
Nanostructured cobalt/copper catalysts for efficient electrochemical carbon dioxide reduction†
Sharon
Abner
and
Aicheng
Chen
 *
*
Electrochemical Technology Centre, Department of Chemistry, University of Guelph, 50 Stone Road East, Guelph, Ontario N1G 2 W1, Canada. E-mail: aicheng@uoguelph.ca; sabner@uoguelph.ca; Fax: +1 519 7661499; Tel: +1 519 8244120 Ext.54764
First published on 3rd June 2024
Abstract
The search for an efficient and stable catalyst for the electrochemical reduction of CO2 to value-added chemicals is especially critical for lowering the atmospheric CO2 concentration. In this study, self-supported cobalt/copper nanostructured catalysts were designed, where the influences of the elemental composition and acid-etching on their efficiency towards the CO2 reduction reaction were studied. The developed Co/Cu catalysts showed superb catalytic activity with a low onset potential at −0.2 V vs. RHE. Gas and liquid product analysis revealed that formate and CO were the main products. It was observed that lower reductive potentials were favourable for formate production, while higher reductive potentials were more favourable for CO formation. In situ electrochemical FTIR studies were further conducted to gain insight into the CO2 reduction mechanism. The novel synthetic procedure reported in this study leads to promising electrocatalysts with high efficiencies for the conversion of CO2 into valuable products.
1. Introduction
As of December 2022, the CO2 concentration in the atmosphere has risen to 419 parts per million (ppm) and is expected to double by 2100.1–3 This alarming rise in atmospheric CO2 concentration is expected to increase the global temperature by 4.0 °C relative to the pre-industrial revolution era (280 ppm) if no action is taken to reduce or control CO2 emissions.4,5 A drastic increase in the global temperature not only causes severe climate change, but also causes irreversible changes to ecosystems that are important for human survival on the Earth.6,7 The international attempt to limit global warming has led 196 United Nations parties to sign the Paris Agreement in 2015. This agreement's goal was to mitigate climate change and limit global warming to only 1.5 °C above the pre-industrial revolution level by 2050.8 Many technologies have been proposed to create a “net-zero” environment such as direct air-capturing of CO2 and sequestration in aquifers.5 In addition to the carbon capture and storage methods, recycling technologies of CO2 have attracted considerable interest over the last decade.9 By converting CO2 to low carbon-containing compounds, such as carbon monoxide, formic acid, and methane, carbon-containing chemicals can be recycled to be used again in various industrial sectors.10 Electrochemical reduction of CO2 is a promising method since it can be coupled with renewable energy sources such as solar, hydro, or wind.11–14 An ideal catalyst for the electrochemical reduction process should have an extensive surface area, high conversion efficiency, tunable product selectivity, and long-term stability.15 Metallic nanocatalysts are especially of interest since their morphology can be designed to maximize surface areas and thus the number of active sites on which carbon dioxide can be adsorbed.16–23 By combining two or more metals, the elemental composition of the catalyst can be optimized, and the binding energies of carbon dioxide and other reaction intermediates can be tuned to direct the reduction pathway to a desired product with high faradaic efficiency.15,24,25A recent study has highlighted the effectiveness of cobalt-oxide catalysts for driving the CO2 reduction reaction (CO2RR), leading to the selective formation of formate. In the study, the cobalt-oxide nanodendrite catalyst demonstrated a substantial current density at a comparatively low onset potential of −0.2 V vs. RHE, while also suggesting a potential reaction mechanism for formate production.11 However, it was evident that the faradaic efficiency (FE) towards formate production needed improvement. On the other hand, copper and copper-oxide catalysts have been shown to have optimal binding energy with adsorbed reaction intermediates to lead to the production of C1 products (compounds containing one carbon atom) and C2 products (compounds containing two bonded carbon atoms) with relatively high efficiencies, while suppressing the hydrogen evolution reaction (HER).12,26–29 Due to this reason, copper and its oxide derivatives also exhibit poor product selectivity.30,31 By integrating cobalt and copper, it is possible to modulate the selectivity and efficiency of the reduction pathway, potentially enhancing formate production.32–34 For instance, Dai et al.35 fabricated a cobalt-decorated copper nanowire catalyst with 80% FE towards formate but at a relatively high potential (−0.65 V vs. RHE). Given the high selectivity of Co nanostructures and the ability of copper-oxide catalysts to prioritize the CO2RR over the HER, this study integrates copper into the synthesis of cobalt nanostructured catalysts, aiming to develop bimetallic catalysts for efficient CO2RR.
Herein, we report a self-supported Co/Cu catalyst grown directly on the surface of the cobalt substrate. Fabricating self-supported catalysts circumvents the need for expensive binders while enhancing the electrical conductivity.36 Copper was introduced through a galvanic replacement reaction and an acid-etching step was used to modify the surface morphology. Minor alterations in the synthetic conditions of metal-based catalysts are known to result in dramatic changes to their morphology and electronic properties.37–40 Consequently, this allows for synthetic control over catalytic efficiency for CO2 reduction. In this work, each synthetic step was optimized to maximize CO2RR over HER by investigating how the incorporation of copper on the cobalt nanostructured surface would affect: (i) the morphology of the surface; (ii) the catalytic activity towards the CO2RR and faradaic efficiencies; and (iii) the product selectivity. Additionally, we were interested in understanding how acid etching of the cobalt surface before the incorporation of copper affects the aforementioned factors. This study also aims to understand how the catalytic reduction mechanisms take place on the surface of the optimized Co/Cu nanostructured catalysts using in situ electrochemical attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy. The innovative synthetic method outlined in this study paves the way for the creation of self-supported electrocatalysts that hold significant promise in efficiently converting CO2 into valuable products.
2. Experimental section
The extended Experimental section, including surface and electrode characterization, bulk electrolysis of CO2, product analysis, and in situ electrochemical FTIR study description can be found in the ESI.†2.1. Chemicals and materials
CuSO4·5H2O (99.999%; trace metals basis), CoSO4·7H2O (99.999%; trace metals basis), K2SO4 (≥99.5%), H2SO4 (≥99.99%), KHCO3 (≥99.0%), K2CO3 (≥99.0%), KOH (≥85.0%), D2O (99.9 atomic% D), tetramethylsilane (99.5%) and HNO3 (99.5%) were obtained from Sigma Aldrich. Cobalt plates (99.9985%, 1 cm × 1 cm × 0.5 mm thickness) were purchased from Sigma Aldrich. High-purity carbon dioxide (99.999%) and argon (99.995%) gas tanks were purchased from Praxair. The ionic exchange membrane (AMI-7001) used in the two-compartment electrochemical cell was purchased from Membranes International Inc. A 3 M KCl Ag/AgCl reference electrode was purchased from Tianjin Ida Tech. Co. Ltd, China. Pure H2O produced from a Nanopure Diamond™ UV ultrapure water purification system (18.2 MΩ cm) was used to prepare all the electrolyte solutions.2.2. Fabrication of the Co/Cu catalysts
