniquely tunable band gaps and optoelectronic properties for photocatalytic hydrogen evolution.308 They investigated the photocatalytic performance of low band gap COFs constructed by condensing the extended π-conjugated 9,10-bis(4-aminophenylethynyl)anthracene (AntT) with benzene-1,3,5-tricarbaldehydes containing varying numbers of hydroxy groups on the aldehyde units (n = 0, 1, 2, 3). This resulted in synthesizing AntTTF, AntTMH, AntTDH, and AntTTH, respectively. The AntT monomer functioned as the photoabsorber, while the β-ketoenamine linkage preserved the planarity of the system through intramolecular hydrogen bonding. AntTTF and AntTTH were synthesized according to the procedure reported in the literature.309 Additionally, thin films of the powder COFs were synthesized, capitalizing on their numerous advantages in photocatalytic applications, such as low light scattering and reusability. The number of hydroxy groups on the 1,3,5-triformylbenzene linker significantly influenced the properties of the COFs. AntTDH, the COF prepared from the linker with two hydroxy groups, showed the lowest band gap, exhibiting efficient charge separation and migration due to the formation of J-type aggregates. Photocatalytic hydrogen evolution was performed under visible light (λ > 420 nm, 300 W) with 2 wt% Pt cocatalyst and ascorbic acid as the sacrificial electron donor. AntTDH achieved the highest HER of 8.4 ± 0.5 mmol g−1 h−1 in the powder state. Interestingly, in the COF films, a steady increase in the hydrogen evolution rate was observed with an increasing –OH group. The COF film with the most hydroxy groups, AntTTH, gave the best HER of 1.6 ± 0.2 mmol m−2 h−1 due to heightened charge carrier mobility (Fig. 16). Notably, post-catalysis, both the COF films and powders displayed a partial loss of crystallinity.
 | ||
| Fig. 16 (a) Calculated band position and band gap of the COF powders vs. normal hydrogen electrode (NHE) along with the potential of H+/H2 and oxidation potentials of L-ascorbic acid, HA˙/H2A and A/H2A at pH 2.6. Time course for photocatalytic HER under visible light (≥420 nm) of (b) COF powders (3 mg of COF with 2 wt% Pt) and (c) COF films (with 2 wt% Pt) in 16 mL 0.1 M ascorbic acid aqueous solution at pH 2.6. (d) Photograph of AntTTH film over quartz at different time intervals of photocatalytic reaction showing the formation of H2 gas bubbles. (e) The image of the AntTTH film is illuminated with diffused sunlight inside a photocatalytic reactor with the corresponding enlarged image showing the formation of H2 gas bubbles.308 Reproduced with permission from ref. 308. Copyright 2024, Royal Society of Chemistry. | ||
Using saddle-shaped cyclooctatetrathiophene derivatives as building blocks, the Wang group has recently developed fully conjugated 3D COFs for metal-free photocatalytic hydrogen evolution.310 Through Schiff-base condensation, they synthesized a series of fully conjugated 3D COFs using COThP-CHO, which was synthesized from the literature method,311 with benzidine (BD), 3,7-diaminodibenzo[b,d]thiophene-5,5-dioxide (SA), 2,2′-dimethyl[1,1′-biphenyl]-4,4′-diamine (MBD), and 3,3′-dimethylbiphenyl-4,4′-diamine (DMB), resulting in BUCT-COF-20, -21, -22, and -23, respectively. The photocatalytic reactions for H2 production were conducted under visible light irradiation (λ > 400 nm, 300 W) using 5 mg of photocatalyst in water, 2.5 μL (8 wt%) H2PtCl6 as cocatalyst, and 0.1 M ascorbic acid as an electron donor. BUCT-COF-20 demonstrated the highest HER of 40.36 mmol g−1 h−1, approximately three times higher than BUCT-COF-21, which had a HER of 14.11 mmol g−1 h−1. BUCT-COF-22 achieved a HER of 18.80 mmol g−1 h−1, while BUCT-COF-23 exhibited the lowest performance at 4.33 mmol g−1 h−1. Under similar reaction conditions, the performance of BUCT-COF-20 was compared with that of the non-conjugated COF-320. COF-320 produced a significantly lower hydrogen evolution rate of 0.79 mmol g−1 h−1. BUCT-COF-20 showed the highest apparent quantum yield of 2.58% at 450 nm, and with lower AQY of 2.34%, 1.88%, 1.14%, and 0.73% at 400 nm, 500 nm, 550 nm, and 600 nm, respectively. The exceptional performance of BUCT-COF-20 can be attributed to its D–A electronic structures, porosity, hydrophobicity, and conjugated structure.
6.2 Water oxidation
Water oxidation for oxygen evolution involves a complex four-electron redox process in the rate-determining step of overall water splitting. This process includes the cleavage of the O–H bond, the formation of the O–O bond, and a large overpotential with slow O–O formation kinetics.312–314 As a result, there has been limited research on using COFs as photocatalysts for photocatalytic water oxidation. Among the COFs used for oxygen evolution are bipyridine-containing COFs due to their capacity to form chelate complexes with transition metals.One of the most recent reports of a bipyridine-containing COF for photocatalytic water oxidation is from the Li group.315 They used a triazine-based COF, TAPT-Bpy-COF, synthesized by a Schiff-base condensation from 2,4,6-tris(4-aminophenyl)-1,3,5-triazine and 2,2′-bipyridyl-5,5′-dialdehyde. TAPT-Bpy-COF was coordinated with Co, Fe, and Ni, resulting in TAPT-Bpy-COF-Co-3, TAPT-Bpy-COF-Fe-3, and TAPT-Bpy-COF-Ni-3, respectively. The photocatalytic water oxidation of these metal-coordinated COFs under light illumination (λ ≥ 420 nm, 300 W Xe lamp) was carried out in a silver nitrite sacrificial system. Among them, TAPT-Bpy-COF-Co-3 achieved the highest oxygen evolution rate of 483 μmol g−1 h−1 with an efficient apparent quantum efficiency (AQE) of 7.6% at 420 nm. The performance of TAPT-Bpy-COF-Co-3 is higher than most bipyridine-containing COFs. HR-STEM-HADDF images confirmed cobalt coordination with the bipyridine units within the framework structure. TAPT-Bpy-COF-Co-3 benefits from its broad visible light absorption range, high surface area, well-ordered structure, and improved charge separation.
Studies have demonstrated that crystalline COFs exhibit superior photocatalytic water oxidation performance compared with their amorphous counterparts, owing to their highly ordered arrangement of functional units.316,317 As a proof of concept, in 2022, Würthner et al. successfully integrated Ru(bda)-based (bda = 2,2′-bipyridine-6,6′-dicarboxylate) dialdehyde into a 3D ordered COF.318 This was achieved through imine condensation between the dialdehyde and tetra-(4-anilyl)methane, forming either crystalline or amorphous 3D imine polymers, depending on the solvents used, referred to as Ru(bda)-COF or Ru(bda)-polymer, respectively. The photocatalytic water oxidation was studied using Ru(bda) polymers, with [Ru(bpy)3]Cl2 serving as the photosensitizer (PS) and sodium persulfate (Na2S2O8) as the sacrificial electron acceptor (SEA). The SEA first oxidizes the photogenerated PS* to PS+, which further oxidizes the Ru water oxidation catalyst (WOC) until it reaches the crucial Ru5+ state. For the photocatalytic water oxidation, measurements were performed at λ ≥ 380 nm using a 150 W Xe lamp. At a low catalyst loading of 0.1 g L−1, Ru(bda)-COF achieved an initial rate of 10.4 μmol L−1 s−1, over 20 times higher than the 0.5 μmol L−1 s−1 rate of Ru(bda)-polymer. This disparity in performance is attributed to differences in their mechanistic pathways. During the O–O bond formation process, Ru WOCs can follow two primary mechanistic pathways: water nucleophilic attack (WNA) or the interaction of two highly oxidized Ru-oxyl radicals (I2M).319,320 Ru(bda)-COF photochemical water oxidation follows the I2M pathway and is second order in COF concentration, suggesting that two COF particles are involved in the rate-determining step (RDS). Extensive mechanistic studies, including H/D kinetic isotope effect (KIE) analysis, indicated that the O–O bond formation in Ru(bda)-polymer follows the WNA pathway. The limited access to the Ru(bda) units at the disoriented polymer surface hinders the efficient dimerization of activated catalysts. Consequently, this restricts the water oxidation catalysis to the less efficient WNA mechanism in Ru(bda)-polymer.
The Wang group has recently examined the effectiveness of boranil COFs (N and O-moiety chelated difluoroboron complexes) in photocatalytic water oxidation reactions.321 Initially, they synthesized a series of metalated porphyrin-based COFs, M-TAPP-COFs (where M = Ni, Zn, Co, Mn), through the Schiff-base condensation of 5,10,15,20-tetrakis(4-aminophenyl)porphyrin metal[II] (M-TAPP) and 2,5-dihydroxyterephthalaldehyde.322 Subsequently, they achieved boranil-functionalized fully π-conjugated ordered D–A COFs, M-TAPP-COF-BF2, by post-synthetic modification of M-TAPP-COFs using an excess of BF2·Et2O.323 The photocatalytic water oxidation experiments were conducted in aqueous solution with 5 mg of the photocatalyst (Ni-TAPP-COF, Ni-TAPP-COF-BF2, Zn-TAPP-COF-BF2, Co-TAPP-COF-BF2, and Mn-TAPP-COF-BF2) and AgNO3 as a sacrificial electron acceptor under light irradiation (300 W Xe, AM 1.5G cut-off filter) at 10 °C. The order of photocatalytic oxygen evolution rates was observed as Ni-TAPP-COF-BF2 > Zn-TAPP-COF-BF2 > Co-TAPP-COF-BF2 > Mn-TAPP-COF-BF2 > Ni-TAPP-COF. Notably, Ni-TAPP-COF-BF2 exhibited the highest performance with 1404 μmol g−1 h−1, significantly surpassing the pristine Ni-TAPP-COF, 123 μmol g−1 h−1. Both experimental and theoretical findings indicated that the push/pull (metalloporphyrin/BF2) mechanism and π-conjugation that is well-regulated are crucial for photoabsorbing properties. The apparent quantum yield of Ni-TAPP-COF-BF2 was 1.27% at 450 nm.
Yan and co-workers have recently designed a coordinated enamine-based COF using cobalt dichloride for photocatalytic oxygen evolution through water oxidation.324 Initially, TpBPy was synthesized via Schiff base condensation of 2,4,6-trihydroxybenzene-1,3,5-tricarbaldehyde (Tp) and [2,2′-bipyridine]-5,5′-diamine (BPy) under solvothermal conditions. Subsequently, CoCl2 was incorporated at the bipyridine sites in an ethanol solution to form CoCl2–TpBPy. Photocatalytic O2 evolution analysis was conducted using a closed glass apparatus with 10 mg of photocatalyst mixed in AgNO3 solution (as a sacrificial reagent), under 300 W xenon lamp illumination (λ = 400 nm) at a temperature of 10 °C. After 1 h, CoCl2–TpBPy demonstrated an O2 evolution rate of 1 mmol g−1 h−1, while TpBPy exhibited almost no O2 evolution. CoCl2–TpBPy had an apparent quantum yield of 1.34% at 425 nm. Using a wavelength of 460 nm yielded about 700 μmol g−1 h−1, and at 500 nm yielded about 200 μmol g−1 h−1. Interestingly, MnCl2 or RuCl3 versions produced no yield. In situ XPS and EPR analyses revealed that high-valence cobalt states were important for water oxidation (Fig. 17). The coordination of CoCl2 into TpBPy maintained the structural integrity and the characteristics of charge recombination. Notably, the COF lost its crystallinity post-catalysis.
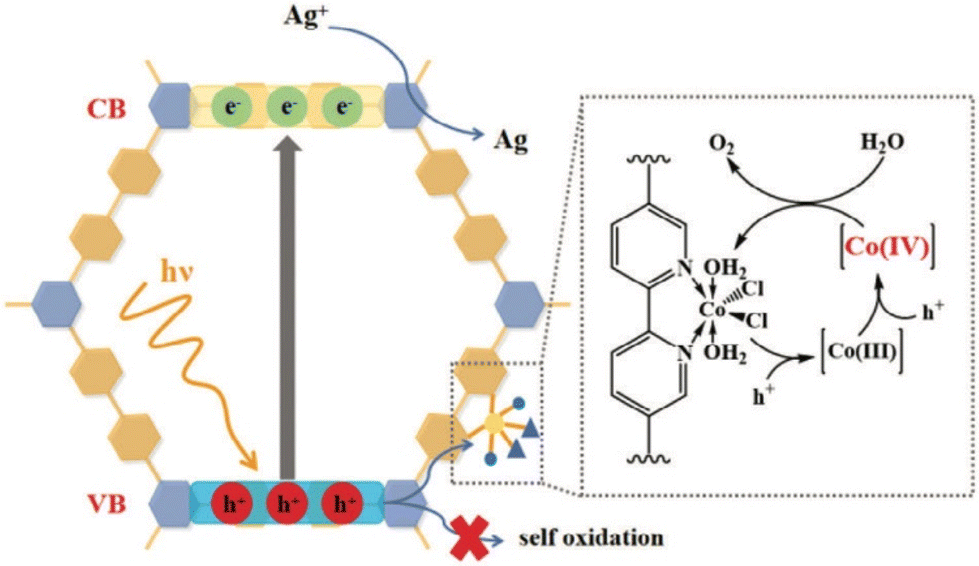 | ||
| Fig. 17 Proposed mechanism for the photocatalytic water oxidation process facilitated by CoCl2–TpBPy.324 Reproduced with permission from ref. 324. Copyright 2024, John Wiley and Sons. | ||
The Wang group recently synthesized an ionic-type COF, CoTPP-CoBpy3, which included a tetraphenyl porphyrin coordinated cobalt (CoTPP) and a bipyridine coordinated cobalt complex {[CoII(bpy)3](OAc)2} for photocatalytic water oxidation.325 This unique COF was created using a one-pot condensation reaction involving 5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphine, 2,2′-bipyridyl-5,5′-dialdehyde, 2,2′-bipyridine, and Co(OAc)2·4H2O. High-resolution cryo-transmission electron microscopy (cryo-TEM) revealed that CoTPP-CoBpy3 COF had a large-sized morphology of over 2 μm, ultra-thin nanosheets with a thickness of approximately 1.25 nm, and high crystallinity. The photocatalytic water oxidation was tested by dispersing 5 mg of the photocatalyst in aqueous AgNO3 solution and illuminating it with a 300 W xenon lamp with an AM 1.5G cut-off filter. After 1 hour, the photocatalytic oxygen production rate reached 7323 μmol g−1 h−1. CoTPP-CoBpy3 COF gave an apparent quantum yield of 4.65% at 450 nm. This performance was attributed to the unique properties of CoTPP-CoBpy3.
6.3 Water splitting
Hydrogen production through water splitting under visible-light irradiation is an area of significant interest in renewable energy research. This method uses solar energy to drive the splitting of water molecules into hydrogen and oxygen, offering a clean and sustainable way to produce hydrogen fuel. This can be challenging due to critical thermodynamics, slow kinetics, dissolved oxygen, reverse and side reactions, etc.326,327 Limited research has been conducted on using COFs for hydrogen production through water splitting under visible-light irradiation.328 Transition metals are mostly used as cocatalysts in the process. The photocatalytic mechanism of water splitting has been previously explored using COFs.328Interestingly, in 2020, Chen and co-workers explored the impact of halogens on a 2D COF for hydrogen production through water splitting under visible-light irradiation by altering the photoactive unit with halogens.329 Photoactive benzothiadiazole (BT) units were strong electron acceptors, facilitating efficient intramolecular charge transfer through alternating D–A skeletons. A series of 2D COFs, Py-XTP-BT-COFs (X = H, F, or Cl), was synthesized by polycondensation of 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetrabenzaldehyde (Py-CHO) with terphenyl-based diamines (XTP-BT-NH2) featuring fluorinated, chlorinated, or non-halogenated BT units under solvothermal conditions. Photocatalytic water-splitting experiments were performed under visible light irradiation (λ > 420 nm) using ascorbic acid as the sacrificial reagent and without a cocatalyst. Py-ClTP-BT-COF, Py-FTP-BT-COF, and Py-HTP-BT-COF achieved HER rates of 44.00, 15.8, and 6.00 μmol h−1, respectively, demonstrating that chlorination and fluorination of the Py-HTP-BT-COF skeleton can enhance photocatalytic activity. Using the same conditions with 5 wt% Pt cocatalyst, Py-ClTP-BT-COF, Py-FTP-BT-COF, and Py-HTPBT-COF showed HER rates of 177.50, 57.50, and 21.56 μmol h−1, respectively. The AQE of Py-ClTP-BT-COF reached 8.45% at 420 nm, one of the highest values reported for COF-based photocatalysts. These findings indicate that halogenation in the BT unit is crucial in modulating the electronic structures of BT-COFs owing to the high electronegativity of chlorine and fluorine atoms. Based on DFT calculations, the adjustment of halogens in the photoactive BT units can effectively inhibit charge recombination and notably lower the energy barrier linked to the creation of H intermediate species (H*) on the polymer surface.
Recently, Zhao and co-workers reviewed functional regulation of COFs for photocatalytic water splitting.330 A prime example comes from the Li group where they synthesized COF-SCAU-2 using 5′-(4-formylphenyl)-[1,1′:3′,1′′-terphenyl]-4,4′′-dicarbaldehyde (TB) and 2,7-diamino-9-fluorenone (F), and this was mechanically exfoliated to afford UCOF-SCAU-2.331 They took these COFs and tested them for photocatalytic water splitting under visible-light irradiation (300 W Xe lamp) with Pt as cocatalyst and ascorbic acid as a sacrificial reagent. The hydrogen evolution rate for COF-SCAU-2 was 3.63 mmol h−1 g−1, and 35.61 mmol h−1 g−1 for UCOF-SCAU-2. The apparent quantum yield of UCOF-SCAU-2 was 10.23% at 420 nm. When tested for overall water splitting COF-SCAU-2 showed no activity, but UCOF-SCAU-2 exhibited hydrogen and oxygen evolution rates of 0.046 and 0.021 mmol h−1 g−1, respectively. However, after five recycles the rates fall to almost half.
In 2023, Lan and co-workers explored a series of β-ketoenamine COFs for photocatalytic water splitting.332 They synthesized TpBD-COF, TpBpy-2-COF, and TpBpy-COF through reversible Schiff base reaction and irreversible enol-to-keto tautomerization. These COFs were then modified and exfoliated through a “top-down” process to create TpBD-NS, TpBpy-2-NS, and TpBpy-NS. To incorporate a cocatalyst, ultra-small Pt nanoparticles (NPs) were generated in situ within the pores of the COFs, resulting in the non-bipyridine Pt@TpBD-NS, and the bipyridine (Bpy)-containing Pt@TpBpy-NS and Pt@TpBpy-2-NS photocatalysts. The reactions were conducted in quartz tubes within a sealed system containing 15 mg of catalyst in H2O and using sodium ascorbate as the sacrificial electron donor. For performance comparison, the non-Bpy Pt@TpBD only performed the H2 half reaction, whereas the Bpy-containing COFs, Pt@TpBpy-NS and Pt@TpBpy-2-NS, demonstrated overall water splitting, producing both H2 and O2. Under visible light, Pt@TpBpy-NS achieved optimal production rates of 9.9 μmol of H2 and 4.8 μmol of O2 in 5 hours while Pt@TpBpy-2-NS produced 3.1 μmol of H2 and 1.4 μmol of O2. With experimental and theoretical calculations, this work proved that the position of the N-sites within the Bpy (Fig. 18), the nanosheet morphology of the COFs, and the incorporation of ultra-small Pt nanoparticles within the COF pores were crucial factors for enhancing water-splitting activity. An apparent quantum yield of 2.8% was measured at 450 nm for Pt@TpBpy-NS.
 | ||
| Fig. 18 DFT calculations and proposed schematic mechanism of TpBpy-NS. (a) UV-Vis absorption spectra of TpBpy-NS compared with TD-DFT calculated fragment. (b) The TD-DFT calculated electronic transition of TpBpy-NS. (c) The possible process of HER on Tp segment and OER via dual-site process on Bpy segment in TpBpy-NS. (d) The comparison of calculated Gibbs free energy change for C2d paths of OER for TpBD-NS, TpBpy-2-NS and TpBpy-NS at pH = 7.332 This image is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/). | ||
Xu and co-workers have recently explored two types of π-conjugated COFs with the same donor–acceptor structure but different linkages for metal-free photocatalytic water splitting.333 The sp2c-Py-BT COF and the imine-Py-BT COF were composed of the electron-donating pyrene (Py) unit and the electron-accepting BT unit, formed through Knoevenagel condensation and Schiff base reaction, respectively. The photocatalytic reactions were conducted by mixing the photocatalyst in pure water without any cocatalyst or sacrificial agent, under visible light irradiation (λ > 420 nm, 300 W Xe lamp). The sp2c-Py-BT COF produced H2 and O2 in a near 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio for overall water splitting, with average production rates of 17.2 and 8.1 μmol h−1 g−1, respectively. Under the same conditions, the imine-Py-BT COF could not reach the overall water-splitting reaction. When adding in triethanolamine, the H2 rate increased to 891.5 μmol h−1 g−1, and when adding in AgNO3, the O2 rate increased to 21.2 μmol h−1 g−1 for sp2c-Py-BT COF. Furthermore, when adding Pt and Co(OH)2 cocatalysts, sp2c-Py-BT COF had increased production rates for H2 and O2 to 71.3 and 30.8 μmol h−1 g−1, respectively. Mechanistic studies indicated that the cyano-vinylene linkage is crucial for adjusting the band structure and enhancing charge separation in COFs, thus enabling overall water splitting. sp2c-Py-BT COF had an apparent quantum yield of 2.53% at 420 nm. Interestingly, the authors explored the intrinsic relationship between crystallinity and photocatalytic activity and found a sixfold improvement for the highly crystalline COF compared with the amorphous polymer, and they saw a linear relationship of increasing rates from an amorphous polymer to a low-crystalline COF to a medium-crystalline COF to a highly crystalline COF.
:1 stoichiometric ratio for overall water splitting, with average production rates of 17.2 and 8.1 μmol h−1 g−1, respectively. Under the same conditions, the imine-Py-BT COF could not reach the overall water-splitting reaction. When adding in triethanolamine, the H2 rate increased to 891.5 μmol h−1 g−1, and when adding in AgNO3, the O2 rate increased to 21.2 μmol h−1 g−1 for sp2c-Py-BT COF. Furthermore, when adding Pt and Co(OH)2 cocatalysts, sp2c-Py-BT COF had increased production rates for H2 and O2 to 71.3 and 30.8 μmol h−1 g−1, respectively. Mechanistic studies indicated that the cyano-vinylene linkage is crucial for adjusting the band structure and enhancing charge separation in COFs, thus enabling overall water splitting. sp2c-Py-BT COF had an apparent quantum yield of 2.53% at 420 nm. Interestingly, the authors explored the intrinsic relationship between crystallinity and photocatalytic activity and found a sixfold improvement for the highly crystalline COF compared with the amorphous polymer, and they saw a linear relationship of increasing rates from an amorphous polymer to a low-crystalline COF to a medium-crystalline COF to a highly crystalline COF.
6.4 Carbon dioxide reduction
The increasing consumption of fossil fuels has led to uncontrolled carbon dioxide (CO2) emissions, resulting in serious environmental issues.334,335 Solutions proposed to address this dilemma include carbon capture utilization and storage (CCUS),336 amine absorption,337 and bioenergy with carbon capture and storage (BECCS).338 Additionally, the photochemical conversion of CO2 into valuable chemicals like methanol, ethane, formic acid, ethanol, and others presents sustainable and renewable solutions for recycling CO2 while addressing global issues.339 Known for their exceptional capacity to adsorb and trap CO2,340 COFs are regarded as a promising platform for catalysing the reduction of CO2 using photocatalysis.25 Most reported COF-based photocatalysts rely on metalation or transition metals as additive cocatalysts to enhance activity.25 Ni-,341 Co-341–344 or Re-coordinated345,346 COFs have gained great attention, particularly in CO2 photoreduction due to their high electron affinity. The mechanism of photocatalytic CO2 reduction using COFs has also been explicated.25Wang et al. have recently reported two Co-coordinated COFs, Co-2,3-DHTA-COF and Co-TP-COF, for photocatalytic CO2 reduction.347 TP-COF and 2,3-DHTA-COF were synthesized using 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT) with 1,3,5-trimethylbenzene (TP) and 2,3-dihydroxybenzene-1,4-dicarboxaldehyde (2,3-DHTA), respectively, in acid-catalysed solvothermal conditions. The COFs were then treated with Co(NO3)2 in water and ethanol, affording Co-TP-COF and Co-2,3-DHTA-COF. Co-TP-COF created Co–O3N sites within its frameworks, whereas Co-2,3-DHTA-COF developed Co–O4 sites. This difference in Co-coordination between the two COFs was due to adjustments to the position of the hydroxyl group in the aldehyde monomer used. The formation of Co–O4 was identified using spectral characterizations such as X-ray absorption fine structure (FT-EXAFS) spectrum and XPS. The photocatalytic CO2 reduction to CO was conducted by mixing the photocatalyst with acetonitrile (MeCN)/water solution under simulated visible light irradiation (λ ≥ 420 nm), using [Ru(bpy)3]Cl2 as the photosensitizer, and TEOA as the electron donor. Under optimized conditions, Co-2,3-DHTA-COF demonstrated a CO production rate of 18000 μmol g−1 h−1 and a selectivity of 95.7% (800 μmol g−1 h−1 H2), whereas Co-TP-COF showed a lower CO production rate of 11600 μmol g−1 h−1 and selectivity of 76.6%. The higher performance with Co-2,3-DHTA-COF was attributed to the Co–O4 sites. Based on in situ and ex situ spectral analyses and DFT calculations, the Co–O4 sites in the COF framework greatly enhanced the charge separation, preventing the recombination of photogenerated electron–hole pairs during the photocatalytic process. The highest apparent quantum yield for Co-2,3-DHTA-COF was 0.47% at 450 nm. Notably, the COF showed a decrease in crystallinity and rate after three cycles.
