
Inducing preferential intercalation of Zn2+ in MnO2 with abundant oxygen defects for high-performance aqueous zinc-ion batteries†
Simin
Dai‡
a,
Xinyan
Zhuang‡
a,
Hongrun
Jin‡
a,
Ruixuan
Yang
b,
Yan
Wang
a,
Bei
Qi
a,
Wenhuan
Guo
a,
Kefeng
Xie
 c,
Zhimi
Hu
a,
Meilin
Liu
d and
Liang
Huang
*a
c,
Zhimi
Hu
a,
Meilin
Liu
d and
Liang
Huang
*a
aWuhan National Laboratory for Optoelectronics, School of Optical and Electronic Information, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: huangliang421@hust.edu.cn
bSchool of Materials Science and Engineering, Xi'an University of Technology, Xi'an 710048, China
cSchool of Chemistry and Chemical Engineering, Lanzhou Jiaotong University, Lanzhou, 730000, China
dSchool of Materials Science and Engineering, Georgia Institute of Technology, 771 Ferst Drive, Atlanta, Georgia 30332-0245, USA
First published on 10th October 2024
Abstract
Unfavorable proton intercalation leading to the generation and shedding of side reaction products is still a major challenge for the performance of manganese-based aqueous zinc-ion batteries (AZIBs). In this study, we present a porous oxygen-deficient MnO2 (Od-MnO2) synthesized through n-butyllithium reduction treatment to induce preferential Zn2+ intercalation, thereby effectively mitigating the adverse consequences of proton intercalation for high-performance AZIBs. Remarkably, Od-MnO2 as a cathode material for AZIBs exhibits a specific capacity of 341 mA h g−1 at 0.1 A g−1 and 139 mA h g−1 at 5 A g−1, and outstanding long-term stability with a capacity retention of 85.4% for over 1200 cycles at 1 A g−1. Moreover, the Zn/Od-MnO2 pouch cell displays decent durability with a capacity retention of ∼90% for over 200 cycles at 1C. Our study opens new opportunities for the rational design of high-performance cathode materials by regulating the electronic structure and optimizing the energy storage process for rechargeable AZIBs.
 Liang Huang | Dr Liang Huang is a principal investigator at the Wuhan National Laboratory for Optoelectronics (WNLO), Huazhong University of Science and Technology. His current interests focus on salt-assisted synthesis of 2D materials for energy conversion and storage. He published over 160 papers in journals including Science Advances, Advanced Materials, Nano Letters, ACS Energy Letters, ACS Nano, Advanced Energy Materials, etc., with an H-index of 63. He is an IAAM fellow, Chutian Scholars of Hubei Province, one of the top 2% scientists selected by Stanford–Elsevier (2019–2024), and has received the 2020 IAAM Young Scientist Medal and Nanomaterials 2022 Young Investigator Award. |
1. Introduction
Given the looming concern of energy crisis and environmental pollution, it is the trend of times to develop renewable and clean energy. Despite the potential of wind, solar, water, and tidal energy, their intermittent nature has spurred the search for low-cost, environmentally friendly and reliable electrochemical energy storage.1–5 Lithium-ion batteries (LIBs) have been at the forefront of energy storage due to their long lifespan and high energy density.6–10 Nevertheless, LIBs face challenges including constrained resource supply, potential safety hazards and environmental problems associated with flammable and toxic organic electrolytes.11–13 Fortunately, benefiting from the high natural abundance,14 high theoretical capacity (820 mA h g−1) of the Zn anode,15–17 good environmental compatibility, and high ionic conductivity of aqueous electrolytes, aqueous zinc-ion batteries (ZIBs) as viable alternatives show promise for significant advancements in next-generation large-scale energy storage systems.18–22To date, manganese-based materials, vanadium-based materials, and Prussian blue analogues stand as the most widely studied cathode materials for ZIBs.19,23–26 Out of these, MnO2 stands out for its high theoretical capacity (616 mA h g−1, contributed capacity of two electron transfer), high operating voltage, polymorphic structures, low cost, and eco-friendliness.27–29 Presently, there are four main reaction mechanisms in Zn-MnO2 batteries: (1) Zn2+ intercalation, (2) H+/Zn2+ co-intercalation, (3) chemical conversion reaction, and (4) deposition–dissolution mechanism.30–34 Among these mechanisms, H+/Zn2+ co-intercalation is the most typical. From the kinetic point of view, proton intercalation is easier than that of Zn2+, but the variation of H+ concentration leads to the formation and disappearance of side reaction products such as Zn4SO4(OH)6·nH2O (ZSH) or Znx(OTf)y(OH)2x−y·nH2O (ZOTH) on the surface of the cathode. This phenomenon is prone to electrolyte depletion and increased interfacial impedance, and in practice, the shedding of ZSH or ZOTH decreases the reversibility of the electrochemical behavior, resulting in faster capacity degradation and a shorter cycle life (Fig. 1a).30,35–39 It is generally believed that increasing the proportion of Zn2+ intercalation and decreasing the proportion of H+ intercalation can effectively reduce the accumulation of by-products and thus enhance the electrode's cycling stability. It is well known that oxygen defects can serve as shallow donors to regulate the electronic structure of metal oxides (especially in charge density distribution, metal valence state, and energy band structure). The optimized charge density distribution is beneficial for adjusting the interaction between metal oxides and transport ions. Furthermore, a change in the electronic structure promotes a deeper degree of electron delocalization, thereby boosting electron activity and enhancing material conductivity.40–42 Up to now, however, there is rare study to discuss the modulation of defect sites on ion selective intercalation, which will be critical for the rational. design of electrode materials.
