
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the cellular antioxidant mechanism against cytotoxic silver nanoparticles: a Raman spectroscopic analysis†
Davide
Redolfi-Bristol
 *acg,
Kenta
Yamamoto
c,
Elia
Marin
ahi,
Wenliang
Zhu
a,
Osam
Mazda
c,
Pietro
Riello
g and
Giuseppe
Pezzotti
*abcdefg
*acg,
Kenta
Yamamoto
c,
Elia
Marin
ahi,
Wenliang
Zhu
a,
Osam
Mazda
c,
Pietro
Riello
g and
Giuseppe
Pezzotti
*abcdefg
aCeramic Physics Laboratory, Kyoto Institute of Technology, Sakyo-ku, Matsugasaki, 606-8585, Kyoto, Japan. E-mail: davide.redolfi@unive.it; pezzotti@kit.ac.jp
bDepartment of Molecular Genetics, Institute of Biomedical Science, Kansai Medical University, 2-5-1 Shinmachi, Hiraka-ta, Osaka 573-1010, Japan
cDepartment of Immunology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, 465 Kajii-cho, Kamigyo-ku, Kyoto 602-8566, Japan
dDepartment of Dental Medicine, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, 465 Kajii-cho, Kamigyo-ku, Kyoto 602-8566, Japan
eDepartment of Orthopedic Surgery, Tokyo Medical University, 6-7-1 Nishi-Shinjuku, Shinjuku-ku, 160-0023 Tokyo, Japan
fDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy
gDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari di Venezia, Via Torino 155, 30172 Venezia, Italy
hDepartment Polytechnic of Engineering and Architecture, University of Udine, 33100, Udine, Italy
iBiomedical Research Center, Kyoto Institute of Technology, Sakyo-ku, Matsugasaki, Kyoto 606-8585, Japan
First published on 2nd May 2024
Abstract
Silver nanoparticles (AgNPs) hold great promise for several different applications, from colorimetric sensors to antimicrobial agents. Despite their widespread incorporation in consumer products, limited understanding of the detrimental effects and cellular antioxidant responses associated with AgNPs at sublethal concentrations persists, raising concerns for human and ecological well-being. To address this gap, we synthesized AgNPs of varying sizes and evaluated their cytotoxicity against human dermal fibroblasts (HDF). Our study revealed that toxicity of AgNPs is a time- and size-dependent process, even at low exposure levels. AgNPs exhibited low short-term cytotoxicity but high long-term impact, particularly for the smallest NPs tested. Raman microspectroscopy was employed for in-time investigations of intracellular molecular variations during the first 24 h of exposure to AgNPs of 35 nm. Subtle protein and lipid degradations were detected, but no discernible damage to the DNA was observed. Signals associated with antioxidant proteins, such as superoxide dismutase (SOD), catalase (CAT) and metallothioneins (MTs), increased over time, reflecting the heightened production of these defense agents. Fluorescence microscopy further confirmed the efficacy of overexpressed antioxidant proteins in mitigating ROS formation during short-term exposure to AgNPs. This work provides valuable insights into the molecular changes and remedial strategies within the cellular environment, utilizing Raman microspectroscopy as an advanced analytical technique. These findings offer a novel perspective on the cytotoxicity mechanism of AgNPs, contributing to the development of safer materials and advice on regulatory guidelines for their biomedical applications.
Introduction
In the ever-expanding landscape of nanotechnology, silver nanoparticles (AgNPs) represent promising candidates for several different applications, which range from colorimetric sensors to antimicrobial agents.1–4 Indeed, AgNPs play a significant role as a constituent in numerous products accessible in the market, including fabrics, healthcare gadgets, cosmetics and antibacterial sprayers.5–9 Additionally, AgNPs find application in the healing of injuries and burns, along with their use in coating for medical implants.10–13 Nevertheless, despite the growing incorporation of AgNPs in consumer goods, there is still limited understanding of the detrimental effects and the cellular responses associated with AgNPs, raising concerns about the potential consequences on human and ecological well-being.5,7,14 Due to the widespread utilization of AgNPs in fabrics, bandages, athletic attire, and other items that directly interact with the skin, it is crucial to thoroughly evaluate their effect during dermal exposure.11,15,16 Researchers have assessed the penetration of AgNPs through the skin, particularly in cases of compromised skin integrity, or after employing dressings coated with AgNPs in the treatment of extensive burns.17–19 This increases the concern about their potential side-effects and therefore the need to deeply investigate their behavior when they come into contact with human cells.The toxicity of AgNPs is mainly due to the generation of reactive oxygen species (ROS), causing oxidative stress and damaging cellular components. Studies have shown that AgNPs undergo a dissolution process upon entering inside the cell, leading to the release of Ag+ ions.20–22 The exogenous metal ions induce the production of ROS, including superoxide radicals and hydrogen peroxide, within the cells.23–25 These elevated levels of ROS can lead to damage of cellular components such as lipids, proteins, and DNA if they are not properly contained by antioxidant agents.26,27 Indeed, enzymatic antioxidants, such as superoxide dismutase (SOD) and catalase (CAT), and non-enzymatic antioxidants, such as glutathione and the family of metallothioneins (MTs), play a crucial role in maintaining cellular homeostasis by counteracting exogenous metal ions and oxidative stress inside cells. However, if the toxic action is persistent and prolonged over time, it can overwhelm the initially effective defense mechanism, ultimately resulting in its failure.25,28,29
Given these reasons, it is essential to achieve a thorough comprehension of the diverse physicochemical changes resulting from exposure to silver nanoparticles. This is crucial to enhance our knowledge of cellular responses, thereby enabling better understanding of exposure limits and potential implications associated with their usage. Moreover, while the literature extensively explores the impact of AgNPs at elevated concentrations for a short period of time, an inherent lack of emphasis on investigations below toxicity thresholds underscores the need to investigate the intricate cellular defense mechanisms. Unraveling these complex interactions between AgNPs and living organisms demands sophisticated analytical techniques capable of providing detailed insights into their behavior at the molecular level.
Raman microspectroscopy stands out as a powerful analytical technique for investigating the effects of nanomaterials on cells due to its unique advantages. Unlike traditional methods, Raman spectroscopy provides label-free and non-invasive probing capabilities, allowing researchers to directly observe molecular changes within living cells.30,31 Indeed, Raman microspectroscopy has been employed for the characterization of biochemical evolution during cellular differentiation,32,33 neuronal cell networking and separation,34 metabolic changes in cancer cells35,36 and apoptosis.37,38 In the nano-toxicology context, this technique can have the advantageous ability to provide detailed information on cellular component alterations, which can be correlated with cytotoxic responses, oxidative stress, or inflammation induced by nanomaterial exposure.39–42 Furthermore, the non-destructive nature and detailed molecular information obtained through Raman spectroscopy make it a helpful tool for advancing the comprehension of nanomaterial–cell interactions in biomedical research and toxicology.31,39 In recent years, a small number of studies have been performed to assess the toxic effects induced by AgNPs through Raman spectroscopy.43–45 However, no study on the temporal evolution of the cellular stress condition has ever been carried out and, in most cases, red blood cells, which are known to be susceptible to oxidative stress due to the absence of a nucleus and other organelles, have been employed as a cellular model.
In this work, we synthesized silver nanoparticles of different sizes and tested their cytotoxicity against human dermal fibroblast (HDF) cells. We employed HDF cells as a model to better explore the consequences of dermal exposure to AgNPs, given their utilization in fabrics and wound dressings. After discovering the concentration limits for which these NPs exhibited low cytotoxicity in the short term but high cytotoxicity in the long term, an in-time investigation into intracellular molecular variation that occurs in the first 24 h of exposure was carried out using Raman microspectroscopy. Raman spectroscopy allowed the detection of slight degradations of proteins and lipids, triggered by the presence of AgNPs inside the cell. Signals deriving from the C–S bond and from the active sites of SOD and CAT proteins increased during the exposure time, caused by the increased production of antioxidant agents. Eventually, fluorescence microscopy was employed to confirm the efficacy of the overexpressed antioxidant proteins in mitigating the ROS formation during short-term exposure to AgNPs. Our work provides an elucidation about the molecular changes and remedial strategies that occur within the cellular environment using the advanced technique of Raman microspectroscopy. This novel approach allowed for real-time monitoring of molecular variations within the cellular environment, providing advanced insights into the dynamic interactions between AgNPs and cellular components. Supported by conventional end-point analyses, the focus on the temporal dynamics of cellular responses detected through Raman microspectroscopy allowed the observation of a significant increase in specific signals associated with antioxidant actions, a phenomenon rarely documented in previous studies, in particular in real-time. By uncovering these novel molecular signatures our results allow understanding, from a different perspective, the mechanism underlying the cytotoxicity of silver nanoparticles, providing valuable insights into the development of safer materials and advice on the regulatory guidelines for their use in biomedical applications.