In 2020, Cooper and co-workers demonstrated the photocatalytic reduction of CO2 into fuels by tethering the rhenium complex Re(bpy)(CO)3Cl onto COF materials to create a heterogeneous photocatalyst.348 They synthesized the 2D sp2c-COF through the Knoevenagel condensation of 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy) and 5,5′-bis(cyanomethyl)-2,2′-bipyridine. To produce the metal-coordinated Re-Bpy-sp2c-COF, they ligated the bipyridine sites in Bpy-sp2c-COF with Re(CO)5Cl. To demonstrate the strong CO2 affinity of Re-Bpy-sp2c-COF, they measured CO2 uptake, finding that the metalated COF adsorbed 1.7 mmol g−1 CO2 at 273 K and 1.1 mmol g−1 at 298 K. Photocatalytic CO2 reduction experiments were conducted in a quartz flask at 1 atm CO2 with MeCN as the solvent and TEOA as the proton source and sacrificial electron donor under visible light irradiation (λ > 420 nm) using a 300 W Xe light source. Re-Bpy-sp2c-COF produced CO at a rate of 1040 μmol g−1 h−1 with 81% selectivity versus H2 resulting in a turnover number (TON) of 18.7 for CO over 17.5 hours. Re-Bpy-sp2c-COF outmatched the homogeneous catalyst Re(bpy)(CO)3Cl under the same conditions, as the latter deactivated after 3 hours with a TON of 10.3. This superior performance is likely due to the strong visible light absorption and high CO2 binding affinity of Re-Bpy-sp2c-COF. The apparent quantum yield was measured at 420 nm, giving 0.5%. When using different wavelengths of light, blue light performed the best (about 400 μmol g−1 h−1) followed by green light (about 100 μmol g−1 h−1), and then orange light (about 50 μmol g−1 h−1). They also compared an amorphous polymer version which demonstrated a lower TON of 2.3 after 12 hours compared with 12.9 for the COF. Notably, after 50 hours of irradiation the COF lost crystallinity.
The photocatalytic reduction of CO2 using metal-free COFs has also gained a lot of attention.349–352 For example, in 2021, Liu and co-workers reported metal-free 2D COFs, TpBb-COF and COF-TpPA-1, and graphitic carbon nitride (g-C3N4) for visible-light-driven photocatalytic reduction of CO2.353 An acid-catalysed Schiff-base reaction was used to synthesize TpBb-COF by the condensation of 2,6-diaminobenzo[1,2-d:4,5-d1]bisthiazole (Bb-NH2) and 1,3,5-triformylphloroglucinol (Tp-CHO). With minor adjustments, COF-TpPA-1 and g-C3N4 were synthesized as described in the literature.354,355 Photocatalytic reduction of CO2 was conducted in a glass-sealed reactor under visible light irradiation within a gas–solid system, utilizing the COFs and g-C3N4 as the photocatalysts. It should also be noted that no cocatalysts, photosensitizers, or sacrificial reagents were used for these measurements. Both COFs and g-C3N4 observed CO as the main product. TpBb-COF demonstrated an optimal average CO production rate of 52.8 μmol g−1 h−1 at 80 °C with a CO selectivity of 99.0%. This rate was more than double that observed for COF-TpPa-1, 18.3 μmol g−1 h−1, and g-C3N4, 23.2 μmol g−1 h−1. TpBb-COF gave a CO production rate of 89.9 μmol g−1 h−1 when the CO2 concentration was changed from pure to 30.0% at 80 °C. The apparent quantum efficiency of TpBb-COF was 0.031% at 420 nm in 30% CO2 and 0.019% in pure CO2. This suggests that lower concentrations of CO2 facilitated the photocatalytic reduction of CO2. The performance, supported by DFT calculations, can be attributed to the more favourable adsorption of H2O on TpBb-COF, which enhanced the adsorption and reduction of CO2. The reaction mechanism was investigated using the rate equation for CO2 photocatalytic reduction. The rate equation derived correlates the CO2 concentration with the CO production rate, aligning with the experimental results and theoretical calculations.
In 2023, Jin and co-workers designed hydrophilic, fully conjugated COF materials that utilized H2O as the hydrogen and electron source for photocatalytic conversion of CO2 (Fig. 19).356 This research converted imine-linkages into 4-carboxyl-quinoline linkages in the COFs producing crystalline, porous, polymeric photocatalysts for CO2 photoreduction. The imine-linked LZU1-COF was synthesized through the Schiff-base reaction by condensing 1,3,5-triformylbenzene and 1,4-diaminobenzene. The quinoline-linked QL-COF was prepared with the one-pot Doebner reaction by reacting 1,3,5-triformylbenzene, 1,4-diaminobenzene, and pyruvic acid. The photocatalytic experiments were conducted using 2 mg of catalyst dispersed in ethanol which was packed onto a glass sheet and placed into a photoreactor containing aqueous solution. This setup was backfilled with pure CO2 for about 1 h at 1 atm pressure using a 300 W Xe lamp for light illumination. The performances of LZU1-COF and QL-COF were evaluated for CO2 photoreduction, and both COFs produced CO as the main product along with minor CH4 production. Compared with LZU1-COF, QL-COF exhibited superior photocatalytic performance, producing CO at a rate of 156 μmol g−1 h−1. LZU1-COF produced 25 μmol g−1 h−1 CO, 6 times less than QL-COF. This high performance is consistent with QL-COF's great H2O and CO2 capture capacities and higher carrier transfer efficiency. The inclusion of 4-carboxyl-quinoline in QL-COF improved its light absorption properties and the separation and transfer of photogenerated charge carriers, leading to its excellent performance in CO2 reduction.
 | ||
| Fig. 19 Natural photosynthesis, and artificial photosynthesis using the hydrophilic 4-carboxyl-quinoline COF (QL-COF).356 Reproduced with permission from ref. 356. Copyright 2023, Royal Society of Chemistry.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
In 2022, Zhang and colleagues synthesized 3D COFs using an 8-connected porphyrin-based building block arranged in a cubic configuration.357 These frameworks could be interwoven into an exceptional interpenetrated pcb topology. These distinctive 3D COFs were employed for the photocatalytic reduction of CO2, resulting in the formation of CO and CH4. To synthesize the 3D COFs, NUST-5 or NUST-6, 5,10,15,20-tetrayl(tetrakis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarbaldehyde)))-porphyrin (TTEP) was reacted with p-phenylenediamine (PDA) or dimethyl-p-phenylenediamine (PDA-Me) using a Schiff-base reaction under solvothermal conditions, respectively. CO2 photoreduction experiments were performed with the respective COFs dispersed in a mixture of MeCN, triethanolamine, and H2O, in a ratio of 3:1:1 under 1 atm CO2. The mixture was purged with CO2 for 30 min while stirring and then illuminated with a 225 W Xe lamp (420 nm cut-off filter). After 10 h, CO production was 54.7 μmol g−1 for NUST-5 and 76.2 μmol g−1 for NUST-6. Moreover, NUST-5 and NUST-6 displayed low CH4 production rates of 17.2 μmol g−1 and 12.8 μmol g−1, respectively.
6.5 Hydrogen peroxide production
Hydrogen peroxide has become essential in many commercial products such as antiseptics, disinfectants, and as a bleaching agent. Moreover, it is commonly used in synthetic chemistry as a relatively safe oxidant, producing only water and oxygen as by-products. However, at the industrial scale, synthesis of hydrogen peroxide is done through the hydrogenation of anthraquinone using a Pd catalyst followed by oxidation using molecular oxygen.358 Therefore, new methods to create hydrogen peroxide in less energy-intensive ways have been investigated.359–362 In recent years, COFs have been expansively used for photocatalytic H2O2 production, including the elucidation of the mechanism.363 Herein, we report on recent cases using COFs to photocatalytically synthesize hydrogen peroxide.Van Der Voort and Thomas were the first to use COFs for H2O2 production.364 COFs TAPD-(Me)2 and TAPD-(OMe)2 were constructed from N,N,N′,N′-tetrakis(4-aminophenyl)-1,4-phenylenediamine and either 2,5-dimethylbenzene-1,4-dicarboxaldehyde or 2,5-dimethoxybenzene-1,4-dicarboxaldehyde (Fig. 20). The COFs were then applied as photocatalysts for H2O2 production under visible light from an oxygen-saturated water/ethanol mixture, where ethanol acted as the electron and proton donor. TAPD-(Me)2 and TAPD-(OMe)2 produced 25.3 and 22.6 μmol of H2O2, respectively, over 16 hours. When TAPD-(Me)2 COF was used, and the ethanol was increased, the production increased to 57.2 μmol. In a five-hour window, the production of hydrogen peroxide was 97 ± 10 and 91 ± 10 μmol h−1 gcat−1 for TAPD-(Me)2 and TAPD-(OMe)2, respectively.
 | ||
| Fig. 20 Synthesis of TAPD-(Me)2 and TAPD-(OMe)2 COFs forming a dual pore kagome lattice.364 Reproduced with permission from ref. 364. Copyright 2020, American Chemical Society. | ||
Van Der Voort et al. also synthesized four pyrene COFs from 1,3,6,8-tetrakis(4-formylphenyl)pyrene (Py-CHO) with either 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetraaniline (Py-NH2), 1,4-diaminobenzene (Da-NH2), 2,2′-bipyridine-5,5′-diamine (Bpy-NH2), or N,N,N′,N′-tetrakis(4-aminophenyl)-1,4-phenylenediamine (TAPD) giving Py–Py-COF, Py-Da-COF, Py-Bpy-COF, and Py-TAPD-COF.365 DFT studies showed that the pyrene unit was the most favoured location for O2 adsorption comparatively to the N atoms on the bpy or TAPD units. The COFs were tested for hydrogen peroxide production under visible light in O2 saturated water. Py-Da-COF performed the best at 461 μmol g−1 followed by Py-Bpy-COF and Py-TAPD-COF with 241 and 142 μmol g−1, respectively. Interestingly, Py–Py-COF only garnered 47 μmol g−1 H2O2. When extended to three hours 868, 480, 361, and 85 μmol g−1 was produced for Py-Da-COF, Py-Bpy-COF, Py-TAPD-COF, and Py–Py-COF, respectively. When ethanol was added to the mixture, the production of hydrogen peroxide increased for Py-Da-COF and Py-Bpy-COF with a rate of 682 μmol g−1 and 452 μmol g−1, respectively. Notably, when benzyl alcohol (BA) was added a two-phase mixture was created that helped to prevent H2O2 decomposition and acted as a hole scavenger. The Py-Da-COF rate increased to an astonishing 3670 μmol g−1 in a 1:1 v/v BA/water mixture. The AQY of Py-Da-COF was measured at 2.4% in water/ethanol and 4.5% in water/BA at 420 nm. Overall, the authors noted that porosity played a key role in the COF activity in addition to O2 binding sites and reaction conditions.
Wang and co-workers synthesized three benzotrithiophene-based COFs with benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene-2,5,8-tricarbaldehyde (Btt) and 4,4,4-triaminotriphenylamine (Tpa), 1,3,5-tris(4-aminophenyl)benzene (Tapb), or 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (Tapt) affording TpaBtt, TapbBtt, and TaptBtt.366 The COFs were used as photocatalysts for hydrogen peroxide production using a Xe lamp light source in a water solution and under air atmosphere. TaptBtt exhibited the highest production of 31.67 μmol followed by TapbBtt with 12.54 μmol and finally TpaBtt with 5.68 μmol in 90 min. The difference was attributed to the D–A ability of the COFs. When tuning COF loading, 1.5 g L−1 gave the best results, increasing hydrogen peroxide production to 1407 μmol g−1 h−1 using TaptBtt. The authors noted that too much COF material would end up inhibiting light absorption. Furthermore, they synthesized TaptBtt with varying degrees of crystallinity and illustrated that a more crystalline COF gave higher yields. TaptBtt had an AQY of 4.6% at 450 nm. DFT studies revealed the imine linkage was important as a binding site for intermediates. Notably, the COF showed a slight decrease in crystallinity after recycling.
Ni and cohort constructed three β-ketoenamine-linked COFs using 1,3,5-triformylphloroglucinol with melamine, 1,3,5-triphenyltriazine, 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline, or 4′,4′′′,4′′′′′-(1,3,5-triazine-2,4,6-triyl)tris(([1,1′-biphenyl]-4-amine)) giving COF-N31, COF-N32, and COF-N33.367 They took these COFs and analysed them for photocatalytic hydrogen peroxide production and how adding phenyl units would affect the activity. The experiments were conducted in pure water under visible light using a Xe lamp for twelve hours. COF-N32 displayed 7092 μmol g−1 (or 605 μmol g−1 h−1) followed by COF-N31 with 4316 μmol g−1 followed by COF-N33 with 1736 μmol g−1. Notably, COF-N31 exhibited a loss of crystallinity and lowering yields when reused opposed to the other COFs. COF-N32 showed a superior AQY of 6.2% at 459 nm. COF-N32 was also used ex situ and in situ for disinfection and degradation of diclofenac. Aside from ultrapure water, the photosynthesis of hydrogen peroxide using COF-N32 could also be achieved in tap water, river water, and sea water with rates of 667, 648, and 554 μmol g−1 h−1 for three hours, respectively. The COFs were also used in membrane filter reactors, garnering about 30 μmol H2O2 in two hours for four cycles.
Shen et al. synthesized a multi-linker COF, TDB-COF, from 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde (TFPT), 2,5-bis(2-(ethylthio)ethoxy)terephthalohydrazide (BETH), and 2,5-diethoxyterephthalohydrazide (DETH).368 TDB-COF was used as a photocatalyst for hydrogen peroxide production in water using visible light under air or oxygen atmosphere. In air, 231 μmol g−1 h−1 was achieved, and under O2 723.5 μmol g−1 h−1 was achieved. When adding 10% ethanol as a sacrificial reagent the yield increased to 1000.1 μmol g−1 h−1. TDB-COF also performed better in acidic conditions than basic conditions. When TFPT-COF, the analogous COF without any thioether monomer, was used the H2O2 production decreased to 79.2 μmol g−1 h−1, showing the importance of the thioether moieties. The AQY values of TDB-COF were 1.4% at 365 nm and 1.0% at 400 nm. Notably, TDB-COF lost its crystallinity after recycling.
Wang and co-workers constructed two olefin-linked COFs from benzotrithiophene-2,5,8-tricarbaldehyde (BTT) with 2,4,6-trimethylbenzene-1,3,5-tricarbonitrile (TBTN) or 2,4,6-trimethyl-1,3,5-triazine (TMT) giving TBTN-COF and TMT-COF, respectively.369 These COFs were then used for the photosynthesis of hydrogen peroxide in water using visible light by a xenon lamp under an oxygen atmosphere. In the first 10 minutes, TBTN-COF exhibited 11013 μmol g−1 h−1 H2O2 comparatively to TMT-COF with 6392.3 μmol g−1 h−1. The authors attributed this difference to the cyano group playing a key role in the catalysis. The AQY of TBTN-COF was 7.59% at 420 nm. Interestingly, when p-benzoquinone was added as a scavenger, only TBTN-COF was affected, indicating that *OOH was a mechanistic intermediate for TBTN-COF but not for TMT-COF, which was also supported by DRIFTS. Also, when air was used instead of pure oxygen, the production rates decreased significantly for the COFs. DFT calculations and DRIFTS supported an O2 Yeager-type binding over Pauling-type binding in TBTN-COF.
Lan et al. synthesized two 3D COFs, TAA-CTP-COF and TAB-CTP-COF, with hexa-(4-formyl-phenoxy)-cyclotriphosphazene (CTP) and 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAA) or 1,3,5-tris(4-aminophenyl)benzene (TAB) (Fig. 21).370 The COFs were then studied for their photocatalytic performance in hydrogen peroxide production in water using a Xe lamp under an oxygen-saturated atmosphere. TAA-CTP-COF was able to furnish 1041 μM h−1 while TAB-CTP-COF furnished 818 μM h−1. TAA-CTP-COF was explained to perform better as the triazine unit helped increase electron–hole separation. When isopropanol was added as a sacrificial reagent 2221 μM h−1 was achieved with TAA-CTP-COF. When pH tests were performed, increasing the acidity or alkalinity decreased the performance.
 | ||
| Fig. 21 Schematic representation of COFs with different topological structures synthesized by CTP.370 Reproduced with permission from ref. 370. Copyright 2024, American Chemical Society.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
The Dong group constructed COF-Tfp-BDDA and COF-Tfp-BP by triacetaldehydemesitylphenol (Tfp) and 4,4′-(buta-1,3-diyne-1,4-diyl)diphenylamine (BDDA) or 4,4′-biphenylenediamine (BP).371 The two COFs were employed as photocatalysts in hydrogen peroxide production using visible light irradiation in pure water under an oxygen atmosphere. COF-Tfp-BDDA showed a hydrogen peroxide yield of 880 μmol g−1 h−1 while COF-Tfp-BP only furnished 100 μmol g−1 h−1. This demonstrated the importance of the diacetylene units. DRIFTS studies revealed an increase in the alkynyl band indicating oxygen binding, and the mechanism proceeded through an oxygen reduction reaction versus a water oxidation reaction. Notably, COF-Tfp-BDDA lost almost all crystallinity after 100 h of cycling.
Chen and cohort synthesized DVA-COF and PDA-COF from 1,3,5-tris(4-aminophenyl)benzene (TAPB) with either 2,5-divinylterephthalaldehyde (DVA) or terephthaldehyde (PDA).372 The COFs were used for the photosynthesis of H2O2 using LEDs in a water/benzyl alcohol two-phase mixture. DVA-COF produced 84.5 μmol of hydrogen peroxide after 1 hour, while PDA-COF produced only 8.6 μmol, indicating the importance of the vinyl moieties. The authors proposed that the vinyl groups increased O2 absorption from Yeager-type absorption. The vinyl groups also enhanced charge separation and transfer efficiency. Notably, benzaldehyde was produced in near equal amounts from the benzyl alcohol additive. The AQY of DVA-COF was 2.84% at 420 nm.
Yu et al. constructed six COFs using 1,3,5-triformylphloroglucinol (Tp) with ortho or para diamines with core benzene (Bda), pyridine (Pda) and pyrazine (Pzda) units, named o- or p-COF-TpBda, -TpPda and -TpPzda.373 These COFs were tested for their activity in the photocatalytic production of H2O2 under visible light in O2-saturated water. The production rate for o-COF-TpBda was 1030 μmol g−1 h−1 in one hour, o-COF-TpPda was 1947 μmol g−1 h−1, and o-COF-TpPzda was an astonishing 4396 μmol g−1 h−1. This demonstrated that adding pyridinic nitrogen atoms in the linker greatly enhanced the photoactivity. o-COF-TpPzda was also used in natural sunlight and air-saturated water and still achieved 1733 μmol g−1 h−1. The p-COFs were then tested, with rates of 282, 794 and 6434 μmol g−1 h−1 for p-COF-TpBda, p-COF-TpPda and p-COF-TpPzpa, respectively. Only p-COF-TpPzpa performed better than its o-COF counterpart; however, it showed severe structural damage by PXRD. DFT and DRIFTS showed that the pyridinic N atoms were important for binding H atoms to help induce the 2e− oxygen reduction reaction (ORR) while the β-ketoenamine linkages were important for the 4e− water oxidation reaction (WOR) process.
Zhu and co-workers synthesized COFs with aliphatic linkers, TAH-COF and BAH-COF, using tartaric acid dihydrazide (TAH) or butanedioic acid dihydrazide (BAH) and 1,3,5-triformylphloroglucinol.374 These COFs were then applied as photocatalysts for hydrogen peroxide production and hydrogen evolution under visible light using a Xe lamp. For hydrogen peroxide production in pure water, BAH-COF exhibited an impressive 1297 μmol h−1 g−1 and TAH-COF showed an outstanding 6003 μmol h−1 g−1. TAH-COF has the largest reported hydrogen peroxide production rate even with the absence of any cocatalyst, sacrificial reagent, or biphasic solvent mixture. The authors attributed the high rates to the hydroxyl groups allowing for hydrogen bonding. The AQY values of TAH-COF were measured at 7.12% at 475 nm, 7.72% at 500 nm, 7.39% at 520 nm, 3.94% at 550 nm, and 2.22% at 600 nm. TAH-COF was also used for HER, garnering 31.4 mmol h−1 g−1 with 3 wt% Pt cocatalyst and ascorbic acid as a sacrificial reagent. The authors attributed the success of these systems to the aliphatic linker leading to increased hydrophilicity, high crystallinity, and polarization of the knot which lowers the band gap. This work demonstrates the utility of using aliphatic linkers versus the more common rigid π-backbone monomers.
6.6 Aryl boronic acid oxidation
Phenolic compounds are useful for the synthesis of many organic compounds and intermediates in pharmaceuticals.375,376 Thus their synthesis has become prevalent in research. One route to synthesize these phenolic compounds has been through the oxidation of boronic acids, and performing this through benign means such as photocatalysis has been investigated.377–381 Herein, we report the use of COFs as photocatalysts for the oxidative conversion of boronic acids to phenols.Wang et al. constructed highly stable benzoxazole-linked COFs, LZU-190, LZU-191, and LZU-192, using 2,5-diamino-1,4-benzenediol dihydrochloride with 1,3,5-triformylbenzene, 2,4,6-tris(4-formylphenyl)-1,3,5-triazine, and 1,3,6,8-tetrakis(4-formylphenyl)pyrene, respectively.59 These COFs were then used as photocatalysts for the oxidation of boronic acids. The reaction was optimized on 4-carboxyphenylboronic acid where all three COFs showed 99% yield using white LEDs in air over 48 hours. LZU-190 was then applied to a substrate scope of 10 other aryl boronic acids, affording yields of 55–99%. The reaction was also successful in the presence of large substrates such as pyrenyl, naphthyl, and quinolinyl boronic acids with longer reaction times. The authors stated that the photocatalytic activity was derived from the benzoxazole units.
Zhang and co-workers synthesized three olefin-linked COFs using tricyanomesitylene with 4,4′′-diformyl-p-terphenyl(DFPTP), 4,4′-diformyl-1,1′-biphenyl (DFBP), or 1,3,5-tris-(4-formylphenyl)benzene (TFPB) to give COF-p-3Ph, COF-p-2Ph, and COF-m-3Ph, respectively.382 These COFs were then applied for the photocatalytic oxidative hydroxylation of boronic acids. The reaction conditions were optimized on 2-naphthylboronic acid, where COF-p-3Ph performed the best with 99% yield using a Xe lamp and oxygen with triethylamine as a sacrificial electron donor. The conditions were then used on 8 other boronic acids, garnering yields of 56–99%. The activity was attributed to excellent π-delocalization and great light absorption.
Jiang and co-workers synthesized porphyrin COFs from 2,5-dihydroxyterephthalaldehyde (Dha) and 5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphine to give COFs DhaTph-M (M = Zn or Ni).383 These COFs were used as photocatalysts in a multitude of oxidation reactions. First, they were tested for the oxidation of α-terpinene using a xenon lamp as the light source under an oxygen atmosphere. The Zn-metalated COF achieved the highest yield and selectivity of ascaridole in 93%, and 7% yield of p-cymene, whereas the Ni-metalated version gave 83% yield of ascaridole and 17% p-cymene. The difference in yields was attributed to the different mechanistic pathways and reactive oxygen species that were generated. Notably, when using air, instead of pure oxygen, conversion decreased and selectivity towards p-cymene increased. DhaTph-M COFs were then applied to the photocatalytic oxidation of phenylboronic acid to phenol using a Xe lamp under an oxygen atmosphere with triethylamine as an electron donor. DhaTph-Ni displayed 99% yield, but DhaTph-Zn only yielded 35%. The authors stated that DhaTph-Ni had higher charge separation leading to higher photocatalytic activity. The authors then used the COFs for the oxidation of thioanisole to form the methyl phenyl sulfoxide product using a Xe lamp under an oxygen atmosphere. DhaTph-Zn afforded 82% yield while DhaTph-Ni afforded only 20% yield. Again, this discrepancy was attributed to varying catalytic pathways. Overall, the authors conducted experiments illustrating that Zn metalation increases triplet oxygen formation and Ni metalation induces exciton dissociation to charge carriers.
Mondal et al. constructed two benzothiazole COFs from imine-linked COFs; the imine-linked COFs were synthesized from tris(4-formylphenyl)amine (TPA) and benzene-1,3,5-tricarbaldehyde (BCA) with 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (Tt), and then using elemental sulfur to form the benzothiazole linkage, making BTZ-BCA-COF and BTZ-TPA-COF.384 These COFs were then used for the photocatalytic oxidation of phenylboronic acid using oxygen and blue LEDs with triethylamine as a sacrificial reagent. While both COFs exhibited quantitative yields, BTZ-TPA-COF performed the reaction in two-thirds of the time of BTZ-BCA-COF due to its D–A system and smaller band gap. BTZ-TPA-COF was used on five other substrates, furnishing yields of 90–99%. The catalytic performance was due to superior π-delocalization and optoelectronic properties derived from the benzothiazole linkage.
Wen and co-workers prepared COF-JLU25 from 1,3,6,8-tetrakis(4-aminophenyl)pyrene (PyTA) and 4-[4-(4-formylmethyl)-2,5-dimethoxyphenyl]benzaldehyde (TpDA).385 They took this COF and used it as a photocatalyst for oxidative hydroxylation of aryl boronic acids. The optimized parameters of using white LEDs, air as oxidant, and N,N-diisopropylethylamine as a sacrificial electron donor were able to achieve a quantitative yield of the 4-(methoxycarbonyl)phenylboronic acid substrate. COF-JLU25 was also used as a photocatalyst using these conditions on 12 other compounds, attaining yields between 25 and 99%. Notably, aryl boronic acids with slightly electron-withdrawing or any kind of electron-donating group gave poor yields, but increasing the reaction times from 12 to 72 hours produced yields ranging from 90 to 99%. The catalytic activity was attributed to connecting boron atoms to electron-poor π-electron systems to increase the tendency of the boron atom to accept an electron. Notably, the COF had a significant decrease in crystallinity after 7 cycles.
The Huo group made a D–A COF, BTT-BTDDA-COF from benzotrithiophene (BTT) and 4,4′-(2,1,3-benzothiadiazole-4,7-diyl)dianiline (BTDDA).386 Using this COF as a photocatalyst, they were able to convert phenylboronic acid to phenol using blue LEDs, air as the oxidant, and triethylamine as the electron donor in 93% yield. When applying these conditions to a substrate scope of 10 other compounds, high yields of 82–96% were obtained. The photoelectric performance was created by the wide light absorption and narrow band gap energy from efficient intramolecular charge transfer as well as good charge separation. The authors proposed a single-electron transfer mechanism and superoxide radical as the reactive oxygen species (Fig. 22).
 | ||
| Fig. 22 Possible reaction mechanism.386 Reproduced with permission from ref. 386. Copyright 2022, Royal Society of Chemistry.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
Recently, Thomas et al. constructed CzDA-TAPT-COF from 4,4-(9-butyl-9H-carbazole-2,7-diyl)dibenzaldehyde and tris(4-aminophenyl)benzene.387 CzDA-TAPT-COF was then employed as a photocatalyst for the oxidative hydroxylation of boronic acids. Using 4-formylphenylboronic acid as a benchmark compound, 92% yield was garnered using blue LEDs, air as the oxidant, and tetramethylethylenediamine (TMEDA) as the sacrificial reagent. Applying this on a broad substrate scope, all substrates gave yields between 53 and 94% with the exception of a triphenylamine-based boronic acid that gave low yield. The high activity was attributed to efficient charge transfer and separation.
