
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular polymerization of [6]helicene-based cyano-luminogens: on the overall efficiency of self-assembled circularly polarized emitters†
Lucia
López-Gandul
 a,
Rafael
Rodríguez
*bd,
Nicolas
Vanthuyne
c,
Jeanne
Crassous
*d and
Luis
Sánchez
*a
a,
Rafael
Rodríguez
*bd,
Nicolas
Vanthuyne
c,
Jeanne
Crassous
*d and
Luis
Sánchez
*a
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria, s/n, 28040-Madrid, Spain. E-mail: lusamar@ucm.es
bCentro Singular de investigación en Química Biolóxica e Materiais Moleculares (CiQUS) e Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782, Santiago de Compostela, Spain. E-mail: rafael.rodriguez.riego@usc.es
cAix Marseille Univ, CNRS, Centrale Med, FSCM, Marseille, France
dUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226 35000, Rennes, France. E-mail: jeanne.crassous@univ-rennes1.fr
First published on 19th June 2024
Abstract
The synthesis of the [6]helicene-based luminophores 1 and 2 is reported. These chiral systems, endowed with cyano-stilbene fragments, form supramolecular polymers by the operation of intermolecular H-bonding interactions between the amides present in the peripheral side chains. The dissimilar disubstitution of 1 and 2 plays a crucial role in their self-assembling features. Thus, 1 does not show an efficient π-stacking of the central aromatic moiety. Instead, its self-assembling process results in a zig-zag arrangement of the monomeric units to form the aggregated species. On the other hand, 2 presents an efficient overlap of the aromatic backbones that affords a co-facial arrangement of the monomeric units. The solvent-dependent studies indicate that both [6]helicenes self-assemble following a cooperative supramolecular polymerization mechanism with a higher degree of cooperativity and stability for compound 2. The enantioenriched samples of both 1 and 2 display a rich dichroic pattern that changes when the supramolecular polymerization takes place. Furthermore, the presence of the cyano-stilbene moieties gives rise to an aggregation induced emission effect. The inherent chirality of both the monomeric and aggregated species of 1 and 2 provides the systems with CPL-emitting properties, presenting a remarkable overall CPL-efficiency, quantified by the BCPL parameter, that increases upon supramolecular polymerization.
Introduction
The development of optical technologies relies on the availability of tailored optically active materials.1 The chemical structure of a large number of these materials consists of π-conjugated chromophores, in which the substitution pattern plays a relevant role to bias their optical features. Importantly, a variety of π-conjugated systems -like π-conjugated oligomers, polycyclic aromatic hydrocarbons, rylenes, BODIPYs, porphyrins, etc.- can experience a controlled self-assembly to generate highly organized supramolecular ensembles, whose optical properties differ from those shown by the isolated systems.2 Supramolecular polymers, macromolecular species formed by the non-covalent interactions of monomeric units, are an excellent benchmark to investigate the formation of highly organized structures with outstanding optical properties.3 Furthermore, the decoration of such monomeric units with elements of asymmetry yields chiral supramolecular polymers able to amplify the asymmetry to the final aggregated species, showing remarkable chiroptical features.4 A variety of π-conjugated scaffolds self-assemble into H-type aggregated species, in which the emission of the monomeric units is deactivated by an aggregation caused quenching (ACQ) effect that limits their potential applicability in optical technologies.5 To circumvent this drawback, two different strategies can be found in the literature. The first one is the self-assembly of monomeric units into J-type aggregates, characterized by a bathochromic shift in both the absorption and emission wavelength maxima and an increase in the intensity of emission of the aggregated species in comparison to the monomers.5 The second one is the formation of H-type aggregates displaying aggregation induced emission (AIE). In these H-type aggregates showing AIE, the restricted intramolecular rotation of the aromatic moieties constitutive of the chromophores or the planarization of the aromatic backbones induced by the aggregation process contributes to increase the emission intensity.6 At the same time, a selected group of chiral supramolecular polymers has been reported to behave as circularly polarized light (CPL) emitters with remarkable values of luminescence dissymmetry factor (glum),7 comparable to those reported for molecular systems.8The enhanced emissive features of those systems exhibiting AIE provides an effective way to build up new CPL-emitters. Polymers, liquid crystals9 or very few small molecules have been reported to behave as CPL active AIE materials.10 However, in these examples, it is difficult to achieve and predict an ordered supramolecular organization of the chromophores that favours an effective exciton coupling of the pendant luminophores.11 The supramolecular polymerization of tailored monomeric units constitutes a useful tool to attain this specific arrangement of emissive scaffolds.12 Helicenes, non-planar aromatic units with helical chirality,13 exemplify this chiral requirement to achieve efficient CPL-emitters and have been scarcely utilized as building blocks for supramolecular polymers, exhibiting not only CPL-activity but also additional properties like spin filtering capabilities.14
Our research groups have investigated the CPL features of [6]helicenes as isolated systems,15 as well as taking part of supramolecular polymers.14 On the other hand, we have also reported on the efficient generation of CPL upon the supramolecular polymerization of cyano-luminogens as building blocks that experience AIE and behave as CPL-emitters.16 Herein, we report on the synthesis and self-assembling features of 2,15- (compound 1, Fig. 1a) and 4,13-bis-ethynyl-carbo[6]helicene (compound 2, Fig. 1a) endowed with p-phenylene cyano-benzamide segments. These segments favour the supramolecular polymerization of [6]helicenes 1 and 2 by the operation of intermolecular H-bonding interactions and the π-stacking of the lateral p-phenylene cyano moieties (Fig. 1a). The π-stacking of these luminogenic segments gives rise to a remarkable AIE effect (Fig. 1b). Furthermore, the presence of the [6]helicene core provides the asymmetry element required to generate CPL-emitters. Interestingly, and despite compounds 1 and 2 present glum values of ∼0.001, the overall CPL efficiency of these [6]helicenes 1 and 2, determined by the parameter CPL brightness (BCPL),17 undergoes a remarkable increase upon the corresponding supramolecular polymerization, providing values for the BCPL among the highest reported for [6]helicenes.17 We demonstrate the strong influence of the substitution pattern of [6]helicenes not only in the self-assembling features, but also in the final overall efficiency (BCPL1 = 11.6 M−1 cm−1; BCPL2 = 40.6 M−1 cm−1). The studies presented herein complement those initiated for [6]helicenes decorated with TPE fragments18 and contribute to expanding the establishment of structure–function relationships for CPL-emitting materials.
