
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiophysical insights into sugar-dependent medium acidification promoting YfaL protein-mediated Escherichia coli self-aggregation, biofilm formation and acid stress resistance†
Yankel
Chekli
a,
Stanislas
Thiriet-Rupert
a,
Céline
Caillet
 b,
Fabienne
Quilès
c,
Hélène
Le Cordier
b,
Emilie
Deshayes
a,
Benjamin
Bardiaux
d,
Thierry
Pédron
e,
Marie
Titecat‡
e,
Laurent
Debarbieux
e,
Jean-Marc
Ghigo
a,
Grégory
Francius
c,
Jérôme F. L.
Duval
b and
Christophe
Beloin
*c
b,
Fabienne
Quilès
c,
Hélène
Le Cordier
b,
Emilie
Deshayes
a,
Benjamin
Bardiaux
d,
Thierry
Pédron
e,
Marie
Titecat‡
e,
Laurent
Debarbieux
e,
Jean-Marc
Ghigo
a,
Grégory
Francius
c,
Jérôme F. L.
Duval
b and
Christophe
Beloin
*c
aInstitut Pasteur, Université Paris Cité, Genetics of Biofilms Laboratory, 75015 Paris, France
bUniversité de Lorraine, CNRS, Laboratoire Interdisciplinaire des Environnements Continentaux (LIEC), F-54000 Nancy, France
cUniversité de Lorraine, CNRS, LCPME UMR 7564, F-54000 Nancy, France. E-mail: christophe.beloin@pasteur.fr
dInstitut Pasteur, Université Paris Cité, Bacterial Transmembrane Systems Unit, CNRS UMR 3528, Paris, France
eInstitut Pasteur, Université Paris Cité, Bacteriophage Bacterium Host, 75015 Paris, France
First published on 17th August 2024
Abstract
The ability of bacteria to interact with their environment is crucial to form aggregates and biofilms, and develop a collective stress resistance behavior. Despite its environmental and medical importance, bacterial aggregation is poorly understood and mediated by few known adhesion structures. Here, we identified a new role for a surface-exposed Escherichia coli protein, YfaL, which can self-recognize and induce bacterial autoaggregation. This process occurs only under acidic conditions generated during E. coli growth in the presence of fermentable sugars. These findings were supported by electrokinetic and atomic force spectroscopy measurements, which revealed changes in the electrostatic, hydrophobic, and structural properties of YfaL-decorated cell surface upon sugar consumption. Furthermore, YfaL-mediated autoaggregation promotes biofilm formation and enhances E. coli resistance to acid stress. The prevalence and conservation of YfaL in environmental and clinical E. coli suggest strong evolutionary selection for its function inside or outside the host. Overall, our results emphasize the importance of environmental parameters such as low pH as physicochemical cues influencing bacterial adhesion and aggregation, affecting E. coli and potentially other bacteria's resistance to environmental stress.
1 Introduction
Bacterial colonization of environments and host tissues depends on bacteria ability to adhere to surfaces and withstand disturbances. This adhesion is primarily facilitated by cell surface appendages called adhesins,1 which promote cell adhesion to various surfaces and enable interactions leading to the formation of aggregates.2Bacterial aggregation occurs in two ways: autoaggregation, where identical adhesins on the same type of bacteria interact,2,3 and co-aggregation, where the interaction occurs between different adhesins or surface structures expressed by genetically different bacteria.4,5 Bacterial aggregation is crucial for forming multicellular communities and developing collective functions in both environmental and pathogenic contexts.6,7 For example, during starvation, soil-dwelling myxobacteria form large aggregates called fruiting bodies to ensure their survival.8–11 Similarly, opportunistic pathogens like Staphylococcus aureus,12Streptococcus pyogenes13 and Pseudomonas aeruginosa14,15 form aggregates during infections, which better protects them against antibiotics compared to planktonic cells.16,17
Escherichia coli, a Gram-negative bacterium with a diderm envelope, can colonize diverse environments, including the mammalian digestive tract, soil and water.18 It possesses various adhesins such as chaperone-usher fimbriae19 and surface proteins secreted by the type V secretion system including trimeric autotransporters, two-partner system adhesins and autotransporters.20 Autotransporters (or ATs) are the largest group of type V secreted proteins and share specific structural and functional features: an N-terminal signal sequence that directs the protein to the general secretion system, a passenger or alpha domain that provides protein function, and a C-terminal translocator or β-barrel domain.20 On the other hand, Chaperone–Usher fimbriae (or CU-fimbriae) are linear heteropolymers composed of different subunits, including pilins and a tip adhesin directly involved in bacterial adhesion.19 While some adhesins like Type 1 fimbriae, Yad CU-fimbriae or Ag43 autotransporter are well-studied,21–26 others remain uncharacterized, and most of the corresponding encoding genes are cryptic under laboratory conditions.27–29 Our laboratory has shown that, when produced, most E. coli K12 ATs and CU-fimbriae can promote adhesion and biofilm formation on various abiotic surfaces,27,29 though their specificities and functions, and ability to promote bacteria–bacteria interactions are not well understood yet.
Here, we investigated the homotypic and heterotypic interaction abilities of six ATs (Ag43, YpjA, YcgH, YdhQ, YcgV and YfaL), one inverted AT (YeeJ) and eight CU-fimbriae (Type 1 fimbriae, Yad, Ycb, Yfc, Yeg, Sfm, Yra and Ybg) in E. coli K12. We found that YfaL, a highly conserved and prevalent AT adhesin, promotes bacterial autoaggregation through homotypic interactions. We showed that an acidic environment, acquired during cell growth in the presence of sugar, is a necessary condition to induce YfaL post-transcriptional regulation and YfaL-dependent autoaggregation, which, in turn, enhances both biofilm formation and E. coli survival under low pH conditions. Our findings emphasize the critical role of environmental pH in modulating autoaggregation and bacterial resistance to environmental stress.
2 Results
2.1 YfaL mediates bacterial autoaggregation when grown in the presence of arabinose
To investigate the interactions between the eight CU-fimbriae and the seven type V secreted proteins encoded in the E. coli K12 genome, which are mostly cryptic under laboratory growth conditions,27,28 we created two sets of isogenic strains. We placed CU-fimbriae operons under the control of the constitutive promoter λPR (PcL),27 and the genes encoding the type V secreted proteins under the control of the arabinose-inducible pBAD promoter.29 Each strain was tagged with either blue (CFP) or yellow (YFP) fluorescent reporters at the lambda phage attachment site (λATT), and we deleted the ag43 gene to prevent Ag43-dependent autoaggregation (the E. coli Δag43 mutant will therefore be referred to as our wild-type strain). We monitored homotypic and heterotypic interactions by mixing CFP and YFP strains in Miller's Lysogeny Broth (LB).Among all interaction assays conducted to detect aggregation, only Ag43 (our positive control) and YfaL – a highly conserved AT adhesin – induced homotypic autoaggregation (Fig. 1A and B). However, expression of yfaL under the constitutive PcL promoter instead of the arabinose-inducible pBAD promoter did not lead to autoaggregation of the PcLyfaL strain, unless arabinose was added to the growth medium (Fig. 1C and D). This dependency on arabinose was confirmed in a strain where yfaL expression occurred from the permissive chromosomal λATT site (Fig. 1D). These findings indicated that YfaL-mediated autoaggregation was arabinose-dependent under the tested experimental conditions.
