DOI:
10.1039/D4NR00111G
(Paper)
Nanoscale, 2024,
16, 7154-7166
Proceeding of catalytic water splitting on Cu/Ce@g-C3N4 photocatalysts: an exceptional approach for sunlight-driven hydrogen generation†
Received
9th January 2024
, Accepted 29th February 2024
First published on 1st March 2024
Abstract
Increasing energy demands and environmental problems require carbon-free and renewable energy generation systems. For this purpose, we have synthesized efficient photocatalysts (i.e., g-C3N4, Cu@g-C3N4, Ce@g-C3N4 and Cu/Ce@g-C3N4) for H2 evolution from water splitting. Their optical, structural and electrochemical properties were investigated by UV-Vis-DRS, PL, XRD, FTIR, Raman and EIS methods. Their surface morphologies were evaluated by AFM and SEM analyses. Their chemical characteristics, compositions and stability were assessed using XPS, EDX and TGA techniques. Photoreactions were performed in a quartz reactor (150 mL/Velp-UK), whereas hydrogen generation activities were monitored using a GC-TCD (Shimadzu-2014/Japan). The results depicted that Cu/Ce@g-C3N4 catalysts are the most active catalysts that deliver 23.94 mmol g−1 h−1 of H2. The higher rate of H2 evolution was attributed to the active synergism between Ce and Cu metals and the impact of surface plasmon electrons (SPEs) of Cu that were produced during the photoreaction. The rate of H2 production was optimized by controlling various factors, including the catalyst amount, light intensity, pH, and temperature of the reaction mixture. It has been concluded that the current study holds promise to replace the conventional and costly catalysts used for hydrogen generation technologies.
Introduction
80% of the world's energy demands are fulfilled by fossil fuels. The most significant problem that emerges from the burning of fossil fuels is CO2 emission which is the major contributor to global warming.1 At the current moment, there is no technology that can replace fossil fuels. However, scientists are developing materials and strategies to generate clean and renewable energy with minimum environmental consequences.2–4 One potential solution to produce sustainable energy is the concept of the “hydrogen-based economy”. It involves the combustion of molecular hydrogen with no harmful byproduct emission.5,6
Heterogeneous photocatalytic reactions have gained significant attention for addressing environmental and energy challenges.7,8 Currently, hydrogen is being produced through catalytic methane reforming.9 The major drawback associated with this method is that it requires high temperatures. Recently, photocatalytic water splitting has emerged as a promising technology to generate hydrogen by utilizing sunlight.10–12 Various semiconductors such as TiO2,13,14 Ta2O5,15 Zn3V2O8,16 CdS,17 CdSe,18 CeVO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and CdZnS20 have been extensively utilized for photocatalytic hydrogen evolution via water splitting. However, these materials have limitations; for example, TiO2 works only under UV light due to its 3.0–3.2 eV band gap,21 while metal sulphides are unstable due to photocorrosion.22 Graphitic carbon nitride (g-C3N4) has garnered significant attention due to its appealing characteristics, which include a moderate band gap, high thermal stability and a large surface area.23,24 Graphitic carbon nitride photocatalysts can work efficiently under sunlight. Modifications in g-C3N4 can lead to its practical application on a larger scale. The modification strategies include doping, heterojunction formation, bandgap engineering, and the use of cocatalysts. The use of g-C3N4 with effective cocatalysts enables it to progressively produce hydrogen in the absence of sacrificial reagents.25,26 Moreover, the overpotential of g-C3N4 can also be minimized by the use of other metal cocatalysts.27
19 and CdZnS20 have been extensively utilized for photocatalytic hydrogen evolution via water splitting. However, these materials have limitations; for example, TiO2 works only under UV light due to its 3.0–3.2 eV band gap,21 while metal sulphides are unstable due to photocorrosion.22 Graphitic carbon nitride (g-C3N4) has garnered significant attention due to its appealing characteristics, which include a moderate band gap, high thermal stability and a large surface area.23,24 Graphitic carbon nitride photocatalysts can work efficiently under sunlight. Modifications in g-C3N4 can lead to its practical application on a larger scale. The modification strategies include doping, heterojunction formation, bandgap engineering, and the use of cocatalysts. The use of g-C3N4 with effective cocatalysts enables it to progressively produce hydrogen in the absence of sacrificial reagents.25,26 Moreover, the overpotential of g-C3N4 can also be minimized by the use of other metal cocatalysts.27
Various metals have been effectively utilized as cocatalysts to improve the H2 production efficiency of photocatalysts such as Au, Pd, Cu, Ni, and Ag.17,28–30 It has been reported that metal/semiconductor interfaces exhibit a favourable characteristic that potentially decreases the charge recombination during photoreactions.31 Metals such as Au, Ag, and Cu exhibit high work function and possess surface plasmon electrons (SPEs).24,32,33 SPEs have the potential to enhance the photocatalytic activity through the facilitation of higher photon absorption and by providing supplementary hot electrons. Metals such as Ni, Pd, Pt and Ce reduce the activation energy barriers of catalysts by generating Schottky junctions between the metal and the semiconductor support.34–36 The impact of these metals on photocatalysts revealed that they effectively decrease the band gap energy. Additionally, the Pt and Pd metals exhibit a tendency to penetrate into the g-C3N4 structure to promote the transfer of charges.37
In this study, we have used Ce and Cu metal cocatalysts to boost the H2 evolution activity of g-C3N4 catalysts. These metal cocatalysts can overcome the energy barriers that hinder the H2 generation performance. We strategically utilized Cu to enhance light absorption via SPEs, while Ce effectively quenches electrons and facilitates cycling between the Ce3+/Ce4+ couple. Moreover, Ce has the ability to release electrons because of its low electron affinity for H+ ion reduction. This approach (active synergism between Ce and Cu) will effectively boost the photocatalytic performance of g-C3N4.
Experimental
All chemicals used during the synthesis of catalysts are of analytical grade: melamine (C3H6N6), copper nitrate (Cu(NO3)2·5H2O), ammonium cerium(IV) nitrate (NH4)2Ce(NO3)6 and deionized water. The synthesis process is illustrated in Fig. 1 and briefly described below.
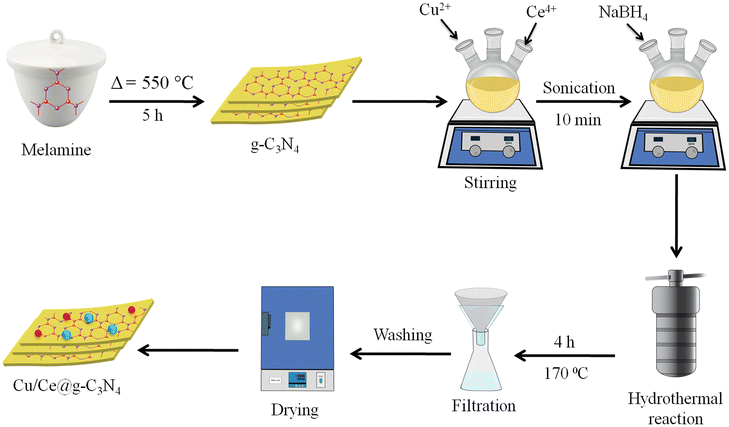 |
| | Fig. 1 Synthesis scheme of the Cu/Ce@g-C3N4 photocatalyst. | |
Synthesis of Cu/Ce@g-C3N4
g-C3N4 was produced by thermal polycondensation of melamine at 550 °C for 5 h in a muffle furnace.38 The process is carried out in a sealed system comprising an alumina crucible covered with a lid wrapped in aluminium foil. The heating rate employed was 5 °C per minute. The obtained material was ground into fine powder using a mortar and pestle. The synthesis scheme of Cu/Ce@g-C3N4 photocatalysts is illustrated in Fig. 1. Briefly, for the synthesis of Cu/Ce@g-C3N4 catalysts, 200 mg of pristine g-C3N4 was sonicated in 50 mL of deionized water. Then 7.6 mg of Cu(NO3)2·5H2O and 7.8 mg of (NH4)2Ce(NO3)6 were used to prepare metal precursor solutions. These solutions (i.e., Cu/Ce in 1:1 overall 2 wt%) were transferred into g-C3N4 slurry. An optimized amount of NaBH4 solution (reducing agent) was added dropwise to reduce the Cu2+ and Ce3+ metal ions (note: Ce being highly electropositive gets oxidized again on contact with air). The precursor slurry was then transferred into a Teflon-lined autoclave for a hydrothermal reaction at 170 °C for 4 h. The obtained catalysts were collected and dried at 90 °C for 8 h. The monometallic catalysts (Ce@g-C3N4 and Cu@g-C3N4) were synthesized using a similar approach with a single metal solution (either Ce or Cu).