Scheme 1 illustrates the fabrication procedures of the annealed Co/Cu (Co/Cu–A) and Co/Cu–Acid Treated & Annealed (Co/Cu–AA) catalysts used for the electrochemical reduction of CO2. Smooth cobalt substrates (1.0 cm2) were chemically etched in 35% HNO3 for 1 min and thoroughly rinsed with pure H2O. The substrates were cleaned with acetone using an ultrasonic bath for 10 min followed by sonication in pure H2O for an additional 10 min. A 1 cm × 2 cm Co plate was used as the anode in the electrochemical (EC) deposition and was also cleaned using the above procedure. The Co TF was fabricated by EC deposition of Co on the substrates in a three-electrode cell containing a 0.1 M CoSO4·7H2O solution and using Ag/AgCl (3 M KCl) as the reference electrode. Chronoamperometry was conducted at −1.2 V vs. Ag/AgCl for 20 min. This resulted in the deposition of a 617 μm layer of cobalt on the smooth cobalt substrate. The Co/Cu–A catalyst was fabricated by drop-casting 50 μL of 0.1 M CuSO4·5H2O solution on the freshly produced Co TF and annealing at 600 °C for 2 h under atmospheric air conditions. The catalyst was left in the oven overnight to cool down to room temperature. Thereafter, the catalyst was electrochemically reduced (EC treatment) at −1.2 V vs. Ag/AgCl (−0.6 V vs. RHE) for 10 min in CO2-saturated 1 M KOH solution (pH = 8.00). To generate the Co/Cu–AA catalyst, 20 μL of 0.1 M H2SO4 was dropped on the Co TF surface in increments of 10 μL, left to air-dry, and then washed thoroughly with pure water. The acid-treated surface was electrochemically treated to reduce the oxide formed during the acid-etching process. Then 50 μL of 0.1 M CuSO4·5H2O solution was drop-casted on the reduced surface to facilitate the galvanic replacement of Co(0) atoms on the surface by Cu(0) atoms. The surface was then annealed at 600 °C for 2 h in atmospheric air and followed by the EC treatment before any electrochemical experiment was conducted. | ||
| Scheme 1 Schematics of the fabrication pathways of the Co/Cu catalysts. | ||
3. Results and discussion
3.1. Surface and electrochemical characterization of the Co/Cu catalysts
In this study, two kinds of electrocatalysts were developed and tested for their catalytic activity in the electrochemical CO2 reduction reaction (CO2RR): Co/Cu–A and Co/Cu–AA. The fabrication procedure is illustrated in Scheme 1. The first step of the fabrication process was the electrodeposition of cobalt on the surface of a pristine cobalt plate. This was done to create a malleable cobalt layer that can be manipulated to achieve various surface morphologies. The volume of 0.1 M CuSO4 solution drop-cast on the surface of the Co thin film (Co TF) to obtain Co/Cu–A was optimized by measuring the electrocatalytic activities of six electrodes prepared with 5 to 120 μL of 0.1 M CuSO4 solution in the CO2RR cell. CO2-saturated 1 M KOH (pH 8.0) electrolyte was used for this purpose. Fig. S1† represents linear sweep voltammograms (LSVs) and corresponding chronoamperometric (CA) profiles of the six electrodes compared to those obtained using a smooth cobalt plate and Co thin film (TF). In the LSV plot, an earlier onset (less negative potential) indicates a catalyzed reaction with lower overpotential, while a higher measured reductive current (more negative current density) indicates a faster rate of reaction. As can be seen, the Co plate had the latest onset (−0.5 V vs. RHE) and the lowest reductive current density (−42 mA cm−2 at −0.9 V) compared to Co TF and the six electrodes of Co TF dropcast with various amounts of 0.1 M CuSO4 solution. As for the Co TF catalyst, the reaction onset shifted to −0.4 V vs. RHE, and a higher reductive current was measured (−86 mA cm−2 at −0.9 V). This result indicated that when cobalt was electrodeposited on the smooth cobalt plate, there was an increase in the catalytic activity towards the CO2RR. Since no copper was added during the initial cobalt electrodeposition, the catalytic improvement was attributed to an increase in surface roughness. This was also seen in our previous work using cobalt-based catalysts.11 Between the six electrodes with the added 0.1 M CuSO4 solution on the Co TF surface, there was an increase in the measured reductive current as the volume of CuSO4 increased from 5 to 50 μL. However, there was no significant difference between the electrodes prepared with 50, 100, or 120 μL CuSO4 solution. It was concluded that Co TF drop-casted with 50 μL 0.1 M CuSO4 solution provided the electrode with the optimal catalytic activity. Similar trends can be seen in the steady-state measurements at −0.6 V and −0.8 V vs. RHE (Fig. S1b†).To check if the initial cobalt electrodeposition step was necessary, a control electrode was prepared by drop-casting 50 μL of 0.1 M CuSO4 solution directly on a pristine Co plate and its catalytic activity was tested in the electrochemical cell. The LSV plots at 20 mV s−1 scan rate and CA at constant potentials of −0.6 V and −0.8 V vs. RHE are shown in Fig. S2.† The solid lines represent the electrochemical measurements obtained during CO2RR in CO2-saturated 1 M KOH (pH 8.0). These plots show that the measured reductive current densities at steady-state conditions for the Co plate + 50 μL of 0.1 M CuSO4 solution (red solid lines) were approximately −7.5 mA cm−2 at −0.6 V and −33 mA cm−2 at −0.8 V (Fig. S2b and Fig. S2c†). These measured current densities are less negative than those obtained during the CO2RR using the Co TF + 50 μL of 0.1 M CuSO4 solution, which were approximately −35 mA cm−2 at −0.6 V and −92 mA cm−2 at −0.8 V (Fig. S1b†). These results indicate that the Co TF electrode had a higher exposed surface area in which cobalt atoms could have been galvanically substituted with copper atoms in comparison to the smooth cobalt plate. The activity of Co plate + 50 μL of 0.1 M CuSO4 was also tested under experimental conditions where only the hydrogen evolution reaction (HER) could take place. The dashed lines in Fig. S2† represent the electrochemical measurements conducted in Ar-saturated 0.5 M K2SO4. To maintain comparable pH conditions, the pH of the bulk electrolyte was adjusted to 8.0 using a few drops of 0.1 M KOH. It is interesting to note that the catalytic activity of the Co plate + 50 μL of 0.1 M CuSO4 electrode towards the HER (red dashed lines) was comparable to the catalytic activity of a pristine cobalt plate in both the HER (the black dashed lines) and CO2RR (the black solid lines). This indicates that the galvanic replacement of cobalt with copper atoms on the smooth electrode surface improved the catalytic activity towards CO2 reduction as opposed to HER.
Next, the Co TF + 50 μL of 0.1 M CuSO4 electrode was annealed at 600 °C in the air for 2 h. The annealing temperature was selected based on previous experiments conducted by our group and compared to those found in the literature with similar experimental designs. Dondapati et al.41,42 annealed 1 cm2 acid-etched Co plates (1.0 mm thickness) in various temperatures in the range of 250 to 550 °C in atmospheric air. They found that at lower annealing temperatures, amorphous Co3O4 was formed on the surface while at high annealing temperatures, a crystalline Co3O4 was formed with spinal structures.41 Dubale et al.43 annealed Cu films at temperatures varying from 350 to 650 °C. Their X-ray diffraction (XRD) patterns revealed that at 650 °C CuO phases were more dominant than Cu2O or Cu(0).43 In this study, 600 °C was selected to achieve a crystalline mixture of Co3O4 and CuO. After annealing, the surface was electrochemically (EC) treated by applying −0.6 V vs. RHE for 600 s to maintain a uniform morphology with high oxygen defects. Field emission scanning electron microscopic (FE-SEM) images of the resulting Co/Cu–A catalyst are presented in Fig. 1a–c at different magnifications. A uniform layer of sharp-edged nanoparticles was observed.
 | ||
Fig. 1 Scanning electron microscopy (SEM) images of Co/Cu–A (a–c) and Co/Cu–AA (d–f) at 1000× (a and d), 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× (b and e), and 100000× (c and f) magnifications. 000× (b and e), and 100000× (c and f) magnifications. | ||
The Co/Cu–AA catalyst was prepared by acid etching Co TF before casting 50 μL of 0.1 M CuSO4 solution on the surface. It has been shown that low concentrations (0.1–0.5 M) of sulfuric acid can act as an etching agent on the surface of cobalt or copper metals and thus can help in the synthesis of various nanostructures.11,44 To reduce the oxide formation on the surface during the etching process and to ensure that the maximum amount of Co(0) atoms was present, the acid-etched surface was EC treated. It was observed that in the absence of this EC treatment between drop-casting 20 μL of 0.1 M H2SO4 and 50 μL of 0.1 M CuSO4 solutions on the Co TF surface, the reproducibility of the electrochemical results was inconsistent. The acid-etching process had caused the formation of Co(0) on the surface of Co TF to oxidize and therefore prevented the direct displacement of the Co(0) atoms with Cu(0), resulting in inconsistent catalytic activity. Once the surface was acid-etched, EC treated, and then dropcast with 0.1 M CuSO4 solution, it was thermally annealed at 600 °C for 2 h and followed by another EC treatment. FE-SEM images of the resulting Co/Cu–AA catalyst are presented in Fig. 1d–f at different magnifications. A uniform layer of flower-like particles was observed.