 | ||
| Fig. 1 The schematic diagram of the ion intercalation process of (a) MnO2 and (b) Od-MnO2. | ||
In this work, we found that oxygen-deficient sites have stronger interactions with Zn2+ and may induce the preferential intercalation of Zn2+. In this way, an increase in the proportion of Zn2+ intercalation corresponds to a decrease in the proportion of H+ intercalation, which inhibits the generation and shedding of by-products and the accompanying cathode dissolution problems, and thus increases the reversibility of the electrochemical behavior (Fig. 1b). Then, δ-MnO2 with abundant oxygen defects (named Od-MnO2) was rationally designed and synthesized by the molten salt method followed by n-butyllithium treatment. The Od-MnO2 based cathode for AZIBs exhibits an outstanding rate capability (341 mA h g−1 at 0.1 A g−1 and 94 mA h g−1 at 10 A g−1) and remarkable cycling stability with a capacity of 275 mA h g−1 after 200 cycles at a low current density of 0.2 A g−1 (94.4% capacity retention), and retains 190.8 mA h g−1 even after 1200 cycles at 1 A g−1 (85.4% capacity retention).
2. Results and discussion
Firstly, we calculated the adsorption energies of Zn2+ and H+ on pristine MnO2 and Od-MnO2 using DFT calculations. As shown in Fig. S1a–e (ESI†), the Zn2+ adsorption energy of Od-MnO2 (−3.51 eV) is significantly higher than that of MnO2 (−3.06 eV), while the H+ adsorption energy is almost unchanged. This result verifies that oxygen-deficient sites have stronger interactions with Zn2+ than H+, which can induce preferential intercalation of Zn2+, and thus reduce the by-products generated by proton intercalation. Therefore, based on the simulation results, MnO2 with abundant oxygen defects is expected to exhibit superior electrochemical performance as a promising ZIB cathode material.As depicted in the schematic diagram of synthesis (Fig. 2a), MnO2 was first synthesized by the molten salt method and then treated with n-butyllithium to obtain Od-MnO2 nanosheets (detailed synthesis process is described in the ESI†). The X-ray diffraction (XRD) patterns of MnO2 and Od-MnO2 are shown in Fig. S2a and S2b (ESI†) which indicate that these two samples exhibit similar crystallographic structures of layered δ-MnO2 (JCPDS no. 52-0556).43,44 Compared with MnO2, the broader and weaker XRD peak features of Od-MnO2 indicate its poor crystallinity, which may be attributed to oxygen defects. The scanning electron microscopy (SEM) images (Fig. 2b and Fig. S3, ESI†) show that both MnO2 and Od-MnO2 exhibit an ultrathin two-dimensional nanosheet morphology, which can effectively shorten ion diffusion paths. The transmission electron microscopy (TEM) images (Fig. 2c and Fig. S4a, ESI†) show that numerous nanopores with sizes of 2–10 nm (Fig. S4b, ESI†) are uniformly dispersed on the Od-MnO2 nanosheet. In the high-resolution transmission electron microscopy (HRTEM) image (Fig. 2d), lattice fringes with a d-spacing of 0.24 nm can be clearly observed, corresponding to the (012) crystal plane of δ-MnO2. The HRTEM magnification and inverse fast Fourier transformation (FFT) diagrams related to the lattice spacing calculation of Od-MnO2 are shown in Fig. S5a–c, ESI.† The corresponding selected area electron diffraction (SAED) pattern (Fig. S6, ESI†) presents typical polycrystalline diffraction rings indexed to the (012), (015) and (110) crystal planes, matching well with the XRD results. The nitrogen adsorption–desorption measurement (Fig. 2e, Fig. S7a and b, ESI†) indicates that Od-MnO2 exhibits a higher Brunauer–Emmett–Teller (BET) specific surface area (150.5 m2 g−1) than MnO2 (92.2 m2 g−1), with nanopores mainly ranging from 2 to 10 nm, providing copious ion diffusion channels and active sites and promoting electrolyte penetration. The presence of abundant oxygen defects is verified by X-ray photoelectron spectroscopy (XPS) and electron paramagnetic resonance (EPR) spectroscopy. The high-resolution O 1s spectrum can be fitted into three peaks at 529.8, 531.4 and 532.9 eV assigned to the Mn–O–Mn bond, oxygen defects (Mn–O–H) and H–O–H bond, respectively (Fig. 2f).10,45–48 The ratio of oxygen defects (calculated from the integrated area in the fitted spectra) for Od-MnO2 (41.5%) is substantially higher than that of pristine MnO2 (18.8%), demonstrating the enhanced concentration of oxygen defect sites after n-butyllithium treatment. The Mn 2p spectrum in Fig. S8a (ESI†) demonstrates the negative shift of the Mn 2p peak position of Od-MnO2 compared to pristine MnO2, suggesting a decrease in Mn valence due to the introduction of oxygen defects. Moreover, the high-resolution Mn 2p spectrum of Od-MnO2 reveals the co-existence of Mn3+ (641.6 eV and 653.1 eV) and Mn4+ (642.8 eV and 654.5 eV) deconvoluted from the Mn 2p3/2 and Mn 2p1/2 orbit, respectively (Fig. S8a, ESI†).49,50 Meanwhile, as shown in Fig. S8b (ESI†), the Mn 3s multistate splitting energy (ΔE) in Od-MnO2 (5.01 eV) is larger than that of MnO2 (4.72 eV), further demonstrating that oxygen defects lower the Mn valence. The EPR spectrum (Fig. 2g) exhibits a stronger electron spin resonance signal at g = 2.0 for Od-MnO2 compared to MnO2, which proves the presence of rich oxygen defects in Od-MnO2.