Characterization
To obtain the diffraction patterns, a Bruker D2-Phaser was used that was outfitted with a high-performance LYNXEYE XE-T detector. The Cu X-ray source (λ = 1.54 Å) was operated at 40 Kv and 40 mA, and the instrument scanned a 2θ range of 10–80° at a scan rate of 2° min−1. A Raman spectrometer RMP-500/JASCO was employed to obtain the Raman spectrum of Cu/Ce@g-C3N4 catalysts in the 0–1800 cm−1 range. A Bruker Alpha Platinum spectrometer was used to obtain the FTIR spectrum of the catalysts in ATR mode in the 400–4000 cm−1 range to identify the presence of functional groups. Thermal analyses of melamine and synthesised Cu/Ce@g-C3N4 catalysts were conducted on a Q500/Q50 TGA instrument up to 800 °C at a heating rate of 10 °C min−1. TGA provides valuable insights into the thermal stability and decomposition behaviour of melamine and the synthesised catalysts. The optical properties were measured in the range of 300–750 nm, and for this purpose, UV-Vis/DRS was carried out on a PerkinElmer diffuse reflectance spectrophotometer UV-2550/Shimadzu equipped with a tungsten–halogen source. The spectra were recorded using BaSO4 as the reference sample. An Agilent 5500/AFM was used in tapping mode to acquire high-resolution topography images of Cu/Ce@g-C3N4 catalysts, revealing surface features with nanometre resolution. The photoluminescence spectra of catalysts were obtained using a PerkinElmer LS-45 spectrometer with an excitation wavelength of 375 nm. SEM analyses were conducted using an FEI-Nova NanoSEM-450 operating at 10–30 kV electron energy and secondary electrons (SE) were detected using an ETD/TLD detector. The elemental compositions of the Cu/Ce@g-C3N4 catalysts were analyzed using SEM equipped with an EDX detector. An acceleration voltage of 20 kV and an acquisition time of 100 seconds per spot were employed. XPS studies were performed using a ULVAC-PHI XPS system with an Al Kα X-ray source under ultrahigh vacuum (10−6 mbar) to determine the elemental composition and chemical state. A Micromeritics’ Tristar II 3020 analyzer was used to determine the surface area of catalysts using the Brunauer–Emmett–Teller (BET) method via N2 gas for the adsorption and desorption processes. Degassing of N2 was carried out at an elevated temperature (120 °C) and under vacuum conditions (1 × 10−3 Torr). The Solartron impedance analyzer-1260 was used to perform electrochemical impedance spectroscopy (EIS) on a three-electrode system. A Mott–Schottky (M–S) curve was obtained by exploring the flat band gap of the sample, utilizing a scanning voltage range from −1.5 V to 1.0 V.
Photocatalytic hydrogen generation experiments
All the synthesized catalysts were used to estimate the rates of H2 production under sunlight irradiation in a Pyrex reactor (150 mL). Photocatalytic experiments were conducted in September 2023 between 10.00 am and 4.00 pm on bright sunny days with clear sky in Bahawalpur (29°22′42′′ N and 71°45′53′′ E Punjab, Pakistan). Solar irradiance was measured using Extech's Light Meter (LT 300) and was within the range of 600–800 mW m−2. The reaction temperature was 25–35 °C throughout the experiments. Before starting the photoreactions, the dissolved oxygen content was removed by purging N2 at a rate of 10 mL min−1 for 10 min. Purging is necessary to prevent the consumption of excited electrons of g-C3N4 by molecular oxygen leading to the formation of highly reactive superoxide radicals. 10 mg of catalysts was dispersed in 50 mL of deionized water. No sacrificial agents were used to confirm true photocatalytic water splitting. The duration of each photoreaction was 6 h. At regular intervals, the H2 evolution rate was monitored by extracting gas headspace samples (0.5 mL) and introducing them into a gas chromatograph (Shimadzu 2014, Japan). This chromatograph was fitted with a Thermal Conductivity Detector (TCD) and employs a molecular sieve capillary column (25 meters in length; with an inner diameter of 0.32 mm and an average thickness of 0.50 μm). Quantification of the produced H2 was performed using the calibration curve of the GC system. To ensure accuracy, each sample was tested at least three times.
Results and discussion
The as-prepared photocatalysts g-C3N4, Cu@g-C3N4, Ce@g-C3N4 and Cu/Ce@g-C3N4 were characterized by XRD, FTIR, SEM, UV-Vis-DRS, PL, Raman spectroscopy, AFM, and EIS analyses and nitrogen adsorption–desorption isotherms for specific surface area measurement.
XRD
The X-ray diffraction results of g-C3N4, Cu@g-C3N4, Ce@g-C3N4 and Cu/Ce@g-C3N4 are shown in Fig. 2a. The pronounced diffractions of g-C3N4 were revealed at 13.02°, 27.21°, and 57.2° and were assigned to the (100), (002) and (004) planes, respectively. The diffraction patterns observed at the (002) plane were assigned to the 2D layered arrangement, and specifically the (100) plane emerged from the spacing between the tri-s-triazine ring units.38 In Cu/Ce@g-C3N4, no obvious peaks were observed for the Ce metal due to the low loading. However, a small peak was observed at 43.3° which corresponds to the Cu (111) plane. These results suggest that no structural changes were observed in g-C3N4 by the introduction of the Cu/Ce cocatalyst. However, Cu@g-C3N4, Ce@g-C3N4, and Cu/Ce@g-C3N4 exhibit a reduced intensity in the high-intensity peak (002), due to the incorporation of metals over the g-C3N4 surface.39
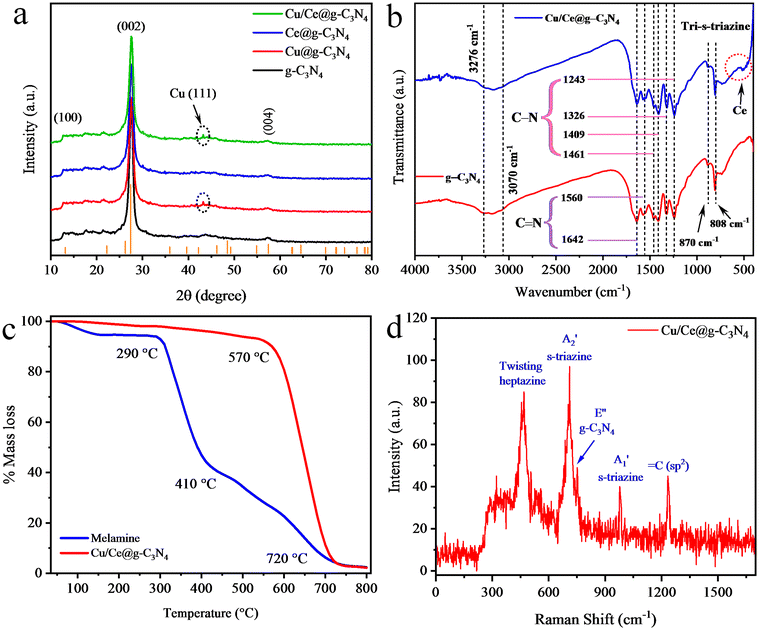 |
| | Fig. 2 (a) XRD patterns, (b) FTIR spectra of g-C3N4 and Cu/Ce@g-C3N4, (c) TGA curves of melamine and Cu/Ce@g-C3N4, and (d) Raman spectrum of Cu/Ce@g-C3N4. | |
FTIR
The FTIR technique was utilized to examine the functional groups and purity of prepared catalysts.40,41 The results of pristine and Cu/Ce loaded g-C3N4 catalysts are shown in Fig. 2b. The strong vibrations observed at 810 and 870 cm−1 were ascribed to monomers of s-triazine that is characteristic of the g-C3N4 structure.42 The aromatic C–N stretching was depicted at 1243, 1326, 1409, and 1461 cm−1, while the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching was found at 1560 and 1642 cm−1. The broad vibration at 3070 cm−1 was assigned to the O–H bond of absorbed H2O.43 During the g-C3N4 formation, trace amounts of N–H bonds persisted which appeared at 3276 cm−1. However, the FTIR results of the Cu/Ce@g-C3N4 catalyst demonstrated that the Ce metal was present in the form of oxides (CeO2) and its vibrations were identified at 500 cm−1.44