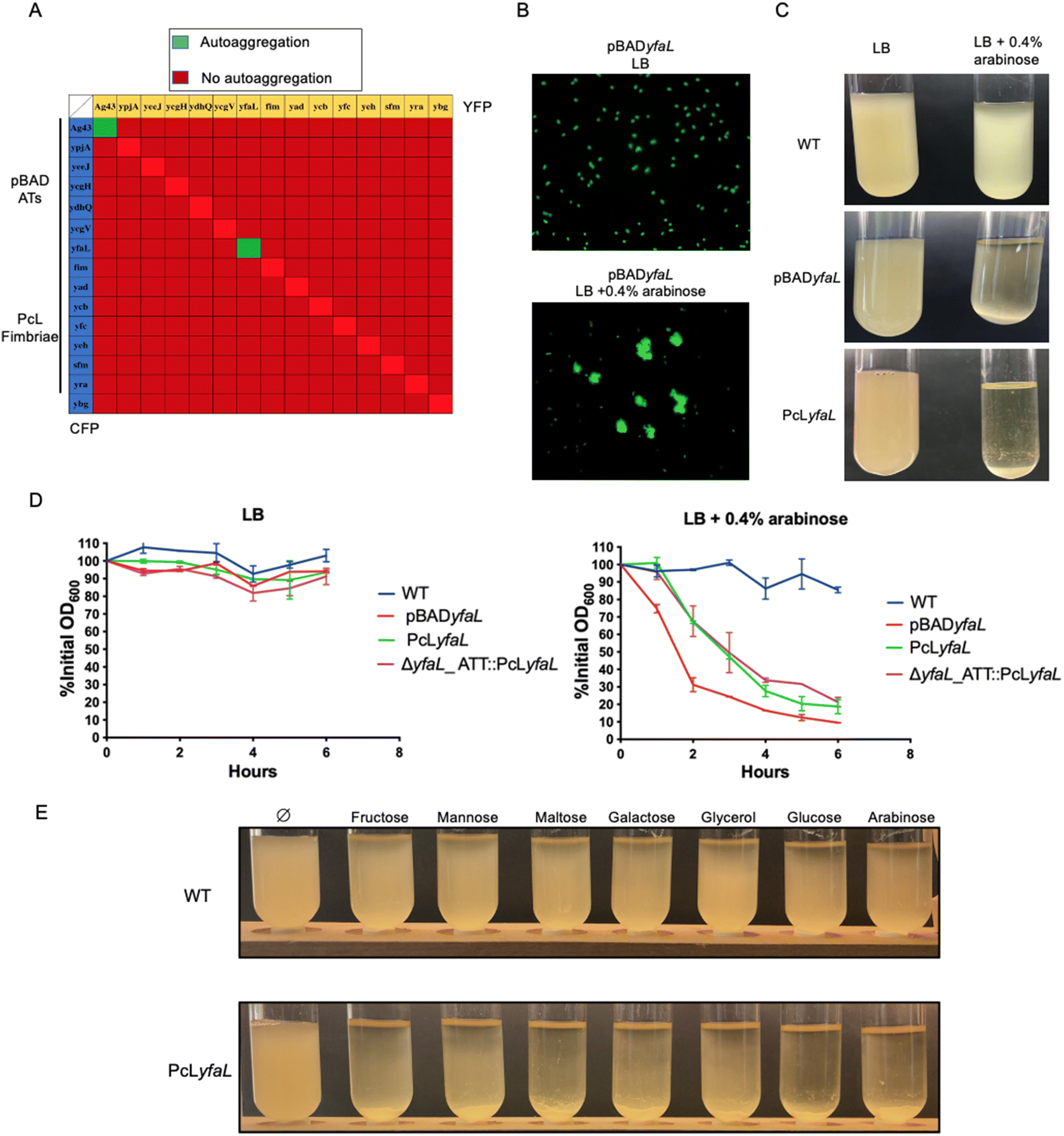 | ||
| Fig. 1 YfaL can mediate E. coli autoaggregation through homotypic interactions in the presence of fermentable sugars. (A) Grid representing the different interaction scenarios tested: the diagonal represents the homotypic interactions tested while squares outside that diagonal represent the tested heterotypic interactions. (B) Epifluorescence microscopy of the pBADyfaL YFP strain grown in LB or LB + 0.4% arabinose, magnification ×1000. (C) Macroscopic observation of aggregate formation by pBADyfaL and PcLyfaL strains after 5 hours on the bench. (D) Aggregation curves of bacterial cultures. Results are expressed as a percentage of the OD measured at the top of the tube at T0, the time at which cultures were homogenized. A 100% OD measurement means the absence of aggregation, and a decrease in % OD indicates the formation of aggregates. Plotted data represent the mean ± standard deviation of 3 biological replicates (n = 3) (each biological replicate is the mean of 2 technical replicates). (E) Picture of aggregation tubes containing LB supplemented with different carbon sources after overnight culture. The tubes in the upper panel correspond to the WT strain and tubes in the lower panel correspond to the strain PcLyfaL which expresses yfaL constitutively. | ||
2.2 Sugar uptake induces medium acidification driving YfaL-mediated cell autoaggregation
To further investigate YfaL-mediated arabinose-dependent autoaggregation, we supplemented LB growth medium with various simple carbohydrate sugars (fructose, mannose, maltose, galactose, glycerol and glucose). E. coli PcLyfaL cultures exhibited autoaggregation under all tested conditions (Fig. 1E), indicating that YfaL-mediated aggregation is not specific to arabinose and that sugar addition impacts YfaL activity. E. coli sugar metabolism in the LB medium causes medium acidification through mixed fermentation by-products.30 Accordingly, the pH of the culture dropped from 7 without added sugars to 4.5–5 with sugar supplements. Higher sugar concentrations led to more acidic conditions and faster YfaL-mediated autoaggregation (ESI Fig. S1†). Buffering of LB medium to pH 7 with MOPS prevented YfaL-mediated aggregation, regardless of the presence of sugar (Fig. 2A and B). | ||
| Fig. 2 YfaL-mediated auto-aggregation is operative under acidic conditions. (A) Macroscopic observation of aggregate formation by the PcLyfaL strain grown in LB or LB-MOPS supplemented with 0.2% glucose (Glc) or 0.4% arabinose (Ara), after 5 hours on the bench. (B, C and D) Aggregation curves of bacterial cultures. In (B), the aggregation was monitored in cultures grown in the presence or absence of MOPS and adjusted with their corresponding spent growth medium. In (C) and (D), after growth the bacteria were resuspended in PBS medium adjusted to different pH values instead of the spent growth medium. Results are expressed as a percentage of the OD measured at the top of the tube at T0, the time at which cultures were homogenized. For each strain a measurement is taken at the exact same position in the tube over time. A 100% OD measurement corresponds to the absence of aggregation, and a decrease in % OD indicates aggregate formation. Plotted data represent the mean ± standard deviation of 3 biological replicates (n = 3, each replicate is the mean of 2 technical replicates). | ||
To further explore the impact of medium acidification on YfaL-mediated autoaggregation, we transferred stationary phase E. coli PcLyfaL cultures grown in absence of sugar directly into PBS adjusted to pH = 4, 5, 6 or 7. No aggregation of PcLyfaL bacteria was observed (Fig. 2C), thereby suggesting that gradual acidification is necessary for YfaL-mediated autoaggregation to occur. When PcLyfaL bacteria grown in the presence of sugar acidifying the medium were resuspended in PBS at different pH values, aggregation occurred under all conditions, with faster aggregation of cells resuspended at lower pH (Fig. 2D). These findings indicated that medium acidification following bacterial sugar metabolism is essential to induce YfaL-dependent aggregation. Subsequently adjusting pH to 7 necessarily increased cell surface charge and slowed aggregation but did not prevent it entirely, which suggests that aggregation is influenced by factors other than cell surface charge reduction with decreasing pH.
2.3 Sugar-dependent medium acidification increases YfaL protein levels and induces changes in the YfaL conformation
Large E. coli surface adhesins, such as type 1 fimbriae or flagella, can mask smaller structures such as Ag43 (10 nm for Ag43, https://www.rcsb.org/structure/4KH3) and hinder Ag43-mediated autoaggregation.31,32 We hypothesized that YfaL-mediated autoaggregation in an acidified medium could occur due to the reduced production of type 1 fimbriae, flagella or curli. However, deleting the genes encoding these large adhesins in a PcLyfaL strain did not result in aggregation without sugar supplementation (ESI Fig. S2†).Alternatively, sugar-mediated acidification of the medium could alter YfaL protein level and structure, favoring YfaL-mediated autoaggregation. To test this hypothesis, we performed denaturing protein gel electrophoresis followed by immunodetection using antibodies against the passenger domain of YfaL, as well as immunofluorescence to detect cell-surface YfaL (Fig. 3). These analyses revealed that sugar and the resulting pH reduction increased the amount of YfaL on the cell surface (Fig. 3C and D). This effect was specific to YfaL, as Ag43 level did not change upon sugar supplementation (ESI Fig. S3†). Moreover, growth in the presence of sugars led to an increase of cell size. Consistent with an impaired YfaL-mediated aggregation when the medium was buffered with MOPS (see Fig. 2A and B), immunodetection showed much lower YfaL levels (Fig. 3E and F). This suggested that medium acidification in the presence of sugars increases the export of YfaL proteins to the bacterial surface. This finding was unexpected since yfaL was under the control of a constitutive promoter. The higher YfaL protein levels observed with sugar could result from the stabilization of yfaL mRNA and/or post-translational modifications that stabilize the protein. Quantitative RT-PCR (qRT-PCR) showed that the yfaL mRNA levels were slightly higher with sugar supplementation than those measured in the absence of sugar (Fig. 3G). Additionally, yfaL mRNA levels in LB-MOPS were much lower than in LB without sugar, indicating that medium buffering negatively impacts yfaL mRNA levels regardless of the presence of sugar. Finally, the addition of sugar to LB-MOPS restored mRNA production at levels comparable to those measured in LB without sugar (Fig. 3G). These results suggested that sugar-dependent acidification stabilizes yfaL mRNA, but sugar itself contributes to yfaL mRNA stabilization independently of pH.
 | ||
Fig. 3 The presence of sugar and medium acidification modify YfaL migration properties, enhance the level of YfaL exposed at the E. coli surface as well as the yfaL mRNA level. (A) Domain organization of YfaL. Molecular weight was estimated with Expasy.33 The diagram is not to scale. (B) Structural model of YfaL predicted by alpha-fold (https://alphafold.ebi.ac.uk/entry/P45508![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34,35). The β-domain and the passenger domain are in blue and red, respectively. (C) Western blot of whole cell extracts from WT and PcLyfaL strains, using a primary rabbit antibody directed against the passenger domain of YfaL or mouse antibody against αRNAP and secondary antibodies HRP-linked anti-rabbit or anti-mouse, Glc = glucose, Ara = arabinose. The YfaL full-length protein is indicated by an arrow. (D) Immunofluorescence of whole cells from WT and PcLyfaL strains, using a primary rabbit antibody directed against the passenger domain of YfaL and a secondary Alexa 488 conjugated anti-rabbit antibody, scale bar = 2 μm. (E) Western blot of whole cell extracts from PcLyfaL strain grown in LB or LB-MOPS supplemented with 0.2% glucose (Glc) or 0.4% arabinose (Ara), using primary rabbit antibodies directed against the passenger domain of YfaL or primary mouse antibodies against alpha-RNAP, and secondary antibodies HRP-linked anti-rabbit or anti-mouse, Glc = glucose, Ara = arabinose. The YfaL full-length protein is indicated by an arrow. (F) Immunofluorescence on whole cells from PcLyfaL strain grown in LB MOPS or LB MOPS ara, using primary rabbit antibodies directed against the passenger domain of YfaL and secondary Alexa 488 conjugated anti-rabbit antibodies, scale bar = 2 μm. (G) qRT-PCR performed with yfaL specific primers. Six biological replicates (n = 6) of the PcLyfaL strain grown in LB, LB ara, LB MOPS and LB MOPS ara have been performed. Each biological replicate is the mean of 3 technical replicates. *p < 0.05; **p < 0.005, non-parametric two-tailed Mann–Whitney test. Only significantly different samples are indicated. 34,35). The β-domain and the passenger domain are in blue and red, respectively. (C) Western blot of whole cell extracts from WT and PcLyfaL strains, using a primary rabbit antibody directed against the passenger domain of YfaL or mouse antibody against αRNAP and secondary antibodies HRP-linked anti-rabbit or anti-mouse, Glc = glucose, Ara = arabinose. The YfaL full-length protein is indicated by an arrow. (D) Immunofluorescence of whole cells from WT and PcLyfaL strains, using a primary rabbit antibody directed against the passenger domain of YfaL and a secondary Alexa 488 conjugated anti-rabbit antibody, scale bar = 2 μm. (E) Western blot of whole cell extracts from PcLyfaL strain grown in LB or LB-MOPS supplemented with 0.2% glucose (Glc) or 0.4% arabinose (Ara), using primary rabbit antibodies directed against the passenger domain of YfaL or primary mouse antibodies against alpha-RNAP, and secondary antibodies HRP-linked anti-rabbit or anti-mouse, Glc = glucose, Ara = arabinose. The YfaL full-length protein is indicated by an arrow. (F) Immunofluorescence on whole cells from PcLyfaL strain grown in LB MOPS or LB MOPS ara, using primary rabbit antibodies directed against the passenger domain of YfaL and secondary Alexa 488 conjugated anti-rabbit antibodies, scale bar = 2 μm. (G) qRT-PCR performed with yfaL specific primers. Six biological replicates (n = 6) of the PcLyfaL strain grown in LB, LB ara, LB MOPS and LB MOPS ara have been performed. Each biological replicate is the mean of 3 technical replicates. *p < 0.05; **p < 0.005, non-parametric two-tailed Mann–Whitney test. Only significantly different samples are indicated. | ||
In addition to increased YfaL protein levels, we also observed changes in the apparent molecular weight of YfaL in the presence of sugar, suggesting protein modifications. Without sugar, we detected two bands using an anti-YfaL antibody in a strain constitutively expressing yfaL (PcLyfaL): a band between 100 and 130 kDa, likely monomers, and a band at about 250 kDa, likely dimers of the full-length protein. These bands were absent in WT strain, confirming the cryptic state of the yfaL gene under laboratory conditions (Fig. 3C). With glucose or arabinose, we also detected at least three additional bands between 130 and 250 kDa in the strain PcLyfaL, potentially representing modified YfaL forms or dimers of YfaL passenger domains (Fig. 3C).