 | ||
| Fig. 2 Material preparation and characterization. (a) The illustration of the synthesis procedures, (b) SEM image, (c) TEM image, and (d) HRTEM image of Od-MnO2. (e) The nitrogen adsorption–desorption isotherms, (f) O 1s high-resolution XPS spectra, and (g) EPR spectra of MnO2 and Od-MnO2. | ||
To investigate the effect of oxygen defects on the reaction kinetics, a coin cell was assembled using pristine MnO2 or Od-MnO2 as the cathode (electrode preparation details are given in the ESI†), Zn foil as the anode, and 2 M Zn (CF3SO3)2 + 0.1 M MnSO4 solution as the electrolyte. All the electrochemical tests in this paper were done in a two-electrode configuration. Fig. 3a displays the cyclic voltammetry (CV) curves of Zn/MnO2 and Zn/Od-MnO2 batteries where the pristine MnO2 or Od-MnO2 electrode is discharged first and then charged in the potential range from 0.8 V to 1.9 V at a scan rate of 0.1 mV s−1, in which two pairs of redox peaks at 1.184/1.598 V and 1.36/1.651 V for MnO2 and 1.203/1.596 V and 1.364/1.646 V for Od-MnO2 corresponding to the insertion/extraction processes of Zn2+ and H+ can be observed, respectively.48,51 It is clear that Od-MnO2 presents higher peak current densities and smaller overpotential gaps than MnO2, suggesting the improved reaction kinetics due to oxygen defects. The basic coincidence of CV curves (Fig. S9†) proves the high reversibility of Od-MnO2. The galvanostatic charge/discharge (GCD) curves of Zn/MnO2 and Zn/Od-MnO2 batteries at a current density of 0.1 A g−1 are displayed in Fig. 3b, demonstrating two obvious charging and discharging voltage plateaus, in agreement with the CV results. In general, it is believed that the first plateau in the MnO2 discharge curve corresponds to the intercalation of H+, while the second plateau is mainly generated by the intercalation of Zn2+. It is known that the capacity contribution of Zn2+ of Od-MnO2 is improved (Fig. 3b), which suggests that oxygen defects can promote the process of Zn2+ intercalation. According to the reaction equation:31,41,52
| xZn2+ + 2xe− + MnO2 → MnOOZnx | (1) |
 | ||
| Fig. 3 Electrochemical kinetics of Zn/MnO2 and Zn/Od-MnO2 batteries. (a) CV curves at 0.1 mV s−1. (b) Galvanostatic charge/discharge curves at 0.1 A g−1. (c) The percentage of Zn2+ intercalation contribution calculated by GCD and ICP. (d) CV curves at different scan rates, (e) Determination of the b value using the relationship between the peak current and scan rate, and (f) the corresponding percent of the pseudocapacitive contribution of the Zn/Od-MnO2 battery. (g) EIS spectra, with a larger view of the equivalent series resistance in the inset. (h) GITT curves and (i) the corresponding ion diffusion coefficients. | ||
To further analyze the electrochemical kinetics of MnO2 and Od-MnO2, the CV experiment at various scan rates from 0.1 to 0.5 mV s−1 was performed as illustrated in Fig. 3d and Fig. S11a (ESI†). The peak current (i) and scan rate (v) follow the relationship below:53,54
| i = avb | (2) |
| i = k1v + k2v1/2 | (3) |