Recently, Abbaspourrad and co-workers synthesized unique tetrazine-based vinylene-linked COFs from 3,6-dimethyl-tetrazine (DMTAZ) with 1,3,5-tris(4-formylphenyl)benzene (TFPB) and 1,3,5-tris(4-formylbiphenyl)benzene giving TA-COF-1 and TA-COF-2, respectively.388 These COFs were employed as photocatalysts for the oxidation of arylboronic acids to phenols using blue LEDs, air as the oxidant, and N,N-diisopropylethylamine as a sacrificial electron donor. Excellent yields of 92–99% were obtained using a wide substrate scope in only three hours. The superior activity was attributed to the tetrazine moiety being the active center, as it can accept an electron to form a radical anion to make a very reactive superoxide radical facilely. The COFs were also shown to be highly active for photocatalytic oxidative coupling of benzylamines, furnishing yields of 90–99% of five different benzylamines substrates. The COFs also were utilized for sunlight-driven photolysis of 5-nitro-1,2,4-triazol-3-one (NTO). Tetrazine is able to be in a stable protonated state, thus also allowing for protonation of TA-COFs (Fig. 23). In fact, TA-COF-1-H+ and TA-COF-2-H+ completely degraded NTO within 20 and 25 min, respectively.
 | ||
| Fig. 23 (a) Fluorescence spectrum of acid-treated TA-COF-1 suspension (0.1 mg mL−1) in 1,4-dioxane under excitation at 465 nm (acid concentration: 0–59.6 mmol L−1). (b) Correlation curve of fluorescence intensity versus acid concentration. (c) PL decay spectra of TA-COF-1 monitored upon excitation at 365 nm. (d) Possible protonation and deprotonation sites on TA-COF. (e) Naked-eye photographs of TA-COF powders upon exposure of HCl and NH3 vapors.388 Reproduced with permission from ref. 388. Copyright 2024, John Wiley and Sons.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
6.7 Sulfide oxidation
Sulfoxides have become prevalent in many drug molecules and pesticides.389–392 Furthermore, the conversion of sulfides to sulfoxides has become a viable method to detoxify harmful sulfur-based warfare agents.393–395 However, selective oxidation of organic sulfides to sulfoxides can be problematic due to sulfur being easily overoxidized to the sulfone, which has drastically different properties. Conventional methods rely on metals, harsh oxidants and/or thermal conditions, so more environmentally friendly methods such as photocatalysis have been explored to perform this oxidation more easily and selectively.396–398 Here, we report on using COFs as efficient photocatalysts for sulfide oxidation.Bai et al. synthesized AQ-COF from 2,6-diaminoanthraquinone and 1,3,5-triformylphloroglucinol.399 They also made AQ-COFDMF which had the same framework but different morphology and structure due to entrapped DMF molecules. These COFs were used as a photocatalyst for the oxidation of sulfides using oxygen as the oxidant and a Xe lamp as the light source. In the presence of thioanisole, the AQ-COF demonstrated 99% conversion with 97% selectivity for the sulfoxide while AQ-COFDMF only gave 73% conversion with 99% selectivity. The AQY of AQ-COF was 4.34% and 2.70% for AQ-COFDMF at 420 nm. This work demonstrated that the morphology and structure of a COF is important in photocatalytic applications due to a difference in the optical and electronic character. Using AQ-COF, 99% conversion was achieved with selectivity of 94–99% on a variety of aryl sulfides. However, 4-nitrothioanisole and diphenyl sulfide achieved lower yields of 77% and 37%, respectively.
Jin and co-workers constructed crystalline CTFs using p-phthalaldehyde and 4-(hydroxymethyl)-benzaldehyde affording CTF-HUST-D1 and CTF-HUST-E1, respectively.400 The CTFs were then used for photocatalytic oxidation of sulfides using blue LEDs using oxygen in the air as the oxidant. CTF-HUST-D2 achieved conversions of 91–99% and selectivity of 81–96% for the sulfoxide on a variety of sulfide substrates. Notably, ethylphenylsulfide and thiane gave less than 90% selectivity of 81% and 85%, respectively. The authors noted that putting the electron-donating methoxy groups on the CTF backbone enhanced its photoelectron properties.
Zhao and cohort synthesized a unique CPTPA-COF from 4,4′,4′′-nitrilotrisbenzaldehyde (NTBA) and hexakis(4-aminophenyl)-cyclotriphosphazene (HACP) (Fig. 24).401 The CPTPA-COF was then applied as a photocatalyst for the oxidation of sulfides using air as the oxidant and using LED lights. This system was able to quantitatively convert ten various sulfides in four hours with near perfect selectivity for the sulfoxide including 2-chloroethyl ethyl sulfide, a nerve agent simulant. However, 4-bromothioanisole, 2-(methylthio)naphthalene, and ethylphenylsulfide had poor selectivity of around 70%. EPR measurements showed that the COF was an effective photocatalyst for producing O2˙−.
 | ||
| Fig. 24 Schematic diagram for the synthesis of CPTPA-COF.401 Reproduced with permission from ref. 401. Copyright 2022, American Chemical Society.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
Dong and co-workers synthesized a rare chiral COF, (R)-DTP-COF-QA, from 2,5-dimethoxyterephthaldehyde (DMTP), 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAPP), and N,N-diethyl-N-(4-ethynylbenzyl)ethanaminium bromide (PA-QA) using a Cu catalyst with a chiral ligand.402 Using LEDs and air as the oxidant, the chiral COF was used as a photocatalyst for the oxidation of thioanisole. Using white LEDs gave appreciable yield of 52% and green LEDs gave a higher 87% yield. When red LED light was used, a high yield of 94% was produced with 99% (R) enantiomeric excess (ee). Furthermore, when the (S)-DTP-COF-QA was used, a similar yield of 98% and 94% (S) ee was afforded. Both chiral COFs were then used to synthesize the drug molecule (R/S)-modafinil, respectively. A yield of 90% with 88% ee was achieved for (R)-modafinil, while (S)-modafinil obtained an 88% yield with 86% ee. This work demonstrated a rare example of using red light to efficiently promote a photocatalytic reaction, and the utilization of a chiral COF to directly synthesize a drug molecule in high yield and enantiomeric excess.
Wang and co-workers constructed a nanoparticle-based COF using 1,3,5-tris(4-aminophenyl)benzene (TAPB) and [2,2′-bipyridine]-5,5′-dicarbaldehyde (Bp) and Co(NO3)2.403 The Co(NO3)2/COF was then used to photocatalytically convert thioanisole to the sulfone product under an air atmosphere with a Xe lamp as the light source. Compared with the pristine COF, the nanoparticle-based COF showed 99% yield and 100% selectivity for methyl phenyl sulfone in six hours. The authors attributed this activity to good separation of photogenerated excitons and low charge recombination. This work illustrated a rare example of making the sulfone versus sulfoxide product.
Lang and cohort synthesized a thiazole-linked COF, TTT-COF, from PSM of the imine-linked TTI-COF from 1,3,5-tris(4-aminophenyl)-triazine (TA) and 2,4,6-tris(4-formylphenyl)-1,3,5-triazine (TF).404 They took these COFs and employed them for the photocatalytic oxidation of aryl sulfides using blue LEDs and oxygen as the oxidant. The imine-linked TTI-COF showed yields lower than 20% while the thiazole-linked TTT-COF afforded yields ranging from 50 to 85% in only 25 min. They also tested the change in photocatalytic activity with varying LEDs. Purple and blue LEDs afforded high yields, green LEDs only afforded 20%, and yellow or red LEDs afforded trace yields. Applying TTT-COF on a larger substrate scope, 17 different sulfides were converted above 90% with near perfect selectivity of the sulfoxide, with the exception of di-nbutyl sulfide, in 8–135 min. The authors attributed the photocatalytic performance to the highly photoactive thiazole linkage of the TTT-COF. Notably, the imine-linked TTI-COF lost crystallinity when recycled.
Lang and co-workers designed three olefin-linked COFs from 2,4,6-trimethyl-1,3,5-triazine (TMT) with 1,3,5-triformylbenzene (TFB), 1,3,5-tris(4-formylphenyl)benzene (TFPB), and 1,3,5-tris(4-formylphenylethynyl)benzene (TFPEB) giving TMT-TFB-COF, TMT-TFPB-COF, and TMT-TFPEB-COF, respectively (Fig. 25).405 These COFs were then used for photocatalytic oxidation of sulfides using blue LEDs under an oxygen atmosphere. TMT-TFPEB-COF possessed much stronger opto-electronic properties compared with the other COFs due to the presence of the alkynyl units. On an initial small substrate scope the TMT-TFPEB-COF performed well, achieving 90% and greater conversion rates, while TMT-TFPB-COF gave much smaller 15–55% conversions, while the TMT-TFB-COF afforded conversions below 10%. The authors also tested the use of green LEDs but only furnished about 15% yield, and yellow or red LEDs resulted in no yield. TMT-TFPEB-COF was employed on ten substrates affording high (>70%) conversion and near perfect selectivity of the sulfoxide products in 30 min. However, 4-nitrothioanisole and diphenylsulfide only gave 13 and 14% conversion, respectively. Overall, the authors illustrated that expanding the π-conjugation with alkynyl units increased the photocatalytic activity of the material. Interestingly, when probing the importance of crystallinity, a highly crystalline TMT-TFPEB-COF performed better than a low crystalline TMT-TFPEB-COF and much better than an amorphous TMT-TFPEB polymer.
 | ||
| Fig. 25 Schematic illustration of the construction of TMT-TFB-COF, TMT-TFPB-COF, and TMT-TFPEB-COF.405 Reproduced with permission from ref. 405. Copyright 2024, American Chemical Society.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
6.8 Benzyl alcohol oxidation
The oxidation of aromatic compounds is a critical transformation in organic chemistry with significant applications in pharmaceuticals and the chemical industries.406–409 Photocatalytic oxidation of alcohols has also been explored as a greener method.410 COFs have been used as photocatalysts in alcohol oxidation reactions; however, there are a limited number of reports. There has also been a report utilizing a MOF–COF hybrid as a photocatalyst for benzyl alcohol oxidation.411In 2021, Lotsch and co-workers incorporated an alloxazine chromophore unit into a COF framework for metal-free photocatalytic oxidation reactions.412 FEAx-COF, the first COF featuring an alloxazine unit, was synthesized by condensing 1,3-diethyl-6,9-bis-(4-formylphenyl)alloxazine (FEAx) with 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT) under solvothermal conditions. The alcohol oxidation reaction was carried out by irradiating a reaction mixture containing 4-methoxybenzyl alcohol (MBA) and FEAx-COF in oxygenated acetonitrile with blue light at a wavelength of 463 nm for 17 hours. Under these reaction conditions, MBA was oxidized to 4-methoxybenzaldehyde (MBAld) with a yield of 70% and a selectivity of 96%. Furthermore, the authors explored the use of other LEDs and observed yields were decreased in green light and there was no yield in orange light. Investigation into the reductive quenching of FEAx-COF, based on the proposed mechanism and quantum chemical calculations of a molecular model representing the COF, demonstrated that MBA was photooxidized by the flavin.
In 2020, Palkovits and colleagues used semi-crystalline thiophene-based covalent triazine frameworks (CTFs) for metal-free photocatalytic oxidation of aromatic alcohols.413 They synthesized four distinct CTFs, using the solvothermal amidine-aldehyde method, interconnected by a robust Kongming lock.414 Two of these CTFs contained sulfur. The synthesized CTFs were CTF-Ph (formed from terephthalaldehyde and terephthalamidine), CTF-Th (formed from 2,5-thiophenedicarboxaldehyde and terephthalamidine), CTF-BiPh (formed from 1,4-phthalaldehyde and [1,1′-biphenyl]-4,4′-bis(carboximidamide)), and CTF-PhTh (formed from 2,5-thiophenedicarboxaldehyde and [1,1′-biphenyl]-4,4′-bis(carboximidamide)). The CTFs incorporating thiophene units, enhanced by sulfur, demonstrated a smaller band gap with greater thermal stability in comparison with the phenyl-based CTFs. When the reaction was performed in the conditions of benzyl alcohol, acetonitrile, and CTF photocatalyst under UV light (λ = 365 nm, 0.1 W cm−2) and under O2 (balloon), all of the CTFs showed benzaldehyde selectivity of 100% after 1 h. CTF-PhTh and CTF-BiPh exhibited high catalytic activity in the photocatalytic oxidation of benzaldehydes, achieving nearly 100% selectivity at or near an 80% conversion rate at a concentration of 1 mg mL−1, 1 h time and a light source of 0.8 W LED UV light (λ = 365 nm). This performance ranks among the most effective metal-free photocatalysts and is comparable to some metal-based counterparts. They also used the CTFs on a small substrate scope of five other aromatic alcohols, showing conversions of 12–100% with selectivity of 42–100%, still showing the need for improved CTF and COF photocatalysts.
In 2020, Huang and co-workers synthesized a series of COF/CdS photocatalysts by varying the cadmium sulfide content ratio and testing these photocatalysts for their photocatalytic activity in the oxidation of aromatic alcohols.415 This study used terephthalaldehyde and 1,3,5-tris(4-aminophenyl)benzene as precursors with acetic acid as the catalyst to synthesize the COF. COF/CdS composites were obtained by varying the ratio of CdS in the composites. The final products were designated as COF/CdS-1, COF/CdS-2, COF/CdS-3, and COF/CdS-4 with 8.63 wt%, 17.03 wt%, 26.81 wt% and 29.70 wt% CdS, respectively. The reaction was carried out using 20 mg COF catalyst, in trifluorotoluene and acetonitrile using a white light LED lamp of 5.0 W power (λ = 420–780 nm, power density 150 mW cm−2), at 1 atm O2. The photocatalytic results for selective oxidation of benzyl alcohol to benzaldehyde were formed with no other by-products such as benzoic acid, indicating excellent selectivity. When pure CdS and COF were used separately as photocatalysts, they demonstrated conversion rates of 38.3% and 6.1%, respectively, for benzyl alcohol. For COF/CdS, the conversion of benzyl alcohol rose from 41.3% for COF/CdS-1 to 97.1% for COF/CdS-3, showing high performance for the COF/CdS combination. This demonstrated that depositing an appropriate amount of CdS nanoparticles on a COF can increase their photocatalytic oxidation activity. The authors showed evidence for the use of superoxide radicals to perform this reaction and provided a mechanism (Fig. 26). They also tested COF/CdS-3 on six other aromatic alcohols, achieving conversions of 74–100% with excellent selectivity.
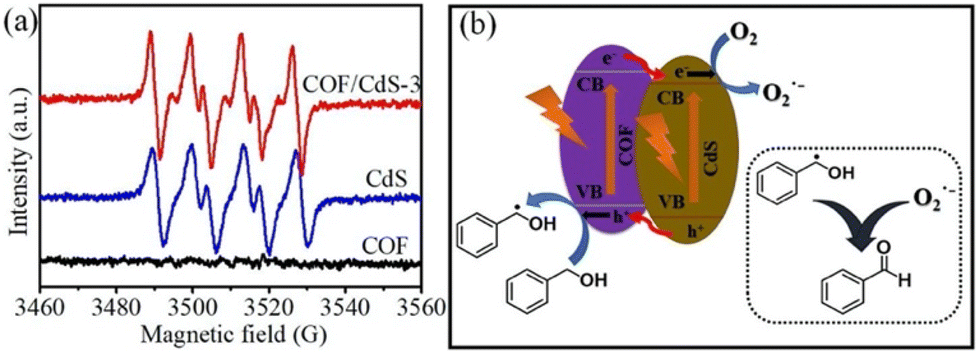 | ||
| Fig. 26 (a) ESR spectra of benzyl alcohol radicals and O2˙− radical species trapped by DMPO. (b) Proposed catalytic mechanism of the photocatalytic oxidation of benzyl alcohols over COF/CdS-3.415 Reproduced with permission from ref. 415. Copyright 2021, Elsevier.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
Yang and co-workers recently introduced a COF–COF heterojunction designed to enhance exciton dissociation for visible-light-driven alcohol oxidation.416 This hybrid structure was composed of two COFs, Tp-TTA COF, synthesized from Tp (1,3,5-triformylphloroglucinol) and TTA (4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline) and QH-COF, created via a one-pot Povarov cascade imine formation and cycloaddition reaction. A COF–COF heterojunction was formed by the deposition of nano Tp-TTA colloids onto QH-COF. The reactions were conducted in benzotrifluoride bubbled with oxygen for 20 min before being exposed to light from a Xe lamp equipped with a UV-cut-off filter (λ ≥ 420 nm). When tested individually, Tp-TTA and QH-COF achieved lower conversions of 29% and 55%, respectively, for the photocatalytic oxidation of benzyl alcohol (BA) to benzaldehyde (BAD) after 6 hours. This could be attributed to their poor charge separation efficiency. Interestingly, the Tp-TTA/QH-10 heterojunction effectively catalysed the oxidation of BA, resulting in a high conversion rate of 92% with a BAD selectivity of 87%. Notably, the Tp-TTA/QH-10 heterojunction showed a decrease in crystallinity when recycled.
Yang and co-workers also constructed QH-COF@TiO2 and TiO2@QH-COF hybrids by doping QH-COF with TiO2 or growing the COF on TiO2, respectively.417 For the photocatalytic oxidation of aromatic alcohols, the reaction was conducted in benzotrifluoride at 1 atm O2 using a 300 W Xe lamp (λ ≥ 420 nm). Using benzyl alcohol as a model compound, QH-COF alone achieved 56% conversion and 95% selectivity, and TiO2 alone showed no conversion. The QH-COF@TiO2(10%) hybrid showed the best performance at 84% conversion and 93% selectivity for benzaldehyde. This was extended to a substrate scope of aromatic, aliphatic, and cyclic alcohols, garnering conversions of 35–89% and selectivity of 59–95%. However, the QH-COF@TiO2(10%) hybrid lost crystallinity when recycled.
6.9 Oxidative amine coupling
The utilization of COFs as heterogeneous photocatalysts for oxidative amine coupling has garnered a lot of attention due to their use in synthesizing dyes, pharmaceutical drugs, and agrochemicals.418,419 This transformation is traditionally done using transition metals or organoboron.420–422In 2022, Tan and colleagues enhanced photogenerated carrier transport and separation efficiency for photocatalytic oxidative amine reactions by utilizing unique donor–acceptor pairs within a COF framework through a push–pull electronic effect.423 They synthesized the donor–acceptor COF through the polycondensation of electron-donating 1,3,5-triformylphloroglucinol (Tp) and electron-accepting 1,3,5-triazine-2,4,6-triamine (Tt) monomers. The reactions involved the COF photocatalyst in acetonitrile conducted in a cylindrical quartz vessel irradiated with a 35 W halogen lamp (λ > 420 nm) under 1 atm of air. For benzylamine, TpTt-COF displayed complete conversion and excellent selectivity, and in contrast the non-donor–acceptor TfTa-COF was less efficient, producing only a 33% yield of N-benzylidenebenzylamine. The TpTt-COF was also highly effective in the visible-light-driven oxidative coupling of nine other amines with yields ranging from 71 to 99% with great selectivity. DFT calculations confirmed that the internal donor–acceptor pairs in TpTt-COF facilitated high light harvesting and charge separation, leading to superior performance, and the authors proposed a mechanism based on these calculations and UV-Vis data (Fig. 27).
 | ||
| Fig. 27 (A) Proposed mechanism for the visible light-driven oxidative coupling of benzylamine (BA) to N-benzylidenebenzylamine with air over TpTt-COF. (B) Changes in the UV-vis spectra of the reaction system upon the addition of DPD.423 Reproduced with permission from ref. 423. Copyright 2022, Royal Society of Chemistry. | ||
In 2020, Wang and co-workers synthesized fully conjugated D–A COFs for photocatalytic oxidative amine coupling and thioamide cyclization.424 By incorporating benzothiadiazole units into the frameworks, they achieved high stability and improved charge separation. The Py-BSZ-COF was created through a base-catalysed polycondensation of 1,3,6,8-tetrakis(4-formylphenyl)pyrene and 4,4-(benzothiadiazole-4,7-diyl)diacetonitrile. For the photocatalytic oxidation of amines, the COF photocatalyst was dispersed in acetonitrile at 1 atm air under visible light irradiation with a 15 W 520 nm LED bulb (5 mW cm−2). After 12 hours, N-benzyl-1-phenylmethanimine was formed with a 99% conversion rate. The performance of Py-BSZ-COF was compared with sp2c-COF-3 and COF-JLU22. Under identical reaction conditions, sp2c-COF-3 achieved a 67% conversion, while COF-JLU22 reached 90%. Py-BSZ-COF was also used on eight other benzylamines, achieving conversions of 52–99% in 98 or 99% selectivity. The superior performance of Py-BSZ-COF was attributed to its production of superoxide radical anions (O2˙−).
In 2022, the Wu group developed a series of hydrophilic 2D COFs for photocatalytic oxidative amine reactions in aqueous conditions.425 The three COFs, TFB-XX-DMTH, were synthesized through the polycondensation of 2,5-dimethoxyterephthalohydrazide (DMTH), 1,3,5-triformylbenzene (TFB), and 2-hydroxy-1,3,5-benzenetricarbaldehyde (SOH) under solvothermal conditions. The “XX” denotes the different molar ratios of the aldehyde unit (TFB/SOH = 1:2, 1:1, 2:1) used, resulting in TFB-33-DMTH, TFB-50-DMTH, and TFB-66-DMTH, respectively. These COFs were employed for metal-free photocatalytic aerobic oxidative coupling of amines. The reactions were conducted under visible light irradiation from a 30 W blue LED lamp, in air, and at room temperature in water. TFB-33-DMTH, which had the highest number of hydroxyl groups, exhibited the highest conversion rate, selectivity, and excellent recyclability. On nine different benzylamines, conversions were observed at 95–99% with 93–100% selectivity. This performance was attributed to the complementary effect of the extended π-units and the hydroxyl groups, which enhanced the push–pull effect, thereby improving visible-light absorption and the efficiency of charge separation.
Porphyrin-based COFs have been greatly investigated for photocatalytic oxidative amine coupling due to their light absorption capabilities and unique electronic properties.426–430 In 2022, Jiang and co-workers developed a photoresponsive porphyrin-based COF that converts to its photoisomer under UV irradiation for photocatalytic oxidative amine reactions.431 This COF incorporated light-harvesting porphyrin photosensitizer units and photochromatic diarylethene with photoswitchable properties, enabling the formation of o-COF for the generation of singlet oxygen. The imine-linked photoresponsive ring-opened o-COF was synthesized by the reaction between dithienylethene–dialdehyde (o-BBTP) and 5,10,15,20-tetra(p-aminophenyl)-porphyrin (H2TAPP) in an acid-catalysed solvothermal reaction. c-COF, the ring-closed photoisomer, was obtained by irradiating o-COF with UV light. Photocatalytic oxidation of amines was conducted using COF photocatalyst in acetonitrile with irradiation provided by a 25 W blue LED light under an air atmosphere. o-COF exhibited excellent photocatalytic performance, achieving 99% conversion to N-benzylidenebenzylamine after 60 min of irradiation in both air and O2 environments. Interestingly, c-COF showed lower efficiency with only 63% conversion, which was attributed to its reduced capability in generating photoinduced singlet oxygen (1O2), proving different energy transformations for both COFs. To confirm the involvement of singlet oxygen in the reaction mechanism, triethylenediamine (DABCO), a scavenger for 1O2, was introduced, resulting in a significantly reduced product yield of only 9%. This experiment strongly supports the role played by 1O2 in facilitating the oxidative coupling of benzylamine. o-COF was also tested on seven other benzylamines, achieving full conversions in 30–60 min.
β-Ketoenamine COFs have been investigated for their photocatalytic oxidative coupling of amines due to their tunable properties and high stability.432,433 In 2023, the Lang group synthesized a series of β-ketoenamine COFs, TpPa-COF, TpBD-COF, and TpDT-COF, featuring different linker lengths for photocatalytic oxidative amine coupling.434 TpPa-COF and TpBD-COF were prepared following slightly modified procedures from the literature.435 TpDT-COF was synthesized using 1,3,5-triformylphloroglucinol and the diamine, 4,4′′-p-terphenyldiamine, through a solvothermal condensation reaction. For the photocatalytic oxidation of amine coupling, benzylamine and 5 mg of the photocatalyst were dispersed in acetonitrile in a Pyrex photoreactor, stirred, and irradiated with violet LEDs (3 W × 4). Under the same reaction conditions, TpBD-COF achieved a 78% conversion of benzylamine, while TpPa-COF and TpDT-COF achieved 22% and 61% conversion, respectively. With its moderate linker length, TpBD-COF demonstrated the highest photocatalytic activity for photocatalytic aerobic oxidation of amines, highlighting the significance of linker length in designing more effective COF photocatalysts.
Liu and co-workers constructed diketopyrrolopyrrole-based COFs, TpDPP-Py COF and DPP-Py COF, from 5,5′-(2,5-bis(2-ethylhexyl)-3,6-dioxo-2,3,5,6-tetrahydropyrrolo[3,4-c]pyrrole-1,4-diyl)bis(thiophene-2-carbaldehyde) (TpDPP) or (4,4′-(2,5-bis(2-ethylhexyl)-3,6-dioxo-2,3,5,6-tetrahydropyrrolo[3,4-c]pyrrole-1,4-diyl)dibenzaldehyde) (DPP) and 1,3,6,8-tetrakis(4-aminophenyl)pyrene (Py).436 TpDPP-Py COF exhibited a very small band gap of 1.38 eV due to the very strong acceptor and donor units. For the photocatalytic amine coupling, the reaction took place in a sealed Pyrex vessel in acetonitrile that was bubbled with O2 for 30 min and irradiated by a 300 W Xe lamp with AM 1.5G solar intensity. TpDPP-Py COF exhibited superior catalytic efficacy for the conversion of benzylamine, achieving 100% conversion and 100% selectivity in only 20 min, the best of any reported COF photocatalyst, and most photocatalysts in general. DPP-Py COF took 40 min to achieve the same outcome. TpDPP-Py COF was also used as a photocatalyst on six other aromatic amines, garnering 100% conversion with 100% selectivity in 15–40 min. The authors attribute the success to enhanced absorption capacity and multi-photon absorption effects.
