
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoresponsive arylazopyrazole surfactant/PDADMAC mixtures: reversible control of bulk and interfacial properties†
Michael
Hardt
a,
Christian
Honnigfort
a,
Javier
Carrascosa-Tejedor
 bc,
Marius G.
Braun
a,
Samuel
Winnall
bc,
Dana
Glikman
a,
Philipp
Gutfreund
b,
Richard A.
Campbell
c and
Björn
Braunschweig
*a
bc,
Marius G.
Braun
a,
Samuel
Winnall
bc,
Dana
Glikman
a,
Philipp
Gutfreund
b,
Richard A.
Campbell
c and
Björn
Braunschweig
*a
aInstitute of Physical Chemistry and Center for Soft Nanoscience, University of Münster, Corrensstraße 28/30, 48149 Münster, Germany. E-mail: braunschweig@uni-muenster.de
bInstitut Laue-Langevin (ILL), 71 avenue des Martyrs, CS 20156, 38042 Grenoble Cedex 9, France
cDivision of Pharmacy & Optometry, University of Manchester, Manchester M13 9PT, UK
First published on 25th April 2024
Abstract
In many applications of polyelectrolyte/surfactant (P/S) mixtures, it is difficult to fine-tune them after mixing the components without changing the sample composition, e.g. pH or the ionic strength. Here we report on a new approach where we use photoswitchable surfactants to enable drastic changes in both the bulk and interfacial properties. Poly(diallyldimethylammonium chloride) (PDADMAC) mixtures with three alkyl-arylazopyrazole butyl sulfonates (CnAAP) with -H, -butyl and -octyl tails are applied and E/Z photoisomerization of the surfactants is used to cause substantially different hydrophobic interactions between the surfactants and PDADMAC. These remotely controlled changes affect significantly the P/S binding and allows for tuning both the bulk and interfacial properties of PDADMAC/CnAAP mixtures through light irradiation. For that, we have fixed the surfactant concentrations at values where they exhibit pronounced surface tension changes upon E/Z photoisomerization with 365 nm UV light (Z) and 520 nm green (E) light and have varied the PDADMAC concentration. The electrophoretic mobility can be largely tuned by photoisomerisation of CnAAP surfactants and P/S aggregates, which can even exhibit a charge reversal from negative to positive values or vice versa. In addition, low colloidal stability at equimolar concentrations of PDADMAC with CnAAP surfactants in the E configuration lead to the formation of large aggregates in the bulk which can be broken up by irradiation with UV light when the surfactant's alkyl chain is short enough (C0AAP). Vibrational sum-frequency generation (SFG) spectroscopy reveals changes at the interface similar to the bulk, where the charging state at air–water interfaces can be modified with light irradiation. Using SFG spectroscopy, we interrogated the O–H stretching modes of interfacial H2O and provide qualitative information on surface charging that is complemented by neutron reflectometry, from which we resolved the surface excesses of PDADMAC and CnAAP at the air–water interface, independently.
Introduction
Polyelectrolyte/surfactant (P/S) mixtures play a major role in various applications ranging from personal care products to drug delivery systems and paints.1–3 That is because of their versatile physical properties that can be used to stabilise the relevant soft matter interfaces, e.g. in foams or emulsions, while they can also control bulk properties like viscosity and electrophoretic mobilities.4–7 Owing to their widespread use in applications, the properties of P/S mixtures with classical surfactants like dodecylsulfate or alkylammonium in bulk solutions and at interfaces are studied extensively to the present day.5–12 Using different P/S mixing ratios allows one to tailor soft matter properties such as the size of the P/S aggregates that form after mixing or the stability of aqueous foams.However, these properties are notoriously difficult to change after P/S mixtures have been prepared, where only drastic changes in chemical composition e.g., pH values, or in the ionic strengths are useful triggers.
In this work, we will present results on an alternative approach where we report on the use of poly(diallyl dimethylammonium chloride) (PDADMAC) mixtures with different alkyl-arylazopyrazole (CnAAP) sulfonates. Here the CnAAPs photoisomerize between linear (E state) and bended (Z state) configurations (Fig. 1) and allow for remote control of both interfacial and bulk properties through light irradiation without the need for changes in the sample compostion.13,14 In that respect, light is a powerful tool as it can be localized in both time and space.
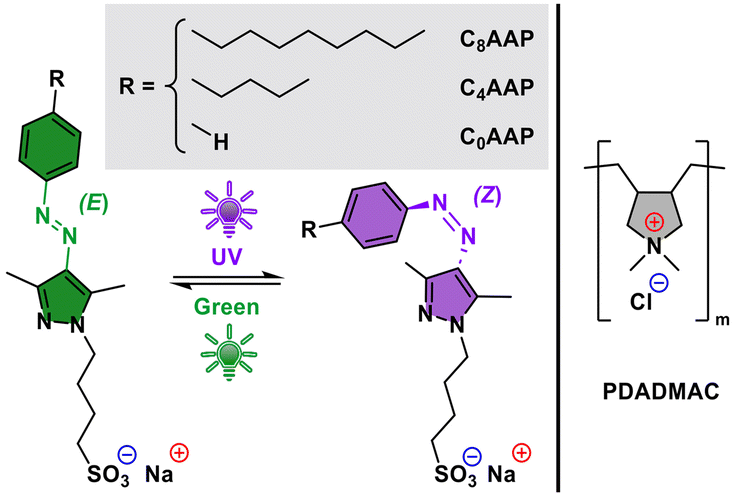 | ||
| Fig. 1 Molecular structures and photoisomerizaton of CnAAP surfactants used in this study and the PDADMAC polyelectrolyte, where n = 8, 4 or 0 in the R groups corresponds to octyl, butyl and H tails, respectively. | ||
It may be noted that the P/S mixtures that we report in this work are conceptionally similar to PDADMAC/SDS mixtures that have been frequently studied in the past,15–18 but the CnAAP surfactants used in our study can regulate the P/S interactions through the E/Z photoisomerization of the surfactants. Through this additional control by light irradiation, we can modify the size of the PADMAC/CnAAP aggregates, their electrophoretic mobility and the interfacial properties to a large extent and with high reversibility. In addition, we explore how the latter depends on the length of the alkyl tail at the CnAAP centre (with n being 8, 4 and 0) as it can alter the hydrophobic interactions with the PDADMAC chains considerably.
Experimental
Materials and sample preparation
To ensure the high purity required for all measurements, a strict protocol has been followed: all glassware was precleaned with an Alconox (Sigma-Aldrich) aqueous solution and subsequently rinsed multiple times with ultra-pure water (Millipore Reference A+; 18 MΩ cm; total oxidizable carbon <5 ppb), dried in a stream of N2 gas and stored for at least 12 h in concentrated sulfuric acid (98%, p.a., Carl Roth) with Alnochromix (Alconox Inc., USA) to remove all organic impurities. The acid-cleaned parts were then rinsed with copious amounts of ultra-pure water and dried again with nitrogen gas (99.999%, Westfalen).PDADMAC (20 wt%, 400–500 kDa, Sigma-Aldrich) was used as received. The CnAAP surfactants have been synthesized and purified as described previously.19 For the preparation of the mixtures, both components were dissolved in ultra-pure water and shaken gently with an orbital shaker (KS-15; Edmund Bühler GmbH). The P/S mixtures were prepared by mixing equal volumes of both components rapidly to minimise the formation of kinetically trapped aggregates.20 The PDADMAC concentration was systematically varied in the experiments and is reported throughout this work with respect to the monomer concentration which can be also directly compared to the CnAAP concentration. Different from the approach with a varying polyelectrolyte concentration, the surfactant concentration was fixed at a value where the surfactants exhibit the largest changes in surface tension in the absence of PDADMAC (see tensiometry results in Fig. S1 and SFG spectra in Fig. S14 in the ESI†) that is 10 mM for the C0AAP, 1 mM for C4AAP and 0.1 mM for C8AAP surfactant.19,21
For the irradiation of the samples, LEDs emitting green (520 nm, Roschwege, LSC-G) and UV (365 nm, Roschwege, Star-UV365-05-00-00) light were used. If not stated otherwise, their intensities were set to 2.9 and 10.2 mW cm−2 for the 520 nm and 365 nm LEDs, respectively.