Materials and methods
Materials
Silver Nitrate (AgNO3), sodium citrate tribasic dihydrate, tannic acid and 2′,7′-dichlorofluorescein diacetate (DCFH-DA) were purchased from Sigma-Aldrich (Merck KGaA, Germany). Dulbecco's modified Eagles’ medium (DMEM), fetal bovine serum (FBS), MEM non-essential amino acid solution, L-sodium pyruvate, penicillin–streptomycin mixed solution, superoxide dismutase from bovine erythrocytes, catalase from bovine liver, an MTT cell count kit, radio-immuno-precipitation assay (RIPA) buffer and phosphate-buffered saline (PBS) solutions were purchased from Nacalai Tesque (Japan). tert-Butyl hydroperoxide (TBHP), was acquired from FUJIFILM Wako Pure Chemical Corporation (Japan). TaKaRa BCA protein assay kit was purchased from TaKaRa Bio Inc. (Japan). Superoxide dismutase (SOD) inhibitory activity assay was acquired from DOJINDO (Japan).Synthesis of silver nanoparticles (AgNPs25–50nm)
Silver nanoparticles (AgNPs) were prepared following a modified method from that reported by Bastus et al.46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm per 15 min) in MilliQ water.
000 rpm per 15 min) in MilliQ water.
Characterization of AgNPs
Cell culture
Normal human dermal fibroblast (HDF) cells derived from a 22-year-old black female were purchased from Toyobo Life Science (Osaka, Japan). HDF cells were cultured in Dulbecco's modified Eagles’ medium (DMEM) containing phenol red, supplemented with 10% v/v fetal bovine serum (FBS), 1% MEM nonessential amino acid solution, 1% L-sodium pyruvate and 1% penicillin–streptomycin mixed solution (complete medium) in a humidified incubator at 37 °C and under 5% CO2 conditions.Cell viability quantification
Cell viability quantification was performed by MTT assay (MTT cell count kit, Nacalai Tesque, Japan) based on the cleavage of a tetrazolium salt by metabolically active cells to form a water-insoluble formazan dye. Briefly, HDF cells were seeded in 24-well plates (2 × 104 cells per well) and incubated overnight. AgNPs were added to the wells to final concentrations of 0.2, 2 and 20 μg mL−1, and incubated for additional periods of 24 and 72 h. At the end of the treatment, the medium from each well was removed and the cells were washed once with PBS to remove possible interference from AgNPs. The cells were incubated with 500 μL of complete culture media and 50 μL of the MTT solution for 3 h in the incubator. Subsequently, 500 μL of the solubilization solution was added and the precipitated formazan was dissolved by pipetting. Two aliquots of 100 μL were collected from each well and placed in a 96-well plate. Eventually, the absorbance at 550 nm was measured using an Infinite F50 Plus microplate reader (Tecan, Switzerland). The cell viability of each group is expressed as a percentage of the mean value of the control. The measurements were carried out in triplicate.Raman microspectroscopy
For Raman studies, HDF cells were grown in a complete growth medium in an incubator at 37 °C and under 5% CO2 conditions for 24 h on CaF2 dishes, to minimize the background signal as reported in the literature.48 Subsequently, the cells were treated with 10 μg mL−1 of AgNPs35nm diluted in supplemented DMEM for 4, 8, 12 and 24 h. Prior to Raman microspectroscopy, the cells were fixed with formaldehyde solution. Briefly, the cells were washed twice with PBS, then treated with 4% formaldehyde solution for 5 minutes and finally washed three times with PBS. Raman microspectroscopy measurements were performed while CaF2 dishes were kept in PBS.Raman spectra of HDF cells were collected with a dedicated Raman device (RAMANtouch, Nanophoton Co., Osaka, Japan). The RAMANtouch spectroscope was operated with an excitation source of 532 nm (excitation power density = 576 mW μm−2), a 300 gr mm−1 grating and a 60× immersion objective lens (NA = 1.0). The spectral resolution was about 3 cm−1 and was recorded in the range of 400 to 1800 cm−1. The RAMANtouch spectroscope was operated in “point mode”, acquiring 6 spectra per cell (3 from the cytoplasm and 3 from the nucleus), analyzing ten cells per sample. Around 60 spectra were therefore acquired per sample. The exposure time for each measurement was 1 s and the acquisition was averaged five times. All the experiments were performed in triplicate. Reference Raman spectra of SOD and CAT were also collected with the RAMANtouch spectroscope after depositing the sample (SOD powder; CAT water solution) on the CaF2 substrate.
Raman data were processed averaging around 60 spectra per sample through RAMAN Viewer software (Nanophoton Co., Osaka, Japan), excluding those that showed too large intensity caused by the surface enhanced Raman scattering (SERS) phenomenon. On the averaged spectrum background subtraction, smoothing (Savitsky–Golay smoothing; degree 2, size 7, height 11) and baseline correction (manually selecting the points representative of the background) were performed by means of LabSpec software (version 5.5, Horiba, Japan). Subsequently, the standard normal variate (SNV) normalization was performed using SpectraGryph software (F. Menges, “Spectragryph – optical spectroscopy software”, version 1.2.16.1, 2022, https://www.effemm2.de/spectragryph/). Finally, an average of the resulting Raman spectrum for each replicate was obtained using Origin software (version 9.8.0.200, OriginLab, Massachusetts, USA). More details about the data processing are reported in Fig. S2.†
SOD inhibitory activity assay
HDF cells were seeded in 6-well plates (2 × 105 cells per well) and grown in complete growth medium in an incubator at 37 °C and under 5% CO2 conditions for 24 h. Subsequently, the cells were treated with AgNPs35nm at 10 μg mL−1 for 8, 12, 24 and 72 h. After the treatment, the cells were collected, and their proteins were extracted using diluted RIPA buffer. The amount of protein in each sample was quantified using a TaKaRa BCA protein assay kit, and subsequently equalized prior to SOD activity quantification. The SOD inhibitory activity assay was performed following the manufacturer's instructions. Briefly, 20 μL of each sample solution was added to a well of a 96-well plate. Then, 200 μL of WST-1 working solution was added to each well, and finally 20 μL of enzyme (xanthine oxidase) working solution was introduced. The plate was incubated at 37 °C for 20 min and eventually the final absorbance was measured at 450 nm using a microplate reader. The measurements were carried out in triplicate.Fluorescence microscopy for ROS quantification
HDF cells were seeded in 24-well plates (2 × 104 cells per well) and grown in complete growth medium in an incubator at 37 °C and under 5% CO2 conditions for 24 h. Subsequently, the cells were treated with AgNPs35nm at 10 μg mL−1 for 8, 12, 24 and 72 h. The complete growth medium and tert-butyl hydroperoxide (100 μM) were used as the negative and positive control, respectively. After the treatment, the cells were washed twice with PBS and then stained with DCFH-DA solution diluted in culture media for 30 min, keeping them in the dark inside the incubator chamber. Then, ROS production was evaluated using a fluorescence microscope (BZ-X810, KEYENCE, Japan) and BZ-II Analyzer software (KEYENCE, Japan). The fluorescence intensity of each group was expressed as a percentage of the mean value of the control. The measurements were carried out in triplicate.Results and discussion
Silver nanoparticles of different dimensions were prepared following a modified procedure reported by Bastus et al.46 AgNPs25nm seeds were initially synthesized through AgNO3 reduction by means of sodium citrate and tannic acid. The addition of reducing reagents to the silver nitrate solution resulted in its change of color from transparent to light yellow in a few seconds, confirming the formation of silver nanoparticles. The progressive growth steps performed by adding additional sodium citrate, tannic acid and AgNO3, led to the darkening of the solution and its color turning towards yellow-green, which indicated an increase in the size of the NPs. The produced samples were thoroughly characterized by UV-vis spectroscopy, analytical centrifugation (AC), dynamic light scattering (DLS) and scanning electron microscopy (SEM).Fig. 1a shows the UV-visible spectra of AgNPs25nm seeds and their subsequent growth steps. As can be seen from the absorption bands, it is possible to observe a red-shift in the maximum absorbance as the size of the nanoparticles increases. This fact is correctly correlated with the color change of the solutions during the different growth steps. The precise maximum absorbance values are reported in Table 1.