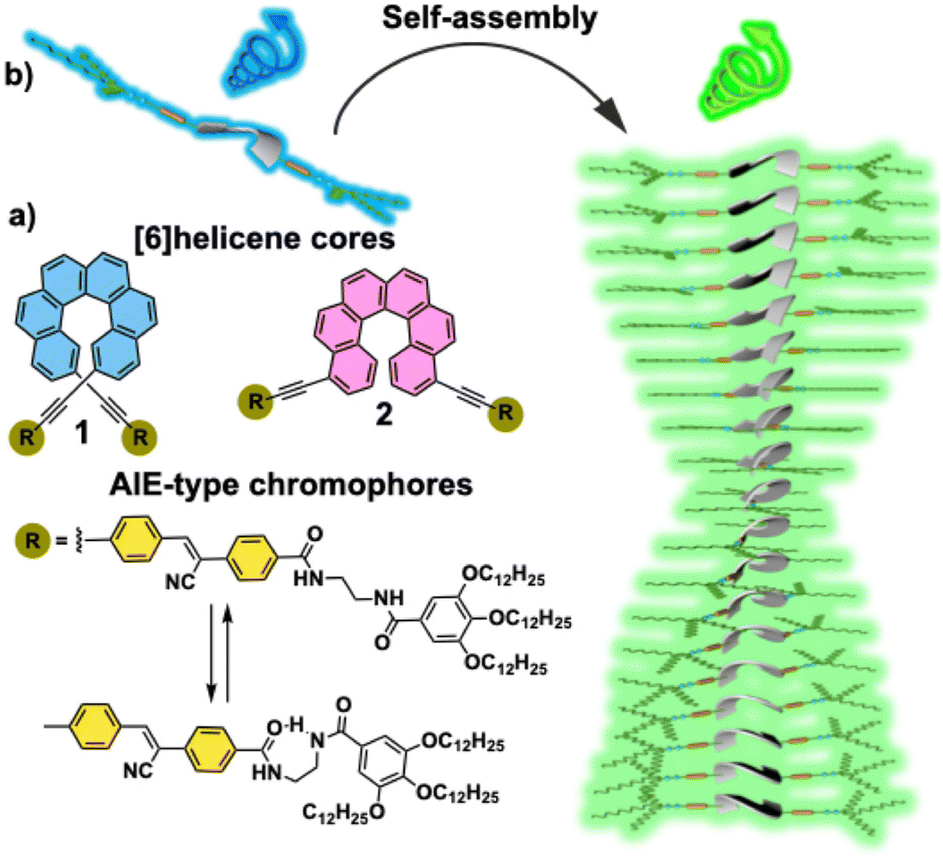 | ||
| Fig. 1 (a) Chemical structure of the [6]helicene-based derivatives 1 and 2 presented in this work. (b) Schematic illustration of the modulation of the CPL emission upon supramolecular of helicene-AIE like molecules. | ||
Results and discussion
Synthesis and self-assembly
The targeted [6]helicenes 1 and 2 were synthesized starting from enantiopure bisalkynyl helicene derivatives following a Sonogashira-Hiyara Pd catalized cross-coupling reaction with the corresponding (Z)-4-(1-cyano-2-(4-iodophenyl)vinyl)-benzamide moieties (Fig. 1 and S7–S9† for details). These benzamide moieties are endowed with two amide functional groups that give rise to an array on H-bonding interactions in the appropriate experimental conditions.We have previously demonstrated the influence of the substitution pattern on the self-assembling features of 2,15- and 4,13-disubstituted [6]helicenes. The more contorted geometry of the 2,15-disubstituted [6]helicenes prevents an efficient π-stacking of the aromatic backbones. Therefore, the self-assembly of the 2,15-disubstituted [6]helicene takes place in a zig-zag arrangement of the aromatic segments and the formation of an intermolecular array of H-bonds between the amide functional groups. In contrast, the 4,13-disubstitution of the [6]helicene core results in a more accessible aromatic surface that favours not only the above mentioned operation of intermolecular H-bonding interactions but also the π-stacking of the [6]helicene fragments.14a Taking into account the resemblance between the chemical structure of compounds 1 and 2, and the previously reported self-assembling [6]helicenes, we have investigated the supramolecular polymerization of both 1 and 2 by using different spectroscopic techniques. Firstly, we have registered 1H NMR spectra of (M)-1 in CDCl3 and different concentrations. Despite CHCl3 is a good solvent that favours the solvation of the monomeric units, at relatively high concentrations it is possible to see the first clues of self-assembly. Thus, most of the aromatic resonances and all the aliphatic protons of (M)-1 experience negligible shifts upon increasing the concentration (Fig. S1†). This is not the case of the resonances ascribable to the amide protons of (M)-1, that shift downfield upon increasing the concentration, diagnostic of the formation of intermolecular H-bonding interactions.19 Noteworthy, the singlet ascribable to the vinylogous proton at the cyano-stilbene moiety, even if weakly, shields upon increasing the concentration, which could be indicative of the π-stacking of the peripheral luminophores in (M)-1 (Fig. S1†). ROESY experiments of a concentrated solution of (M)-1 in CDCl3, total concentration cT = 20 mM, display cross-peaks between the aliphatic protons and the aromatic resonances that can only be justified by considering a zig-zag arrangement of the monomeric units upon self-assembly (Fig. 2a).