The electrochemically active surface area (EASA) values were calculated using the double-layer capacitance of each electrode and compared with the specific capacitance of the pristine Co substrate. A one-compartment cell was used for this purpose with a CO2-saturated 1 M KOH electrolyte (adjusted pH 8.00). The potential window of 0.00 to −0.20 V vs. RHE was selected since no faradaic processes have been observed in this range. Cyclic voltammograms of the various catalysts, using scan rates of 10 to 100 mV s−1, are presented in Fig. S3a.† The calculated capacitance values, coefficients of determinations (R2), and EASA values are summarized in Table S1.† The Co electrodeposition on the pristine surface has led to an increase of EASA by 5.5 times. This evidence also confirms our previous conclusion that the initial Co electrodeposition was a necessary step in the synthesis process. Drop-casting of 0.1 M CuSO4 on the Co FT surface has led to a slight increase in the EASA value, indicating that there were no major changes to the morphology of the surface during the galvanic replacement of cobalt atoms on the surface with copper atoms. When the catalyst was annealed, such as in the case of Co/Cu–A and Co/Cu–AA, the double-layer capacitance significantly increased (24.28 mF cm−2 and 32.67 mF cm−2, respectively), resulting in the increase of the EASA by a factor of 14.5 and 19.4, respectively. Due to the increased resistance and thus deviation from linearity at higher scan rates using the Co/Cu–AA catalyst, only the Δj points measured at low scan rates (10–60 mV s−1) were used to calculate the capacitance of the catalyst.45 The acid-etching step caused the Co/Cu–AA catalyst to have a rougher surface in comparison to the Co/Cu–A catalysts. EASA-corrected LSV curves of Co/Cu–A and Co/Cu–AA are shown in Fig. S4.†
Fig. 2a presents energy-dispersive X-ray (EDX) spectroscopic profiles of the various catalysts examined in this study and Table S2† summarizes the calculated atomic percentages of cobalt, copper, and oxygen in each catalyst. There was a very slight variation in the elemental composition between the Co substrate (black line) and Co TF (red line) due to an increase in surface area and exposure to atmospheric oxygen. When 50 μL of 0.1 M CuSO4 was dropcast on Co TF (blue line), the atomic percent (at%) of Co decreased to 28.8% while Cu was 68.6%, meaning that almost 70% of the surface Co(0) atoms were galvanically replaced with Cu(0) with minimum oxide formation. The Co/Cu–A catalyst (turquoise line) showed a further decrease in the at% of Co (12.7%) and an increase in the at% of Cu (76.2%). Even though the catalyst was EC reduced before these EDX profiles were collected, the at% of oxygen increased to 11.1%. The increase in the oxygen present in the catalyst is attributed to the oxide formation during the annealing process and the exposure of the sample to air during the transportation into the EDX instrument. The elemental composition of the Co/Cu–AA catalyst (orange line) showed a lower at% of both cobalt and copper (11.4% and 73.4%, respectively) and a higher at% of oxygen (15.2%) in comparison to Co/Cu–A. This might be the result of a greater surface area being exposed to air and thus more surface-oxide formation.
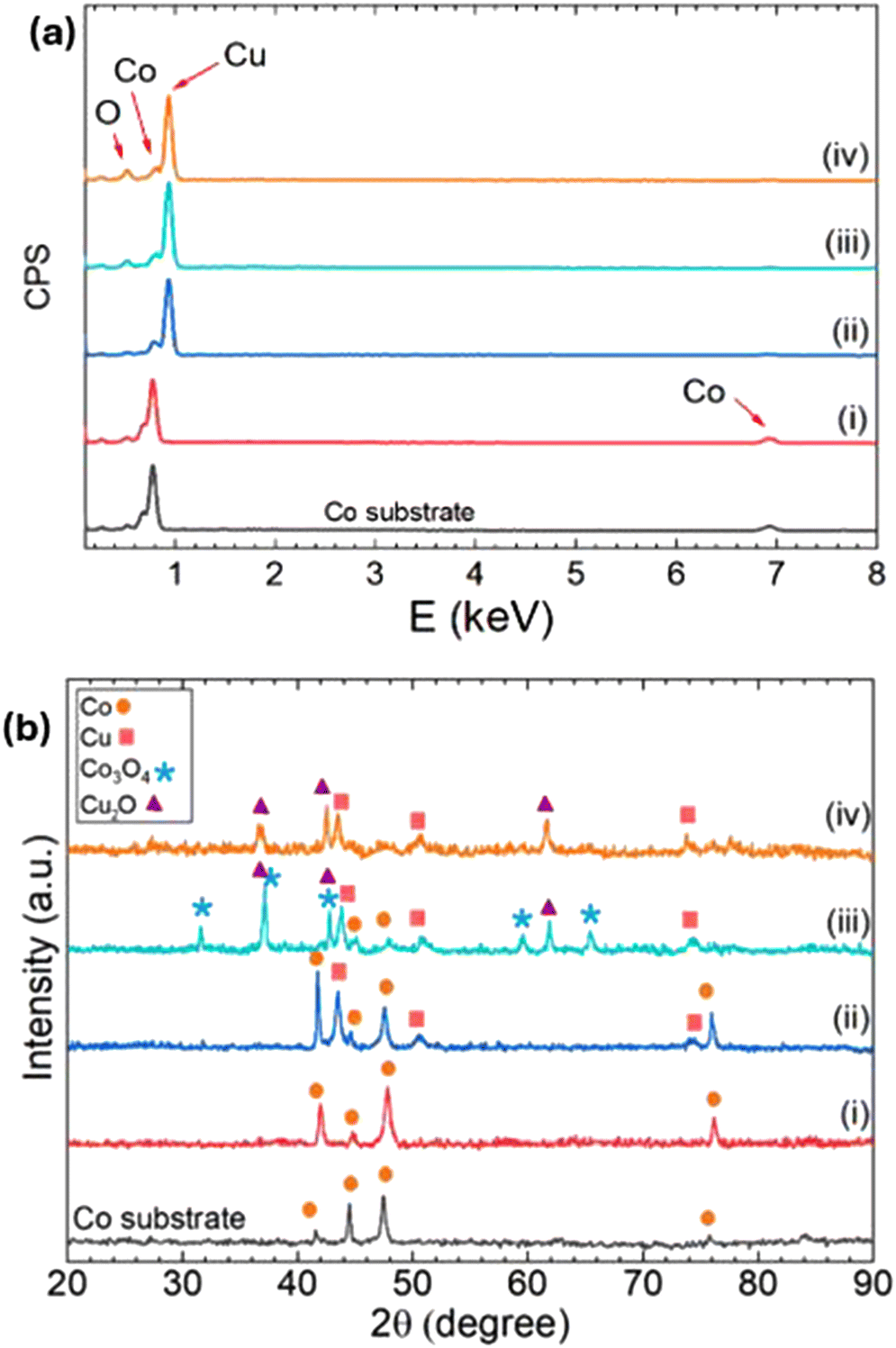 | ||
| Fig. 2 (a) Energy dispersive X-ray (EDX) spectroscopy profiles and (b) X-ray diffraction patterns of the Co substrate, (i) Co TF, (ii) Co TF + 0.1 M CuSO4, (iii) Co/Cu–A, and (iv) Co/Cu–AA catalysts. | ||
Fig. 2b compares the X-ray diffraction (XRD) patterns of the two Co/Cu catalysts to the patterns of the pristine Co substrate and Co TF. The assigned hkl planes, 2θ angles, and peak intensities are listed in Table S3.† The 2θ peaks located at 42.0°, 44.5°, 47.4°, and 75.8° were assigned to Co(0), a hexagonal close-packed (hcp) crystal system, with planes (100), (002), (101), and (110).11,46,47 These peaks were observed in the patterns of the Co substrate (black line), Co TF (red line), and Co TF + 0.1 M CuSO4 (blue line) surfaces. Additional planes were observed in the pattern of Co TF + 0.1 M CuSO4 at 2θ angles = 43.5°, 50.6°, and 74.5°, corresponding to the Miller indices of face-centred cubic (fcc) Cu(0): (111), (020), and (022) planes, respectively.35,48,49 This confirms the substitution of Co(0) atoms on the surface of Co TF with Cu(0) when 0.1 M CuSO4 solution was introduced. The XRD pattern of the Co/Cu–A catalyst (turquoise line) showed a mixture of crystalline phases of which some of the peaks overlapped. When compared to patterns found in the XRD database, the most dominant phases were those corresponding to Cu2O (cubic, Pn-3m space group) and Co3O4 (cubic, Fd-3 m space group). The peaks located at 37.5°, 42.8°, and 61.9° were assigned to Cu2O planes (111), (200), and (220).43,50,51 The peaks located at 31.6°, 37.0°, 42.0°, 59.6°, and 65.3° were assigned to Co3O4 planes (220), (311), (400), (511), and (440), respectively.51,52 This confirms that the thermal annealing of the Co/Cu catalyst at 600° C for 2 h in air produced a crystalline mixture of Cu2O and Co3O4 as was synthetically desired. However, the XRD pattern of the Co/Cu–AA catalyst (orange line) did not show any of the peaks corresponding to Co3O4, instead only the Cu2O crystalline phases were present along with the peaks belonging to Cu(0). The absence of Co3O4 peaks in the pattern might suggest that the acid-etching step (drop-casting 20 μL of 0.1 M H2SO4) selectively dissolved Co atoms from the catalyst. Thus, the acid treatment affected the morphology of the surface as such that during the annealing, no significant amount of the cobalt spinal (Co3O4) structure was formed on the surface of the Co/Cu–AA catalyst to be detected by the XRD.