N stretching was found at 1560 and 1642 cm−1. The broad vibration at 3070 cm−1 was assigned to the O–H bond of absorbed H2O.43 During the g-C3N4 formation, trace amounts of N–H bonds persisted which appeared at 3276 cm−1. However, the FTIR results of the Cu/Ce@g-C3N4 catalyst demonstrated that the Ce metal was present in the form of oxides (CeO2) and its vibrations were identified at 500 cm−1.44
TGA
Thermogravimetric analyses were conducted to examine the thermal stability, % mass decomposition and preparation mechanism of g-C3N4 catalysts.45 TGA curves of the precursor melamine and the as-prepared Cu/Ce@g-C3N4 are shown in Fig. 2c. In melamine, by increasing the temperature up to 100 °C, a slight weight loss was noticed, signifying the loss of adsorbed water molecules.46 The rapid weight loss in the range of 290–410 °C is due to melamine sublimation and thermal condensation.47 This weight loss was attributed to the polymerization process that releases volatile products (i.e., ammonia).46 Beyond 410 °C, the weight loss becomes slow, ascribed to the polymerization of tri-s-triazine units. It is worth mentioning that g-C3N4 was formed in between 410 and 720 °C. To confirm this claim, the TGA results of Cu/Ce@g-C3N4 catalysts were studied, which demonstrate that the synthesized g-C3N4 catalysts are stable up to 570 °C. After that, the rapid weight loss was observed, attributed to the decomposition of g-C3N4, which reaches completion at 720 °C. The residual mass left behind is due to the metal and carbon content in Cu/Ce@g-C3N4 catalysts.48
Raman spectroscopy
Raman spectroscopy was employed to examine the structural properties of the synthesized materials. Fig. 2d exhibits the Raman results of Cu/Ce@g-C3N4, revealing five distinct Raman vibrations. The twisting (in-plane) vibrations of heptazine heterocyclic bonds were noticed at 468.43 cm−1. The breathing modes (i.e., A2′ and A1′) of s-triazine rings were detected at 711.03 and 979.46 cm−1, respectively. The layer-to-layer deformation vibrations of C–N were examined at 753.88 cm−1 and the C(sp2) bending vibrations were observed at 1233.61 cm−1, respectively.49,50
SEM and EDX
Morphological analysis of the as-prepared Cu/Ce@g-C3N4 catalysts was conducted using secondary electron mode in SEM. The morphology of the precursor material (i.e., melamine) is depicted in Fig. 3a, showing larger irregularly shaped particles with smooth surfaces.51 The SEM results of g-C3N4 are shown in Fig. 3b and confirm the successful synthesis of g-C3N4 catalysts.52 The SEM micrographs of Cu/Ce@g-C3N4 were obtained at 2 μm and 200 nm, respectively (Fig. 3c and d). The SEM results depict the 2D irregular lamellar stacking of the g-C3N4 catalyst. Moreover, the particle size has been significantly reduced as compared to melamine, which ensures a large surface area. This morphology ensures a higher rate of hydrogen production. The comparative result reveals the morphological changes during the synthesis of the g-C3N4 and Cu/Ce@g-C3N4 catalysts.53 The identification of elements within the sample was carried out using energy dispersive X-ray (EDX) analysis (Fig. 3c). The EDX analysis of Cu/Ce@g-C3N4 confirmed the presence of carbon, nitrogen, copper and cerium.54 The trace amounts of oxygen (O) detected in the analysis indicate the presence of Ce in an oxide form; our EDX results match with the FTIR findings. The EDX examination verified the successful synthesis of Cu/Ce@g-C3N4.
 |
| | Fig. 3 SEM results of (a) melamine, (b) g-C3N4, and (c) Cu/Ce@ g-C3N4 at 2 μm and (d) 200 nm. (e) EDX of the Cu/Ce@ g-C3N4 photocatalyst. | |
AFM
Atomic force microscopy was used to study the topographical features at the nanometre scale. Fig. 4a and b represent the 2D images of the Cu/Ce@g-C3N4 photocatalyst. The scan area taken was 4.26 × 4.26 μm. Fig. 4c depicts a horizontal histogram where the y-axis denotes the height (nm) and the x-axis denotes the percentage of the entire particle population. The average thickness of Cu/Ce@g-C3N4 was observed to be 35 nm. The 3D images are illustrated in Fig. 4d and e. The root mean square and average roughness were found to be 4.43 and 3.97 nm, respectively.13
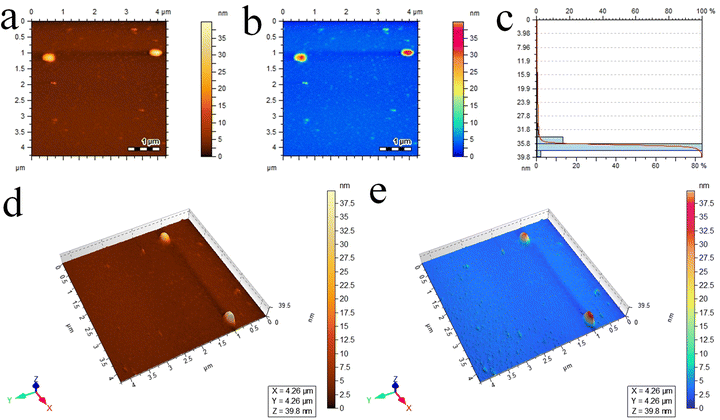 |
| | Fig. 4 AFM results of the Cu/Ce@g-C3N4 photocatalyst: (a and b) 2D images, (c) size distribution histogram and (d and e) the corresponding 3D images. | |
XPS
The chemical composition and oxidation states of the 2% Cu/Ce@g-C3N4 catalyst were examined by XPS. According to the XPS results, the C, N, Cu and Ce elements were identified. In the XPS results of carbon (Fig. 5a), binding energies at 284.7 and 288.2 eV correspond to the C–N–C and CN bonds, respectively.55 Meanwhile, the two binding energies (398.6 and 399.8 eV) of the N 1s, which are shown in Fig. 5b, correspond to the nitrogen in the CN–C and C–N–C bonds. The Ce 3d results (Fig. 5c) exhibit two pairs of doublets; one at 904.1 and 899.9 eV attributes to Ce 3d3/2 while the other at 885.4 and 881.4 eV corresponds to Ce 3d5/2. These four binding energies confirm the presence of cerium in the form of an oxide (Ce2O3) with a Ce3+ oxidation state. Cu 2p results demonstrate two binding energies at 933 and 952.5 eV that correspond to 2p3/2 and 2p1/2, respectively, and confirm the presence of metallic copper (Fig. 5d).56,57
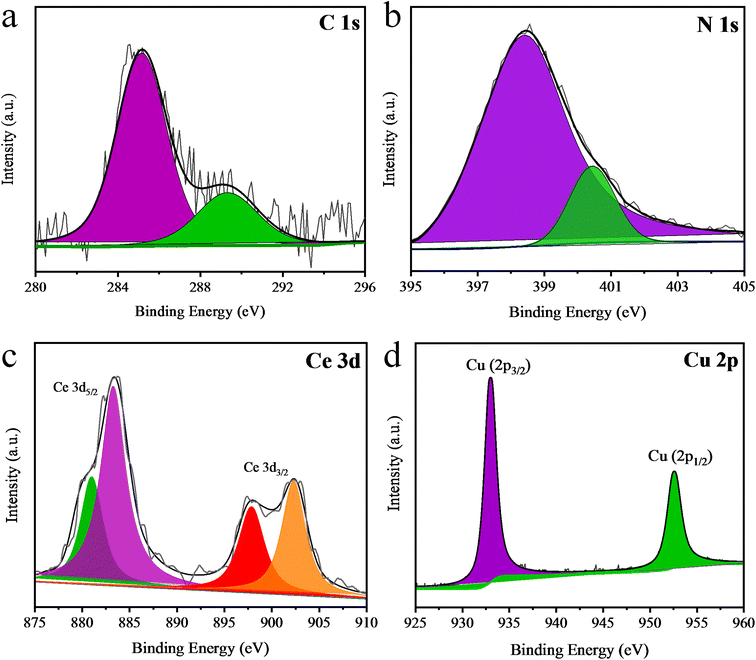 |
| | Fig. 5 XPS results of (a) C 1s, (b) N 1s and (c) Ce 3d and (d) the Cu 2p results. | |
UV-Vis-DRS
The UV-Vis-DRS technique was employed to examine the photon absorption (i.e., optical) properties of catalysts. Fig. 6a shows the obtained UV-Vis-DRS results of the synthesized catalysts. The pristine g-C3N4 exhibits the absorption edge at around 460 nm.25 For all the abovementioned catalysts, the absorbance was enhanced in the visible range. Cu loaded g-C3N4 catalysts showed enhanced absorbance (560–600 nm) due to the LSPR of Cu nanoparticles. Moreover, a significant red shift was also observed. In the case of Ce@g-C3N4 catalysts, the LSPR wasn't detected, confirming the existence of the Ce cocatalyst in an oxide form. The catalysts loaded with a bimetallic cocatalyst exhibited even greater light absorption. The bandgap values were calculated using the Tauc plot method, as illustrated in Fig. S1.†58 Crystalline sizes and band gap values of all synthesized catalysts are tabulated in Table S1.† From the calculated band gap values, it is confirmed that Cu/Ce loading lessened the band gap energy of pristine g-C3N4 photocatalysts.59 These results depict that loading of Cu/Ce on g-C3N4 captures more photons and promotes water splitting reactions.