To gain a comprehensive insight into the energy storage mechanism of Zn/Od-MnO2 batteries, ex situ measurements including XRD, Raman, XPS, and TEM elemental mapping were performed to analyze the changes in the chemical composition and electronic structure of Od-MnO2 electrodes. Fig. 4a shows the GCD profiles of the first and second cycles at 0.1 A g−1, along with the selected charge–discharge status for ex situ measurements. Ex situ XRD patterns of Od-MnO2 and MnO2 are displayed in Fig. 4b and Fig. S13 (ESI†). It is apparent that MnO2 shows obvious peaks of the Znx(OTf)y(OH)2x−y·nH2O (ZOTH) phase (Fig. S13, ESI†). While the ZOTH peaks of Od-MnO2 are much weaker than that of MnO2 during discharge, and the ZOTH peaks gradually decrease and almost disappear in Od-MnO2 when charging to 1.9 V (Fig. 4b), which further proves the preferential intercalation of Zn2+, thereby reducing the ZOTH produced by H+ intercalation.55,56 Moreover, the appearance/disappearance of the ZnMn2O4 phase (JCPDS no. 71-2499) indicates the reversible Zn2+ insertion/extraction.54,56 In addition, the morphology of Od-MnO2 nanosheets remains unchanged before and after cycling (Fig. S14, ESI†), which further demonstrates that zinc ions are reversibly intercalated/de-intercalated into Od-MnO2. The XRD characteristic peaks of Od-MnO2 remain (Fig. S15a†) and clear lattice fringes attributed to the (012) and (015) crystal planes can still be observed in HRTEM (Fig. S15b†) after discharge, proving that there are no obvious structural changes. Moreover, the high-resolution O 1s spectra (Fig. S16†) indicate that there are still abundant oxygen defects in Od-MnO2 after discharge, ensuring the effectiveness of the preferential Zn2+ intercalation induced by oxygen defects. In ex situ Raman spectra (Fig. 4e), a band of around 650 cm−1 assigned to the symmetric stretching vibration (Mn–O) of the MnO6 groups remains almost unchanged, suggesting the structural stability of Od-MnO2. Upon discharging and charging, the emergence and disappearance of a pair of bands between 300 and 400 cm−1 associated with Zn–O vibrations demonstrate the reversible insertion/extraction of Zn2+.51,57 The intercalation mechanism is further unveiled by ex situ XPS (Fig. 4d and e). The high-resolution Mn 2p analysis (Fig. 4d) reveals the change in the valence state of Mn during charging and discharging. The peak area of Mn3+ and Mn2+ apparently enlarges during discharge, implying the intercalation of Zn2+ into the Od-MnO2 electrode and the reduction of Mn4+.19 Then the peak area of Mn4+ gradually increases during charging along with the manganese which is oxidized to the initial state.57–60 The high-resolution Zn 2p spectra (Fig. 4e) reveal prominent peaks in the discharged state due to the intercalation of Zn2+ into the Od-MnO2 electrode, which weaken after charging caused by Zn2+ extraction.54,61 These phenomena are consistent with the elemental mapping results of Od-MnO2 in fully discharged/charged states (Fig. S14, ESI†), which shows obvious Zn signals together with Mn and O in the discharged state (Fig. S17a†) while showing very weak Zn signals in the charged state (Fig. S17b†), verifying successful Zn2+ insertion/extraction into Od-MnO2.
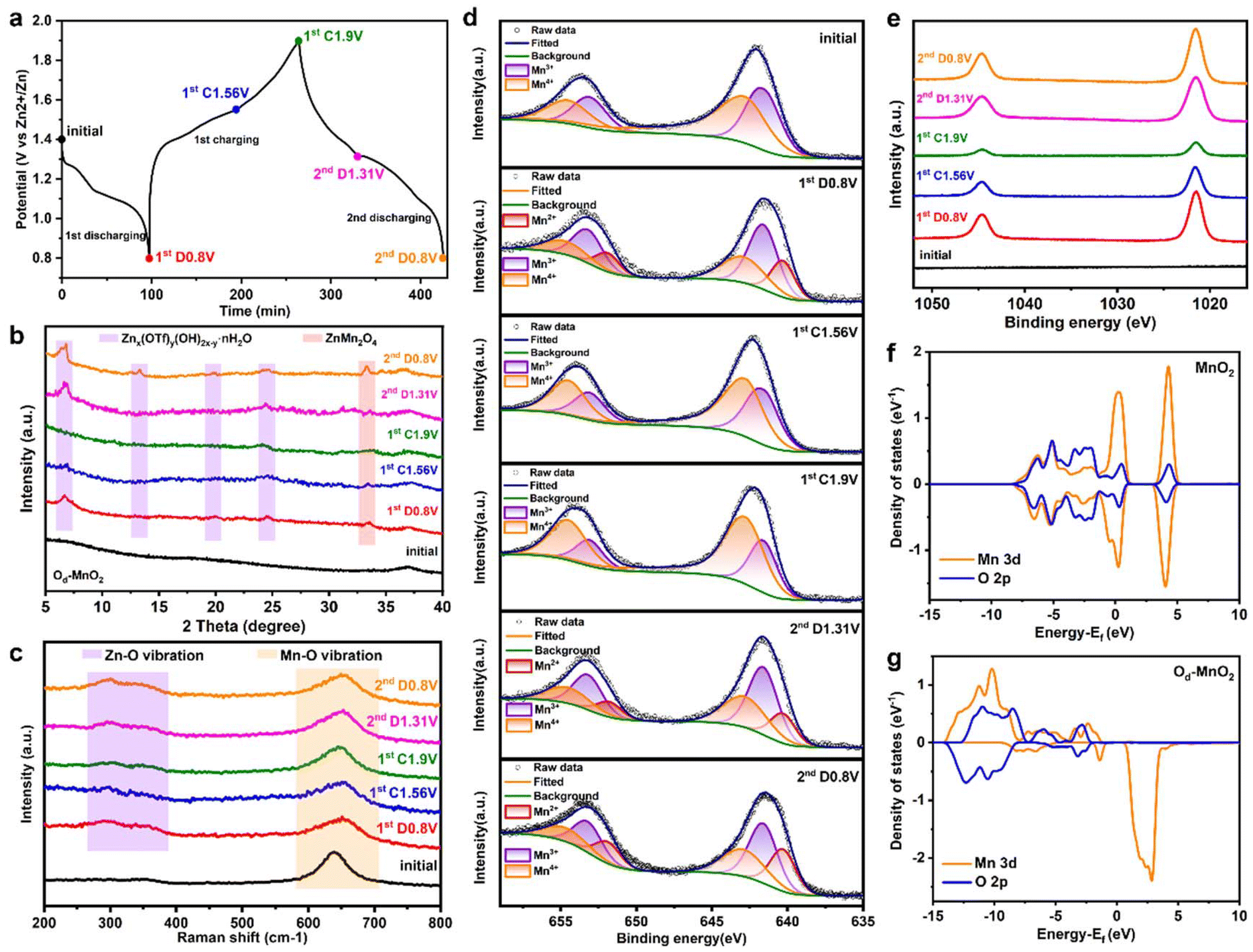 | ||
| Fig. 4 Energy storage mechanism of the Zn/Od-MnO2 battery. (a) GCD profiles of the first and second cycles at 0.1 A g−1. (b) The ex situ XRD patterns. (c) The ex situ Raman spectra. The ex situ high-resolution XPS spectra of (d) Mn 2p and (e) Zn 2p. The partial DOS of (f) MnO2 and (g) Od-MnO2. | ||