Altogether, these results evidenced that medium acidification with sugar leads to changes in the YfaL cell-surface quantity and structure, thereby contributing to bacterial autoaggregation.
2.4 Sugar-dependent acidification of the medium alters the hydrophobic features of cells producing YfaL
Medium acidification upon E. coli metabolism of sugars could favor bacterial autoaggregation by altering the hydrophilic/hydrophobic surface properties of YfaL-expressing bacteria. To investigate this, we used Chemical Force Microscopy (CFM) of cells grown with or without sugar. AFM-tips were modified with 1-dodecanethiol to create hydrophobic probes. We measured the adhesion force between the probes and the bacterial surface by retracting the probes after contact. For WT and PcLyfaL strains grown without sugar, the tip-to-cell retraction force–distance curves showed multiple rupture events over distances up to 350 nm with values of the rupture (or adhesion) forces of 0.1–0.4 nN, indicating the stretching or unfolding of hydrophobic macromolecules on the cell surface (Fig. 4A and C). For cells grown with sugar in acidified medium, unlike the WT strain, the shape of the force–distance curves was dramatically modified only for the PcLyfaL strain, with an increased number of rupture or adhesive events that spanned over a longer rupture distance (up to 600 nm) and with an increase in the magnitude of the adhesion force (0.2–0.8 nN) (Fig. 4B and D). The analysis of multiple retraction force curves collected on several individual bacteria (at least 10 per examined condition) confirmed these effects and the increase of both the adhesive hydrophobic force and the tip-to-cell rupture distance (or molecular cell surface stretching) only for PcLyfaL strain grown with sugar (Fig. 4E, F and ESI Fig. S4, Table S3†). This increase in the PcLyfaL surface hydrophobicity likely results from a larger exposure of YfaL hydrophobic residues due to structural changes induced by sugar-mediated acidification. It also correlated with the higher YfaL protein levels observed by immunoblotting and immunofluorescence (Fig. 3C and D). The changes in YfaL stretching properties detected by CFM could be due to the decrease of polar interactions along the protein structure caused by pH changes, increasing its stretchability. The violin plots displayed in Fig. 4F clearly evidence bimodal distributions of rupture distances for both WT and PcLyfaL cells grown in the presence or absence of sugar in the culture medium. Accordingly, this bimodality of the distributions cannot be explained by, nor connected with, the only structure properties of YfaL and its related folding/unfolding features (ESI Fig. S4†). | ||
| Fig. 4 Presence of fructose increases the hydrophobicity and the unfolding of surface material of cells expressing yfaL. Representative retraction force curves recorded from CFM experiments performed on E. coli WT and E. coli PcLyfaL cultures grown in LB (A and C, respectively) or in LB supplemented with 0.3% fructose (B and D, respectively). Force curves were measured using 1-dodecanthiol coated AFM-tip in PBS buffer with a retraction rate of 1 μm s−1 and a dwell time of 100 ms. (E) CFM measurements of the hydrophobic adhesion force (nN) between the hydrophobic AFM probe (coated by –CH3) and the E. coli WT or PcLyfaL strains grown in LB or in LB supplemented with 0.3% fructose. Violin plots with box plot overlay indicate the shift in median adhesion force (nN) for bacterial cells grown in LB (control) and LB with 0.3% fructose. (F) As in (E) but for the rupture distance. The latter distance is defined as the distance at which the last rupture event is detected in the cell-to-tip force curve upon retraction of the tip from the cell surface. Reported hydrophobic forces and rupture distances were evaluated on each collected retraction curve (n curves). *p < 0.05; **p < 0.005, ****p < 0.00005 non-parametric two-tailed Mann–Whitney test. Only significantly different samples are indicated. | ||
We also recorded infrared (IR) spectra of WT and PcLyfaL strains grown overnight in LB with or without sugar (cf. ESI Fig. S5† and the corresponding supplementary discussion). The spectra of both strains grown in LB with sugar show acetate production, consistent with medium acidification and the ability of E. coli to ferment sugars into acetate/acetic acid.33 For both strains grown with sugar, the spectra also indicated higher nucleic acid content compared to proteins, suggesting increased metabolic activity in this medium. In the amide I and II regions (1700–1500 cm−1), specific bands assigned to β-sheet secondary structures of proteins34 appeared only for the PcLyfaL strain grown with sugar. This increase in the number of β-sheets correlates with the higher amount of YfaL on the surface of PcLyfaL strain in the presence of sugar (Fig. 3D). Additionally, the spectra showed lower production of extracellular polysaccharides (EPS) in PcLyfaL with sugar, which may also enhance YfaL exposure on the cell surface.
Taken together, CFM and IR experiments confirmed that sugar-induced medium acidification increases the YfaL presence on the cell surface. The CFM results also revealed enhanced hydrophobic surface properties, which may contribute to cell–cell aggregation through YfaL self-interactions.
2.5 Electrophoresis reveals marked changes in the electrohydrodynamic properties of YfaL-producing bacteria grown in sugar-dependent acidified medium
To further examine the effect of sugar-mediated acidification on the physicochemical surface properties of yfaL-expressing cells, we measured the electrophoretic mobility μ35–38 of WT and PcLyfaL grown overnight in the absence and presence of sugar in 1 to 150 mM KNO3 electrolyte (concentration denoted as cKNO3) at pH 4.6. This pH corresponds to the pH measured in stationary phase culture grown with sugar (Fig. 5A). The effect of sugar-mediated acidification was further analyzed by plotting the mobility difference (μ0.3% fructose − μno fructose)/μno fructose, where μ0.3% fructose and μno fructose are the cell mobilities with and without sugar, respectively (Fig. 5B). | ||
| Fig. 5 The presence of sugar during PcLyfaL strain growth leads to significant changes in both the electrostatic and electroosmotic flow-permeability properties of the cell surface. Dependence of the electrophoretic mobility, μ, of WT and PcLyfaL on KNO3 electrolyte concentration, cKNO3, at pH 4.6 after growth in the presence or absence of 0.3% fructose (indicated). In (A), dotted lines are fits of electrophoretic data using Ohshima eqn (1)–(4) valid at sufficiently large cKNO3 (see details in the Methods section). In (B), symbols represent relative changes in the electrophoretic mobility of WT and PcLyfaL grown in the presence of fructose as compared to the situation where fructose is absent from the growth medium. Dotted lines in (B) are guides to the eye. Each data point in this figure corresponds to a triplicate measurement (n = 3). | ||
μ was negative for both WT and PcLyfaL strains across all cKNO3 values, which is consistent with the expected negative charge of bacterial surfaces (Fig. 5A). The magnitude of μ decreases with increasing cKNO3 due to electrolyte ions screening the bacterial surface charge. As reported in previous studies,35–38μ asymptotically approaches a non-zero plateau value at high cKNO3, indicating the penetration of electroosmotic flow within the soft bacterial surface structure.39–41
Electrophoretic properties of WT and PcLyfaL strains grown without sugar are similar (Fig. 5A), indicating that YfaL contribution to the electrophoretic properties of the PcLyfaL strain is minimal under such conditions. However, unlike for the WT strain, the addition of sugar severely reduces the dependence of μ on cKNO3 for the PcLyfaL strain, decreasing μ at cKNO3 < 30 mM and increasing it at cKNO3 > 30 mM (in absolute value) (Fig. 5A). Fig. 5B confirms that these changes in PcLyfaL mobility μ in the presence of sugar are most pronounced at high cKNO3 (>20–30 mM), where bacterial surface electrostatics is most efficiently screened, and that WT strain's mobility in this range of cKNO3 values is unaffected by the presence of sugar. According to the theory for soft surface electrophoresis (SSE),40,41 these findings suggest that the presence of sugar during the growth of the PcLyfaL strain significantly changes the electrostatic and electroosmotic flow-permeability properties of the YfaL-decorated PcLyfaL strain surface. This is supported by evaluating the effective concentration of charged groups on the cell surface (|ρ0|/F, where ρ0 is the negative density of cell surface charges and F is the Faraday number) and the electroosmotic flow penetration within the cell surface, reflected by the Brinkman length (1/λ0, see the Methods section and eqn (1)–(4) therein) (ESI Table S4†). The addition of sugar impacts |ρ0|/F and 1/λ0 only for the PcLyfaL strain, with an ∼10 fold decrease of |ρ0| and ∼4 fold increase of 1/λ0. These changes indicate significant physicochemical modifications to the YfaL-surface coating of the PcLyfaL strain potentially involving a decrease in the number of dissociable groups carried by a given YfaL, changes in their protonation/deprotonation properties, variations in the total surface amount of YfaL, and/or changes in the YfaL conformation. Changes in the YfaL surface concentration and/or conformation would affect the overall friction exerted by the cell surface on the electroosmotic flow (a property that is subsumed in the value of 1/λ0), altering the YfaL surface layer thickness and, in turn, the density ρ0 of the charges this layer carries.