Dynamic light scattering (DLS) and electrophoretic mobility
DLS experiments were performed with a Zetasizer Nano ZSP (Malvern Panalytical) using the internal HeNe Laser (10 mW, 633 nm) and backscattering detector at an angle of 173°. Before each measurement, the samples were irradiated with either green or UV light for at least 10 min. Due to the different experimental setup, the intensity of the LEDs used for irradiating the samples was higher and 6.5 mW cm−2 (green) and 40 mW cm−2 (UV) were used. First the samples were irradiated with green light and measured in the Zetasizer and then irradiated with UV light and were measured again. This order of the DLS experiments was followed for all P/S mixing ratios.Vibrational sum-frequency generation (SFG) spectroscopy
Vibrational SFG spectroscopy, as a non-linear optical technique, allows for the investigation of non-centrosymmetric environments. The necessary break in bulk symmetry at interfaces, makes this technique inherently interface-specific for materials with centrosymmetry. Consequently, SFG spectroscopy is a powerful tool to investigate air–water interfaces on a molecular level. For that, two ultrashort laser pulses, an IR and a visible pulse, are combined spatially and temporally at the interface, where a third pulse with the sum frequency of the incoming beams is generated. The SFG intensity can be expressed as follows:22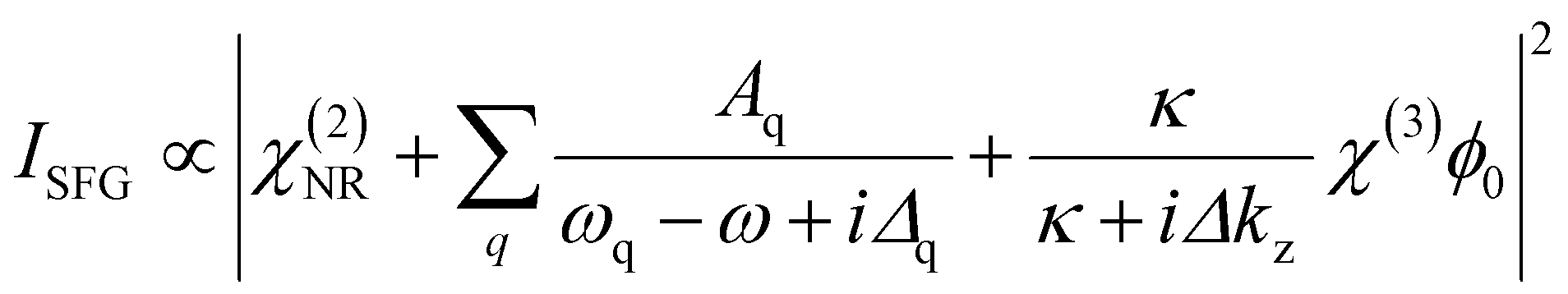 | (1) |
Here, χ(2)NR is a non-resonant contribution, while the Lorentzian terms originate from a resonant contribution to the second-order electric susceptibility. In case, of charged interfaces a third term becomes relevant that depends on the third-order electric susceptibility χ(3) and the surface potential ϕ0. The spectrum of χ(3) is mostly similar to that of the resonant contribution, which depends on the frequency ω of the IR beam, the eigenfrequencies ωq of vibrational modes from interfacial molecules and their bandwidth Δq. The amplitude Aq of a vibrational mode mainly depends on the number and the orientation of the molecules at the interface. In the bulk solution, Aq ∝ N〈β(2)q〉 is zero as the orientational average 〈⋯〉 of the molecules’ hyper-polarizability 〈β(2)q〉 is zero. At the interface, the molecules’ net orientation causes nonzero amplitudes and, therefore, noticeable SFG signals. In case the surface charge and consequently the surface potential changes, further modification of the SFG intensity can take place. In particular, the intensity of the O–H stretching modes from interfacial water molecules are largely affected by the χ(3) term in eqn (1), which can be used to analyze the change in the interfacial charging state qualitatively.22–24
For SFG spectroscopy we have applied a tunable broadband IR pulse with 70 fs pulse duration and >300 cm−1 bandwidth and a time-asymmetric narrowband visible pulse with a pulse duration of 1–2 ps and a bandwidth of <5 cm−1. The IR and Vis beams were overlapped at the interface at angles normal to the interface of 60° and 55°, respectively. The reflected SFG beam was spectrally dispersed in a Kymera (Andor, Oxford Instruments) spectrograph and detected with a Newton (Andor, Oxford Instruments) EMCCD. Additional details on the spectrometer and fitting procedures can be found elsewhere24 and in the ESI.† The samples have been equilibrating for >60 min under green light before recording the spectra. Subsequently, the samples were irradiated for 10 min with UV light and spectra of the interface with surfactants in the predominant Z state were measured.
Neutron reflectometry (NR)
NR can provide details on both the structure and composition of the P/S aggregates at the air–water interface.25,26 The specular reflectivity R of neutrons is measured as a function of momentum transfer perpendicular to the interface QZ, which can be written as:27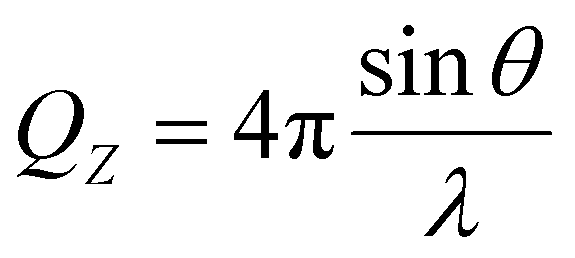 | (2) |
The resulting neutron reflectivity profiles were measured at the FIGARO instrument (Institut Laue-Langevin (ILL), France) with a wavelength λ range of 2 to 20 Å (angle of incidence, θ = 0.62°) and 2 to 30 Å (θ = 2.0° and θ = 3.8°).28 The experiments were conducted using different isotopic contrasts: PDADMAC mixtures with hydrogenous or deuterated surfactant in air contrast matched water (ACMW, 8.1% D2O in H2O) or D2O. The deuterated surfactants d6-C8AAP and d13-C4AAP, which are shown in the ESI,† were synthesized similar to the hydrogenous surfactants, but with deuterated educts instead of their hydrogenous analogues.19 Note, that most concentrations were measured in 0.8% D2O in H2O instead of ACMW due to an unfortunate erroneous calculation of the concentrations. The concentrations measured in the correct ACMW were C4AAP mixtures with 0.01 mM, 0.5 mM and 1 mM PDADMAC. The different backgrounds of the solvents have been considered in the analysis. Also, due to limited experimental time some concentrations could be only measured in two overall contrasts. Within the available sample changer, six samples were loaded in the PTFE troughs and measured in a sequence which optimized the available measurement time at the NR experiment. All samples were equilibrated for approximately 90 min before the structural measurements were started and required a measurement time of about 4 h per set of six samples. The samples were constantly irradiated with green or UV light throughout the NR measurement time using the intensities stated in the section above on sample preparation.
In general, NR experiments were started by irradiating the samples first with green light and subsequently with UV light. Only mixtures of 1 mM C4AAP with 0.5 and 1 mM PDADMAC as well as mixtures of 0.1 mM C8AAP with 0.1, 0.15 and 3 mM PDADMAC were first irradiated with UV. This different sequence was chosen explicitly to avoid aggregation and sedimentation that can occur at these concentrations under green irradiation later affecting the measurements under UV irradiation (see discussion below). Measurements of the solvents were used to subtract the solvent background from the neutron reflectivity profiles using the software COSMOS.29 Subsequent to the NR experiments, an alignment problem of the samples’ height was found which required a careful analysis of the obtained data (see ESI†). Unfortunately, it was not possible to correct all data, especially in the low-Q for mixtures of C8AAP with 0.01 mM PDADMAC. As a consequence, these data were omitted in the analysis.