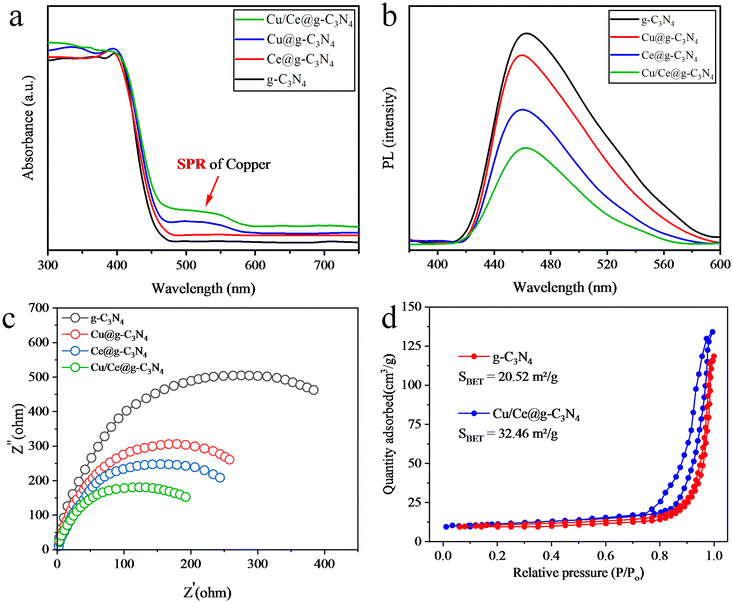 |
| | Fig. 6 (a) UV-Vis-DRS, (b) photoluminescence, (c) EIS and (d) BET results of the as-synthesized catalysts. | |
PL
The photoluminescence (PL) technique was employed to explore the separation efficiencies of photogenerated charges.25 The PL peaks occur due to the emission of radiation that came from a recombination of the charges between different energy states. Fig. 6b shows the PL results of pristine g-C3N4, Cu@g-C3N4, Ce@g-C3N4 and Cu/Ce@g-C3N4 catalysts. In g-C3N4, a higher emission intensity was observed at 460 nm and is due to a higher recombination of photogenerated charges. However, the metal-loaded catalysts exhibit low PL intensities endorsed by the significant quenching of photogenerated electrons.59 The bimetallic Cu/Ce@g-C3N4 catalysts exhibit the lowest charge recombination rate ascribed to the development of a heterojunction between g-C3N4 and metal cocatalysts. Therefore, the photogenerated electrons efficiently transferred to metal active sites ensure a higher H2 evolution activity.60
EIS
Electrochemical impedance spectroscopy (EIS) was employed to examine the charge transport at the interface of Cu/Ce@g-C3N4 catalysts. EIS measures the impedance of an electrochemical system as a function of frequency and presents the data in a Nyquist plot. The semicircle diameter within this plot directly signifies the charge transfer resistance.61 A smaller diameter means reduced impedance, suggesting enhanced charge transfer, improved conductivity, and lower recombination rates of photogenerated charges. The observed sequence of semicircle curves was as follows: g-C3N4 > Ce@g-C3N4 > Cu@g-C3N4 > Cu/Ce@g-C3N4, as shown in Fig. 6c. Notably, Cu/Ce@g-C3N4 displayed the smallest diameter, which indicates that the introduction of Cu and Ce metals synergistically enhanced charge separation and transfer mechanisms, effectively decreasing the recombination rates of photogenerated charges.62
BET analysis
The nitrogen adsorption–desorption isotherm method was utilized to estimate the specific surface areas of the catalysts. The pristine g-C3N4 and Cu/Ce@g-C3N4 catalysts exhibit SBET of 20.52 and 32.46 m2 g−1, respectively (Fig. 6d). Heat treatment provided for hydrothermal deposition of Cu and Ce is the reason for the increased surface area of Cu/Ce@g-C3N4.63 Moreover, it is also evident from our SEM results that the particle size was reduced after metal deposition, which also caused the increase in the surface area.