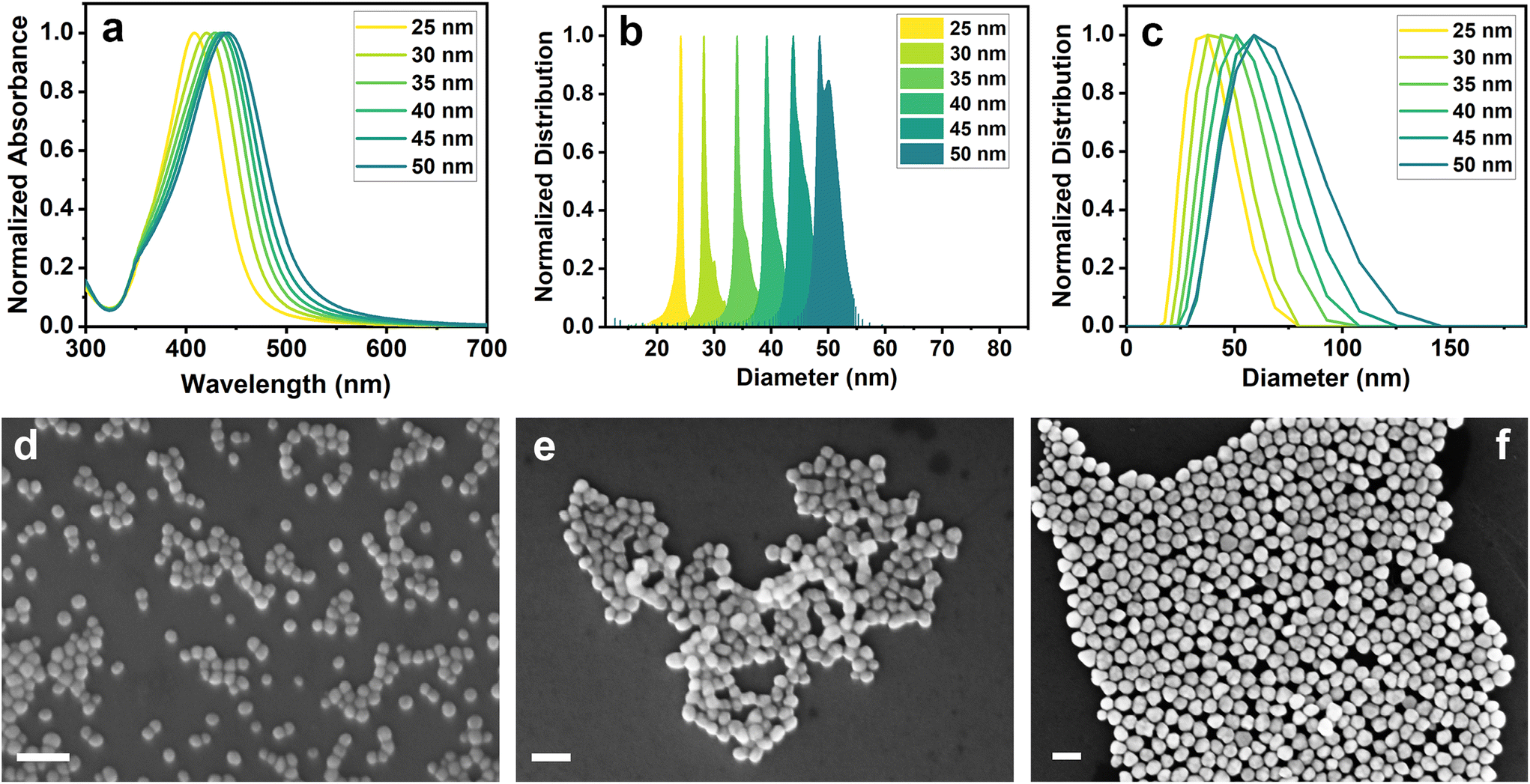 | ||
| Fig. 1 (a) UV-visible spectra, (b) analytical centrifugation and (c) DLS distribution of AgNPs. SEM images of (d) AgNPs25nm, (e) AgNPs35nm and (f) AgNPs50nm (scale bar: 100 nm). | ||
| Sample name | UV-visible (Absmax, nm) | AC – number distribution (diameter, nm) | DLS – intensity distribution (diameter, nm) | SEM – number distribution (diameter, nm) |
|---|---|---|---|---|
| AgNPs25nm | 408 | 24 ± 2 | 33.2 (PDI = 0.1) | 24 ± 3 |
| AgNPs30nm | 421 | 30 ± 3 | 37.5 (PDI = 0.1) | 31 ± 3 |
| AgNPs35nm | 429 | 34 ± 4 | 42.7 (PDI = 0.1) | 37 ± 4 |
| AgNPs40nm | 434 | 40 ± 3 | 46.8 (PDI = 0.1) | 44 ± 5 |
| AgNPs45nm | 438 | 45 ± 3 | 53.1 (PDI = 0.1) | 47 ± 5 |
| AgNPs50nm | 442 | 49 ± 7 | 55.7 (PDI = 0.1) | 52 ± 6 |
Analytical centrifugation and DLS analysis were performed to assess the hydrodynamic diameter of AgNPs, and the results are reported in Fig. 1b and c and Table 1. AC measurements show a narrow distribution with an increase in the size of the NPs of approximately 5 nm after each growth step. These results were confirmed by DLS analysis, which also revealed a growth of around 5 nm between the steps. However, DLS results showed a broader distribution with a larger average diameter size. The slight differences in the size estimation among the techniques may be due to the different solvation effects that the particles encounter during the measurements. Fig. 1d–f shows three representative SEM images, respectively, of AgNPs25nm seeds, AgNPs35nm and AgNPs50nm obtained through different growth steps. SEM images of other growth steps are reported in Fig. S1.† Silver nanoparticles show a spherical shape with a homogeneous distribution that increases by about 5 nm between the steps (Table 1). This confirms the correct growth of the nanoparticles and that the differences in the measured size are only caused by the different characterization techniques.
After the synthesis, the AgNPs were washed and concentrated through centrifugation. Before testing the cytotoxicity, the amount of silver in the colloidal solutions was quantified through ICP-OES. This was done to properly verify that the quantity of silver tested for viability assay was the same for all the different NP types. ICP-OES results are reported in Table 2 and they highlight a large difference between the expected concentrations of the final solutions and that of the real one, except for AgNPs50nm. During the synthesis, a large quantity of silver ions did not react or, during the concentration process, a large number of nanoparticles remained in the supernatant, thus resulting in a lower concentration than expected. More studies would be necessary to discern these two possibilities.
| Sample name | Expected concentration (μg mL−1) | Measured concentration (μg mL−1) |
|---|---|---|
| AgNPs25nm | 540 | 2.9 ± 0.1 |
| AgNPs30nm | 1050 | 32.5 ± 0.2 |
| AgNPs35nm | 1580 | 159.1 ± 0.3 |
| AgNPs40nm | 2110 | 354.3 ± 0.3 |
| AgNPs45nm | 2650 | 1093 ± 1.0 |
| AgNPs50nm | 3250 | 3521 ± 2.0 |
AgNPs35nm and AgNPs50nm were selected to study the toxicity of the nanoparticles against HDF cells for 24 and 72 h at 0.2, 2 and 20 μg mL−1. These two sizes were chosen because of their distinct distribution and homogeneous diameters, while also being abundant enough to be used in toxicological tests. In addition, the concentrations were selected because they demonstrated reduced toxicity for similar NP sizes in previous studies on HDF cells.49,50 Quantification of living cells was performed by MTT assay, as reported in previous studies.49,50
The results reported in Fig. 2 indicate that nanoparticles exhibited cytotoxicity against HDF cells following prolonged exposure of 72 h, especially for AgNPs35nm; however, no discernible toxicity was observed during shorter exposures up to 24 h. After 24 h, AgNPs35nm displayed a low cytotoxicity for the concentration tested, with a slightly larger reduction in cell viability at 20 μg mL−1. An even lower cytotoxicity was observed for cells treated with AgNPs50nm, showing a maximum viability reduction of about 9% at the highest concentration tested for 24 h. In contrast, after 72 h, the toxic effect of the nanoparticles significantly increased, exhibiting a low cellular viability of 10% for AgNPs35nm and 63% for AgNPs50nm at 20 μg mL−1. As reported in the literature, it is clear that cytotoxicity is size- and time-dependent, when the same concentration in μg mL−1 is used to test them.50,51 Larger nanoparticles seem to be internalized at a slower rate when compared to smaller ones.52 Moreover, at an equivalent mass concentration (expressed in μg mL−1), their number concentration (expressed in N° of NPs mL−1) and their global surface area are lower, further diminishing their presence in the medium and within the cell. The prolonged exposure to nanoparticles can instead exert a long-lasting toxic and anti-proliferative effect. This phenomenon can be due to the fact that the AgNPs, remaining inside the cells, undergo a degradation process whose kinetics is expected to be subject to the dimension of AgNPs (smaller nanoparticles dissolve more rapidly than larger ones), ultimately leading to a size-dependent release of Ag+ ions.21,22,53 The release of Ag+ ions represents the primary cause of cytotoxicity associated with silver nanoparticles, as their presence within the cell gives rise to the generation of an excess of ROS.21,25 A longer permanence of the nanoparticles inside the cell therefore causes a continuous release of Ag+ ions and, consequently, a more prolonged cytotoxic effect over time.