Additional measurements performed at pH 5.7 (ESI Fig. S6 and Table S4†) show the same electrokinetic pattern as that discussed at pH 4.6 (refer to the discussion below ESI Fig. S6†). However, at this higher pH, sugar in the growth medium results in less pronounced changes of |ρ0| and 1/λ0, decreasing and increasing them by only a factor of ∼2, respectively. This aligns with Fig. 2, which indicates that medium alcalinisation after growth in the presence of sugar decreases only slightly the capability of PcLyfaL to autoaggregate.
In conclusion, electrokinetic experiments demonstrated significant changes of the electrostatic and hydrodynamic properties (ρ0 and 1/λ0, respectively) of the PcLyfaL strain following sugar-induced acidification of the growth medium.
2.6 YfaL mediated autoaggregation correlates with enhanced biofilm formation and tolerance to low pH
The presence of bacterial aggregates can enhance biofilm formation and increase tolerance to environmental stresses.2 Therefore, we assessed whether sugar-induced, YfaL-mediated autoaggregation displayed such properties (Fig. 6A). As previously shown, the expression of yfaL promoted biofilm formation29 even without sugar, suggesting that YfaL may enhance biofilm formation by increasing initial adhesion. Moreover, whilst adding fructose during bacterial growth did not affect the WT strain's biofilm formation, it moderately but significantly increased biofilm formation in the PcLyfaL strain by 1.6 times compared to LB-without-fructose conditions. These results indicated that, in addition to YfaL's inherent ability to promote cell adhesion, YfaL-driven autoaggregation correlates with a ca. 40% increase in biofilm formation (Fig. 6A). | ||
| Fig. 6 YfaL-mediated autoaggregation correlates with increased biofilm formation and resistance to acid medium conditions. (A) Boxplot representing 96 well-plate crystal violet biofilm formation assay after 24 hours of growth of WT and PcLyfaL strains in LB and LB + 0.3% fructose. Mean of the data pertaining to WT in LB was adjusted to 100%. For each strain and tested condition, 6 biological replicates (n = 6) have been realized, each biological replicate is the mean of 2 technical replicates. *p < 0.05; **p < 0.01, non-parametric two-tailed Mann–Whitney test. Only significantly different samples are indicated. (B) Boxplot representing the CFU mL−1 for WT and PcLyfaL strains after incubation for 1 h in media adjusted to pH 4.5 (pH value measured at the end of culture in the presence of 0.3% fructose), 2, 1.5 and 1 (indicated in each panel). Red-blue colors in (B) is the same as in (A). For each strain tested, 6 biological replicates (n = 6) (each replicate is the mean of 2 technical replicates) have been performed. *p < 0.05; **p < 0.01; non-parametric two-tailed Mann–Whitney test. Only significantly different samples are indicated. | ||
Given that YfaL-mediated autoaggregation occurs under acidic conditions, we hypothesized that these aggregates could be protected against prolonged acidic stress. To test this, we compared the acid stress resistance of non-aggregated WT bacteria to that of aggregates formed by the PcLyfaL strain after 1 hour in media at pH 4.5, 2, 1.5 and 1. Both strains showed similar survival at pH 4.5, 2 and 1.5, with the WT strain having a slight advantage at pH 4.5 and 2. However, almost all WT bacteria died at pH 1, whereas PcLyfaL aggregates survived (Fig. 6B). This indicates that under extreme acid conditions (pH = 1), YfaL-mediated aggregation correlates with significant improvement in bacterial survival.
2.7 In vivo relevance of YfaL-mediated aggregate formation
During the colonization of the gastrointestinal tract, E. coli must survive a wide range of pH conditions, including the highly acidic environment of the stomach (pH=1–2), which could promote YfaL-dependent aggregate formation. To test the role of YfaL-dependent autoaggregation in the survival of bacteria during stomach passage and subsequent gut colonization, we compared the in vivo colonization capacities of WTΔyfaL and PcLyfaL strains. We used a conventional streptomycin-treated mouse model for in vivo mono- and mixed-culture competition experiments with WTΔyfaL and PcLyfaL streptomycin-resistant strains grown in LB with sugar. First, we verified that both strains could colonize the gut of mice at similar levels in mono-colonization experiments (ESI Fig. S7,† WTΔyfaL and PcLyfaL panels). We then conducted in vivo competition experiments by colonizing mice with a 1:1 mix of both strains. Analysis of the feces over 8 days showed that both strains were present in similar concentrations (ESI Fig. S7,† Mix1 and Mix2 panels), indicating equivalent colonization capacities. This analysis demonstrated that under the tested conditions, YfaL-mediated aggregation does not significantly enhance the colonization capacity of E. coli K12.
2.8 YfaL is highly prevalent and conserved in the E. coli species, mostly diverging in its alpha-domain
Given this apparent lack of impact of YfaL-mediated aggregation on the gut colonization capacity of E. coli K12, we investigated its potential significance by assessing YfaL prevalence and conservation within the E. coli genus.First, we compared the prevalence of YfaL to that of 12 AIDA-I ATs (AatA, Ag43, EhaA, UpaB, UpaC, UpaE, UpaH, YcgV, YdeK, YejO, YfaL and YpjA,42) in 2053 E. coli genomes. YfaL was the second most prevalent (88.6%, Fig. 7A and ESI Table S5A, B†) and was present in all phylogroups (Fig. 7B and ESI Fig. S8, Table S5C†), indicating its potentially conserved function across different E. coli habitats.
 | ||
| Fig. 7 AIDA-I ATs prevalence in 2053 genomes of E. coli and sequence analysis of YfaL. (A) Prevalence of each AIDA-I ATs in the 2053 E. coli genomes analyzed. Prevalence is expressed as the percentage of all genomes in which a given AT is identified. (B) Heatmap showing the prevalence of each AIDA-I ATs in the same set of 2053 E. coli genomes detailed for each E. coli phylogroup. (C) Bar plot showing, for each phylogroup, the mutation frequency of YfaL alpha- and beta-domain relative to the reference strain MG1655 and compared to each other using the Wilcoxon test. | ||
To gain more insights into YfaL diversity, we analyzed the yfaL DNA sequence in these 2053 E. coli genomes. We then screened the corresponding protein sequences for mutations relative to E. coli K12 MG1655 YfaL, noting the mutation frequency of the alpha and beta-domains (Fig. 7C and ESI Table S6A†). Overall, the alpha-domain is significantly more variable than the beta-domain, especially in the phylogroups B2, F and G, with the phylogroups A and B1 showing the opposite trend (Fig. 7C and ESI Table S6B†). Additionally, the phylogroup distribution along YfaL protein phylogeny mirrored that of the E. coli species, suggesting that YfaL sequence evolution follows the phylogroup evolution rather than being driven by environmental factors or selection pressures on individual strains (ESI Fig. S8†). However, the higher mutation accumulation in the alpha-domain for the phylogroups B2, C, D, E, F, and G suggests a potential fine-tuning of the YfaL function, possibly influencing YfaL-mediated aggregation.
3 Discussion
The E. coli core and accessory genome contains many genes encoding fimbrial and afimbrial adhesins, allowing the bacteria to colonize diverse habitats.27,29,42,43 However, these genes often remain cryptic, and their functions are not well understood. Besides contributing to non-specific surface adhesion and biofilm formation, some adhesins may also mediate specific bacteria–bacteria interactions and aggregation, leading to bacterial stress tolerance.2 We tested 7 type V secreted proteins and 8 CU-fimbriae for their ability to mediate E. coli K12 aggregation through homotypic and heterotypic interactions. We found that, in addition to Ag43, a well-known mediator of E. coli aggregation, another common AT named YfaL, can also mediate autoaggregation, but only in low pH environments induced by the use of sugars during cell growth.Only a few adhesins are known to mediate bacterial aggregation in E. coli, most of them being type V secretion system proteins, such as the self-associating autotransporter (SAAT) group, which includes AIDA-I, TibA, EhaA and Ag43,26,44–46 or trimeric autotransporters like EhaG/UpaG or UpaJ.47–49 This limited number of known aggregation-mediating structures might be due to the need for specific environmental conditions to produce certain adhesins, and initiate or enhance their activity. For example, Yad fimbriae better stabilize at 30 °C compared to 37 °C.25 Type 1 fimbriae expression increases under oxygen-limited conditions50 and is completely suppressed under anaerobic conditions.51 Our study identified that sugar-dependent medium acidification is the environmental factor that reveals YfaL's autoaggregation function.
We evidenced that both sugar and pH condition are crucial in this autoaggregation process. Sugars increase cell size, potentially expanding the biosurface area for interactions, and stabilize YfaL mRNA, independently of medium acidification. pH affects the amount and the conformation/structure of the protein. While Ag43,24,52 AIDA45 and TibA44 can induce autoaggregation at neutral pH, their optimal aggregation pH is around 4, indicating that pH also influences their properties.44 However, the specific effects of pH on the aggregation of these ATs were not determined.
We showed that medium acidification changes YfaL's gel migration pattern, with new bands possibly indicating different protein conformations and/or modifications (phosphorylation, acetylation, glycosylation) likely responsible for the observed YfaL-mediated aggregation. To support this hypothesis, mass spectrometry and further biophysical analyses on the purified passenger domain of YfaL are needed to determine any structural and/or chemical modification triggered by sugar-mediated acidification.
Probing physicochemical surface properties at the individual cell level (Chemical Force Microscopy – CFM) and at the population scale (electrokinetics) showed that medium acidification significantly changes the hydrophobic/hydrophilic balance of the surface of the PcLyfaL strain. CFM results indicated an increase in hydrophobicity, while electrokinetics revealed a strong decrease in the cell surface charge/electrostatics in the peripheral cell surface structure. This was inferred from variations in cell flow-permeability properties depending on the presence/absence of sugar in the growth medium. Additionally, a larger surface amount of YfaL in the PcLyfaL strain grown in the presence of sugar suggests a thicker YfaL-surface coating and a higher tendency for electroosmotic flow to enter that coating, consistent with the increase of the Brinkman length 1/λ0. Infrared microscopy confirmed these surface properties by showing enhanced detection of β-sheet secondary structures. CFM further supported this point with a measured increase in the overall rupture force magnitude and the number of tip-to-cell rupture events when sugar was present in the growth medium. Overall, these surface property changes induced by acidification during growth with fermentable sugars likely reduce the repulsive cell–cell interaction energy barrier. This favors the recognition of YfaL proteins of neighboring cells, enhancing attractive interactions between hydrophobic cell surface YfaL components. To fully decipher the molecular mechanisms driving YfaL-mediated self-aggregation of PcLyfaL cells grown in the presence of sugar, future work should analyse the specific YfaL–YfaL protein interactions as a function of solution pH by measuring the force curves between AFM probes functionalized with purified YfaL passenger domain or anti-YfaL monoclonal antibodies, and the PcLyfaL strain grown or not in the presence of sugar.