DFT calculations were done to unravel the underlying mechanism of improving electrochemical kinetics. The calculated partial density of states (PDOS) and charge density distribution of pristine MnO2 and Od-MnO2 are illustrated in Fig. 4f and g. Pristine MnO2 exhibits a significant bandgap at the Fermi level (Fig. 4f), indicating its semiconducting feature. The PDOS of Od-MnO2 (Fig. 4g) shifts to low energy levels and the bandgap narrows after introducing oxygen defects, corresponding to easier electron transition from the valence band to the conduction band, which effectively increases the conductivity and results in lower ohmic polarization and improved rate capability. Moreover, the charge density difference analysis (Fig. S18a and S18b, ESI†) reveals an altered charge density distribution with the introduction of oxygen defects, thereby accelerating electron transfer kinetics.40,62
The electrochemical performance was further elucidated with coin cells and pouch cells using pristine MnO2 or Od-MnO2 as a cathode, Zn foil as an anode, and 2 M Zn(CF3SO3)2 + 0.1 M MnSO4 solution as an electrolyte. Od-MnO2 displays remarkable rate capability, demonstrating high discharge capacities of 341, 306, 254, 217, 187, and 139 mA h g−1 at current densities of 0.1, 0.2, 0.5, 1, 2 and 5 A g−1, respectively (Fig. 5a and Fig. S19, ESI†). Even at an ultrahigh current density of 10 A g−1, Od-MnO2 can still deliver a capacity of 94 mA h g−1 As expected, Od-MnO2 exhibits considerably higher capacities than that of MnO2 at each current density. Upon reverting to a current density of up to 0.1 A g−1, 97.5% of the discharge capacity (332.4 mA h g−1) of Od-MnO2 is recovered. The superior rate performance could be attributed to the stabilization and excellent kinetics of Od-MnO2. To highlight the superiority and practical realization of the Od-MnO2 electrode, Ragone plots reflecting the relationship between the energy density and power density are shown in Fig. 5b. The Zn/Od-MnO2 battery delivers a maximum power density of 16 kW kg−1 and a maximum energy density of 532 W h kg−1 (based on the active mass of the Od-MnO2 electrode), which are better than those of most reported cathode materials for aqueous ZIBs, such as α-MnO2,63 CuHCF,64 Mn2O3,65 δ-MnO2,66 ZnHCF,67 β-MnO2,55 Zn0.25V2O5·nH2O,2 VS2,68 Todorokite,69 Od-Mn3O4@C,54 and K0.8Mn8O16,53 and is promising for energy storage applications. Furthermore, Od-MnO2 exhibits outstanding long-term cycling stability (Fig. 5c) with a discharge capacity of 186.7 mA h g−1 and a high-capacity retention of 85.4% for over 1200 cycles at a current density of 1 A g−1, while the Zn/MnO2 battery shows rapid capacity fading after 300 cycles. Remarkably, even at low current densities of 0.2A g−1 and 0.5A g−1, Od-MnO2 exhibits impressive durability for over 400 cycles (84.7% retention) and over 800 cycles (81.8% retention) with high depths of discharge (DODs), respectively, as shown in Fig. S20a and S20b (ESI†). To further evaluate the practicability of the Od-MnO2 cathode, pouch cells with a size of 4 cm × 4 cm were assembled. As can be seen in Fig. S21 (ESI†), the Zn/Od-MnO2 pouch cell presents similar charge–discharge curves to those in the coin cell, demonstrating the uniformity of electrochemical performance when the battery is scaled up. At a rate of 1C, the Zn/Od-MnO2 pouch cell exhibits a high reversibility of ≈100% CE and negligible capacity decay for over 100 cycles. Moreover, the specific capacity (based on the mass of the active materials of the cathode) after 500 cycles is 126.2 mA h g−1, about 69.8% retention compared with the maximum value (180.7 mA h g−1). These results underscore the potential of a Zn/Od-MnO2 system utilizing a mild aqueous electrolyte for high performance, long life, and environmentally friendly energy storage.