 | ||
| Fig. 2 Quantification of living cells after incubation with AgNPs35nm and AgNPs50nm for 24 and 72 h. *Significant differences from the negative control (P < 0.05). | ||
In light of the cytotoxicity results, we decided to investigate the cellular and antioxidant mechanisms that occur inside the cells during the first 24 h of exposure to AgNPs. Raman microspectroscopy was conducted over time on HDF cells treated with AgNPs35nm at a concentration of 10 μg mL−1. This size and concentration were chosen as the optimal compromise between the rapidity of cytotoxic action and the degree of toxicity between the two previously tested sizes. At different time points (0, 4, 8, 12 and 24 h) the cells were washed and fixed with formaldehyde prior to Raman microspectroscopy analysis. Although it is acknowledged that cell fixation introduces some changes to the resulting Raman spectrum, this step was performed to enable a more accurate and extensive analysis of the cells. This approach minimizes potential distortions arising from temporal gaps between measurements and from external environmental factors (e.g., temperature fluctuations and CO2 levels). Moreover, formaldehyde fixation was chosen based on previous studies indicating that it is the method that induces the least alteration to the final spectrum.54–57
The Raman spectra reported in Fig. 3a were obtained by averaging the signals from around 60 spectra per sample, followed by a final averaging per three replicates. The cytoplasmic and nuclear spectra were combined, as no significant differences relevant to our study were observed when analyzed separately. The points from which spectra were acquired were carefully chosen avoiding nanoparticle aggregates that could have given rise to exceedingly intense SERS phenomena, in order to prevent misinterpretations or spectral alterations. More details about the data processing are reported in Fig. S2.† The Raman bands are associated with diverse vibrations of specific biochemical molecules found within the cell and the assignment of these signals is presented in Table 3.
 | ||
| Fig. 3 (a) Time-dependent normalized Raman spectra collected from HDF cells exposed to AgNPs35nm (10 μg mL−1) and (b) normalized intensities of the vibrations corresponding to the 1572 cm−1 and 638 cm−1 bands, collected as a function of time from the HDF cells exposed to AgNPs35nm (10 μg mL−1). *Significant differences from the “0 hour” (P < 0.05). | ||
| Wavenumber (cm−1) | Biological molecule | Tentative assignment of bond vibration | Ref. |
|---|---|---|---|
| 638 | Proteins | –C–S– bond | 58 and 59 |
| 705–720 | Lipids | Phospholipid C–N stretch | 60 |
| 740–760 | Protein | Tryptophan (ring breathing) | 58, 60 and 61 |
| 770–785 | DNA/RNA | DNA backbone O–P–O, uracil, cytosine and thymine (ring breathing) | 58, 60 and 61 |
| 810–825 | RNA | RNA backbone O–P–O | 58, 60 and 61 |
| 920–965 | Proteins | –C–C– backbone | 58, 60 and 61 |
| 990–1005 | Proteins | Phenylalanine (symmetric ring breathing) | 58, 60 and 61 |
| 1020–1035 | Proteins | Phenylalanine (C–H in plane bending) | 58, 60 and 61 |
| 1075–1095 and 1115–1125 | Proteins and lipids | C–N stretching in proteins and C–C stretching in lipids | 58 and 61 |
| 1235–1270 | Proteins | Amide III | 36, 58 and 61 |
| 1280–1340 | Proteins and lipids | Amide III and CH2 and CH3 vibrations of lipids | 58 and 61 |
| 1430–1460 | Proteins and lipids | C–H vibration | 33, 58 and 61 |
| 1572 | Proteins | Superoxide dismutase and catalase | 62–67 |
| 1600–1670 | Proteins | Amide I | 58 and 61 |
In Fig. 3a, it is possible to observe that the Raman bands exhibit variations in intensity at different time points. Nevertheless, they maintain their characteristic shape, confirming that the formaldehyde fixing process enables a consistent and reproducible comparison between the samples. The bands at around 940, 995, 1030, 1082, 1120 and 1440 cm−1 show a small but general decrease in intensity with time, compared to the initial state (Fig. S3†). All these bands belong mainly to protein and lipid molecular structures.58,60,61 As reported in the literature, this fact indicates that in the cell a deterioration process occurs, which is caused by the presence of exogenous and toxic substances.68–70 The AgNPs that penetrate inside the cells undergo a dissolution process, releasing hazardous Ag+ ions, which impairs the regular functions of cells.21,22 The introduction of NPs and external metal ions therefore stimulates the generation of free radicals within the cellular environment, eventually contributing to the breakdown of proteins and lipids.21,22,26 However, in our case, this process seems to occur to a smaller extent and with a slower velocity. This fact could therefore be due to the action of anti-oxidation and detoxification mechanisms, that take place inside the cell to prevent the detrimental effect of AgNPs.
Over time, in Raman spectra, the appearance of a band at ∼638 cm−1 and the intensification of the one at ∼1572 cm−1 can be observed (Fig. 3). The band at ∼638 cm−1 corresponds to the vibrations of C–S bonds in peptides and proteins, originating from sulfur-containing amino acids such as methionine and cysteine.58,59 Biological molecules rich in sulfur-containing amino acids are usually involved in antioxidant and detoxification processes, and are synthesized by cells in order to reduce the harmful action of toxic compounds and ROS. The most important antioxidant molecules produced by the cells are the two enzymes superoxide dismutase (SOD) and catalase (CAT), the small molecule glutathione (GSH) and the cysteine-rich, low molecular weight family of proteins named metallothionein (MT; involved in the binding of metal ions to prevent them from catalyzing the production of ROS).25,28,71 All these molecules are rich in sulfur-containing amino acids, particularly cysteine, which are crucial for their structure and function. An increase in intensity relative to the C–S bond band can therefore be attributed to their increased synthesis due to the activation of a cellular defensive mechanism against the presence of exogenous AgNPs.22,29,72–75 The band at ∼1572 cm−1 can be instead attributed to the vibrations of bonds in the active sites of the two antioxidant enzymes SOD and CAT, which have been observed with similar laser sources. For the SOD enzyme it originates from the metal-bridging imidazole ring of histidine,65–67 while for CAT it comes from one vibration of the porphyrin skeletal structure.62–64 Therefore, the growth of this band during time additionally confirms the increased synthesis of the two antioxidant enzymes. Eventually, the temporal evolution of the two bands, fitted with an exponential curve, aligns well with the characteristic trends exhibited by biological processes.76 This reinforces the hypothesis regarding the correct assignment of signals to the ongoing biological processes.
To verify the contribution of the band at ∼1572 cm−1, we acquired the Raman spectra under 532 nm excitation of pure superoxide dismutase and catalase. The comparison between the Raman spectrum of the region at around 1572 cm−1 collected from the HDF cells exposed to AgNPs35nm for 12 h and the spectrum of the enzymes is shown in Fig. S4.† Although the enzymes are non-human-derived, for both proteins it is possible to observe a band at around 1572 cm−1, especially for the CAT enzyme, which further supports the hypothesis that this signal derives from the two antioxidants.