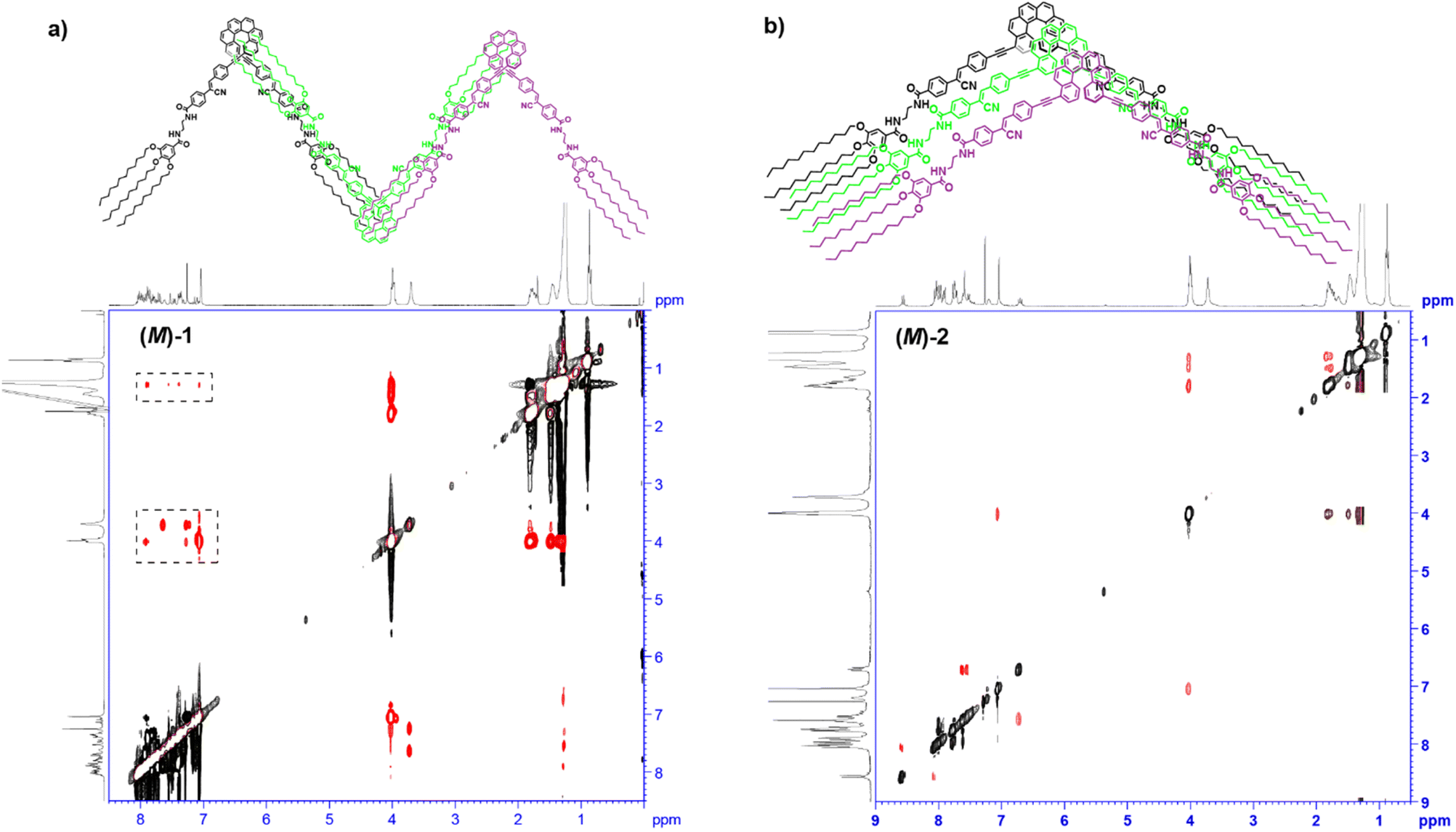 | ||
| Fig. 2 (a) ROESY NMR spectra (CDCl3, 300 MHz, cT = 20 mM; 293 K) of (a) (M)-1 and (b) (M)-2. The dotted rectangles depict the intermolecular through-space coupling signals. The upper part of panels (a) and (b) depicts a schematic illustration of the binding mode experienced by the reported [6]helicenes 1 and 2 upon self-assembly. | ||
Further evidence of the operation of intermolecular H-bonds between the amide functional groups has been extracted from the FTIR in solution at cT = 1 mM. In CHCl3, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O Amide II stretching band appears centered at 1652 cm−1, while two bands at 3451 and 3337 cm−1, ascribable to the stretching NH bands, are observed for (M)-1. The wavenumber of these stretching bands is indicative of the presence of the molecularly dissolved species presenting both free NH and intramolecular 7-membered hydrogen-bonded pseudocycles (Fig. 1a and 3a).20 In MCH, however, these bands appear shifted at 1636 and 3295 cm−1, which implies the intermolecularly hydrogen-bonded supramolecular structures (Fig. 1b and 3a).20
O Amide II stretching band appears centered at 1652 cm−1, while two bands at 3451 and 3337 cm−1, ascribable to the stretching NH bands, are observed for (M)-1. The wavenumber of these stretching bands is indicative of the presence of the molecularly dissolved species presenting both free NH and intramolecular 7-membered hydrogen-bonded pseudocycles (Fig. 1a and 3a).20 In MCH, however, these bands appear shifted at 1636 and 3295 cm−1, which implies the intermolecularly hydrogen-bonded supramolecular structures (Fig. 1b and 3a).20
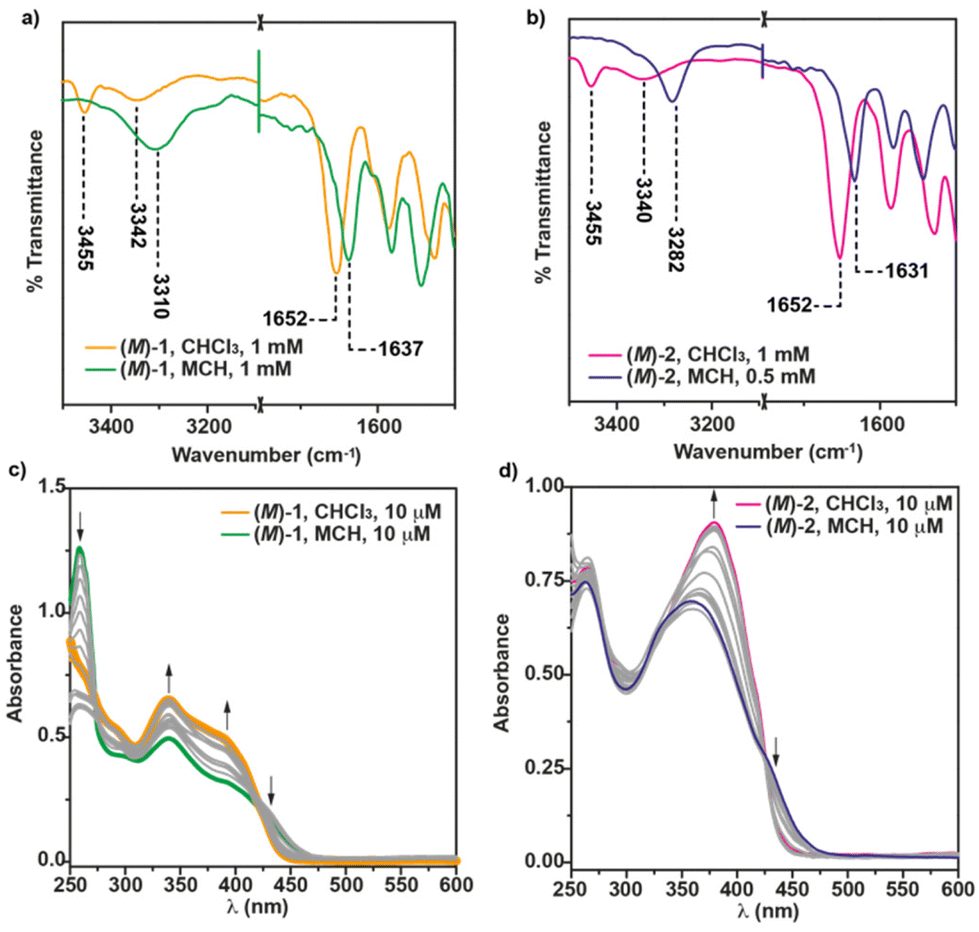 | ||
| Fig. 3 FTIR spectra of (M)-1 (a) and (M)-2 (b) in CHCl3 and MCH. The dotted lines show the wavenumber values for the NH and Amide I stretching bands; UV-Vis spectra of (M)-1 (c) and (M)-2 (d) in CHCl3, MCH and in different MCH/CHCl3 mixtures. Arrows in panel (c) and (d) show the absorption changes upon increasing the amount of the good solvent (CHCl3). | ||
The substitution of the [6]helicene core at the 4 and 13 positions has been reported to favour the π-stacking of this aromatic backbone upon self-assembly.14a In this case, compound 2, showing this 4,13- substitution pattern, also presents a more efficient π-stacking of the aromatic units upon self-assembly. Thus, the concentration dependent 1H NMR experiments of the (M) enantiomer of 2 in CDCl3 show a slight upfield shift of the aromatic resonances, especially relevant for the proton at the double bond of the p-phenylenevynylene unit, diagnostic of the above mentioned π-stacking. As occurs for (M)-1, a deshielding of the amide protons, due to the formation of H-bonds between the amide functional groups, is also observed (Fig. S2†). Unlike 1, the ROESY experiment of (M)-2 does not show any cross-peak between the peripheral aliphatic protons and the aromatic moieties, which could be indicative of a co-facial arrangement of the monomeric units in the aggregated state of (M)-2 (Fig. 2b). The formation of an array of intermolecular H-bonding interactions in the formation of (M)-2 supramolecular polymers has been also corroborated by registering the corresponding FTIR spectra in solution. In the good solvent CHCl3, the NH stretching bands appear at 3455 and 3340 cm−1, which implies the presence of both the free and the intramolecularly H-bonded pseudocycle (Fig. 1a and 3b). The wavenumber of the Amide I band, observed at 1652 cm−1, corroborates that these carbonyls are not participating as H-bonds acceptors.20 The NH and Amide I stretching bands wavenumber change by using MCH as solvent and these bands are observed at 3282 and 1631 cm−1, respectively. These wavenumber values are associated to the formation of intermolecular H-bonding interactions between the NH and the CO of the adjacent amide functional groups (Fig. 1a and 3b).20 Previously described self-assembling cyano-stilbenes have been reported to show intermolecular CN⋯H interactions in the aggregated state, since the stretching band ascribable to the cyano group shifts to lower wavenumbers (from 2216 to 2203 cm−1) upon aggregation.21 In this case, the shift of the stretching cyano band is of only 2 cm−1 which implies that this non-covalent interaction is not playing a relevant role in the self-assembly of the reported [6]helicenes (Fig. S3†).