 | ||
| Fig. 5 Electrochemical performance of Zn/MnO2 and Zn/Od-MnO2 batteries. (a) Rate capability of Zn/MnO2 and Zn/Od-MnO2 coin cells from 0.1 to 10 A g−1. (b) Comparison of the Ragone plots (based on the mass of the cathode) of Od-MnO2 with some previous cathode materials for AZIBs. Long-term cycling performance of (c) Zn/MnO2 and Zn/Od-MnO2 coin cells at 1 A g−1 and (d) Zn/MnO2 and Zn/Od-MnO2 pouch cells at 1C (1C = 0.2 A g−1). | ||
3. Conclusions
In summary, we have designed and synthesized oxygen-defective porous MnO2 through n-butyllithium treatment, where the oxygen defects can induce preferential Zn2+ intercalation and thus reduce the negative impacts of by-products from H+ co-intercalation. In the meantime, the oxygen defects can regulate the electrochemical activity, facilitate ion diffusion, and increase the electrical conductivity of MnO2. Benefiting from the synergetic effects, the aqueous Zn/Od-MnO2 battery exhibits excellent rate capability with a reversible capacity of 94 mA h g−1 at a high current density of 10 A g−1 and a long cycle life of up to 1200 cycles with a high-capacity retention of 85.4% at 1 A g−1, and thus shows impressive energy density and power density. This study introduces a strategy of defect engineering for regulating the electronic structure of MnO2 and optimizing the H+/Zn2+ co-intercalation process, which sheds light on the advancement of MnO2 cathodes for high-performance aqueous zinc-ion batteries.Data availability statements
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant No. 2022YFA1203503), the National Natural Science Foundation of China (52272111 and 62161160311), and the Innovation Fund of WNLO and Shccig-Qinling Program. The authors also thank the facility support from the Center for Nanoscale Characterization & Devices, WNLO of Huazhong University of Science and Technology (HUST) and the Analytical and Testing Center of HUST. The authors extend their thanks to Engineer Jun Su at the Optoelectronic Micro&Nano Fabrication and Characterizing Facility, Wuhan National Laboratory for Optoelectronics of the Huazhong University of Science and Technology for support with the XRD test. Computations were done at the National Supercomputing Center in Shenzhen, P. R. China.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 Search PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 Search PubMed.
- T. Gu, D. Zhang, Y. Yang, C. Peng, D. Xue, C. Zhi, M. Zhu and J. Liu, Adv. Funct. Mater., 2023, 33, 2212299 Search PubMed.
- T. Gu, J. Shen, Z. Sun, F. Li, C. Zhi, M. Zhu and J. Liu, Small, 2024, 20, 2308355 Search PubMed.
- L. Zhao, T. Gu, Z. Liang and J. Liu, Sci. China: Technol. Sci., 2022, 65, 2221–2245 Search PubMed.
- J. He, C. Lu, H. Jiang, F. Han, X. Shi, J. Wu, L. Wang, T. Chen, J. Wang, Y. Zhang, H. Yang, G. Zhang, X. Sun, B. Wang, P. Chen, Y. Wang, Y. Xia and H. Peng, Nature, 2021, 597, 57–63 Search PubMed.
- R. F. Service, Science, 2021, 372, 890–891 Search PubMed.