Photocatalytic activities
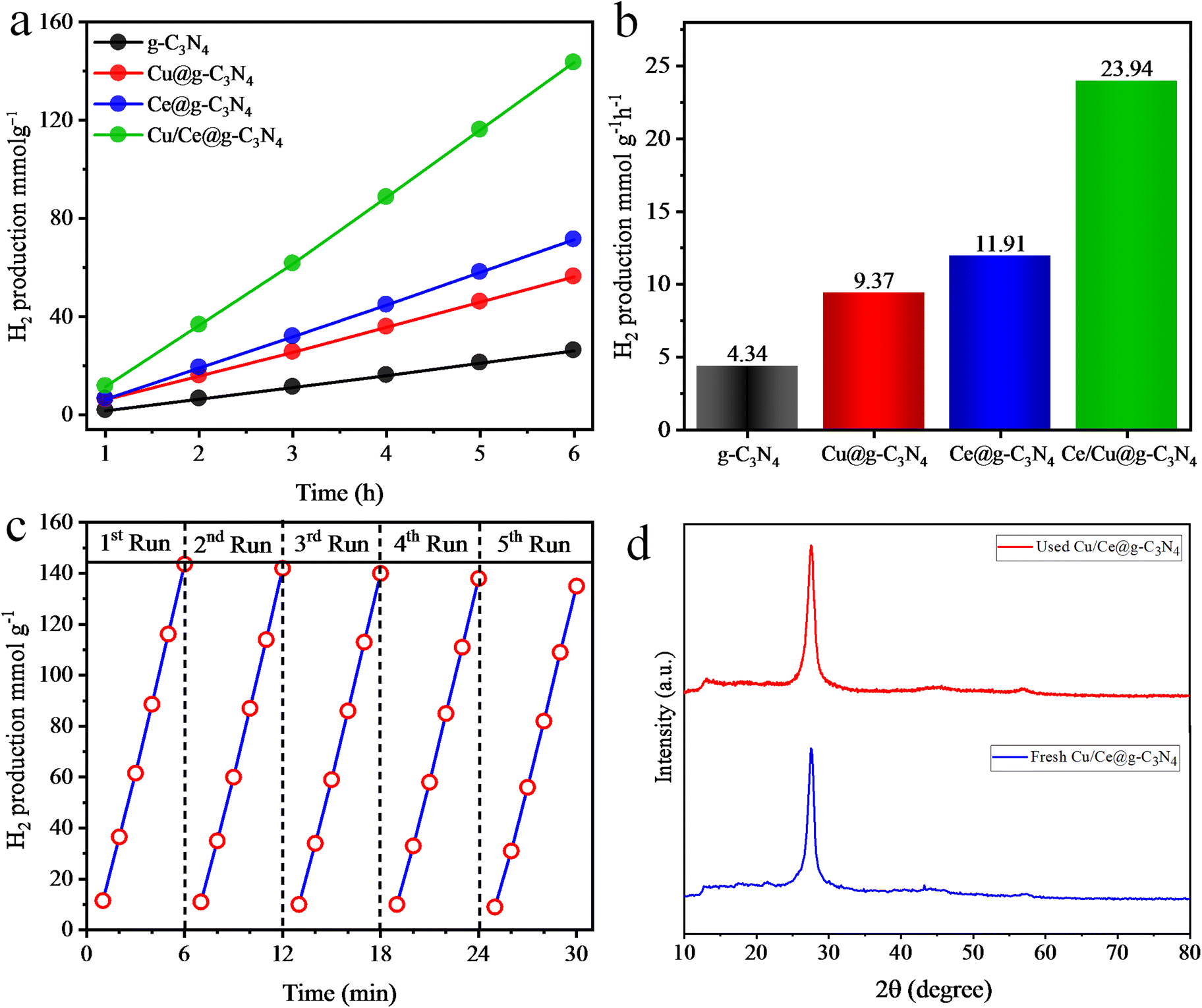
The hydrogen evolution rates of the synthesized photocatalysts are presented in Fig. 7a and b, calculated in mmol g−1 and mmol g−1 h−1 units. The pristine g-C3N4 catalysts demonstrated a minimal hydrogen production rate of 4.34 mmol g−1 h−1 owing to high recombination rates of photogenerated charges. Upon each 1 wt% of Cu and Ce metal loading on g-C3N4, the H2 production significantly escalated to 9.37 and 11.91 mmol g−1 h−1, respectively. However, Cu/Ce@g-C3N4 catalysts surpassed the individual performances of Cu and Ce loaded g-C3N4 catalysts, exhibiting the hydrogen evolution activity at 23.94 mmol g−1 h−1. The hydrogen evolution performances of the as-synthesized catalysts are tabulated in Table S2.† These improved activities were ascribed to the (a) active synergism between Cu and Ce metals, (b) Schottky effect between g-C3N4 and Cu64, (c) heterojunction formation between Ce2O3 and g-C3N4, (d) the supplemental SPEs of Cu metal, (e) improved light absorption, (f) large active site and (g) increased surface area.65 A comparison of the H2 evolution activities of the reported catalysts in this study is provided in Table 1.
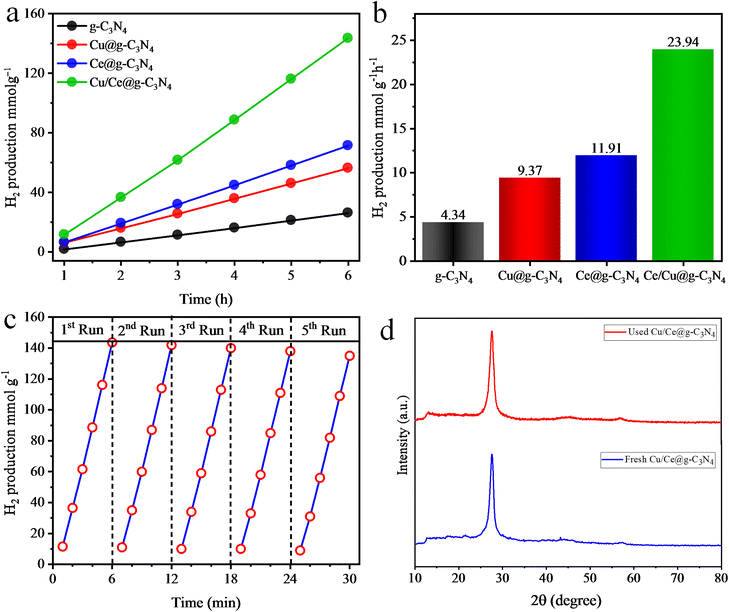 |
| | Fig. 7 H2 production activities of g-C3N4, Cu@g-C3N4, Ce@g-C3N4, and Cu/Ce@g-C3N4 in (a) mmol g−1 and (b) mmol g−1 h−1, (c) H2 generation efficiencies for 5 consecutive runs and (d) the XRD patterns of fresh and used Cu/Ce@g-C3N4 photocatalysts. | |
Table 1 Comparison of H2 production activities of reported catalysts with those in this study
| Catalysts |
Synthesis method |
Catalyst amount (mg) |
Light source |
Sacrificial reagent |
Activity (H2) (mmol g−1 h−1) |
Ref. |
| Au/g-C3N4 |
Impregnation-annealing |
— |
λ > 420nm |
Triethanolamine |
0.159 |
66
|
| Pt/g-C3N4 |
Thermolysis followed by chemisorption |
50 |
425 nm LED |
Triethanolamine |
11.00 |
67
|
| Pd–Ag/g-C3N4 |
Chemical reduction |
10 |
Direct sunlight |
Triethanolamine |
1.25 |
68
|
| Au/Pd/g-C3N4 |
Chemical deposition |
50 |
300 W Xe arc lamp |
Triethanolamine |
0.326 |
69
|
| Na doped g-C3N4 |
Calcined at 600 °C for 1 h under N2 |
50 |
350 W Xe lamp |
Triethanolamine |
18 |
70
|
| Li doped g-C3N4 |
Calcined at 600 °C for 1 h under N2 |
50 |
350 W Xe lamp |
Triethanolamine |
12.5 |
70
|
| K doped g-C3N4 |
Calcined at 600 °C for 1 h under N2 |
50 |
350 W Xe lamp |
Triethanolamine |
13.5 |
70
|
| Eu doped g-C3N4 |
Calcination at 550 °C for 4 h |
50 |
300 W Xe arc lamp |
Triethanolamine |
0.40 |
71
|
| Fe doped g-C3N4 |
Polycondensation at 550 °C for 4h |
100 |
300 W Xe lamp |
10% methanol/0.05 M H2PO4 |
0.53 |
72
|
| P-doped g-C3N4/MoS2 |
Sonication followed by ventilation under N2 |
100 |
300 W Xe lamp |
— |
0.121 |
73
|
| C-doped g-C3N4 |
Calcined at 550 °C, 4 h |
50 |
300 W Xe lamp |
Triethanolamine |
0.30 |
74
|
| B and P doped g-C3N4 |
Calcined at 400 °C, 2 h |
— |
Visible light |
— |
4.00 |
75
|
| Cu/Ce@g-C3N4 |
calcination followed by chemical reduction and hydrothermal methods |
10 mg |
Sunlight |
— |
23.94 |
This study |
Stability of the photocatalysts
To assess whether the catalyst retains its activity, structural identity, and functionality after being used multiple times, the recyclability tests of Cu/Ce@g-C3N4 catalysts were performed. In every cycle, the reaction mixture underwent argon gas purging, removing the previously generated H2 content. Fig. 7c demonstrates the H2 evolution rates in each photocatalytic run. A slight decrease (5%) in H2 evolution was observed after five cyclic runs. Therefore, the Cu/Ce@g-C3N4 photocatalysts exhibit good stability for reuse. Fig. 7d shows the XRD patterns of fresh and used Cu/Ce@g-C3N4 photocatalysts. No changes in the diffraction pattern were observed, suggesting that the structure and morphology of the photocatalyst remained unaffected after 5 consecutive runs of photoreactions.76,77
Factors affecting photocatalytic performances
Effect of pH.