To further unravel the non-covalent forces involved in the self-assembly of [6]helicenes 1 and 2, we have registered UV-Vis spectra in the good solvent CHCl3 and the bad solvent MCH. In the case of the 2,15-disubstituted [6]helicenes, the UV-Vis spectrum of (M)-1 in CHCl3 shows several consecutive maxima, centered at 430, 410, 393, 344 and 251 nm, corresponding to the monomeric state (Fig. 3c). In the bad solvent MCH, the aggregation provokes noticeable changes in the UV-Vis spectrum of (M)-1, the maximum being at 450 nm and accompanied with bands at 399, 342, 302 nm (Fig. 3c). Interestingly, and unlike the previous self-assembling 2,15-disubstituted [6]helicenes,14a the UV-Vis spectra in these two solvents and in MCH/CHCl3 mixtures present crossing points at ∼275 and ∼420 nm, diagnostic of the interaction between the aromatic units. In good analogy with the UV-Vis spectra reported for referable self-assembled cyanostilbenes,16,22 the molecularly dissolved species of (M)-2 features a broad band centered at λ = 379 nm that shifts both hypsochromically and hypochromically upon self-assembly (λ = 360 nm), which can be ascribed to the formation of H-type aggregates, in which the whole aromatic surface, both the [6]helicene and the conjugated cyano-p-phenylene,23 participates (Fig. 3d).
Chiroptical and emissive properties: supramolecular polymerization mechanism and overall efficiency of the CPL-emitters
The chiroptical properties of enantioenriched samples of [6]helicenes 1 and 2 have been investigated by circular dichroism (ECD) spectroscopy, both in the monomeric and aggregated species. The mirror-image ECD spectra of (M)-1 and (P)-1 in CHCl3 show a rich dichroic pattern with maxima at λ = 425 and 403 nm, a zero-crossing point at λ = 366 nm, and four bands centered at λ = 340, 313, 291, 273 and 248 nm (Fig. 4a). This dichroic pattern, associated to the monomeric state, experiences noticeable changes upon the addition of MCH. Thus, the ECD spectra of (M)-1 and (P)-1 in pure MCH are also mirror-images, displaying a new band centered at λ = 450 nm—which is in keeping with the UV-Vis experiments—followed by bands at λ = 433, 406, 391, 357, 318, 274 and 254 nm and a zero-crossing point at λ = 339 nm (Fig. 4a). The dissimilarity on the ECD spectra of the molecularly dissolved and aggregated species has been utilized to derive the thermodynamic parameters associated to the supramolecular polymerization of (M)-1, by applying the solvent denaturation (SD) protocol described by Meijer and co-workers. This method consists of adding aliquots of a solution of the investigated compound in CHCl3 to a solution of the same compound and at the same concentration in the bad solvent MCH (Fig. 4b).24 This model has been successfully applied to derive the thermodynamic parameters of a number of supramolecular polymers.14a,16,19b,20d Plotting the variation of the degree of aggregation, α, calculated from the dichroic response at λ = 425 nm,25versus the molar fraction of the good solvent, CHCl3, and applying eqn (1), displays a non-sigmoidal curve that can be fitted to the SD model to provide the released free Gibbs energy (ΔG) of the process, the m coefficient, which relates the stability of the supramolecular polymer to the solvent in such a way that increasing the m-value results in a decreasing value for the critical chloroform volume fraction, and the degree of cooperativity, σ (Fig. 4c). The calculated values are collected in Table 1.| ΔG′ = ΔG + mX | (1) |