Analysis of the neutron reflectivity profiles was done using Motofit30 (Igor Pro 8.0.3.3) and by applying a 4-layer model. The model was based on a previously reported three-layer model,13 involving surfactant chains protruding into the air phase (layer 1), solvated surfactant heads mixed with the polyelectrolyte chains (layer 2), and solvated polyelectrolyte chains protruding into the bulk (layer 3). However, a supplementary layer of highly solvated surfactant structures (layer 4), which has been recently reported in related systems,9,27,31 improved the quality of the model fit. Consistent with related studies, a small Kiessig fringe in the reflectivity profiles for the deuterated surfactants corroborates our assumption of extended structures (see Fig. S5 of the ESI†). Each layer is defined by different parameters which are the inter-layer roughness r, the layer thickness d, as well as the composition of the layer which we describe by the volume fractions Φi of the moieties present in the layer. In order to minimise the number of free parameters, some restrictions were applied: (i) the roughness was fixed to 3.5 Å with respect to the capillary waves resulting from the surface tension.32 (ii) The volume fraction of air in layer 1 was kept constant for both systems (Φ1,air = 0.9). Note that this value is slightly higher than the value previously reported by Honnigfort et al.21 who studied the pure C4AAP surfactant monolayers in the absence of polyelectrolyte at the air–water interface. The higher value assumed in the present work resulted, however, in much better fits to the neutron reflectivity profiles of CnAAP/PADMAC mixtures at the air–water interface. (iii) The volume fraction of the surfactant heads in the second layer Φ2,heads was constrained so that the surface excesses of chains in layer 1 and heads in layer 2 were equal. (iv) The thickness of the second layer was constrained between 4 and 6 Å to respect reasonable physical boundaries, similar to the value used by Honnigfort et al.21 (v) The fourth layer was included in the model only if the volume fraction of surfactants exceeded a threshold of 2% while its thickness was restricted to twice the length of the surfactants as reported in previous studies.27 The model fit was optimized until a global minimum of the chi2 was found. Further details on the fitting procedure are described in the ESI.†
Results and discussion
CnAAP/PDADMAC mixtures in the bulk phase
We start by addressing the size and the electrophoretic mobility of aggregates in P/S mixtures of cationic polyelectrolytes PDADMAC and anionic CnAAP surfactants. These aggregates form spontaneously in the bulk solution and can spread material at the air–water interface.9 For classical surfactants the phase behaviour of oppositely charged P/S mixtures has been extensively studied in the past and can be roughly divided into three regions. At high excess of the polyelectrolyte, ‘undercharged’ complexes are formed through binding of surfactant monomers or micelles to individual polyelectrolyte chains,33,34 while overcharged and kinetically stabilised aggregates form when the surfactant is in large excess. This leads in many cases to a highly charged surfactant shell surrounding a core of P/S aggregates.35 At intermediate mixing ratios, there exists an equilibrium two-phase region where the net charge of the P/S complexes is minimised that causes low colloidal stability and drives the formation of large P/S aggregates that can sediment over time and, thus, give rise to a loss of surface-active material.5,20Fig. 2 depicts the hydrodynamic diameter dh and the electrophoretic mobility uζ of the PDADMAC/CnAAP mixtures as a function of the polyelectrolyte concentration, while the AAP concentration was kept fixed as explained in the experimental details. Close inspection of Fig. 2 reveals that for each of the CnAAP surfactants, uζ changes drastically from negative to positive values as the PDACMAC concentration increases, which is consistent with the general behaviour of P/S mixtures with classical surfactants.36,37 Interestingly, the charge reversal for the CnAAP surfactants in the E configuration (irradiation with green light) takes place at a monomer concentration of PDADMAC equal to the surfactant concentration as indicated by the vertical lines in Fig. 2a.
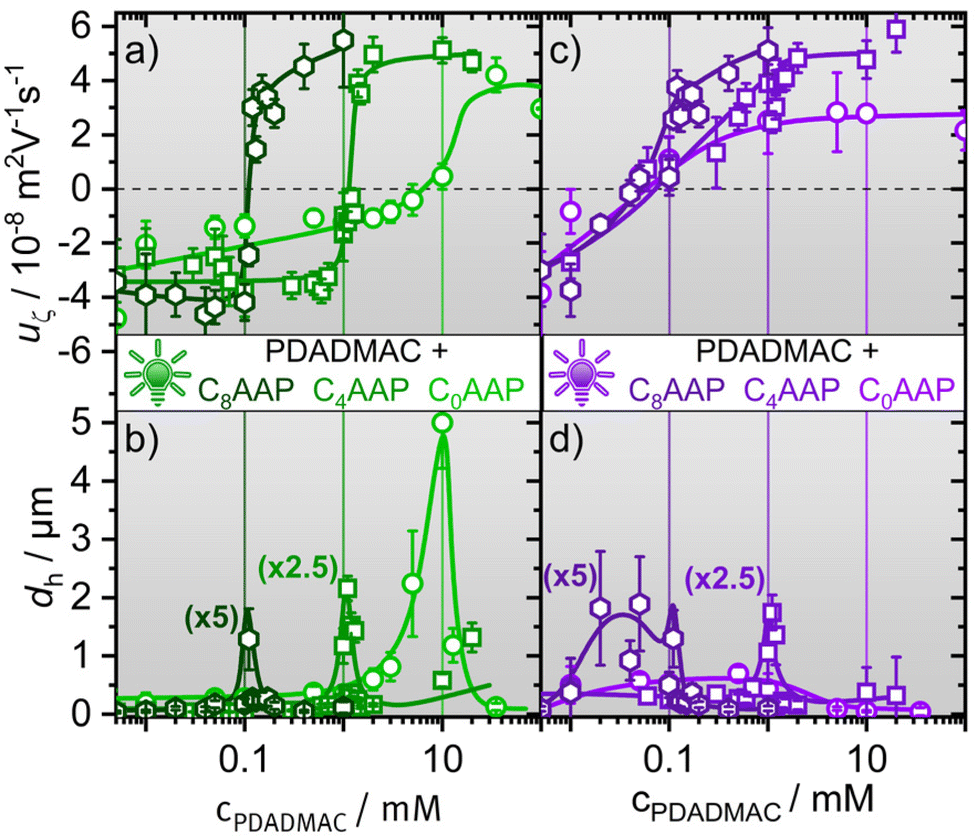 | ||
| Fig. 2 Electrophoretic mobilities uζ of CnAAP/PDADMAC mixtures with the surfactants in (a) the E state (green iradiation) and (c) the Z state (UV irradiation). (b) and (d) present the hydrodynamic diameter dh of CnAAP/PDADMAC mixtures for E and Z isomers of CnAAP surfactants, respectively. The vertical lines represent the concentration of the surfactants which were fixed to 0.1, 1 and 10 mM for C8AAP, C4AAP and C0AAP, respectively. For a better comparision of the data, some hydrodynamic diameters were multiplied by a constant factor as indicated in the figure. | ||
Equal molar concentration of surfactants and oppositely charged polyelectrolytes at the point of zero net charge (negligible uζ) are indicative of high binding affinities of the surfactants to the PDADMAC chains.10 The steepness of the charge reversal is largely affected by the terminal chain of the surfactants: while for PDADMAC mixtures with C8AAP as well as C4AAP, the change from negative to positive uζ is observed in a narrow concentration range, the charge reversal for mixtures of PDADMAC with C0AAP takes place in a much wider range of concentrations (Fig. 2a). These apparent differences are presumably caused by a variation of the hydrophobic interaction between the AAP surfactants and PDADMAC, where the CnAAP surfactants with butyl and octyl terminal groups have a higher binding affinity as compared to C0AAP.36,38
In Fig. 2b we present the hydrodynamic diameters dh of the P/S mixtures as a function of the PDADMAC concentration.
At concentrations with small or negligible uζ, aggregates are formed which exhibit only small electrostatic disjoining pressures between aggregates promoting the formation of larger aggregates in the bulk as compared to concentrations where |uζ| and, thus, the electrostatic repulsion is high. This behaviour is not unique for PDADMAC/CnAAP mixtures but well-known also for other P/S systems with classical surfactants.16,18,36,37 Although, larger aggregates with PDADMAC formed for all CnAAP surfactants, their sizes increased substantially with decreasing chain length (correlated with increasing bulk concentration), which can be associated with the total ionic strength in the system. PDADMAC mixtures with C0AAP exhibited the highest ionic strength (20 mM) where the mixtures with C8AAP had the lowest (0.2 mM). Consequently, charge screening for PDADMAC/C0AAP mixtures is much more pronounced, reduces the electrostatic disjoining pressure between aggregates and, thus, promotes the formation of very large aggregates with dh of ∼5 μm, while dh was for the other surfactant mixtures well below 1 μm outside the equilibrium two-phase region.
As can be seen from the data in Fig. 2c and d, E → Z photoisomerization caused by UV irradiation of the samples leads to a drastic shift in the point of zero net charge (or uζ, see Fig. 2c) to much lower PDADMAC concentrations in case of C4AAP and C0AAP. This is also visible in the trends of the hydrodynamic diameters where the sharp maxima vanish and, instead, broad maxima at lower concentrations appear (Fig. 2d).
Irradiation with UV light causes E → Z photo-isomerization of the surfactants and results in a reduced binding affinity to the PDADMAC chain. This is due to a change in hydrophobic interactions between both molecules, that varies with the surfactants’ configuration (E or Z, see Fig. 1). Hydrophobic interactions are reduced when the surfactants are in the Z state, which was already observed in a previous work on a cationic AAP surfactant with an ammonium head group and the anionic polystyrene sulfonate.13 As a result of the reduced binding affinity, a much larger excess of anionic CnAAP surfactants is needed to overcharge the cationic PDADMAC leading to negative electrophoretic mobilities and net charges of the aggregates. This shifts the point of zero net charge to lower P/S ratios (or polyelectrolyte concentrations) consistent with the results in Fig. 2a and c. This is, particularly, pronounced for PDADMAC/C0AAP mixtures where the point of zero net charge is shifted from a P/S ratio of ∼1 (10 mM PDADMAC) for the E isomer to ∼0.01 (0.1 mM PDADMAC) for the Z isomer.