The effect of pH on hydrogen generation activity was investigated by adjusting the solution's pH from 2 to 12 using 0.01 M HCl/NaOH. The experiments were conducted at a constant temperature of 35 °C to better estimate the pH effect. The results are displayed in Fig. S2a,† and they indicate a notable rise in the rate of hydrogen production under acidic conditions. The acidic condition improves the proton availability for the reduction reaction, specifically at pH 5 (optimum pH), and the Cu/Ce@g-C3N4 catalysts exhibited the highest hydrogen production rate. However, beyond this pH level, H2 production declined. g-C3N4 is stable under illumination across pH 2–12 due to strong carbon–nitride bonds.78 The outcomes of varying pH on hydrogen evolution performances of Cu/Ce@g-C3N4 photocatalysts are tabulated in Table S3.† The increased H2 evolution originated from the formation of a novel electron trapping site situated 0.3 eV below the g-C3N4 conduction band. This trapping site reduces the photoexcited charge recombination.79 So, acidic conditions improve H2 production due to enhanced charge separation. Under basic conditions, metals form hydroxides that hinder the catalytic performances. Additionally, OH− reacted with photogenerated H+ to form water that slows down the hydrogen production.
Effect of temperature.
The impact of temperature on hydrogen production was investigated across different temperatures from 30–60 °C in a reactor set at the optimized pH 5. The results are depicted in Fig. S2b† and tabulated in Table S4.† The results revealed a lower hydrogen production rate at room temperature as compared to higher temperatures. This finding is consistent with the existing literature, which suggests a higher rate of charge carrier recombination at room temperature.80 According to the Varshni equation (1), the band gap of a semiconductor decreases with increasing temperature, allowing for improved charge carrier mobility and conductivity on light absorption:81| | 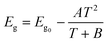 | (1) |
Effect of light intensity.
The hydrogen production rate correlates with light intensity: a higher intensity yields more electrons/holes, boosting hydrogen generation in the photocatalyst. During the photoreactions, the light intensities were controlled (100–1000 W m−2) and the hydrogen production rate was monitored (Fig. S2c† and Table S5†). The highest hydrogen production rate was observed at 700 W m−2. Despite increasing the light intensity, no further increase in the H2 production was observed because the number of active sites remained constant.17 Moreover, the higher light intensity results in a more pronounced enhancement of the electromagnetic field around the Cu metal experiencing LSPR, which is in agreement with the literature. This intensified field can facilitate more efficient charge separation and transfer.82
Effect of the catalyst dosage.
The effect of photocatalyst dosage on the rate of hydrogen generation was quantified using 2–10 mg of Cu/Ce@g-C3N4 catalysts in a Pyrex-150 mL/UK photo-reactor. The results demonstrate that 5 mg of catalysts is the optimal amount for photocatalytic hydrogen production (Fig. S2d† and Table S6†). This quantity optimizes the number of active sites available for catalytic reactions. Additionally, this photocatalyst dose achieves excellent dispersion within the reaction mixture, minimizing agglomeration and maximizing their exposure to sunlight. Moreover, this quantity of the catalyst also promotes favourable water molecule adsorption and facilitates easy desorption of H2 molecules from the catalyst's surfaces. Beyond 5 mg, hydrogen production increased but didn't align with dosage increments due to excessive catalyst hindering light absorption within the solution's inner particles.2,59
Mechanism
Water splitting using solar energy is required to overcome a thermodynamic barrier of approximately 1.23 eV. A photocatalyst with a minimum CB potential of 0.0 eV and a VB potential greater than 1.23 eV can achieve water splitting.14,83 g-C3N4 is an n-type semiconductor with a bandgap energy of 2.72 eV. The CB potential of g-C3N4 was measured via a Mott–Schottky plot to be −0.72 eV (Fig. S3†). The VB potential of g-C3N4 was determined using the equation EG = EVB – ECB, resulting in a value of 2.02 eV. Upon visible light irradiation, pristine g-C3N4 catalysts have a higher charge recombination rate. Therefore, pristine g-C3N4 cannot be employed for photocatalytic water splitting. In order to increase its effectiveness, Cu and Ce metals play a crucial role. It has been observed that Cu improves the photon absorption capability of g-C3N4 due to d–d transitions and provides SPEs during photoreactions.17 The highly energetic electrons (known as hot electrons or SPEs) transfer in a continuous flow from copper sites towards the CB of g-C3N4.84 Note: electrons always flow from a material of low work function towards those of higher work functions.85 In this case, the work function of the Cu metal (4.7 eV) is lower than that of the g-C3N4 semiconductor (5.22 eV), which means that electrons flow from the surface of the Cu metal to the g-C3N4 surfaces,86 where they are quenched by Ce cocatalysts.87 Moreover, the photogenerated electrons of g-C3N4 cross the potential energy barrier formed between Ce cocatalysts and g-C3N4 interfaces. These electrons reduce Ce4+ to Ce3+ species,88 which then participate in catalytic processes (proton reduction) to produce molecular hydrogen. Simultaneously, Ce3+ species are oxidized back to Ce4+,89 the photogenerated holes were transferred to the VB of g-C3N4, where oxidation of water completes the photocatalytic cycle. This process prolongs the lifetime of these charge carriers available for hydrogen evolution during the photoreaction (Fig. 8). Additionally, due to Ce metal cocatalysts, chances of recombination reactions between electron–hole (e−/h+) pairs become quite low. The combined utilization of SPR mediated by Cu and the electron scavenging ability of the Ce3+/Ce4+ redox couple manifest as a synergistic effect.90,91 The Cu and Ce co-catalysts work synergistically to facilitate the transfer of electron–hole pairs of g-C3N4 for enhancing the H2 generation performances of g-C3N4. This synergistic effect is pivotal in affirming the substantial enhancement of the photocatalytic activity of dual metal loaded Cu/Ce@g-C3N4 catalysts. The presence of Cu and Ce on the g-C3N4 system holds promise as a fresh approach for researchers toward H2 evolution from water splitting reactions (Fig. S2†).
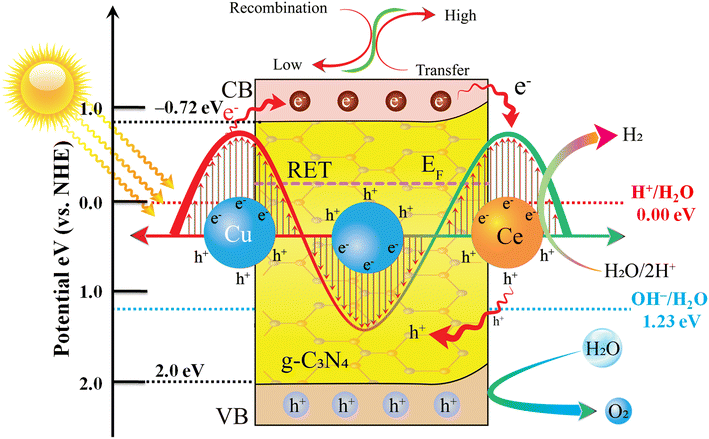 |
| | Fig. 8 Synergistic effect of Cu and Ce on the g-C3N4 photocatalyst for H2 generation. | |
Conclusion
This study investigates the photocatalytic hydrogen evolution by Cu/Ce supported g-C3N4 catalysts via water splitting reactions. The photocatalysts (Cu@g-C3N4, Ce@g-C3N4, and Cu/Ce@g-C3N4) were successfully synthesized using chemical reduction followed by the hydrothermal method. The catalysts were thoroughly characterized by XRD, FTIR, Raman, TGA, SEM, EDX, AFM, UV-Vis-DRS, PL, EIS and BET techniques. The results demonstrate the structural, optical, morphological and compositional features that support the contributions of the Cu/Ce cocatalysts to improve the H2 evolution performances. The Cu/Ce@g-C3N4 photocatalysts exhibit 5.5 times higher hydrogen evolution performances as compared to pristine g-C3N4 catalysts. The improved activity was ascribed to the active synergism between Cu and Ce metals. Moreover, hot electrons of the Cu metal and the Schottky junction at the metal/semiconductor interface are responsible for the enhanced H2 production performance. Despite the challenges, this study has laid a promising foundation for future hydrogen energy technologies.
Conflicts of interest
The authors state no conflict of interest.