- H. Wang, Z. Yu, X. Kong, W. Huang, Z. Zhang, D. G. Mackanic, X. Huang, J. Qin, Z. Bao and Y. Cui, Adv. Mater., 2021, 33, 2008619 Search PubMed.
- H. Duan, Y.-X. Yin, Y. Shi, P.-F. Wang, X.-D. Zhang, C.-P. Yang, J.-L. Shi, R. Wen, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2018, 140, 82–85 Search PubMed.
- Y. Zeng, Z. Lai, Y. Han, H. Zhang, S. Xie and X. Lu, Adv. Mater., 2018, 30, 1802396 Search PubMed.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 Search PubMed.
- Y. Q. Lv, Y. Xiao, L. T. Ma, C. Y. Zhi and S. M. Chen, Adv. Mater., 2022, 34, 2106409 Search PubMed.
- X. Gao, H. Zhang, X. Liu and X. Lu, Carbon Energy, 2020, 2, 387–407 Search PubMed.
- Y. Yuan, R. Sharpe, K. He, C. Li, M. T. Saray, T. Liu, W. Yao, M. Cheng, H. Jin, S. Wang, K. Amine, R. Shahbazian-Yassar, M. S. Islam and J. Lu, Nat. Sustainable, 2022, 5, 890–898 Search PubMed.
- S. Liu, R. Zhang, J. Mao, Y. Zhao, Q. Cai and Z. Guo, Sci. Adv., 2022, 8, eabn5097 Search PubMed.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 Search PubMed.
- L. Ma, S. Chen, N. Li, Z. Liu, Z. Tang, J. A. Zapien, S. Chen, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 1908121 Search PubMed.
- D. Chao, C. Ye, F. Xie, W. Zhou, Q. Zhang, Q. Gu, K. Davey, L. Gu and S.-Z. Qiao, Adv. Mater., 2020, 32, 2001894 Search PubMed.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 Search PubMed.
- G. Nam, C. Hwang, H. Jang, N. Kane, Y. Ahn, M.-J. Kwak, Z. Luo, T. Li, M.-G. Kim, N. Liu and M. Liu, Small, 2023, 2306919 Search PubMed.
- G. Zhang, T. Wu, H. Zhou, H. Jin, K. Liu, Y. Luo, H. Jiang, K. Huang, L. Huang and J. Zhou, ACS Energy Lett., 2021, 6, 2111–2120 Search PubMed.
- P. Ruan, X. Chen, L. Qin, Y. Tang, B. Lu, Z. Zeng, S. Liang and J. Zhou, Adv. Mater., 2023, 35, 2300577 Search PubMed.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 Search PubMed.
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610–16751 Search PubMed.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 Search PubMed.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 Search PubMed.
- G. J. Li, L. Sun, S. L. Zhang, C. F. Zhang, H. Y. Jin, K. Davey, G. M. Liang, S. L. Liu, J. F. Mao and Z. P. Guo, Adv. Funct. Mater., 2024, 34, 2301291 Search PubMed.
- V. Mathew, B. Sambandam, S. Kim, S. Kim, S. Park, S. Lee, M. H. Alfaruqi, V. Soundharrajan, S. Islam, D. Y. Putro, J.-Y. Hwang, Y.-K. Sun and J. Kim, ACS Energy Lett., 2020, 5, 2376–2400 Search PubMed.
- B. Yao, S. Chandrasekaran, J. Zhang, W. Xiao, F. Qian, C. Zhu, E. B. Duoss, C. M. Spadaccini, M. A. Worsley and Y. Li, Joule, 2019, 3, 459–470 Search PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 Search PubMed.
- B. Sambandam, V. Mathew, S. Kim, S. Lee, S. Kim, J. Y. Hwang, H. J. Fan and J. Kim, Chem, 2022, 8, 924–946 Search PubMed.
- X. H. Chen, P. C. Ruan, X. W. Wu, S. Q. Liang and J. Zhou, Acta Phys.-Chim. Sin., 2022, 38, 2111003 Search PubMed.
- N. Zhang, Y.-R. Ji, J.-C. Wang, P.-F. Wang, Y.-R. Zhu and T.-F. Yi, J. Energy Chem., 2023, 82, 423–463 Search PubMed.
- N. Zhang, J.-C. Wang, Y.-F. Guo, P.-F. Wang, Y.-R. Zhu and T.-F. Yi, Coord. Chem. Rev., 2023, 479, 215009 Search PubMed.
- B. Lee, H. R. Seo, H. R. Lee, C. S. Yoon, J. H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, ChemSusChem, 2016, 9, 2948–2956 Search PubMed.
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. G. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 Search PubMed.
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Nat. Commun., 2023, 14, 3067 Search PubMed.
- X. Liu, H. Euchner, M. Zarrabeitia, X. Gao, G. A. Elia, A. Groß and S. Passerini, ACS Energy Lett., 2020, 5, 2979–2986 Search PubMed.