In order to test the reversibility of the aggregate formation and breaking up of aggregates upon E/Z photoisomerization, we have performed several consecutive E/Z switching cycles and present the results for the hydrodynamic diameter and the electrophoretic mobility in Fig. 3, where equimolar concentrations of CnAAP and PDADMAC were used (for additional results using other molar ratios see ESI†).
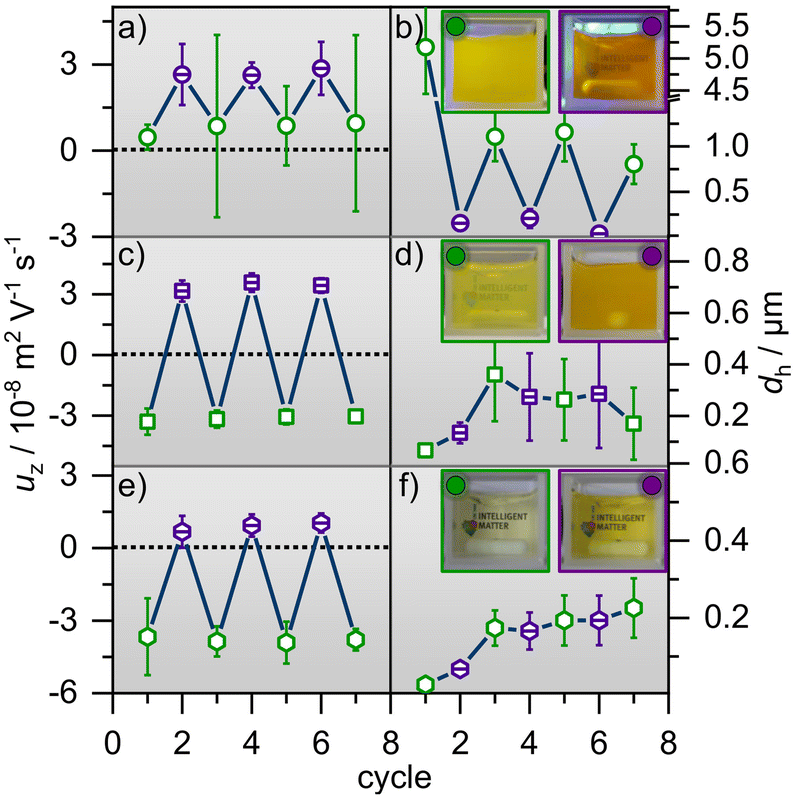 | ||
| Fig. 3 Electrophoretic mobilities uζ and the hydrodynamic diameters dh after several irradiation cycles of green (open symbols) and UV light (dashed symbols) for mixtures. (a) and (b) show uζ and dh for equimolar mixtures of PDADMAC with 10 mM C0AAP wheras (c) and (d) as well as (e) and (f) present uζ and dh for 1 mM C4AAP and 0.1 mM C8AAP, respectively. Photos of the mixtures under green and subsequent UV irradiation in (b), (d) and (f) demonstrate the influence of the alternating light conditions on the sample turbidity. More information on turbidity can be found in Fig. S7 or the video in the ESI.† | ||
C0AAP mixtures with PDADMAC show remarkable differences from the other P/S mixtures (Fig. 3): uζ and dh of C0AAP/PDADMAC aggregates can be changed nearly reversibly (Fig. 3a and b) through E/Z photoisomerization of the C0AAP surfactants evidencing control of the aggregate properties over several irradiation cycles.
On the one hand, this is indeed consistent with previous work13 where mixtures of anionic polystyrene sulfonate and cationic surfactants similar to C0AAP were investigated. On the other hand, the data from PDADMAC mixtures with C4AAP and C8AAP are distinct, as reversible control of uζ (Fig. 3c and e) through E/Z photo-isomerization is observed, but reversible control of the aggregate size is not, because the latter increases with increasing number of cycles (Fig. 3d and f). Instead of switching the size from large to small aggregates and vice versa, the aggregate size increases with every switching cycle. This can have several causes like the higher hydrophobic interactions of C4AAP and C8AAP having longer alkyl chains. Also, the fact that the electrophoretic mobility needs to cross zero values might affect the reproducibility. The high reversibility of aggregates’ electrophoretic mobility is pointing to a charging state of the aggregates which is determined by surfactant ad- and desorption at their outer shell, as was previously proposed by Mészáros et al., who studied PEI/SDS mixtures.20 The missing reversibility of the aggregate sizes for PDADMAC mixtures with C4AAP and C8AAP can be also demonstrated through visual inspection of the solutions’ turbidity (Fig. 3 and ESI†).
CnAAP/PDADMAC mixtures at the interface
For the investigation of CnAAP/PDADMAC binding at the air–water interface, we have combined vibrational SFG spectroscopy and NR. Fig. 4 presents SFG spectra of CnAAP mixtures with PDADMAC for different mixing ratios and for both E and Z configurations of the surfactants. The spectra show vibrational bands at ∼2850 and ∼2880 cm−1 due to symmetric stretching vibrations of methylene (d+) and methyl (r+) groups, respectively.19,39,40 Whereas the antisymmetric stretching bands are centred at ∼2930 (d−) and ∼2965 cm−1 (r−), bands at ∼3030 cm−1 and ∼3060 cm−1 are attributed to the C–H stretching modes of the aromatic ring. Broad bands around 3200 and 3450 cm−1 derive from O–H stretching modes of H-bonded interfacial water molecules. Because C–H bands can arise from both CnAAP and PDADMAC moieties at the interface, their analysis in terms of the individual contributions is more complex and we, thus, concentrate on the analysis of the O–H bands from interfacial water molecules. Although, interpretations of O–H stretching bands in SFG spectroscopy can be also rather complex we assume that the O–H intensity is largely dominated by the third-order contribution of the interfacial double layer and thus the surface potential ϕ0 (see eqn (1)),22–24 while i.e. changes in hydrogen-bonding and interfacial water structure may contribute as well but to a far smaller extend. This assumption is corroborated by complementary results from NR, discussed below, and allows us to perform a qualitative analysis on the interfacial charging state from the differences in O–H intensity.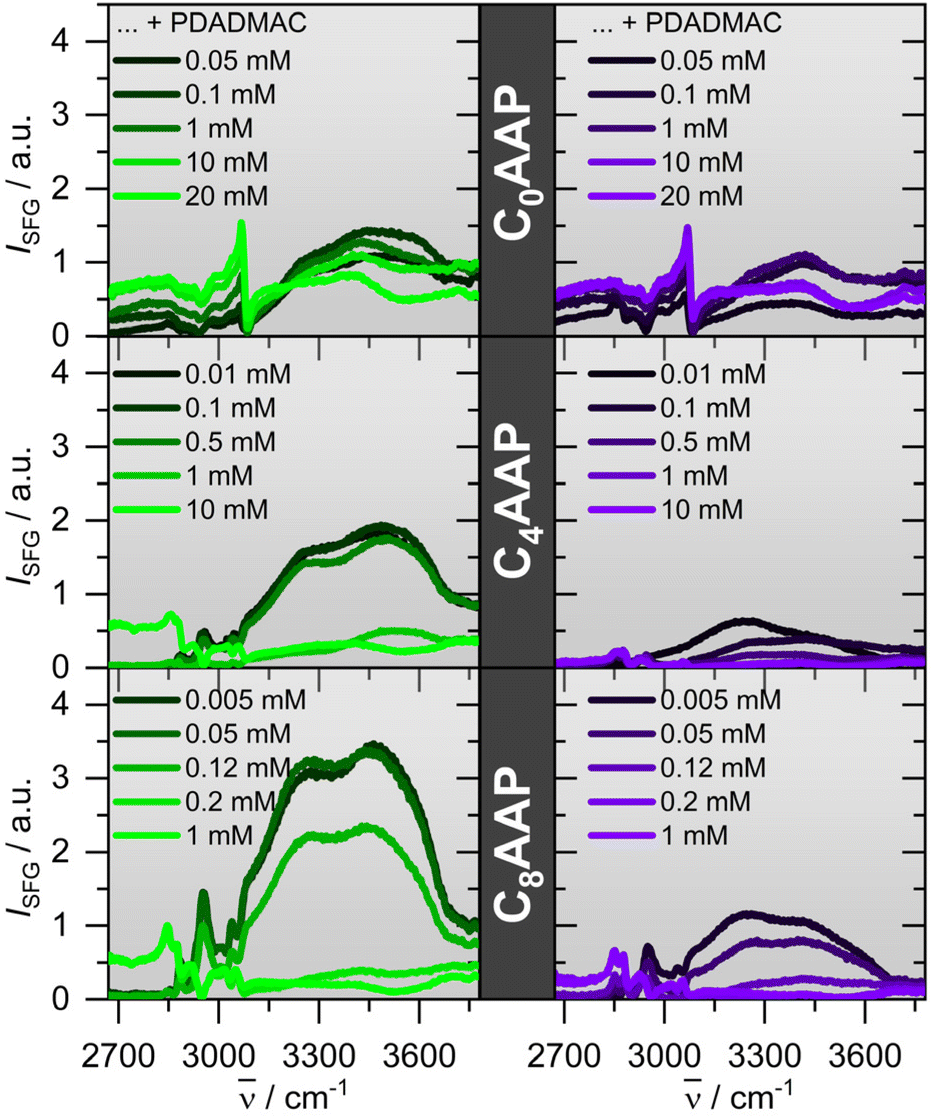 | ||