Fig. S5a† shows the X-ray photoelectron spectroscopic (XPS) survey spectra of the various Co/Cu catalysts and the corresponding atomic percentages (at%) of cobalt, copper, and oxygen determined by XPS are summarized in Table S4.† Due to the high sensitivity of the technique to surface species, the at% of oxygen is much higher than those recorded using EDX (Table S2†). Despite the catalysts undergoing EC reduction before the XPS measurement, the introduction of high surface-oxygen content occurred during the transportation of the samples to the instrument. Similar to the trends observed from the EDX results, the Co at% decreases after the galvanic replacement reaction and continues to drop as the catalysts are acid-treated and annealed. High-resolution spectra of Co 2p and Cu 2p peaks were obtained for the various catalysts and the curve-fitting results are shown in Tables S5 and S6,† respectively. The curve fitting results of the high-resolution Co 2p spectra (Table S5†) of CoTF suggest the formation of 53% Co3O4 and 47% Co(OH)2. After the galvanic replacement reaction took place, the Co3O4 content decreased to only 10% and none was detected on the surface after the annealing or acid treatments of Co/Cu–A and Co/Cu–AA catalysts. As for the curve-fitting result using the Cu 2p spectra (Fig. S5c†), Cu with 0 and +1 oxidation states (Cu(0) and Cu(I), respectively) are not easily resolved due to the overlap of their energy peaks.53–55 Table S6† summarizes the results of the curve-fitting and the suggested percentages of Cu(0) + Cu(I) and Cu(II) in the Co/Cu–A and Co/Cu–AA catalysts. Because the XRD analysis shows a mixture of both Cu(0) and Cu2O bulk structures, it was deduced that the surface of the Co/Cu – A (45.7 %) and Co/Cu – AA (42.1 %) catalysts likely comprised a mixture of both Cu(0) and Cu(I) species. While the Cu(II) species present on the surface of Co/Cu–A (54.3%) and Co/Cu–AA (57.9%) catalysts could be a combination of CuO or Cu(OH)2.
3.2. Catalytic performance evaluation and product analysis
The catalytic efficiencies of the Co/Cu surfaces for the electrochemical conversion of CO2 to value-added chemicals were evaluated using linear sweep voltammetry (LSV) and chronoamperometry (CA) methods. The solid lines in Fig. 3a represent the LSV curves of the Co substrate (black), Co TF (red), and Co TF + 0.1 M CuSO4 (blue), obtained during the CO2RR at 20 mV s−1 scan rate in a CO2-saturated 1 M KOH (pH 8.0) electrolyte. The dashed lines in Fig. 3a represent the LSV curves of each catalyst, obtained during the hydrogen evolution reaction (HER) at 20 mV s−1 scan rate in an Ar-saturated 0.5 M K2SO4 electrolyte. The pH of the Ar-saturated 0.5 M K2SO4 electrolyte was adjusted to 8.0 using a few drops of 0.1 M KOH to ensure comparable pH conditions. For coherency, the LSV curves obtained during the CO2RR (solid lines) and the HER (dashed lines) using Co/Cu–A (turquoise) and Co/Cu–AA (orange) catalysts are presented separately in Fig. 3d. As presented in Fig. 3a, Co TF and Co TF + 0.1 M CuSO4 catalysts showed an improvement in current density (−92 and −115 mA cm−2 at −0.9 V vs. RHE, respectively) compared to the pristine Co substrate (−42 mA cm−2). This improvement was attributed to the increase in surface roughness after Co was electrodeposited on the pristine surface, and improved electronic properties after the substitution of cobalt with copper atoms on the surface, such as in the case of Co TF + 0.1 M CuSO4. The Co/Cu–A and Co/Cu–AA catalysts produced significantly higher reductive current densities towards the CO2RR compared to the other catalysts, with −137 and −209 mA cm−2 at −0.9 V vs. RHE, respectively. In addition, the two Co/Cu catalysts exhibited an earlier onset potential (−0.2 V vs. RHE) in comparison to the other catalysts that were not annealed. Since Co/Cu–AA had a better performance than Co/Cu–A, it can be concluded that the acid-etching step caused an increase in the EASA which resulted in improved catalytic activity. Fig. S4† presents the LSVs recorded during the CO2RR and the HER using Co/Cu–A and Co/Cu–AA using the corrected EASA values instead of the geometric areas. It is important to note that all measured reductive currents during the HER were much lower than for the CO2RR, indicating that the catalysts were selective toward the reduction of CO2 molecules as opposed to water reduction (i.e., HER). This can also be seen in the CA plots under steady-state conditions. Fig. 3c and f display the measured current densities at −0.8 V vs. RHE for 10 min. The catalytic activity towards the CO2RR improved in this order: Co substrate (−21 mA cm−2) < Co TF (−71 mA cm−2) < Co TF + 0.1 M CuSO4 (−92 mA cm−2) < Co/Cu–A (−100 mA cm−2) < Co/Cu–AA (−170 mA cm−2). The highest current efficiency was measured when Co/Cu–AA (93%) was used as the catalyst at −0.4 V, while Co/Cu–A had the second highest current efficiency (90%) at the same potential. | ||
| Fig. 3 (a & d) LSVs of (a) the Co substrate (black), Co TF (red), CoTF + 50 μL of 0.1 M CuSO4 (blue), Co/Cu–A (turquoise), and Co/Cu–AA (orange) electrodes recorded at 20 mV s−1 scan rate. Dashed lines represent HER experiments conducted in Ar-saturated 0.5 M K2SO4 (adjusted pH 8.0) and solid lines represent CO2RR experiments conducted in CO2-saturated 1 M KOH (pH 8.0); (b & e) corresponding CA curves of the electrodes measured at −0.6 V vs. RHE for 600 s; and (c & f) corresponding CA curves of the electrodes measured at −0.8 V vs. RHE for 600 s. | ||
To verify that the trends in the catalytic activities are consistent at different pH levels, the LSV experiments were repeated in a CO2-saturated 0.5 M K2SO4 electrolyte with pH 6.7 and were compared to those obtained in Ar-saturated 0.5 M K2SO4 electrolyte, where the pH was adjusted to 6.7. Fig. S6† presents the LSVs during the CO2RR (solid lines) and the HER (dashed lines) using Co/Cu–A (turquoise) and Co/Cu–AA (orange) catalysts. Both catalysts were found to be more efficient for CO2RR as opposed to HER since their reductive current densities were higher (more negative) when the electrolyte was purged with CO2, and an earlier onset potential has been observed. It should be noted that the reductive current densities recorded during this electrochemical test were much lower than those recorded using CO2-saturated 1 M KOH electrolyte. This was attributed to the lower solubility of CO2 gas molecules in the 0.5 M K2SO4 electrolyte in comparison to the 1 M KOH. Regardless of the lower measured current densities, the trend in the catalytic activity was consistent between the two electrochemical tests. Co/Cu–AA resulted in a higher reductive current (−92 mA cm−2) than Co/Cu–A (−82 mA cm−2).
Faradaic efficiencies were calculated by collecting the gas and liquid products after 1 h of chronoamperometry at potentials −0.4 to −0.9 V vs. RHE (Fig. S7†) in CO2-saturated 1 M KOH electrolyte. Gas chromatography (GC) analysis showed that CO and H2 were the only gas products detected during the CO2RR using Co/Cu–A and Co/Cu–AA catalysts. Proton NMR revealed that only formate ions were present in the liquid products (Fig. S9†). Fig. 4a and b show the faradaic efficiencies of Co/Cu–A and Co/Cu–AA towards the production of formate ions and carbon monoxide. The numeric results are summarized in Table S7.† As the reduction potential increased from −0.4 to −0.9 V, formate faradaic efficiencies decreased from 28.56% to 6.27% for Co/Cu–A and from 31.65% to 3.93% for Co/Cu–AA. A different trend was observed during CO production. When the Co/Cu–A catalyst was used, CO faradaic efficiencies initially increased from 24.25% at −0.4 V to 59.46% at −0.7 V and remained relatively constant at −0.8 and −0.9 V with 57.66% and 57.64%, respectively. When the Co/Cu–AA catalyst was used, CO efficiencies increased from 44.54% at −0.4 V to 74.36% at −0.9 V. This indicated that lower reductive potentials were favourable for formate production while the higher reductive potentials were more favourable for CO production. Overall, the Co/Cu–AA catalyst has been shown to have higher combined faradaic efficiencies, reaching 78.29% at −0.9 V in comparison to Co/Cu–A with the highest combined faradaic efficiency of 70.31% at −0.7 V. Fig. 4c & d present the calculated production rate of formate and carbon monoxide as a function of applied potential. Both Co/Cu–A and Co/Cu–AA catalysts resulted in a maximum formate yield between −0.5 to −0.6 V, and slowly decreased as the reductive potential increased. When Co/Cu–A was used as the catalyst, there was a small increase in CO production rate between −0.4 to −0.6 and a drastic increase between −0.7 to −0.9 V. When Co/Cu–AA was used as the catalyst, there was a close-to-linear increase in CO production rate as a higher reductive potential was applied. A close inspection of Fig. 4d showed that there was a switch between the reduction products at −0.6 V. At −0.4 and −0.5 V, the formate production rates were higher than those of CO, while at −0.8 and −0.9 V CO was the dominant product. This result is interesting since the HER is usually more dominant at high negative potentials and a drop in the production rates of both formate and CO was expected at −0.9 V vs. RHE. It can be concluded that the electronic and morphological properties of the Co/Cu–AA catalyst favoured the reduction of CO2 over the HER even at high reductive potentials. Lastly, stability tests were conducted at −0.6 and −0.8 V during the CO2RR using Co/Cu–A or Co/Cu–AA catalysts for 12 h (Fig. S8†). Both catalysts have shown relatively stable current densities for the duration of the electrochemical tests.
