
Low-temperature synthesis of porous organic polymers with donor–acceptor structure and β-ketoenamine for photocatalytic oxidative coupling of amines†
Rusong
Sang
ab,
Yezi
Hu
a,
Zewen
Shen
a,
Guixia
Zhao
 *a,
Junrong
Yue
*b and
Xiubing
Huang
*c
*a,
Junrong
Yue
*b and
Xiubing
Huang
*c
aMOE Key Laboratory of Resources and Environmental System Optimization, College of Environmental Science and Engineering, North China Electric Power University, Beijing 102206, PR China. E-mail: guixiazhao@ncepu.edu.cn
bState Key Laboratory of Multi Phase Complex System, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, PR China. E-mail: jryue@ipe.ac.cn
cBeijing Advanced Innovation Center for Materials Genome Engineering, Beijing Key Laboratory of Function Materials for Molecule & Structure Construction, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, PR China. E-mail: xiubinghuang@ustb.edu.cn
First published on 15th April 2024
Abstract
In light of the widespread use of fossil fuels and the resulting environmental pollution, it is crucial to develop efficient photocatalysts for renewable energy applications that utilize visible light. Organic photocatalysts based on β-ketoenamine offer several advantages, including facile preparation, high stability, structural controllability, and excellent photovoltaic properties. However, in previous studies, the synthesis of porous organic polymers (POPs) often involved long, high-temperature processes. In this study, POPs with donor (D)–acceptor (A) structure were constructed by utilizing various branched bridging groups and 2,4,6-triformylphloroglucinol, across multiple temperature gradients. Through adjustments in hydrothermal temperature, we successfully synthesized a series of POPs with varying enol–keto structure ratios. Among these POPs, the dimethoxybenzidine-POPs (DMDPOPs) with methoxy electron-rich branched chains exhibited superior photovoltaic performance, electron transfer rate, and photocatalytic activity compared to the dihydroxybenzidine-POPs (DHDPOPs) with electron-deficient hydroxyl branched chains. Notably, DMDPOP-30 demonstrated outstanding performance, achieving a conversion rate of 98% within 3 h. Additionally, other POPs exhibited favorable conversions (90%), further confirming the feasibility of this synthetic approach. Moreover, the synthesis of DMDPOP-30 was achieved under mild conditions at room temperature, highlighting its significant potential for practical applications.
1. Introduction
Selective oxidation of raw organic materials into high-value chemicals plays a crucial role in the production of biomedicine, dyes, chemical intermediates, perfumes, and various industrial products.1 To relieve the substantial consumption of fossil energy and the resulting environmental pollution, there is a growing need for cleaner and more environmentally sustainable production methods.2–4 Especially, in response to the increasing importance of renewable solar energy research, scientists have made significant progress in developing a wide range of high-performance, stable, and recyclable photocatalysts for organic synthesis.5,6 In particular, imines, which serve as crucial intermediates in the pharmaceutical and biological domains, have attracted considerable interest, particularly in the context of photocatalytic aerobic oxidative coupling of amines.7–9 While traditional semiconductor materials such as TiO2 and C3N4 have been applied in these areas, they have certain limitations, including low light utilization efficiency, rapid recombination of electron–hole pairs, and limited structural adaptability.10,11 Porous organic polymers (POPs) represent a class of meticulously designed porous polymeric materials formed through covalent bonding. POPs offer the advantage of versatile organic building block designs, enabling the achievement of a substantial surface area, a tailored pore structure, and specific functionalization.12,13 As a result, they have garnered extensive attention and become a focal point of research.14–16Organic photocatalysts featuring β-ketoenamine as their core have gained significant attention from researchers due to their ease of synthesis, high stability, tunable structure, and impressive photovoltaic properties.17–19 This structural motif has found widespread application in covalent organic frameworks (COFs), which are metal-free crystalline organic porous materials. Research on the photocatalytic capabilities of these COFs is growing, with interest in various applications such as photocatalytic organic degradation, CO2 reduction, hydrogen production, and organic conversion.20–24 However, the conventional synthesis of COFs typically involves solvothermal reactions at elevated temperatures (120–150 °C) often under vacuum or inert atmospheres.25,26 In light of the increasing emphasis on sustainable synthesis and cost-efficiency, there is a pressing need to identify catalysts that can be prepared under low temperatures and straightforward conditions.
Enhancing photocatalytic performance involves constructing a Donor (D)–Acceptor (A) structure within the photocatalyst, facilitating the spontaneous transfer of charges from D to A.27,28 Previous research has demonstrated that D–A structures effectively reduce the band gap, enhance charge transfer rates, and prolong carrier lifetimes.29–31 Additionally, incorporating π units can impact the electron acceptor domain, expediting charge transfer and the migration of photoexcited electrons.32,33 The aim of this study was to develop β-ketoenamine-based POPs with distinct electron-rich and electron-deficient branched π-units to enhance benzylamine oxidative coupling. Our approach involved modulating thermodynamic and kinetic factors, which represents a significant stride towards efficient and sustainable photocatalytic systems.