| Fig. 4 Vibrational SFG spectra of CnAAP/PDADMAC mixtures at the air–water interface for selected PDADMAC concentrations as indicated in the figure. Note that similar to the bulk characterization, the concentrations of C0AAP, C4AAP and C8AAP surfactants were fixed to 10, 1 and 0.1 mM, respectively. SFG spectra of interaces with the surfactants in predominant E and Z states are indicated by green and violet-colored solid lines, respectively. | ||
The spectra of the C8AAP and C4AAP surfactants with very low PDADMAC concentrations show strong O–H contributions to the SFG intensity. Close comparison of the SFG spectra at low PDADMAC concentrations, but 0.1 mM, 1 mM and 10 mM concentrations of C0AAP, C4AAP and C8AAP surfactants, respectively, indicates that the O–H intensities are decreasing with the chain length. This is related to effectiveness of the surfactants and the total ionic strength in the system being additionally determined by the surfactant concentration. In fact, the required concentrations to establish densely-packed surfactant monolayers and to reach the maximum switchability between E and Z configurations as well as the surfactants’ critical micelle concentration (CMC) are about an order of magnitude different between C0AAP and C8AAP.19 For that reason, largely different concentrations of 10, 1 and 0.1 mM for C0AAP, C4AAP and C8AAP and consequently largely different ionic strength were needed. As a result, charge screening at the interface is much more substantial for mixtures with C0AAP and causes a drastic decrease of the O–H intensity even when PADMAC is absent. The decrease of O–H intensities with ionic strength is well known for SFG spectroscopy at charged interfaces and caused by the third-order contribution to the SFG intensity of O–H bands (eqn (1)) which decreases as the ionic strength increases because of charge screening and the smaller Debye length of the electric double layer.22–24
For C0AAP the SFG intensity is dominated by the C–H modes and a non-resonant contribution which causes the apparent offset in the SFG spectra (Fig. 4). An increase of the PDADMAC concentration results in an additional decrease in the O–H intensity and is attributed to the adsorption of PDADMAC/CnAAP aggregates at the air–water interface. Particularly, aggregates with low net charge can efficiently reduce the interfacial charge and the resulting surface potential ϕ0 (eqn (1)). Further manipulation of the interfacial charge can be realized by UV irradiation: here, E → Z isomerisation of the CnAAP surfactants causes a further decrease in the O–H intensity when the surfactants are in the Z state as compared to the E state which can be also re-established by irradiation with green light.
In order to obtain more quantitative information from SFG, the spectra in Fig. 4 were fitted using Lorentzian functions for the narrow CH bands, and Voigt profiles for the inhomogenously broadened O–H bands (details in the ESI†). The results for the O–H amplitudes are shown in Fig. 5a.
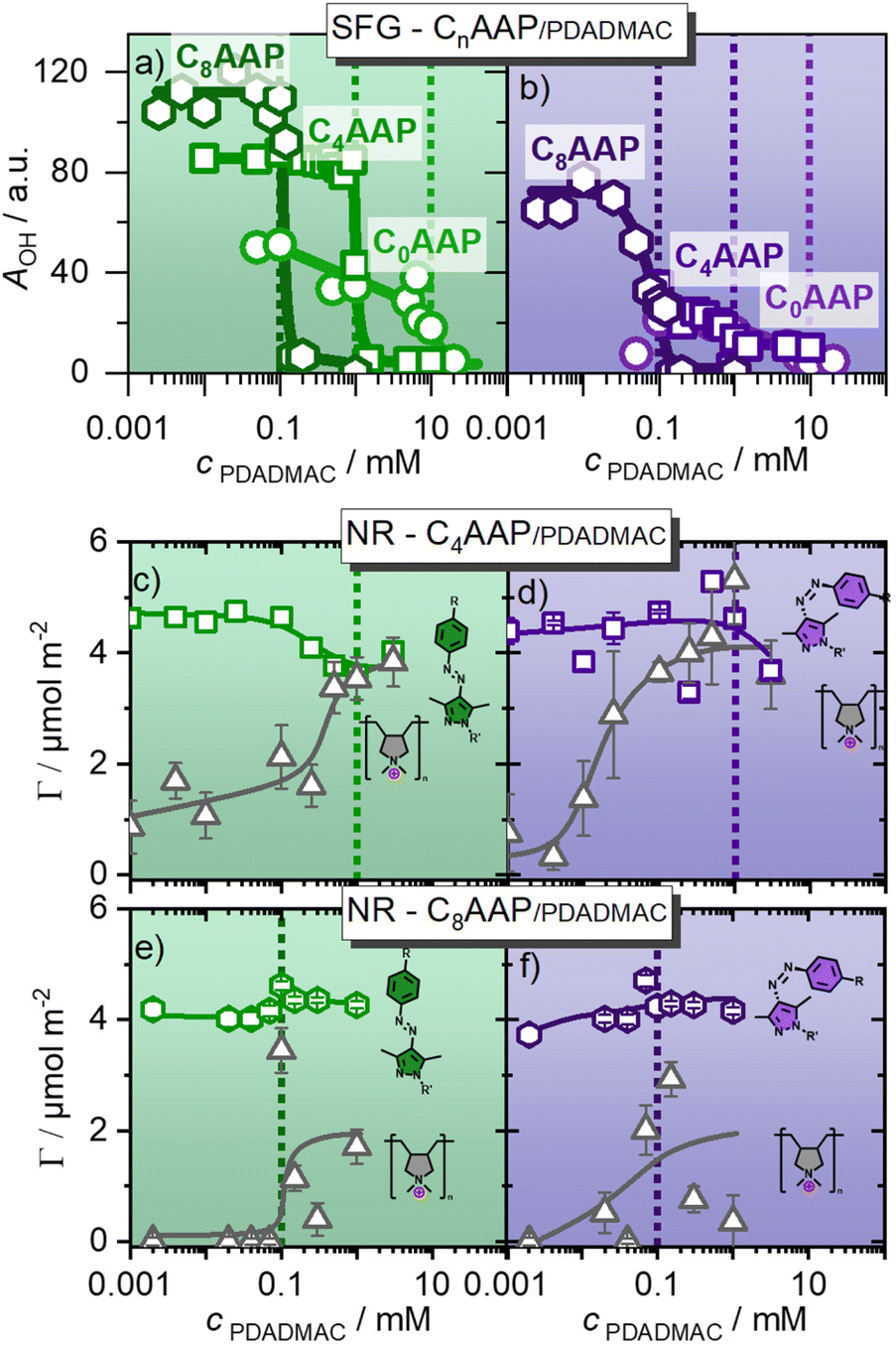 | ||
| Fig. 5 SFG amplitudes of the O–H stretching modes of interfacial water molecules that report qualitatively on the charging state (the surface potential ϕ0 see eqn (1)), for CnAAP/PDADMAC mixtures at the air–water interface with the surfactants in (a) E and (b) Z configurations. (c) and (d) as well as (e) and (f) present the surface excess of C8AAP/PDADMAC and C4AAP/PDADMAC mixtures at the air–water interface, which was resolved using NR. (c) and (e) show the results for C4AAP and C8AAP surfactants in E configurations (green irradiation), while in (d) and (f) the results for the surfactants in the Z state are presented. Dashed lines indicate equimolar ratios of polyelectrolytes and surfactants. Symbols in grey colour represents the PDADMAC surface excess, while green and violet colored symbols indicate the surface excesses of the E and Z isomers of CnAAP surfactants. | ||
The trend of the O–H amplitudes follows the trend in electrophoretic mobilities (Fig. 2): in case the surfactants are in the E state (irradiation with green light), O–H bands exhibit a high amplitude which is strongly decreased at equal CnAAP/PDADMAC concentrations.