Being that SOD is the more abundant and ubiquitous antioxidant enzyme in cells,77,78 we quantified its total amount in cells after their exposure to AgNPs using the SOD activity assay. The SOD activity assay bases its function on the conversion of the tetrazolium salt WST-1 into a water-soluble formazan dye, upon reduction with the superoxide anion (O2−). The rate of reduction of WST-1 with the superoxide anion is linearly related to xanthine oxidase (XO) activity (the enzyme used to generate O2−), and this reduction is inhibited by the presence of SOD (Fig. 4). Therefore, a lower reduction of WST-1 into the formazan dye in the treated sample indicates the presence of a larger amount of the SOD enzyme, as SOD can catalyze the dismutation of O2− and inhibit WST-1 reduction. Fig. 4 shows the decrease with time of the conversion levels of WST-1 into the formazan dye in the samples treated with AgNPs. This observation indicates an increasing presence of SOD enzymes in the cells, which consequently prevents the formation of the superoxide anion and the conversion of WST-1. However, further investigations with immunoassay tests would be necessary to precisely quantify the overproduction of SOD and CAT enzymes.
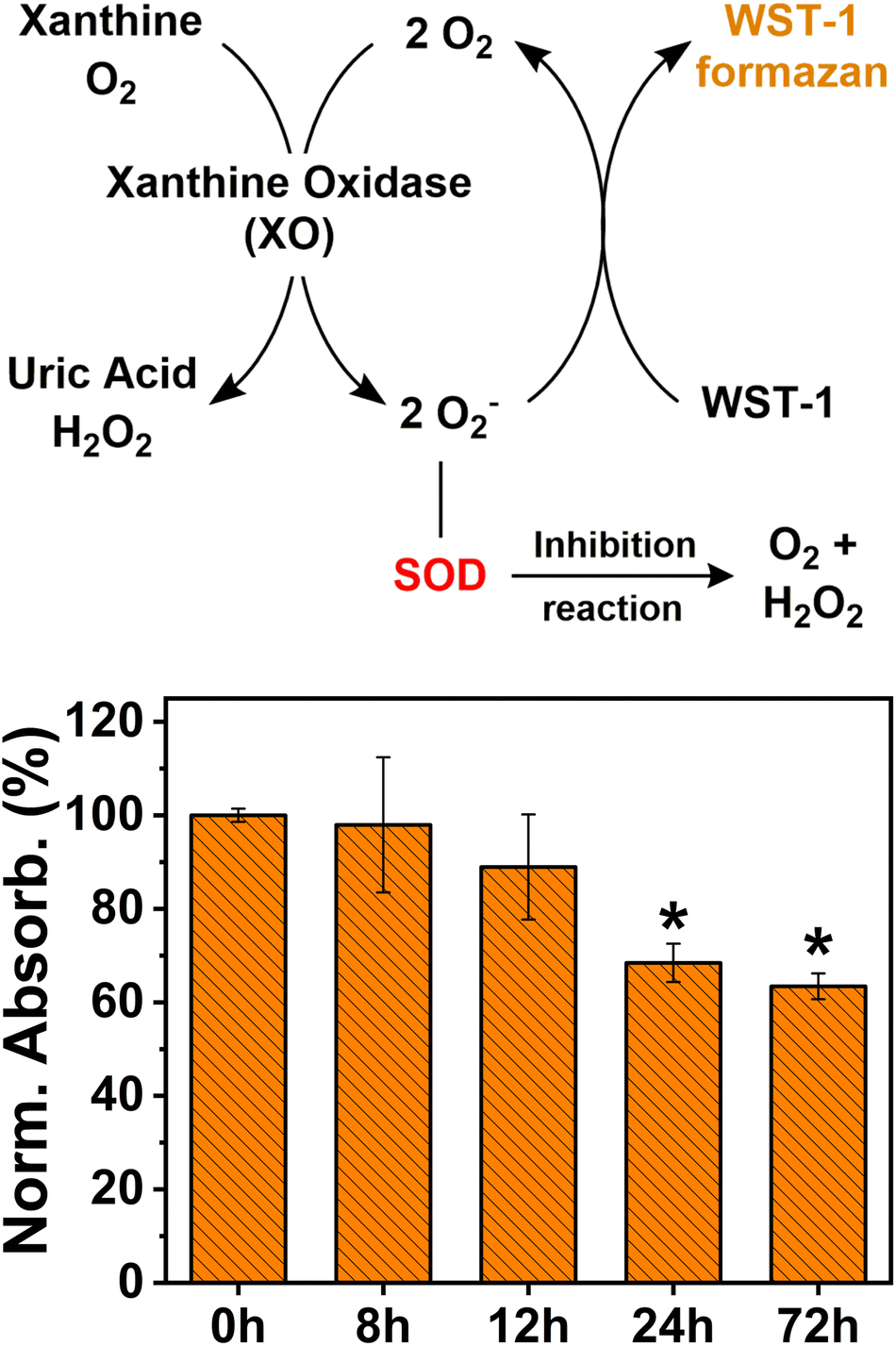 | ||
| Fig. 4 Working mechanism of the SOD assay and levels of SOD expressed as the normalized absorbance intensity profile with respect to the non-treated sample (0 h). | ||
In addition, despite the high levels of SOD in the samples treated for 72 h, the viability of the cells was greatly reduced, as seen from the results of the MTT assay. This finding suggests that the cytotoxicity of AgNPs towards human cells may result from mechanisms other than ROS overproduction.29,79
In the literature, it has been reported that the band at 1572 cm−1 can result from vibrations of dissolved oxygen in the culture medium.48 However, during data processing, the average dissolved oxygen signal in culture media is uniformly subtracted from each sample through background subtraction. In contrast, other works assign this band to the vibrations of purine nucleobases, guanine and adenine. However, the absence of intensity variation for the band at ∼780 cm−1, related to the DNA phosphate backbone and pyrimidine bases (uracil, cytosine, and thymine),58,60,61 suggests that their vibrational contribution is only partial and that the increase in the intensity of the ∼1572 cm−1 band cannot be related to variations in the structure of the DNA/RNA. Furthermore, the fact that the ∼780 cm−1 band remains stable over time suggests that no genotoxic effect is induced by the presence of AgNPs35nm during the first 24 h of exposure. The absence of genotoxicity may also be attributed to the reduced presence of nanoparticle aggregates within the cell nuclei, which are mainly observed in the vesicles of the cytoplasmic space (Fig. 5).