According to AlphaFold predictions,53 the passenger domain of YfaL folds into a β-helix structure, similar to Ag43, with β-strands forming an extended, pseudo-repetitive helical shape (Fig. 3B). The YfaL passenger domain surface is mainly acidic, creating a highly electronegative surface at pH 7, unlike the more balanced Ag43 passenger domain at the same pH (ESI Fig. S9†). AlphaFold models and PROPKA analysis54 show that the pI of the YfaL passenger domain is 3.3, lower than Ag43's pI, which ranges from 4.8 to 5.3 based on available X-ray structures (4KH3, 7KO9, 7KOB, and 7KOH). This difference largely explains the predicted free energy of folding, with a minimum pH value of around 3.9 for YfaL. At pH 4.5, which is the pH of the culture after growth in the presence of sugar, YfaL's passenger domain shows a more balanced electrostatic surface potential, with a few electropositive patches. Despite the limits of this estimation, it explains the effect of pH on YfaL's surface electrostatics at the molecular level and possibly its structural organization. Thus, the sugar/pH dependent YfaL-mediated autoaggregation could be due to (i) a more favorable surface electrostatics at low pH as compared to neutral pH where negative surface charges prevent autoaggregation, (ii) an excess of negative charges at neutral pH, causing partial unfolding of the passenger domain structure, or (iii) combination of both. By analogy with the oligomeric structures of Ag4355 and H. influenzae Hap adhesin,56 whose passenger domains also fold as β-helices and auto-associate via lateral interactions, the repulsive coulombic interactions at pH 7 would prevent YfaL passenger domain from folding into a structure compatible with auto-association.
Throughout this study, we confirmed that YfaL promotes biofilm formation, and that YfaL-mediated aggregation increases E. coli biofilm formation and survival in an acidic environment. These aggregates could protect E. coli against other stresses, such as antibiotics, predation, chemical stresses or the immune system, as seen for other bacterial aggregates.2,16,17,57–60 Further studies are needed to determine whether YfaL-mediated aggregates can withstand such stresses. YfaL-mediated aggregation does not affect E. coli ability to colonize the gastro-intestinal tracts of mice after passing through acidic stomach. While we cannot exclude that YfaL-mediated aggregation plays a role in the host under different low pH conditions, such as during phagocytosis or in the presence of other chemical or biological stresses, one can envisage that it also plays a role in the environment where E. coli could face certain stressful episodes.
Our study reveals that YfaL is the second most prevalent autotransporter in E. coli, supporting and expanding on findings from previous reports on fewer strains or specific pathotypes of E. coli.49,61 YfaL shows high conservation (more than 80% identity), suggesting a strong selection pressure to maintain this protein and highlighting its potential specific functions and importance in E. coli physiology and behavior from both environmental and host contexts. However, we also found evidence that YfaL functions may have evolved, as indicated by the significant polymorphism in its passenger domain. This variability is particularly notable in the phylogroups B2, F and G, associated with pathogenic strains,62,63 where the passenger domain mutation frequency is more than three times higher than that of the β-domain. Conversely, the passenger domain is more conserved than the β-domain in the phylogroups A and B1, associated with commensal and generalist strains. This pattern extends to other ATs, where the distribution of AIDA-I ATs similarly clusters phylogroups by their pathogenic potential, distinguishing them from more generalist strains. Comparing YfaL with its orthologs, UpaI in uropathogenic E. coli or EhaC in entero-hemorrhagic E. coli, reveals functional differences. Both UpaI and YfaL promote biofilm formation, whereas EhaC does not.46,49 UpaI also exhibits self-aggregation capabilities,49 albeit weaker and slower compared to the relatively rapid aggregation observed with YfaL. In contrast, EhaC does not promote autoaggregation in the presence of sugar, possibly due to experimental conditions limiting growth to an insufficiently low pH.46 Despite YfaL's high sequence identity with EhaC (95.5%) and UpaI (79.3%), these functional discrepancies likely stem from both genetic polymorphism and differences in the experimental design aimed at studying aggregation dynamics.
4 Conclusions
In conclusion, our study demonstrates that among the numerous adhesins encoded in the E. coli K12 genome, only two – Ag43 and YfaL – are capable of self-recognition and mediation of homotypic bacterial autoaggregation. Furthermore, we found that YfaL, the second most widespread AT in E. coli, facilitates bacterial aggregation specifically under low pH conditions following growth in a medium supplemented with fermentable sugar. These findings underscore how environmental conditions can profoundly influence adhesin properties, enabling bacteria to swiftly adapt to varying environments.5 Methods
5.1 Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in ESI Table S1.† Bacteria were grown in Miller's Lysogeny Broth (LB) (Corning) supplemented with kanamycin (50 μg mL−1), streptomycin (100 μg mL−1), ampicillin (100 μg mL−1) or chloramphenicol (25 μg mL−1) when needed. When specified, cultures were also supplemented with sugar, glucose (0.2% final volume), arabinose (0.4%), fructose (0.3%), mannose (0.2%), maltose (0.2%), galactose (0.2%) or glycerol (0.2%). Liquid cultures were incubated at 37 °C with 180 rpm shaking. Solid cultures were performed on LB with 15% agar supplemented with the appropriate antibiotics. Bacteria were streaked on LB agar from glycerol stock prior to liquid cultures. Chemicals and media were purchased from Sigma-Aldrich.All experiments and genetic construction were done in E. coli K12 MG1655 (F−, λ−, rph-1) obtained from the E. coli genetic stock center CGSC#6300.
5.2 Strain construction
Insertion of the CFP and YFP cassettes as well as yfaL deletion were done by P1vir phage transduction from the existing strain in the laboratory collection or from the Keio collection.64 Insertions of the cassettes carrying the resistance genes for deletion or insertion of different promoters were done as follows: first, the recipient strain was transformed with the pKOBEGA plasmid, coding for the λ-Red operon under the control of the pBAD arabinose inducible promoter. After the induction of the λ-Red genes, electro-competent cells were prepared from this strain. In parallel, the cassettes carrying the resistance genes for deletion and the cassettes coding for the different promoters were amplified by PCR (PCR master mix, Thermo Scientific, F548) using long floating primers at each end 40 bp of homology with the insertion region. The CmPcL, KmPcL and resistance genes cassettes were amplified from existing strains in the laboratory collection. The PCR products were then dialyzed on a 0.025 μm porosity filter and electroporated into the recipient pKOBEGA strain. After electroporation, the bacteria were spread on LB plates + the appropriate antibiotic and incubated overnight at 37 °C. Once the mutants obtained were verified by PCR, the pKOBEGA plasmid was removed by plating at 42 °C and the constructs were transduced using P1vir into a clean genetic background. The strains were then verified one more time by Sanger sequencing.Primers used in this study are described in ESI Table S2.†
5.3 Adhesin interaction assay
Bacteria expressing different CU-fimbriae and type V secreted proteins were grown as described above until OD600 = 0.8. From these pre-cultures, new overnight cultures were then launched, with the strains expressing adhesins, whose interaction was to be tested, at a 1:1 ratio. For each interaction test, one strain carried the CFP tag and the other strain carried the YFP tag in order to differentiate homotypic and heterotypic interactions. After overnight cultures, the tubes were left for aggregation during 5 hours at room temperature. Aliquots were taken from the bottom of the tubes, spotted on a microscopy glass slide (Superfrost Plus, Thermo Fisher Scientific) and covered with a coverslip prior to observation with an epifluorescence microscope at ×1000 magnification.
5.4 Aggregation curves
Cultures were grown as described above, and 1 mL aliquots were diluted to an OD600 = 3 in spent growth medium (i.e. LB or LB + sugar) allowing us to maintain the generated acidic conditions while avoiding further bacterial growth during the aggregation process. Once diluted, all tubes were vortexed and an aliquot of 50 μL was removed 1 cm below the top of the culture, and diluted with 50 μL of LB prior to OD600 measurement. The bacteria were then left to aggregate at room temperature for 6 hours, while repeating the measurement of OD600 1 cm below the top of the culture every hour.5.5 Western blot
Aliquots of 1 mL at OD600 = 2 from overnight cultures were centrifuged (6000 rpm for 10 min), resuspended in 100 μL of Laemmli buffer 1× (Bio-Rad #1610747), and boiled for 10 min at 95 °C. 10 μL of these raw cell extracts were separated by SDS-PAGE using TGX 4–15% gradient precast gels (Bio-Rad). Then, the proteins were transferred onto a 0.2 μm nitrocellulose membrane using Trans-Blot Turbo Transfer System (Bio-Rad). The membranes were washed with PBS containing 0.05% tween (Tween20 Sigma) (PBST) and blocked with PBST supplemented with 5% skimmed milk powder at room temperature for 1 hour. The membranes were then washed in PBST twice, before incubation with the primary antibodies that were either rabbit anti-alphaYfaL (a gift from Prof M. Schembri), rabbit anti-alphaAg43 or mouse anti-RNAPalpha (BioLegend #663104) diluted 1/5000 in PBST supplemented with 1% skimmed milk powder for 1 hour at room temperature. The membranes were blocked again with PBST milk for 1 hour at room temperature and washed twice in PBST before incubation with secondary antibodies that were either goat anti-rabbit HRP-linked (Abcam, ab6013) or goat anti-mouse HRP-linked (Invitrogen G21040) diluted 1/7500 in PBST for 1 hour at room temperature. The membranes were then washed 4 times in PBST for 15 min. The membranes were then revealed using an ECL kit (GE Healthcare) and the iBright™ CL1500 system (Thermo Fisher).5.6 Immunofluorescence
Glass slides containing 12 wells (MP Biomedicals™) were successively washed with water, 70% EtOH and 100% EtOH and then quickly flamed. Each well was treated with poly-L-lysine (Sigma P8920) for 2 min, washed 3 times with PBS and left to dry.Aliquots of 1 mL at OD600 = 1 from overnight cultures were centrifuged (6000 rpm, 10 min) and washed twice with 1 mL of PBS. 50 μL of the different samples were then placed in the wells and left for 5 min at room temperature. Bacteria were then fixed with a solution of 4% paraformaldehyde (PFA) (Sigma P6148) for 10 min at room temperature. The wells were then washed 3 times with PBS, coated with 50 mM NH4Cl (in PBS) for 3 min at room temperature before being washed again 3 times with PBS. Each well was then saturated with a solution of PBS containing 0.5% BSA (Sigma A7888) for 15 min at room temperature. The excess of BSA was removed, and the wells were covered with primary antibody anti-alphaYfaL diluted at 1/500 in 0.5% PBS-BSA for 45 min. The wells were then washed 3 times with PBS, before incubation with a solution of secondary antibody anti-rabbit-Alexa488 diluted at 1/300 (Invitrogen Molecular Probe) mixed with DAPI diluted at 1/100 (Invitrogen Molecular Probe D1306) for 45 min at RT. The wells were then washed 3 times with PBS and once with 1 mL of water and left to dry. Finally, Dako fluorescent mounting medium (Dako S3023) was added on top of each well before adding the coverslip and the slides were left for 30 min at room temperature to solidify before observation with an epifluorescence microscope (EVOS M7000, Invitrogen).