Mixtures with lower total ion concentrations due to, e.g. lower CnAAP concentration like C8AAP exhibit higher O–H intensities which is caused by a reduced charge screening as noted above. The effect of UV irradiation is a further decrease in SFG amplitude (Fig. 5a and b) consistent with a change in electrophoretic mobilities of bulk aggregates (Fig. 2a and c). However, opposed to the electrophoretic mobilities, no strong charge reversal is visible at the interface which would give rise to a different phase of the water bands and thus different spectral shape. This conclusion is corroborated by the NR results discussed below.
Using surface tensiometry and SFG spectroscopy, we have also tested the reversibility of the changes at the air–water interface. In order to identify whether a similar reversibility as seen for the aggregates in the bulk solution is possible (see Fig. 3 and its discussion), we have studied the air–water interface after several E/Z switching cycles and present the results in Fig. S12 (ESI†). As expected, the spreading of P/S aggregates at the air–water interface is not reversible and the surface tension changes between E and Z isomers are negligible. However, the massive change in O–H intensity as indicated in Fig. 4 and 5 is partially reversible (Fig. S12†). Particularly, for PDADMAC mixtures with C0AAP the reversibility in O–H intensity is very good whereas for C4AAP and C8AAP the O–H intensity is reduced with every switching cycle.
Next, we discuss our results from NR, in order to explain how the interfacial composition in terms of the surface excesses of PDADMAC and CnAAP at the interface is changing with mixing ratio and E/Z configuration of the surfactants. This can be also used to infer qualitatively the interfacial charging state and allows for a comparison with the results from SFG spectroscopy. Neutron reflectivity profiles were recorded only for C4AAP and C8AAP mixtures with PDADMAC. The corresponding profiles and their analysis are shown and discussed in the ESI.† In the following, the main focus is on the resulting surface excesses Γ of CnAAP surfactants and PDADMAC polyelectrolyte (Fig. 5c–f). The remaining parameters are stated in the ESI.†
The results for the C4AAP/PDADMAC system complement the SFG results: at low PDADMAC concentrations, an interfacial excess of anionic surfactants is consistent with a highly negatively charged interface, i.e. with relatively intense O–H bands in SFG spectroscopy (Fig. 4 and 5a, b). Increasing the PDADMAC concentration leads to an increase in the surface excess of PDADMAC, as expected, while Γ slightly decreases for C4AAP at high PDADMAC concentrations. Consequently, the net charge at the air–water interface and, thus, the surface potential is driven by the increasing PDADMAC surface excess while the surface excess of the oppositely charged AAP stays roughly the same.
For the C4AAP mixtures with the surfactant in the E state, already at a low polyelectrolyte concentration (0.001 mM), PDADMAC is present at the interface. Surface tension experiments support the occurrence of interfacial aggregates even at very low PDADMAC concentrations as a lower surface tension compared to the C4AAP solution without PDADMAC was observed (Fig. S3, ESI†). The analysis of the neutron reflectometry profiles yields more details on the molecular picture of the changes at the air–water interface (NR parameters in Table S2 in the ESI†). Here we focus particularly on the trends of the PDADMAC in layer 3, that is below the surfactant layers at the interface which can change significantly depending on the E/Z configurations of CnAAP surfactants. The thickness and the surface excess of the surfactants in layer 1 (tails) and 2 (heads) remain similar to the values of the surfactant monolayers with no or little effect of the PDADMAC concentration (Tables S2 and S3, ESI†).21 The thickness of layer 3 which is dominated by PDADMAC is higher (7–18 nm) for mixtures with C4AAP at relatively low polyelectrolyte concentrations. It becomes more compact with a thickness of <0.9 nm at higher concentrations when the surfactants are in the E state. Conversely, for the Z state, the thickness of the PDADMAC dominated layer 3 remains relatively independent (with 1–3 nm) of the polymer concentration (Table S2†). This is different from PDADMAC/C8AAP mixtures where layer 3 is about 4 to 7 nm thick independent on the polymer concentration and the choice of the surfactant (E/Z) isomer (Table S3, ESI†).
A notable characteristic of the PDADMAC/C4AAP mixtures with the Z isomer is the observation of clear Kiessig fringes for some concentrations e.g. 1 mM PDADMAC, i.e. a minimum at approx. QZ = 0.08 Å−1 for d-surfactants in ACMW (Fig. S5†). Recent NR studies of several related systems attribute these Kiessig fringes to the existence of extended structures at low coverage, e.g. hemimicelles or bilayer patches.9,27,31 For our systems, only the mixtures with the C4AAP where Z-isomer showed the necessity of the extended structures. We hypothesize that this might be related to the rather special behaviour of C4AAP in the Z state as the pure surfactant can form bilayer structures at the air–water interface while C8AAP and C0AAP do not.19,21
Under green irradiation, the surface excesses of PDADMAC and C4AAP approach each other at equimolar P/S ratios (Fig. 5c and d), which causes a negligible interfacial charge consistent with the observations from SFG spectroscopy. UV irradiation shifts this concentration of charge neutralisation to lower PDADMAC concentrations due to the reduced binding affinity compared to the E isomers. This result is consistent with previous results on mixtures of an AAP triethylammonium bromide surfactant with sodium polystyrene sulfonate where the reduced binding affinity of Z isomer decreased the necessary polyelectrolyte concentration for, e.g. the charge reversal of the electrophoretic mobility.13 PDADMAC mixtures with C8AAP surfactants demonstrate a similar behaviour as the C4AAP mixtures. The surface excesses of PDADMAC and C8AAP approach each other at equimolar ratio for the E and lower PDADMAC concentrations for the Z isomer, but the overlap between the surface excesses is, compared to C4AAP, less pronounced for C8AAP (Fig. 5). A close inspection of Fig. 5 reveals differences between NR and SFG results that are likely to be associated with the layer structure of PDADMAC which is extending into the bulk phase, but is still contributing to the surface excess. Conclusions on reduced interfacial charge from SFG experiments are based on the water molecules’ reduced net orientation perpendicular to the interface. The common explanation of this reduction is an also reduced electric field of interfacial molecules (eqn (1)). In this system, however, positively charged loops and chains of PDADMAC in layer 3 penetrate into the bulk phase, and water molecules at these charged sites do not contribute to the SFG spectra as the signal from water at these sites is cancelled due to local inversion symmetry. PDADMAC/C4AAP mixtures at equimolar concentrations in the interfacial layers are much more compact (thickness of PADADMAC layer ∼0.7 nm, Table S2†) and drive the positive charges of PDADMAC much closer to the negative charges of the surfactant layer which reduces the local electric field at the very interface and, consequently, the SFG signals from interfacial H2O to a large extent. This is different for PDADMAC/C8AAP mixture where the thickness of the PDADMAC layer 3 expands to about 5 nm (Table S3†) and can, thus, less efficiently screen the negative charges of the surfactants at the topmost interface (layer 2).