 | ||
| Fig. 5 Raman maps of the distribution of AgNPs inside cells, acquired at the respective time intervals (scale bar 20 μm). Raman maps were acquired with a RAMANtouch spectroscope, operated with a linear detector (400 pixels) with an exposure of 10 s for each line, performing the measurements singularly. Red spots represent the location of AgNP aggregates inside vesicles from which SERS signals are derived; only a few spots can be seen within the central nuclei of the cells (marked by blue dotted lines). | ||
Interestingly, no appearance of the band for the S–S bond at around ∼500 cm−1 occurs during the analysis. This band is generally associated with the formation of the disulfide bond between glutathione molecules, occurring during cellular antioxidative processes. This phenomenon could be caused by different factors: (i) the oxidation of glutathione is not among the predominant mechanisms for the reduction of oxidative stress; (ii) the GSH action occurs at times lower or higher than those taken into consideration.80
To confirm the action of the antioxidant molecules to regulate the formation of ROS, fluorescence microscopy was performed on HDF cells exposed to 10 μg mL−1 of AgNPs35nm for 8, 12, 24 and 72 h, using DCFH-DA as the fluorescent reporter molecule (Fig. 6). As expected, no ROS formation was observed for the cells treated with only culture media (negative control), while an increased signal resulted from those cultured in the presence of TBHP (100 μM) for 60 minutes. The cells treated with AgNPs35nm at the initial time points (8 and 12 h) showed the same conversion of DCFH-DA into the DCF fluorescent molecule as for control samples. This fact confirms that in the early stages of exposure to low concentration levels of AgNPs, the cellular antioxidant mechanism is able to control the detrimental effect of Ag+ ions and the overproduction of ROS. After 24 h, a slight increase in fluorescence intensity was observed, indicating that already at this time point the production of ROS begins to no longer be contained. Eventually, after 72 h it is possible to observe a large intensification in fluorescence, which indicates greater formation of ROS and consequently their potentially harmful effect. Nevertheless, the large standard deviation recorded for this time point suggests that several cells are not affected by the presence of AgNPs and thus do not produce ROS. As reported in the literature, ROS action could therefore not be the only mechanism that causes cytotoxicity of AgNPs towards human cells, and more studies should be carried out to confirm these hypotheses.29,79
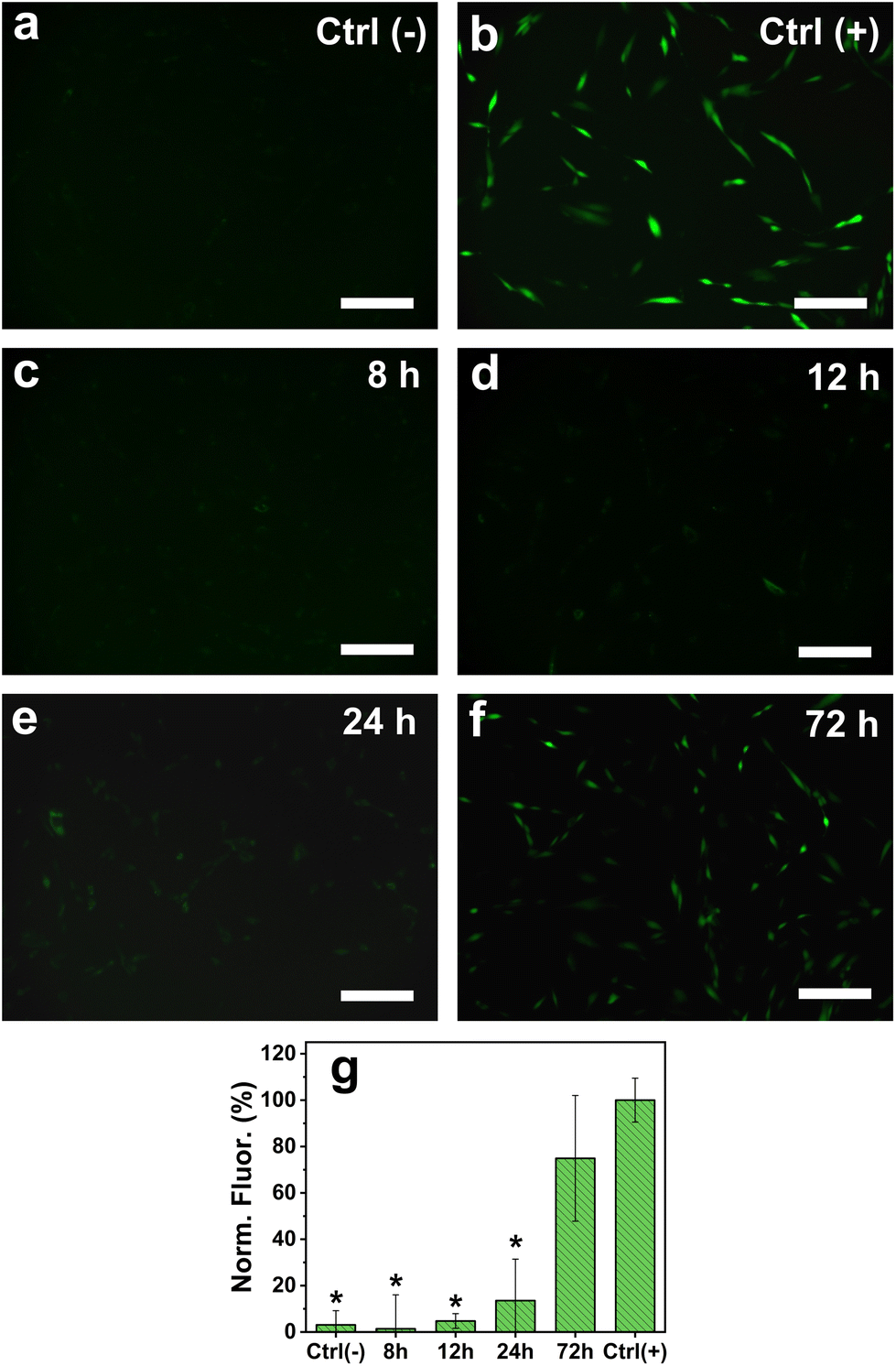 | ||
| Fig. 6 Fluorescence microscopy images of HDF cells (a) untreated (negative control), (b) treated with TBHP (100 μM) for 60 minutes, and exposed to AgNPs35nm (10 μg ml−1) for (c) 8 h, (d) 12 h, (e) 24 h and (f) 72 h (scale bar 200 μm); (g) normalized fluorescence intensity profile with respect to Ctrl(+) (positive control sample). *Significant differences from positive control (P < 0.05). | ||
A proposed defense mechanism of HDF cells in the presence of sub-lethal concentrations of AgNPs for short exposure times can be seen in Fig. 7 where: (i) initially, AgNPs are taken up by the cells and they enter the cellular environment; (ii) within the first 24 hours, the cells trigger their antioxidant system wherein metallothioneins, superoxide dismutase and catalase collaborate to effectively suppress the overproduction of ROS generated by the presence of AgNPs and Ag+ ions. This combined action successfully mitigates oxidative stress, contains the deterioration of proteins and lipids, and prevents damage to the DNA; (iii) as time progresses beyond 24 h, the physiological and cellular environments perpetrate the dissolution of the AgNPs, leading to an always increasing presence of Ag+ ions.22,81 Consequently, this sustained presence of Ag+ ions can induce an amplified generation of ROS. The increasingly hostile cellular environment overwhelms the initially effective antioxidant mechanism, ultimately resulting in its failure; (iv) after 72 hours of chronic exposure, the cumulative effect of heightened ROS production, coupled with the compromised defense system, is manifested in high cytotoxicity. In summary, the cytotoxic action of low concentration levels of AgNPs involves an initial defense response that succumbs to their prolonged exposure, leading to a cascade of events culminating in cytotoxicity after 72 h of chronic exposure.
 | ||
| Fig. 7 Possible antioxidant mechanism against the cytotoxicity of AgNPs during the first 24 h of exposure. Initially, AgNPs enter the cells and trigger a defense response involving MT, SOD, and CAT to contain Ag+ ions and suppress ROS overproduction. Beyond 24 h, AgNPs dissolve, leading to an increasing presence of Ag+ ions. After 72 h, the sustained Ag+ ions amplify ROS generation, overwhelming the antioxidant mechanism leading to high cytotoxicity. | ||
Conclusions
In our work, we investigated the short- and long-term cytotoxicity of silver nanoparticles and the defense mechanisms that cells activate in order to reduce their harmful effects. During short-term exposure to AgNPs, Raman microspectroscopy has shown that only a small portion of proteins and lipids exhibit a deterioration process, while no damage seems to occur at the DNA level. Concurrently, an antioxidant protein like SOD, and to minor extents CAT and MTs, result in being overexpressed to hamper the toxic action of Ag+ ions and suppress the formation of ROS. Fluorescence microscopy confirmed the efficacy of this defensive process, revealing that ROS production in HDF cells treated with AgNPs is comparable to that of untreated samples during short exposure periods. These findings prove that the advanced technique of Raman microscpectroscopy is a valuable tool to observe subtle variations during cellular biological processes, thus making it a valid and novel alternative for nanotoxicology studies. In addition, these findings enhance our understanding of cellular detoxification processes in the presence of toxic nanomaterials, revealing that, despite the apparent non-toxicity of AgNPs during short-term and low-concentration exposures, prolonged contact can ultimately result in elevated toxicity levels.Abbreviations
| HDF | Human dermal fibroblast |
| AgNPs | Silver nanoparticles |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| CAT | Catalase |
| MT | Metallothionein |
| GSH | Glutathione |
| SERS | Surface enhanced Raman scattering |
| TBHP | tert-Butyl hydroperoxide |
Author contributions
Conceptualization: D. R.-B.; data curation: D. R.-B.; formal analysis: D. R.-B.; funding acquisition: P. R. and G. P.; investigation: D. R.-B.; methodology: D. R.-B.; project administration: P. R. and G. P.; resources: O. M., P. R., and G. P.; supervision: P. R. and G. P.; validation: D. R.-B., E. M., P. R., and G. P.; visualization: D. R.-B.; writing – original draft: D. R.-B.; writing – review & editing: D. R.-B., E. M., K. Y., O. M., W. Z., P. R., and G. P.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Kyoto Prefectural University of Medicine for the use of laboratories and the fluorescence microscope. The authors acknowledge Prof. Kohji Maeda for the ICP-OES quantification analysis.References
- G. Alberti, C. Zanoni, L. R. Magnaghi and R. Biesuz, Chemosensors, 2021, 9, 108 CrossRef CAS.