Acknowledgements
The research was supported by HEC-Pakistan, Islamabad (grant #377/IPFP-II/Batch-I/SRGP/NAHE/HEC/2022/27). Synthesis of catalysts and their hydrogen evolution performances were carried out at Inorganic Materials Laboratory 52S, Institute of Chemistry, IUB-Pakistan. Dr E. H. thanks the Carnegie Mellon University of USA for instrumental access that was mandatory to complete this work.
References
- R. J. Batrice and J. C. Gordon, RSC Adv., 2021, 11, 87–113 RSC.
- M. Sabir, K. Rafiq, M. Z. Abid, U. Quyyum, S. S. A. Shah, M. Faizan, A. Rauf, S. Iqbal and E. Hussain, Fuel, 2023, 353, 129196 CrossRef CAS.
- M. Z. Rahman, J. Zhang, Y. Tang, K. Davey and S.-Z. Qiao, Mater. Chem. Front., 2017, 1, 562–571 RSC.
- S. Sun, X. Gou, S. Tao, J. Cui, J. Li, Q. Yang, S. Liang and Z. Yang, Mater. Chem. Front., 2019, 3, 597–605 RSC.
- J. Andrews and B. J. P. E. Shabani, Procedia Eng., 2012, 49, 15–25 CrossRef CAS.
- W. Wang, S. Wang, L. Guo, X. Wang, H. Liu, Z. Li and W. Zhou, Mater. Chem. Front., 2023, 7, 4547–4552 RSC.
- A. Habibi-Yangjeh and K. Pournemati, Crit. Rev. Environ. Sci. Technol., 2024, 54, 290–320 CrossRef CAS.
- A. Akhundi, A. Zaker Moshfegh, A. Habibi-Yangjeh and M. Sillanpää, ACS ES&T Eng., 2022, 2, 564–585 Search PubMed.
- K. Harun, S. Adhikari and H. Jahromi, RSC Adv., 2020, 10, 40882–40893 RSC.
- A. Fujishima and K. J. N. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12(4), 237–268 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- Z. Abid, K. Rafiq, A. Rauf, R. Hamed Althomali and E. Hussain, Mater. Adv., 2024 10.1039/D3MA00710C.
- E. Hussain, I. Majeed, M. A. Nadeem, A. Badshah, Y. Chen, M. A. Nadeem and R. Jin, J. Phys. Chem. C, 2016, 120, 17205–17213 CrossRef CAS.
- C. Zhou, L. Shang, H. Yu, T. Bian, L.-Z. Wu, C.-H. Tung and T. Zhang, Catal. Today, 2014, 225, 158–163 CrossRef CAS.
- M. Jalil, K. Rafiq, M. Z. Abid, A. Rauf, S. Wang, S. Iqbal and E. Hussain, Nanoscale Adv., 2023, 5, 3233–3246 RSC.
- F. Saleem, M. Z. Abid, K. Rafiq, A. Rauf, K. Ahmad, S. Iqbal, R. Jin and E. Hussain, Int. J. Hydrogen Energy, 2024, 52, 305–319 CrossRef CAS.
- R. Shi, Y. Cao, Y. Bao, Y. Zhao, G. I. N. Waterhouse, Z. Fang, L.-Z. Wu, C.-H. Tung, Y. Yin and T. Zhang, Adv. Mater., 2017, 29, 1700803 CrossRef PubMed.
- M. Jalil, K. Rafiq, M. Z. Abid, M. Rafay, A. Rauf, R. Jin and E. Hussain, Catal. Sci. Technol., 2024, 14, 850–862 RSC.
- A. Shahzad, K. Rafiq, M. Z. Abid, N. A. Khan, S. S. A. Shah, R. H. Althomali, A. Rauf and E. Hussain, J. Catal., 2024, 429, 115210 CrossRef CAS.
- I. Rabani, H.-N. Jang, Y.-J. Park, M. S. Tahir, Y.-B. Lee, E.-Y. Moon, J. W. Song and Y.-S. Seo, RSC Adv., 2022, 12, 33653–33665 RSC.
- E. Ö. Alagöz, H. Jahangiri and S. Kaya, Mater. Adv., 2024, 5, 1513–1522 RSC.
- P. Hemmati-Eslamlu and A. Habibi-Yangjeh, FlatChem, 2023, 100597 Search PubMed.
- Z. Zhang, Y. Xu, Q. Zhang, S. Fang, H. Sun, W. Ou and C. Su, Sci. Bull., 2022, 67, 71–78 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- M. Caux, F. Fina, J. T. Irvine, H. Idriss and R. Howe, Catal. Today, 2017, 287, 182–188 CrossRef CAS.
- Y. Chen, J. Ma, J. Fu, L. Sun, J. Cheng and J.-F. Li, Int. J. Hydrogen Energy, 2024, 51, 1145–1152 CrossRef CAS.
- N. Wajid, K. Rafiq, M. Z. Abid, A. Ilyas, T. Najam, A. Rauf and E. Hussain, Mater. Chem. Phys., 2023, 306, 128062 CrossRef CAS.
- E. Hussain, I. Majeed, M. A. Nadeem, A. Badshah, Y. Chen, M. A. Nadeem and R. Jin, J. Phys. Chem. C, 2016, 120, 17205–17213 CrossRef CAS.
- Z. Abid, A. Ilyas, K. Rafiq, M. A. Nadeem, A. Rauf, A. Wasim and E. Hussain, Environ. Sci.: Water Res. Technol., 2023, 9(9), 2238–2252 RSC.
- F. Fina, H. Ménard and J. T. S. Irvine, Phys. Chem. Chem. Phys., 2015, 17, 13929–13936 RSC.
- X. Zhang, Y. L. Chen, R.-S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- A. V. Zhurenok, D. B. Vasilchenko and E. A. Kozlova, Int. J. Mol. Sci., 2022, 24, 346 CrossRef PubMed.
- A. Hayat, M. Sohail, J. A. S. Syed, A. G. Al-Sehemi, M. H. Mohammed, A. A. Al-Ghamdi, T. Taha, H. S. AlSalem, A. M. Alenad and M. A. Amin, Chem. Rec., 2022, 22, e202100310 CrossRef CAS PubMed.
- A. Hayat, J. A. S. Syed, A. G. Al-Sehemi, K. S. El-Nasser, T. Taha, A. A. Al-Ghamdi, M. A. Amin, Z. Ajmal, W. Iqbal and A. Palamanit, Int. J. Hydrogen Energy, 2022, 47, 10837–10867 CrossRef CAS.
- J. Wu, L. Wang, S. Xu, Y. Cao, Z. Han and H. Li, RSC Adv., 2023, 13, 2024–2035 RSC.
- F. Fina, S. K. Callear, G. M. Carins and J. T. S. Irvine, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS.
- A. Durairaj, T. Sakthivel, A. Obadiah and S. Vasanthkumar, J. Mater. Sci.: Mater. Electron., 2018, 29, 8201–8209 CrossRef CAS.
- A. Aslam, M. Z. Abid, K. Rafiq, A. Rauf and E. Hussain, Sci. Rep., 2023, 13, 6306 CrossRef CAS PubMed.
- M. Z. Abid, K. Rafiq, A. Rauf, S. S. A. Shah, R. Jin and E. Hussain, Nanoscale Adv., 2023, 5, 3247–3259 RSC.
- Y. Wang, C. Peng, A. Liu, J. Song, X. Li and T. Jiang, J. Mater. Chem. C, 2023, 11, 1165–1172 RSC.
- B. Salah, A. Abdelgawad, Q. Lu, A. K. Ipadeola, R. Luque and K. Eid, Green Chem., 2023, 25, 6032–6040 RSC.
- X. Cao, L. Kong, Z. Gu and X. Xu, RSC Adv., 2022, 12, 23183–23192 RSC.
- U. Quyyum, K. Rafiq, M. Z. Abid, F. Ahmad, A. Rauf and E. Hussain, Environ. Sci.: Water Res. Technol., 2023, 9, 1147–1160 RSC.
- P. Makoś-Chełstowska, E. Słupek and A. Małachowska, J. Hazard. Mater., 2022, 425, 127972 CrossRef PubMed.