CnAAP/PDADMAC mixtures for stabilization of aqueous foam
The molecular changes at the air–water interface can drive macroscopic property changes of aqueous foams, as demonstrated in our previous works.21 The foam stability for selected P/S mixing ratios is presented in Fig. 6. Both C4AAP and C8AAP surfactants form stable foams without PDADMAC when they are in the E configuration and remain stable for several hours (Fig. 6). Switching to the Z isomer with UV irradiation causes rapid foam decay with half lifetimes of about 1 h and 350 s for C8AAP and C4AAP, respectively. PDADMAC/CnAAP mixtures show a decreased foam stability, but with more drastic differences between E and Z isomers, where the PDADMAC/CnAAP stabilized foams are quite stable for the E isomer (∼1 h) while the Z isomer leads to very poor stability of less than 1 min.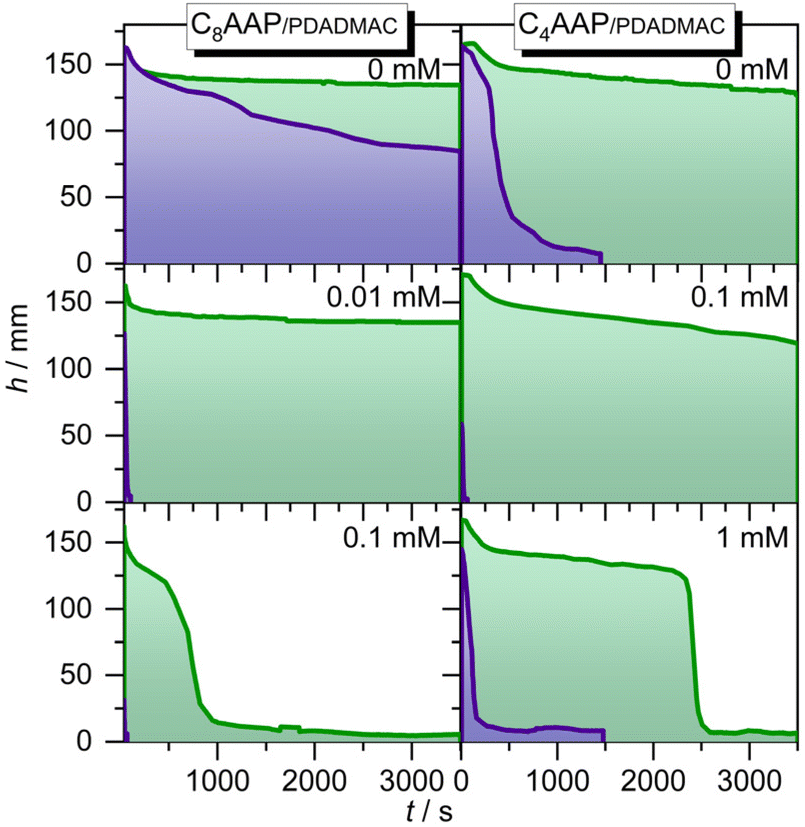 | ||
| Fig. 6 Foam height as a function of time after formation of the foams for different PDADMAC mixtures with C4AAP and C8AAP surfactants. The surfactant concentrations are fixed to 0.1 mM (C8AAP) and 1 mM (C4AAP) whereas the PDADMAC concentration was varied as is indicated in the figure. | ||
Foams of P/S aggregates are mainly stabilized through electrostatic disjoining forces41 or steric repulsion8,9 as well as due to Pickering effects with larger particles.13 When the surfactants are in the E state the disjoining pressure between gas bubbles is sufficiently high but is reduced drastically for the Z isomer where surfactant desorption from the interface (Fig. 5 and discussion) causes a reduction of the electrostatic disjoining pressure. This conclusion on the dominant influence of the electrostatic disjoining pressure on the foam stability is also corroborated by the decrease in foam stability with increasing PDADMAC/CnAAP mixing ratio (Fig. 6). The latter leads to a lower net charge at the interface as we have discussed above and which can be inferred from the results shown in Fig. 5.
Since P/S mixtures like 0.1 mM PDADMAC with 0.1 mM C8AAP exhibit larger particles in the bulk solution (Fig. 2) but show no improved foam stabilities there is, for PDADMAC/CnAAP mixtures, no beneficial effect from the presence of the particles in the bulks solution which could lead to Pickering stabilization (Fig. 6). Using PDADMAC/CnAAP mixtures is, however, advantageous when the difference in foam stability with the surfactants in the E and Z state needs to be maximized i.e., the foam half-life time with the surfactants in the state can be reduced from >1 h to less than 1 min (Fig. 6), whereas the half-life time of the foams with P/S mixtures and the surfactants in the E state is comparable to foams stabilized by C8AAP or C4AAP only (Fig. 6). This improves the applicability of CnAAP/PDADMAC mixtures as candidates for highly responsive photoswitchable foams.
Conclusions
In this work, we report on a new approach to render polyelectrolyte/surfactant (P/S) mixtures of the cationic PDADMAC with anionic surfactants responsive to light. Unlike the well-known P/S mixtures with classical surfactants we are using photoswitchable arylazopyrazole amphiphiles (CnAAP) with three different terminal groups -octyl, -butyl, and -H in order to tune remotely the surfactant CMC and the binding affinity of the surfactant to the polyelectrolyte chain. In addition, we can show that through E/Z photoisomerization of the surfactants we are able to change their binding to PDADMAC substantially, which causes different bulk as well as interfacial behaviour. In the bulk, P/S aggregates form and their size as well as electrophoretic mobilities uζ change with mixing ratio as expected from earlier reports with classic surfactants, but they also change drastically and partly reversibly through the irradiation with UV and green light. For instance, uζ can be driven from negative to positive values through light irradiation, while larger particles in the form of P/S aggregates can be broken up and reversibly formed again depending on the nature of the CnAAP surfactant. Similar effects at the air–water interface are observed for two of the three CnAAP/PDADMAC mixtures, where the surface composition and net charge depend on the P/S mixing ratio and can be changed after formation of the interface through light irradiation. The interfacial structures including extended structures involving bilayer patches in contact with single P/S layers were resolved using a model with up to four layers to fit the results from NR. The binding of PDADMAC with the CnAAP surfactants increases the polyelectrolytes’ surface excess and, consequently, leads near equimolar concentrations to efficient charge screening at the interface. The latter has been consistently determined from vibrational SFG spectroscopy and NR as two complementary methods that report on the interfacial structure and are highly useful for explaining the stability of aqueous foams and their responsiveness to light. We could indeed show that the application of photo-responsive P/S mixtures in foams allows for remote control of the interfacial properties (binding affinity, interfacial structure) as well as mesoscopic and macroscopic properties such as the electrophoretic mobility, particle size and foam stability. Further, we expect our results can be used to derive other soft matter materials i.e., emulsions responsive to light.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project-ID 433682494-SFB 1459 Intelligent Matter. We also thank the Institute Laue-Langevin (Grenoble, France) for allocating neutron beam time on FIGARO (https://doi.org/10.5291/ILL-DATA.9-11-1984 and https://doi.org/10.5291/ILL-DATA.9-12-652).References
- M. Gradzielski, Polyelectrolyte–Surfactant Complexes As a Formulation Tool for Drug Delivery, Langmuir, 2022, 38, 13330–13343 CrossRef CAS PubMed.
- S. Llamas, E. Guzmán, F. Ortega, N. Baghdadli, C. Cazeneuve, R. G. Rubio and G. S. Luengo, Adsorption of Polyelectrolytes and Polyelectrolytes-Surfactant Mixtures at Surfaces: A Physico-Chemical Approach to a Cosmetic Challenge, Adv. Colloid Interface Sci., 2015, 222, 461–487 CrossRef CAS PubMed.
- G. S. Luengo, A.-L. Fameau, F. Léonforte and A. J. Greaves, Surface Science of Cosmetic Substrates, Cleansing Actives and Formulations, Adv. Colloid Interface Sci., 2021, 290, 102383 CrossRef CAS PubMed.
- E. Weißenborn, J. Droste, M. Hardt, D. Schlattmann, C. Tennagen, C. Honnigfort, M. Schönhoff, M. R. Hansen and B. Braunschweig, Light-Induced Switching of Polymer-Surfactant Interactions Enables Controlled Polymer Thermoresponsive Behaviour, Chem. Commun., 2021, 57, 5826–5829 RSC.
- M. Gradzielski and I. Hoffmann, Polyelectrolyte-surfactant complexes (PESCs) composed of oppositely charged components, Curr. Opin. Colloid Interface Sci., 2018, 35, 124–141 CrossRef CAS.
- B. K. Schabes, E. J. Hopkins and G. L. Richmond, Molecular Interactions Leading to the Coadsorption of Surfactant Dodecyltrimethylammonium Bromide and Poly(styrenesulfonate) at the Oil/Water Interface, Langmuir, 2019, 35, 7268–7276 CrossRef CAS PubMed.
- L. Fernández-Peña, E. Guzmán, C. Fernández-Pérez, I. Barba-Nieto, F. Ortega, F. Leonforte, R. G. Rubio and G. S. Luengo, Study of the Dilution-Induced Deposition of Concentrated Mixtures of Polyelectrolytes and Surfactants, Polymers, 2022, 14, 1335 CrossRef PubMed.
- L. Braun and R. von Klitzing, When Bulk Matters: Disentanglement of the Role of Polyelectrolyte/Surfactant Complexes at Surfaces and in the Bulk of Foam Films, Langmuir, 2023, 39, 111–118 CrossRef CAS PubMed.
- L. Braun, M. Uhlig, O. Löhmann, R. A. Campbell, E. Schneck and R. von Klitzing, Insights into Extended Structures and Their Driving Force: Influence of Salt on Polyelectrolyte/Surfactant Mixtures at the Air/Water Interface, ACS Appl. Mater. Interfaces, 2022, 14, 27347–27359 CrossRef CAS PubMed.
- I. Varga and R. A. Campbell, General Physical Description of the Behavior of Oppositely Charged Polyelectrolyte/Surfactant Mixtures at the Air/Water Interface, Langmuir, 2017, 33, 5915–5924 CrossRef CAS PubMed.
- E. Tran, A. N. Mapile and G. L. Richmond, Peeling Back the Layers: Investigating the Effects of Polyelectrolyte Layering on Surface Structure and Stability of Oil-In-Water Nanoemulsions, J. Colloid Interface Sci., 2021, 599, 706–716 CrossRef CAS PubMed.
- E. Guzmán, A. Maestro, F. Ortega and R. G. Rubio, Association of oppositely charged polyelectrolyte and surfactant in solution: equilibrium and nonequilibrium features, J. Phys.: Condens.Matter, 2023, 35, 323001 CrossRef PubMed.