- S. Zhang, Y. Tang and B. Vlahovic, Nanoscale Res. Lett., 2016, 11, 1–8 CrossRef CAS PubMed.
- A. Roy, O. Bulut, S. Some, A. K. Mandal and M. D. Yilmaz, RSC Adv., 2019, 9, 2673–2702 RSC.
- M. Park, J. Im, M. Shin, Y. Min, J. Park, H. Cho, S. Park, M. B. Shim, S. Jeon, D. Y. Chung, J. Bae, J. Park, U. Jeong and K. Kim, Nat. Nanotechnol., 2012, 7, 803–809 CrossRef CAS PubMed.
- B. Nowack, H. F. Krug and M. Height, Environ. Sci. Technol., 2011, 45, 1177–1183 CrossRef CAS PubMed.
- S. McLaughlin, M. Ahumada, W. Franco, T. F. Mah, R. Seymour, E. J. Suuronen and E. I. Alarcon, Nanoscale, 2016, 8, 19200–19203 RSC.
- T. M. Benn and P. Westerhoff, Environ. Sci. Technol., 2008, 42, 4133–4139 CrossRef CAS PubMed.
- J. Hedberg, M. Eriksson, A. Kesraoui, A. Norén and I. Odnevall Wallinder, Environ. Sci. Pollut. Res., 2021, 28, 12968–12979 CrossRef CAS PubMed.
- A. Boyadzhiev, C. Trevithick-Sutton, D. Wu, N. Decan, M. Bazin, G. M. Shah and S. Halappanavar, Chem. Res. Toxicol., 2020, 33, 1266–1278 Search PubMed.
- J. Butler, R. D. Handy, M. Upton and A. Besinis, ACS Nano, 2023, 17, 7064–7092 CrossRef CAS PubMed.
- K. Kalantari, E. Mostafavi, A. M. Afifi, Z. Izadiyan, H. Jahangirian, R. Rafiee-Moghaddam and T. J. Webster, Nanoscale, 2020, 12, 2268–2291 RSC.
- C. M. Xie, X. Lu, K. F. Wang, F. Z. Meng, O. Jiang, H. P. Zhang, W. Zhi and L. M. Fang, ACS Appl. Mater. Interfaces, 2014, 6, 8580–8589 CrossRef CAS PubMed.
- N. Duraipandy, R. Lakra, K. V. Srivatsan, U. Ramamoorthy, P. S. Korrapati and M. S. Kiran, J. Mater. Chem. B, 2015, 3, 1415–1425 RSC.
- S. W. P. Wijnhoven, W. J. G. M. Peijnenburg, C. A. Herberts, W. I. Hagens, A. G. Oomen, E. H. W. Heugens, B. Roszek, J. Bisschops, I. Gosens, D. Van De Meent, S. Dekkers, W. H. De Jong, M. Van Zijverden, A. J. A. M. Sips and R. E. Geertsma, Nanotoxicology, 2009, 3, 109–138 CrossRef CAS.
- C. Saweres-Argüelles, I. Ramírez-Novillo, M. Vergara-Barberán, E. J. Carrasco-Correa, M. J. Lerma-García and E. F. Simó-Alfonso, Eur. J. Pharm. Biopharm., 2023, 182, 128–140 CrossRef PubMed.
- N. Hadrup, A. K. Sharma and K. Loeschner, Regul. Toxicol. Pharmacol., 2018, 98, 257–267 CrossRef CAS PubMed.
- F. F. Larese, F. D'Agostin, M. Crosera, G. Adami, N. Renzi, M. Bovenzi and G. Maina, Toxicology, 2009, 255, 33–37 CrossRef CAS PubMed.
- M. Trop, M. Novak, S. Rodl, B. Hellbom, W. Kroell and W. Goessler, J. Trauma: Inj., Infect., Crit. Care, 2006, 60, 648–652 CrossRef PubMed.
- Y. K. Tak, S. Pal, P. K. Naoghare, S. Rangasamy and J. M. Song, Sci. Rep., 2015, 5, 16908 CrossRef CAS PubMed.
- D. McShan, P. C. Ray and H. Yu, J. Food Drug Anal., 2014, 22, 116–127 CrossRef CAS PubMed.
- M. Qi, X. Wang, J. Chen, Y. Liu, Y. Liu, J. Jia, L. Li, T. Yue, L. Gao, B. Yan, B. Zhao and M. Xu, ACS Nano, 2023, 17, 8851–8865 CrossRef CAS PubMed.
- L. Wang, T. Zhang, P. Li, W. Huang, J. Tang, P. Wang, J. Liu, Q. Yuan, R. Bai, B. Li, K. Zhang, Y. Zhao and C. Chen, ACS Nano, 2015, 9, 6532–6547 CrossRef CAS PubMed.
- J. T. Buchman, N. V. Hudson-Smith, K. M. Landy and C. L. Haynes, Acc. Chem. Res., 2019, 52, 1632–1642 CrossRef CAS PubMed.
- S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC.
- J. K. Tee, C. N. Ong, B. H. Bay, H. K. Ho and D. T. Leong, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 414–438 CAS.
- C. Andrés Juan, J. Manuel, J. M. Pérez de la Lastra, F. J. Plou, E. Pérez-Lebeña and S. Reinbothe, Int. J. Mol. Sci., 2021, 22, 4642 CrossRef PubMed.
- Z. Yu, Q. Li, J. Wang, Y. Yu, Y. Wang, Q. Zhou and P. Li, Nanoscale Res. Lett., 2020, 15, 1 CrossRef PubMed.
- P. Babula, M. Masarik, V. Adam, T. Eckschlager, M. Stiborova, L. Trnkova, H. Skutkova, I. Provaznik, J. Hubalek and R. Kizek, Metallomics, 2012, 4, 739–750 CrossRef CAS PubMed.
- Y. Hayashi, P. Engelmann, R. Foldbjerg, M. Szabó, I. Somogyi, E. Pollák, L. Molnár, H. Autrup, D. S. Sutherland, J. Scott-Fordsmand and L. H. Heckmann, Environ. Sci. Technol., 2012, 46, 4166–4173 CrossRef CAS PubMed.
- C. Krafft, M. Schmitt, I. W. Schie, D. Cialla-May, C. Matthäus, T. Bocklitz and J. Popp, Angew. Chem., 2017, 129, 4458–4500 CrossRef.
- H. J. Byrne, F. Bonnier, E. Efeoglu, C. Moore and J. McIntyre, Front. Bioeng. Biotechnol., 2020, 8, 544311 CrossRef PubMed.
- G. Pezzotti, T. Adachi, F. Boschetto, W. Zhu, M. Zanocco, E. Marin, B. S. Bal and B. J. McEntire, Int. J. Mol. Sci., 2019, 20, 17 Search PubMed.
- X. Dou, Y. Zhao, M. Li, Q. Chen and Y. Yamaguchi, Spectrochim. Acta, Part A, 2020, 224, 117438 CrossRef CAS PubMed.
- G. Pezzotti, S. Horiguchi, F. Boschetto, T. Adachi, E. Marin, W. Zhu, T. Yamamoto, N. Kanamura, E. Ohgitani and O. Mazda, ACS Chem. Neurosci., 2018, 9, 3038–3048 CrossRef CAS PubMed.
- G. Cutshaw, N. Hassan, S. Uthaman, X. Wen, B. Singh, A. Sarkar and R. Bardhan, Anal. Chem., 2023, 95, 13172–13184 CrossRef CAS PubMed.
- V. Notarstefano, A. Belloni, P. Mariani, G. Orilisi, G. Orsini, E. Giorgini and H. J. Byrne, Analyst, 2023, 148, 4365–4372 RSC.
- O. Jonas, J. W. Kang, S. P. Singh, A. Lammers, F. T. Nguyen, R. R. Dasari, P. T. C. So, R. Langer and M. J. Cima, Analyst, 2018, 143, 4836–4839 RSC.
- Z. Farhane, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2017, 409, 1333–1346 CrossRef CAS PubMed.