 | ||
| Fig. 4 (a & b) Calculated faradaic Efficiencies of carbon monoxide (CO) and formate ion (HCOO−) obtained after 60 min of the applied potential using (a) Co/Cu–A and (b) Co/Cu–AA catalysts. (c & d) Measured CO and HCOO− production rates during the CO2RR at each applied potential using (c) Co/Cu–A and (d) Co/Cu–AA catalysts. | ||
3.3. Potential-dependent in situ electrochemical FTIR
In situ electrochemical ATR-FTIR experiments were conducted to detect and monitor intermediates and products formed during the CO2RR using the Co/Cu–A and Co/Cu–AA catalysts. In this study, the SNIFTIR technique was applied (eqn (S10)†) and the spectra were plotted in terms of ΔR/R versus wavenumber (cm−1) at each applied potential. CO2-saturated 0.5 M K2SO4 solution made with D2O was used for these experiments. These in situ experiments were conducted in the Otto configuration in which approximately 10 μm thick electrolyte solution was trapped between the catalyst and the ZnSe window. This configuration limited the mass transport between the bulk electrolyte and the thin layer of solution in the vicinity of the electrode. This issue was mitigated by withdrawing the working electrode away from the ATR crystal to allow the electrolyte to mix after each applied potential (E2). E2 was applied for 100 s during which 64 interferograms were collected and averaged.
Fig. 5a and d show stacked plots of the spectra collected at −0.1 to −0.9 V vs. RHE during the CO2RR using Co/Cu–A and Co/Cu–AA catalysts. The spectrum recorded at 0.0 V vs. RHE was used as the baseline (R(E1)). Using this method, positive (upwards) directed peaks correspond to consumed species or species that are no longer detected in the thin layer of solution trapped between the catalyst and the ZnSe window. Negative (downwards) directed peaks correspond to the produced species that can be either adsorbed intermediates or CO2RR products detected in the vicinity of the electrode. For example, the positive peak at 2340 cm−1 in Fig. 5a & d corresponds to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO asymmetric stretch vibrational mode of soluble carbon dioxide molecules in the electrolyte solution (νas(CO2)). This peak is directed upwards because its concentration was lower when E2 was applied than when the base potential (E1) was applied. This means that CO2 molecules were consumed as a more reductive potential was applied. Fig. S10† compares the integrated areas of the CO2 peaks in Fig. 5a and d as a function of the applied potential for Co/Cu–A and Co/Cu–AA. νas(CO2) peak area increased between −0.1 and −0.5 V at a relatively similar rate for both catalysts, but it was slightly higher for Co/Cu–AA than for the Co/Cu–A catalyst. Between −0.5 and −0.9 V, the integrated peak areas of νas(CO2) dropped by 60% for Co/Cu–A and only 30% for Co/Cu–AA. This shows that at a high reductive current (−0.5 to −0.9 V), CO2 reduction still takes place on the surface of both catalysts, but it was more prominent on the surface of Co/Cu–AA. The peak at 1095 cm−1 was assigned to the symmetric stretch of the SO42− molecules in the electrolyte. SO42− ions were not consumed or produced during the CO2RR; however, a positively directed peak was observed. This was the result of a drop in concentration of SO42− ions in the vicinity of the electrode as other negative ions (such as OH−, HCO3−, and CO32−) accumulated in the thin layer of solution as a more reductive potential was applied. This phenomenon has also been observed in our previous studies.11,12,56
CO asymmetric stretch vibrational mode of soluble carbon dioxide molecules in the electrolyte solution (νas(CO2)). This peak is directed upwards because its concentration was lower when E2 was applied than when the base potential (E1) was applied. This means that CO2 molecules were consumed as a more reductive potential was applied. Fig. S10† compares the integrated areas of the CO2 peaks in Fig. 5a and d as a function of the applied potential for Co/Cu–A and Co/Cu–AA. νas(CO2) peak area increased between −0.1 and −0.5 V at a relatively similar rate for both catalysts, but it was slightly higher for Co/Cu–AA than for the Co/Cu–A catalyst. Between −0.5 and −0.9 V, the integrated peak areas of νas(CO2) dropped by 60% for Co/Cu–A and only 30% for Co/Cu–AA. This shows that at a high reductive current (−0.5 to −0.9 V), CO2 reduction still takes place on the surface of both catalysts, but it was more prominent on the surface of Co/Cu–AA. The peak at 1095 cm−1 was assigned to the symmetric stretch of the SO42− molecules in the electrolyte. SO42− ions were not consumed or produced during the CO2RR; however, a positively directed peak was observed. This was the result of a drop in concentration of SO42− ions in the vicinity of the electrode as other negative ions (such as OH−, HCO3−, and CO32−) accumulated in the thin layer of solution as a more reductive potential was applied. This phenomenon has also been observed in our previous studies.11,12,56
 | ||
| Fig. 5 (a and d) Potential-dependent FTIR spectra recorded during the CO2 reduction using (a) Co/Cu–A and (d) Co/Cu–AA catalysts in a CO2-saturated 0.5 M K2SO4 solution in D2O in the potential range of −0.1 to −0.9 V vs. RHE; (b and e) overlayed FTIR spectra in the range of 1800–1200 cm−1 using (b) Co/Cu–A and (e) Co/Cu–AA catalysts. (c–f) absolute values of the corresponding peak intensities calculated for (c) Co/Cu–A and (f) Co/Cu–AA catalysts as a function of the applied potential. | ||
It was also noted in Fig. 5a and d that peaks corresponding to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O vibrations were not detected in the spectra during the CO2RR using either Co/Cu–A or Co/Cu–AA catalysts. Linearly adsorbed or bridged carbon monoxide peaks were expected to appear in the wavenumber region of 1900 to 2100 cm−1.57–59 Since carbon monoxide has been detected and quantified using GC analysis, it was concluded that the ATR-FTIR in this configuration was not sufficiently sensitive to detect adsorbed CO on the surface of the catalysts. Unlike formate ions that are in the liquid form and soluble, the CO gas molecules migrated from the thin-layer solution to the bulk of the electrolyte. The peaks in the region of 1800 to 1200 cm−1 were assigned to the asymmetric stretch of deuterated formate (νas(DCOO−)) at 1625 cm−1, C–O asymmetric stretch ν3 of CO32− ion at 1410 cm−1, and symmetric stretch of deuterated formate (νss(DCOO−)) at 1365 cm−1. This assignment was based on our previous FTIR studies and other studies reported in the literature with comparable experimental designs.11,12,56,60–63 Overlayed SNIFTIRS plots focusing on the wavenumber region 1800 to 1200 cm−1 are presented in Fig. 5b and e using Co/Cu–A and Co/Cu–AA catalysts, respectively. To track changes in the intensities of the peaks with a cathodic potential being applied, the ΔR/R values at 1625, 1410, and 1365 cm−1 were plotted against the applied potential as shown in Fig. 5c and f. There were major differences between the trends in peak evolution during the CO2RR using Co/Cu–A and Co/Cu–AA catalysts. Starting with Co/Cu–A (Fig. 5b and c), the two formate peaks (νas(DCOO−) and νss(DCOO−)) were directed downwards between −0.2 to −0.7 V, which means that formate ions were produced in this potential window. Between −0.4 to −0.7 V, the formate peaks reached a plateau in which the consumption of formate ions was constant. At −0.8 V, the peak at 1625 cm−1 was directed upwards, which would mean that formate ions were seemingly consumed. However, since each spectrum at E2 is represented relative to the base potential E1 at which no formate ions are present, the possibility of this positive peak appearing at −0.8 V corresponding to the formate is very slim. This peak is more likely to correspond to the asymmetric stretch of deuterated bicarbonate ions in solution (νas(DCO32−)) with the infrared active vibrational mode appearing at 1620 cm−1.61,64,65 This peak was directed in the positive direction since bicarbonate ions were consumed to produce carbonate ions (CO32−) according to Reaction 1. As can be seen in Fig. 5a and b, the growth of νas(DCO32−) in the positive direction was accompanied by the growth of the ν3(CO32−) peak in the negative direction. The growth of the ν3(CO32−) peak in the negative direction indicated the accumulation