- M. Schnurbus, M. Hardt, P. Steinforth, J. Carrascosa-Tejedor, S. Winnall, P. Gutfreund, M. Schönhoff, R. A. Campbell and B. Braunschweig, Responsive Material and Interfacial Properties through Remote Control of Polyelectrolyte–Surfactant Mixtures, ACS Appl. Mater. Interfaces, 2022, 14, 4656–4667 CrossRef CAS PubMed.
- H. A. Ritacco, Polyelectrolyte/Surfactant Mixtures: A Pathway to Smart Foams, ACS Omega, 2022, 7, 36117–36136 CrossRef CAS PubMed.
- V. V. Lyadinskaya, A. G. Bykov, R. A. Campbell, I. Varga, S. Y. Lin, G. Loglio, R. Miller and B. Noskov, Dynamic surface elasticity of mixed poly(diallyldimethylammonium chloride)/sodium dodecyl sulfate/NaCl solutions, Colloids Surf., A, 2014, 460, 3–10 CrossRef CAS.
- A. Akanno, E. Guzmán, L. Fernández-Peña, S. Llamas, F. Ortega and R. G. Rubio, Equilibration of a Polycation-Anionic Surfactant Mixture at the Water/Vapor Interface, Langmuir, 2018, 34, 7455–7464 CrossRef CAS PubMed.
- R. A. Campbell, M. Yanez Arteta, A. Angus-Smyth, T. Nylander and I. Varga, Multilayers at Interfaces of an Oppositely Charged Polyelectrolyte/Surfactant System Resulting from the Transport of Bulk Aggregates under Gravity, J. Phys. Chem. B, 2012, 116, 7981–7990 CrossRef CAS PubMed.
- L. Patel, O. Mansour, H. Bryant, W. Abdullahi, R. M. Dalgliesh and P. C. Griffiths, Interaction of Low Molecular Weight Poly(diallyldimethylammonium chloride) and Sodium Dodecyl Sulfate in Low Surfactant–Polyelectrolyte Ratio, Salt-Free Solutions, Langmuir, 2020, 36, 8815–8825 CrossRef CAS PubMed.
- M. Hardt, F. Busse, S. Raschke, C. Honnigfort, J. Carrascosa-Tejedor, P. Wenk, P. Gutfreund, R. A. Campbell, A. Heuer and B. Braunschweig, Photo-Responsive Control of Adsorption and Structure Formation at the Air-Water Interface with Arylazopyrazoles, Langmuir, 2023, 39, 5861–5871 CrossRef CAS PubMed.
- R. Mészáros, L. Thompson, M. Bos, I. Varga and T. Gilányi, Interaction of Sodium Dodecyl Sulfate with Polyethyleneimine: Surfactant-Induced Polymer Solution Colloid Dispersion Transition, Langmuir, 2003, 19, 609–615 CrossRef.
- C. Honnigfort, R. A. Campbell, J. Droste, P. Gutfreund, M. R. Hansen, B. J. Ravoo and B. Braunschweig, Unexpected Monolayer-To-Bilayer Transition of Arylazopyrazole Surfactants Facilitates Superior Photo-Control of Fluid Interfaces and Colloids, Chem. Sci., 2020, 11, 2085–2092 RSC.
- P. E. Ohno, H.-F. Wang and F. M. Geiger, Second-Order Spectral Lineshapes From Charged Interfaces, Nat. Commun., 2017, 8, 1032 CrossRef PubMed.
- G. Gonella, C. Lütgebaucks, A. G. F. de Beer and S. Roke, Second Harmonic and Sum-Frequency Generation from Aqueous Interfaces Is Modulated by Interference, J. Phys. Chem. C, 2016, 120, 9165–9173 CrossRef CAS.
- N. García Rey, E. Weißenborn, F. Schulze-Zachau, G. Gochev and B. Braunschweig, Quantifying Double-Layer Potentials at Liquid-Gas Interfaces from Vibrational Sum-Frequency Generation, J. Phys. Chem. C, 2019, 123, 1279–1286 CrossRef PubMed.
- M. W. Skoda, Recent Developments in The Application of X-Ray and Neutron Reflectivity to Soft-Matter Systems, Curr. Opin. Colloid Interface Sci., 2019, 42, 41–54 CrossRef CAS.
- T. Narayanan, H. Wacklin, O. Konovalov and R. Lund, Recent Applications of Synchrotron Radiation and Neutrons in the Study of Soft Matter, Crystallogr. Rev., 2017, 23, 160–226 CrossRef CAS.
- J. Carrascosa-Tejedor, A. Santamaria, A. Tummino, I. Varga, M. Efstratiou, M. J. Lawrence, A. Maestro and R. A. Campbell, Polyelectrolyte/surfactant films: from 2D to 3D structural control, Chem. Commun., 2022, 58, 10687–10690 RSC.
- R. A. Campbell, H. P. Wacklin, I. Sutton, R. Cubitt and G. Fragneto, FIGARO: The new horizontal neutron reflectometer at the ILL, Eur. Phys. J. Plus, 2011, 126, 107 CrossRef.
- P. Gutfreund, T. Saerbeck, M. A. Gonzalez, E. Pellegrini, M. Laver, C. Dewhurst and R. Cubitt, Towards generalized data reduction on a chopper-based time-of-flight neutron reflectometer, J. Appl. Crystallogr., 2018, 51, 606–615 CrossRef CAS.
- A. Nelson, Co-refinement of multiple-contrast neutron/X-ray reflectivity data using \it MOTOFIT, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- J. Carrascosa-Tejedor, L. M. Miñarro, M. Efstratiou, I. Varga, M. W. A. Skoda, P. Gutfreund, A. Maestro, M. J. Lawrence and R. A. Campbell, Control of the structure and morphology of polypeptide/surfactant spread films by exploiting specific interactions, Nanoscale, 2023, 15, 11141–11154 RSC.
- A. Braslau, M. Deutsch, P. S. Pershan, A. H. Weiss, J. Als-Nielsen and J. Bohr, Surface Roughness of Water Measured by X-Ray Reflectivity, Phys. Rev. Lett., 1985, 54, 114–117 CrossRef CAS PubMed.
- R. Mészaros, I. Varga and T. Gilanyi, Effect of polymer molecular weight on the polymer/surfactant interaction, J. Phys. Chem. B, 2005, 109, 13538–13544 CrossRef PubMed.
- L. Chiappisi, I. Hoffmann and M. Gradzielski, Complexes of oppositely charged polyelectrolytes and surfactants – recent developments in the field of biologically derived polyelectrolytes, Soft Matter, 2013, 9, 3896 RSC.
- A. Mezei, K. Pojják and R. Mészáros, Nonequilibrium Features of the Association between Poly(vinylamine) and Sodium Dodecyl Sulfate: The Validity of the Colloid Dispersion Concept, J. Phys. Chem. B, 2008, 112, 9693–9699 CrossRef CAS PubMed.
- F. Schulze-Zachau and B. Braunschweig, CnTAB/Polystyrene Sulfonate Mixtures at Air-Water Interfaces: Effects of Alkyl Chain Length on Surface Activity and Charging State, Phys. Chem. Chem. Phys., 2019, 21, 7847–7856 RSC.
- F. Schulze-Zachau and B. Braunschweig, Structure of Polystyrenesulfonate/Surfactant Mixtures at Air-Water Interfaces and Their Role as Building Blocks for Macroscopic Foam, Langmuir, 2017, 33, 3499–3508 CrossRef CAS PubMed.
- E. Guzmán, S. Llamas, A. Maestro, L. Fernández-Peña, A. Akanno, R. Miller, F. Ortega and R. G. Rubio, Polymer-Surfactant Systems in Bulk and at Fluid Interfaces, Adv. Colloid Interface Sci., 2016, 233, 38–64 CrossRef PubMed.
- E. Tyrode and J. Hedberg, A Comparative Study of the CD and CH Stretching Spectral Regions of Typical Surfactants Systems Using VSFS: Orientation Analysis of the Terminal CH3 and CD3 Groups, J. Phys. Chem. C, 2012, 116, 1080–1091 CrossRef CAS.
- R. N. Ward, D. C. Duffy, P. B. Davies and C. D. Bain, Sum-Frequency Spectroscopy of Surfactants Adsorbed at a Flat Hydrophobic Surface, J. Phys. Chem., 1994, 98, 8536–8542 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Burlington, MA, 3rd edn, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tensiometry, analysis of neutron reflectivity profiles, SFG details, reversibility of E/Z switching, details on foam formation. See DOI: https://doi.org/10.1039/d3nr05414d |
| This journal is © The Royal Society of Chemistry 2024 |