- E. Efeoglu, M. A. Maher, A. Casey and H. J. Byrne, Anal. Bioanal. Chem., 2018, 410, 1631–1646 CrossRef CAS PubMed.
- H. Salehi, I. Calas-Bennasar, J. C. Durand, E. Middendorp, J. Valcarcel, C. Larroque, K. Nagy, K. K. Turzó, I. Dekany and F. J. G. Cuisinier, J. Raman Spectrosc., 2014, 45, 807–813 CrossRef CAS.
- E. Efeoglu, M. A. Maher, A. Casey and H. J. Byrne, Analyst, 2017, 142, 3500–3513 RSC.
- E. Fazio, A. Speciale, S. Spadaro, M. Bonsignore, F. Cimino, M. Cristani, D. Trombetta, A. Saija and F. Neri, Colloids Surf., B, 2018, 170, 233–241 CrossRef CAS PubMed.
- S. Barkur, J. Lukose and S. Chidangil, ACS Omega, 2020, 5, 1439–1447 CrossRef CAS PubMed.
- P. Cronholm, H. L. Karlsson, J. Hedberg, T. A. Lowe, L. Winnberg, K. Elihn, I. O. Wallinder and L. Möller, Small, 2013, 9, 970–982 CrossRef CAS PubMed.
- A. Bankapur, S. Barkur, S. Chidangil and D. Mathur, PLoS One, 2014, 9, 7 CrossRef PubMed.
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836–2846 CrossRef.
- S. Tadjiki, M. D. Montaño, S. Assemi, A. Barber, J. Ranville and R. Beckett, Anal. Chem., 2017, 89, 6056–6064 CrossRef CAS PubMed.
- A. F. Palonpon, J. Ando, H. Yamakoshi, K. Dodo, M. Sodeoka, S. Kawata and K. Fujita, Nat. Protoc., 2013, 8, 677–692 CrossRef CAS PubMed.
- A. Galandáková, J. Franková, N. Ambrožová, K. Habartová, V. Pivodová, B. Zálešák, K. Šafářová, M. Smékalová and J. Ulrichová, Hum. Exp. Toxicol., 2016, 35, 946–957 CrossRef PubMed.
- A. Avalos, A. I. Haza, D. Mateo and P. Morales, Int. Wound J., 2016, 13, 101–109 CrossRef PubMed.
- B. Dalzon, C. Aude-Garcia, H. Diemer, J. Bons, C. Marie-Desvergne, J. Pérard, M. Dubosson, V. Collin-Faure, C. Carapito, S. Cianférani, M. Carrière and T. Rabilloud, Environ. Sci.: Nano, 2020, 7, 2032–2046 RSC.
- J. Dolai, K. Mandal and N. R. Jana, ACS Appl. Nano Mater., 2021, 4, 6471–6496 CrossRef CAS.
- T. S. Peretyazhko, Q. Zhang and V. L. Colvin, Environ. Sci. Technol., 2014, 48, 11954–11961 CrossRef CAS PubMed.
- A. J. Hobro and N. I. Smith, Vib. Spectrosc., 2017, 91, 31–45 CrossRef CAS.
- J. W. Chan, D. S. Taylor and D. L. Thompson, Biopolymers, 2009, 91, 132–139 CrossRef CAS PubMed.
- F. Draux, C. Gobinet, J. Sulé-Suso, A. Trussardi, M. Manfait, P. Jeannesson and G. D. Sockalingum, Anal. Bioanal. Chem., 2010, 397, 2727–2737 CrossRef CAS PubMed.
- A. D. Meade, C. Clarke, F. Draux, G. D. Sockalingum, M. Manfait, F. M. Lyng and H. J. Byrne, Anal. Bioanal. Chem., 2010, 396, 1781–1791 CrossRef CAS PubMed.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493–541 CrossRef CAS.
- G. Zhu, X. Zhu, Q. Fan and X. Wan, Spectrochim. Acta, Part A, 2011, 78, 1187–1195 CrossRef PubMed.
- H. G. Schulze, S. O. Konorov, J. M. Piret, M. W. Blades and R. F. B. Turner, Analyst, 2013, 138, 3416–3423 RSC.
- D. W. Shipp, F. Sinjab and I. Notingher, Adv. Opt. Photonics, 2017, 9, 315 CrossRef.
- K. D. Sharma, L. A. Andersson, T. M. Loehrs, J. Terner and H. M. Goff, J. Biol. Chem., 1989, 264, 12772–12779 CrossRef CAS PubMed.
- W. J. Chuang, J. Heldt and H. E. Van Wart, J. Biol. Chem., 1989, 264, 14209–14215 CrossRef CAS PubMed.
- W.-J. Chuang, S. Johnson and H. E. Van Wart, J. Inorg. Biochem., 1988, 34, 201–219 CrossRef CAS PubMed.
- S. Hashimoto, K. Ono and H. Takeuchi, J. Raman Spectrosc., 1998, 29, 969–975 CrossRef CAS.
- A. Toyama, Y. Takahashi and H. Takeuchi, Biochemistry, 2004, 43, 4670–4679 CrossRef CAS PubMed.
- D. Wang, X. Zhao, M. Vargek and T. G. Spiro, J. Am. Chem. Soc., 2000, 122, 2193–2199 CrossRef CAS.
- J. M. Surmacki, I. Quiros-Gonzalez and S. E. Bohndiek, Antioxidants, 2022, 11, 3 CrossRef PubMed.
- W. T. Chang, H. L. Lin, H. C. Chen, Y. M. Wu, W. J. Chen, Y. T. Lee and I. Liau, J. Raman Spectrosc., 2009, 40, 1194–1199 CrossRef CAS.
- R. Buckmaster, F. Asphahani, M. Thein, J. Xu and M. Zhang, Analyst, 2009, 134, 1440–1446 RSC.
- S. Dekkers, T. D. Williams, J. Zhang, J. Zhou, R. J. Vandebriel, L. J. J. De La Fonteyne, E. R. Gremmer, S. He, E. J. Guggenheim, I. Lynch, F. R. Cassee, W. H. De Jong and M. R. Viant, Environ. Sci.: Nano, 2018, 5, 1506–1517 RSC.
- H. Zhang, X. Wang, M. Wang, L. Li, C. H. Chang, Z. Ji, T. Xia and A. E. Nel, Small, 2015, 11, 3797–3805 CrossRef CAS PubMed.
- G. Veronesi, A. Deniaud, T. Gallon, P. H. Jouneau, J. Villanova, P. Delangle, M. Carrière, I. Kieffer, P. Charbonnier, E. Mintz and I. Michaud-Soret, Nanoscale, 2016, 8, 17012–17021 RSC.
- E. M. Luther, M. M. Schmidt, J. Diendorf, M. Epple and R. Dringen, Neurochem. Res., 2012, 37, 1639–1648 CrossRef CAS PubMed.
- M. Minghetti, W. Dudefoi, Q. Ma and J. G. Catalano, Environ. Sci.: Nano, 2019, 6, 2948–2957 RSC.
- R. K. Hobbie and B. J. Roth, in Intermediate Physics for Medicine and Biology, ed. R. K. Hobbie and B. J. Roth, Springer New York, New York, NY, 2007, 31–47 Search PubMed.
- C. J. Weydert and J. J. Cullen, Nat. Protoc., 2010, 5, 51–66 CrossRef CAS PubMed.
- O. M. Ighodaro and O. A. Akinloye, Alexandria J. Med., 2018, 54, 287–293 CrossRef.
- P. Orlowski, M. Krzyzowska, R. Zdanowski, A. Winnicka, J. Nowakowska, W. Stankiewicz, E. Tomaszewska, G. Celichowski and J. Grobelny, Toxicol. in Vitro, 2013, 27, 1798–1808 CrossRef CAS PubMed.
- S. R. Panikkanvalappil, S. M. Hira and M. A. El-Sayed, Chem. Sci., 2016, 7, 1133–1141 RSC.
- C. Graf, D. Nordmeyer, C. Sengstock, S. Ahlberg, J. Diendorf, J. Raabe, M. Epple, M. Köller, J. Lademann, A. Vogt, F. Rancan and E. Rühl, Langmuir, 2018, 34, 1506–1519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional description of Raman spectra acquisition and processing steps, and magnification of specific parts of the time-dependent Raman spectra. See DOI: https://doi.org/10.1039/d4nr00462k |
| This journal is © The Royal Society of Chemistry 2024 |