of CO32− ions in the vicinity of the catalyst and subsequently increase in pH in the thin layer of solution. At −0.9 V, the νss(DCOO−) peak no longer appears in the spectra, indicating that no formate ions are produced at this potential using the Co/Cu–A catalyst. This also might be due to the accelerated production of CO on the surface of Co/Cu–A between −0.7 and −0.9 V (Fig. 4c). As for Co/Cu–AA (Fig. 5e and f), the two formate peaks remained directed downwards in a wider potential window (−0.2 to −0.8 V) in comparison to the Co/Cu–A catalyst. The switch to the positive direction occurred between −0.8 and −0.9 V, at which bicarbonate ions started to deplete significantly in the thin layer of the solution. The ν3(CO32−) peak did not change in intensity between −0.1 and −0.5 V. At −0.6 to −0.9 V, CO32− ions started to accumulate noticeably. The accelerated production of CO32− ions could also be the result of the HER taking place in this potential window (−0.7 to −0.9 V) in addition to the CO2RR since during both reduction reactions hydroxide ions were produced. The increase in hydroxide ions (OH−) in the vicinity of the electrode caused the pH to increase and thus an equilibrium shift from bicarbonate ions (HCO3−) to bicarbonate ions (CO32−) took place.25,66
O vibrations were not detected in the spectra during the CO2RR using either Co/Cu–A or Co/Cu–AA catalysts. Linearly adsorbed or bridged carbon monoxide peaks were expected to appear in the wavenumber region of 1900 to 2100 cm−1.57–59 Since carbon monoxide has been detected and quantified using GC analysis, it was concluded that the ATR-FTIR in this configuration was not sufficiently sensitive to detect adsorbed CO on the surface of the catalysts. Unlike formate ions that are in the liquid form and soluble, the CO gas molecules migrated from the thin-layer solution to the bulk of the electrolyte. The peaks in the region of 1800 to 1200 cm−1 were assigned to the asymmetric stretch of deuterated formate (νas(DCOO−)) at 1625 cm−1, C–O asymmetric stretch ν3 of CO32− ion at 1410 cm−1, and symmetric stretch of deuterated formate (νss(DCOO−)) at 1365 cm−1. This assignment was based on our previous FTIR studies and other studies reported in the literature with comparable experimental designs.11,12,56,60–63 Overlayed SNIFTIRS plots focusing on the wavenumber region 1800 to 1200 cm−1 are presented in Fig. 5b and e using Co/Cu–A and Co/Cu–AA catalysts, respectively. To track changes in the intensities of the peaks with a cathodic potential being applied, the ΔR/R values at 1625, 1410, and 1365 cm−1 were plotted against the applied potential as shown in Fig. 5c and f. There were major differences between the trends in peak evolution during the CO2RR using Co/Cu–A and Co/Cu–AA catalysts. Starting with Co/Cu–A (Fig. 5b and c), the two formate peaks (νas(DCOO−) and νss(DCOO−)) were directed downwards between −0.2 to −0.7 V, which means that formate ions were produced in this potential window. Between −0.4 to −0.7 V, the formate peaks reached a plateau in which the consumption of formate ions was constant. At −0.8 V, the peak at 1625 cm−1 was directed upwards, which would mean that formate ions were seemingly consumed. However, since each spectrum at E2 is represented relative to the base potential E1 at which no formate ions are present, the possibility of this positive peak appearing at −0.8 V corresponding to the formate is very slim. This peak is more likely to correspond to the asymmetric stretch of deuterated bicarbonate ions in solution (νas(DCO32−)) with the infrared active vibrational mode appearing at 1620 cm−1.61,64,65 This peak was directed in the positive direction since bicarbonate ions were consumed to produce carbonate ions (CO32−) according to Reaction 1. As can be seen in Fig. 5a and b, the growth of νas(DCO32−) in the positive direction was accompanied by the growth of the ν3(CO32−) peak in the negative direction. The growth of the ν3(CO32−) peak in the negative direction indicated the accumulation of CO32− ions in the vicinity of the catalyst and subsequently increase in pH in the thin layer of solution. At −0.9 V, the νss(DCOO−) peak no longer appears in the spectra, indicating that no formate ions are produced at this potential using the Co/Cu–A catalyst. This also might be due to the accelerated production of CO on the surface of Co/Cu–A between −0.7 and −0.9 V (Fig. 4c). As for Co/Cu–AA (Fig. 5e and f), the two formate peaks remained directed downwards in a wider potential window (−0.2 to −0.8 V) in comparison to the Co/Cu–A catalyst. The switch to the positive direction occurred between −0.8 and −0.9 V, at which bicarbonate ions started to deplete significantly in the thin layer of the solution. The ν3(CO32−) peak did not change in intensity between −0.1 and −0.5 V. At −0.6 to −0.9 V, CO32− ions started to accumulate noticeably. The accelerated production of CO32− ions could also be the result of the HER taking place in this potential window (−0.7 to −0.9 V) in addition to the CO2RR since during both reduction reactions hydroxide ions were produced. The increase in hydroxide ions (OH−) in the vicinity of the electrode caused the pH to increase and thus an equilibrium shift from bicarbonate ions (HCO3−) to bicarbonate ions (CO32−) took place.25,66
| HCO3− + OH− ⇌ CO32− + H2O | (1) |
3.4. Time-dependent in situ electrochemical FTIR study
Fig. 6a and b present the in situ FTIR spectra in the region of 1800 to 1200 cm−1, at −0.4, −0.6, and −0.8 V vs. RHE, recorded during the CO2RR for 100 s using Co/Cu–A and Co/Cu–AA catalysts, respectively. In Fig. S11,† the peaks’ ΔR/R values were monitored and plotted against time (s). The peaks of interest were assigned as follows: νas(DCOO−) at 1625 cm−1, ν3(CO32−) at 1410 cm−1, and νss(DCOO−) at 1365 cm−1. At −0.4 V, the two formate peaks appeared directed downwards using either Co/Cu–A or Co/Cu–AA catalysts. The ν3(CO32−) peak did not change when −0.4 V was applied for either catalyst. As −0.6 V was applied to the Co/Cu–A catalyst, the νas(DCOO−) and νss(DCOO−) peaks increased in intensity in the downward direction (between 20 to 70 s), and then their growth slowed down. The ν3(CO32−) peak increased in intensity in the negative direction linearly between 50 to 100 s, which indicated the accumulation of CO32− ions in the thin layer solution. As −0.6 V was applied to the Co/Cu–AA catalyst, the two formate peaks remained directed downwards and the ν3(CO32−) peak was only noticeable in the spectra after 70 s. At −0.8 V, the transition of the formate ion peaks from the negative to the positive direction occurred after 30 s when Co/Cu–A was used. As was seen in the potential-dependent experiments, the increase in the intensity of the νas(DCO32−) peak in the positive direction was accompanied by the growth of the ν3(CO32−) peak in the negative direction. In the case of Co/Cu–AA, the ν3(CO32−) peak remained unnoticeable in the spectra until after 60 s at −0.8 V. At this point, the formate peak intensities decreased (became more positive) due to the accelerated production of CO that caused the decrease of formate ion concentration in the thin layer solution, and the depletion of DCO32− ions that caused the peak at 1620 cm−1 to approach the zero line. | ||
| Fig. 6 Time-dependent FTIR spectra in the range of 1800–1200 cm−1 were recorded during the CO2RR using (a) Co/Cu–A and (b) Co/Cu–AA catalysts in a CO2-saturated 0.5 M K2SO4 solution in D2O at −0.4, −0.6, and −0.8 V vs. RHE. | ||
Bicarbonate-carbonate equilibria play a significant role in CO2 reduction. Many have been investigating the involvement of these ions in the reaction mechanism. In this study, the accelerated accumulation of CO32− ions and the depletion of DCO3− ions in the thin layer of solution trapped between the ATR crystal and the catalyst can provide an estimation of when the HER is taking place. Using the Co/Cu–A catalyst, this transition took place between −0.7 and −0.8 V vs. RHE, and when −0.9 V was applied, no formate peaks were observed. According to the product analysis, the major CO2RR product was CO in addition to the HER product H2. While the Co/Cu–AA was used as the catalyst, the in situ results showed that formate was continually being produced even at high cathodic potentials of −0.8 and −0.9 V vs. RHE where the major CO2RR product was CO.