2. Experimental section
2.1. Reagents and materials
All reagents were purchased from commercial sources. Specifically, 2,4,6-triformylphloroglucinol, 3,3′-dimethoxybenzidine, 3,3′-dihydroxybenzidine, benzylamine, acetonitrile, chlorobenzene, silver nitrate and KI were purchased from Aladdin, whereas N-benzylidenebenzylamine, homotrimethylbenzene, 1,4-dioxane and tert-butanol were purchased from Macklin. All reagents were of analytical grade and were used as received without further purification.2.2. Preparation
2.3. Photocatalytic tests
In the photocatalytic reaction, the reactor was initially purged with oxygen for 30 minutes to displace the air and ensure an oxygen-rich environment for the reaction. Subsequently, a mixture containing 5 mg of the as-prepared POPs, 0.2 mmol of benzylamine, 1 mL of acetonitrile as the solvent, and 10 μl of chlorobenzene as an internal standard was subjected to 5 minutes of ultrasonication to achieve thorough mixing. This prepared mixture was then introduced into the reactor for the reaction. The reaction was conducted under the illumination of a blue LED (460 ± 15 nm, 10 W) lamp as the light source.2.4. Characterizations
Powder X-ray diffraction (PXRD) measurements were conducted using an Empyrean instrument, and the spectra were collected in the range of 2.5 to 30° at room temperature. Fourier transform infrared spectroscopy (FTIR) data was acquired across the range of 600 to 4000 cm−1 using a TENSOE 27 FTIR spectrometer with KBr pellets. All spectra were collected under ambient atmospheric conditions, and the transmittance (%) was plotted against wavenumbers. Solid-state NMR experiments were conducted using a Bruker WB Avance II 400 MHz spectrometer. The 13C CP/MAS NMR spectra were obtained using a 4 mm double-resonance MAS probe, which operated at a sample spinning rate of 10.0 kHz. X-ray photoelectron spectroscopy (XPS) is tested using the AXIS SUPRA + X-ray photoelectron spectrometer model. The N2 isotherms and specific surface areas were determined at 77 K using a Micromeritics ASAP 2020 HD88 instrument employing the Brunauer–Emmett–Teller (BET) model. Before N2 physisorption, the samples were degassed in a vacuum (<1 × 10−5 bar) at 200 °C for 12 hours in the Micromeritics system. Additionally, porosity distributions were determined using the nonlocal density functional theory (NLDFT) method. Thermal field emission scanning electron microscopy (TFE-SEM) measurements were performed using a JSM-7610F instrument, operating at an acceleration voltage ranging between 5 and 10 kV. The UV–vis diffuse reflectance spectra (UV–vis DRS) of the POPs were measured within the range of 300 to 800 nm using a Cary5000 UV–vis spectrophotometer (Agilent) equipped with a diffuse reflectance measurement accessory, employing BaSO4 as a reflectance standard. Photoelectron Spectroscopy (UPS) is tested using the Thermo ESCALAB XI+ spectrometer model. Thermogravimetric analysis (TGA) spectra were recorded using a TG DTA7300 thermogravimeter under an N2 atmosphere, with a heating rate of 10 °C min−1 from 30 °C to 800 °C. Upon completion, the reaction mixture underwent analysis via gas chromatography (GC), followed by product identification utilizing gas chromatography–mass spectrometry (GC–MS) using an Agilent 7890A instrument.2.5. Electrochemical measurements
Electrochemical measurements were performed using an HC instrument in a three-electrode electrochemical cell connected to an electrochemical workstation. To prepare the working electrodes, 2 mg of the catalyst was dissolved in 1.0 mL of absolute ethanol, followed by the addition of a 50 μL mixed solution containing Nafion. The solution was then sonicated for 30 minutes, and 50 μL of the resulting mixture was drop-cast onto the ITO surface, covering an illuminated area of π × (1)2 cm2. Afterward, the samples were dried at room temperature with supplemental infrared irradiation. Subsequently, 0.1 M Na2SO4 was introduced as the aqueous electrolyte. The reference and counter electrodes were the Ag/AgCl electrode and platinum wire, respectively. In addition, twelve blue LEDs emitting light at a wavelength of 460 ± 15 nm were positioned 2 cm away from the photoelectrochemical cell as the light source. Photocurrent measurements were conducted with light on–off cycles of 30-second intervals and a scan rate of 100 mV s−1. Technical terminology was defined upon initial mention. Mott–Schottky measurements were performed under dark conditions at frequencies of 500 Hz and 1000 Hz. Furthermore, electrochemical impedance spectroscopy (EIS) was conducted under light irradiation at a bias potential of +0.5 V.3. Results and discussion
Scheme 1 illustrates the synthesis of the crystalline framework using 3,3′-dimethoxybenzidine with electron-rich side chains and 3,3′-dihydroxybenzidine with electron-deficient side chains as bridging units, reacting with 2,4,6-triformylphloroglucinol. This synthetic process involves a reversible Schiff base reaction that generates the crystalline framework and an irreversible enol–keto tautomeric isomerization, enhancing stability by forming enol–keto structures within the POPs. The synthesis of distinct POPs under various temperature gradients was achieved through controlling reaction temperature. | ||
| Scheme 1 Designing POP structures with electron-rich π-electron (i.e., DMDPOP-X) and electron-deficient π-electron (DHDPOP-X) systems. | ||
XRD analysis was performed to investigate the crystallinity of different POP catalysts. In the case of DMDPOP-30, a prominent diffraction peak is observed at 2θ = 2.98°, corresponding to the (100) crystallographic plane (Fig. 1a). This indicates a high degree of crystallinity and a well-ordered structural sequence for efficient electron transport and transfer. The Materials Studio software was used to create a simulated material with an AA stacking structure. The lattice parameters were set to a = b = 30.8 Å, c = 3.48 Å, and α = β = 90°, γ = 120° to enable a detailed analysis of the crystal structure. Table S1† provides comprehensive crystal structure information and XRD pattern comparisons between experimental and simulated data, demonstrating their high consistency. The experimental peaks at 3.12° and 14.2° were matched with the simulated crystal faces, corresponding to the (100) and (210) planes, respectively. Generally, all POPs exhibit characteristic peaks around 5°, confirming their crystalline nature as polymers (Fig. 1a). Additionally, the presence of stable diffraction peak spectra suggests that all POPs possess a stable and ordered structure. The formation of packet peaks around 26°, attributed to the π–π stacking interaction, further indicates an organic polymeric structure formation. However, the presence of packet peaks also suggests that the POPs are not highly crystalline.