6.10 C–H activation and functionalization
C–H activation and functionalization has become a very important method to target a wide array of compounds from abundant alkane building blocks.437–439 Directly activating and functionalizing C–H bonds bypasses intermediate functional group installations and conversions, leading to fewer steps in otherwise long synthetic pathways, and increases overall atom economy. However, stoichiometric reagents and directing groups, harsh oxidants, and precious metals are typically required due to the inertness of C–H bonds. In addition, poor selectivity is common due to C–H bonds possessing similar bond dissociation energies and reactivity.437–439 Performing C–H activation and functionalization photocatalytically has been explored,440–442 and more recently using heterogeneous photocatalysts.443 The emergence of COFs as efficient heterogeneous photocatalysts for C–H activation has garnered substantial interest in the scientific community by enabling the selective and sustainable light-driven functionalization of organic molecules.444–447Dong and co-workers synthesized a highly crystalline donor–acceptor (D–A) COF, TPPy-PBT-COF, from 7-(4-formylphenyl)benzo[c][1,2,5]thiadiazole-4-carbaldehyde (PBT) and 1,3,6,8-tetrakis(4-aminophenyl)pyrene (TPPy) via imine condensation.448 This COF was used for photocatalytic aerobic cross-dehydrogenative coupling reactions including Mannich and aza-Henry reactions. The reaction was performed in a quartz cell in methanol with L-proline additive under O2 with LED lights or sunlight. Using N-phenyl-1,2,3,4-tetrahydroisoquinoline and acetone as the model reaction, white light, blue light, green light, or sunlight all achieved similar yields between 77 and 84%. Changing either the tetrahydroisoquinoline or the nucleophile, yields of 52–86% were achieved using sunlight. The reaction was also performed on a gram scale using sunlight and still obtained 77% yield. Based on EPR measurements, the authors proposed a mechanism where upon light irradiation TPPy-PBT-COF photogenerates electrons that react with oxygen to form superoxide radicals. The superoxide radicals then abstract a hydrogen from the substrate followed by a second abstraction by the resulting hydroperoxide radical. The imine cation then undergoes nucleophilic addition to form the product with hydrogen peroxide as a by-product.
Yang and co-workers synthesized four imine-linked 2D-COFs and one olefin-linked 2D-COF from previous reports.444 Specifically, the olefin-linked COF, 2D-COF-2, constructed from 2,4,6-trimethyltriazine and [1,1′-biphenyl]-4,4′-dicarbaldehyde, was used as a photocatalyst for decarboxylative alkylation of heterocycles with N-hydroxyphthalimide (NHPI) esters.449 The reactions were conducted in N,N-dimethylacetamide, stoichiometric trifluoracetic acid, under blue LED lights and under an argon atmosphere. On the model reaction between isoquinoline with cyclohexyl N-hydroxyphthalimide, 2D-COF-2 outperformed its imine counterparts, achieving a yield of 85%, and notably the imine COFs decomposed in the reaction conditions. The reaction was conducted on an impressive 55 substrates obtaining yields from 43 to 97%. They also tested the reaction on the drug molecules lithocholic acid and ibuprofen and achieved yields of 71–78%, and even increased to a 0.5 g scale for lithocholic acid and still garnered 68% yield. The authors suggested a mechanism where the photoexcited 2D-COF-2 reduces the NHPI ester to give an alkyl radical, with the loss of carbon dioxide, that attacks the TFA-activated quinoline which eventually forms the product after oxidization and deprotonation. Notably, the COF was recyclable but showed decreased crystallinity.
Lin and co-workers constructed an anthraquinone-based 2D COF, DAAQ-COF, from 1,3,5-triformylphloroglucinol (TFP) and 2,6-diaminoanthraquinone (DAAQ).450 DAAQ-COF was used for photocatalytic C–H functionalization as a direct hydrogen transfer reagent as well as the photocatalyst. The coupling between diethyl azodicarboxylate (DEAD) and tetrahydrofuran (THF) as the model reaction under compact fluorescent light (CFL) irradiation was performed, obtaining 94% yield. The reaction was tested on 15 substrates for C–N coupling with DEAD with yields ranging from 50–98%. The authors proposed the photoexcited COF abstracts hydrogens using the anthraquinone units, which creates a radical that reacts with DEAD to eventually form the product. They also found DAAQ-COF could promote C–H pyridylation using activated pyridines, garnering yields of 53–96% on eight substrates.
Cui and co-workers synthesized COF-1 and COF-2 from 4′,4′′′,4′′′′′,4′′′′′′-tetraaldehyde (ETBC) and triamine NBC or BADA, respectively, with two-fold interpenetrated ffc networks (Fig. 28).451 These COFs were used for photocatalytic cross-dehydrogenative coupling (CDC) and the asymmetric α-alkylation of aldehydes. The CDC reactions were conducted under blue LEDs in air at 40 °C in acetonitrile, and α-alkylations were performed under blue LEDs under a nitrogen atmosphere at −10 °C in N,N′-dimethylformamide with 2,6-lutidine additive and Macmillan chiral co-catalyst. On 8 substituted tetrahydroisoquinoline derivatives, CDC reactions using either COF-1 or COF-2 achieved yields of 50–85%, showing almost identical yields between the two. Interestingly, when comparing the COFs with their amorphous counterparts the COFs performed about 10% better, indicating that crystallinity improved the photocatalytic performance. On 6 aldehydes, α-alkylations using COF-1 or COF 2 obtained yields of 51–88% with 83–94% ee. Interestingly, when white light was used instead of blue light, the reaction suffered, with a yield of 73% with only 36% ee using COF-1. Notably, the COFs lost their crystallinity after these reactions but could be restored by taking the resulting amorphous materials and resubjecting them to the initial synthesis conditions.
 | ||
| Fig. 28 Structural representations of the COFs. (a) Single ffc network of COF-1; (b) twofold interpenetrated ffc network of COF-1; (c) twofold interpenetrated ffc network of COF-2; (d) space-filling models of the 3D structure of COF-2.451 Reproduced with permission from ref. 451. Copyright 2020, Royal Society of Chemistry. | ||
In 2023, Xiang and co-workers designed non-substituted quinoline-linked COFs (NQ-COFs).452 The NQ-COFs were able to be post-synthetically oxidized by m-CPBA to yield the quinoline N-oxide form (NQ-COF-O). Specifically, NQ-COFE5-O was used for photocatalytic C–H activation using potassium persulfate as the oxidant, trifluoroacetic acid, and acetonitrile as solvent using blue LEDs under an argon atmosphere. The model reaction of isoquinoline and toluene was first explored, achieving 93% yield using the aforementioned conditions. Low yields were afforded when other oxidants, acids, or solvent mixtures were used, and using a homogeneous analog or the non-oxidized version of the COF provided yields below 15%. This reaction was scaled up to 12 mmol, and with increased reaction time, could still achieve 84% yield. When other substituted quinolines, a benzothiazole, and a benzoxazole were tested, yields ranging from 67 to 93% were obtained. This work demonstrated the utility of embedding hydrogen atom transfer moieties within a COF for photocatalytic C–H activation. The authors proposed a mechanism in which the photogenerated holes oxidize the quinoline N-oxide units of the COF to create N-oxy radicals, which abstracts a hydrogen atom from the C–H substrates to afford alkyl radicals that undergo a Minisci-type reaction.
In 2024, Zhang and co-workers constructed four vinylene-linked COFs (V-COFs) based on 2,4,6-trimethyl-1,3,5-triazine (TMTA) with 1,4-diformylbenzene (DFB) (V-COF-1), DFB and anthracene-9,10-dicarbaldehyde (AN) (V-COF-AN), DFB and benzo[c][1,2,5]thiadiazole-4,7-dicarbaldehyde (BT) (V-COF-BT), or a combination of DFB, AN, and BT (V-COF-AN-BT).453 V-COF-1 was used as a control with no oxidation or reduction sites, V-COF-AN was used as a control only bearing oxidation sites, V-COF-BT was used as a control only bearing reductive sites, and V-COF-AN-BT was the main COF containing both oxidative and reductive sites. They then applied these COFs for photocatalytic direct C–H difluoromethylation of heterocycles using NaSO2CF2H as the fluorine source, oxygen as the oxidant, and DMSO as the solvent using blue LEDs. Using 1-methylquinoxolin as the model substrate, V-COF-AN-BT performed the best, yielding 91% product, with the other V-COFs yielding 55% or less, showing the importance of the dual-active-center approach. Based on experiments, the authors proposed that upon light irradiation, the photogenerated electrons react with oxygen to form superoxide radicals and photogenerated holes react with NaSO2CF2H to produce the CF2H radical. The CF2H radical reacts with the substrate, undergoes a 1,2-H shift, and then is oxidized by the superoxide radical to form the product with H2O2 as a by-product. They performed a substrate scope using 12 different substituted quinoxalin-2(1H)-ones, heterocycles, and five bioactive molecules, for example xanthine derivatives and uracil, obtaining yields of 53–95%. The model reaction was also conducted on a gram scale, achieving 71% yield.
Banerjee and co-workers synthesized three COFs from 1,3,5-triformylphloroglucinol (Tp) and 4,4′-azodianiline (Azo), 2,8-diamino(6-phenylphenanthridine) (Dpp), or 1,3,5-tris(4-aminophenyl)benzene (Tab) giving TpAzo, TpDpp, and TpTab, respectively.454 These COFs were used as photocatalysts for C–H borylation using trimethylamine-borane in acetic acid under a nitrogen atmosphere with blue LEDs and using ammonium persulfate as a sacrificial electron acceptor. On the model substrate, 4-methylquinoline, TpAzo obtained 90% yield while TpDpp and TpTab only garnered 21 and 33%, respectively. Notably, the COF lost crystallinity upon five recycles and there was a decrease to 77% yield in the fifth run. On 12 substrates, TpAzo achieved yields ranging from 18 to 96%. The authors proposed that upon light irradiation, the photogenerated electron from the COF reacts with persulfate to form a sulfate radical that abstracts a hydrogen from trimethylamine-borane. The resulting boryl radical reacts with the protonated quinoline in a Minisci addition. Finally, this radical cation formation undergoes rearomatization through oxidation by the photogenerated hole of the COF, and deprotonation to afford the product.
6.11 C–N cross-coupling
C–N cross-coupling has shown prominence in the synthesis of natural products and drug molecules.455–458 Like most cross-coupling reactions, this is typically mediated by precious metals and researchers have investigated using sustainable ways to promote C–N cross-coupling reactions.459–463 Here, we summarize some reports of COFs enabling photocatalytic C–N cross-coupling reactions.Maji and Banerjee synthesized TpBpy COF from 1,3,5-triformylphloroglucinol (Tp) and 5,5′-diamino-2,2′-bipyridine (Bpy).464 This COF was dual metalated using [Ir(ppy)2(CH3CN)2]PF6 and then NiCl2. The Ir complex acted as a photosensitizer while the Ni acted as the catalytic center. The Ni–Ir@TpBpy COF was used as a photocatalyst for visible-light-mediated C–N bond formation in the presence of 1,1,3,3-tetramethylguanidine (TMG) base in acetonitrile at 40 °C using blue LEDs under an N2 atmosphere. The Ni–Ir@TpBpy COF was successfully applied as a photocatalyst for C–N coupling on over 60 substrates with yields ranging from 42 to 95%, and this was also used on over 10 drug precursors and bio-active molecules garnering 46–81% yields. The authors attributed the high activity to the short distances between the Ir and Ni centers and the lack of multinuclear deactivation. Furthermore, an amorphous version of the COF showed only 31% yield of the standard reaction, showing the importance of crystallinity.
Thomas et al. used 2,6-diaminoacridine (Acr) and 1,3,5-triformylbenzene-based monomers to construct different β-ketoenamine/imine COFs with 0, 1, and 2 imine bonds ratios giving Tp-Acr, DHTA-Acr and HTA-Acr COFs, respectively.465 These COFs were used for photocatalytic C–N coupling of 4-bromobenzotrifluoride and pyrrolidine with NiBr2 additive in DMA using blue LEDs where Tp-Acr showed the highest activity, 91% yield, due to the larger amount of β-ketoenamine linkages and thus metal binding sites. When using green LEDs instead, only 38% yield was obtained even when tripling the reaction time. They also showed a substrate scope of five other aryl bromides and achieved yields from 66 to 94%. The authors attributed the high catalytic activity of the COF to the broad range light absorption and to high charge carrier separation. Notably, the COF lost crystallinity over recycling, and addition of nickel catalyst was needed in each cycle.
Yang and cohort manufactured an imine COF from 5,5′,5′′-(benzene-1,3,5-triyl)tripicolinaldehyde with p-phenylenediamine followed by a Povarov reaction to yield a quinoline-linked COF, Q-COF, and metalated it with NiCl2 and either [Ir(ppy)2(MeCN)2]OTf or [Ir(coum)2(MeCN)2]OTf to give [Ir(ppy)2(N^N) + NiCl2]@Q-COF or [Ir(coum)2(N^N) + NiCl2]@Q-COF.466 The [Ir(coum)2(N^N) + NiCl2]@Q-COF was employed as a photocatalyst for both C–S and C–N coupling using blue LEDs in acetonitrile with TMG or pyridine as the base, achieving 92 and 90% yields, respectively, while the [Ir(ppy)2(N^N) + NiCl2]@Q-COF only gave 47 and 44% yield, respectively, showing the importance of the Ir photosensitization ability. They also performed the reaction on 35 C–S substrates and 10 C–N substrates, affording yields of 65–99%. The authors proposed a mechanism going through Ni(I) and Ni(III) based on calculations and CV data (Fig. 29).
 | ||
| Fig. 29 (a) Proposed mechanism of [Ir(coum)2(N^N) + NiCl2]@Q-COF catalyzed C–S cross-coupling. (b) The binding energies between different substrates (iodobenzene left and thiophenol right) and COF. (c) Comparative Stern–Volmer quenching experiment in C–S cross-coupling. (d) CVs of model complexes.466 Reproduced with permission from ref. 466. Copyright 2024, Royal Society of Chemistry.279 Reproduced with permission from ref. 279. Copyright 2020, American Chemical Society. | ||
Hou and co-workers synthesized an imine-linked COF using 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) and 2,6-diformylpyridine (DFP) and then metalated with NiCl2 giving Ni SAS/TD-COF.467 This Ni SAS/TD-COF was applied as a photocatalyst for a C–N coupling reaction using 4-bromobenzotrifluoride and pyrrolidine using DABCO as the base in DMA and using blue LEDs, giving 99% yield. The optimized conditions were applied on a large substrate scope of over 30 compounds and afforded yields of 71–99%.
Zhang and co-workers constructed three bipyridine, pyrene-based cyano-vinylene COFs from TFPPY, TFPPY-F, and TFPPY-OCH3 with BPY-CN and metalated with NiCl2 to make sp2c-COFdpy-Ni, sp2c-F-COFdpy-Ni, sp2c-OCH3-COFdpy-Ni, respectively.468 They applied these COFs for both C–N and C–O coupling photocatalytically using blue LEDs. They achieved optimal yields of 99% for both reactions when using the fluorinated version, with drastically reduced yields using the unsubstituted or methoxy versions, illustrating the importance of monomer substitution. The authors attributed this discrepancy to fluorine enhancing the migration of photogenerated electrons. The authors also displayed a substrate scope of C–O coupling on 15 other compounds, obtaining yields of 60–99% and a substrate scope for C–N coupling of 13 other compounds with yields varying between 80 and 99%.
Thomas and co-workers synthesized 8 COFs using a mix of 2,6-acridinediamine (Acr) and 2,2′-bipyridine-5,5′-diamine (Bpy) with benzene-1,3,5-tricarbaldehydes with 0–3 alcohols substituents which were then metallated with NiCl2.469 Acr2-Tf-Bpy1 COF was used as a photocatalyst for the C–S coupling of 4-iodobenzotrifluoride with methyl 3-mercaptopropanoate using blue LEDs and N,N-tert-butylisopropylamine as a base in acetonitrile, achieving a quantitative yield. Using green light gave 74% yield, and a much longer reaction time of 48 hours using red light gave quantitative yield. When the other COFs were applied the authors saw that decreasing the number of hydroxy groups, and thus keto tautomerization, caused an increase in product with yields from 4 to 86%. The COFs were also tested on the coupling of 4-iodobenzotrifluoride and sodium p-toluenesulfinate in DMA getting yields of 25–55%. These were then tested on the C–N coupling between pyrrolidine and 4-bromobenzotrifluoride in DMA in which the opposite trend was observed, where increasing the number of hydroxy groups, and thus keto tautomerization, caused an increase in product with yields from 4 to 80%. The authors described the discrepancy as being caused by a mechanistic difference. In the C–S coupling, the catalytic center is the Ni binding at the bipyridine sites and thus is aided from high charge-carrier mobility from imine-based linkages. In the C–N coupling, the active catalyst is Ni(pyrrolidine)n which is sensitized by the COF that benefits from the linkage which creates good charge separation of excitons and stabilizes electrons on the acridine moiety. Notably, the COF lost crystallinity under these photocatalytic conditions.
6.12 C–O cross-coupling
C–O bonds are highly prevalent in natural products and many drug molecules, making their formation key to obtaining high-value chemicals.470–472 Furthermore, C–O bonds have been investigated as coupling partners due to their abundance in nature and lesser toxicities. However, C–O bonds have high dissociation energies which require high temperature and catalyst loading, so methods to perform these coupling reactions under more ambient conditions, such as photocatalysis, have been examined.461,462,473 Here, we present research that has been reported on using COFs as photocatalysts for C–O cross-coupling reactions.Wang and co-workers synthesized LZU-713@Ni (Fig. 30) using a metalated phenanthroline monomer, Phen-CHO@Ni, and 1,3,6,8-tetrakis(4-aminophenyl)pyrene (PyTTA).474 They used this COF for photocatalytic C–O cross-coupling of 4-bromobenzonitrile and methanol, obtaining a high yield of 97% using blue light in DMA and triethylamine as the base. They applied the reaction conditions to 20 substrates and achieved yields ranging from 69 to 98%, but the reaction did not work when substituting methanol for water or phenol. The authors also used this COF for C–S cross-coupling of aryl halides and aryl thiols which afforded yields of 80–99% on 20 substrates and using 2,6-lutidine as the base instead. Even more, the COF was applied to C–P cross-coupling of aryl iodides and phosphine oxides which garnered yields of 56–99% on 18 substrates, and using caesium carbonate as the base and methanol as the solvent instead. The authors attributed the catalytic activity to easily accessible catalytic sites in the COF and fast transfer of photogenerated electrons. Notably, the COF had a decrease in crystallinity post-reaction and a total loss of crystallinity after four runs.
 | ||
| Fig. 30 Bottom-up synthesis of LZU-713@Ni.474 Reproduced with permission from ref. 474. Copyright 2023, John Wiley and Sons. | ||
Chen and co-workers synthesized sp2-COFdpy from 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy) and 2,2′-([2,2′-bipyridine]-5,5′-diyl)diacetonitrile (BPYDAN) which was then metalated using NiCl2 to give sp2-COFdpy-Ni.475 They used this COF for C–O photocatalytic cross-coupling of 4-bromobenzonitrile and methanol using blue LEDs and triethylamine as the base, achieving a yield of 99%. Interestingly, when using green LEDs (520–525 nm) 91% yield was attained, but no yield was obtained while using red LEDs. When adding in NiCl2in situ with the pristine COF versus the metalated version 99% yield was also obtained. The optimized conditions were applied to a 25-compound substrate scope, getting yields of 55–99%. The authors also showed that water could be used instead of methanol, but the reaction had to be heated at 50 °C. Furthermore, they showed the reaction could be scaled up to 10 mmol and get 98% yield. The authors believed that the catalytic activity was due to bimolecular photoinduced electron transfer to create the active COF-NiI species.
Zhang and cohort synthesized two hydrazone-linked COFs using 1,3,5-tris(4-formylbiphenyl)benzene (TFBPB) or 1,3,5-tri(3-hydroxy-4-formyl-ethynylphenyl)-benzene (THFEB) with 1,3,5-benzene tricarbohydrazide (BTH) to make BPh-COF and AC-COF, respectively.476 They used these COFs as photocatalysts for C–O cross-coupling of 4-chlorobenzonitrile and methanol using blue LEDs with NiCl2 added in situ and NaOH as the base. AC-COF achieved 90% yield while BPh-COF only afforded 45% yield. They attributed this difference to the AC-COF having better light absorption than BPh-COF. When using white LEDs instead, only 20% yield was obtained for AC-COF. They then took AC-COF and used it for the C–O coupling on 15 substrates of aryl chlorides and their bromide counterparts, giving 15–99% yields. The aryl chlorides typically performed around a half to two-thirds as well as the bromide versions. They also showed that ethanol and benzyl alcohol could be used instead of methanol. Notably, this system was shown to be not very recyclable, losing about 45% yield over five cycles when adding in more Ni every time, and all the way down to 10% when not adding fresh Ni in. ICP-OES analysis showed large amounts of Ni leaching. The authors believed this was the result of having to use a strong base, NaOH, to perform this reaction, versus the typically used triethylamine, which caused the metal to leach off. Additionally, the COF showed a decrease in crystallinity over the recycling process. The COFs were also used for photocatalytic oxidative coupling of amines where AC-COF showed quantitative yields of 8 benzylamines and BPh-COF showed yields of 32–82%, again due to smaller range light absorption.
Zhang et al. constructed bpy-COF from 4′,4′,4′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) and 5,5′-dialdehyde-2,2′-bipyridine (BPDA) which was metalated using NiBr2 to give Ni(II)-bpy-COF.477 This COF was used as a photocatalyst for the hydroxylation of aryl chlorides. The reaction was optimized for C–O coupling of 4-chlorobenzonitrile and H2O in DMF/MeCN with triethylamine as the base and using blue LEDs at 40 °C, achieving 99% yield. This system was applied successfully to 14 other substrates in yields of 47–98%. Interestingly, when applied to a biphenyl-based substrate the yield fell to 27% and when applied to a terphenyl-based substrate no yield was observed, which the authors stated was likely due to poor substrate diffusion into the pores and steric hindrance. Furthermore, the authors found that the COF allowed for efficient transfer of photogenerated electrons to the Ni centers and the photogenerated holes generated highly reactive hydroxyl radicals from their reaction with water.
6.13 C–C cross-coupling
C–C cross-coupling reactions are used to construct many diverse organic molecules for a wide variety of applications.458,478–481 This includes the use of the widely known Suzuki, Sonogashira, Heck, Hiyama, Stille, Kumada, and Negishi coupling reactions. While these reactions are typically conducted at high temperatures, researchers have investigated performing these reactions photochemically in the presence of a suitable catalyst.462,482–485 Here, we report the use of COFs as photocatalysts to perform C–C cross-coupling reactions.Cai and co-workers were the first to report a COF for cross-coupling photocatalytically.486 They synthesized TTI-COF from 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde, and then used sulfur to post-synthetically convert the imine-linkage to a benzothiazole-linkage, making TTT-COF. The TTT-COF was then treated with Na2PdCl2 and NaBH4 to form Pd nanoparticles in the COF, namely Pd NPs@TTT-COF (Fig. 31). This hybrid COF was then used as a photocatalyst for the Suzuki coupling of 4-iodotoluene and phenylboronic acid using K2CO3 as the base in ethanol and water using a Xe lamp, affording quantitative yield. This was applied to a small substrate scope, garnering yields ranging from 82 to 99%. Moreover, this system was impressively used for Stille, Heck, and Sonogashira couplings with substrate scopes obtaining yields of 39–99%. The authors attributed the success of the reactions to the benzothiazole linkage, which can bind the nanoparticles and provides high conduction efficiency of the photocarriers.
 | ||
| Fig. 31 Graphical representation of the synthesis of Pd NPs@TTT-COF.486 Reproduced with permission from ref. 486. Copyright 2020, Royal Society of Chemistry. | ||
Islam and co-workers mechanochemically made TpPa-1 from phenylenediamine and 2,4,6-triformylphloroglucinol (TFP), and this COF was used as a photocatalyst alongside Ni(dmg)2 for the carboxylation of aryl halides using CO2.487 The reaction was optimized on iodobenzene using blue LEDs and triethylamine (TEA) as a sacrificial reagent, achieving a yield of 86%. Interestingly, when p-terphenyl was used instead of Ni(dmg)2 a 56% yield was obtained. The optimized conditions were used on 12 substrates including aryl bromides and chlorides, garnering yields of 70–91%. This COF was also used as a photocatalyst for the C–H activation of benzene, nitrobenzene, fluorobenzene, and anisole, obtaining yields of 60–70%.
Mas-Ballesté and co-workers constructed Phen-COF using a phenanthroline monomer and 1,3,5-tris(4-aminophenyl)benzene (TAPB) which was then metalated with [(dF(CF3)ppy)2-Ir-μ-Cl]2 and NiCl2·glyme to form Ir,Ni@Phen-COF.488 This COF was employed as a photocatalyst for the cross-coupling of potassium benzyltrifluoroborates and aryl bromides. Optimized conditions were performed using potassium benzyltrifluoroborate and 4-bromotoluene using 2,6-lutidine as the base in acetone/MeOH and blue LEDs, achieving 90% yield. The reaction conditions were applied to 8 other compounds, obtaining yields of 34–99%, with electron-donating substituents markedly giving lower yields. To test the utility of this COF as a photocatalyst, the authors also showed it was active for the cross-couplings of potassium tert-butyltrifluoroborate and 4-bromobenzonitrile, organic silicate and 4-bromobenzonitrile, and Boc-protected proline and 4-bromobenzonitrile, obtaining yields of 76, 92, and 86%, respectively. The authors attributed the catalytic activity to the phenanthroline stabilizing the coordination of the metal centers.
Maji and cohort synthesized Bpy-sp2c-COF from 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy) and 2,2′-([2,2′-bipyridine]-5,5′-diyl)diacetonitrile (BPDAN) which was then metalated using NiCl2 to give Ni@Bpy-sp2c-COF.489 They applied this COF as a photocatalyst in eight C–X (X = B, C, N, O, P, S) coupling reactions, achieving yields ranging from 61 to 99%. The C–C coupling reaction was performed on allyl acetate and 2-naphthaldehyde using blue LEDs at 40 °C, obtaining a yield of 61%. C–O coupling was investigated using methyl 4-iodobenzoate and Boc-protected proline, obtaining 91% yield at 60 °C using compact fluorescence light. The C–O coupling reaction was performed on about 50 compounds, giving yields of 42–86% including drug functionalization and derivatizing naproxen using multiple aryl iodides. C–S coupling was analysed using iodobenzene and thiophenol using blue LEDs at 40 °C and obtained quantitative yield. This was furthered by testing methyl 4-iodobenzoate with sodium benzenesulfonate and getting the sulfonate product in 94% yield but using purple LEDs instead. The C–S coupling reaction was also performed on about 50 substrates, with yields garnering 31–93% including functionalization of bioactive derivatives. C–N coupling was investigated using methyl 4-iodobenzoate and p-toluene-benzenesulfonamide or sodium azide, achieving yields of 91 and 89%, respectively, using blue LEDs at 40 °C. However, using azides required the use of triethanolamine as a sacrificial reagent. C–B coupling was studied using 4-iodobenzonitrile with bis(pinacolato)diboron, getting 96% yield with purple LEDs at 40 °C. C–P coupling was investigated using methyl 4-iodobenzoate and diphenylphosphine oxide, obtaining 73% yield using blue LEDs at 40 °C. Notably this is the only reaction where the homogeneous catalyst performed better, garnering 81% yield. The reactions could also be performed on a gram scale.