- J. T. Huang, J. Zhou and S. Q. Liang, Acta Phys.-Chim. Sin., 2021, 37, 2005020 Search PubMed.
- A. Q. Zhang, R. Zhao, Y. H. Wang, J. J. Yang, C. Wu and Y. Bai, Energy Environ. Sci., 2023, 16, 3240–3301 Search PubMed.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y.-W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 Search PubMed.
- S. Yao, S. Wang, R. Liu, X. Liu, Z. Fu, D. Wang, H. Hao, Z. Yang and Y.-M. Yan, Nano Energy, 2022, 99, 107391 Search PubMed.
- Z. Hu, X. Xiao, H. Jin, T. Li, M. Chen, Z. Liang, Z. Guo, J. Li, J. Wan, L. Huang, Y. Zhang, G. Feng and J. Zhou, Nat. Commun., 2017, 8, 15630 Search PubMed.
- L. Liu, Y.-C. Wu, L. Huang, K. Liu, B. Duployer, P. Rozier, P.-L. Taberna and P. Simon, Adv. Energy Mater., 2021, 11, 2101287 Search PubMed.
- Q. Zhao, A. Song, W. Zhao, R. Qin, S. Ding, X. Chen, Y. Song, L. Yang, H. Lin, S. Li and F. Pan, Angew. Chem., Int. Ed., 2021, 60, 4169–4174 Search PubMed.
- Y. Li, X. Li, H. Duan, S. Xie, R. Dai, J. Rong, F. Kang and L. Dong, Chem. Eng. J., 2022, 441, 136008 Search PubMed.
- J. Xia, Y. Zhou, J. Zhang, T. Lu, W. Gong, D. Zhang, X. Wang and J. Di, Small, 2023, 19, 2301906 Search PubMed.
- A. Zhang, R. Zhao, Y. Wang, J. Yue, J. Yang, X. Wang, C. Wu and Y. Bai, Angew. Chem., Int. Ed., 2023, 62, e202313163 Search PubMed.
- H. Ren, J. Zhao, L. Yang, Q. Liang, S. Madhavi and Q. Yan, Nano Res., 2019, 12, 1347–1353 Search PubMed.
- Y. Zhang, Y. Liu, Z. Liu, X. Wu, Y. Wen, H. Chen, X. Ni, G. Liu, J. Huang and S. Peng, J. Energy Chem., 2022, 64, 23–32 Search PubMed.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 Search PubMed.
- Y. Jin, L. Zou, L. Liu, M. H. Engelhard, R. L. Patel, Z. Nie, K. S. Han, Y. Shao, C. Wang, J. Zhu, H. Pan and J. Liu, Adv. Mater., 2019, 31, 1900567 Search PubMed.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 Search PubMed.
- Q. Tan, X. Li, B. Zhang, X. Chen, Y. Tian, H. Wan, L. Zhang, L. Miao, C. Wang, Y. Gan, J. Jiang, Y. Wang and H. Wang, Adv. Energy Mater., 2020, 10, 2001050 Search PubMed.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 Search PubMed.
- Y. Dai, J. Li, L. Chen, K. Le, Z. Cai, Q. An, L. Zhang and L. Mai, ACS Energy Lett., 2021, 6, 684–686 Search PubMed.
- Y. Jiang, D. Ba, Y. Li and J. Liu, Adv. Sci., 2020, 7, 1902795 Search PubMed.
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S.-Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 Search PubMed.
- H. Tang, W. Chen, N. Li, Z. Hu, L. Xiao, Y. Xie, L. Xi, L. Ni and Y. Zhu, Energy Storage Mater., 2022, 48, 335–343 Search PubMed.
- S. Wang, Z. Yuan, X. Zhang, S. Bi, Z. Zhou, J. Tian, Q. Zhang and Z. Niu, Angew. Chem., Int. Ed., 2021, 60, 7056–7060 Search PubMed.
- G. Cui, Y. Zeng, J. Wu, Y. Guo, X. Gu and X. W. Lou, Adv. Sci., 2022, 9, 2106067 Search PubMed.
- A. Q. Zhang, R. Gao, L. Y. Hu, X. G. Zang, R. Yang, S. Y. Wang, S. Y. Yao, Z. Y. Yang, H. G. Hao and Y. M. Yan, Chem. Eng. J., 2021, 417, 129186 Search PubMed.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 Search PubMed.
- R. Trócoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 Search PubMed.
- B. Jiang, C. Xu, C. Wu, L. Dong, J. Li and F. Kang, Electrochim. Acta, 2017, 229, 422–428 Search PubMed.
- N. Qiu, H. Chen, Z. Yang, S. Sun and Y. Wang, Electrochim. Acta, 2018, 272, 154–160 Search PubMed.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 Search PubMed.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 Search PubMed.
- J. Lee, J. B. Ju, W. I. Cho, B. W. Cho and S. H. Oh, Electrochim. Acta, 2013, 112, 138–143 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03100h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |