
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and optical control through anion and cation exchange processes for Sn-halide perovskite nanostructures†
Kushagra
Gahlot
 ,
Julius
Meijer
and
Loredana
Protesescu
*
,
Julius
Meijer
and
Loredana
Protesescu
*
Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, Groningen, 9747AG, The Netherlands. E-mail: l.protesescu@rug.nl
First published on 6th February 2024
Abstract
Metal halide perovskite nanostructures, characterized by their ionic nature, present a compelling avenue for the tunability of dimensions and band gaps through facile compositional transformations involving both cationic and anionic exchange reactions. While post-synthetic ion-exchange processes have been extensively explored in Pb-halide perovskite nanocrystals, the inherent instability of Sn2+ has limited the exploration of such processes in Sn-halide perovskite nanostructures. In this study, we present a straightforward cation exchange process wherein 2D [R–NH3]2SnX4 Ruddlesden–Popper (RP) nanostructures with n = 1 transition to 3D ASnX3 nanocrystals at room temperature with the addition of A-cation oleate. In addition, anion exchange processes have been demonstrated for both 2D [R–NH3]2SnX4 RP nanostructures and 3D nanocrystals, showcasing transitions between iodide and bromide counterparts. Furthermore, we have fabricated a thin film of 2D [R–NH3]2SnX4 RP nanostructures for cation exchange, wherein A-cation diffusion through a liquid–solid interface facilitates the transformation into a 3D ASnX3 crystal. This investigation underscores the versatility of ion exchange processes in engineering the composition of Sn-halide perovskite nanostructures and, consequently, modulating their optical properties.
 Loredana Protesescu | Loredana Protesescu, PhD recipient from ETH Zurich (2016) and MIT post-doc, has served as an assistant professor at the Zernike Institute for Advanced Materials, University of Groningen, since 2019. She specializes in the chemistry of novel nanomaterials, emphasizing their structural features and surface chemistry for self-assembly. Her expertise extends to the development of tailored semiconductor nanocrystals, notably contributing to lead and tin halide perovskites with 2D and 3D structures. With over 50 publications and two patents, she advises the Sweden-based startup Peafowl Plasmonics. Her honors include the Nanomaterials 2020 Young Investigator Award and a 2019 Veni NWO grant. |
Introduction
Ion exchange processes in semiconductor nanocrystals (NCs) have emerged as potent chemical methods to easily modulate their composition, crystal structure, and hence, properties, forming new high-quality colloidal nanomaterials inaccessible via the conventional direct synthetic methodology.4–7 The conventional semiconductor NCs (quantum dots, QDs, III–V, II–VI, and IV–VI) as well as metal halide perovskite NCs demonstrate these ion exchange capabilities for both cations and anions while maintaining the structural and morphological integrity of the NCs.8 The ion exchange processes in QDs need various reaction conditions, depending on the ions to be exchanged and the nature of their bonding. For example, to form III–V, Alivisatos and co-workers reported cation exchange of Cd2+ in Cd2As3 NCs 4.55 nm via In3+/Ga3+ at a temperature of 270 °C.9 Moreover, for II–VI NCs, an excess concentration of the exchange cation can drive the reaction at room temperature completely exchanging Ag+ in 4.2 nm CdSe NCs to form Ag2Se NCs and vice versa.5,10,11 The diffusion of the cation into the crystal structure while keeping the anion sub-lattice intact has led to high quality NCs in various QDs while the anion exchange processes have usually yielded poor-quality materials owing to the low mobility of anions.4,12In halide perovskite NCs with the chemical formula AMX3, where M = Pb2+ and Sn2+ and halide X = Cl−, Br−, and I−, with the A cation being Cs+ or FA+, the ionic nature of bonding facilitates these exchange processes under relatively milder reaction conditions.13 Nedelcu et al.6 and Akkerman et al.14 studied the post-synthetic anion exchange in Pb-halide perovskite NCs at room temperature to achieve compositional tunability (I, Br, and Cl) and hence, emission tunability over the full visible region.6 Following these reports, for Pb-halide perovskite NCs, cation (A-site) exchange was also successfully achieved at room temperature in both bulk thin films and NCs (in solution and thin films).14–20 For example, in mixed Pb–Sn perovskite bulk thin films, the exchange of longer organic cations (such as phenyl ethylammonium) with small organic cations (such as formamidinium) by just dipping the thin film in the exchange cation solution was reported.16 In this case, the A cation exchange also led to a change in the dimensionality of the perovskite structure from 2D to 3D.
While reports are abundant for Pb-based perovskites in all forms, the Sn halide perovskite chemistry has remained unexplored. This is majorly due to the lack of synthetic reports on Sn-halide perovskite NCs owing to their ambient environment instability, a result of the oxidation process (Sn2+ → Sn4+ + 2e−) impairing the existence of the desired ASnX3 chemical composition.
In this work, we took advantage of the earlier reported synthesis of high-quality CsSnI3 NCs,21 and demonstrated the cation exchange and anion processes for the Sn-halide perovskite nanostructures of the 2D [R–NH3]2SnX4 Ruddlesden–Popper (RP) perovskite as well as 3D ASnX3 NCs. Scheme 1 outlines the successfully demonstrated ion exchange processes in these materials. We showed that the cation exchange of [R–NH3]+ in 2D [R–NH3]2SnX4 with the Cs+/FA+ cation increases the dimensionality of the nanostructures from 2D to 3D at room temperature. The cation exchange reactions can be performed both in solution and in thin films taking advantage of the solid–liquid interface. Also, directly synthesized 2D [R–NH3]2SnX4 RP nanostructures and 3D CsSnX3 NCs can exchange the anions at room temperature, specifically bromide to iodide and vice versa by utilizing benzoyl halides (BzX) as the anion source. The anion exchange on directly synthesized CsSnI3 and CsSnBr3 NCs was performed to achieve partial and complete exchange for bromide and iodide, respectively. The utilization of excess benzoyl iodide (BzI) led to the oxidation of Sn2+, resulting in the double perovskite structure Cs2SnI6.
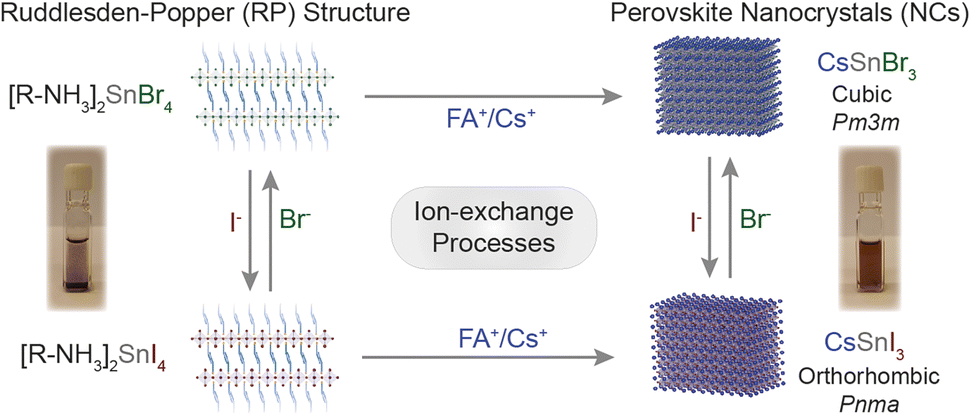 | ||
| Scheme 1 Overview of the ion exchange processes successfully achieved to Sn-halide perovskite nanostructures. | ||
Results and discussion
2D oleyl ammonium-based RP n = 1 Sn-halide perovskite nanostructures
2D RP Sn-halide perovskite structures have the general chemical formula R2An−1SnnX3n+1 (n = 1); here R is a monovalent long-chain organic cation and A is a small organic or inorganic cation, in our case cesium (Cs+) and formamidinium (FA+). For this study, we employed oleyl ammonium (C18H38N+–[R–NH3]+) to synthesize n = 1 2D [R–NH3]2SnX4 RP nanostructures. SnX2 salts were complexed with oleyl amine (OLA) and oleic acid (OA) in a non-coordinating solvent, i.e. octadecene (ODE), to form a transparent coloured solution above 160 °C. This transparent solution self-assembled to form n = 1 2D [R–NH3]2SnX4 RP structures when cooled to room temperature with the help of ice-water (detailed description in the Experimental section). Fig. 1(a) shows the powder X-ray diffraction (XRD) pattern of the 2D [R–NH3]2SnBr4 (green) and [R–NH3]2SnI4 (brown) nanosheets with an aspect ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 in both cases. As [R–NH3]+ has a C18 chain, the n = 1 RP nanosheets both with I and Br had d-spacings as high as 3.6 to 4 nm, which were calculated by using the 2θ periodicity of diffraction peaks observed at low angles of the diffraction pattern.22 These 2D RP nanosheets are highly ordered, identified by the presence of secondary interference peaks at higher 2θ degrees (inset in Fig. 1(a)).23 We also observed that the d-spacings are dependent on the quenching rate of the reaction mixture as the organic moiety is flexible in nature as shown in Fig. S1.† We further analyzed the morphology and size of these 2D RP nanostructures using scanning electron microscopy (SEM) performed in the transmission mode at 18 kV as shown in Fig. 1(b) and (c), which showed the presence of nanosheets (≈4–6 μm in length), with an in-plane aspect ratio of 15 to 20 for iodide and bromide counterparts, respectively. Since these 2D nanostructures are a monolayer of [SnX6]4− octahedra inter-coordinated with [R–NH3]+, they showed a strong quantum confinement effect which can be observed from their optical properties. Fig. 1(d) shows the UV–visible absorption (dotted line) and photoluminescence (PL) (solid line) spectra for 2D [R–NH3]2SnBr4 (green) and [R–NH3]2SnI4 (brown) RP nanosheets. The absorption spectra showed a narrow excitonic peak at 450 nm for 2D [R–NH3]2SnBr4 and at 605 nm for 2D [R–NH3]2SnI4 with a relatively sharp PL peak at 488 and 638 nm, respectively (FWHM of 0.2 eV and 0.15 eV, respectively). These self-assembled 2D RP Sn-halide perovskite nanostructures are not colloidally stable as can be observed via the snapshots in Fig. 1(e), and from the scattering observed in the UV–visible spectroscopy absorption spectra.
:5 in both cases. As [R–NH3]+ has a C18 chain, the n = 1 RP nanosheets both with I and Br had d-spacings as high as 3.6 to 4 nm, which were calculated by using the 2θ periodicity of diffraction peaks observed at low angles of the diffraction pattern.22 These 2D RP nanosheets are highly ordered, identified by the presence of secondary interference peaks at higher 2θ degrees (inset in Fig. 1(a)).23 We also observed that the d-spacings are dependent on the quenching rate of the reaction mixture as the organic moiety is flexible in nature as shown in Fig. S1.† We further analyzed the morphology and size of these 2D RP nanostructures using scanning electron microscopy (SEM) performed in the transmission mode at 18 kV as shown in Fig. 1(b) and (c), which showed the presence of nanosheets (≈4–6 μm in length), with an in-plane aspect ratio of 15 to 20 for iodide and bromide counterparts, respectively. Since these 2D nanostructures are a monolayer of [SnX6]4− octahedra inter-coordinated with [R–NH3]+, they showed a strong quantum confinement effect which can be observed from their optical properties. Fig. 1(d) shows the UV–visible absorption (dotted line) and photoluminescence (PL) (solid line) spectra for 2D [R–NH3]2SnBr4 (green) and [R–NH3]2SnI4 (brown) RP nanosheets. The absorption spectra showed a narrow excitonic peak at 450 nm for 2D [R–NH3]2SnBr4 and at 605 nm for 2D [R–NH3]2SnI4 with a relatively sharp PL peak at 488 and 638 nm, respectively (FWHM of 0.2 eV and 0.15 eV, respectively). These self-assembled 2D RP Sn-halide perovskite nanostructures are not colloidally stable as can be observed via the snapshots in Fig. 1(e), and from the scattering observed in the UV–visible spectroscopy absorption spectra.
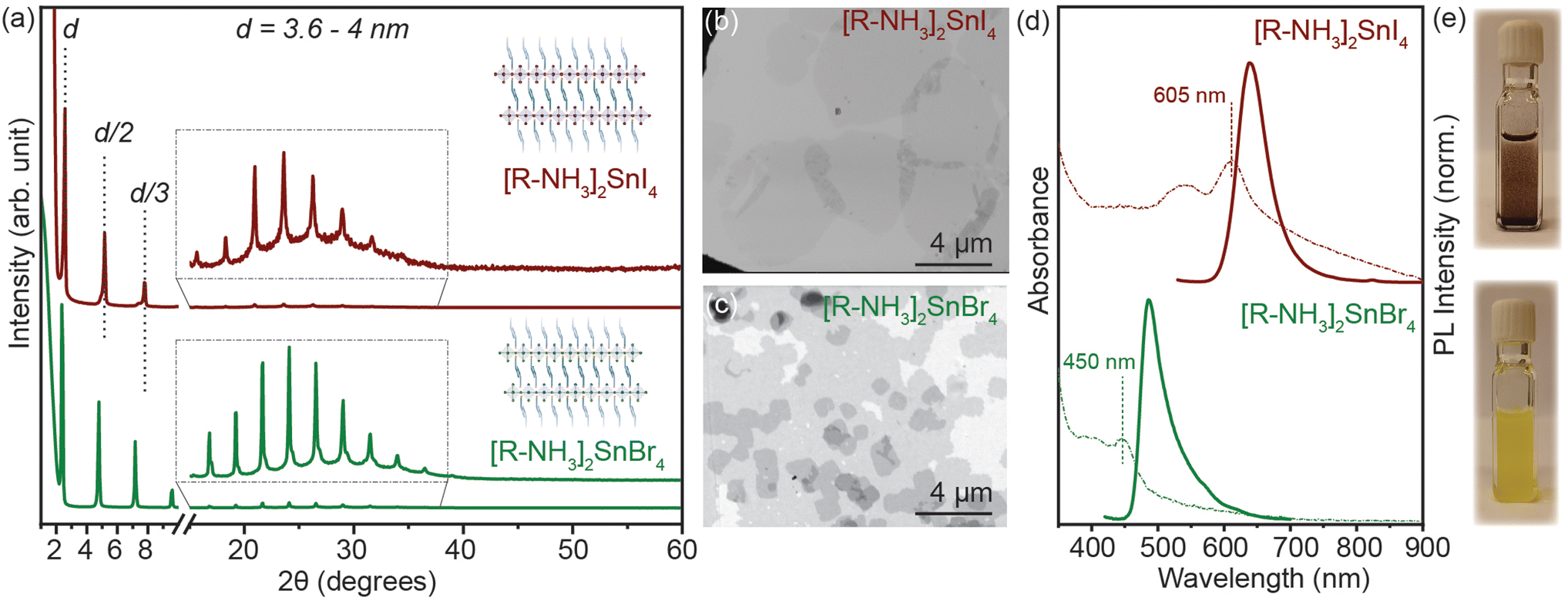 | ||
| Fig. 1 (a) Powder XRD pattern of the 2D [R–NH3]2SnI4 (brown) and [R–NH3]2SnBr4 (green) RP nanostructures respectively. (b and c) SEM Images of the 2D [R–NH3]2SnI4 (brown) and [R–NH3]2SnBr4 (green) RP nanostructures showing the nanosheet morphology. (d) UV–visible absorption (dotted line) and PL (solid line) spectra of the 2D [R–NH3]2SnI4 (brown) and [R–NH3]2SnBr4 (green) RP nanostructures, respectively. (e) Photographs of 2D [R–NH3]2SnI4 (dark brown suspension) and [R–NH3]2SnBr4 (yellow suspension) RP nanostructures in visible light. | ||
Cation exchange in a 2D RP perovskite suspension
Generally, in metal halide perovskites, the cation exchange process has a high activation energy of ≈0.65 eV for exchange between 3D cations (Cs+ and FA+). For larger cations, this can even cause the disintegration of lattice structures undergoing exchange reactions.24 Nonetheless, post-synthetic cation exchange processes have been demonstrated in Pb-halide perovskite NCs and in thin films under precisely controlled physical and chemical parameters,24–27 but not yet fully understood for Sn halide perovskite nanostructures. We further probed how efficient this process can be for our synthesized nanostructures. Oleate complexes of an organic cation, i.e. FA+, and an inorganic cation, i.e. Cs+, were utilized for the cation exchange reaction to obtain 2D [R–NH3]2SnBr4 (green in Fig. 1) and [R–NH3]2SnI4 (brown in Fig. 1) RP perovskite nanostructures. In short, a colloidal suspension of 2D [R–NH3]2SnX4 RP perovskite nanostructures was injected with the FA(Ol) and Cs(Ol) precursors under vigorous stirring at room temperature; the solution became homogeneous and colloidally stable. These cation-exchanged solutions were then purified following the protocol mentioned in the Experimental section. We observed that cation exchange with a small-sized cation, which can form the 3D perovskite structure, increased the dimensionality of the perovskite from 2D to 3D in the form of NCs because of the remaining oleate in the system. The oleate together with extra R–NH3+ will ensure the colloidal stability of the newly formed NCs. We tracked this transformation using absorption and PL spectroscopy (Fig. 2(c) and (f)). Here, we noted a red shift of the excitonic peak from 605 nm to 675 nm and from 450 nm to 488 nm for both the halide systems in FASnX3 NCs.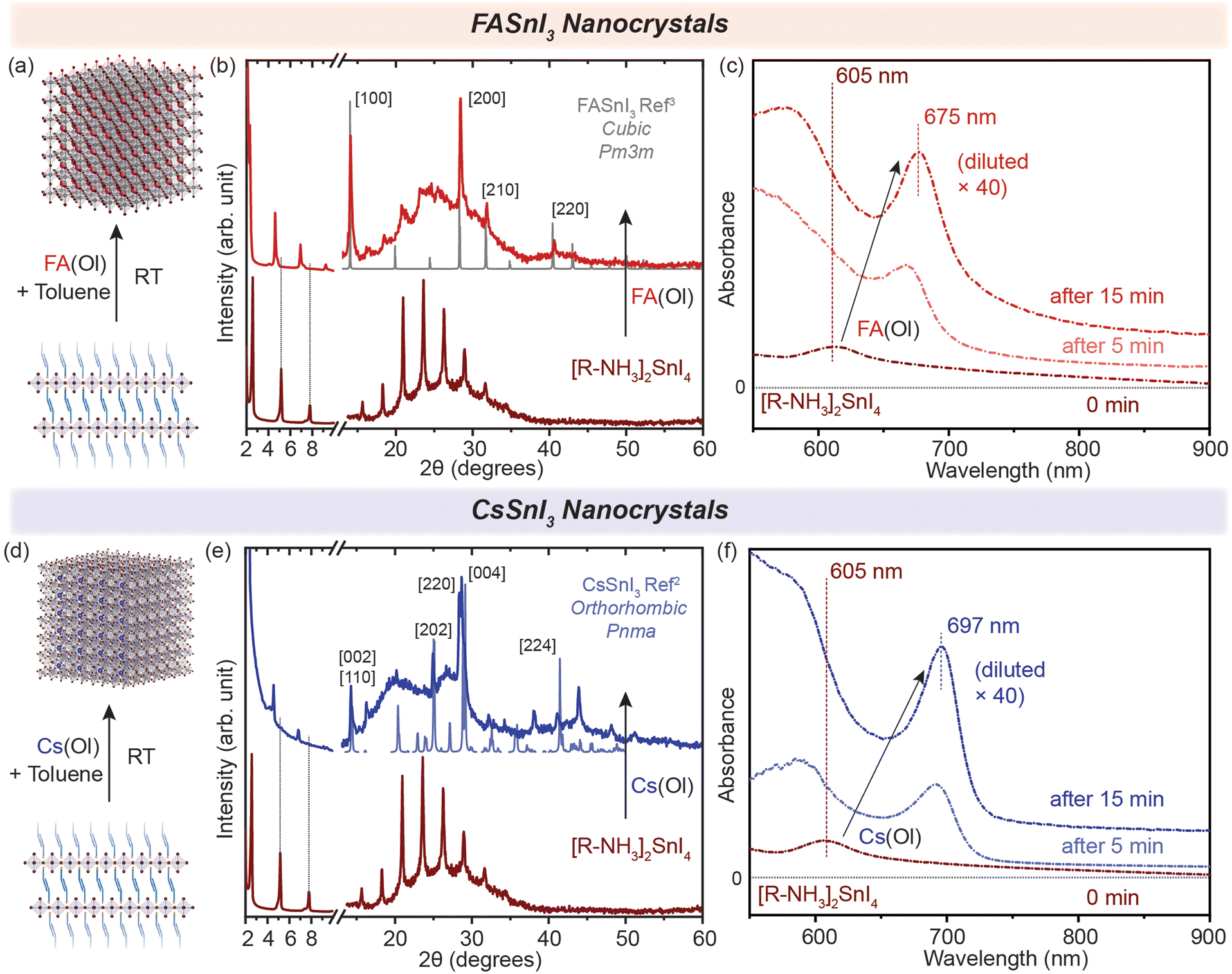 | ||
| Fig. 2 (a and d) Schematic representation of the conversion of 2D [R–NH3]2SnI4 into 3D FASnI3 and CsSnI3 NCs, respectively. (b and e) Powder XRD pattern of the converted 3D FASnI3 (red) and CsSnI3 (blue) NCs from 2D [R–NH3]2SnI4 nanosheets, respectively. (c and f) In situ UV–visible absorption spectra showing the formation of 3D FASnI3 (red) and CsSnI3 (blue) NCs from the 2D [R–NH3]2SnI4 RP nanosheet structure with the addition of A cation. The XRD references are adapted from ref. 3 for FASnI3 (cubic, Pm3m) and ref. 2 for CsSnI3 (orthorhombic, Pnma). | ||
The cation exchange process is illustrated in Fig. 2(a) and (d). In the case of the X = I system, the XRD pattern in Fig. 2(b) and (e) clearly shows the presence of diffraction peaks at higher 2θ values, which correspond to the FASnI3 and CsSnI3 NCs in the cubic crystal structure (Pm3m) and the orthorhombic crystal structure (Pnma), respectively. The diffraction peaks at lower angles and secondary interferences belonging to the 2D [R–NH3]2SnI4 (brown) RP perovskite diminished, showing the conversion of 2D [R–NH3]2SnI4 into both FASnI3 and CsSnI3. Similarly, for the cation exchange in 2D [R–NH3]2SnBr4 (green), the injection of the FA(Ol) and Cs(Ol) precursor solution led to the formation of FASnBr3 and CsSnBr3 NCs, which crystallized in the cubic perovskite crystal structure (Pm3m) as can be observed in Fig. S2(b) and (e)† with the presence of 2D [R–NH3]2SnBr4 in the Cs case. Note that the diffraction measurements were performed under an inert atmosphere using a dome that cannot be revolved; therefore the relative intensities of the diffraction peaks toward a certain lattice plane are preferred over others. Fig. S3† shows the SEM images demonstrating the formation of 3D FASnI3 NCs with an average edge length of 15–20 nm as an example. This change in the dimensionality of the Sn-halide perovskite nanostructures from 2D to 3D is better represented by their optical properties. Fig. 2(c) shows the evolution of UV–visible absorption spectra with the addition of FA(Ol) in a non-colloidal suspension of 2D [R–NH3]2SnI4. We observed that the excitonic peak began to red shift from 605 nm and remained at 675 nm after 15 min. At that point, the solution became colloidally stable and saturated the detector. Hence, we used a 40 times diluted solution to perform the absorption measurements. These optical features are in a similar range as reported earlier for FASnI3 NCs.28,29 For Cs(Ol) addition, the excitonic peak remained at 697 nm after 15 min for half the concentration when compared to FA(Ol) (Fig. 2(f)). Fig. S2(c) and (e)† plot the temporal change of UV–visible spectra for the bromide case, where a similar red shift was observed; in the case of FA(Ol), the peak remained at 488 nm and in the case of Cs(Ol), the peak remained at 530 nm after 15 min for the same concentration of the cation. The bulk band gap for FASnBr3 and CsSnBr3 for the cubic perovskite crystal structure (Pm3m) was previously reported to be 2.4 eV (≈516 nm) and 1.92 (≈645 nm).30–32 Thus, our observations are in good agreement with the formed nanostructures being within the confinement range of these materials. We also kept the cation exchange reaction for extended times up to 20 h (Fig. S4†), which led to more bulk-like structures. Furthermore, it was essential to know the effect of the ratios of FA(Ol) and Cs(Ol) with respect to 2D [R–NH3]2SnX4 RP perovskite nanostructures on the cation exchange process and the final product obtained.
To test this, we started by adding 120 μL of 0.05 M FA(Ol), and within 30 min, we observed that the excitonic peak remained stable at 645 nm (Fig. 3(a)). However, when the ratio of FA:[R–NH3]2SnI4 was increased, and we doubled the volume of FA(Ol) added to 240 μL of 0.05 M solution, the wavelength of the excitonic peak quickly increased from 605 nm to be stabilized as a broad feature at 650 nm. We concluded that the concentration and volume of the FA(Ol) solution dictate the steady transformation into 3D NCs over a period of time. For the chemical transformation of [R–NH3]2SnI4 nanosheets into CsSnI3 NCs, similar evolution was observed in the absorption spectra, with the excitonic peak stable at 699 nm, as displayed in Fig. 3(b). Furthermore, the bromide counterparts demonstrated similar evolution of absorbance spectra with excitonic peaks stabilizing at 540 nm and 600 nm for FASnBr3 and CsSnBr3, respectively (Fig. S5†). For the cation exchange from [R–NH3]+ to FA+ or Cs+, three simultaneous processes are involved in this process, namely, the cation exchange chemical reaction, a structural reconstruction that is not thermodynamically favorable, and a morphology change. Therefore, the cation exchange reactions are highly sensitive to the reaction time and concentration and require a precise supply of exchange cations to recrystallize into 3D halide perovskite NCs.33,34
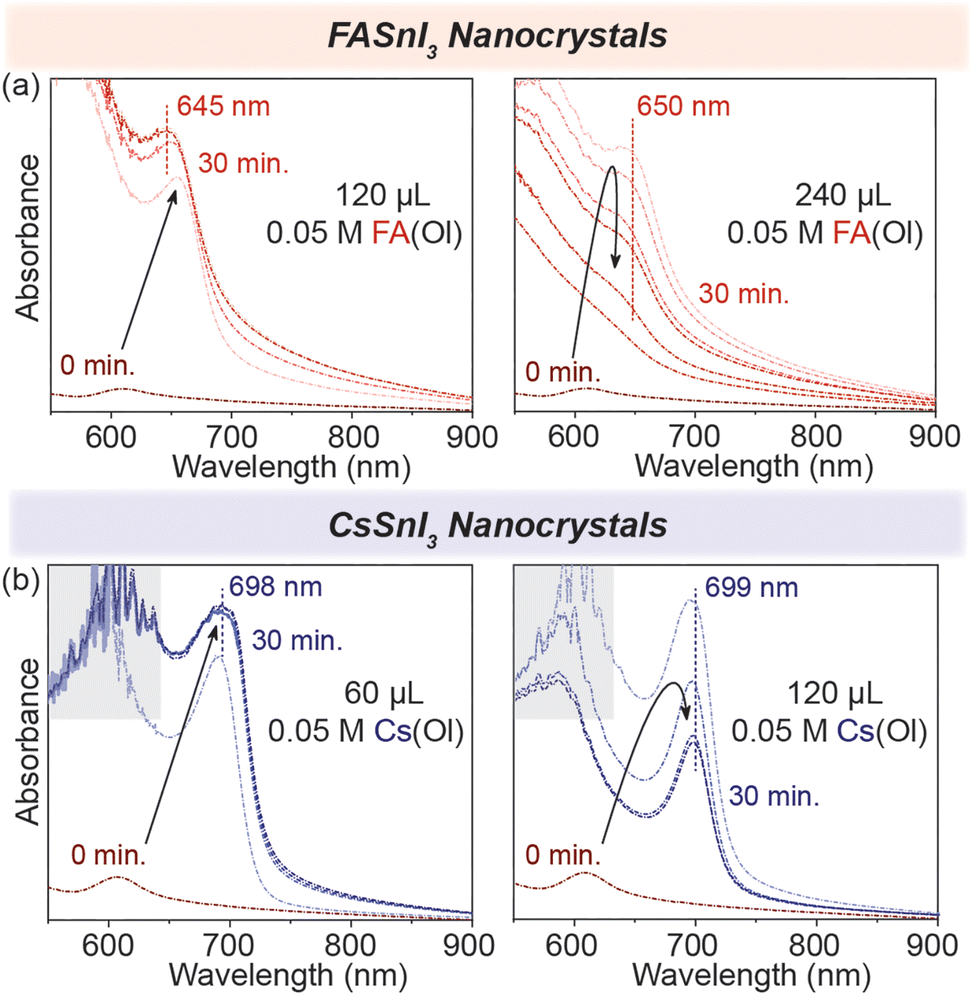 | ||
| Fig. 3 Concentration dependence in situ UV–visible absorption spectroscopy. (a) The formation of the 3D FASnI3 (red) NCs with the addition of FA(Ol) in the 2D [R–NH3]2SnI4 RP structures for 30 min. (b) The formation of the 3D CsSnI3 (blue) NCs with the addition of Cs(Ol) in the 2D [R–NH3]2SnI4 RP structures for 30 min. The grey area highlights the detector saturation region in the above plots. | ||
Cation exchange in 2D RP perovskite thin films
We demonstrated that the cation exchange reactions can be successfully performed in solution converting the non-colloidal suspension of 2D [R–NH3]2SnX4 RP nanostructures into 3D ASnX3 NCs at room temperature. While this process can have advantages in precisely controlling the synthesis of ASnX3 NCs, for most optoelectronic applications, those transformations are desired in thin films.35,36 So far, a few reports have successfully demonstrated facile cation exchange reactions on Sn-halide and mixed Pb–Sn halide perovskite bulk thin films, which transforms the dimensionality of the perovskite structure from 2D to 3D.16,34,37 However, there are no similar reports showcasing the dimensionality transformation with the existence of a strong confinement effect indicative of newly fabricated NCs. To fabricate high-quality thin films of the non-colloidal suspension, we utilized the modified drop-casting methodology illustrated in Fig. 4(a), which helped in the uniform outspread of the 2D [R–NH3]2SnX4 RP perovskite nanostructures over a glass substrate, taking advantage of the buoyant force.38–40 Toluene is used to wet the surface of the substrate for a homogeneous spread, which is then naturally allowed to dry over a period of time (≈20–30 min). When fully dried, we dropped 30 μL of 0.05 M FA(Ol) and 20 μL of 0.05 M Cs(Ol) precursor solutions on the 2D [R–NH3]2SnX4 RP perovskite thin film, which quickly changed its colour from deep red to dark brown as a result of the cation exchange reaction. We also tried the spin-coating procedure but found it to be unsuitable for fabricating a thin film of the non-colloidal suspension.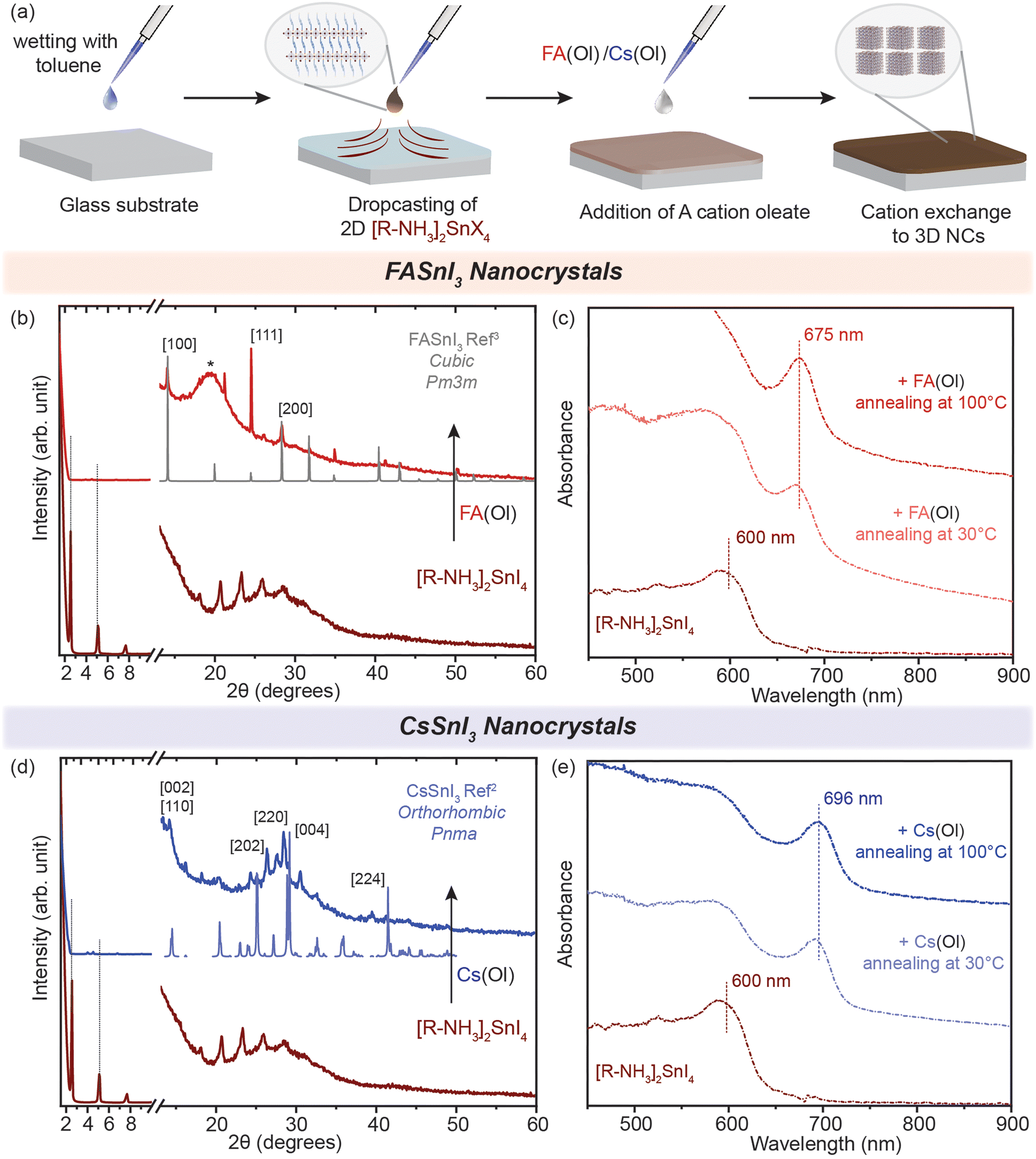 | ||
| Fig. 4 Cation exchange in thin films. (a) Schematic of the methodology utilized to fabricate the thin films. (b and d) Powder XRD patterns of the converted 3D FASnI3 (red) and CsSnI3 (blue) NC thin film from the 2D [R–NH3]2SnI4 RP perovskite, respectively. (c and e) UV–visible spectroscopy of the formation of the 3D FASnI3 (red) and CsSnI3 (blue) nanocrystal thin film from the 2D [R–NH3]2SnI4 RP structure annealed at 30 °C and 100 °C for 1 h. * marks the peak arising from the glass substrate in the dome XRD sample holder. The XRD references are adapted from ref. 3 for FASnI3 (cubic, Pm3m) and ref. 2 for CsSnI3 (orthorhombic, Pnma). | ||
The morphology of these 2D [R–NH3]2SnX4 RP perovskite thin films before cation exchange can be seen in Fig. S6(a) and (b)† for the bromide and iodide cases, respectively. After naturally drying the 2D [R–NH3]2SnX4 RP perovskite thin film, the FA(Ol)/Cs(Ol) precursor solution (0.05 M) was slowly dropped on it and annealed for an hour at different temperatures (from 30 °C to 100 °C) for the recrystallization process. The converted thin films showed negligible diffraction peaks pertaining to the 2D [R–NH3]2SnX4 RP perovskite 2θ values from 1 to 10 (Fig. 4(b) and (d)). The cation exchange with FA+ led to a cubic crystal structure (Pm3m) while with Cs+ it led to an orthorhombic crystal phase (Pnma) (as measured using a bench-top XRD system with reasonable resolution). This observation is consistent with the crystal phases observed in cation exchange in the non-colloidal suspension (Fig. 2). However, we noticed that in the case of FA+, the FASnI3 NC thin film had a preferred ordering along the [111] lattice planes. In Fig. 4(c) and (e), we show the absorbance spectra of the obtained thin films recorded in the transmission mode. Both FA+ and Cs+ systems displayed a similar red shift as observed for solutions with an insignificant effect of the annealing temperature. The excitonic peak corresponding to FASnI3 and CsSnI3 NCs in thin films was observed at 675 and 696 nm. We used various concentrations (from 0.002 to 0.05 M) and annealing temperatures (from 30 to 100 °C) for the conversion of 2D [R–NH3]2SnBr4 (green) and [R–NH3]2SnI4 (brown) with the FA+ cation (Fig. S7†). A higher annealing temperature led to the broadening of the excitonic feature towards the bandgap of the bulk materials, which is consistent with the formation of more bulk in the thin films. For [R–NH3]2SnX4, the increase in the concentration of the Cs+ cation led to the degradation of the nanostructures to form a white coloured thin film corresponding cesium halides (Fig. S8†).
Anion exchange in Sn-halide perovskite nanostructures
We have shown that the cation exchange processes offer a large playground for the Sn-halide perovskite chemistry. Next, we aimed to probe the mobility of anions in our systems. Although anion exchange processes have been demonstrated to be fast and facile in the Pb-halide perovskite NCs, these processes are kinetically driven under a concentration gradient of the exchange anion.6,17,18,41 For Sn-halide perovskite NCs, there is no post-synthetic anion exchange report.For our systems, we used benzoyl halide as an exchange halide source. In a typical experiment, we loaded a glass vial with 100 μL of 2D RP nanostructures (C ≈ 3 mg mL−1) or 200 μL of 3D NCs (C ≈ 20 mg mL−1) and diluted it with 1.5 mL of toluene. To this system, a BzX stock solution (50 to 300 μL) was added with constant stirring, and the mixture was kept for 10 min and then purified as mentioned in the Experimental section. We observed an anion exchange reaction in 2D RP Sn-halide nanostructures and in 3D CsSnX3 NCs within a few minutes as illustrated in Fig. 5(a) and (d). The XRD pattern shown in Fig. 5(b) confirmed the presence of 2D RP nanostructures after the anion exchange for each halide, with the lattice spacing varying from 3.6 to 4 nm depending on the extent of the nanostructures towards the [100] plane in the exchanged perovskite. The changes in the d-spacing were relatively smaller after the halide exchange for the exchanged structure (≈4 times the difference between the Sn–X bond lengths). Thus, the XRD pattern only confirms the presence of 2D RP nanostructures after the anion-exchange. The steady-state PL of the directly synthesized 2D RP nanostructures as can be observed from Fig. 5(c) shows a peak at 488 nm for [R–NH3]2SnBr4 and at 635 nm for [R–NH3]2SnI4. After the bromide exchange, the PL peak of the exchanged 2D [R–NH3]2SnBr4 was blue shifted to 488 nm, presenting a similar band gap to the directly synthesized (no anion exchange) 2D [R–NH3]2SnBr4. Similarly, for the iodide exchange, the PL peak of the exchanged 2D [R–NH3]2SnI4 was red shifted to 630 nm. The restoration of the PL peak to the directly synthesized 2D RP nanostructures confirms the anion exchange reaction.
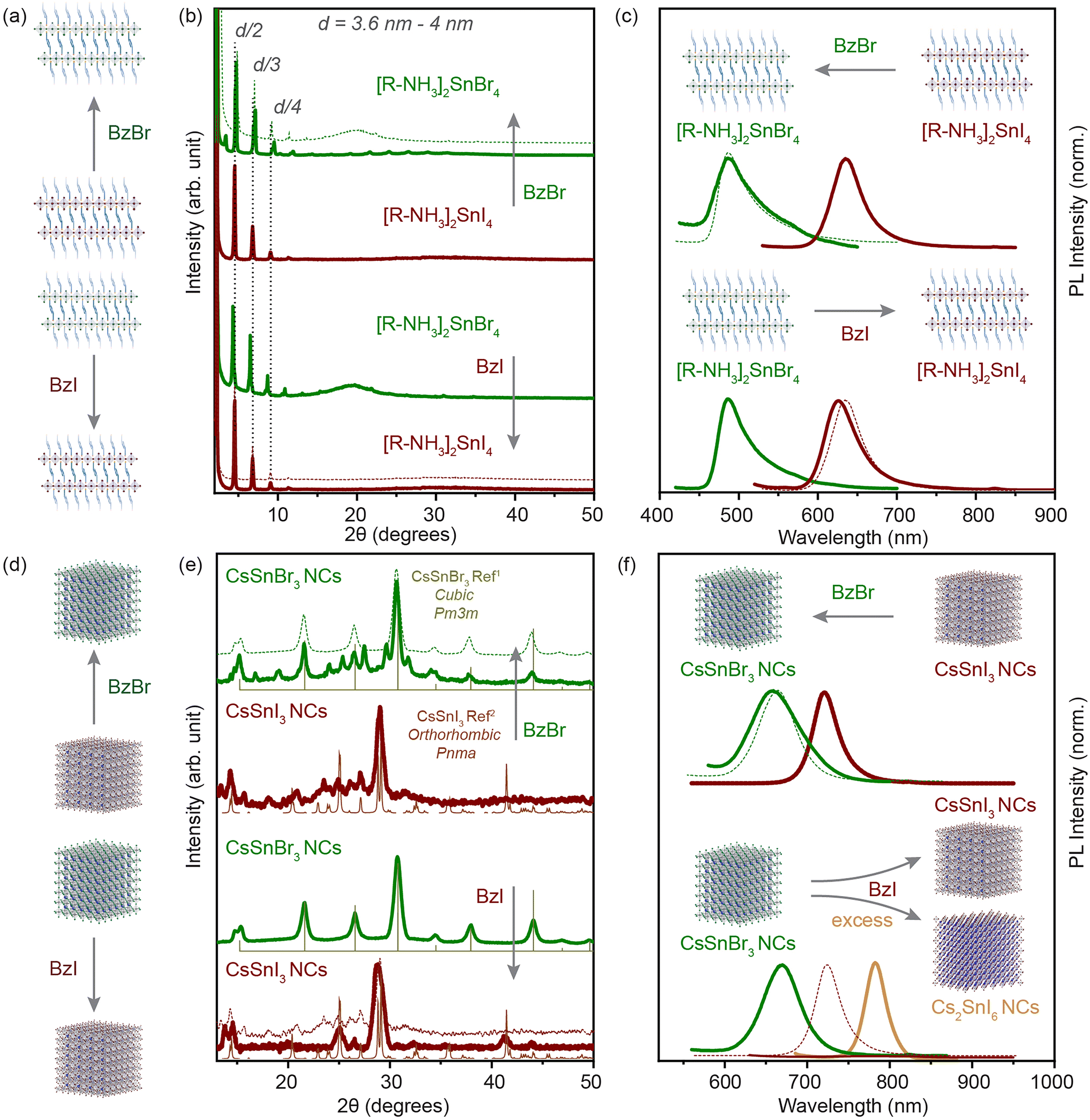 | ||
| Fig. 5 Anion-exchange processes in solution. (a) and (d) illustrate the direction of the anion exchange reaction in the 2D RP nanostructures and the 3D NCs. (b and c) The powder XRD pattern and PL spectra of the 2D RP nanostructures in comparison with the anion-exchanged product for iodide (brown) and bromide (green). (e and f) The XRD pattern and PL spectra of the 3D NCs in comparison with the anion-exchanged product for iodide (brown) and bromide (green). The XRD references are plotted for CsSnBr3 cubic Pm3m (light green) and CsSnI3 orthorhombic Pnma (light brown).1,2 Dotted lines in each plot shows the feature of the directly synthesized product for comparison. | ||
We applied a similar approach to the 3D CsSnX3 NCs. Fig. 5(e) shows the XRD pattern for the directly synthesized 3D NCs in comparison with the anion-exchanged NCs (experimental details in the Experimental section). The iodide exchanged CsSnI3 NCs show full transformation into an orthorhombic Pnma crystal structure from apparently cubic Pm3m CsSnBr3 NCs. However, the well-defined PL peak disappeared after the anion exchange process most probably due to low PLQY, a result of self p-doping, which is a result of oxidation of the Sn2+ cation especially on the surface of NCs (Fig. 5(f)) as shown also in previous studies.42 Note that if the BzI used for the iodide exchange was added in an excessive amount or it was synthesized for more than a week in advance, the obtained material would be correlated to the XRD pattern of Cs2SnI6 in a cubic Fm3m (Fig. S11†). This observation was in good agreement with the PL spectroscopy where we identified a shift of the initial peak to 780 nm (Fig. 5(f)). Fig. S12† shows the evolution of the crystal structure from CsSnI3 (orthorhombic, Pnma) to Cs2SnI6 (cubic, Fm3m) when an increasing amount of BzI was added. The bromide exchanged CsSnI3 NCs showed the transformation into the cubic Pm3m crystal phase majorly with the negligible presence of the CsSnI3 orthorhombic Pnma diffraction peaks, which does not indicate a complete halide exchange process. (Note that we have associated the crystal structure after analyzing bench-top XRD measurements in a protected environment.) However, the PL peak blue shifted to 665 nm, which correlated well with the directly synthesized CsSnBr3 NCs. In addition, the increase in FWHM (≈0.02 eV) indicated the increase in size-dispersity of the exchanged NCs. We further probed the anion exchange from CsSnBr3 NCs to CsSnCl3 NCs via BzCl. The blue shift was observed in the absorbance and PL spectra after the addition of BzCl, however, due to the instability of the crystal structure, we were unable to perform any structural characterization (Fig. S9–S11†).
Conclusions
The ion exchange processes within Sn-halide perovskite nanostructures facilitated facile compositional tuning, inducing systematic alterations in band gap characteristics through either the modulation of dimensionality, as observed in cation exchange, or the modification of the crystal structure, as evidenced in anion exchange. Notably, cation exchange transitioned seamlessly, resulting in the transformation of two-dimensional [R–NH3]2SnX4 RP perovskite nanostructures into three-dimensional nanocrystals (3D NCs) at room temperature, which is particularly evident in the iodide counterparts in comparison with bromide. The thin films composed of 2D [R–NH3]2SnX4 RP perovskite nanostructures exhibited diffusion of smaller cations through a liquid–solid interface, culminating in the conversion to higher-dimensional 3D NCs. Anion exchange processes, utilizing benzoyl halide sources, were conducted on both 2D [R–NH3]2SnX4 RP nanostructures and 3D NCs. The anion exchange to bromide-based 3D NCs induced a crystalline structural transition from orthorhombic to cubic, while the iodide exchange manifested the reverse transformation. This study exemplifies the ion-exchange capabilities inherent in Sn-halide perovskite nanostructures, demonstrating their capacity to traverse inter-dimensional and inter-crystallographic phases under controlled physical and chemical parameters.Experimental section
Materials
Cesium carbonate (Cs2CO3, 99.9%), formamidine acetate (FAAc, 99%), tin(II) iodide (anhydrous beads, 99.99%), benzoyl bromide (BzBr, 97%), benzoyl chloride (BzCl, 99%), sodium iodide (NaI, 99.999%), octadecene (ODE, Tech. grade, 90%), oleic acid (OA, tech. grade, 90%) and oleyl amine (OLA, tech. grade, 80–90%) were purchased from Sigma Aldrich. Tin(II) bromide (SnBr2, 99.9%) was purchased from TCI Europe. Toluene (extra dry, 99.85%) was purchased from ACROS organics.Synthesis of the formamidium oleate precursor (0.25 M)
Formamidinium acetate (0.26 g, 2.5 mmol), OlAc (1.25 ml, 4 mmol), and ODE (10 ml) were loaded into a 25 ml 3-neck round-bottom flask and vigorously stirred under vacuum for 1 h at 50 °C and then the temperature was increased slowly to 105 °C. The reaction mixture was degassed for 1 h. Now under an N2 flow, the reaction mixture was heated up to 160 °C to assure the complete conversion of formamidine acetate salt into formamidinium oleate. The obtained formamidinium oleate solution was carefully transferred to the glove box when cooled to room temperature. This solution was diluted with toluene to obtain a 0.05 M FA(Ol) precursor solution.Synthesis of cesium oleate precursor (0.22 M)
Cesium carbonate (0.408 g, 1.25 mmol), OlAc (1.25 ml, 4 mmol), and ODE (10 ml) were loaded into a 25 ml 3-neck round-bottom flask and vigorously stirred under vacuum for 1 h at 120 °C. Furthermore, the reaction mixture was heated up to 180 °C under an N2 flow to ensure the complete conversion of cesium carbonate into cesium oleate. The obtained cesium oleate solution was carefully transferred to the glove box when cooled to room temperature. This solution was diluted with toluene to obtain a 0.05 M Cs(Ol) precursor solution and heated to produce a clear solution before the usage.Synthesis of the 2D [R–NH3]2SnX4 RP perovskite nanostructures
SnI2/SnBr2 (2 mmol) along with dried OA (0.63 mL, 2 mmol), OLA (0.66 mL, 2 mmol), and ODE (5 mL) were loaded into a 25 mL 3-neck flask inside a N2 glovebox. The reaction mixture was carefully transferred to a Schlenk line and stirred under vacuum for 10 min at room temperature, after which the temperature was increased to 105 °C and the mixture vigorously stirred for 45 min. The temperature was further increased to 200 °C under an N2 flow, and the reaction was then cooled to room temperature using an ice-water bath after 5 s. Note: Avoid the shaking of the round-bottom flask at this point as the reaction mixture quickly crystallizes at the cooler surfaces of the flask. The reaction mixture turns dark red in the case of iodide and yellow in the case of bromide. The reaction flask was carefully transferred to the glovebox for the purification step. The crude solution was equally divided into two centrifuge tubes and centrifuged at 5000 rpm (7082 rcf) for 3 min. The clear supernatant was discarded, and the precipitate was redispersed in toluene (5 mL) followed by centrifugation at 13000 rpm (18412 rcf) for 5 min. The supernatant was discarded again and the colloidally unstable yellow precipitate was stored in 5 mL of toluene in the glovebox for further experiments.
Cation exchange of the 2D [R–NH3]2SnX4 RP perovskite nanostructure suspension
The stock solution of the 2D [R–NH3]2SnX4 RP perovskite nanostructures was prepared in toluene with a concentration of ≈3 mg mL−1. A magnetic stirrer was placed in a 4 mL glass vial containing 200 μL of the stock solution with 1.5 mL of toluene, and this vial was then injected with the required volumes of 0.05 M FA(Ol)/Cs(Ol) under vigorous stirring to observe the changes in the UV–visible spectra. For a complete cation exchange, 240 μL of 0.05 M FA(Ol) and 120 μL of 0.05 M Cs(Ol) precursor solution injection followed by 30 min of stirring was utilized. For the concentration dependence study, different volumes of the 0.05 M FA(Ol)/Cs(Ol) precursor solution were employed.Cation exchange of the 2D [R–NH3]2SnX4 RP perovskite nanostructure thin film
The soda-lime glass substrates (10 mm × 10 mm) were cleaned with soap water and washed using distilled water. The substrates were then subjected to sonication for 15 min in ethanol, dried, and sonicated for another 15 min in acetone, followed by drying with a strong airflow. The cleaned substrates were transferred to the glove box. For the thin-film fabrication of 2D [R–NH3]2SnX4 RP perovskite nanostructures, 20 μL of anhydrous toluene was used to wet the surface of the substrate. Then 3 mg mL−1 solution of 2D [R–NH3]2SnX4 was dropcast with the assistance of toluene. This thin film was then dried naturally within 20–30 min. After the film was dried, 30 μL of a 0.05 M FA(Ol) or 20 μL Cs(Ol) solution was dropped on the thin film, which made the appearance of the film darker and more homogeneous. This thin film was then kept for annealing for 1 h at different temperatures to provide time for cation diffusion through liquid–solid interface. After 1 h, the thin film can be washed with the help of 20 μL of anhydrous toluene over a spin-coater.Synthesis of the 3D CsSnX3 perovskite NCs
The 3D CsSnX3 NCs were synthesized utilizing the protocol from Gahlot et al.21 SnI2/SnBr2 (2 mmol) along with dried OA (0.63 mL, 2 mmol), OLA (0.66 mL, 2 mmol), and ODE (5 mL) were loaded into a 25 mL 3-neck flask inside a N2 glovebox. The reaction mixture was carefully transferred to a Schlenk line and stirred under vacuum for 10 min at room temperature, after which the temperature was increased to 105 °C and the mixture vigorously stirred for 45 min. The temperature was further increased to 200 °C under an N2 flow, and 2.8 mL (0.622 mmol) of 0.222 M cesium oleate was swiftly injected. The reaction was stopped after 25 s by quickly immersing the reaction flask in an ice-water bath. The reaction flask was carefully transferred to the glovebox for the purification step. The crude solution was equally divided into two centrifuge tubes and centrifuged at 5000 rpm (7082 rcf) for 3 min. The supernatant was discarded, and the precipitate was redispersed in toluene (5 mL) followed by centrifugation at 13000 rpm (18412 rcf) for 5 min. The supernatant was discarded and the precipitate was re-dispersed in 5 mL of toluene, and stored in the glovebox for further experiments.
Synthesis of benzoyl iodide (BzI)
Benzoyl iodide was synthesized following the modified procedure from Theobald and Smith.43 In the glove box, benzoyl chloride (2.8 mL) was added to a 20 mL glass vial containing 6 g of sodium iodide. This reaction mixture was vigorously stirred overnight at 80 °C for 6 h. The dark orange-red solution was diluted with 3 mL toluene and filtered with a 0.45 μm PTFE filter. This solution was then utilized for anion exchange in 2D [R–NH3]2SnX4 RP perovskite nanostructures and 3D CsSnX3 perovskite NCs. It would be best to utilize the fresh BzI solution.Anion exchange of the 2D [R–NH3]2SnX4 RP perovskite nanostructures
The stock solution of 2D [R–NH3]2SnX4 RP perovskite nanostructures was prepared in toluene with a concentration of ≈3 mg mL−1. 100 μL of the stock solution was mixed with 1.5 mL of toluene, 100 μL of BzI was added to 2D [R–NH3]2SnBr4/100 μL of BzBr (v/v – 1/9 in toluene) was added to 2D [R–NH3]2SnI4 for an anion exchange reaction. The reaction was stirred for 10 min. After 10 min, the reaction mixture was transferred to a centrifuge tube. The reaction mixture was centrifuged at 13000 rpm (18412 rcf) for 5 min, the supernatant was discarded, and the precipitate was redissolved in 3 ml of toluene, which was again centrifuged using the previous parameters. More supernatant was discarded, and the precipitate was redissolved in 3 ml of toluene and utilized for further characterization.
Anion exchange of CsSnI3 perovskite NCs
A stock solution of 20 mg mL−1 of freshly prepared CsSnI3 NCs in toluene was prepared. The BzBr and BzCl precursor solutions were prepared by dissolving 100 μL in 5 mL toluene. For an anion exchange experiment, 200 μL of CsSnI3 NCs was mixed with 1.5 mL of toluene in a 4 mL glass vial with a magnetic stirrer. 300 μL of the BzBr solution was added and stirred for 1 h. The reaction mixture was then centrifuged at 13000 rpm (18412 rcf) for 5 min. The supernatant was discarded and the precipitate was redissolved in 1.5 mL of toluene. Then for a Cl exchange, the same solution, added with 300 μL of BzCl, was stirred for 1 h. The reaction mixture was then centrifuged at 13000 rpm (18412 rcf) for 5 min. The supernatant was discarded and the precipitate was redissolved in 1.5 mL of toluene. For further structural and optical characterization, the precipitate obtained was re-dissolved in 2 ml of toluene and the solution was centrifuged at 2000 rpm for 3 min. The precipitate was discarded and the supernatant was used. These anion exchange processes are air and time-sensitive in nature.
Anion exchange of CsSnBr3 perovskite NCs
A stock solution of 20 mg mL−1 CsSnBr3 NCs in toluene was prepared. The solutions of freshly prepared BzI and BzCl were used in the same concentration mentioned above. For an anion exchange experiment, 200 μL of CsSnBr3 NCs was mixed with 1.5 mL of toluene in a 4 mL glass vial with a magnetic stirrer. 50 μL of the BzI solution and 100 μL of the BzCl solution were added and the mixture was stirred for 1 h to obtain CsSnI3 and CsSnCl3 NCs, respectively. The reaction mixture was then centrifuged at 13000 rpm (18412 rcf) for 5 min. The supernatant was discarded and the precipitate was redissolved in 1.5 mL of toluene. The precipitate obtained was re-dissolved in 2 ml of toluene and centrifuged at 2000 rpm for 3 min. The precipitate was discarded and the supernatant was used for characterization. These anion exchange processes are air and time-sensitive in nature.
Characterization
All measurements were performed in an air-free environment. Powder X-ray diffraction measurements were performed in an inert dome sample holder in a Bruker D8 Advanced diffractometer with Bragg–Brentano geometry using Cu Kα radiation (λ = 1.54 Å) and a Lynxeye detector. SEM images were acquired via a FEI Helios G4 CX electron microscope in the transmission mode operated at 18 kV with 1.4 nAmp beam current using an ultrathin grid with 400 mesh, Cu (Ted Pella, Inc. 01822-F) for NCs while the lens detector mode (TLD) was used for thin films. The absorption measurements were performed on a table-top Avantes UV–vis spectrophotometer using a tungsten and halogen filament lamp as the excitation source. The steady-state photoluminescence measurements were performed on a Horiba Scientific Jobin Yvon spectrometer equipped with a PMT detector.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Dutch Research Council (NWO) via VENI grant number VI.Veni.192.048 and by the Advanced Materials research program of the Zernike National Research Centre under the Bonus Incentive Scheme of the Dutch Ministry for Education, Culture and Science. The authors acknowledge the technical support from Jacob Baas and Dr Peter Bramwell.References
- D. H. Fabini, G. Laurita, J. S. Bechtel, C. C. Stoumpos, H. A. Evans, A. G. Kontos, Y. S. Raptis, P. Falaras, A. Van der Ven, M. G. Kanatzidis and R. Seshadri, J. Am. Chem. Soc., 2016, 138, 11820–11832 CrossRef CAS PubMed.
- I. Chung, J.-H. Song, J. Im, J. Androulakis, C. D. Malliakas, H. Li, A. J. Freeman, J. T. Kenney and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 8579–8587 CrossRef CAS PubMed.
- S. Kahmann, O. Nazarenko, S. Shao, O. Hordiichuk, M. Kepenekian, J. Even, M. V. Kovalenko, G. R. Blake and M. A. Loi, ACS Energy Lett., 2020, 5, 2512–2519 CrossRef CAS.
- S. Gupta, S. V. Kershaw and A. L. Rogach, Adv. Mater., 2013, 25, 6923–6944 CrossRef CAS PubMed.
- D. H. Son, S. M. Hughes, Y. Yin and A. Paul Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- X. Li, M. Ji, H. Li, H. Wang, M. Xu, H. Rong, J. Wei, J. Liu, J. Liu, W. Chen, C. Zhu, J. Wang and J. Zhang, Matter, 2020, 2, 554–586 CrossRef.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 19977–19980 CrossRef CAS PubMed.
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef CAS PubMed.
- P. K. Jain, L. Amirav, S. Aloni and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 9997–9999 CrossRef CAS PubMed.
- M. Saruyama, R. Sato and T. Teranishi, Acc. Chem. Res., 2021, 54, 765–775 CrossRef CAS PubMed.
- H. Jiang, S. Cui, Y. Chen and H. Zhong, Nano Sel., 2021, 2, 2040–2060 CrossRef CAS.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- A. Greco, A. Hinderhofer, M. I. Dar, N. Arora, J. Hagenlocher, A. Chumakov, M. Grätzel and F. Schreiber, J. Phys. Chem. Lett., 2018, 9, 6750–6754 CrossRef CAS PubMed.
- J. Xi, H. Duim, M. Pitaro, K. Gahlot, J. Dong, G. Portale and M. A. Loi, Adv. Funct. Mater., 2021, 31, 2105734 CrossRef CAS.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Chem. Mater., 2018, 30, 4887–4891 CrossRef CAS.
- N. Livakas, S. Toso, Y. P. Ivanov, T. Das, S. Chakraborty, G. Divitini and L. Manna, J. Am. Chem. Soc., 2023, 145, 20442–20450 CrossRef CAS PubMed.
- G. E. Eperon and D. S. Ginger, ACS Energy Lett., 2017, 2, 1190–1196 CrossRef CAS.
- Y. Zhang, D. Lu, M. Gao, M. Lai, J. Lin, T. Lei, Z. Lin, L. N. Quan and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12648–12653 CrossRef CAS PubMed.
- K. Gahlot, S. de Graaf, H. Duim, G. Nedelcu, R. M. Koushki, M. Ahmadi, D. Gavhane, A. Lasorsa, O. De Luca, P. Rudolf, P. C. A. van der Wel, M. A. Loi, B. J. Kooi, G. Portale, J. Calbo and L. Protesescu, Adv. Mater., 2022, 34, 2201353 CrossRef CAS PubMed.
- S. Ghimire, K. Oldenburg, S. Bartling, R. Lesyuk and C. Klinke, ACS Energy Lett., 2022, 7, 975–983 CrossRef CAS.
- S. Toso, D. Baranov, C. Giannini and L. Manna, ACS Nano, 2021, 15, 20341–20352 CrossRef CAS PubMed.
- A. Hazarika, Q. Zhao, E. A. Gaulding, J. A. Christians, B. Dou, A. R. Marshall, T. Moot, J. J. Berry, J. C. Johnson and J. M. Luther, ACS Nano, 2018, 12, 10327–10337 CrossRef CAS PubMed.
- C. Otero-Martínez, M. Imran, N. J. Schrenker, J. Ye, K. Ji, A. Rao, S. D. Stranks, R. L. Z. Hoye, S. Bals, L. Manna, J. Pérez-Juste and L. Polavarapu, Angew. Chem., Int. Ed., 2022, 61, e202205617 CrossRef PubMed.
- M. Hao, Y. Bai, S. Zeiske, L. Ren, J. Liu, Y. Yuan, N. Zarrabi, N. Cheng, M. Ghasemi, P. Chen, M. Lyu, D. He, J.-H. Yun, Y. Du, Y. Wang, S. Ding, A. Armin, P. Meredith, G. Liu, H.-M. Cheng and L. Wang, Nat. Energy, 2020, 5, 79–88 CrossRef CAS.
- M. Suri, A. Hazarika, B. W. Larson, Q. Zhao, M. Vallés-Pelarda, T. D. Siegler, M. K. Abney, A. J. Ferguson, B. A. Korgel and J. M. Luther, ACS Energy Lett., 2019, 4, 1954–1960 CrossRef CAS.
- L. Dai, Z. Deng, F. Auras, H. Goodwin, Z. Zhang, J. C. Walmsley, P. D. Bristowe, F. Deschler and N. C. Greenham, Nat. Photonics, 2021, 15, 696–702 CrossRef CAS.
- D. N. Dirin, A. Vivani, M. Zacharias, T. V. Sekh, I. Cherniukh, S. Yakunin, F. Bertolotti, M. Aebli, R. D. Schaller, A. Wieczorek, S. Siol, C. Cancellieri, L. P. H. Jeurgens, N. Masciocchi, A. Guagliardi, L. Pedesseau, J. Even, M. V. Kovalenko and M. I. Bodnarchuk, Nano Lett., 2023, 23, 1914–1923 CrossRef CAS PubMed.
- W. Ke, C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2019, 31, 1803230 CrossRef CAS PubMed.
- A. Pisanu, A. Mahata, E. Mosconi, M. Patrini, P. Quadrelli, C. Milanese, F. De Angelis and L. Malavasi, ACS Energy Lett., 2018, 3, 1353–1359 CrossRef CAS.
- B. Li, R. Long, Y. Xia and Q. Mi, Angew. Chem., Int. Ed., 2018, 57, 13154–13158 CrossRef CAS PubMed.
- M. Pitaro, E. K. Tekelenburg, S. Shao and M. A. Loi, Adv. Mater., 2022, 34, 2105844 CrossRef CAS PubMed.
- M. M. Byranvand, C. Otero-Martínez, J. Ye, W. Zuo, L. Manna, M. Saliba, R. L. Z. Hoye and L. Polavarapu, Adv. Opt. Mater., 2022, 10, 2200423 CrossRef CAS.
- M. G. Panthani and B. A. Korgel, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 287–311 CrossRef CAS PubMed.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- E. Aktas, N. Rajamanickam, J. Pascual, S. Hu, M. H. Aldamasy, D. Di Girolamo, W. Li, G. Nasti, E. Martínez-Ferrero, A. Wakamiya, E. Palomares and A. Abate, Commun. Mater., 2022, 3, 104 CrossRef CAS.
- C. Y. Watson, G. M. DeLoid, A. Pal and P. Demokritou, Small, 2016, 12, 3172–3180 CrossRef CAS PubMed.
- P. Estellé, D. Cabaleiro, G. Żyła, L. Lugo and S. M. S. Murshed, Renewable Sustainable Energy Rev., 2018, 94, 931–944 CrossRef.
- A. A. Zhumekenov, V. M. Burlakov, M. I. Saidaminov, A. Alofi, M. A. Haque, B. Turedi, B. Davaasuren, I. Dursun, N. Cho, A. M. El-Zohry, M. De Bastiani, A. Giugni, B. Torre, E. Di Fabrizio, O. F. Mohammed, A. Rothenberger, T. Wu, A. Goriely and O. M. Bakr, ACS Energy Lett., 2017, 2, 1782–1788 CrossRef CAS.
- E. Scharf, F. Krieg, O. Elimelech, M. Oded, A. Levi, D. N. Dirin, M. V. Kovalenko and U. Banin, Nano Lett., 2022, 22, 4340–4346 CrossRef CAS PubMed.
- D. Ricciarelli, D. Meggiolaro, F. Ambrosio and F. De Angelis, ACS Energy Lett., 2020, 5, 2787–2795 CrossRef CAS.
- D. W. Theobald and J. C. Smith, Chem. Ind., 1958, 1007–1009 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06075f |
| This journal is © The Royal Society of Chemistry 2024 |