6.14 Pollution degradation
Water treatment has become a huge field with the development of more pharmaceuticals, pesticides, textile dyes, and other organic pollutants that have been released into water sources around the world.490–492 Furthermore, water treatment to kill harmful bacteria and viruses such as Salmonella, Campylobacter, and Norovirus is also important.493 Common methods to treat water include disinfection with chlorine or chloramines, ultraviolet disinfection, ozonation, Fenton reactions, adsorption, coagulation and flocculation, and membrane filtering.490–493 One of the most promising methods is photocatalytic degradation due to the potential of using visible light under ambient conditions. Here, we summarize some key reports of COFs used as photocatalysts to eliminate contaminants.Tong and co-workers synthesized two donor–acceptor COFs with β-ketoenamine linkages using 1,3,4-thiadiazole-3,5-diamine (TD1) or 1,2,4-thiadiazole-2,5-diamine (TD2) with 1,3,5-triformylphloroglucinol (Tp) giving COF-TD1 and COF-TD2, respectively.494 They used these COFs for photodegradation of paracetamol, diclofenac, bisphenol A, naproxen, and tetracycline hydrochloride using a xenon arc lamp with a UV cut-off filter. COF-TD1 showed 98% degradation of paracetamol in 60 min, and 97% within 120 min for the other contaminants. Interestingly, 97% of paracetamol could also be degraded in 60 min using natural sunlight. This process was scaled up using the COF on ITO glass with simulated sunlight, degrading 92% of paracetamol in 4 hours. The authors attributed this activity to both intermolecular charge transfer and proficient electron–hole separation from the push–pull effect of the D–A system.
Bhaumik and co-workers constructed C6-TRZ-TPA COF from tris(4-formylphenyl)amine (TPA-aldehyde) and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TRZ-amine).495 This COF was employed as a photocatalyst to degrade rose bengal (RB) and methylene blue (MB) dyes as well as iodine vapor capture under visible light irradiation. The COF showed 99% degradation of RB in 80 min and even 90% within 30 min of a 250 ppm solution. It also degraded 97% of a 20 ppm MB solution within 120 min. The COF also exhibited high iodine vapor uptake capacity of 4832 mg g−1. The activity of the COF was due to efficient electron–hole charge transfer from the D–A system.
Jiang and co-workers constructed TFPB-TAPT-COF from 1,3,5-tris(p-formylphenyl)benzene (TFPB) and 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT), which was mixed with several emulsifiers including Triton X-100, sodium dodecyl sulfate (SDS), or polyvinyl alcohol type 1788 (PVA).496 These COF–emulsifier matrixes were used in the photodegradation of 10 ppm solutions of methylene blue (MB) and sodium fluorescein (SF) under simulated natural light conditions. The pristine COF only degraded 26% of MB in 50 min while COF-Triton X-100 degraded 98% in the same amount of time. Furthermore, 72% of SF dye could be degraded in 90 min using the COF and 82% using COF-Triton X-100. The authors credited the emulsifier with increasing the surface area of the COF, and thus creating more oxygen vacancies on the particle surface and therefore more active sites.
Wang and co-workers synthesized HDU-26 from benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene-2,5,8-tricarbaldehyde (BTT) and 4,4′-(benzo[c][1,2,5]thiadiazole-4,7-diyl)dianiline (BT).497 HDU-26 was used as a photocatalyst for the reduction of Cr(VI). This was done by exposing the COF and a 10 ppm K2Cr2O7 solution to simulated visible light using a Xe lamp. The COF demonstrated 99% reduction after 60 min of irradiation. They also studied the effects of initial pH, catalyst dosage, and Cr(VI) concentration. HDU-26 showcased removal rates of rhodamine B (RhB), methylene blue (MB) and methyl orange (MO) of 82.6, 56.4, and 99.1%, respectively. Notably, when the COF was employed in a one-batch system, including the dyes and Cr(VI), the dye removal rates were increased to 97.2, 62.3, and 99.2%, respectively. The authors attributed this to the COF generating electron–hole pairs for the oxidation of organics with a synergistic effect of Cr(VI) reduction.
Van Der Voort and cohort made three olefin-linked COFs, TMT-TT-COF, TMT-N-COF, and TMT-BT-COF, from 2,4,6-trimethyl-1,3,5-triazine (TMT) and bithiophene (TT), naphthalene (N), and benzothiadiazole (BT) monomers.498 These COFs were used as photocatalysts for bisphenol A (BPA) degradation and H2O2 production. TMT-TT-COF and TMT-N-COF illustrated exceptional BPA degradation of 96% and 94.7% of a 25 ppm solution under visible light within 60 min while TMT-BT-COF only showed 40% degradation. When tested for H2O2 production TMT-TT-COF, TMT-N-COF, and TMT-BT-COF generated 1952, 1742, and 748 μmol g−1 h−1, respectively, using a Xe lamp under an O2 atmosphere. Under an Ar atmosphere, they displayed much lower rates of 234, 117, and 13 μmol g−1 h−1, respectively. The authors attributed these results to TMT-TT-COF and TMT-N-COF being made from D–A building units that promote efficient charge transfer and separation. Moreover, the thiophene moiety in TMT-TT-COF induces a narrower bandgap, better intramolecular charge carrier transport, and less charge recombination.
Li and co-workers made two isomeric D–A COFs from pyrene (Py) and benzo[c][1,2,5]thiadiazole (BT) units containing either aldehyde or amino functional groups to form Py-C-BT-COF and Py-N-BT-COF (Fig. 32).499 They took these COFs and applied them in rhodamine B degradation and H2 evolution photocatalytically. Using visible light, 95% of rhodamine B was degraded within 60 min by Py-N-BT-COF, while it was only 70% degraded with Py-C-BT-COF. The opposite activity was seen for H2 evolution. Under visible light with sodium ascorbate as sacrificial reagent and 5 wt% Pt cocatalyst, Py-C-BT-COF revealed an average H2 evolution rate of 253.1 mmol g−1 h−1 while Py-N-BT-COF showed little activity. This work showed that changing the isomerism and atomic positioning in a COF can change the band gap and optical properties of the material, which influences the photocatalytic activity.
 | ||
| Fig. 32 Schematic representation of the synthesis of the Py-BT-COF isomers.499 Reproduced with permission from ref. 499. Copyright 2024, Royal Society of Chemistry. | ||
Xi and co-workers constructed three COFs from different combinations of 2,4,6-tris(4-formyl-phenyl)-1,3,5-benzene (TFPB), 2,4,6-tris(4-amino-phenyl)-1,3,5-benzene (TAPB), 2,4,6-tris(4-formyl-phenyl)-1,3,5-triazine (TFPT) and 2,2′-(5′-(4-(cyanomethyl)phenyl)-[1,1′:3′,1′′-terphenyl]-4,4′′-diyl)diacetonitrile (TCPB) to make TFPB-TCPB COF, TFPT-TCPB COF, and TFPT-TAPB COF.500 The cyano-olefin linkage of TFPT-TCPB COF was further oxidized to a carboxylic acid to make TFPT-TCPB COF-COOH. These were used as photocatalysts to degrade 200 ppm solutions of rhodamine B (RhB) dye using a Xe lamp. First, they were tested for pure absorption of the dye with no light. TFPT-TCPB COF, TFPB-TCPB COF, and TFPT-TCPB COF-COOH achieved around 35% RhB removal in 90 min; however, TFPT-TAPB COF had about 50% removal in the same time frame, likely due to its much larger surface area according to the authors. In the presence of light 97% of RhB could be removed, but no degradation was shown. When peroxymonosulfate (PMS) was added, the RhB dye could be degraded with light. Using TFPT-TCPB-COF-COOH 62.2% of RhB was mineralized in 50 min due to its activation of PMS.
El-Mahdy and co-workers synthesized 3D, D–A hetero[6]radialene-based COFs from 2,4,6-triformylphloroglucinol and (4,4′,4′′,4′′′-pyrene-1,3,6,8-tetrayl)tetraaniline (PyTA-4NH2) or 4,4′,4′′,4′′′-([9,9′-bifluor-enylidene]-3,3′,6,6′-tetrayl)tetraaniline (BFTB-4NH2), giving TFP-Py 3D COF and TFP-BF 3D COF, respectively.501 They used these COFs for absorption of rhodamine B dye. On an 18 mg L−1 sample, complete absorption occurred in 10 minutes for TFP-Py 3D COF and 60 minutes for TFP-BF 3D COF. They then tested the photodegradation of a 10 mg L−1 rhodamine B sample under visible light using the COFs. The TFP-BF 3D COF and TFP-Py 3D COFs achieved complete removal of rhodamine B within 150 minutes and 165 minutes, respectively, with photodegradation efficiencies of 96.60% and 95.90%, respectively. The authors also tested these COFs for photocatalytic hydrogen evolution using a Xe lamp with UV and visible light with or without Pt cocatalyst and ascorbic acid as a sacrificial reagent. TFP-Py and TFP-BF 3D COFs without the inclusion of Pt cocatalyst were 4.96 and 21.04 mmol g−1 h−1, respectively. When adding 1 wt% Pt, HER for TFP-BF 3D COF achieved 30.72 mmol g−1 h−1. AQYs for TFP-BF 3D COF were 8.70, 10.12, and 15.73% at wavelengths of 420, 450, and 600 nm, and AQYs of 2.54, 4.21, and 0.49% for TFP-Py 3D COF at the same wavelengths, respectively.
6.15 Other applications
Thanks to their tunable photoelectronic properties, COFs have been designed and leveraged to accomplish various transformations. While COFs have been mainly applied as photocatalysts for the reactions previously discussed, they have also been utilized for a plethora of other organic reactions. Herein, recent reports of various non-related photoapplications are detailed that use COFs photocatalytically.Banerjee et al. synthesized a β-ketoenamine-linked COF, TpTt, using melamine, or 1,3,5-triazine-2,4,6-triamine (Tt), and 2,4,6-triformylphloroglucinol (Tp).502 They took the TpTt COF and used it for E to Z photoisomerization of olefins. Optimizing conditions, using blue LEDs in DMF, the authors achieved 90% yield of cis-stilbene from trans-stilbene. They performed this on seven other substrates, garnering yields of 42–71%. The authors also conducted mechanistic studies and DFT calculations and showed the COF absorbs light and is excited, undergoes intersystem crossing to the triplet state, then transfers energy to the substrate (trans-stilbene) which transforms into a biradical triplet intermediate state which is converted to the product (cis-stilbene).
Gu and co-workers constructed an anionic Ti-based COF, Ti-COF-1, from Na2Ti(2,3-DHTA)3 (DHTA = 2,3-dihydroxyterephthalaldehyde) and 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetraaniline (PyTTA), (Fig. 33).92 Ti-COF-1 was then used as a photocatalyst for a Meerwein addition reaction using white LEDs. Using different arenediazonium salts and styrenes, yields of 49–75% were achieved with 132–202 turnover number. The authors attribute the success to the anionic skeleton of Ti-COF-1 being an excellent adsorbent for cationic arenediazonium salts which can then easily reduce them by the photogenerated electrons to form reactive aryl radicals.
 | ||
| Fig. 33 (a) Synthesis of Na2Ti(2,3-DHTA)3. (b) X-ray single-crystal structure of Na2Ti(2,3-DHTA)3. All H atoms, Na+ and solvent molecules have been omitted for clarity. C – gray, O – red, Ti – yellow. (c) Synthesis of Ti-COF-1.92 Reproduced with permission from ref. 92. Copyright 2021, John Wiley and Sons. | ||
Zhang and cohort made TPT-COF-Cu from 4,4′,4′′-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tribenzaldehyde (TPT-CHO) and [2,2′-bipyridine]-5,5′-diamine (Bpy-NH2) using a deep eutectic solvent followed by metalation by CuCl2.503 TPT-COF-Cu was then used as a photocatalyst for phosphorylation of terminal alkynes. Optimized conditions were performed on the model reaction of phenylacetylene and diethyl phosphite. Using blue LEDs, triethylamine base, and 2-MeTHF as solvent 95% yield was obtained. Notably, using white or green light achieved yields of 83 and 90%, respectively. They also used these conditions on an expansive substrate scope, 32 compounds, obtaining yields ranging from 85 to 95%. When increased to a gram scale 92% yield was isolated.
Chen and co-workers mimicked graphitic carbon nitride (g-C3N4) by making heptazine COFs from (1,3,3a1,4,6,7,9-heptaazaphenalene-2,5,8-triyltris(benzene-4,1-diyl))tris(methanetriyl)hexaacetate (HEP-OAc) and 1,3,5-tris(4-aminophenyl)triazine (TAPT-NH2) or 1,3,5-tris(4-aminophenyl)benzene (TAPB-NH2), giving HEP-TAPT-COF and HEP-TAPB-COF, respectively.504 The COFs were then used for photocatalytic oxidations of benzylic C–H bonds and sulfides using O2 as the oxidant and a xenon lamp as the light source. For the test reaction on isochroman, HEP-TAPT-COF performed better than HEP-TAPB-COF or g-C3N4, achieving 100% conversion and 90% selectivity for isochromanone. Notably, smaller yields were obtained when using an amorphous version of the COF or a heptazine-based amorphous polymer, Cy-pip, indicating crystallinity is important for charge separation. Four other isochroman derivatives and three other benzylic hydrocarbons achieved yields of 70–99% of the ketone product. HEP-TAPT-COF was also able to selectively oxidize six sulfides to the corresponding sulfoxides in quantitative yields. The authors believe the heptazine moieties played a crucial role in generating the reactive 1O2 for the oxidations.
Zhao and co-workers synthesized several COFs, COFX-Au (X = 1–5, based on monomer units), from D2-symmetric porphyrins (1–4 = proximal H, 5 = proximal pentafluorobenzene) and benzothiadiazole (1), pyrazine (2), benzene (3), or dimethoxybenzene (4) linkers and metalated with KAuCl4.505 The COFs were then used for photocatalytic nitrogen fixation using a xenon lamp as the light source and K2SO3 as a sacrificial reagent. COF1-Au, the COF with electron-withdrawing benzothiadiazole units, achieved a rate of 333.0 μmol g−1 h−1. When using COF4-Au, the COF with electron-donating dimethoxybenzene units, a lower rate of 196.1 μmol g−1 h−1 was achieved. When COF5-Au, the COF with the pentafluorobenzene-substituted porphyrin, was used the rate increased to 427.9 μmol g−1 h−1. N2H4 was also tested as a product but only 0.5 μmol g−1 h−1 was observed. Furthermore, when the reaction was done in basic conditions instead, a small rate of 7.8 μmol g−1 h−1 was achieved. The authors attributed the success of COF1-Au and COF5-Au to higher charge separation efficiency and lower charge transfer resistance from the electron-withdrawing moieties.
Wang and cohort synthesized a D–A COF, PTZ-TTA-COF (Fig. 34), using N-(4-formylphenyl)-phenothiazine-3,7-dicarbaldehyde (PTZ-CHO) and 1,3,5-tris(4-aminophenyl)triazine (TTA-NH2).506 They used PTZ-TTA-COF and a control TPA-TTA-COF (TPA = triphenylamine) as photocatalysts for amine homocoupling and cyclization to thioamides using blue LEDs and air as oxidant. On 14 substrates, PTZ-TTA-COF garnered yields of 75–99% for the oxidative amine coupling while the TPA-TTA-COF gave much lower yields of 36–56%. For the cyclization of five different thioamides PTZ-TTA-COF performed well, achieving 60–72% yield, and again TPA-TTA-COF achieving lower yields of 25–37%. The authors illustrated the importance of donor strength for creating efficient D–A systems by reducing the band gap of the photocatalyst.
 | ||
| Fig. 34 (a) Synthesis of the donor–acceptor PTZ-TTA-COF. Top (b) and side views (c) of the AA stacking mode as well as the top view (d) of the AB stacking mode for the PTZ-TTA-COF (carbon, gray; nitrogen, blue; sulphur, yellow; hydrogen, white).506 Reproduced with permission from ref. 506. Copyright 2023, Royal Society of Chemistry. | ||
Li and co-workers constructed an imidazole-linked COF, PyPor-COF, using pyrene-4,5,9,10-tetraone, 5,10,15,20-tetrakis-(4-benzaldehyde)porphyrin, and ammonium acetate.507 This COF impressively demonstrated light absorption over the entire visible light spectrum and exhibited a super low band gap of 1.75 eV. To test its singlet oxygen generation, PyPor-COF was illuminated with red light in the presence of 9,10-anthracenediyl-bis(methylene)-dimalonic acid (ABDA), which showed a fluorescence quantum yield of 10.3%, and with the addition of rose bengal photosensitiser the quantum yield increased to 77.8%. PyPor-COF was then used as a photocatalyst for the oxidative cleavage of olefins to carbonyls using red light and air as the oxidant. On the model substrate 1,1-diphenylethylene 96% yield of benzophenone was obtained. When using purple, blue, or green LEDs instead, respective yields of 94, 90, and 89% were obtained, showing the great light-absorbing and photocatalytic properties of the COF. This was then tested on nine styrene derivatives, achieving yields of 71–95%. PyPor-COF was also tested for the oxidation of 14 thioanisole derivatives to their corresponding sulfoxides, garnering yields of 73–99%. This work demonstrated a rare example of a COF photocatalyst using red light for organic transformations.
Zhao and co-workers synthesized TFPA-TAPT-COF-Q and TFPA-TPB-COF-Q using tris(4-formylphenyl)amine (TFPA) and 1,3,5-tris-(4-aminophenyl)triazine (TAPT) or 1,3,5-triphenylbenzene (TPB) and converting the imine linkage to a quinoline linkage using phenylacetylene and BF3.508 The quinoline-linked versions showed further red light absorption and much lower band gaps by about 0.5 eV. These COFs were then used photocatalytically for the oxidative decarboxylation of arylacetic acids using 1,1,3,3-tetramethylguanidine (TMG) as the cocatalyst and air as the oxidant (Fig. 35). The reaction was optimized using 4-methoxyphenylacetic acid and achieved 100% conversion and 86% selectivity for 4-methoxybenzaldehyde, with 4-methoxybenzyl alcohol as the side product. This was conducted on six other arylacetic acids, garnering conversions of 64–100% with selectivity of 80–92%. The COFs were also used for oxidative amine homocoupling, achieving conversions of 65–100% and selectivity of 53–100%. Furthermore, TFPA-TAPT-COF-Q was used to photosynthesize hydrogen peroxide using a Xe lamp as the light source with pure oxygen as the oxidant in a water/benzyl alcohol (10:1) solution, obtaining a production of 295900 μmol g−1. Notably, the imine-linked versions exhibited considerably deceased yields for each of these reactions. This work illustrated the importance of the linkage for enhancing photochemical properties of COFs.
 | ||
| Fig. 35 Proposed mechanism for the decarboxylation of arylacetic acids.508 This image has been adapted showing only part (e). This image is licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/). | ||
Zeng and cohort constructed bipyridine, ionic bipyridine, and ionic imidazole COFs named PY-BPY-COF, ion-PY-BPY-COF, and im-PY-BPY-COF, respectively, from 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)-tetraaniline (PY) with [2,2′-bipyridine]-5,5′-dicarbaldehyde (BPY).509 Through PSM of the bipyridine, the ionic bipyridine COF was made through reaction with bromoethane, and the ionic imidazole COF was made through cycloaddition with paraformaldehyde. These COFs were used as photocatalysts for the oxygen reduction reaction (ORR). The im-PY-BPY-COF achieved the highest TOF of 0.0198 s−1 at a potential of 0.7 V, which was 5.4 and 3.2 times greater than the values exhibited by PY-BPY-COF (0.0037 s−1) and ion-PY-BPY-COF (0.0061 s−1), respectively. The mass activity for im-PY-BPY-COF was 0.57 A mg−1, which was higher than PY-BPY-COF (0.17 A mg−1) and ion-PY-BPY-COF (0.19 A mg−1). The im-PY-BPY-COF was also the most selective, with only H2O2 by-product yields of 10–18% compared with that of PY-BPY-COF of 12–50% and ion-PY-BPY-COF of 20–30%. Using DFT calculations and DRIFTS, the authors showed the important reactive oxygen species was OOH*. This work exhibited a rare example of using a COF photocatalyst for ORR.
7 Conclusions and outlook
Covalent organic frameworks have emerged as a prosperous class of crystalline, porous polymers. The properties of COFs have lent them the ability to be used as effective photocatalysts for a plethora of photoinduced transformations, in addition to other applications. In this review, the design, synthesis, characterization, and photocatalytic application of COFs have been presented. Numerous linkages of COFs were discussed with an emphasis on their effect on stability and structural properties. Furthermore, the dimensionality and morphology of COFs were summarized. The various synthetic methods were also outlined including solvothermal, microwave-assisted, ionothermal, mechanochemical, and more. Instrumentation, techniques and methods to characterize COFs as well as photocatalytic products and intermediates were also broadly explored. Moreover, approaches for enhancing COF photoelectric properties and photocatalytic activity, such as extending light absorption, band gap engineering, doping, composites, and others, were assessed. Finally, the use of COFs as photocatalysts in a myriad of organic reactions was reviewed.This review has illustrated that COFs are well positioned for photocatalytic applications. Owing to their capacity to integrate countless organic building blocks and diverse covalent bonds they can be tailored to integrate a high degree of functionality. The inherent in-plane conjugation and interlayer π-stacking of COFs enhance their light-absorbing capability. Furthermore, they can host high light-absorbing chromophore units and photosensitizers for superior performance in light-driven processes. COFs also feature high surface areas and porosity, enabling suitable diffusion of reactants and products through the pores and easy access to the active sites. Their rigidity and use of donor–acceptor systems permits for high charge carrier mobility and separation of photogenerated holes and electrons. Furthermore, their heterogeneous nature can reduce or altogether avoid deactivation pathways seen in homogeneous systems such as the formation of inactive nanoparticles or complexes, irreversible oxidation or reduction, excited state side reactions, and collisional and aggregation-based quenching.
However, there are some drawbacks and areas for further improvement in the use of COFs in photocatalytic applications.
(1) Most reports use blue light or white light to perform the catalysis but have not studied what wavelength or range is suitable for the specific catalytic reaction. Studies that have examined the usage of different wavelengths of light illustrated a high dependence on blue light and/or, less often, green light. New COF systems need to be designed to absorb further into the red-light region of the visible spectrum or increase the molar absorption coefficient to perform these transformations using green and longer wavelength light.
(2) A fair amount of COF photocatalysts in the literature require metals to perform the photocatalytic transformation, or at least to perform it efficiently. Strategies to incorporate superior organic units, or a combination of units, need to be addressed to move towards more sustainable practices to accomplish these transformations without the need for metals.
(3) The use of photosensitizers as additives or directly anchored to the COF is common, especially in photocatalytic coupling reactions. These photosensitizers are typically Rh, Ru, or Ir-based and are expensive and toxic. Again, for true metal-free systems these reagents need to be removed. Future work needs to be conducted to enable COFs to be more photoactive or absorb enough light to omit the use of photosensitizers.
(4) While COFs have been designed to lower band gaps by incorporating specific moieties or through the use of D–A systems, precise control over the band gap is not fully realized. Reducing the band gap is simpler in practice, but dictating the precise location of the valence and conduction bands is extremely difficult. The location of the conduction band is of key importance as it can control product distribution in photocatalytic applications. For instance, reducing carbon dioxide can result in many products (i.e. CO, HCOOH, MeOH, CH4, and other hydrocarbons), and positioning the conduction band between the reduction energy levels of various products can limit what products can form, and the COF literature typically shows formation of only CO. Studies need to be performed in order to understand the construction of COFs to precisely control valence band and conduction band energy positions.
(5) Although COFs are constructed through relatively stable covalent bonds and tested in aqueous, acidic, alkaline, and organic solvent conditions, their photochemical stability has not been well established. Most COF materials are not analysed for morphology or crystallinity change when subjected to high-intensity light (more than ambient) and, as discussed, some COFs were reported to lose crystallinity over time. Thus, more research is necessary to determine the long-term photostability of COFs after catalysis for prolonged periods. In the same vein, COFs with boron-based and C–N based linkages are quite dynamic in nature and result in high crystallinities, whereas olefin and ring linkages are more irreversible and typically have a partial or low degree of crystallinity. In saying this, studies need to be conducted to determine the relationship between amorphous, semi-crystalline, poly-crystalline, and single-crystalline COFs in respect to their photocatalytic activity.
(6) Similarly, COFs are lauded for their tunability and capacity to be constructed with different dimensionality and topology and morphology. However, there are limited examples showing a clear link to the structure–property relationship in COFs. As previously mentioned, there have been some studies on 2D versus 3D COFs, but more work is needed to look at differences in 3D versus 3D topologies (i.e. bor versus ctn), 2D AA versus AB stacking, and overall morphology (e.g. rods versus spheres versus wires, etc.) and how it affects the photocatalytic performance of COFs. Furthermore, there is an abundant need to make and prove how pore size, geometry, and chirality can make selective products in photocatalytic applications. For example, chiral COFs have been used in many catalytic applications, but chiral COF use in photocatalysis is rare.
(7) In general, there is a lack of research into the large-scale application of COFs. One reason is that the complex monomer design can make monomer synthesis difficult and low yielding, and therefore limit the monomer available for the COF synthesis. This is also partially due to the fact that the synthesis of COFs is often limited to smaller scale reactions to afford a crystalline material. Designing simpler yet effective systems as well as better synthetic methods can aid in large-scale reactions. Furthermore, when COFs have been employed as photocatalysts they are done on a small laboratory scale, and this needs to be extended to larger scale reactions including the use of larger batch reactors, flow reactors, and membrane reactors in order to make photocatalytic COFs industrially feasible.
Author contributions
J. G. D. developed the concept and structured the paper, screened the literature, made illustrations, and wrote Sections 1, 2, 4, 5, 6.5–6.7, 6.10–6.15, and 7. B. L. co-wrote Section 4 and wrote 6.1–6.4, 6.8 and 6.9. A. S. wrote Section 3. All authors contributed to revising the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Chenhao Dang for making the generic COF stick structure that is in the TOC graphic.References
- Solar Irradiance, https://sunclimate.gsfc.nasa.gov/article/solar-irradiance, accessed June 2024.