3.5. CO2RR mechanism on Co/Cu-based catalysts
The utilization of in situ electrochemical FTIR spectroscopy enabled real-time monitoring of the chemical processes occurring at the Co/Cu–A and Co/Cu–AA catalysts’ surfaces during the electrochemical reduction of CO2. Based on the observed spectral changes and reaction kinetics, a comprehensive analysis was conducted, culminating in the proposal of a reaction mechanism elucidating the conversion of CO2 into formate and CO. The first step in the mechanism and the rate-determining step is the binding of the CO2 molecule to the surface.67–69 This step involves an electron transfer from the metal surface to CO2 to bind the molecule to the surface and the bending of the CO2 molecule to form an adsorbed intermediate *COO−. The electron transfer reaction is followed by a proton transfer from the electrolyte to form the *COOH intermediate.47 In contrast to previous studies conducted using Co-based catalysts,11 peaks belonging to the adsorbed intermediate *COO− were not observed in the presented FTIR results using Co/Cu–A and Co/Cu–AA catalysts. The asymmetric stretch νas of the adsorbed intermediate *COO− was expected to appear at 1565 cm−1 while its symmetric counterpart νss usually appears at 1410–1400 cm−1. By observing the presence of these two peaks in the FTIR spectra, the orientation of the adsorbate *COO− on the surface can be determined. According to surface selection rules, if the *COO− is bound to the surface through the two oxygen atoms in a bidentate orientation, only the symmetric stretch of *COO− appears in the spectra. If the molecule is attached to the surface through the carbon atom in a monodentate orientation, two peaks appear in the spectra that correspond to the symmetric and asymmetric stretches of the adsorbate.63,70,71 The absence of these two peaks suggests that the adsorbed intermediate *COO− did not last long enough on the surface of the Co/Cu catalysts to be detected by the FTIR before it was protonated to *COOH or desorbed from the surface as formate following a second electron transfer reaction. This might suggest that the two steps happened in a concerted step called a proton-coupled electron transfer reaction (PCET). Based on the spectral observation, it is impossible to know in which orientation the adsorbate is attached to the surface. However, based on theoretical energy calculations and modelling presented in the literature concerning the production of formate and CO, the *COO− intermediate is most likely attached to the Co/Cu catalysts’ surfaces through the C atom in a monodentate orientation.67,71–75Depending on the binding strength of the *COOH intermediate to the surface, this molecule can either be desorbed from the surface following a second PCET reaction to form formic acid, or it can undergo a dehydration reaction alongside the second PCET reaction to form the adsorbed *CO molecule. The *CO molecule desorbs from the surface as a gas molecule and escapes the thin solution of the electrolyte trapped between the catalyst and the ATR crystal. The product analysis showed that at a reductive potential greater than −0.6 V vs. RHE, the main CO2RR product was CO. This means that using Co/Cu–AA, 74% of the adsorbed *COOH intermediates undergo the later process.
4. Conclusions
Given that each fabrication step can introduce variations in the catalyst morphology and metal distribution on the surface, it can subsequently affect the binding affinity of reaction intermediates, thereby impacting the selectivity and efficiency of the catalysts. In this study, cobalt nanoparticles decorated with copper catalysts have been systematically optimized and studied for their efficiency in the electrochemical reduction of CO2. The self-supported catalysts were prepared using electrochemical deposition, galvanic replacement reaction, acid treatments, and thermal annealing techniques. The novel synthesis method discussed in this study can be scaled up for large-scale production without the use of surfactants or capping agents. Both Co/Cu–A and Co/Cu–AA catalysts have been shown to exhibit a large EASA (×14 and ×19 greater than the specific capacitance of the pristine Co substrate, respectively), high stability, and selective catalytic conversion of CO2 to formate and carbon monoxide. GC and NMR spectroscopy were employed to identify and quantify the gas and liquid products. While using the Co/Cu–AA catalyst, formate was the more favourable product with 31% FE at lower reductive potentials (−0.4 V vs. RHE) and carbon monoxide was more favourable with 74% FE at high reductive potential (−0.9 V vs. RHE). Through the inclusion of copper in the nanostructure, the overall FE was improved with the predominant production of carbon monoxide gas alongside a minor quantity of formate. The advantage lies in the distinct phases of the two resulting products, facilitating their relatively straightforward separation. The in situ electrochemical ATR-FTIR spectroscopy study presented in this study offered a unique insight into the reaction mechanisms of the Co/Cu nanocatalysts. It was revealed that using Co/Cu–A, the formate peaks were at maximum at −0.4 and −0.5 V and diminished between −0.8 and −0.9 V. When Co/Cu–AA was used as the catalyst, formate was continually being produced even at high reductive potentials where CO was the major CO2RR product and the HER was also taking place. The synthetic approach described in the present study has provided a viable strategy for the development of similar efficient nanostructured catalysts for the realization of industrial-scale CO2 reduction applications. This study also highlights the importance of methodically refining each step in the fabrication process of the nanocatalyst, not only to enhance the CO2 conversion efficiency, but also to steer the reduction pathway toward the desired product. Despite the advancements presented in this study, there is a strong demand for experiments utilizing surfaces with meticulously controlled morphologies to grasp the influence of active site distribution within the bimetallic structure on catalytic activity and selectivity. For instance, employing Co/Cu nanoparticles of consistent size and elemental composition with exposed distinct facets could be pivotal in elucidating how activity is affected. Moreover, employing in situ electrochemical FTIR spectroscopy can provide deeper insights into the reaction mechanisms involving various exposed facets of the Co/Cu nanocatalysts.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2022-04238). A. C. acknowledges NSERC and the Canada Foundation for Innovation (CFI) for the Canada Research Chair Award in Electrochemistry and Nanoscience.References
- Trends in Atmospheric Carbon Dioxide, https://gml.noaa.gov/ccgg/trends/global.html.
- F. Aldred, R. J. H. Dunn, N. Gobron, J. B. Miller and K. M. Willett, Bull. Am. Meteorol. Soc., 2022, 103, S11–S142 Search PubMed.
- J. A. Augustine and A. Capotondi, J. Geophys. Res.: Atmos., 2022, 127, 1–18 Search PubMed.
- T. Ahmad and D. Zhang, Energy Rep., 2020, 6, 1973–1991 CrossRef.
- N. Thonemann, L. Zacharopoulos, F. Fromme and J. Nühlen, J. Cleaner Prod., 2022, 332, 130067 CrossRef CAS.
- S. J. Bograd, M. G. Jacox, E. L. Hazen, E. Lovecchio, I. Montes, M. Pozo Buil, L. J. Shannon, W. J. Sydeman and R. R. Rykaczewski, Ann. Rev. Mar. Sci., 2023, 15, 1–26 CrossRef PubMed.
- A. Mirzabaev, L. Olsson, R. B. Kerr, P. Pradhan, M. G. R. Ferre and H. Lotze-Campen, Science and innovation for food systems transformation: Climate Change and Food Systems, Springer, Cham, 2021 Search PubMed.
- F. Babonneau, A. Haurie and M. Vielle, Oper. Res. Lett., 2023, 51, 33–39 CrossRef.
- Z. Hao, M. H. Barecka and A. A. Lapkin, Energy Environ. Sci., 2022, 15, 2139–2153 RSC.
- H. L.-V. Assche and T. Compernolle, Clean Technol. Environ. Policy, 2022, 24, 467–491 CrossRef.
- S. Abner and A. Chen, Appl. Catal., B, 2022, 301, 120761 CrossRef CAS.
- A. Salverda, S. Abner, E. Mena-Morcillo, A. Zimmer, A. Elsayed and A. Chen, J. Phys. Chem. C, 2023, 127, 7151–7161 CrossRef CAS.
- M. N. Hossain, R. M. Choueiri, S. Abner, L. D. Chen and A. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 51889–51899 CrossRef CAS PubMed.
- W. Yang, J. H. Zhang, R. Si, L. M. Cao, D. C. Zhong and T. B. Lu, Inorg. Chem. Front., 2021, 8, 1695–1701 RSC.
- A. Kaliyaperumal, P. Gupta, Y. S. S. Prasad, A. K. Chandiran and R. Chetty, ACS Eng. Au, 2023, 3, 403–425 CrossRef CAS.
- A. Mustafa, B. G. Lougou, Y. Shuai, Z. Wang, S. Razzaq, E. Shagdar, J. Zhao and J. Shan, J. Mater. Chem. A, 2021, 9, 4558–4588 RSC.
- D. Wu, G. Huo, W. Y. Chen, X. Z. Fu and J. L. Luo, Appl. Catal., B, 2020, 271, 118957 CrossRef CAS.
- D. Siltamaki, S. Chen, F. Pakravan, J. Lipkowski and A. Chen, J. Electrochem., 2021, 27, 278–290 CAS.
- F. Zhang, C. Chen, S. Yan, J. Zhong, B. Zhang and Z. Cheng, Appl. Catal., A, 2020, 598, 117545 CrossRef CAS.
- H. Wang, H. Chuai, X. Chen, J. Lin, S. Zhang and X. Ma, ACS Appl. Mater. Interfaces, 2023, 15, 1376–1383 CrossRef CAS PubMed.
- C. Rettenmaier, A. Herzog, D. Casari, M. Rüscher, H. S. Jeon, D. Kordus, M. L. Luna, S. Kühl, U. Hejral, E. M. Davis, S. W. Chee, J. Timoshenko, D. T. L. Alexander, A. Bergmann and B. R. Cuenya, EES Catal., 2024, 2, 311–323 RSC.
- X. Hu, Y. Gao, X. Luo, J. Xiong, P. Chen and B. Wang, Nanoscale, 2024, 16, 4909–4918 RSC.
- Y. Chen, F. Hu, Y. Hao, Y. Wang, Y. Xie, H. Wang, L. Yin, D. Yu, H. Yang, J. Ma, D. Kai, L. Li and S. Peng, Nano Res., 2022, 15, 3283–3289 CrossRef CAS.
- H. Liao, A. Fisher and Z. J. Xu, Small, 2015, 11, 3221–3246 CrossRef CAS PubMed.
- H. Ma, E. Ibáñez-Alé, R. Ganganahalli, J. Pérez-Ramírez, N. López and B. S. Yeo, J. Am. Chem. Soc., 2023, 145, 24707–24716 CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, 1–18 Search PubMed.
- M. Wu, C. Zhu, K. Wang, G. Li, X. Dong, Y. Song, J. Xue, W. Chen, W. Wei and Y. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 11562–11569 CrossRef CAS PubMed.
- H. Lu, L. Li, Q. Wu, S. Mu, R. Zhao, X. Zheng, C. Long, Q. Li, H. Liu and C. Cui, ACS Appl. Mater. Interfaces, 2023, 15, 13228–13237 CrossRef CAS PubMed.
- Z. Liu, M. N. Hossain, J. Wen and A. Chen, Nanoscale, 2021, 13, 1155–1163 RSC.
- Y. Gao, Y. Guo, Y. Zou, W. Liu, Y. Luo, B. Liu and C. Zhao, ACS Appl. Energy Mater., 2023, 6, 1340–1354 CrossRef CAS.
- J. Wang, D. Deng, Q. Wu, M. Liu, Y. Wang, J. Jiang, X. Zheng, H. Zheng, Y. Bai, Y. Chen, X. Xiong and Y. Lei, ACS Nano, 2023, 17, 18688–18705 CrossRef CAS PubMed.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- D. J. Siltamaki, S. Chen, F. Rahmati, J. Lipkowski and A. Chen, J. Electrochem., 2021, 27, 278–290 CAS.
- F. Rahmati, N. Sabouhanian, J. Lipkowski and A. Chen, Nanomaterials, 2023, 13, 778 CrossRef CAS PubMed.
- C. Dai, L. Sun, J. Song, H. Liao, A. C. Fisher and Z. J. Xu, Small Methods, 2019, 3, 1–7 Search PubMed.
- Y. Hao, Y. Sun, H. Wang, J. Xue, J. Ren, A. A. S. Devi, M. Y. Maximov, F. Hu and S. Peng, Electrochim. Acta, 2023, 449, 142213 CrossRef CAS.
- H. Wang, Y. Hao, Y. Sun, J. Pan, F. Hu, D. Kai and S. Peng, Catal. Lett., 2023, 153, 2115–2124 CrossRef CAS.
- V. S. S. Mosali, A. M. Bond and J. Zhang, Nanoscale, 2022, 14, 15560–15585 RSC.
- M. Bernal, A. Bagger, F. Scholten, I. Sinev, A. Bergmann, M. Ahmadi, J. Rossmeisl and B. R. Cuenya, Nano Energy, 2018, 53, 27–36 CrossRef CAS.
- Y. Wang, F. Hu, Y. Chen, H. Wang, A. E. Fetohi, Y. Hao, L. Li, K. M. El-Khatib and S. Peng, J. Mater. Sci., 2022, 57, 7276–7289 CrossRef CAS.
- J. S. Dondapati, M. Govindhan and A. Chen, Chem. Commun., 2022, 58, 11127–11130 RSC.
- B. Sidhureddy, J. S. Dondapati and A. Chen, Chem. Commun., 2019, 55, 3626–3629 RSC.
- A. A. Dubale, C. J. Pan, A. G. Tamirat, H. M. Chen, W. N. Su, C. H. Chen, J. Rick, D. W. Ayele, B. A. Aragaw, J. F. Lee, Y. W. Yang and B. J. Hwang, J. Mater. Chem. A, 2015, 3, 12482–12499 RSC.
- M. N. Hossain, S. Chen and A. Chen, Appl. Catal., B, 2019, 259, 118096 CrossRef.
- B. Serapinienė, L. Gudavičiūtė, S. Tutlienė, A. Grigucevičienė, A. Selskis, J. Juodkazytė and R. Ramanauskas, Coatings, 2023, 13, 1335 CrossRef.
- S. R. Chung, K. W. Wang, S. R. Sheen, C. T. Yeh and T. P. Perng, Electrochem. Solid-State Lett., 2007, 10, A155–A158 CrossRef CAS.
- J. Huang, X. Guo, G. Yue, Q. Hu and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 44403–44414 CrossRef CAS PubMed.
- K. Lejaeghere, V. Van Speybroeck, G. Van Oost and S. Cottenier, Crit. Rev. Solid State Mater. Sci., 2014, 39, 1–24 CrossRef CAS.
- O. J. Rutt, G. R. Williams and S. J. Clarke, Chem. Commun., 2006, 88, 2869–2871 RSC.
- R. S. Hyam, J. Lee, E. Cho, J. Khim and H. Lee, J. Nanosci. Nanotechnol., 2012, 12, 8396–8400 CrossRef CAS PubMed.
- M. Rüsing, G. Berth, K. Lischka and A. Pawlis, J. Nanomater., 2013, 2013, 714853–714859 Search PubMed.
- D. D. M. Prabaharan, K. Sadaiyandi, M. Mahendran and S. Sagadevan, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 1–6 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- S. Chen and A. Chen, J. Phys. Chem. C, 2019, 123, 23898–23906 CrossRef CAS.
- M. Papasizza and A. Cuesta, ACS Catal., 2018, 8, 6345–6352 CrossRef CAS.
- L. Lukashuk, N. Yigit, R. Rameshan, E. Kolar, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, K. Föttinger and G. Rupprechter, ACS Catal., 2018, 8, 8630–8641 CrossRef CAS PubMed.
- Y. Hao, F. Hu, S. Zhu, Y. Sun, H. Wang, L. Wang, Y. Wang, J. Xue, Y. F. Liao, M. Shao and S. Peng, Angew. Chem., Int. Ed., 2023, 62, e202304179 CrossRef CAS PubMed.
- M. Moradzaman and G. Mul, ACS Catal., 2020, 10, 8049–8057 CrossRef CAS.
- B. Innocent, D. Pasquier, F. Ropital, F. Hahn, J. M. Léger and K. B. Kokoh, Appl. Catal., B, 2010, 94, 219–224 CrossRef CAS.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- A. R. Davis and B. G. Oliver, J. Solution Chem., 1972, 1, 329–339 CrossRef CAS.
- B. G. Oliver and A. R. Davis, Can. J. Chem., 1973, 51, 698–702 CrossRef CAS.
- T. Wu, H. Bu, S. Tao and M. Ma, Nanoscale, 2024, 16, 3926–3935 RSC.
- Y. Katayama, F. Nattino, L. Giordano, J. Hwang, R. R. Rao, O. Andreussi, N. Marzari and Y. Shao-Horn, J. Phys. Chem. C, 2019, 123, 5951–5963 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- T. Tsujiguchi, Y. Kawabe, S. Jeong, T. Ohto, S. Kukunuri, H. Kuramochi, Y. Takahashi, T. Nishiuchi, H. Masuda, M. Wakisaka, K. Hu, G. Elumalai, J. Fujita and Y. Ito, ACS Catal., 2021, 11, 3310–3318 CrossRef CAS.
- C. H. Kim, S. W. Han, T. H. Ha and K. Kim, Langmuir, 1998, 14, 6113–6120 CrossRef.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- X. Zhi, A. Vasileff, Y. Zheng, Y. Jiao and S. Z. Qiao, Energy Environ. Sci., 2021, 14, 3912–3930 RSC.
- L. Fu, Z. Qu, L. Zhou and Y. Ding, Appl. Catal., B, 2023, 339, 123170 CrossRef CAS.
- Z. Liu, C. Liu, J. Zhang, S. Mao, X. Liang, H. Hu and X. Huang, Appl. Catal., B, 2024, 341, 123274 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, electrochemical ATR-FTIR schematics, supporting electrochemical experiments, EASA plots, XPS spectra,12 h stability tests, supporting FTIR spectra, time-dependent FTIR spectra, capacitance, and EASA summarized results, EDX atomic percentages, XRD peak assignments, a summary of faradaic efficiencies and rates of production, and comparison of Co- and Cu-based electrocatalysts for CO2RR. See DOI: https://doi.org/10.1039/d4nr00909f |
| This journal is © The Royal Society of Chemistry 2024 |