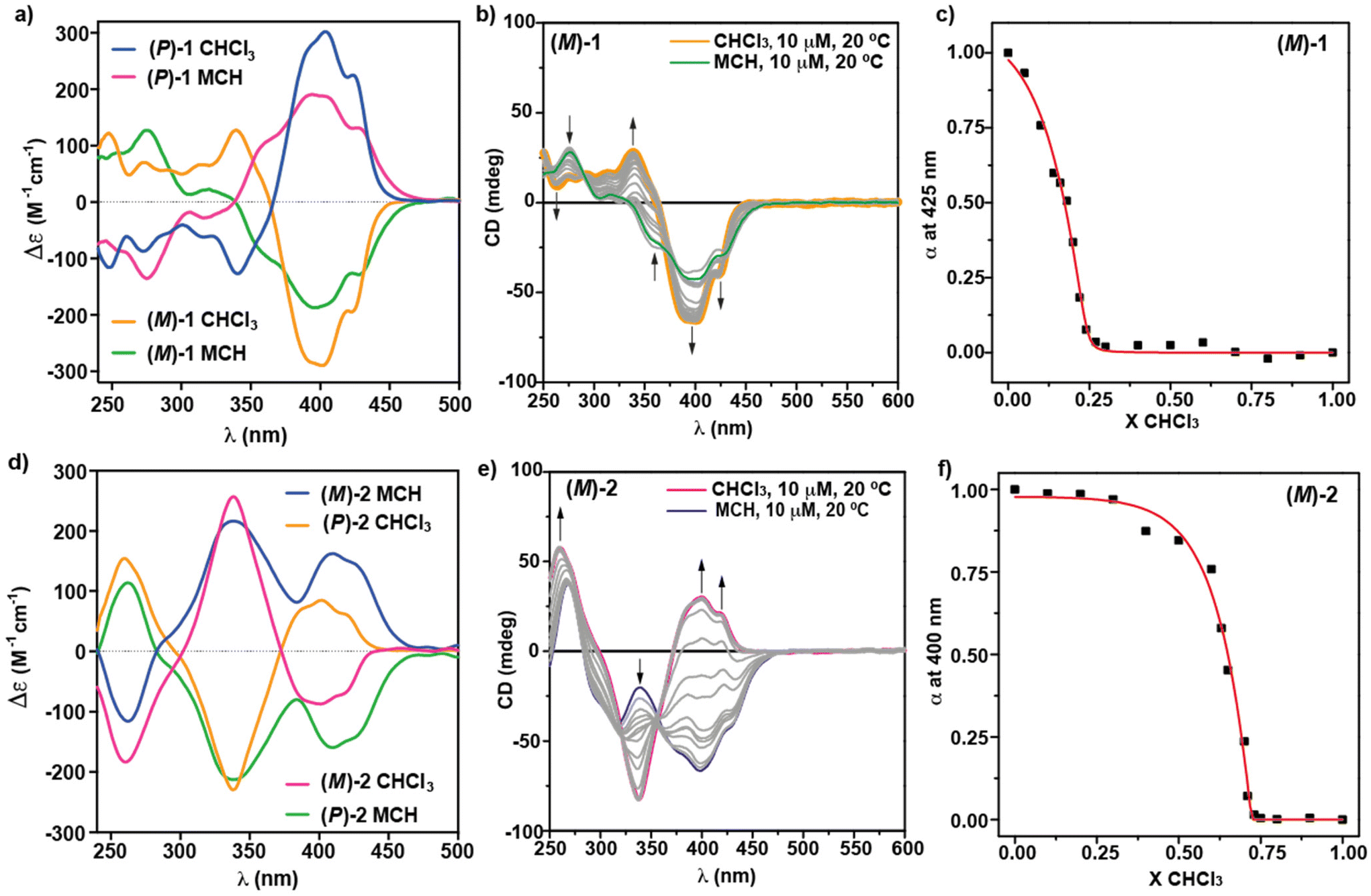 | ||
| Fig. 4 (a) CD spectra of the M and P enantiomers of [6]helicene 1 in CHCl3 and MCH; (b) CD spectra of (M)-1 in MCH/CHCl3 mixtures; (c) plot of the variation of the degree of aggregation α at λ = 422 nm versus the molar fraction of the good solvent CHCl3; (d) CD spectra of the M and P enantiomers of [6]helicene 2 in CHCl3 and MCH; (b) CD spectra of (M)-2 in MCH/CHCl3 mixtures; (c) plot of the variation of the degree of aggregation α at λ = 400 nm versus the molar fraction of the good solvent CHCl3. Arrows in panels (b) and (e) represents the changes in the dichroic response upon increasing the ratio of the good solvent CHCl3. The red lines in panels (c) and (f) correspond to the fit to the SD model. Experimental conditions: cT = 10 μM; 20 °C. | ||
| Compound | ΔG′ (kJ mol−1) | m | σ |
|---|---|---|---|
| (M)-1 | −33.9 ± 0.9 | 23.1 | 1.6 × 10−3 |
| (M)-2 | −45.5 ± 3.0 | 24.1 | 9.7 × 10−6 |
The dichroic pattern of [6]helicenes 2 is also very rich. Thus, the ECD spectra of the molecularly dissolved species of the monomeric M and P enantiomers of 2 present several intense dichroic bands at λ = 423, 402, 386, 340 and 260 nm, and zero-crossing points at λ = 373 and 300 nm (Fig. 4d). The supramolecular polymerization of these enantiomers, monitored by registering the ECD spectra in increasing molar fractions of MCH, occurs with a slight depletion of the intensity of the bands at λ = 340 and 260 nm. However, ECD experiments show a remarkable stereomutation and increase in intensity of the region distinctive to the CN-stilbene unit, i.e., from −86 to +162 M−1 cm−1 at λ = 409 nm for (M)-2 and viceversa for (P)-2 (Fig. 4d and e). Therefore, the ECD spectra of the supramolecular polymers formed by compounds 2 present two maxima at λ = 428, 400 and 342 nm, a zero-crossing point at λ = 284 nm and a minimum at λ = 263 nm (Fig. 4d and e).
The remarkable changes observed in the ECD spectra of the monomeric and aggregated species of (M)-2 allows the straightforward application of the above-mentioned SD model to calculate the stability of the supramolecular polymers formed. Thus, the addition of increasing amounts of a solution of (M)-2 in CHCl3 at cT = 10 μM and at 20 °C to a solution of (M)-2 in MCH at the same experimental conditions provokes the gradual depletion of the dichroic band at λ = 400 nm, ascribable to the aggregated species, and the appearance of the bands at λ = 428 and 409 nm. Furthermore, similar changes are observed for the bands at λ = 342 nm (Fig. 4e). A clear non-sigmoidal curve arises by plotting the degree of aggregation α at λ = 400 nm versus the molar fraction of the good solvent CHCl3, that is fitted to the SD model (Fig. 4f). This SD model brings to light the values for ΔG, the m coefficient and the degree of cooperativity σ collected in Table 1. Interestingly, the comparison between the denaturation curves obtained for (M)-1 and (M)-2 clearly demonstrates that the supramolecular polymers formed by the 4,15-disubstituted [6]helicenes 2 are more stable than those formed by the 2,13-disubstituted [6]helicenes 1. Whilst the former needs a molar fraction of CHCl3X = 0.7 to achieve a complete disassembly, the later only needs a molar fraction of CHCl3X = 0.23. This higher stability is well represented by the larger ΔG value derived for (M)-2 in comparison to (M)-1 (Table 1). Interestingly, the calculated degree of cooperativity for both (M)-1 and (M)-2 implies the cooperative character of the supramolecular polymerization of both systems, the latter being more cooperative than the former. These mechanistic differences could be accounted for by considering the more efficient π-stacking of the whole aromatic core in (M)-2 in comparison to (M)-1.
The formation of supramolecular polymers from (M)-1 has been visualized by Atomic Force Microscopy (AFM) imaging. The AFM images of a spin-coated diluted solution of this [6]helicene (cT = 10 μM) onto highly oriented pyrolytic graphite (HOPG) show the formation of long fibrillar aggregates with heights of ∼4 nm and thicker fibers of ∼11 nm that could be constituted by intertwined thin fibers (Fig. 5a, b and S4†). In good analogy to (M)-1, the AFM images of (M)-2, obtained by spin-coating a 10 μM solution of this [6]helicene onto HOPG, also reveal the formation of rope-like fibrillar structures with heights of around 4 nm and thicker fibers of ∼10 nm height (Fig. 5c, d and S5†).
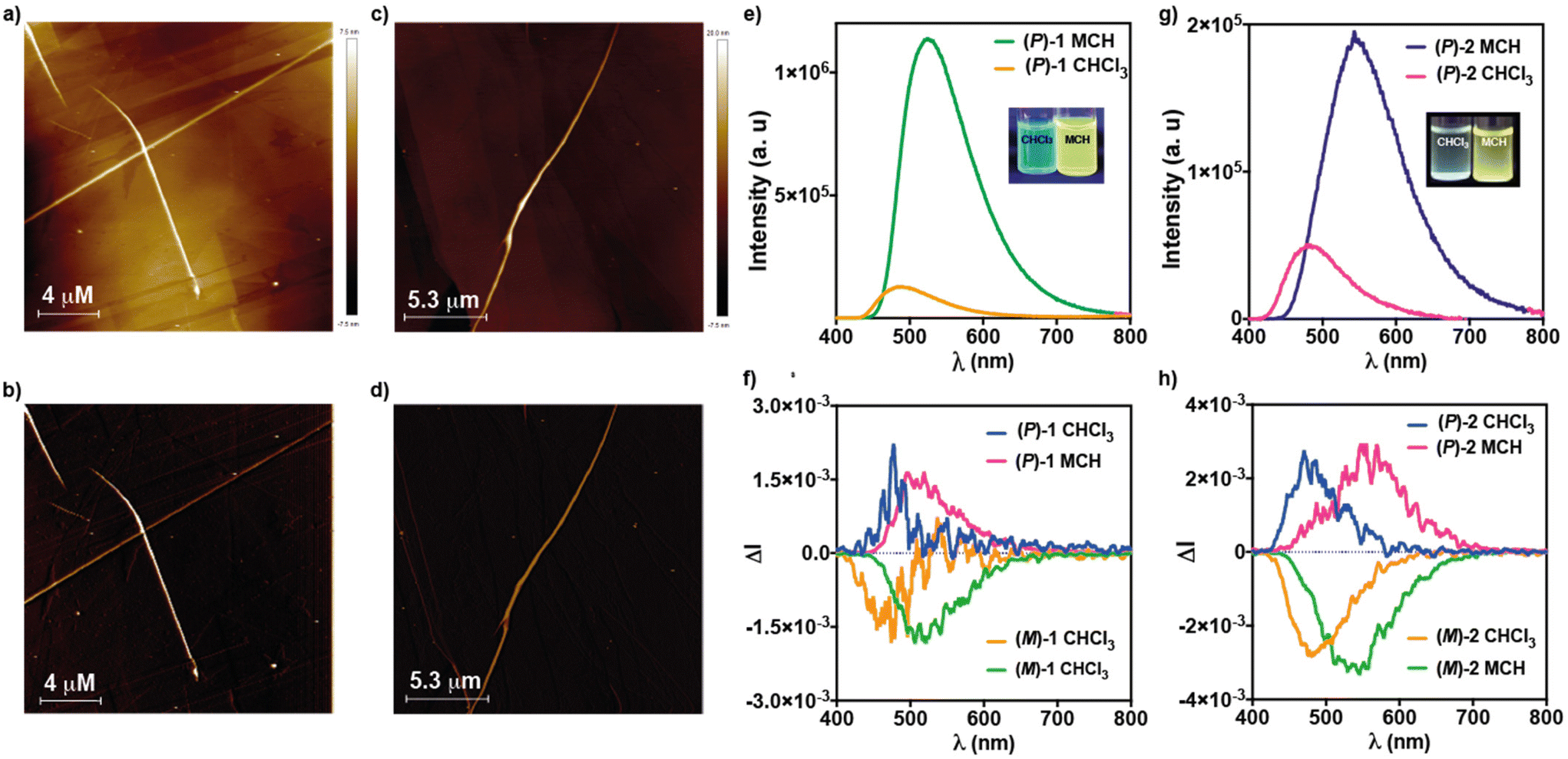 | ||
| Fig. 5 Height (a and c) and phase (b and d) AFM images of the fibrillar aggregates formed by (M)-1 (a and b) and (M)-2 (c and d) upon spin-coating a 10 μM solution of the corresponding helicene onto HOPG. Emission spectra of (M)-1 (e) and (M)-2 (g) in CHCl3 and MCH (cT = 10 μM, λexc = 365 nm); CPL spectra of (M)-1, (P)-1 (f) and (M)-2 and (P)-2 (h) CHCl3 and MCH (cT = 10 μM, λexc = 365 nm). The inset in panel (e) and (g) show the pictures of the solutions of (M)-1 and (M)-2 in CHCl3 and MCH upon excitation at λ = 365 nm. | ||
Previous reports on cyano-p-phenylene demonstrate that the self-assembly of these contorted units can be utilized to provoke an AIE effect and, hence, to increase the emission efficiency. Taking into account the relationship between the emission and CPL-activity, this effect could be beneficious for achieving remarkable CPL activity. Consequently, the presence of the cyano-p-phenylene moieties in both [6]helicenes 1 and 2, that show dissimilar self-assembling features, can be utilized for achieving biased luminescent and CPL-emitters. The emission spectrum of 1 in a molecularly dissolved state shows a broad, blue emission with maxima at λ = 494 nm (Fig. 5e). The supramolecular polymerization of 1 provokes a bathochromic shift in the emission maxima (λ = 532 nm) and an intense increase in the emission intensity, the intensity in MCH being >2000 times higher than in CHCl3 (Fig. 5e). This increase in emission was gauged by quantum yield measurements (ϕPL) moving from ΦPL = 0.04 to ΦPL = 0.21 in molecularly dissolved and aggregated states, respectively, that reveal the operation of an AIE effect (Table 2, see also the emission lifetimes in Fig. S6†).
| Compound | Solvent |
ε ×104![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (M−1 cm−1) a (M−1 cm−1) |
Φ |
g
abs ×10−3b |
g
lum ×10−3c |
B lum (M−1 cm−1) |
|---|---|---|---|---|---|---|
| a ε was measured at λ = 365 nm for both (M)-1 and (M)-2 in CHCl3 and MCH. b g abs of (M)-1 was measured at λ = 426 and 432 nm for CHCl3 and MCH, respectively; gabs of (M)-2 was measured at λ = 420 and 431 nm for CHCl3 and MCH, respectively. c g lum of (M)-1 was measured at λ = 480 and 520 nm for CHCl3 and MCH, respectively; glum of (M)-2 was measured at λ = 480 and 550 nm for CHCl3 and MCH, respectively. | ||||||
| (M)-1 | CHCl3 | 8.2 | 0.04 | −5.21 | −1.51 | 2.8 |
| (M)-1 | MCH | 6.1 | 0.21 | −4.50 | −1.81 | 11.6 |
| (M)-2 | CHCl3 | 9.6 | 0.04 | −1.22 | −2.80 | 5.4 |
| (M)-2 | MCH | 8.4 | 0.31 | +4.55 | −3.12 | 40.6 |
Given the archetypal nature of helicenes as chiral luminophores, CPL experiments were carried out. Fig. 5f displays the CPL spectra of (P)- and (M)-1 in both molecularly dissolved and aggregated states. According to PL experiments, the aggregation process promotes a red shift in the emission maxima, while maintaining the dissymmetry factor glum—defined as glum = 2·(IL − IR)/(IL + IR)—in the range of glum = ±1.5 × 10−3 (Fig. 5f and Table 2).
Similar findings have been extracted by investigating the emissive features of compounds 2. The emission spectra of 2 in CHCl3 show a broad, blue emission with maxima at λ = 494 nm (Fig. 5g). The supramolecular polymerization of 2 produces a clear bathochromic shift, the maxima being at λ = 532 nm, and an increment of the fluorescence (PL) intensity (Fig. 5g). The PL intensity of 2 in MCH is ∼4 times more intense than in CHCl3. This moderate increment of the emission intensity of 2 in comparison to 1 could be justified by considering the more efficient overlap of the aromatic backbones. However, the calculated ϕPL values for the monomeric and aggregated states in 2 are in the same range to those calculated for 1 thus confirming the operation of an AIE effect in the former (Table 2). Finally, we have also measured the CPL activity of the (M) and (P) enantiomers of [6]helicene 2 in both molecularly dissolved and aggregated states. In CHCl3, compounds 2 show a glum = +2.4 × 10−3 and −2.8 × 10−3 for the (P) and (M) enantiomers, respectively, centered at λ = 480 nm. In MCH, and unlike in the corresponding ECD spectra, no changes in the sign of the CPL activity are observed, suggesting different absorption and emission excited states. Furthermore, the CPL spectra appears 70 nm red-shifted in comparison to CHCl3—the maxima being at λ = 550 nm— showing glum values of = +2.89 × 10−3 and −3.20 × 10−3 (Fig. 5h and Table 2).
The glum values measured for all the investigated luminophores 1 and 2 are in the range of 10−3 regardless of the species being molecularly dissolved or in an aggregated state. Apparently, the supramolecular polymerization of these [6]helicenes endowed with cyano-p-phenylenes yields an efficient AIE but does not improve the CPL-activity. These findings demonstrate that an increase in the emission intensity cannot exert a beneficial influence on the CPL-activity. In fact, the glum values for the 2,15-disubstituted [6]helicene 1 is lower than those reported for referable self-assembled congeners.14 This trend confirms the rule observed for luminophores constituted by [6]helicenes endowed with a TPE moiety at 2 position connected by an ethynylene linker.18 However, the CPL-activity of the investigated compounds 1 and 2 are in the same range of some other self-assembled scaffolds showing an AIE effect like Pt(II) complexes bearing phenylisoxazole peripheral units,26 1,3,5-benzenetricarboxamides-diaminocyclohexane conjugates,27 self-assembling [6]helicenes14 and several cyano-stilbene luminophores.16,28,29
Very recently, L. Arrico, L. di Bari and F. Zinna, in an attempt to accurately describe the efficiency of a CPL-emitter, have defined the brightness fluorescence parameter (BCPL).17 This parameter, that relates the molar extinction coefficient, the emission quantum yield and the glum by eqn (2), can be utilized as a simple and effective tool to evaluate the overall efficiency in emission of chiral luminophores.
| BCPL = (ελ·Φpl·|glum|)/2 | (2) |
Considering the derived values of the ε at the emission maxima, the ϕPL and the glum values for 1 and 2 in CHCl3 and in MCH, we have calculated the BCPL values by applying eqn (2). In the case of the monomeric state, both [6]helicenes 1 and 2 present very similar values (2.8 and 5.4 M−1 cm−1 for 1 and 2, respectively; Table 2). The supramolecular polymerization, and the concomitant AIE effect, produces a clear increase in the BCPL values, especially for compounds 2, that reach a BCPL = 40.6 M−1 cm−1, that lay among the highest for other reported helicenes (Table 2).17
Conclusions
The synthesis of two [6]helicene-based luminophores 1 and 2, endowed with two cyano-stilbene fragments, is reported. Both compounds 1 and 2 form supramolecular polymers in a cooperative manner by the operation of intermolecular H-bonding interactions between the amide functional groups. Importantly, the 2,15- and 4,13-disubstitution of the reported [6]helicenes plays a relevant role in the self-assembling features of the investigated luminophores. Thus, compound 1, with a 2,15-disubstitution pattern, self-assembles in a zig-zag fashion with a negligible interaction between the [6]helicene cores but with the π-stacking of the cyano-stilbene fragments. The more accessible π-surface of compound 2, in which the peripheral substituents are in the 4 and 13 positions, yields a highly stable supramolecular polymers with a high degree of cooperativity due to the efficient π-stacking of the whole aromatic units. The enantioenriched samples of the (M) and (P) enantiomers of both 1 and 2 display rich CD spectra both in the molecularly dissolved and in the aggregated states. The changes observed in the CD spectra in MCH and CHCl3 allow deriving the thermodynamic parameters associated to the supramolecular polymerization. Interestingly, the formation of the supramolecular polymers is accompanied of a clear AIE effect that increases the emission quantum yield. Furthermore, the chiral character of the investigated compounds due to the presence of the [6]helicene core affords CPL activity for all the investigated compounds. The overall CPL efficiency of compounds 1 and 2, determined by the BCPL parameter, undergoes a remarkable increase upon the corresponding supramolecular polymerization providing values for the BCPL among the highest reported for [6]helicenes. The studies presented herein contribute to expand the establishment of structure–function relationship for CPL-emitting materials and depicts a new rare example of chiral supramolecular polymer from helicenic cores.Author contributions
L. López-Gandul (first author), synthesis, spectroscopic characterization, supramolecular polymerization mechanisms: investigation, data curation, formal analysis, visualization and revising the original draft; R. Rodríguez: chiroptical and emissive properties and writing; N. Vanthuyne: isolation of enantiomers; J. Crassous and L. Sánchez: conceptualization, supervision, validation, writing – review & editing and funding.Data availability
The data that supports the findings of this study have been included in the main text and ESI† and are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the MCIN/AEI of Spain (PID2020-113512GB-I00, TED2021-130285B-I00 and Juan de la Cierva Incorporación (IJC2020-042689-I) for R. R.) and Xunta de Galicia (Centro Singular de Investigación de Galicia acreditación 2019-2022, ED431G 2019/03), and the European Regional Development Fund (ERDF) is acknowledged. J. C. and N. V. thank the CNRS for financial support (GDR CHIRAFUN).References
- (a) H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620 CrossRef CAS PubMed; (b) M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Chem. Rev., 2018, 118, 8889 CrossRef CAS PubMed.
- (a) J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38 RSC; (b) G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145 CrossRef CAS PubMed; (c) A. Mateo-Alonso, Chem. Mater., 2023, 35, 1467 CrossRef CAS.
- (a) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed; (b) M. Wehner and F. Würthner, Nat. Rev., 2020, 4, 38 CAS.
- (a) F. García, R. Gómez and L. Sánchez, Chem. Soc. Rev., 2023, 52, 7524 RSC; (b) Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Chem. – Eur. J., 2019, 25, 5848 CrossRef CAS PubMed; (c) A. R. A. Palmans, E. W. Meijer, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (c) F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192 CrossRef PubMed.
- (a) E. E. Greciano, R. Rodríguez, K. Maeda and L. Sánchez, Chem. Commun., 2020, 56, 2244 RSC; (b) Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1 CrossRef PubMed; (c) J. S. Kang, S. Kang, J.-M. Suh, S. M. Park, D. K. Yoon, M. H. Lim, W. Y. Kim and M. Seo, J. Am. Chem. Soc., 2022, 144, 2657 CrossRef CAS PubMed.
- (a) E. S. Gauthier, L. Abella, N. Hellou, B. Darquié, E. Caytan, T. Roisnel, N. Vanthuyne, L. Favereau, M. Srebro-Hooper, J. A. G. Williams, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 8394 CrossRef CAS PubMed; (b) E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346 CrossRef PubMed; (c) M. A. Medel, R. Tapia, V. Blanco, D. Miguel, S. P. Morcillo and A. G. Campaña, Angew. Chem., Int. Ed., 2021, 60, 6094 CrossRef CAS PubMed.
- J. Roose, B. Z. Tang and K. S. Wong, Small, 2016, 12, 6495 CrossRef CAS PubMed.
- (a) S. Zhang, Y. Wang, F. Meng, C. Dai, Y. Cheng and C. Zhu, Chem. Commun., 2015, 51, 9014 RSC; (b) Y. Sheng, D. Shen, W. Zhang, H. Zhang, C. Zhu and Y. Cheng, Chem. – Eur. J., 2015, 21, 13196 CrossRef CAS PubMed.
- (a) G. H. Wagnière, in Comprehensive Chiroptical Spectroscopy, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, John Wiley & Sons, Inc., Hoboken, 2012, vol. 2, pp. 3–34 Search PubMed; (b) K. Dhbaibi, L. Favereau, M. Srebro-Hooper, M. Jean, N. Vanthuyne, F. Zinna, B. Jamoussi, L. Di Bari, J. Autschbach and J. Crassous, Chem. Sci., 2018, 9, 735 RSC.
- (a) Z. Xie, V. Stepanenko, K. Radacki and F. Würthner, Chem. – Eur. J., 2012, 18, 7060 CrossRef CAS PubMed; (b) J. Buendía, E. E. Greciano and L. Sánchez, J. Org. Chem., 2015, 80, 12444 CrossRef PubMed; (c) M. Wehner, M. I. S. Röhr, V. Stepanenko and F. Würthner, Nat. Commun., 2020, 11, 5460 CrossRef CAS PubMed.
- (a) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS PubMed; (b) M. Gingras, Chem. Soc. Rev., 2013, 42, 968 RSC.
- (a) R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. Kumar Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709 CrossRef PubMed; (b) R. Rodríguez, C. Naranjo, A. Kumar, K. Dhbaibi, P. Matozzo, F. Camerel, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, Chem. – Eur. J., 2023, 29, e202302254 CrossRef PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846 CrossRef CAS PubMed.
- L. López-Gandul, C. Naranjo, C. Sánchez, R. Rodríguez, R. Gómez, J. Crassous and L. Sánchez, Chem. Sci., 2022, 13, 11577 RSC.
- L. Arrico, L. Di Bari and F. Zinna, Chem. – Eur. J., 2021, 27, 2920 CrossRef CAS PubMed.
- C. Shen, F. Gan, G. Zhang, Y. Ding, J. Wang, R. Wang, J. Crassous and H. Qiu, Mater. Chem. Front., 2020, 4, 837 RSC.
- (a) F. García, P. M. Viruela, E. Matesanz, E. Ortí and L. Sánchez, Chem. – Eur. J., 2011, 17, 7755 CrossRef PubMed; (b) I. Helmers, M. S. Hossain, N. Bäumer, P. Wesarg, B. Soberats, L. S. Shimizu and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202200390 CrossRef CAS PubMed.
- (a) M. Wehner, M. I. S. Röhr, M. Böhler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092 CrossRef CAS PubMed; (b) E. E. Greciano, S. Alsina, G. Ghosh, G. Fernández and L. Sánchez, Small Methods, 2020, 4, 1900715 CrossRef CAS; (c) C. Naranjo, S. Adalid, R. Gómez and L. Sánchez, Angew. Chem., Int. Ed., 2023, 62, e202218572 CrossRef CAS PubMed; (d) B. Matarranz, S. Díaz-Cabrera, G. Ghosh, I. Carreira-Barral, B. Soberats, M. García-Valverde, R. Quesada and G. Fernández, Angew. Chem., Int. Ed., 2023, 62, e202218555 CrossRef CAS PubMed.
- T. Dünnebacke, K. K. Kartha, J. M. Wiest, R. Q. Albuquerque and G. Fernández, Chem. Sci., 2020, 11, 10405 RSC.
- (a) F. Aparicio, S. Cherumukkil, A. Ajayaghosh and L. Sánchez, Langmuir, 2016, 32, 284 CrossRef CAS PubMed; (b) L. Ji, Y. Sang, G. Ouyang, D. Yang, P. Duan, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 844 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS PubMed.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343 CrossRef CAS PubMed.
- T. Ikeda, K. Hirano and T. Haino, Mater. Chem. Front., 2018, 2, 468 RSC.
- Y. Sang, D. Yang, P. Duan and M. Liu, Chem. Commun., 2019, 55, 11135 RSC.
- Y. Xue, C. Zhang, T. Lv, L. Qiu and F. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300972 CrossRef CAS PubMed.
- K. Y. Kim, S. H. Jung, S. Lee, C. J. Moon, Y. Choi, M. Y. Choi and J. H. Jung, J. Phys. Chem. C, 2018, 122, 22143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02110j |
| This journal is © The Royal Society of Chemistry 2024 |