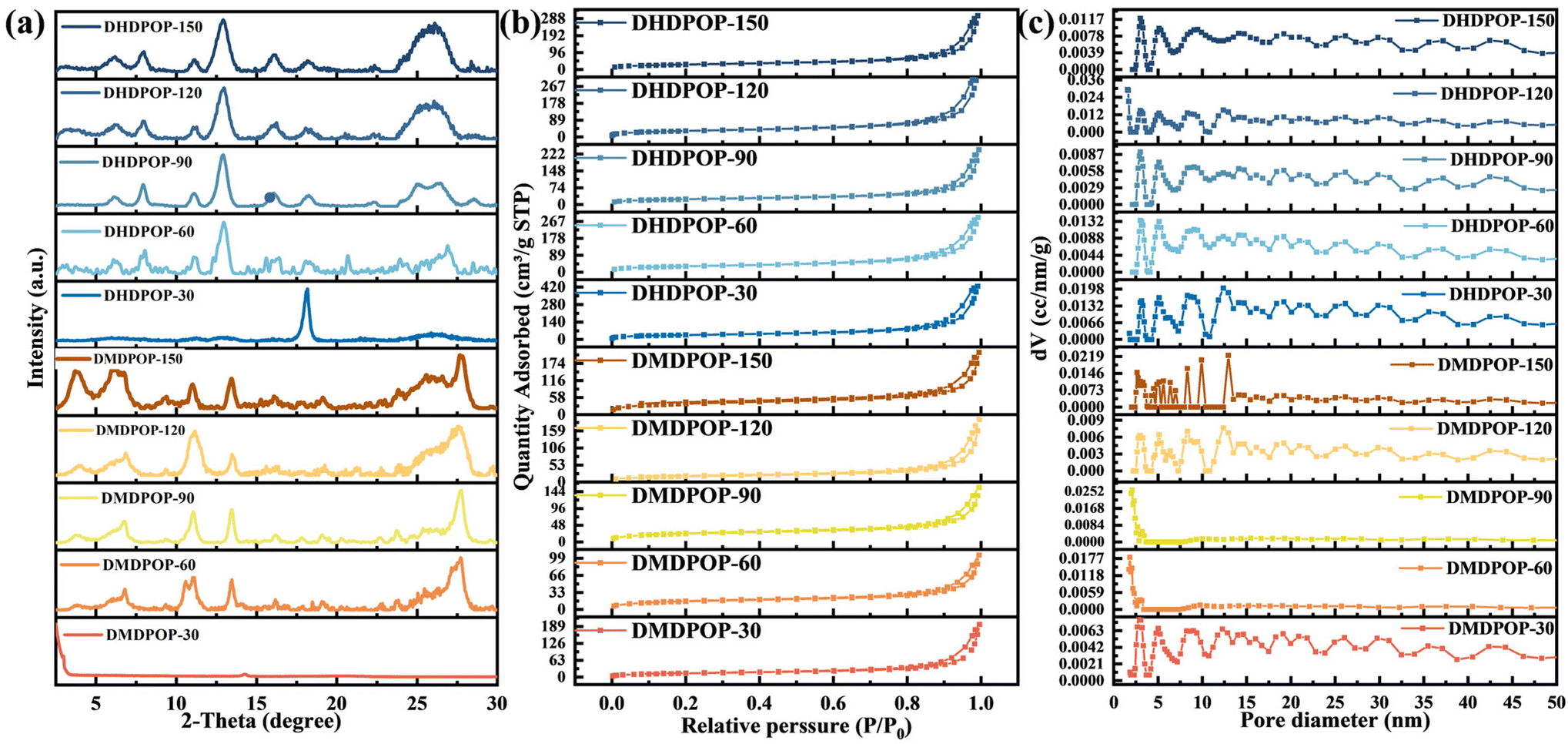 | ||
| Fig. 1 XRD patterns of DMDPOP-X and DHDPOP-X (a); N2 adsorption–desorption isotherms of DMDPOP-X and DHDPOP-X (b); pore size distribution of DMDPOP-X and DHDPOP-X (c). | ||
The Brunauer–Emmett–Teller (BET) specific surface areas of DMDPOP-X were 52.4, 57.1, 86.9, 214.7, and 331.8 m2 g−1, respectively (Fig. 1b). Synthesizing DMDPOP-X under different temperature gradients revealed a correlation between higher temperatures and larger specific surface areas. This suggests that thermodynamically driven synthesis of electron-rich branched POPs yields more microporous structures with increased active sites. The lower specific surface area of DMDPOP-30 could be attributed to the collapse of the pore structure caused by constant stirring during synthesis. The specific surface areas of DHDPOP-X with the electron-deficient hydroxyl group as the branched chain were 129, 110, 85, 115.3, and 101 m2 g−1, respectively (Fig. 1b). These values exhibited similar curves and a relatively balanced distribution of specific surface areas compared to those of POPs with electron-rich branched chains. This suggests that different branched chains influence the microstructure of POPs in terms of relative specific surface area and microporous structure.34 The synthesized series of POPs in this study exhibited lower specific surface areas. This was primarily due to constraints on crystal growth, such as synthesis temperature and reaction time. Lower synthesis temperatures and shorter reaction periods limited crystal growth, resulting in reduced surface areas. Furthermore, the specific surface areas of the materials were significantly impacted by their chemical composition and structural properties. The comparison between DMDPOPs and DHDPOPs illustrated this. To increase the surface area, it is recommended to use strategies such as raising the reaction temperature, extending the reaction time, selecting specific bridging groups, and performing post-synthesis steps such as washing, drying, and activation. These measures offer a viable pathway for enhancing the surface area of POPs. Additionally, the pore size distribution diagram indicates that the polymer's pore channels primarily consist of micropores and mesopores (Fig. 1c).
FTIR analysis provides further insights into the structure and chemical composition of DMDPOP-X and DHDPOP-X. Characteristic peaks at 1268, 1580 and 1612 cm−1 indicate the presence of C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and CO stretching bonds, confirming the existence of the keto form (Fig. 2a).35,36 Additionally, the bending vibration peak at 1297 cm−1 corresponding to C–OH suggests the presence of the enol form.37 The weakening of the N–H stretching band at 3500–3300 cm−1 indicates the condensation of amino groups in aniline as a bridging group. These signals confirm the successful synthesis of β-ketoenamine linkages and the formation of the fundamental bridging skeleton of POPs. The peak for stretching of aromatic CC and C–H are at 1450 cm−1 and 798 cm−1, respectively.38 Furthermore, a slight vibration at 2885 cm−1 is observed, indicating the asymmetric stretching of the methoxy C–H bond in DMDPOP. DMDPOP-30 was synthesized at room temperature and exhibits remarkable catalytic properties, holding great potential for various applications. The solid-state 13C NMR spectrum of DMDPOP-30 is presented in Fig. 2b. Notably, the keto form displays a characteristic peak at 180.8 ppm, representing the typical CO bond,38 while the enol form exhibits a characteristic peak at 175 ppm, indicating the presence of the structural C–OH bond.39,40 These findings further support the notion that the fundamental framework of POPs comprises the keto–enol form, which aligns with the results obtained from the FTIR analysis.
C, and CO stretching bonds, confirming the existence of the keto form (Fig. 2a).35,36 Additionally, the bending vibration peak at 1297 cm−1 corresponding to C–OH suggests the presence of the enol form.37 The weakening of the N–H stretching band at 3500–3300 cm−1 indicates the condensation of amino groups in aniline as a bridging group. These signals confirm the successful synthesis of β-ketoenamine linkages and the formation of the fundamental bridging skeleton of POPs. The peak for stretching of aromatic CC and C–H are at 1450 cm−1 and 798 cm−1, respectively.38 Furthermore, a slight vibration at 2885 cm−1 is observed, indicating the asymmetric stretching of the methoxy C–H bond in DMDPOP. DMDPOP-30 was synthesized at room temperature and exhibits remarkable catalytic properties, holding great potential for various applications. The solid-state 13C NMR spectrum of DMDPOP-30 is presented in Fig. 2b. Notably, the keto form displays a characteristic peak at 180.8 ppm, representing the typical CO bond,38 while the enol form exhibits a characteristic peak at 175 ppm, indicating the presence of the structural C–OH bond.39,40 These findings further support the notion that the fundamental framework of POPs comprises the keto–enol form, which aligns with the results obtained from the FTIR analysis.
 | ||
| Fig. 2 FT-IR spectra of DMDPOP-X and DHDPOP-X (a); 13C CP-MAS solid-state NMR spectra of DMDPOP-30 (b). | ||
XPS was employed to investigate the conformational ratio of keto to enol in the prepared DMDPOP-X samples. The β-ketoenamine POPs tested are composed of C, N and O elements as shown by the total XPS spectra (Fig. 3a). Fig. 3b–f illustrate the peaks observed at approximately 400.3 eV and 399.8 eV, which correspond to the keto form (–C–NH–) and the enol form (–CN–),33 respectively. By analyzing the peak intensities, it is possible to calculate the relative contents of the keto form and enol form in a semi-quantitative manner. Kinetic control during the room temperature stirring synthesis of DMDPOP-30 results in nearly equal proportions of the keto form and enol form (Fig. 3b). As the hydrothermal synthesis temperature increases from 60 °C to 120 °C, the proportion of the keto form in DMDPOPs gradually increases. However, at a hydrothermal temperature of 150 °C, the proportion of the keto form decreases and approaches equilibrium with the enol form (Fig. 3c–f). The C 1s XPS spectrum of DMDPOP-X exhibits six distinct peaks at specific binding energies: 284.6 eV (CC), 285.2 eV (C–C), 285.8 eV (CN), 286.2 eV (C–N), 287.0 eV (CO), and 288.4 eV (C–O),26 as detailed in Fig. S1.† Furthermore, the O 1s peaks predominantly arise from methoxy branched chain C–O–C bonds at around 533.6 eV, enol–imine C–OH bonds at around 533.2 eV, and ketoenamine CO bonds at around 530.7 eV (Fig. S2†).41 These observations suggest that the relative contents of the keto form and enol form are influenced by both thermodynamics and kinetics, eventually reaching a relative equilibrium state at high temperatures.
 | ||
| Fig. 3 (a) Full XPS and high-resolution XPS N 1s spectra of DMDPOP-30 (b), DMDPOP-60 (c), DMDPOP-90 (d), DMDPOP-120 (e) and DMDPOP-150 (f). | ||
The surface morphologies of the β-ketoenamine POPs were examined using SEM. The results indicate distinct morphologies for different POPs. DMDPOP-30 exhibits a coral-like shape; DMDPOP-60 shows a stacked lamellar structure; DMDPOP-90 and DMDPOP-120 display a fine-fibrous state; and DMDPOP-150 exhibits a mixed growth of lamellar and fibrous structures (Fig. 4a–e). These observations suggest a correlation between the microstructure of electron-rich branched POPs during hydrothermal synthesis and the enol–keto structures. Specifically, the enol-form tends to adopt a lamellar structure, while the keto-form tends to exhibit a fibrous morphology (Fig. 4f–j). On the other hand, the morphologies of electron-deficient branched POPs are more similar, both of which have a fiber-interlaced microstructure. This similarity in microstructural features also corresponds to the close specific surface area results. The variance in microscopic morphology between the two types of POPs may be ascribed to the effect of the nature of the branched chains on π–π superpositions.
 | ||
| Fig. 4 SEM images of DMDPOP-30 (a), DMDPOP-60 (b), DMDPOP-90 (c), DMDPOP-120 (d), DMDPOP-150 (e), DHDPOP-30 (f), DHDPOP-60 (g), DHDPOP-90 (h), DHDPOP-120 (i) and DHDPOP-150 (j). | ||
UV-vis diffuse reflectance spectroscopy (UV-vis DRS) is a technique used to assess the light absorption capacity of POPs. The absorption of visible light in the POPs is attributed to intramolecular charge transfer and the conjugated structure of π–π stacking.42,43 Both DMDPOP-X and DHDPOP-X exhibit significant absorption capabilities in the visible light range. Notably, DMDPOP-30 demonstrates exceptional absorption in the 500–800 nm wavelength range compared to other electron-rich branched POPs, enabling it to adapt to more complex lighting conditions (Fig. 5a and b). By employing the Kubelka–Munk function, the energy gaps (Eg) of DMDPOP-X are determined as follows: 1.83, 2.03, 2.12, 2.00, and 2.07 eV, individually. These results indicate that the keto-form in electron-rich branched POPs can widen the band gap. Conversely, the energy gaps of DHDPOP-X are 1.74, 1.87, 2.03, 1.83 and 2.06 eV, respectively (Fig. 5c and d). Comparatively, POPs with hydroxyl-deficient electron-branched chains exhibit narrower band gaps and superior absorbance, which can be attributed to keto–enol tautomerism and the electron push–pull effect. Upon analysis of the Tauc plots, a linear relationship between (αhν)2 and hν was observed, confirming that the POPs in question possess the characteristics of direct bandgap semiconductors.44 This property enhances the absorption of visible light, making them useful for applications in the fields of photovoltaics and photocatalysis. Fig. S3† shows the valence band top energies of the materials under vacuum conditions, which were determined through UPS spectra analysis. The valence band top energies for the DMDPOP-X and DHDPOP-X series are 4.41, 5.18, 4.39, 4.25, 4.86, 4.15, 4.43, 4.08, 4.96, and 4.66 eV, respectively. However, it is worth noting that UPS measurements are conducted in a high-vacuum environment to prevent interactions between electrons and air molecules. In contrast, electrochemical tests are performed in electrolyte solutions, which more closely simulate actual application conditions. Therefore, future studies will continue to use electrochemical methods to determine the band structure of these materials.
 | ||
| Fig. 5 UV-vis spectra (a) and Tauc plots (b) of DMDPOP-X; UV-vis spectra (c) and Tauc plots(d) of DHDPOP-X; the transient photocurrent densities (e) and EIS spectra (f) of POPs. | ||
To investigate the impact of electron-rich units and electron-deficient units on the charge separation and migration, we examined the optoelectronic properties of DMDPOP-X and DHDPOP-X, among which, DMDPOP-X demonstrated superior photocurrent response and cyclic stability, indicating its excellent ability to generate and separate photoexcited electron–hole (e−–h+) pairs (Fig. 5e).29,30 Additionally, in electrochemical impedance spectroscopy (EIS), DMDPOP-X exhibited smaller semicircle diameters, suggesting a higher efficiency of charge migration (Fig. 5f). The aforementioned observations may be ascribed to the electron-donating impact of the methoxyl groups, which promotes intensive electron delocalization and facilitates charge migration.
To estimate the conduction band (CB) positions of POPs, electrochemical tests were conducted to obtain Mott–Schottky curves (Fig. S4†). All the curves exhibited positive slopes, indicating that the POPs are n-type semiconductors with flat band potentials (Vfb) relative to Ag/AgCl. The normalized hydrogen electrode (NHE) was calculated using the equation (ENHE = EAg/AgCl + 0.22). The CB levels of DMDPOP-X and DHDPOP-X were determined to be −0.15, −0.04, −0.02, −0.08, −0.12, −0.02, −0.04, −0.40, −0.05 and −0.22 V (vs. NHE), individually. The corresponding VBs were measured as 1.68, 1.99, 2.10, 1.92, 1.95, 1.72, 1.83, 1.63, 1.78, and 1.84 V (vs. NHE), as summarized in Fig. 6. The holes generated by DMDPOP-X and DHDPOP-X are sufficient for the oxidation of benzylamine.
 | ||
| Fig. 6 Calculated band structures of the ten POPs. | ||
Benzylamines find extensive applications as valuable intermediates in the pharmaceutical and biological fields. Photocatalytic selective oxidative coupling of Benzylamines represents a green and mild production method. In this study, we conducted photocatalytic experiments using DMDPOP-X and DHDPOP-X catalysts further to investigate their differences (Fig. 7a). The electron-rich branched DMDPOP-X catalyst exhibited a relatively higher average conversion compared to DHDPOP-X. Additionally, all POPs demonstrated a selectivity of over 97% in generating oxidative coupling products. This can be attributed to the superior photocurrent response and charge migration efficiency of the electron-rich branched POPs. Notably, DMDPOP-30, DMDPOP-60, and DMDPOP-90 displayed exceptional conversion rates of 98%, 90%, and 90%. The high conversion rates observed are independent of the particular surface area and pore configuration. From a chemical structure perspective, the conversion rates are related to keto–enol tautomerism and the electron push–pull effect. When the ratio of keto–enol tautomerism is more balanced or the keto-form dominates, it facilitates electron transfer, thereby promoting the reaction.
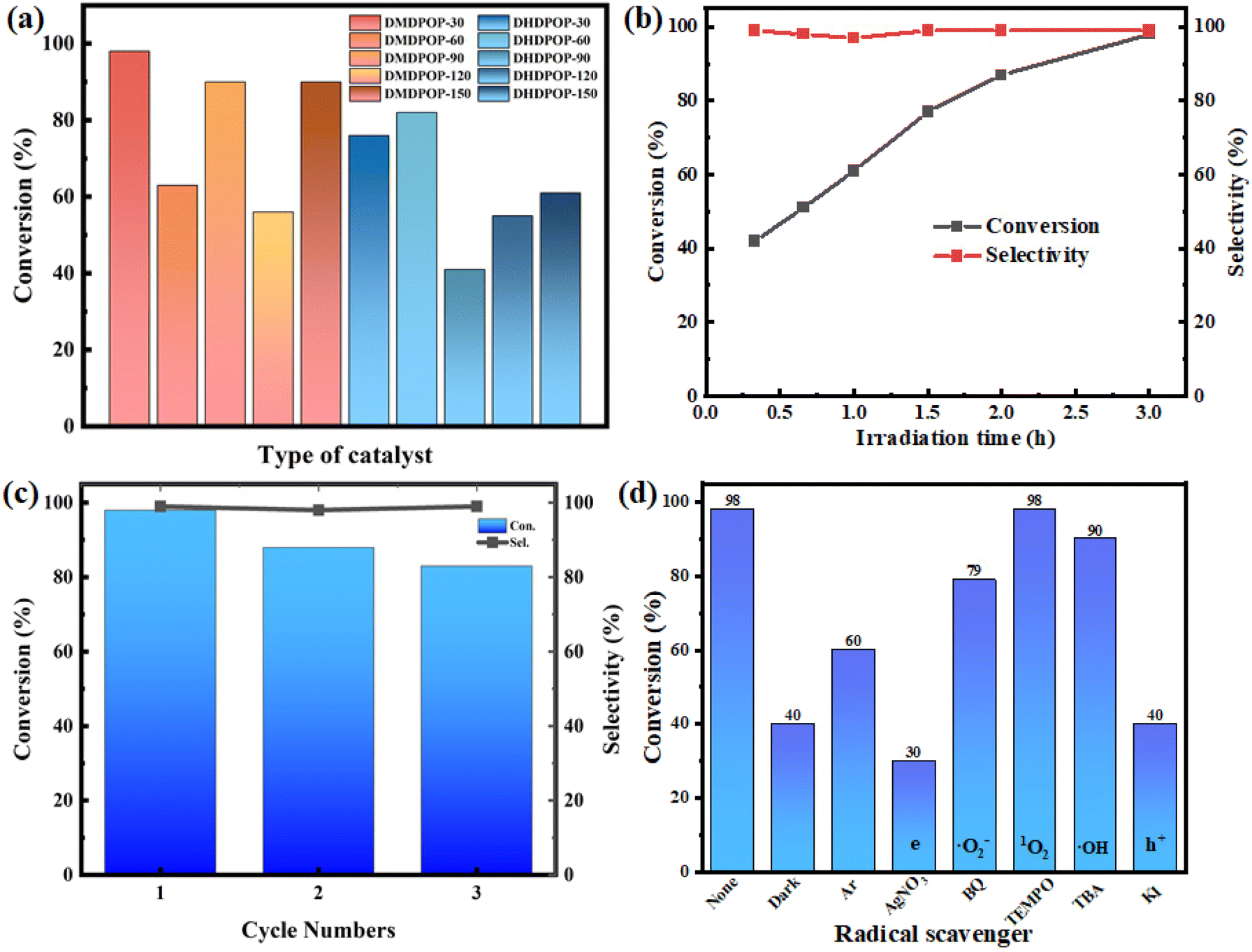 | ||
| Fig. 7 (a) Photocatalytic reaction conversion; (b) time-dependent curve; (c) the recycling results of DMDPOP-30 photocatalyst; (d) quenching experiments of photocatalytic aerobic oxidation of benzylamine. Reaction conditions: DMDPOP-30 (5 mg), benzylamine (0.2 mmol), blue LEDs (460 ± 15 nm), 1 atm O2, CH3CN (1 mL), 3 h. | ||
DMDPOP-30 is highly regarded for its mild synthesis conditions and exceptional photoelectric and catalytic properties. In the current study, the reaction kinetics were investigated using DMDPOP-30 as a catalyst. As depicted in Fig. 7b, the conversion consistently increases with reaction time, aligning with a level 0 kinetic model. Furthermore, Fig. 7c demonstrates that even after three cycles, DMDPOP-30 maintains a conversion rate of over 80%, indicating its commendable cycling performance and reaction stability. The FT-IR and SEM images post-cycling reveal minimal alterations in the chemical structure and microstructure compared to pre-cycling (Fig. S5 and S6†). In essence, the observations indicate that DMDPOP-30 exhibits commendable chemical stability. Additionally, the thermal stability of DMDPOP-30 is evident in Fig. S7,† where the catalyst experiences a weight loss of only approximately 10% when heated to around 300 °C, showing its favorable thermal stability. Considering its high crystallinity, mild synthesis conditions, excellent performance, good cycling performance, and thermal stability, DMDPOP-30 exhibits significant potential for various applications.
To elucidate the mechanism of selective oxidation using DMDPOP-30, a series of comparative experiments were conducted (Fig. 7d). Under dark conditions, the conversion of the reaction significantly decreased, highlighting the importance of light in the reaction. The conversion of 60% in the absence of oxygen implies that oxygen is not a prerequisite for the reaction to occur. To investigate the role of reactive oxygen species (ROS), various agents were added to the reaction. The addition of 1,4-benzoquinone (BQ, O2˙− scavenger), TEMPO (1O2 scavenger), and tertbutyl alcohol (TBA, ˙OH scavenger) did not effectively inhibit the reaction, suggesting that reactive oxygen species do not primarily drive the reaction. However, the addition of AgNO3 and KI, which act as photogenerated electron and hole scavengers respectively, effectively inhibited the reaction. Fig. 8 illustrates the proposed mechanism of selective oxidative coupling of benzylamine by DMDPOP-30 photocatalyst. Within the valence band (VB), benzylamine undergoes oxidation, leading to the formation of nitrogen-centered radical cations. Through a proton transfer process, the hydrogen protons within these nitrogen-centered radical cations readily undergo cleavage, resulting in the generation of carbon-centered radicals. These carbon-centered radicals then react with the existing nitrogen-centered free radicals, ultimately yielding N-benzylidene benzylamines.
 | ||
| Fig. 8 Proposed mechanism of photocatalytic benzylamine oxidation over DMDPOP-30. | ||
To expand the application potential of DMDPOP-30, the oxidation of other amines with diverse substituents was also conducted. The results demonstrate excellent conversion for all target substrates, indicating a consistent pattern across different substituents. Particularly, the para-methoxy substituent exhibits superior enhancement to the photocatalytic performance compared to the neighboring methoxy substituent, attributed to the spatial site resistance effect (Table 1, entries 1 and 2). Furthermore, the conversion rate increases with the electronegativity of halogen substituents, owing to the varying electron density electronic effect of different substituents on the benzyl group (Table 1, entries 3–5). Additionally, amines containing heteroatom N also display enhanced conversion (Table 1, entry 6), further emphasizing the universal efficacy of DMDPOP in the photocatalytic oxidative coupling process of diverse amines.
| Entry | Substrate | T (h) | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|
| Reaction conditions: DMDPOP-30 (5 mg), substituted benzylamine (0.2 mmol), blue LEDs (460 ± 15 nm), 1 atm O2, CH3CN (1 mL). | ||||
| 1 | 4-Methoxybenzylamine | 3.5 | 98 | 98 |
| 2 | 2-Methoxybenzylamine | 3.5 | 92 | 97 |
| 3 | 4-Methylbenzylamine | 3.5 | 99 | 97 |
| 4 | 4-Fluorobenzylamine | 3.5 | 99 | 99 |
| 5 | 4-Bromobenzylamine | 3.5 | 92 | 98 |
| 6 | 2-Picolylamine | 3.5 | 84 | 99 |
4. Conclusions
In this study, we successfully synthesized a series of β-ketamine-based POPs with a donor–acceptor structure. The construction of these POPs involved both kinetic and thermodynamic regulation, utilizing different electron-rich and electron-deficient branched bridging motifs. The resulting POPs exhibited diverse microscopic morphologies, varying enol–keto structural ratios, and distinct optoelectronic properties. Comparative analysis revealed that the electron-rich DMDPOPs outperformed the electron-deficient DHDPOPs in terms of catalytic performance, photoelectric response, and charge migration effect. This finding highlights the potential of electron-rich DMDPOPs as an effective platform for constructing and functionalizing POPs. Notably, DMDPOP-30 demonstrated remarkable performance, achieving 98% conversion and 99% selectivity to benzylamine under 3 hours of light exposure at room temperature. Moreover, the simple and mild synthesis conditions of DMDPOP-30 make it highly promising for practical applications. Our experimental results demonstrated that the chemical structure of β-ketoenamine POPs could be modulated by the combined effects of kinetics and thermodynamics. This structural manipulation led to improved photoelectric properties and enhanced electron transfer rates, providing strong support for the environmentally friendly synthesis of POPs for various applications.Author contributions
R. S. drafted the manuscript. G. Z., X. H., and J. Y. edited the manuscript. All authors analyzed the data, and reviewed and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22176054, 22306060), Fundamental Research Funds for the Central Universities (No. FRFEYIT-23-07), the 2022 “Chunhui Program” Cooperative Scientific Research Project of Ministry of Education of China (No. 20220436), and MOE Key Laboratory of Resources and Environmental System Optimization, College of Environmental Science and Engineering, North China Electric Power University (No. KLRE-KF202201).References
- Z. Wu, Y. Li, C. Zhang, X. Huang, B. Peng and G. Wang, Chem Catal., 2022, 2, 1009–1045 CrossRef CAS.
- D. Tang, G. Lu, Z. Shen, Y. Hu, L. Yao, B. Li, G. Zhao, B. Peng and X. Huang, J. Energy Chem., 2023, 77, 80–118 CrossRef CAS.
- X. Huang, K. Zhang, B. Peng, G. Wang, M. Muhler and F. Wang, ACS Catal., 2021, 11, 9618–9678 CrossRef CAS.
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11, 2003216 CrossRef CAS.
- G. Lu, F. Chu, X. Huang, Y. Li, K. Liang and G. Wang, Coord. Chem. Rev., 2022, 450, 214240 CrossRef CAS.
- M.-Y. Qi, M. Conte, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2022, 16, 17444–17453 CrossRef CAS PubMed.
- F. Chu, Y. Hu, K. Zhang, X. Li, G. Zhao, X. Huang and G. Wang, J. Colloid Interface Sci., 2023, 634, 159–168 CrossRef CAS PubMed.
- S. Wu, Y.-F. Zhang, H. Ding, X. Li and X. Lang, J. Colloid Interface Sci., 2022, 610, 446–454 CrossRef CAS PubMed.
- X. Li, K. Zhang, X. Huang, Z. Wu, D. Zhao and G. Wang, Nanoscale, 2021, 13, 19671–19681 RSC.
- X. Li, S. Lyu and X. Lang, Environ. Res., 2021, 195, 110851 CrossRef CAS PubMed.
- Y. Gao, T. Song, X. Guo, Y. Zhang and Y. Yang, Green Carbon, 2023, 1, 105–117 CrossRef.
- K. Zhang, X. Dong, B. Zeng, K. Xiong and X. Lang, J. Colloid Interface Sci., 2023, 651, 622–632 CrossRef CAS PubMed.
- Y. Zhang, G. Lu, D. Zhao and X. Huang, Mater. Chem. Front., 2023, 7, 4782–4809 RSC.
- H. Chen, H. S. Jena, X. Feng, K. Leus and P. Van Der Voort, Angew. Chem., Int. Ed., 2022, 61, e202204938 CrossRef CAS PubMed.
- Y. Liu, H. Tan, Y. Wei, M. Liu, J. Hong, W. Gao, S. Zhao, S. Zhang and S. Guo, ACS Nano, 2023, 17, 5994–6001 CrossRef CAS PubMed.
- W. Huang, Y. Hu, Z. Qin, Y. Ji, X. Zhao, Y. Wu, Q. He, Y. Li, C. Zhang, J. Lu and Y. Li, Natl. Sci. Rev., 2023, 10, nwac171 CrossRef CAS PubMed.
- Y.-N. Gong, X. Guan and H.-L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- G.-B. Wang, K.-H. Xie, H.-P. Xu, Y.-J. Wang, F. Zhao, Y. Geng and Y.-B. Dong, Coord. Chem. Rev., 2022, 472, 214774 CrossRef CAS.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- Y. Yang, X. Chu, H.-Y. Zhang, R. Zhang, Y.-H. Liu, F.-M. Zhang, M. Lu, Z.-D. Yang and Y.-Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed.
- M. Zhang, X. Wu, Y. Xie, X. Hao, Q. Wang, Y. Zhao, J. Wu and X. Pan, Mater. Chem. Front., 2023, 7, 5399–5405 RSC.
- M. Ma, Y. Yang, Z. Huang, F. Huang, Q. Li and H. Liu, Nanoscale, 2024, 16, 1600–1632 RSC.
- Q. Zhang, M. Chen, Y. Zhang, Y. Ye, D. Liu, C. Xu, Z. Ma, B. Lou, R. Yuan and R. Sa, Nanoscale, 2023, 15, 16030–16038 RSC.
- Y. Chen, J. Qiu, X.-G. Zhang, H. Wang, W. Yao, Z. Li, Q. Xia, G. Zhu and J. Wang, Chem. Sci., 2022, 13, 5964–5972 RSC.
- Z. Wu, X. Huang, X. Li, G. Hai, B. Li and G. Wang, Sci. China: Chem., 2021, 64, 2169–2179 CrossRef CAS.
- C.-X. Liu, D.-L. Pan, Y. Seo, S. Park, J.-L. Kan, J. Y. Koo, W. Choi and E. Lee, ACS Appl. Energy Mater., 2023, 6, 1126–1133 CrossRef CAS.
- X. Zhong, Q. Ling, P. Kuang and B. Hu, Chem. Eng. J., 2023, 467, 143415 CrossRef CAS.
- H. Che, P. Wang, J. Chen, X. Gao, B. Liu and Y. Ao, Appl. Catal., B, 2022, 316, 121611 CrossRef CAS.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y.-B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- F. Yu, Z. Zhu, S. Wang, Y. Peng, Z. Xu, Y. Tao, J. Xiong, Q. Fan and F. Luo, Chem. Eng. J., 2021, 412, 127558 CrossRef CAS.
- Z. Wei, L. Kong, Q. Wang, J. Xu and L. Li, Opt. Mater., 2023, 138, 113704 CrossRef CAS.
- F. Chu, G. Hai, D. Zhao, S. Liu, Y. Hu, G. Zhao, B. Peng, G. Wang and X. Huang, ACS Catal., 2023, 13, 13167–13180 CrossRef CAS.
- C. Li, H. Li, C. Li, X. Ren and Q. Yang, Chin. J. Catal., 2021, 42, 1821–1830 CrossRef CAS.
- W. Zhang, S. Wang, N. Wen, J. Zhao, W. Guo, S. Wu, P. Zhang, Q. Lin, J. Xu and J. Long, Mater. Today Chem., 2022, 26, 101150 CrossRef CAS.
- J. Ming, A. Liu, J. Zhao, P. Zhang, H. Huang, H. Lin, Z. Xu, X. Zhang, X. Wang, J. Hofkens, M. B. J. Roeffaers and J. Long, Angew. Chem., Int. Ed., 2019, 58, 18290–18294 CrossRef CAS PubMed.
- S. Wu, C. Li, Y. Wang, Y. Zhuang, Y. Pan, N. Wen, S. Wang, Z. Zhang, Z. Ding, R. Yuan, W. Dai, X. Fu and J. Long, Angew. Chem., Int. Ed., 2023, 62, e202309026 CrossRef CAS PubMed.
- S. Yang, X. Li, Y. Qin, Y. Cheng, W. Fan, X. Lang, L. Zheng and Q. Cao, ACS Appl. Mater. Interfaces, 2021, 13, 29471–29481 CrossRef CAS PubMed.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed.
- B. P. Biswal, H. A. Vignolo-González, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- W.-R. Cui, C.-R. Zhang, R.-H. Xu, X.-R. Chen, W. Jiang, Y.-J. Li, R.-P. Liang, L. Zhang and J.-D. Qiu, Appl. Catal., B, 2021, 294, 120250 CrossRef CAS.
- X. Li, Q. Gao, J. Aneesh, H.-S. Xu, Z. Chen, W. Tang, C. Liu, X. Shi, K. V. Adarsh, Y. Lu and K. P. Loh, Chem. Mater., 2018, 30, 5743–5749 CrossRef CAS.
- G.-H. Ning, Z. Chen, Q. Gao, W. Tang, Z. Chen, C. Liu, B. Tian, X. Li and K. P. Loh, J. Am. Chem. Soc., 2017, 139, 8897–8904 CrossRef CAS PubMed.
- B. D. Viezbicke, S. Patel, B. E. Davis and D. P. Birnie, Phys. Status Solidi, 2015, 252, 1700–1710 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00391h |
| This journal is © The Royal Society of Chemistry 2024 |