- F. Hassanzadeh-Afruzi, H. Dogari, F. Esmailzadeh and A. Maleki, Appl. Organomet. Chem., 2021, 35, e6363 CrossRef CAS.
- P. Basyach, A. K. Guha, S. Borthakur, L. Kalita, P. Chetia, K. Sonowal and L. Saikia, J. Mater. Chem. A, 2020, 8, 12774–12789 RSC.
- P. Jiménez-Calvo, C. Marchal, T. Cottineau, V. Caps and V. Keller, J. Mater. Chem. A, 2019, 7, 14849–14863 RSC.
- J. Jiang, L. Ou-yang, L. Zhu, A. Zheng, J. Zou, X. Yi and H. Tang, Carbon, 2014, 80, 213–221 CrossRef CAS.
- X. Han, L. Tian, H. Jiang, L. Kong, J. Lv, J. Shan, J. Wang and X. Fan, RSC Adv., 2017, 7, 14372–14381 RSC.
- M. Ikram, M. Shazaib, A. Haider, A. Shahzadi, S. Baz, M. M. Algaradah, A. Ul-Hamid, W. Nabgan, H. S. Abd-Rabboh and S. Ali, RSC Adv., 2023, 13, 25305–25315 RSC.
- Y. Wang, S. Zhong, Z. Niu, Y. Dai and J. Li, Chem. Commun., 2023, 59, 10883–10911 RSC.
- S. Parveen, E. W. Cochran, S. Zulfiqar, M. A. Amin, M. F. Warsi and K. Chaudhary, RSC Adv., 2023, 13, 26822–26838 RSC.
- N. Thangavel, K. Pandi, A. M. Shaheer and B. Neppolian, Catal. Sci. Technol., 2020, 10, 8015–8025 RSC.
- L. Yang, J. Huang, L. Shi, L. Cao, Q. Yu, Y. Jie, J. Fei, H. Ouyang and J. Ye, Appl. Catal., B, 2017, 204, 335–345 CrossRef CAS.
- L. Huang, Y. Li, H. Xu, Y. Xu, J. Xia, K. Wang, H. Li and X. Cheng, RSC Adv., 2013, 3, 22269–22279 RSC.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- A. Ilyas, K. Rafiq, M. Z. Abid, A. Rauf and E. Hussain, RSC Adv., 2023, 13, 2379–2391 RSC.
- J. Yu and J. Ran, Energy Environ. Sci., 2011, 4, 1364–1371 RSC.
- N. O. Laschuk, E. B. Easton and O. V. Zenkina, RSC Adv., 2021, 11, 27925–27936 RSC.
- T. R. Chetia, M. S. Ansari and M. Qureshi, J. Mater. Chem. A, 2016, 4, 5528–5541 RSC.
- K. Chen, R. Pathak, A. Gurung, K. M. Reza, N. Ghimire, J. Pokharel, A. Baniya, W. He, J. J. Wu and Q. Q. Qiao, J. Mater. Chem. A, 2020, 8, 1911–1919 RSC.
- N. A. Khan, C. M. Kim and S. H. Jhung, Chem. Eng. J., 2017, 311, 20–27 CrossRef CAS.
- S. Nikolaev, E. Golubina, I. Krotova, M. Shilina, A. Chistyakov and V. Kriventsov, Appl. Catal., B, 2015, 168, 303–312 CrossRef.
- Z. Guo, F. Dai, H. Yin, M. Zhang, J. Xing and L. Wang, Colloid Interface Sci. Commun., 2022, 48, 100615 CrossRef CAS.
- D. Vasilchenko, A. Zhurenok, A. Saraev, E. Gerasimov, S. Cherepanova, S. Tkachev, P. Plusnin and E. Kozlova, Chem. Eng. J., 2022, 445, 136721 CrossRef CAS.
- I. Majeed, U. Manzoor, F. K. Kanodarwala, M. A. Nadeem, E. Hussain, H. Ali, A. Badshah, J. A. Stride and M. A. Nadeem, Catal. Sci. Technol., 2018, 8, 1183–1193 RSC.
- C. Han, L. Wu, L. Ge, Y. Li and Z. Zhao, Carbon, 2015, 92, 31–40 CrossRef CAS.
- J. Jiang, S. Cao, C. Hu and C. Chen, Chin. J. Catal., 2017, 38, 1981–1989 CrossRef CAS.
- Y. Li, C. Lai, J. Zhong and J. Li, Int. J. Hydrogen Energy, 2023, 48(63), 24356–24368 CrossRef CAS.
- J. Gao, Y. Wang, S. Zhou, W. Lin and Y. Kong, ChemCatChem, 2017, 9, 1708–1715 CrossRef CAS.
- X. Zhou, P. Wang, M. Li, M. Wu, B. Jin, J. Luo, M. Chen, X. Zhou, Y. Zhang and X. Zhou, J. Mater. Sci. Technol., 2023, 158, 171–179 CrossRef CAS.
- G. Liu, M. Xue, Q. Liu, H. Yang and Y. Zhou, Int. J. Hydrogen Energy, 2019, 44, 25671–25679 CrossRef CAS.
- X. Liu, L. Yan, X. Hu, H. Feng, B. Guo, X. Ha and H. Xu, Int. J. Hydrogen Energy, 2023, 48, 13181–13188 CrossRef CAS.
- Z. Liu, F. Wang, Z. Zhang and S. Min, Int. J. Hydrogen Energy, 2020, 45, 31678–31688 CrossRef CAS.
- J. Li, Y. Chen, L. Zhu, L. Liao, X. Wang, X. Xu, L. Qiu, J. Xi, P. Li and S. Duo, RSC Adv., 2022, 12, 32480–32487 RSC.
- S. Yan, Z. Li and Z. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- S. Fang, K. Lv, Q. Li, H. Ye, D. Du and M. Li, Appl. Surf. Sci., 2015, 358, 336–342 CrossRef CAS.
- Z. Zhang, L. Ren, H. Li, D. Jiang, Y. Fang, H. Du, G. Xu, C. Zhu, H. Li and Z. Lu, Small, 2023, 2207173 CrossRef CAS PubMed.
-
D. Wolpert, P. Ampadu, D. Wolpert and P. Ampadu, Managing temperature effects in nanoscale adaptive systems, 2012, pp. 15–33 Search PubMed.
- X. Xing, S. Tang, H. Hong and H. Jin, Int. J. Hydrogen Energy, 2020, 45, 9612–9623 CrossRef CAS.
- M. Z. Abid, K. Rafiq, A. Aslam, R. Jin and E. Hussain, J. Mater. Chem. A, 2024 10.1039/D3TA06548K.
- J. Zhao, Y. Bai, Z. Li, J. Liu, W. Wang, P. Wang, B. Yang, R. Shi, G. I. Waterhouse and X. D. Wen, Angew. Chem., Int. Ed., 2023, 62, e202219299 CrossRef CAS PubMed.
- A. Dey, P. Ghosh, J. Bowen, N. S. J. Braithwaite and S. Krishnamurthy, Phys. Chem. Chem. Phys., 2020, 22, 7685–7698 RSC.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- S. Anandan and M. Miyauchi, Phys. Chem. Chem. Phys., 2011, 13, 14937–14945 RSC.
- Z. Cui, W. Wang, C. Zhao, C. Chen, M. Han, G. Wang, Y. Zhang, H. Zhang and H. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 31394–31403 CrossRef CAS PubMed.
- A. Joseph, A. Ayyappan, T. Subair, M. Pandibayal, S. Nair, R. Ramany, M. R. Varma and S. Thomas, ChemistrySelect, 2023, 8, e202301020 CrossRef CAS.
- F. Sun, J. Song, H. Wen, X. Cao, F. Zhao, J. Qin, W. Mao, X. Tang, L. Dong and Y. Long, Inorg. Chem., 2023, 62, 17961–17971 CrossRef CAS PubMed.
- J. Wu, X. Cao, Y. Ji, F. Zhang, X. Huang, G. F. Ouyang and J. Yu, Adv. Funct. Mater., 2024, 34, 2309825 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Aysha
Tanveer
a,
Khezina
Rafiq
a,
Aysha
Tanveer
a,
Khezina
Rafiq