5.7 RNA extraction and qRT-PCR
150 μL samples from overnight cultures, in LB, LB ara, LB MOPS and LB MOPS ara were washed with 2 volumes of RNA protect (Qiagen) and left for 5 min at room temperature before centrifugation at 5000g. The pellets were kept at −80 °C during the preparation of all biological replicates. Total RNA was extracted from the pellets using FastRNA Pro™ BLUE KIT (MP Biomedicals) following provider's instructions and treated with DNase (Thermo Fischer Scientific, AM1907) for 60 min to remove any DNA contamination. For each condition, 6 biological replicates were performed, and quality and concentration were checked using a Nano Drop instrument.Reverse transcription was performed using the Roche AMV cDNA first strand synthesis kit (Sigma) following the supplier protocol. Briefly, 550 ng of RNA of each sample were mixed with 4 μL MgCl2 25 mM, 2 μL 10× reaction buffer, 1 μL 3′ primer 20 μM, 2 μL dNTP mix at 10 mM each, 0.8 μL AMV reverse transcriptase and 1 μL RNase inhibitor 50 U μL−1, and water (qsp 20 μL). The mix was then successively incubated for 10 min at 25 °C, 60 min at 42 °C and 5 min at 99 °C to inactivate the enzyme. cDNAs were then mixed with SYBR green PCR master mix (Life Technologies) following the provider's instructions, and with the yfaL qPCR primers. The mix was then distributed in a 384-well plate in technical triplicates for each biological replicate. The qPCR was performed with the QuantStudio 6 Flex real-time PCR machine (Thermo Fischer Scientific) and absolute quantification of yfaL mRNA was obtained using the standard-curve quantification method.
5.8 Infrared spectroscopy
Infrared spectra in total attenuated reflection mode (IR-ATR) were recorded between 4000 and 800 cm−1 on a Bruker Tensor 27 spectrometer equipped with a KBr beam splitter and a DTGS detector, driven by the OPUS 7.8 software. The resolution of the single beam spectra was 4 cm−1. A nine-reflection diamond ATR accessory (DurasamplIR™, SensIR Technologies, incidence angle: 45°) was used for spectra acquisition. The number of bidirectional double-sided interferogram scans was 200, which corresponds to a 2 min accumulation. All interferograms were Fourier processed using the Mertz phase correction mode and a Blackman–Harris three-term apodization function. No ATR correction was performed. Measurements were performed at 21 ± 1 °C in an air-conditioned room. A volume of the suspension was centrifuged at 8000 rpm for 5 min and the supernatant was used to remove the spectral background. 50 μL of the bacterial suspension in their culture media was put on the ATR crystal, a cap was laid on the sample to prevent water evaporation. Water vapor subtraction was performed when necessary. One spectrum every 15 minutes was recorded for 4 hours.5.9 Chemical force microscopy
CFM measurements were performed in Force Mapping mode using a MFP3D-BIO (Asylum Research Technology, Atomic Force F&E GmbH, Mannheim, Germany) at room temperature in PBS buffer. The experiment was conducted using 1-dodecanethiol coated gold AFM-tips (NPG-10, Bruker France SAS, Palaiseau, France) with a spring constant of ca. 0.12 nN nm−1. For each sample, 3 force maps were recorded on a 5 × 5 μm2 surface containing at least one bacterium for 32 × 32 measurements (1024 force curves). The hold time was fixed at 100 ms and the retraction rate at 1 μm s−1 for a piezo drive of 1000 nm. Bacterial suspensions were previously immobilized onto PEI-coated glass substrates for which hydrophobic forces of about 1.03 ± 0.21 nN were recorded. Retraction curves were analyzed to determine the number of adhesive events and the amplitude of the corresponding force peak, as well as the rupture or separation distances at which the last adhesive event occurred upon retraction of the AFM probe from the cell surface. Statistical analyses were performed on the number of detected peaks on each retraction curve after excluding all curves recorded onto the PEI-coated substrates.5.10 Electrophoretic mobility
Electrophoretic mobility experiments were performed along the lines detailed elsewhere.35,36,38 Briefly, measurements consisted in monitoring the migration of the selected bacterial strains (cf. details below) in a quartz rectangular capillary under the action of an applied DC electric field. The ensuing trajectories of the bacteria were recorded by Electrophoresis Light Scattering at a 90° detection angle, and electrophoretic velocities were subsequently computed from cell trajectories imaged over time. Electrophoretic mobilities, denoted as μ, were then evaluated from the ratio between electrophoretic velocity and magnitude of the electric field applied between the electrodes immersed in the aqueous medium where bacteria were suspended (here 800 V m−1). The preparation of medium assays for electrophoretic measurements proceeded as follows. After overnight growth in the presence or absence of fructose, the pH of bacterial suspensions was measured, bacteria were then washed twice by centrifugation (4500g for 5 min) and resuspended in 10 mM KNO3 electrolyte before adjustment of OD600nm to 0.2. The corresponding bacterial suspensions were then diluted 10 times in 1 mM to 150 mM KNO3 electrolyte solution batches to get a final bacterial concentration OD600 nm of 0.02 in the quartz electrophoresis measurement cell. Using a Zetaphoremeter IV (CAD Instruments), electrophoretic mobility measurements were subsequently performed in aforementioned KNO3 media at natural pH (5.7) and at pH 4.6 (value corresponding to the final pH of suspension of cells after growth with fructose), at room temperature and in triplicate for each KNO3 concentration tested. For electrophoretic measurements at pH 4.6, rinsing and dilution of cell suspension in the KNO3 electrolyte were carried out with 10 mM and 1 M KNO3 solutions, and ultrapure water previously adjusted to pH 4.6 by the addition of 0.1 M HNO3 aliquots. All salts used for sample preparation and measurements were purchased from Sigma Aldrich with 99% purity. The measurements were performed on the WT and PcLyfaL strains grown (i) without fructose and in the presence of 0.2% fructose at pH 5.7, and (ii) without fructose and in the presence of 0.3% fructose at pH 4.6.The measured dependence of the electrophoretic mobility on the KNO3 electrolyte concentration for a given bacterial strain was quantitatively interpreted on the basis of well-established soft surface electrophoresis (SSE) theory.40,41 This theory goes beyond the conventional zeta-potential and slipping plane concepts, shown to be inappropriate for bacteria featuring soft structures permeable to ions from the electrolyte solution and to electroosmotic flow developed under electrophoresis conditions.40,41 Instead, it explicitly integrates (i) the way cell surface electrostatics is mediated by the density of 3D-distributed cell structural charges, and (ii) the finite penetration of the electroosmotic flow (developed under electrophoresis conditions) within the peripheral cell structure, with both (i) and (ii) affecting cell electrophoretic mobility (cf. e.g. reviews in40 and in65). According to this theory, at sufficiently high electrolyte concentrations (typically above 10 mM–50 mM depending on the bacteria)41 where Donnan electrostatics is established in the electrokinetically active peripheral cell surface layer, the electrophoretic mobility μ of bacterial cells can be approximated by the following expression:41
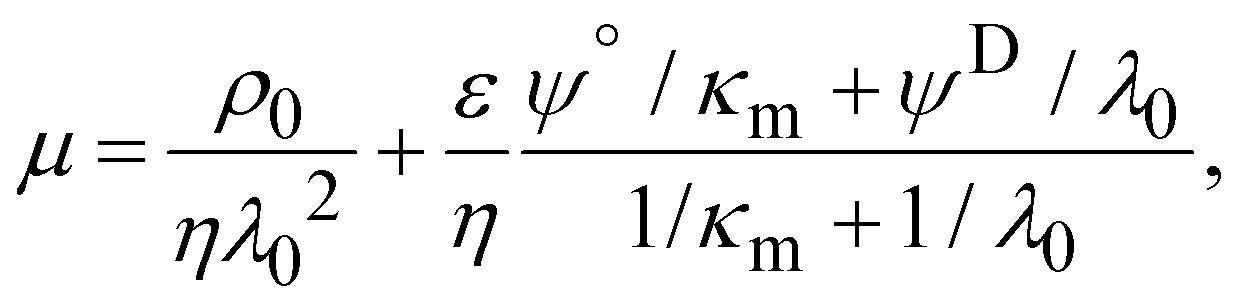 | (1) |
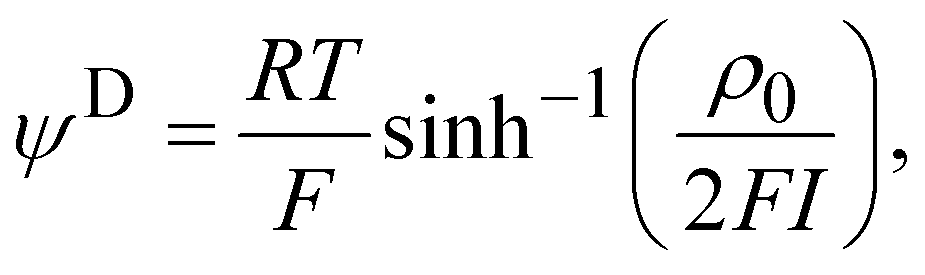 | (2) |
 | (3) |
 | (4) |
5.11 Modeling of the YfaL structure
AlphaFold models for full-length YfaL and Ag43 were generated with a local implementation of ColabFold68 using “ptm” parameters,53 12 recycling, plDDT score for scoring and model relaxation with OpenMM.54 Electrostatic surface potential for the passenger domains (YfaL1–886 and Ag431–654) were generated with ABPS/PDB2PQR,69 using PROPKA70 for pKa prediction at various pH values.5.12 Biofilm formation in 96-well plates
Bacteria were grown overnight as described above in LB. Cultures were diluted to an OD600 = 0.05 in 100 μL LB and LB + 0.3% fructose and inoculated in technical duplicates in polyvinyl chloride (PVC) round bottom 96-well plates (Corning). External wells were filled with 200 μL of water to prevent evaporation. Plates were then incubated at 37 °C for 24 hours. After 24 hours, one plate was resuspended to measure the OD600 using a Tecan Infinite-M200-Pro spectrophotometer. In the other plate, bacteria were fixed using Bouin solution for 15 min and non-attached bacteria were washed away by flicking, wells were then washed with water twice. Biofilms were then stained with 125 μL of crystal violet 1% (V5265; Sigma-Aldrich) for 15 min. Crystal violet was then removed by flicking and biofilms were washed twice with water. Biofilms were then air dried and resuspended using a solution of acetone:ethanol 1:4 mix, and OD at 575 nm was measured using a Tecan Infinite-M200-Pro spectrophotometer.
5.13 pH survival assay
Bacteria were grown overnight as described above, in LB + 0.3% fructose. 1 mL aliquots were then adjusted to an OD600 = 3 using spent media, and left to aggregate on the bench for 5 hours. pH value in each tube was then measured using a pH meter (FiveEasy Plus, METTLER TOLEDO), and adjusted to different values (2, 1.5 and 1) using a 30% HCl solution. HCl was added directly on top of the culture, while constantly monitoring the pH, and mixed carefully to avoid breaking of the aggregates. Tubes containing bacteria in an adjusted-pH medium were then left on the bench for 1 hour. After treatment, bacteria centrifuged at 6000 rpm were washed twice in LB and vigorously vortexed to break the aggregates. Serial dilutions from 10−1 to 10−6 were then performed and distributed on LB agar. Plates were incubated overnight (14–16 hours) at 37 °C before colony forming unit (CFU) counting. Results were plotted and statistical analyses were carried out using R software (version 4.0.2) implemented in Rstudio (version 1.3.1093) using both ggpubr71 and ggplot272 packages.5.14 In vivo colonization experiments
:1 ratio in LB + 0.3% fructose prior to gavage, and this combination was done in duplicate (2 × 3 mice, Mix 1 and Mix 2). The feces were collected before and 6 hours, 1, 2, 3, 6, 7 and 8 days after the gavage. The abundance of each strain in the feces was measured by flow cytometry thanks to the GFP and Mars tag carried by the PcLyfaL and WT strains, respectively. Results were plotted and statistical analyses were done using R software (version 4.0.2) implemented in Rstudio (version 1.3.1093) using both ggpubr71 and ggplot272 packages.
5.15 Bioinformatics analysis
The phylogroup associated with each genome was predicted using ClermonTyping tool.74
The YfaL complete sequence multiple alignment was trimmed using trimal v1.4.176 and used to build a Maximum Likelihood phylogenetic tree with PhyML version 3.177 with 100 bootstraps. The best fit model for the tree reconstruction was inferred using ProtTest version 2.4.78 The phylogenetic tree was then visualized and edited using iTOL v4.79
5.16 Statistics and reproducibility
Data are presented as the mean ± standard deviation (SD) or ±standard error of the mean (SEM) or with individual data points from at least three independent biological experiments. Statistical analyses were performed using Prism 9.5.0 (GraphPad Software Inc.) and correspond to unpaired two-tailed non-parametric Mann–Whitney test between two groups. p-Values <0.05 were defined as the level of statistical significance.Author contributions
Conceptualization: C.B., Y.C., J-M.G., J.F.L.D., G.F., and L.D.; methodology: C.B., Y.C., J-M.G., J.F.L.D., G.F., and L.D.; software: S.T.R. and B.B.; validation: C.B., J-M.G., J.F.L.D., G.F., and L.D.; formal analysis: Y.C., S.T.R., and B.B.; investigation: Y.C., S.T.R., B.B., C.C., F.Q., H.L.C, E.D., T.P., M.T., J.F.L.D., and G.F.; resources: C.B., Y.C., J-M.G., J.F.L.D., G.F., and L.D.; data Curation: S.T.R. and B.B.; writing – original draft: Y.C. and C.B.; writing – review and editing: Y.C., C.B., J-M.G., J.F.L.D., G.F., L.D., B.B., and F.Q.; visualization: Y.C., C.B., S.T.R, J.F.L.D., G.F., L.D., B.B., and F.Q.; supervision: C.B., S.T.R, J.F.L.D., G.F., and L.D.; project administration: C.B.; funding acquisition: C.B., J-M.G., J.F.L.D., G.F., and L.D.Data availability
All the data produced for this study are available within this manuscript with the exception of the scripts used for the bioinformatic analyses that are available at https://github.com/Sthiriet-rupert/AIDA-I_yfaL, and ESI Tables S5, S6 and S7† that are available, respectively, at https://zenodo.org/records/12724448, https://zenodo.org/records/12725303, and https://zenodo.org/records/12725345. A high-resolution version of ESI Fig. S8† is available at https://zenodo.org/records/13284193.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mark Schembri for the generous gift of anti-YfaL antibodies and Nadia Izadi-Pruneyre for critical reading of the manuscript. This work was supported by grants from the French Government's Investissement d'Avenir program, Laboratoire d'Excellence Integrative Biology of Emerging Infectious Diseases (Grant No. ANR-10-LABX-62-IBEID) and the Fondation pour la Recherche Médicale (Grant No. DEQ20180339185). Y. C. was supported by a MENESR (Ministère Français de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche) fellowship. The authors thank the Spectroscopy and Microscopy Service Facility (SMI) of LCPME (https://www.lcpme.ul.cnrs.fr/equipements/smi/) where AFM and infrared experiments were performed. This work was partly carried out using resources from the Pôle de Compétences en Physico-Chimie de l'Environnement, ANATELo, LIEC laboratory, UMR 7360 CNRS – Université de Lorraine and the HPC Core Facility of the Institut Pasteur.References
- C. Berne, C. K. Ellison, A. Ducret and Y. V. Brun, Nat. Rev. Microbiol., 2018, 16, 616–627 CrossRef CAS PubMed.
- T. Trunk, H. S. Khalil and J. C. Leo, AIMS Microbiol., 2018, 4, 140–164 CAS.
- E. Q. A. Nwoko and I. N. Okeke, Biochem. Soc. Trans., 2021, 49, 1147–1157 CrossRef CAS PubMed.
- K. Ochiai, T. Kurita-Ochiai, Y. Kamino and T. Ikeda, J. Med. Microbiol., 1993, 39, 183–190 CrossRef CAS PubMed.
- A. Malik, M. Sakamoto, S. Hanazaki, M. Osawa, T. Suzuki, M. Tochigi and K. Kakii, Appl. Environ. Microbiol., 2003, 69, 6056–6063 CrossRef CAS PubMed.
- J. E. Strassmann, O. M. Gilbert and D. C. Queller, Annu. Rev. Microbiol., 2011, 65, 349–367 CrossRef CAS PubMed.
- D. Wall, Annu. Rev. Microbiol., 2016, 70, 143–160 CrossRef CAS PubMed.
- C. Vassallo, D. T. Pathak, P. Cao, D. M. Zuckerman, E. Hoiczyk and D. Wall, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2939–E2946 CrossRef CAS PubMed.
- D. T. Pathak, X. Wei, A. Dey and D. Wall, PLoS Genet., 2013, 9, e1003891 CrossRef PubMed.
- D. Wall, Mol. Microbiol., 2014, 91, 209–220 CrossRef CAS PubMed.
- P. Cao, A. Dey, C. N. Vassallo and D. Wall, J. Mol. Biol., 2015, 427, 3709–3721 CrossRef CAS PubMed.
- M. Burmølle, T. R. Thomsen, M. Fazli, I. Dige, L. Christensen, P. Homøe, M. Tvede, B. Nyvad, T. Tolker-Nielsen, M. Givskov, C. Moser, K. Kirketerp-Møller, H. K. Johansen, N. Høiby, P. Jensen, S. J. Sørensen and T. Bjarnsholt, FEMS Immunol. Med. Microbiol., 2010, 59, 324–336 CrossRef PubMed.
- I. M. Frick, M. Mörgelin and L. Björck, Mol. Microbiol., 2000, 37, 1232–1247 CrossRef CAS PubMed.
- M. Alhede, K. N. Kragh, K. Qvortrup, M. Allesen-Holm, M. van Gennip, L. D. Christensen, P. Ø. Jensen, A. K. Nielsen, M. Parsek and D. Wozniak, PLoS One, 2011, 6, e27943 CrossRef CAS PubMed.
- T. Bjarnsholt, P. Ø. Jensen, M. J. Fiandaca, J. Pedersen, C. R. Hansen, C. B. Andersen, T. Pressler, M. Givskov and N. Høiby, Pediatr. Pulmonol., 2009, 44, 547–558 CrossRef PubMed.
- J. Haaber, M. T. Cohn, D. Frees, T. J. Andersen and H. Ingmer, PLoS One, 2012, 7, e41075 CrossRef CAS PubMed.
- S. M. Caceres, K. C. Malcolm, J. L. Taylor-Cousar, D. P. Nichols, M. T. Saavedra, D. L. Bratton, S. M. Moskowitz, J. L. Burns and J. A. Nick, Antimicrob. Agents Chemother., 2014, 58, 6851–6860 CrossRef PubMed.
- J. Haaber, M. T. Cohn, D. Frees, T. J. Andersen and H. Ingmer, PLoS One, 2012, 7(7), e41075 CrossRef CAS PubMed.
- S. Geibel and G. Waksman, Biochim. Biophys. Acta, 2014, 1843, 1559–1567 CrossRef CAS PubMed.
- I. Meuskens, A. Saragliadis, J. C. Leo and D. Linke, Front. Microbiol., 2019, 10, 1163 CrossRef PubMed.
- G. Francius, F. Petit, E. Clément, Y. Chekli, J. M. Ghigo, C. Beloin and J. F. L. Duval, Nanoscale, 2021, 13, 1257–1272 RSC.
- M. K. Hospenthal and G. Waksman, Microbiol. Spectrum, 2019, 7 DOI:10.1128/microbiolspec.PSIB-0010-2018.
- A. Jacquot, C. Sakamoto, A. Razafitianamaharavo, C. Caillet, J. Merlin, A. Fahs, J. M. Ghigo, C. Beloin, J. F. L. Duval and G. Francius, J. Biomed. Nanotechnol., 2014, 10, 3361–3372 CrossRef CAS PubMed.
- A. Jacquot, C. Sakamoto, A. Razafitianamarahavo, C. Caillet, J. Merlin, A. Fahs, J. M. Ghigo, J. F. L. Duval, C. Beloin and G. Francius, Nanoscale, 2014, 6, 12665–12681 RSC.
- F. Larsonneur, F. A. Martin, A. Mallet, M. Martinez-Gil, V. Semetey, J. M. Ghigo and C. Beloin, Environ. Microbiol., 2016, 18, 5228–5248 CrossRef CAS PubMed.
- M. W. van der Woude and I. R. Henderson, Annu. Rev. Microbiol., 2008, 62, 153–169 CrossRef CAS PubMed.
- C. G. Korea, R. Badouraly, M. C. Prevost, J. M. Ghigo and C. Beloin, Environ. Microbiol., 2010, 12, 1957–1977 CrossRef CAS PubMed.
- C. G. Korea, J. M. Ghigo and C. Beloin, Bioessays, 2011, 33, 300–311 CrossRef CAS PubMed.
- A. Roux, C. Beloin and J. M. Ghigo, J. Bacteriol., 2005, 187, 1001–1013 CrossRef CAS PubMed.
- J. W. Robbins Jr. and K. B. Taylor, Biotechnol. Bioeng., 1989, 34, 1289–1294 CrossRef CAS PubMed.
- G. C. Ulett, R. I. Webb and M. A. Schembri, Microbiology, 2006, 152, 2101–2110 CrossRef CAS PubMed.
- H. Hasman, T. Chakraborty and P. Klemm, J. Bacteriol., 1999, 181, 4834–4841 CrossRef CAS PubMed.
- D. P. Clark, FEMS Microbiol. Rev., 1989, 5, 223–234 CAS.
- M. Carbonaro and A. Nucara, Amino Acids, 2010, 38, 679–690 CrossRef CAS PubMed.
- J. Chamarande, L. Cunat, C. Caillet, L. Mathieu, J. F. L. Duval, A. Lozniewski, J. P. Frippiat, C. Alauzet and C. Cailliez-Grimal, Microorganisms, 2021, 9, 1602 CrossRef CAS PubMed.
- G. Francius, P. Polyakov, J. Merlin, Y. Abe, J. M. Ghigo, C. Merlin, C. Beloin and J. F. L. Duval, PLoS One, 2011, 6, e20066 CrossRef CAS PubMed.
- C. Pagnout, R. M. Présent, P. Billard, E. Rotureau and J. F. L. Duval, Sens. Actuators, B, 2018, 270, 482–491 CrossRef CAS.
- C. Pagnout, B. Sohm, A. Razafitianamaharavo, C. Caillet, M. Offroy, M. Leduc, H. Gendre, S. Jomini, A. Beaussart, P. Bauda and J. F. L. Duval, Sci. Rep., 2019, 9, 9696 CrossRef PubMed.
- J. F. L. Duval and H. Ohshima, Langmuir, 2006, 22, 3533–3546 CrossRef CAS PubMed.
- J. F. L. Duval and F. Gaboriaud, Curr. Opin. Colloid Interface Sci., 2010, 15, 184–195 CrossRef CAS.
- H. Ohshima, Adv. Colloid Interface Sci., 1995, 62, 189–235 CrossRef CAS.
- P. Marani, S. Wagner, L. Baars, P. Genevaux, J. W. de Gier, I. Nilsson, R. Casadio and G. von Heijne, Protein Sci.: Publ. Protein Soc., 2006, 15, 884–889 CrossRef CAS PubMed.
- S. P. Nuccio and A. J. Bäumler, Microbiol. Mol. Biol. Rev., 2007, 71, 551–575 CrossRef CAS PubMed.
- O. Sherlock, R. M. Vejborg and P. Klemm, Infect. Immun., 2005, 73, 1954–1963 CrossRef CAS PubMed.
- O. Sherlock, M. A. Schembri, A. Reisner and P. Klemm, J. Bacteriol., 2004, 186, 8058–8065 CrossRef CAS PubMed.
- T. J. Wells, O. Sherlock, L. Rivas, A. Mahajan, S. A. Beatson, M. Torpdahl, R. I. Webb, L. P. Allsopp, K. S. Gobius, D. L. Gally and M. A. Schembri, Environ. Microbiol., 2008, 10, 589–604 CrossRef CAS PubMed.
- J. Valle, A. N. Mabbett, G. C. Ulett, A. Toledo-Arana, K. Wecker, M. Totsika, M. A. Schembri, J. M. Ghigo and C. Beloin, J. Bacteriol., 2008, 190, 4147–4161 CrossRef CAS PubMed.
- M. Totsika, T. J. Wells, C. Beloin, J. Valle, L. P. Allsopp, N. P. King, J. M. Ghigo and M. A. Schembri, Appl. Environ. Microbiol., 2012, 78, 2179–2189 CrossRef CAS PubMed.
- I. Zude, A. Leimbach and U. Dobrindt, Int. J. Med. Microbiol., 2014, 304, 243–256 CrossRef CAS PubMed.
- M. C. Lane, X. Li, M. M. Pearson, A. N. Simms and H. L. Mobley, J. Bacteriol., 2009, 191, 1382–1392 CrossRef CAS PubMed.
- K. A. Floyd, J. L. Moore, A. R. Eberly, J. A. Good, C. L. Shaffer, H. Zaver, F. Almqvist, E. P. Skaar, R. M. Caprioli and M. Hadjifrangiskou, PLoS Pathog., 2015, 11, e1004697 CrossRef PubMed.
- P. Klemm, L. Hjerrild, M. Gjermansen and M. A. Schembri, Mol. Microbiol., 2004, 51, 283–296 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L. P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks and V. S. Pande, PLoS Comput. Biol., 2017, 13, e1005659 CrossRef PubMed.
- B. Heras, M. Totsika, K. M. Peters, J. J. Paxman, C. L. Gee, R. J. Jarrott, M. A. Perugini, A. E. Whitten and M. A. Schembri, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 457–462 CrossRef CAS PubMed.
- G. Meng, N. Spahich, R. Kenjale, G. Waksman and J. W. St Geme 3rd, EMBO J., 2011, 30, 3864–3874 CrossRef CAS PubMed.
- M. Alhede, M. Lorenz, B. G. Fritz, P. Ø. Jensen, H. C. Ring, L. Bay and T. Bjarnsholt, Med. Microbiol. Immunol., 2020, 209, 669–680 CrossRef CAS PubMed.
- G. Corno, J. Villiger and J. Pernthaler, Ecology, 2013, 94, 870–881 CrossRef.
- J. Klebensberger, K. Lautenschlager, D. Bressler, J. Wingender and B. Philipp, Environ. Microbiol., 2007, 9, 2247–2259 CrossRef PubMed.
- Y. Chekli, R. J. Stevick, E. Kornobis, V. Briolat, J. M. Ghigo and C. Beloin, Microbiol. Spectrum, 2023, e0069023, DOI:10.1128/spectrum.00690-23.
- T. J. Wells, M. Totsika and M. A. Schembri, Microbiology, 2010, 156, 2459–2469 CrossRef CAS PubMed.
- O. Clermont, O. V. A. Dixit, B. Vangchhia, B. Condamine, S. Dion, A. Bridier-Nahmias, E. Denamur and D. Gordon, Environ. Microbiol., 2019, 21, 3107–3117 CrossRef CAS PubMed.
- O. Tenaillon, D. Skurnik, B. Picard and E. Denamur, Nat. Rev. Microbiol., 2010, 8, 207–217 CrossRef CAS PubMed.
- T. Baba, T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner and H. Mori, Mol. Syst. Biol., 2006, 2, 2006.0008 CrossRef PubMed.
- P. P. Gopmandal and J. F. L. Duval, Curr. Opin. Colloid Interface Sci., 2022, 60, 101605 CrossRef CAS.
- J. F. L. Duval, H. J. Busscher, B. van de Belt-Gritter, H. C. van der Mei and W. Norde, Langmuir, 2005, 21, 11268–11282 CrossRef CAS PubMed.
- F. Gaboriaud, M. L. Gee, R. Strugnell and J. F. L. Duval, Langmuir, 2008, 24, 10988–10995 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G. W. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci.: Publ. Protein Soc., 2018, 27, 112–128 CrossRef CAS PubMed.
- M. H. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- A. Kassambara, ggpubr: “ggplot2” Based Publication Ready Plots, R Package Version 0.4.0, 2020, https://CRAN.R-project.org/package=ggpubr Search PubMed.
- H. Wickham, Media, 2009, 35, 10.1007 Search PubMed.
- C. Camacho, G. Coulouris, V. Avagyan, N. Ma, J. Papadopoulos, K. Bealer and T. L. Madden, BMC Bioinf., 2009, 10, 421 CrossRef PubMed.
- J. Beghain, A. Bridier-Nahmias, H. Le Nagard, E. Denamur and O. Clermont, Microb. Genomics, 2018, 4, e000192 CrossRef PubMed.
- K. D. Yamada, K. Tomii and K. Katoh, Bioinformatics, 2016, 32, 3246–3251 CrossRef CAS PubMed.
- S. Capella-Gutiérrez, J. M. Silla-Martínez and T. Gabaldón, Bioinformatics, 2009, 25, 1972–1973 CrossRef PubMed.
- S. Guindon and O. Gascuel, Syst. Biol., 2003, 52, 696–704 CrossRef PubMed.
- D. Darriba, G. L. Taboada, R. Doallo and D. Posada, Bioinformatics, 2011, 27, 1164–1165 CrossRef CAS PubMed.
- I. Letunic and P. Bork, Nucleic Acids Res., 2019, 47, W256–W259 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01884b |
| ‡ Present address: Université de Lille, INSERM, CHU Lille, U1286-INFINITE-Institute for Translational Research in Inflammation, Lille 59000, France. |
| This journal is © The Royal Society of Chemistry 2024 |