- Reference Air Mass 1.5 Spectra, https://www.nrel.gov/grid/solar-resource/spectra-am1.5.html, accessed June 2024.
- J. Yu, T. Zhang and N. Wu, Sol. RRL, 2021, 5, 2100252 CrossRef.
- N. Serpone and A. V. Emeline, J. Phys. Chem. Lett., 2012, 3, 673–677 CrossRef CAS PubMed.
- A. D. McNaught, A. Wilkinson and A. D. Jenkins, IUPAC Compendium of Chemical Terminology: The Gold Book, International Union of Pure and Applied Chemistry, Research Triangle Park, NC, USA, 2006 Search PubMed.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- X. Yang and D. Wang, ACS Appl. Energy Mater., 2018, 1, 6657–6693 CrossRef CAS.
- L. Wang and J. Yu, in S-scheme Heterojunction Photocatalysts Fundamentals and Applications, ed. J. Yu, L. Zhang, L. Wang and B. Zhu, Interface Science and Technology, Elsevier, Amsterdam, 1st edn, 2023, ch. 1, vol. 35, pp. 1–35 Search PubMed.
- H. Kisch and D. Bahnemann, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef CAS.
- H. M. Chua, Q. Zhu, T. Tang, K. W. Shah and J. Xu, Sol. Energy Mater. Sol. Cells, 2019, 197, 32–75 CrossRef.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 8, 474–475 RSC.
- A. Chaves, J. G. Azadani, H. Alsalman, D. R. da Costa, R. Frisenda, A. J. Chaves, S. H. Song, Y. D. Kim, D. He, J. Zhou, A. Castellanos-Gomez, F. M. Peeters, Z. Liu, C. L. Hinkle, S.-H. Oh, P. D. Ye, S. J. Koester, Y. H. Lee, P. Avouris, X. Wang and T. Low, npj 2D Mater. Appl., 2020, 4, 29 CrossRef CAS.
- E. Yousif and R. Haddad, SpringerPlus, 2013, 2, 398 CrossRef PubMed.
- M. Thangamuthu, Q. Ruan, P. O. Ohemeng, B. Luo, D. Jing, R. Godin and J. Tang, Chem. Rev., 2022, 122, 11778–11829 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yanga and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- C. Xia, K. O. Kirlikovali, T. H. C. Nguyen, X. C. Nguyen, Q. B. Tran, M. K. Duong, M. T. N. Dinh, D. L. T. Nguyen, P. Singh, P. Raizada, V.-H. Nguyen, S. Y. Kim, L. Singh, C. C. Nguyen, M. Shokouhimehr and Q. V. Le, Coord. Chem. Rev., 2021, 446, 214117 CrossRef CAS.
- T. Banerjee, K. Gottschlin, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- B. B. Rath, S. Krause and B. V. Lotsch, Adv. Funct. Mater., 2023, 2309060 Search PubMed.
- Z. Zhou, Y. Xiao, J. Tian, N. Nan, R. Song and J. Li, J. Mater. Chem. A, 2023, 11, 3245–3261 RSC.
- Y.-N. Gong, X. Guan and H.-L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- A. López-Magano, S. Daliran, A. R. Oveisi, R. Mas-Ballesté, A. Dhakshinamoorthy, J. Alemán, H. Garcia and R. Luque, Adv. Mater., 2023, 35, 2209475 CrossRef PubMed.
- L. Qin, C. Ma, J. Zhang and T. Zhou, Adv. Funct. Mater., 2024, 2401562 CrossRef CAS.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- J. R. Hunt, C. J. Doonan, J. D. LeVangie, A. P. Côté and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11872–11873 CrossRef CAS PubMed.
- K. T. Jackson, T. E. Reich and H. M. El-Kaderi, Chem. Commun., 2012, 48, 8823 RSC.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S.-H. Lee and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- Z.-B. Zhou, X.-H. Han, Q.-Y. Qi, S.-X. Gan, D.-L. Ma and X. Zhao, J. Am. Chem. Soc., 2022, 144, 1138–1143 CrossRef CAS PubMed.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- Y. Zhu, D. Huang, W. Wang, G. Liu, C. Ding and Y. Xiang, Angew. Chem., Int. Ed., 2024, 63, e202319909 CrossRef CAS PubMed.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dörfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef PubMed.
- C. Qian, L. Feng, W. L. Teo, J. Liu, W. Zhou, D. Wang and Y. Zhao, Nat. Rev. Chem., 2022, 6, 881–898 CrossRef CAS PubMed.
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2013, 52, 13052–13056 CrossRef CAS PubMed.
- A. Halder, S. Karak, M. Addicoat, S. Bera, A. Chakraborty, S. H. Kunjattu, P. Pachfule, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2018, 57, 5797–5802 CrossRef CAS PubMed.
- A. Halder, M. Ghosh, A. Khayu M, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef CAS PubMed.
- L. M. Lanni, R. W. Tilford, M. Bharathy and J. J. Lavigne, J. Am. Chem. Soc., 2011, 133, 13975–13983 CrossRef CAS PubMed.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- A. Nagai, X. Chen, X. Feng, X. Ding, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 3770–3774 CrossRef CAS PubMed.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Nat. Commun., 2014, 5, 4503 CrossRef PubMed.
- D. Stewart, D. Antypov, M. S. Dyer, M. J. Pitcher, A. P. Katsoulidis, P. A. Chater, F. Blanc and M. J. Rosseinsky, Nat. Commun., 2017, 8, 1102 CrossRef PubMed.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, Polym. Chem., 2016, 7, 4176–4181 RSC.
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 CrossRef CAS PubMed.
- A. Acharjya, P. Pachfule, J. Roeser, F.-J. Schmitt and A. Thomas, Angew. Chem., 2019, 131, 15007–15012 CrossRef.
- D. L. Pastoetter, S. Xu, M. Borrelli, M. Addicoat, B. P. Biswal, S. Paasch, A. Dianat, H. Thomas, R. Berger, S. Reineke, E. Brunner, G. Cuniberti, M. Richter and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 23620–23625 CrossRef CAS PubMed.
- X. Wu, L. Zong and N. Huang, Chem. Commun., 2024, 60, 320–323 RSC.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed.
- B. Zhang, M. Wei, H. Mo, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS PubMed.
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- K. C. Ranjeesh, R. Illathvalappil, S. D. Veer, J. Peter, V. C. Wakchaure, Goudappagouda, K. V. Raj, S. Kurungot and S. S. Babu, J. Am. Chem. Soc., 2019, 141, 14950–14954 CrossRef CAS PubMed.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- Y. Meng, Y. Luo, J.-L. Shi, H. Ding, X. Lang, W. Chen, A. Zheng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 3624–3629 CrossRef CAS PubMed.
- K. Wang, X. Kang, C. Yuan, X. Han, Y. Liu and Y. Cui, Angew. Chem., Int. Ed., 2021, 60, 19466–19476 CrossRef CAS PubMed.
- P. Li, F. Ge, Y. Yang, T. Wang, X. Zhang, K. Zhang and J. Shen, Angew. Chem., Int. Ed., 2024, 63, e202319885 CrossRef CAS PubMed.
- C. Liu, D.-L. Ma, P.-J. Tian, C. Jia, Q.-Y. Qi, G.-F. Jiang and X. Zhao, J. Mater. Chem. A, 2024, 12, 16063–16069 RSC.
- X. Shen and B. Yan, J. Mater. Chem. A, 2024, 12, 6455–6464 RSC.
- K. Lin, J. Wang, S. Qiao and Z. Guo, ACS Sustainable Chem. Eng., 2024, 12, 6719–6727 CrossRef CAS.
- L. Song, W. Gao, S. Jiang, Y. Yang, W. Chu, X. Cao, B. Sun, L. Cui and C.-Y. Zhang, Nano Lett., 2024, 24, 6312–6319 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed.
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- Y. Xie, J. Li, C. Lin, B. Gui, C. Ji, D. Yuan, J. Sun and C. Wang, J. Am. Chem. Soc., 2021, 143, 7279–7284 CrossRef CAS PubMed.
- Y. Liu, J. Li, J. Lv, Z. Wang, J. Suo, J. Ren, J. Liu, D. Liu, Y. Wang, V. Valtchev, S. Qiu, D. Zhang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 9679–9685 CrossRef CAS PubMed.
- X. Kang, X. Han, C. Yuan, C. Cheng, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2020, 142, 16346–16356 CrossRef CAS PubMed.
- H. Li, J. Ding, X. Guan, F. Chen, C. Li, L. Zhu, M. Xue, D. Yuan, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2020, 142, 13334 CrossRef CAS PubMed.
- H. Li, F. Chen, X. Guan, J. Li, C. Li, B. Tang, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2021, 143, 2654–2659 CrossRef CAS PubMed.
- Z. Li, L. Sheng, C. Hsueh, X. Wang, H. Cui, H. Gao, Y. Wu, J. Wang, Y. Tang, H. Xu and X. He, Chem. Mater., 2021, 33, 9618–9623 CrossRef CAS.
- J. Chang, F. Chen, H. Li, J. Suo, H. Zheng, J. Zhang, Z. Wang, L. Zhu, V. Valtchev, S. Qiu and Q. Fang, Nat. Commun., 2024, 15, 813 CrossRef CAS PubMed.
- Y. Lan, X. Han, M. Tong, H. Huang, Q. Yang, D. Liu, X. Zhao and C. Zhong, Nat. Commun., 2018, 9, 5274 CrossRef PubMed.
- H. L. Nguyen, C. Gropp, Y. Ma, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 20335–20339 CrossRef CAS PubMed.
- S. Das, T. Sekine, H. Mabuchi, T. Irie, J. Sakai, Y. Zhao, Q. Fang and Y. Negishi, ACS Appl. Mater. Interfaces, 2022, 14, 48045–48051 CrossRef CAS PubMed.
- F. Jin, E. Lin, T. Wang, S. Geng, T. Wang, W. Liu, F. Xiong, Z. Wang, Y. Chen, P. Cheng and Z. Zhang, J. Am. Chem. Soc., 2022, 144, 5643–5652 CrossRef CAS PubMed.
- Z. Shan, M. Wu, D. Zhu, X. Wu, K. Zhang, R. Verduzco and G. Zhang, J. Am. Chem. Soc., 2022, 144, 5728–5733 CrossRef CAS PubMed.
- S. Das, H. Mabuchi, T. Irie, K. Sasaki, M. Nozaki, R. Tomioka, D. Wen, Y. Zhao, T. Ben and Y. Negishi, Small, 2024, 20, 2307666 CrossRef CAS PubMed.
- W. Liu, K. Wang, X. Zhan, Z. Liu, X. Yang, Y. Jin, B. Yu, L. Gong, H. Wang, D. Qi, D. Yuan and J. Jiang, J. Am. Chem. Soc., 2023, 145, 8141–8149 CrossRef CAS PubMed.
- D. Zhu, Y. Zhu, Y. Chen, Q. Yan, H. Wu, C.-Y. Liu, X. Wang, L. B. Alemany, G. Gao, T. P. Senftle, Y. Peng, X. Wu and R. Verduzco, Nat. Commun., 2023, 14, 2865 CrossRef CAS PubMed.
- Y. Liu, P. Chen, Y. Wang, J. Suo, J. Ding, L. Zhu, V. Valtchev, Y. Yan, S. Qiu, J. Sun and Q. Fang, Angew. Chem., Int. Ed., 2022, 61, e202203584 CrossRef CAS PubMed.
- X. Wang, M. Bahri, Z. Fu, M. A. Little, L. Liu, H. Niu, N. D. Browning, S. Y. Chong, L. Chen, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2021, 143, 15011–15016 CrossRef CAS PubMed.
- O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Fröba, A. Thomas and J. Roeser, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS PubMed.
- Y. Liu, Y. Ma, J. Yang, C. S. Diercks, N. Tamura, F. Jin and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 16015–16019 CrossRef CAS PubMed.
- H.-S. Lu, W.-K. Han, X. Yan, C.-J. Chen, T. Niu and Z.-G. Gu, Angew. Chem., Int. Ed., 2021, 60, 17881–17886 CrossRef CAS PubMed.
- W.-K. Han, H.-S. Lu, J.-X. Fu, X. Liu, X. Zhu, X. Yan, J. Zhang, Y. Jiang, H. Dong and Z.-G. Gu, Chem. Eng. J., 2022, 449, 137802 CrossRef CAS.
- M. Martínez-Abadía, K. Strutyński, B. Lerma-Berlanga, C. T. Stoppiello, A. N. Khlobystov, C. Martí-Gastaldo, A. Saeki, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2021, 60, 9941–9946 CrossRef PubMed.
- X. Xu, P. Cai, H. Chen, H.-C. Zhou and N. Huang, J. Am. Chem. Soc., 2022, 144, 18511–18517 CrossRef CAS PubMed.
- F. Jin, H. L. Nguyen, Z. Zhong, X. Han, C. Zhu, X. Pei, Y. Ma and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 1539–1544 CrossRef CAS PubMed.
- M. Wu, Z. Shan, J. Wang, T. Liu and G. Zhang, Chem. Eng. J., 2023, 454, 140121 CrossRef CAS.
- Reticular Chemistry Structure Resource Search 3-Periodic, https://rcsr.net/nets, accessed 6/30/2024.
- Z. Guo, Z. Zhang and J. Sun, Adv. Mater., 2024, 3, e2312889 CrossRef PubMed.
- R. Roy and A. M. Evans, Commun. Mater., 2024, 5, 102 CrossRef CAS.
- D. Zhu, J.-J. Zhang, X. Wu, Q. Yan, F. Liu, Y. Zhu, X. Gao, M. M. Rahman, B. I. Yakobson, P. M. Ajayan and R. Verduzco, Chem. Sci., 2022, 13, 9655–9667 RSC.
- D. Zhu and R. Verduzco, ACS Appl. Mater. Interfaces, 2020, 12, 33121–33127 CrossRef CAS PubMed.
- B. Diaz de Grenu, J. Torres, J. García-González, S. Muñoz-Pina, R. de Los Reyes, A. M. Costero, P. Amorós and J. V. Ros-Lis, ChemSusChem, 2021, 14, 208–233 CrossRef CAS PubMed.
- A. de la Hoz, A. Díaz-Ortiz and P. Prieto, in Alternative Energy Sources for Green Chemistry, ed. G. Stefanidis and A. Stankiewicz, The Royal Society of Chemistry, Cambridge, 1st edn, 2016, ch. 1, pp. 1–33 Search PubMed.
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Chem. Mater., 2009, 21, 204–206 CrossRef CAS.
- H. Wei, S. Chai, N. Hu, Z. Yang, L. Wei and L. Wang, Chem. Commun., 2015, 51, 12178–12181 RSC.
- S.-T. Yang, J. Kim, H.-Y. Cho, S. Kim and W.-S. Ahn, RSC Adv., 2012, 2, 10179–10181 RSC.
- J. Yoo, S. Lee, S. Hirata, C. Kim, C. K. Lee, T. Shiraki, N. Nakashima and J. K. Shim, Chem. Lett., 2015, 44, 560–562 CrossRef CAS.
- W. Zhao, P. Yan, H. Yang, M. Bahri, A. M. James, H. Chen, L. Liu, B. Li, Z. Pang, R. Clowes, N. D. Browning, J. W. Ward, Y. Wu and A. I. Cooper, Nat. Synth., 2022, 1, 87–95 CrossRef.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed.
- X. Wang, R. Ma, L. Hao, Q. Wu, C. Wang and Z. Wang, J. Chromatogr. A, 2018, 1551, 1–9 CrossRef CAS PubMed.
- N. Brown, Z. Alsudairy, R. Behera, F. Akram, K. Chen, K. Smith-Petty, B. Motley, S. Williams, W. Huang, C. Ingrama and X. Li, Green Chem., 2023, 25, 6287–6296 RSC.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- Y. Peng, G. Xu, Z. Hu, Y. Cheng, C. Chi, D. Yuan, H. Cheng and D. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 18505–18512 CrossRef CAS PubMed.
- S. Kim and H. C. Choi, Commun. Chem., 2019, 2, 60 CrossRef.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- J. Maschita, T. Banerjee, G. Savasci, F. Haase, C. Ochsenfeld and B. V. Lotsch, Angew. Chem., Int. Ed., 2020, 59, 15750–15758 CrossRef CAS PubMed.
- K. S. Song, P. W. Fritz, D. F. Abbott, L. N. Poon, C. M. Caridade, F. Gándara, V. Mougel and A. Coskun, Angew. Chem., Int. Ed., 2023, 62, e202309775 CrossRef PubMed.
- X. Guan, Y. Ma, H. Li, Y. Yusran, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2018, 140, 4494–4498 CrossRef CAS PubMed.
- M. Sawatzki-Park, S.-J. Wang, H. Kleemann and K. Leo, Chem. Rev., 2023, 123, 8232–8250 CrossRef CAS PubMed.
- T. Haggren, H. H. Tan and C. Jagadish, Acc. Mater. Res., 2023, 4, 1046–1056 CrossRef CAS.
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang, C. Lai, D. Jiang, Y. Liu, H. Yi, L. Qin, S. Ye, X. Ren and W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC.
- K. Dey, M. Pal, K. C. Rout, H. S. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- M. Matsumoto, L. Valentino, G. M. Stiehl, H. B. Balch, A. R. Corcos, F. Wang, D. C. Ralph, B. J. Mariñas and W. R. Dichtel, Chem, 2018, 4, 308–317 CAS.
- Q. Hao, C. Zhao, B. Sun, C. Lu, J. Liu, M. Liu, L.-J. Wan and D. Wang, J. Am. Chem. Soc., 2018, 140, 12152–12158 CrossRef CAS PubMed.
- J. I. Feldblyum, C. H. McCreery, S. C. Andrews, T. Kurosawa, E. J. G. Santos, V. Duong, L. Fang, A. L. Ayzner and Z. N. Bao, Chem. Commun., 2015, 51, 13894–13897 RSC.
- Z. Ou, B. Liang, Z. Liang, F. Tan, X. Dong, L. Gong, P. Zhao, H. Wang, Y. Zou, Y. Xia, X. Chen, W. Liu, H. Qi, U. Kaiser and Z. Zheng, J. Am. Chem. Soc., 2022, 144, 3233–3241 CrossRef CAS PubMed.
- Y. Yang, Y. Chen, F. Izquierdo-Ruiz, C. Schäfer, M. Rahm and K. Börjesson, Nat. Commun., 2023, 14, 220 CrossRef CAS PubMed.
- D. Wu, Q. Che, H. He, M. E. El-Khouly, S. Huang, X. Zhuang, B. Zhang and Y. Chen, ACS Mater. Lett., 2023, 5, 874–883 CrossRef CAS.
- X. Shi, D. Ma, F. Xu, Z. Zhang and Y. Wang, Chem. Sci., 2020, 11, 989–996 RSC.
- Y. Qu, Y. Zha, X. Du, S. Xu, M. Zhang, L. Xu and H. Jia, ACS Appl. Polym. Mater., 2022, 4, 7528–7536 CrossRef CAS.
- Y.-B. Zhang, J. Su, H. Furukawa, Y. Yun, F. Gándara, A. Duong, X. Zou and O. M. Yaghi, J. Am. Chem. Soc., 2013, 135, 16336–16339 CrossRef CAS PubMed.
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830–834 CrossRef CAS PubMed.
- C. Kang, K. Yang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, J. Jiang, X. Zou, Z. Huang and D. Zhao, Nat. Commun., 2022, 13, 1370 CrossRef CAS PubMed.
- L. Peng, Q. Guo, C. Song, S. Ghosh, H. Xu, L. Wang, D. Hu, L. Shi, L. Zhao, Q. Li, T. Sakurai, H. Yan, S. Seki, Y. Liu and D. Wei, Nat. Commun., 2021, 12, 5077 CrossRef CAS PubMed.
- Y. Yin, Y. Zhang, X. Zhou, B. Gui, G. Cai, J. Sun and C. Wang, J. Am. Chem. Soc., 2023, 145, 22329–22334 CrossRef CAS PubMed.
- L. Liang, Y. Qiu, W. D. Wang, J. Han, Y. Luo, W. Yu, G.-L. Yin, Z.-P. Wang, L. Zhang, J. Ni, J. Niu, J. Sun, T. Ma and W. Wang, Angew. Chem., Int. Ed., 2020, 59, 17991–17995 CrossRef CAS PubMed.
- H.-S. Xu, Y. Luo, X. Li, P. Z. See, Z. Chen, T. Ma, L. Liang, K. Leng, I. Abdelwahab, L. Wang, R. Li, X. Shi, Y. Zhou, X. F. Lu, X. Zhao, C. Liu, J. Sun and K. P. Loh, Nat. Commun., 2020, 11, 1434 CrossRef CAS PubMed.
- Z. Zhou, L. Zhang, Y. Yang, I. J. Vitorica-Yrezabal, H. Wang, F. Tan, L. Gong, Y. Li, P. Chen, X. Dong, Z. Liang, J. Yang, C. Wang, Y. Hong, Y. Qiu, A. Gölzhäuser, X. Chen, H. Qi, S. Yang, W. Liu, J. Sun and Z. Zheng, Nat. Chem., 2023, 15, 841–847 CrossRef CAS PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed.
- B. Yu, R.-B. Lin, G. Xu, Z.-H. Fu, H. Wu, W. Zhou, S. Lu, Q.-W. Li, Y. Jin, J.-H. Li, Z. Zhang, H. Wang, Z. Yan, X. Liu, K. Wang, B. Chen and J. Jiang, Nat. Chem., 2024, 16, 114–121 CrossRef CAS PubMed.
- J. Han, J. Feng, J. Kang, J.-M. Chen, X.-Y. Du, S.-Y. Ding, L. Liang and W. Wang, Science, 2024, 383, 1014–1019 CrossRef CAS PubMed.
- N. A. Rejali, M. Dinari and Y. Wang, Chem. Commun., 2023, 59, 11631–11647 RSC.
- J. L. Segura, S. Royuela and M. M. Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- I. Romero-Muñiz, A. Mavrandonakis, P. Albacete, A. Vega, V. Briois, F. Zamora and A. E. Platero-Prats, Angew. Chem., Int. Ed., 2020, 59, 13013–13020 CrossRef PubMed.
- J. Hu, H. Mehrabi, Y.-S. Meng, M. Taylor, J.-H. Zhan, Q. Yan, M. Benamara, R. H. Coridan and H. Beyzavi, Chem. Sci., 2021, 12, 7930–7936 RSC.
- X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Nat. Commun., 2018, 9, 1294 CrossRef PubMed.
- P. Sarkar, A. Hazra Chowdhury, S. Biswas, A. Khan and S. M. Islam, Mater. Today Chem., 2021, 21, 100509 CrossRef CAS.
- Y. Li, X. Guo, X. Li, M. Zhang, Z. Jia, Y. Deng, Y. Tian, S. Li and L. Ma, Angew. Chem., Int. Ed., 2020, 59, 4168–4175 CrossRef CAS.
- G. Chen, H.-H. Lan, S.-L. Cai, B. Sun, X.-L. Li, Z.-H. He, S.-R. Zheng, J. Fan, Y. Liu and W.-G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12830–12837 CrossRef CAS.
- X. Kan, J.-C. Wang, J.-L. Kan, J.-Y. Shang, H. Qiao and Y.-B. Dong, Inorg. Chem., 2021, 60, 3393–3400 CrossRef CAS PubMed.
- C. Qian, Q.-Y. Qi, G.-F. Jiang, F.-Z. Cui, Y. Tian and X. Zhao, J. Am. Chem. Soc., 2017, 139, 6736–6743 CrossRef CAS PubMed.
- J. Maschita, T. Banerjee and B. V. Lotsch, Chem. Mater., 2022, 34, 2249–2258 CrossRef CAS PubMed.
- Z.-B. Zhou, P.-J. Tian, J. Yao, Y. Lu, Q.-Y. Qi and X. Zhao, Nat. Commun., 2022, 13, 2180 CrossRef CAS PubMed.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Nat. Commun., 2011, 2, 536 CrossRef PubMed.
- N. Huang, R. Krishna and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7079–7082 CrossRef CAS PubMed.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- D. N. Bunck and W. R. Dichtel, Chem. Commun., 2013, 49, 2457–2459 RSC.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. A. Perman, T. Butts, F.-S. Xiao and S. Ma, Chem, 2018, 4, 1726–1739 CAS.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- S. Rager, M. Dogru, V. Werner, A. Gavryushin, M. Götz, H. Engelke, D. D. Medina, P. Knochel and T. Bein, CrystEngComm, 2017, 19, 4886–4891 RSC.
- P. Nitschke, N. Lokesh and R. M. Gschwind, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 86–134 CrossRef CAS PubMed.
- C. Kranz and M. Wächtler, Chem. Soc. Rev., 2021, 50, 1407–1437 RSC.
- G.-B. Wang, Y.-J. Wang, J.-L. Kan, K.-H. Xie, H.-P. Xu, F. Zhao, M.-C. Wang, Y. Geng and Y.-B. Dong, J. Am. Chem. Soc., 2023, 145, 4951–4956 CrossRef CAS PubMed.
- J. Ø. Duus, C. H. Gotfredsen and K. Bock, Chem. Rev., 2000, 100, 4589–4614 CrossRef CAS PubMed.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- R. M. Maggio, N. L. Calvo, S. E. Vignaduzzo and T. S. Kaufman, J. Pharm. Biomed. Anal., 2014, 101, 102–122 CrossRef CAS PubMed.
- S. Singh, T. Handa, M. Narayanam, A. Sahu, M. Junwal and R. P. Shah, J. Pharm. Biomed. Anal., 2012, 69, 148–173 CrossRef CAS PubMed.
- S. K. Bharti and R. Roy, TrAC, Trends Anal. Chem., 2012, 35, 5–26 CrossRef CAS.
- U. Holzgrabe, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 229–240 CrossRef CAS PubMed.
- Y. Ben-Tal, P. J. Boaler, H. J. A. Dale, R. E. Dooley, N. A. Fohn, Y. Gao, A. García-Domínguez, K. M. Grant, A. M. R. Hall, H. L. D. Hayes, M. M. Kucharski, R. Wei and G. C. Lloyd-Jones, Prog. Nucl. Magn. Reson. Spectrosc., 2022, 129, 28–106 CrossRef CAS PubMed.
- T. Blasco, Chem. Soc. Rev., 2010, 39, 4685–4702 RSC.
- C. Feldmeier, H. Bartling, E. Riedle and R. M. Gschwind, J. Magn. Reson., 2013, 232, 39–44 CrossRef CAS PubMed.
- A. Seegerer, P. Nitschke and R. M. Gschwind, Angew. Chem., Int. Ed., 2018, 57, 7493–7497 CrossRef CAS PubMed.
- D. Lehnherr, Y. Ji, A. J. Neel, R. D. Cohen, A. P. J. Brunskill, J. Yang and M. Reibarkh, J. Am. Chem. Soc., 2018, 140, 13843–13853 CrossRef CAS PubMed.
- Y. Ji, D. A. DiRocco, J. Kind, C. M. Thiele, R. M. Gschwind and M. Reibarkh, ChemPhotoChem, 2019, 3, 984–992 CrossRef CAS.
- X. L. Wang, W. Liu, Y.-Y. Yu, Y. Song, W. Q. Fang, D. Wei, X.-Q. Gong, Y.-F. Yao and H. G. Yang, Nat. Commun., 2016, 7, 11918 CrossRef CAS PubMed.
- H. Parastar, J. R. Radovic, M. Jalali-Heravi, S. Diez, J. M. Bayona and R. Tauler, Anal. Chem., 2011, 83, 9289–9297 CrossRef CAS PubMed.
- P. Jonsson, A. I. Johansson, J. Gullberg, J. Trygg, J. A, B. Grung, S. Marklund, M. Sjöström, H. Antti and T. Moritz, Anal. Chem., 2005, 77, 5635–5642 CrossRef CAS PubMed.
- D. A. Lambropoulou, M. D. Hernando, I. K. Konstantinou, E. M. Thurman, I. Ferrer, T. A. Albanis and A. R. Fernandez-Alba, J. Chromatogr. A, 2008, 1183, 38–48 CrossRef CAS PubMed.
- C. Sahoo and A. K. Gupta, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2015, 50, 1333–1341 CrossRef CAS PubMed.
- B. R. Miller, R. F. Weiss, P. K. Salameh, T. Tanhua, B. R. Greally, J. Muhle and P. G. Simmonds, Anal. Chem., 2008, 80, 1536–1545 CrossRef CAS PubMed.
- J. Hong, W. Zhang, J. Rena and R. Xu, Anal. Methods, 2013, 5, 1086–1097 RSC.
- R. Zaffaroni, D. Ripepi, J. Middelkoop and F. M. Mulder, ACS Energy Lett., 2020, 5, 3773–3777 CrossRef CAS.
- J. Št’ávová, D. C. Stahl, W. S. Seames and A. Kubátová, J. Chromatogr. A, 2012, 1224, 79–88 CrossRef PubMed.
- N. Ragunathan, K. A. Krock, C. Klawun, T. A. Sasaki and C. L. Wilkins, J. Chromatogr. A, 1999, 856, 349–397 CrossRef CAS PubMed.
- C. F. Poole, J. Chromatogr. A, 2015, 1421, 137–153 CrossRef CAS PubMed.
- E. D. Pellizzari, J. Chromatogr. A, 1974, 98, 323–361 CrossRef CAS.
- M. B. Mitchell, in Structure-Property Relations in Polymers, ed. M. W. Urban and C. D. Craver, American Chemical Society, Washington, DC, 1993, ch. 13, vol. 236, pp. 351–375 Search PubMed.
- E. Groppo, S. Rojas-Buzo and S. Bordiga, Chem. Rev., 2023, 123, 12135–12169 CrossRef CAS PubMed.
- I. M. Hill, S. Hanspal, Z. D. Young and R. J. Davis, J. Phys. Chem. C, 2015, 119, 9186–9197 CrossRef CAS.
- F. Zaera, Chem. Soc. Rev., 2014, 43, 7624–7663 RSC.
- F. C. Meunier, React. Chem. Eng., 2016, 1, 134–141 RSC.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- B. Hauchecorne, D. Terrens, S. Verbruggen, J. A. Martens, H. Van Langenhove, K. Demeestere and S. Lenaerts, Appl. Catal., B, 2011, 106, 630–638 CrossRef CAS.
- Q. Zhang, S. Mo, J. Li, Y. Sun, M. Zhang, P. Chen, M. Fu, J. Wu, L. Chen and D. Ye, Catal. Sci. Technol., 2019, 9, 4538–4551 RSC.
- B. N. Choi, J. Y. Seo, Z. An, P. J. Yoo and C.-H. Chung, Chem. Eng. J., 2022, 430, 132807 CrossRef CAS.
- Q. Li, Z.-h. Yue, M.-s. Shan, Y.-l. Li, Y. Liu, Y.-j. Hao and F.-t. Li, Appl. Catal., B, 2023, 330, 122628 CrossRef CAS.
- Y. Fan, Y. Jiang, H. Lin, J. Li, Y. Xie, A. Chen, S. Li, D. Han, L. Niu and Z. Tang, Nat. Commun., 2024, 15, 4679 CrossRef CAS PubMed.
- J. T. Schneider, D. S. Firak, R. R. Ribeiro and P. Peralta-Zamora, Phys. Chem. Chem. Phys., 2020, 22, 15723–15733 RSC.
- A. Trenczek-Zajac, M. Synowiec, K. Zakrzewska, K. Zazakowny, K. Kowalski, A. Dziedzic and M. Radecka, ACS Appl. Mater. Interfaces, 2022, 14, 38255–38269 CrossRef CAS PubMed.
- C. Carlsson, B. Fégeant, E. Svensson, L. Wiklund and M. Jonsson, J. Phys. Chem. C, 2022, 126, 12435–12440 CrossRef CAS.
- L. Ding, Y. Hou, H. Liu, J. Peng, Z. Cao, Y. Zhang, B. Wang, X. Cao, Y. Chang, T. Wang and G. Liu, ACS ES&T Water, 2023, 3, 3534–3543 Search PubMed.
- L. Wang, B. Li, D. D. Dionysiou, B. Chen, J. Yang and J. Li, Environ. Sci. Technol., 2022, 56, 3386–3396 CrossRef CAS PubMed.
- Y. Du and J. Rabani, J. Phys. Chem. B, 2003, 107, 11970–11978 CrossRef CAS.
- G. Žerjav, A. Albreht, I. Vovk and A. Pintar, Appl. Catal., A, 2020, 598, 117566 CrossRef.
- C. D. Georgiou, H. J. Sun, C. P. McKay, K. Grintzalis, I. Papapostolou, D. Zisimopoulos, K. Panagiotidis, G. Zhang, E. Koutsopoulou, G. E. Christidis and I. Margiolaki, Nat. Commun., 2015, 6, 7100 CrossRef CAS PubMed.
- O. A. Badary, R. A. Taha, A. M. G. El-Din and M. H. Abdel-Wahab, Drug Chem. Toxicol., 2003, 26, 87–98 CrossRef CAS PubMed.
- M. Iwamura and N. Inamoto, Bull. Chem. Soc. Jpn., 1967, 40, 703 CrossRef CAS.
- J. R. Harbour, V. Chew and J. R. Bolton, Can. J. Chem., 1974, 52, 3549–3553 CrossRef CAS.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527–6528 CrossRef CAS.
- J. R. Harbour and S. L. Issler, J. Am. Chem. Soc., 1982, 104, 903–905 CrossRef CAS.
- Y. Lion, M. Delmelle and A. Van De Vorst, Nature, 1976, 263, 442–443 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- J. Liu, Y. Zou, B. Jin, K. Zhang and J. H. Park, ACS Energy Lett., 2019, 4, 3018–3027 CrossRef CAS.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2021, 143, 9423–9428 CrossRef CAS PubMed.
- W. Mtangi, F. Tassinari, K. Vankayala, A. V. Jentzsch, B. Adelizzi, A. R. A. Palmans, C. Fontanesi, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2017, 139, 2794–2798 CrossRef CAS PubMed.
- K. Wenderich, W. Kwak, A. Grimm, G. J. Kramer, G. Mul and B. Mei, Sustainable Energy Fuels, 2020, 4, 3143–3156 RSC.
- S. R. Lingampalli, M. M. Ayyub and C. N. R. Rao, ACS Omega, 2017, 2, 2740–2748 CrossRef CAS PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- S. Goldstein, D. Aschengrau, Y. Diamant and J. Rabani, Environ. Sci. Technol., 2007, 41, 7486–7490 CrossRef CAS PubMed.
- K. Sahel, L. Elsellami, I. Mirali, F. Dappozze, M. Bouhent and C. Guillard, Appl. Catal., B, 2016, 188, 106–112 CrossRef CAS.
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., 2012, 124, 808–812 CrossRef.
- L. Wei, J. Zhang and L. Xu, ACS Sustainable Chem. Eng., 2020, 8, 13894–13899 CrossRef CAS.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
- M. Assis, A. F. Gouveia, L. K. Ribeiro, M. A. Ponce, M. S. Churio, O. N. Oliveira, Jr., L. H. Mascaro, E. Longo, R. Llusar, E. Guillamon and J. Andres, Appl. Catal., A, 2023, 652, 119038 CrossRef CAS.
- C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 2205 CrossRef PubMed.
- K. Ohkubo, K. Sugaa and S. Fukuzumi, Chem. Commun., 2006, 2018–2020 RSC.
- C. Zheng, G. He, X. Xiao, M. Lu, H. Zhong, X. Zuo and J. Nan, Appl. Catal., B, 2017, 205, 201–210 CrossRef CAS.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- J. D. Sitter and A. K. Vannucci, J. Am. Chem. Soc., 2021, 143, 2938–2943 CrossRef CAS PubMed.
- P. Bai, X. Tong, Y. Gao and P. Guo, Catal. Sci. Technol., 2019, 9, 5803–5811 RSC.
- Z.-L. Chena and M. H. Huang, J. Mater. Chem. A, 2023, 11, 22198–22205 RSC.
- M. Pavel, C. Anastasescu, R.-N. State, A. Vasile, F. Papa and I. Balint, Catalysts, 2023, 13, 380 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- E. Baur and C. Neuweiler, Helv. Chim. Acta, 1927, 10, 901–907 CrossRef CAS.
- V. Diesen and M. Jonsson, J. Phys. Chem. C, 2014, 118, 10083–10087 CrossRef CAS.
- H. Bader, V. Sturzenegger and A. J. Hoigne, Water Res., 1988, 22, 1109–1115 CrossRef CAS.
- T. Freese, J. T. Meijer, B. L. Feringa and S. B. Beil, Nat. Catal., 2023, 6, 553–558 CrossRef CAS.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- P.-W. Huang and M. C. Hatzell, Nat. Commun., 2022, 13, 7908 CrossRef CAS PubMed.
- A. P. Varghese, B. Neppolian and S. K. Lakhera, Ind. Eng. Chem. Res., 2023, 62, 12530–12537 CrossRef CAS.
- K. A. Othman, R. A. Omer, A. A. A. Mohammad and D. A. Safin, J. Chem. Educ., 2024, 101, 567–577 CrossRef CAS.
- R. Capelle, Anal. Chim. Acta, 1961, 24, 555–572 CrossRef CAS.
- I. Alcón, R. Santiago, J. Ribas-Arino, M. Deumal, I. d. P. R. Moreira and S. T. Bromley, Nat. Commun., 2021, 12, 1705 CrossRef PubMed.
- S. Thomas, H. Li, C. Zhong, M. Matsumoto, W. R. Dichtel and J.-L. Bredas, Chem. Mater., 2019, 31, 3051–3065 CrossRef CAS.
- E. Dautzenberg, M. Lam, T. Nikolaeva, W. M. J. Franssen, B. van Lagen, I. P. A. M. Gerrits-Benneheij, N. Kosinov, G. Li and L. C. P. M. de Smet, J. Phys. Chem. C, 2022, 126, 21338–21347 CrossRef CAS PubMed.
- M. Deng, J. Sun, A. Laemont, C. Liu, L. Wang, L. Bourda, J. Chakraborty, K. V. Hecke, R. Morent, N. De Geyter, K. Leus, H. Chen and P. Van Der Voort, Green Chem., 2023, 25, 3069–3076 RSC.
- Q. Lin, Y. Yusran, J. Xing, Y. Li, J. Zhang, T. Su, L. Yang, J. Suo, L. Zhang, Q. Li, H. Wang, Q. Fang, Z.-T. Li and D.-W. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 5869–5880 CrossRef CAS PubMed.
- N. Singh, D. Yadav, S. V. Mulay, J. Y. Kim, N.-J. Park and J.-O. Baeg, ACS Appl. Mater. Interfaces, 2021, 13, 14122–14131 CrossRef CAS PubMed.
- Y. Chen, X. Luo, J. Zhang, L. Hu, T. Xu, W. Li, L. Chen, M. Shen, S.-B. Ren, D.-M. Han, G.-H. Ning and D. Li, J. Mater. Chem. A, 2022, 10, 24620–24627 RSC.
- X.-R. Chen, W.-R. Cui, R.-P. Liang, C.-R. Zhang, R.-H. Xu, W. Jiang and J.-D. Qiu, ACS Appl. Bio Mater., 2021, 4, 6502–6511 CrossRef CAS PubMed.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S.-W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CrossRef CAS PubMed.
- G.-B. Wang, H.-P. Xu, K.-H. Xie, J.-L. Kan, J. Fan, Y.-J. Wang, Y. Geng and Y.-B. Dong, J. Mater. Chem. A, 2023, 11, 4007–4012 RSC.
- A. Das, Mohit and K. R. J. Thomas, J. Org. Chem., 2023, 88, 14065–14077 CrossRef CAS PubMed.
- R. Liu, Y. Chen, H. Yu, M. Položij, Y. Guo, T. C. Sum, T. Heine and D. Jiang, Nat. Catal., 2024, 7, 195–206 CrossRef CAS.
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. D. Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. Van De Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317 CrossRef CAS PubMed.
- C. Michelin and N. Hoffmann, ACS Catal., 2018, 8, 12046–12055 CrossRef CAS.
- X. Tong, S. Gan, J. Wu, Y. Hu and A. Yuan, Nanoscale, 2020, 12, 7376–7382 RSC.
- C. Yang, H. Hu, C. Qian and Y. Liao, J. Mater. Chem. A, 2023, 11, 25899–25909 RSC.
- S.-Y. Ding, P.-L. Wang, G.-L. Yin, X. Zhang and G. Lu, Int. J. Hydrogen Energy, 2019, 44, 11872–11876 CrossRef CAS.
- Q.-J. Wu, D.-H. Si, S. Ye, Y.-L. Dong, R. Cao and Y.-B. Huang, J. Am. Chem. Soc., 2023, 145, 19856–19865 CrossRef CAS PubMed.
- Y.-Q. Xing and S.-Y. Liu, Chin. J. Struct. Chem., 2022, 41, 2209056–2209068 CAS.
- C. Hu, Z. Zhang, S. Liu, X. Liu and M. Pang, ACS Appl. Mater. Interfaces, 2019, 11, 23072–23082 CrossRef CAS PubMed.
- C. Hu, L. Cai, S. Liu and M. Pang, Chem. Commun., 2019, 55, 9164–9167 RSC.
- L. Li, W. Fang, P. Zhang, J. Bi, Y. He, J. Wang and W. Su, J. Mater. Chem. A, 2016, 4, 12402–12406 RSC.
- Y. He, J. Zhao, Y.-T. Sham, S. Gao, M. Pan, Q. Chen, G. Huang, P. K. Wong and J. Bi, ACS Sustainable Chem. Eng., 2023, 11, 17552–17563 CrossRef CAS.
- Y.-P. Zhang, H.-L. Tang, H. Dong, M.-Y. Gao, C.-C. Li, X.-J. Sun, J.-Z. Wei, Y. Qu, Z.-J. Li and F.-M. Zhang, J. Mater. Chem. A, 2020, 8, 4334–4340 RSC.
- Y. Zhang, Y. Hu, J. Zhao, E. Park, Y. Jin, Q. Liu and W. Zhang, J. Mater. Chem. A, 2019, 7, 16364–16371 RSC.
- P. Sarkar, I. H. Chowdhury, A. Chakraborty, M. Goswami, M. K. Naskar, A. Khan and S. M. Islam, Ind. Eng. Chem. Res., 2024, 63, 5591–5607 CrossRef CAS.
- Z. Liang, R. Shen, P. Zhang, Y. Li, N. Li and X. Li, Chin. J. Catal., 2022, 43, 2581–2591 CrossRef CAS.
- D. Lin, P. Duan, W. Yang, X. Huang, Y. Zhao, C. Wang and Q. Pan, Chem. Eng. J., 2022, 430, 132817 CrossRef CAS.
- Y.-N. Gong, J.-H. Mei, W.-J. Shi, J.-W. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2024, 63, e202318735 CrossRef CAS PubMed.
- H. Xue, Z. Bi, J. Cheng, S. Xiong and Y. Wang, Ind. Eng. Chem. Res., 2021, 60, 8687–8695 CrossRef CAS.
- Y. Peng, M. Zhao, B. Chen, Z. Zhang, Y. Huang, F. Dai, Z. Lai, X. Cui, C. Tan and H. Zhang, Adv. Mater., 2018, 30, 1705454 CrossRef PubMed.
- D. Sun and D.-P. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 20589–20595 CrossRef CAS PubMed.
- M.-Y. Zhang, J.-K. Li, R. Wang, S.-N. Zhao, S.-Q. Zang and T. C. W. Mak, Adv. Sci., 2021, 8, 2101884 CrossRef CAS PubMed.
- G. Yuan, L. Tan, P. Wang, S. Feng, H. Tang, G. Wang, C. Wang and H. Yan, Mater. Lett., 2022, 325, 132863 CrossRef CAS.
- K. S. Fu, M. R. Allen and R. K. Archibald, Sustainability, 2015, 7, 15284–15301 CrossRef.
- S. Wang, W. Liao, H. Su, S. Pang, C. Yang, Y. Fu and Y. Zhang, Energy Fuels, 2023, 37, 1633–1656 CrossRef CAS.
- K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2017, 9, 1523–1544 CrossRef CAS.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- C. Gao, X. Zhang, M. Zhang, X. Wu, R. Chen, C. Zhang, C. Sun, Y. Du, Q. H. Xu and B. Hu, ACS Appl. Energy Mater., 2023, 6, 9427–9433 CrossRef CAS.
- Y. Xiao, K. Wang, W. Dong and L. Li, Polymer, 2024, 300, 126980 CrossRef CAS.
- W. Dong, Y. Xiao, Z. Qin, B. Qiao and L. Li, J. Mater. Chem. A, 2023, 11, 14760–14767 RSC.
- L. Hao, R. Shen, C. Qin, N. Li, H. Hu, G. Liang and X. Li, Sci. China Mater., 2024, 67, 504–513 CrossRef CAS.
- S. Wang, T. Wu, S. Wu, J. Guo, T. He, Y. Wu, W. Yuan, Z. Zhang, Y. Hua and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202311082 CrossRef CAS PubMed.
- J. X. Guo, Z. Y. Wang, C. Q. Han, S. Sun, L. Wang, G. Lu and X. Y. Liu, J. Mater. Chem. C, 2024, 12, 7741–7747 RSC.
- X. Ren, M. Wen, X. Hou, J. Sun, F. Bai and Y. Li, Chem. Commun., 2024, 60, 4423–4426 RSC.
- S. Li, R. Ma, S. Xu, T. Zheng, G. Fu, Y. Wu, Z. Liao, Y. Kuang, Y. Hou, D. Wang, P. S. Petkov, K. Simeonova, X. Feng, L. Z. Wu, X. B. Li and T. Zhang, J. Am. Chem. Soc., 2022, 144, 13953–13960 CrossRef CAS PubMed.
- Y. Zhong, W. Dong, S. Ren and L. Li, Adv. Mater., 2024, 36, 2308251 CrossRef CAS PubMed.
- H. He, R. Shen, P. Zhang, G. Liang and X. Li, J. Mater. Chem. A, 2023, 12, 227–232 RSC.
- G. Fu, D. Yang, S. Xu, S. Li, Y. Zhao, H. Yang, D. Wu, P. S. Petkov, Z. A. Lan, X. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 1318–1325 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- Q. Gao, L. Bai, X. Zhang, P. Wang, P. Li, Y. Zeng, R. Zou and Y. Zhao, Chin. J. Chem., 2015, 33, 90–94 CrossRef CAS.
- R. Gomes and A. Bhaumik, RSC Adv., 2016, 6, 28047–28054 RSC.
- W. Weng and J. Guo, Nat. Commun., 2022, 13, 5768 CrossRef CAS PubMed.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X. Y. Gao, L. Gao, L. Qiao, X. B. Li, L. Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S. W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CrossRef CAS PubMed.
- G. B. Wang, H. P. Xu, K. H. Xie, J. L. Kan, J. Fan, Y. J. Wang, Y. Geng and Y. B. Dong, J. Mater. Chem. A, 2023, 11, 4007–4012 RSC.
- S. Ghosh, H. Küçükkeçeci, R. P. Paitandi, V. Weigelt, V. Dippold, S. Seki and A. Thomas, J. Mater. Chem. A, 2023, 12, 247–255 RSC.
- S. Ghosh, Y. Tsutsui, T. Kawaguchi, W. Matsuda, S. Nagano, K. Suzuki, H. Kaji and S. Seki, Chem. Mater., 2022, 34, 736–745 CrossRef CAS.
- Y. Wang, Z. Qiao, H. Li, R. Zhang, Z. Xiang, D. Cao and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202404726 CrossRef CAS PubMed.
- S. Wang, L. Da, J. Hao, J. Li, M. Wang, Y. Huang, Z. Li, Z. Liu and D. Cao, Angew. Chem., Int. Ed., 2021, 60, 9321–9325 CrossRef CAS PubMed.
- J. W. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem., 2019, 3, 331–341 CrossRef CAS.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- H. Chen, A. M. Gardner, G. Lin, W. Zhao, X. Wang, M. Bahri, N. D. Browning, X. Xu and X. Li, J. Phys. Chem. C, 2023, 127, 14137–14145 CrossRef CAS.
- Z. Xiong, B. Sun, H. Zou, R. Wang, Q. Fang, Z. Zhang and S. Qiu, J. Am. Chem. Soc., 2022, 144, 6583–6593 CrossRef CAS PubMed.
- A. Giri, Y. Khakre, G. Shreeraj, T. K. Dutta, S. Kundu and A. Patra, J. Mater. Chem. A, 2022, 10, 17077–17121 RSC.
- S. Karak, V. Stepanenko, M. A. Addicoat, P. Keßler, S. Moser, F. Beuerle and F. Würthner, J. Am. Chem. Soc., 2022, 144, 17661–17670 CrossRef CAS PubMed.
- D. L. Ashford, B. D. Sherman, R. A. Binstead, J. L. Templeton and T. J. Meyer, Angew. Chem., Int. Ed., 2015, 54, 4778–4781 CrossRef CAS PubMed.
- F. Li, K. Fan, L. Wang, Q. Daniel, L. Duan and L. Sun, ACS Catal., 2015, 5, 3786–3790 CrossRef CAS.
- R. A. Borse, Y. X. Tan, J. Lin, E. Zhou, Y. Hui, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202318136 CrossRef CAS PubMed.
- W. Lin, J. Lin, X. Zhang, L. L. Zhang, R. A. Borse and Y. B. Wang, J. Am. Chem. Soc., 2023, 145, 18141–18147 CrossRef CAS PubMed.
- X. Yan, S. Lyu, X. Q. Xu, W. Chen, P. Shang, Z. Yang, G. Zhang, W. Chen, Y. Wang and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202201900 CrossRef CAS PubMed.
- X. Li, Q. Yang, Y. Yuan, Y. Shama and H. Yan, Small, 2024, 2401168 CrossRef CAS PubMed.
- E. Zhou, X. Zhang, L. Zhu, E. Chai, J. Chen, J. Li, D. Yuan, L. Kang, Q. Sun and Y. Wang, Sci. Adv., 2024, 10, eadk8564 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS.
- Z. Li, C. Liu, Q. Deng and W. Deng, Adv. Funct. Mater., 2024, 2402676 CrossRef CAS.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
- W.-K. Han, W. Yuan, Z.-G. Gu and Y. Zhao, ACS Mater. Lett., 2024, 6, 2276–2294 CrossRef CAS.
- R. Shen, C. Qin, L. Hao, X. Li, P. Zhang and X. Li, Adv. Mater., 2023, 35, 2305397 CrossRef CAS PubMed.
- Y. Yang, X. Chu, H. Y. Zhang, R. Zhang, Y. H. Liu, F. M. Zhang, M. Lu, Z. Di Yang and Y. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed.
- J. Cheng, Y. Wu, W. Zhang, J. Zhang, L. Wang, M. Zhou, F. Fan, X. Wu and H. Xu, Adv. Mater., 2024, 36, 2305313 CrossRef CAS PubMed.
- M. W. Jones, G. P. Peters, T. Gasser, R. M. Andrew, C. Schwingshackl, J. Gütschow, R. A. Houghton, P. Friedlingstein, J. Pongratz and C. Le Quéré, Sci. Data, 2023, 10, 155 CrossRef CAS PubMed.
- A. L. Burrell, J. P. Evans and M. G. De Kauwe, Nat. Commun., 2020, 11, 3853 CrossRef CAS PubMed.
- G. AL-khulaidi, Y. Sun, A. G. Alareqi, A.-W. Ibrahim, A. Magaji and X. Zhang, Energy Fuels, 2024, 38, 8355–8384 CrossRef CAS.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- A. Babin, C. Vaneeckhaute and M. C. Iliuta, Biomass Bioenergy, 2021, 146, 105968 CrossRef CAS.
- J. Lv, J. Xie, A. G. A. Mohamed, X. Zhang, Y. Feng, L. Jiao, E. Zhou, D. Yuan and Y. Wang, Nat. Rev. Chem., 2022, 7, 91–105 CrossRef PubMed.
- H. Li, A. Dilipkumar, S. Abubakar and D. Zhao, Chem. Soc. Rev., 2023, 52, 6294–6329 RSC.
- H. Lin, Y. Liu, Z. Wang, L. Ling, H. Huang, Q. Li, L. Cheng, Y. Li, J. Zhou, K. Wu, J. Zhang and T. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202214142 CrossRef CAS PubMed.
- W. Zhou, X. Wang, W. Zhao, N. Lu, D. Cong, Z. Li, P. Han, G. Ren, L. Sun, C. Liu and W.-Q. Deng, Nat. Commun., 2023, 14, 6971 CrossRef CAS PubMed.
- T. Skorjanc, D. Shetty, M. E. Mahmoud, F. Gandara, J. I. Martinez, A. K. Mohammed, S. Boutros, A. Merhi, E. O. Shehayeb, C. A. Sharabati, P. Damacet, J. Raya, S. Gardonio, M. Hmadeh, B. R. Kaafarani and A. Trabolsi, ACS Appl. Mater. Interfaces, 2022, 14, 2015–2022 CrossRef CAS PubMed.
- Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS PubMed.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- Q. Pan, M. Abdellah, Y. Cao, W. Lin, Y. Liu, J. Meng, Q. Zhou, Q. Zhao, X. Yan, Z. Li, H. Cui, H. Cao, W. Fang, D. A. Tanner, M. Abdel-Hafiez, Y. Zhou, T. Pullerits, S. E. Canton, H. Xu and K. Zheng, Nat. Commun., 2022, 13, 845 CrossRef CAS PubMed.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- P. Sarkar, A. Hazra Chowdhury, S. Riyajuddin, S. Ghosh and S. M. Islam, React. Chem. Eng., 2023, 8, 365–376 RSC.
- Y. Lan, W. Yan, N. Yang, G. Wang, Y. Bi, C. Tian, H. Liu and X. Zhu, Small, 2023, 19, 2208118 CrossRef PubMed.
- K. Lei, D. Wang, L. Ye, M. Kou, Y. Deng, Z. Ma, L. Wang and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS PubMed.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- J. X. Cui, L. J. Wang, L. Feng, B. Meng, Z. Y. Zhou, Z. M. Su, K. Wang and S. Liu, J. Mater. Chem. A, 2021, 9, 24895–24902 RSC.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, R. Banerjee and T. Heine, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- J. Zhou, W. C. Chen, C. Y. Sun, L. Han, C. Qin, M. M. Chen, M. X. L. Wang, E. B. Wang and Z. M. Su, J. Mater. Chem. A, 2018, 6, 21596–21604 RSC.
- X. Yu, K. Gong, S. Tian, G. Gao, J. Xie and X. Jin, J. Mater. Chem. A, 2023, 11, 5627–5635 RSC.
- Z. Shan, M. Wu, D. Zhu, X. Wu, K. Zhang, R. Verduzco and G. Zhang, J. Am. Chem. Soc., 2022, 144, 5728–5733 CrossRef CAS PubMed.
- G. Goor, J. Glenneberg and S. Jacobi, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 7th edn, 2012, pp. 393–427 Search PubMed.
- L. Xie, X. Wang, Z. Zhang, Y. Ma, T. Du, R. Wang and J. Wang, Small, 2023, 19, 2301007 CrossRef CAS PubMed.
- W. Chu, W. Wang, Y. Deng and C. Peng, Environ. Sci.: Water Res. Technol., 2022, 8, 2819–2842 RSC.
- Q. Miao, M. Liu, Y.-Y. Mou, Y.-F. Zhang, Q. Li, Z.-J. Cao and W. Jiang, ACS Sustainable Chem. Eng., 2024, 12, 5596–5607 CrossRef CAS.
- T. Liu, Z. Pan, J. J. M. Vequizo, K. Kato, B. Wu, A. Yamakata, K. Katayama, B. Chen, C. Chu and K. Domen, Nat. Commun., 2022, 13, 1034 CrossRef CAS PubMed.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 CrossRef CAS PubMed.
- C. Krishnaraj, H. S. Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107–20116 CrossRef CAS PubMed.
- J. Sun, H. S. Jena, C. Krishnaraj, K. S. Rawat, S. Abednatanzi, J. Chakraborty, A. Laemont, W. Liu, H. Chen, Y.-Y. Liu, K. Leus, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Angew. Chem., Int. Ed., 2023, 62, e202216719 CrossRef CAS PubMed.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- F. Liu, P. Zhou, Y. Hou, H. Tan, Y. Liang, J. Liang, Q. Zhang, S. Guo, M. Tong and J. Ni, Nat. Commun., 2023, 14, 4344 CrossRef CAS PubMed.
- Z. Zhou, M. Sun, Y. Zhu, P. Li, Y. Zhang, M. Wang and Y. Shen, Appl. Catal., B, 2023, 334, 122862 CrossRef CAS.
- E. Zhou, F. Wang, X. Zhang, Y. Hui and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202400999 CrossRef CAS PubMed.
- J.-P. Liao, M. Zhang, P. Huang, L.-Z. Dong, T.-T. Ma, G.-Z. Huang, Y.-F. Liu, M. Lu, S.-L. Li and Y.-Q. Lan, ACS Catal., 2024, 14, 3778–3787 CrossRef CAS.
- P. Li, H. Zhao, R. Ji, W. Chi, X. Sun, Y. Dong and Y. Zhu, Catal. Sci. Technol., 2024, 14, 2470–2478 RSC.
- H. Yu, F. Zhang, Q. Chen, P.-K. Zhou, W. Xing, S. Wang, G. Zhang, Y. Jiang and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202402297 CrossRef CAS PubMed.
- T. Yang, D. Zhang, A. Kong, Y. Zou, L. Yuan, C. Liu, S. Luo, G. Wei and C. Yu, Angew. Chem., Int. Ed., 2024, 63, e202404077 CrossRef CAS PubMed.
- T. Xu, Z. Wang, W. Zhang, S. An, L. Wei, S. Guo, Y. Huang, S. Jiang, M. Zhu, Y.-B. Zhang and W.-H. Zhu, J. Am. Chem. Soc., 2024, 146, 20107–20115 CrossRef CAS PubMed.
- K. A. Scott, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2022, 65, 7044–7072 CrossRef CAS PubMed.
- F. Shahidi and C.-T. Ho, Phenolic Compounds in Foods and Natural Health Products, ACS Symposium Series, American Chemical Society, Washington, DC, 2005 Search PubMed.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS.
- H.-P. Liang, Q. Chen and B.-H. Han, ACS Catal., 2018, 8, 5313–5322 CrossRef CAS.
- M. H. Muhammad, X.-L. Chen, Y. Liu, T. Shi, Y. Peng, L. Qu and B. Yu, ACS Sustainable Chem. Eng., 2020, 8, 2682–2687 CrossRef CAS.
- S. Bi, P. Thiruvengadam, S. Wei, W. Zhang, F. Zhang, L. Gao, J. Xu, D. Wu, J.-S. Chen and F. Zhang, J. Am. Chem. Soc., 2020, 142, 11893–11900 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- R. Paul, S. C. Shit, H. Mandal, J. Rabeah, S. S. Kashyap, Y. Nailwal, D. B. Shinde, Z. Lai and J. Mondal, ACS Appl. Nano Mater., 2021, 4, 11732–11742 CrossRef CAS.
- G. Xiao, W. Li, T. Chen, W.-B. Hu, H. Yang, Y. A. Liu and K. Wen, Eur. J. Org. Chem., 2021, 3986–3991 CrossRef CAS.
- B. Luo, Y. Zhang, Y. Chen and J. Huo, Mater. Adv., 2022, 3, 4699–4706 RSC.
- K. Verma, M. A. Addicoat and K. R. J. Thomas, ACS Appl. Polym. Mater., 2024, 6, 3909–3917 CrossRef CAS.
- A. Zadehnazari, A. Khosropour, A. A. Altaf, A. S. Rosen and A. Abbaspourrad, Adv. Mater., 2024, 36, 2311042 CrossRef CAS PubMed.
- A. S. Surur, L. Schulig and A. Link, Arch. Pharm. Chem. Life Sci., 2019, 352, e1800248 Search PubMed.
- E. Wojaczyńska and J. Wojaczyńsk, Curr. Opin. Chem. Biol., 2023, 76, 102340 CrossRef PubMed.
- X. Yu, Y. Liu, Y. Li and Q. Wang, J. Agric. Food Chem., 2016, 64, 3034–3040 CrossRef CAS PubMed.
- P. Li, L. Wang and X. Wang, J. Heterocycl. Chem., 2021, 58, 28–39 CrossRef CAS.
- B. Smolkin, N. Levi, N. Karton-Lifshin, L. Yehezkel, Y. Zafrani and I. Columbus, J. Org. Chem., 2018, 83, 13949–13955 CrossRef CAS PubMed.
- S. Zhao, Y. Zhu, H. Xi, M. Han, D. Li, Y. Li and H. Zhao, J. Environ. Chem. Eng., 2020, 8, 104221 CrossRef CAS.
- B. Tanriover, S. M. S. Subasinghe and N. P. Mankad, ACS Catal., 2024, 14, 9323–9327 CrossRef CAS.
- F. Lina, Y. Zhang, L. Wang, Y. Zhang, D. Wang, M. Yang, J. Yang, B. Zhang, Z. Jiang and C. Li, Appl. Catal., B, 2012, 127, 363–370 CrossRef.
- H. Wang, G. W. Wagner, A. X. Lu, D. L. Nguyen, J. H. Buchanan, P. M. McNutt and C. J. Karwacki, ACS Appl. Mater. Interfaces, 2018, 10, 18771–18777 CrossRef CAS PubMed.
- A. Samokhvalov, ChemPhysChem, 2011, 12, 2870–2885 CrossRef CAS PubMed.
- Q. Li, X. Lan, G. An, L. Ricardez-Sandoval, Z. Wang and G. Bai, ACS Catal., 2020, 10, 6664–6675 CrossRef CAS.
- X. Wang, S. Zhang, X. Li, Z. Zhan, B. Tan, X. Lang and S. Jin, J. Mater. Chem. A, 2021, 9, 16405–16410 RSC.
- Y. Li, T.-X. Luan, K. Cheng, D. Zhang, W. Fan, P.-Z. Li and Y. Zhao, ACS Mater. Lett., 2022, 4, 1160–1167 CrossRef CAS.
- X. Kan, J.-C. Wang, Z. Chen, J.-Q. Du, J.-L. Kan, W.-Y. Li and Y.-B. Dong, J. Am. Chem. Soc., 2022, 144, 6681–6686 CrossRef CAS PubMed.
- M. Xu, J. Zhang, L. Liu, X. Cheng, J. Hu, Y. Sha, Z. Su and Y. Wang, Chem. Commun., 2022, 58, 6324–6327 RSC.
- Y. Wang, F. Huang, W. Sheng, X. Miao, X. Li, X.-K. Gu and X. Lang, Appl. Catal., B, 2023, 338, 123070 CrossRef CAS.
- F. Zhang, Y. Wang, H. Zhao, X. Dong, X.-K. Gu and X. Lang, ACS Appl. Mater. Interfaces, 2024, 16, 8772–8782 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, M. Zhang and X. Wang, Chem. Sci., 2013, 4, 3244–3248 RSC.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- Z. Shen, Y. Hu, B. Li, Y. Zou, S. Li, G. W. Busser, X. Wang, G. Zhao and M. Muhler, J. Energy Chem., 2021, 62, 338–350 CrossRef CAS.
- G. Lu, X. Huang, Y. Li, G. Zhao, G. Pang and G. Wang, J. Energy Chem., 2020, 43, 8–15 CrossRef.
- S. Trenker, L. Grunenberg, T. Banerjee, G. Savasci, L. M. Poller, K. I. M. Muggli, F. Haase, C. Ochsenfeld and B. V. Lotsch, Chem. Sci., 2021, 12, 15143–15150 RSC.
- L. Liao, D. Ditz, F. Zeng, M. Alves Favaro, A. Iemhoff, K. Gupta, H. Hartmann, C. Szczuka, P. Jakes, P. J. C. Hausoul, J. Artz and R. Palkovits, ChemistrySelect, 2020, 5, 14438–14446 CrossRef CAS.
- P. L. Wang, S. Y. Ding, Z. C. Zhang, Z. P. Wang and W. Wang, J. Am. Chem. Soc., 2019, 141, 18004–18008 CrossRef CAS PubMed.
- K. Zhang, G. Lu, Z. Xi, Y. Li, Q. Luan and X. Huang, Chin. Chem. Lett., 2021, 32, 2207–2211 CrossRef CAS.
- H. Dai, H. Li and Q. Yang, Microporous Mesoporous Mater., 2022, 342, 112121 CrossRef CAS.
- H. Li, H. Liu, C. Li, J. Liu, J. Liu and Q. Yang, J. Mater. Chem. A, 2020, 8, 18745–18754 RSC.
- S. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443–2465 CrossRef CAS.
- M. Damaceanu, C. Constantin and L. Marin, Dyes Pigm., 2016, 134, 382–396 CrossRef CAS.
- K.-i. Yamada and K. Tomioka, Chem. Rev., 2008, 108, 2874–2886 CrossRef CAS PubMed.
- J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed.
- C. S. Marques and A. J. Burke, ChemCatChem, 2011, 3, 635–645 CrossRef CAS.
- J. Feng, J. Cheng, J. Pang, M. Tang, Z. Liu, C. Rong and R. Tan, Catal. Sci. Technol., 2022, 12, 6865–6874 RSC.
- S. Li, L. Li, Y. Li, L. Dai, C. Liu, Y. Liu, J. Li, J. Lv, P. Li and B. Wang, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS.
- S. Liu, Q. Su, W. Qi, K. Luo, X. Sun, H. Ren and Q. Wu, Catal. Sci. Technol., 2022, 12, 2837–2845 RSC.
- H. He, X. Fang, D. Zhai, W. Zhou, Y. Li, W. Zhao, C. Liu, Z. Li and W. Deng, Chem. – Eur. J., 2021, 27, 14390–14395 CrossRef CAS PubMed.
- P. Li, X. Dong, Y. Zhang, X. Lang and C. Wang, Mater. Today Chem., 2022, 25, 100953 CrossRef CAS.
- R. Chen, J. L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- L. Wang, J. Chakraborty, M. Deng, J. Sun and P. Van Der Voort, ChemCatChem, 2024, e202400200 CrossRef CAS.
- S. Wu, Y. F. Zhang, H. Ding, X. Li and X. Lang, J. Colloid Interface Sci., 2022, 610, 446–454 CrossRef CAS PubMed.
- N. Sun, Y. Jin, H. Wang, B. Yu, R. Wang, H. Wu, W. Zhou and J. Jiang, Chem. Mater., 2022, 34, 1956–1964 CrossRef CAS.
- F. Chu, G. Hai, D. Zhao, S. Liu, Y. Hu, G. Zhao, B. Peng, G. Wang and X. Huang, ACS Catal., 2023, 13, 13167–13180 CrossRef CAS.
- X. Li, S. Yang, F. Zhang, L. Zheng and X. Lang, Appl. Catal., B, 2022, 303, 120846 CrossRef CAS.
- K. Xiong, Y. Wang, F. Zhang, X. Li and X. Lang, Appl. Catal., B, 2023, 322, 122135 CrossRef CAS.
- L. Y. Yin, Y. N. Zhao, Y. M. Xing, H. Q. Tan, Z. L. Lang, W. K. Ho, Y. H. Wang and Y. G. Li, Chem. Eng. J., 2021, 419, 129984 CrossRef CAS.
- Y. Fang, Y. Liu, H. Huang, J. Sun, J. Hong, F. Zhang, X. Wei, W. Gao, M. Shao, Y. Guo, Q. Tang and Y. Liu, Nat. Commun., 2024, 15, 4856 CrossRef CAS PubMed.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- K. M. Altus and J. A. Love, Commun. Chem., 2021, 4, 173 CrossRef PubMed.
- H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed.
- B. Chen, L.-Z. Wu and C.-H. Tung, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS PubMed.
- D. Mazzarella, G. E. M. Crisenza and P. Melchiorre, J. Am. Chem. Soc., 2018, 140, 8439–8443 CrossRef CAS PubMed.
- C. L. Baumberger, V. Z. Valley and M. B. Chambers, Chem. Commun., 2024, 60, 6901–6904 RSC.
- Z. Chai, Angew. Chem., Int. Ed., 2024, 63, e202316444 CrossRef CAS PubMed.
- M. Tian, S. Liu, X. Bu, J. Yu and X. Yang, Chem. – Eur. J., 2020, 26, 369–373 CrossRef CAS PubMed.
- K. Cai, W. Wang, J. Zhang, L. Chen, L. Wang, X. Zhu, Z. Yu, Z. Wu and H. Zhou, J. Mater. Chem. A, 2022, 10, 7165–7172 RSC.
- G. Kumara, R. S. Pillaic, N.-u. H. Khan and S. Neogi, Appl. Catal., B, 2021, 292, 120149 CrossRef.
- B. Ma, X. Yang, J. Yuan, X. Yang, D. Han, K. Zhao, C. Lin, L. Wang, G. Liu and L. Mi, Appl. Catal., A, 2023, 666, 119403 CrossRef CAS.
- F. Yang, C. Li, C. Xu, J. Kan, B. Tian, H. Qu, Y. Guo, Y. Geng and Y. Dong, Chem. Commun., 2022, 58, 1530–1533 RSC.
- M. Tian, Y. Wang, X. Bu, Y. Wang and X. Yang, Catal. Sci. Technol., 2021, 11, 4272–4279 RSC.
- Z. Wang, P. Yeary, Y. Fan and W. Lin, Chem. Sci., 2024, 15, 4920–4925 RSC.
- X. Kang, X. Wu, X. Han, C. Yuan, Y. Liu and Y. Cui, Chem. Sci., 2020, 11, 1494–1502 RSC.
- H. Pang, G. Liu, D. Huang, Y. Zhu, X. Zhao, W. Wang and Y. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202313520 CrossRef CAS PubMed.
- S. Li, W. Wei, K. Chi, C. T. J. Ferguson, Y. Zhao and K. A. I. Zhang, J. Am. Chem. Soc., 2024, 146, 12386–12394 CrossRef CAS PubMed.
- A. Basak, S. Karak and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 7592–7599 CrossRef CAS PubMed.
- R. Emadi, A. B. Nekoo, F. Molaverdi, Z. Khorsandi, R. Sheibani and H. Sadeghi-Aliabadi, RSC Adv., 2023, 13, 18715–18733 RSC.
- J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- J. Rayadurgam, S. Sana, M. Sasikumar and Q. Gu, Org. Chem. Front., 2021, 8, 384–414 RSC.
- C.-H. Lim, M. Kudisch, B. Liu and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 7667–7673 CrossRef CAS PubMed.
- A. Vijeta, C. Casadevall and E. Reisner, Angew. Chem., Int. Ed., 2022, 61, e202203176 CrossRef CAS PubMed.
- T. Gandini, L. Dolcini, L. Di Leo, M. Fornara, A. Bossi, M. Penconi, A. Dal Corso, C. Gennari and L. Pignataro, ChemCatChem, 2022, 14, e202200990 CrossRef CAS.
- J. Li, C.-Y. Huang and C.-J. Li, Chem, 2022, 8, 2419–2431 CAS.
- M. Kuai, Z. Jia, L. Chen, S. Gao and W. Fang, Eur. J. Org. Chem., 2024, e202300933 CrossRef CAS.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- M. Traxler, S. Gisbertz, P. Pachfule, J. Schmidt, J. Roeser, S. Reischauer, J. Rabeah, B. Pieber and A. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202117738 CrossRef CAS PubMed.
- Y.-L. Li, F. Wang, J. J. Vittal, P. Jin, S.-L. Huang and G.-Y. Yang, J. Mater. Chem. A, 2024, 12, 9164–9172 RSC.
- Z. Li, S. Qiu, Y. Song, S. Huang, J. Gao, L. Sun and J. Hou, Sci. Bull., 2022, 67, 1971–1981 CrossRef CAS PubMed.
- C. Song, T. Yan, X. Cui, H. Chen, R. Ye and X. Zhang, Appl. Catal., A, 2023, 661, 119245 CrossRef CAS.
- M. Traxler, S. Reischauer, S. Vogl, J. Roeser, J. Rabeah, C. Penschke, P. Saalfrank, B. Pieber and A. Thomas, Chem. – Eur. J., 2023, 29, e202202967 CrossRef CAS PubMed.
- T. Zhou and M. Szostak, Catal. Sci. Technol., 2020, 10, 5702–5739 RSC.
- H. Zhang, P. Ruiz-Castillo, A. W. Schuppe and S. L. Buchwald, Org. Lett., 2020, 22, 5369–5374 CrossRef CAS PubMed.
- H. Zhang, P. Ruiz-Castillo and S. L. Buchwald, Org. Lett., 2018, 20, 1580–1583 CrossRef CAS PubMed.
- K. Dong, C. Pezzetta, Q.-C. Chen, A. Kaushansky, A. Agosti, G. Bergamini, R. Davidson and L. Amirav, ChemCatChem, 2022, 14, e202200477 CrossRef CAS.
- H.-C. Hu, Z.-P. Wang, L. Liang, X.-Y. Du, T. Li, J. Feng, T.-T. Xiao, Z.-M. Jin, S.-Y. Ding, Q. Liu, L.-Q. Lu, W.-J. Xiao and W. Wang, Chem. – Eur. J., 2024, 30, e202303476 CrossRef CAS PubMed.
- W. Dong, Y. Yang, Y. Xiang, S. Wang, P. Wang, J. Hu, L. Rao and H. Chen, Green Chem., 2021, 23, 5797–5805 RSC.
- Q. Lin, Y. Yusran, J. Xing, Y. Li, J. Zhang, T. Su, L. Yang, J. Suo, L. Zhang, Q. Li, H. Wang, Q. Fang, Z.-T. Li and D.-W. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 5869–5880 CrossRef CAS PubMed.
- K. Wang, H. Jiang, H. Liu, H. Chen and F. Zhang, ACS Catal., 2022, 12, 6068–6080 CrossRef CAS.
- Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC.
- M. Busch, M. D. Wodrich and C. Corminboeuf, ACS Catal., 2017, 7, 5643–5653 CrossRef CAS.
- A. Chen, Y. Han, R. Wu, B. Yang, L. Zhu and F. Zhu, Nat. Commun., 2024, 15, 5228 CrossRef CAS PubMed.
- J. Khamrai, I. Ghosh, A. Savateev, M. Antonietti and B. König, ACS Catal., 2020, 10, 3526–3532 CrossRef CAS.
- X. Zhou, X. Gao, M. Liu, Z. Gao, X. Qin, W. Xu, S. Ye, W. Zhou, H. Fan, J. Li, S. Fan, L. Yang, J. Fu, D. Xiao, L. Lin, D. Ma and S. Yao, Nat. Commun., 2022, 13, 4379 CrossRef CAS PubMed.
- S. Okumura, T. Takahashi, K. Torii and Y. Uozumi, Chem. – Eur. J., 2023, 29, e202300840 CrossRef CAS PubMed.
- T. Wan, L. Capaldo, J. Djossou, A. Staffa, F. J. de Zwart, B. de Bruin and T. Noël, Nat. Commun., 2024, 15, 4028 CrossRef CAS PubMed.
- Y. Yang, H. Niu, W. Zhao, L. Xu, H. Zhang and Y. Cai, RSC Adv., 2020, 10, 29402–29407 RSC.
- P. Chakrabortty, A. Das, A. H. Chowdhury, S. Ghosh, A. Khan and S. M. Islam, New J. Chem., 2021, 45, 4738–4745 RSC.
- A. López-Magano, B. Ortín-Rubio, I. Imaz, D. Maspoch, J. Alemán and R. Mas-Ballesté, ACS Catal., 2021, 11, 12344–12354 CrossRef PubMed.
- A. Jati, S. Dam, S. Kumar, K. Kumar and B. Maji, Chem. Sci., 2023, 14, 8624–8634 RSC.
- S. Sharma and A. Bhattacharya, Appl. Water Sci., 2017, 7, 1043–1067 CrossRef CAS.
- R. Kumar, M. Qureshi, D. K. Vishwakarma, N. Al-Ansari, A. Kuriqi, A. Elbeltagi and A. Saraswat, Case Stud. Chem. Environ. Eng., 2022, 6, 100219 CrossRef CAS.
- A. Saravanan, P. S. Kumar, S. Jeevanantham, S. Karishma, B. Tajsabreen, P. R. Yaashikaa and B. Reshma, Chemosphere, 2021, 280, 130595 CrossRef CAS PubMed.
- Water Treatment, https://www.cdc.gov/healthywater/drinking/public/water_treatment.html, accessed June 2024.
- Y. Hou, F. Liu, B. Zhang and M. Tong, Environ. Sci. Technol., 2022, 56, 16303–16314 CrossRef CAS PubMed.
- S. Ruidas, A. Chowdhury, A. Ghosh, A. Ghosh, S. Mondal, A. D. D. Wonanke, M. Addicoat, A. K. Das, A. Modak and A. Bhaumik, Langmuir, 2023, 39, 4071–4081 CrossRef CAS PubMed.
- X. Li, X. Chen, C. Li, Z. Xu and B. Jiang, New J. Chem., 2023, 47, 2659–2665 RSC.
- W. Lu, C. Wang, Y. Bai, C. Xie, Z. Zhang, W. Song and J. Wang, Environ. Sci.: Nano, 2024, 11, 229–240 RSC.
- M. Deng, L. Wang, Z. Wen, J. Chakraborty, J. Sun, G. Wang and P. Van Der Voort, Green Chem., 2024, 26, 3239–3248 RSC.
- X. Ren, M. Wen, X. Hou, J. Sun, F. Bai and Y. Li, Chem. Commun., 2024, 60, 4423–4426 RSC.
- H. Xu, Y. Xu, Y. Wang, Q. Wang, Y. Zhang, Q. Liao and K. Xi, ACS Appl. Nano Mater., 2024, 7, 3071–3081 CrossRef CAS.
- J. H. Wang, A. E. Hassan, A. M. Elewa and A. F. M. El-Mahdy, J. Mater. Chem. A, 2024, 12, 14005–14021 RSC.
- M. Bhadra, S. Kandambeth, M. K. Sahoo, M. Addicoat, E. Balaraman and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 6152–6156 CrossRef CAS PubMed.
- Y.-X. Chen, M. Zhang, S.-Z. Zhang, Z.-Q. Hao and Z.-H. Zhang, Green Chem., 2022, 24, 4071–4081 RSC.
- D. Chen, W. Chen, G. Zhang, S. Li, W. Chen, G. Xing and L. Chen, ACS Catal., 2022, 12, 616–623 CrossRef CAS.
- T. He, Z. Zhao, R. Liu, X. Liu, B. Ni, Y. Wei, Y. Wu, W. Yuan, H. Peng, Z. Jiang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 6057–6066 CrossRef CAS PubMed.
- Y. Liu, X. Jiang, L. Chen, Y. Cui, Q.-Y. Li, X. Zhao, X. Han, Y.-C. Zheng and X.-J. Wang, J. Mater. Chem. A, 2023, 11, 1208–1215 RSC.
- K.-K. Niu, T.-X. Luan, J. Cui, H. Liu, L.-B. Xing and P.-Z. Li, ACS Catal., 2024, 14, 2631–2641 CrossRef CAS.
- J.-R. Wang, K. Song, T.-X. Luan, K. Cheng, Q. Wang, Y. Wang, W. W. Yu, P.-Z. Li and Y. Zhao, Nat. Commun., 2024, 15, 1267 CrossRef CAS PubMed.
- X. Yang, Q. An, X. Li, Y. Fu, S. Yang, M. Liu, Q. Xu and G. Zeng, Nat. Commun., 2024, 15, 1889 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |