
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reticular synthesis of two-dimensional ionic covalent organic networks as metal-free bifunctional electrocatalysts for oxygen reduction and evolution reactions†
Pampa
Jhariat
 ab,
Arjun
Warrier
b,
Ananta
Sasmal
c,
Subhadip
Das
d,
Shafeeq
Sarfudeen
b,
Priyanka
Kumari
b,
Arpan Kumar
Nayak
e and
Tamas
Panda
*ab
ab,
Arjun
Warrier
b,
Ananta
Sasmal
c,
Subhadip
Das
d,
Shafeeq
Sarfudeen
b,
Priyanka
Kumari
b,
Arpan Kumar
Nayak
e and
Tamas
Panda
*ab
aCentre for Clean Environment, Vellore Institute of Technology, Tamil Nadu 632014, India. E-mail: tamaskumarpanda@vit.ac.in
bDepartment of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore, Tamil Nadu 623014, India
cDepartment of Physics, School of Advanced Sciences, VIT, Vellore, Tamil Nadu 623014, India
dDepartment of chemistry, Chaudhary Ranbir Singh University, Jind, Haryana 126102, India
eDepartment of Energy Engineering, Konkuk University, 120 Neungdong-ro, Seoul, 05029, Republic of Korea
First published on 12th January 2024
Abstract
Bifunctional electrocatalysts for the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER) are the heart of metal–air batteries, fuel cells, and other energy storage systems. Here, we report a series of a novel class of redox-active viologen-based ionic covalent organic networks (vCONs) which are directly used as metal-free bifunctional electrocatalysts towards ORR and OER applications. These vCONs (named vGC, vGAC, vMEL and vBPDP) were synthesized by the well-known Zincke reaction. The installation of redox-active viologen moieties among the extended covalent organic architectures played a crucial role for exceptional acid/base stability, as well as bifunctional ORR and OER activities, confirmed by the cyclic voltammetry (CV) curves. Among all of them, vBPDP showed high ORR efficiency with a half-wave potential of 0.72 V against a reversible hydrogen electrode (RHE) in 1 M KOH electrolyte. In contrast, vMEL demonstrated high OER activity with an overpotential of 320 mV at a current density of 10 mAcm−2 and a Tafel slope of 109.4 mV dec−1 in 1 M KOH electrolyte solution. This work is exceptional and unique in terms of directly used pristine ionic covalent organic networks that are used as bifunctional (ORR and OER) electrocatalysts without adding any metals or conductive materials.
Introduction
Carbon emissions and fossil fuel depletion cause air pollution, global warming and rising sea levels which threaten the world economy and ecosystem. Therefore, renewable energy resource demand has soared.1 Over the past few years, rechargeable aqueous metal–air batteries have attracted significant attention as a resolution for these environmental issues because they are one of the simplest and most effective, renewable energy technologies.2 The ORR and OER play a very crucial role inside the oxygen electrode of metal–air batteries for the overall performance of the device.3 However, high-performing electro catalysts are essential to minimize the overpotentials and maximize the efficiencies of the above electrochemical reactions.4 Traditionally, the noble metal-based electrocatalysts Pt/C and Ir/C served individually as the best choices for the ORR and the OER, respectively. However, their high cost, scarcity, and instability pose various challenges.5–7 In addition, the ORR and OER require different reaction environments, especially electric potentials, which inhibits the catalysis efficiency for both reactions and generates more complications in reducing significant catalytic activity.8,9 Thus, non-precious metals or organic catalysts that perform both ORR and OER operations are very important for aqueous metal–air batteries.10 However, the majority of transition metal-based catalysts have low catalytic activity, poor conductivity, and limited lifetime.11 Subsequently, researchers have studied metal-free catalysts12,13 and their composites with carbon nanotubes,14,15etc. Still, metal-free electrocatalysts for dual ORR and OER applications are limited in number and experimental.Recently, metal-free extended organic networks, such as covalent organic network/polymers (COPs)16 and covalent organic frameworks (COFs),17–20 have received interest as potential electrocatalysts due to their multi-element composition, high atomic accuracy and structural flexibility. Their tunable surface area, adjustable structure, and extended network topology make them excellent single-functional electrocatalysts21 in either OER or ORR applications. Sometimes, these covalent organic network structures are also fabricated by mixing or making composites with highly conductive materials such as graphene, metal nanoparticles carbon nanotubes (CNTs), or transition metal complexes to make them acceptable for bifunctional ORR and OER electrocatalysts.22–25
However, to the best of our knowledge, pristine covalent organic network structures directly used as bifunctional electrocatalysts for the ORR and OER are extremely rare and unprecedented.
Here, we report a series of template-free redox-active viologen-based ionic covalent organic networks (vCONs) synthesized by a solvothermal-assisted Zincke reaction between the Zincke salt L1 (1,1′-bis(2,4-dinitrophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride) and the corresponding triamines [GC: guanidine hydrochloride, GAC: 1,2,3-triaminoguanidine chloride, MEL: melamine and BPDP: N,N′-bis(4-aminophenyl)benzene-1,4-diamine], named vGC, vGAC, vMEL and vBPDP, respectively. All the vCONs are very stable in 6 M HCl and 1 M NaOH aqueous solutions at 25 °C for 72 h. The outstanding chemical stability and inherent redox viologen moieties in the framework architecture of these vCONs showed good efficiency towards bifunctional electrocatalytic ORR and OER applications. Among all of them, vBPDP displayed the highest ORR activity with a half-wave potential of 0.72 V against a reversible hydrogen electrode (RHE) in 1 M KOH electrolyte solution. On the other hand, vMEL showed the highest OER efficiency among all the other vCONs with an overpotential of 320 mV at a current density of 10 mA cm−2 in 1 M KOH solution. This report is unique in terms of directly using pristine covalent organic networks as bifunctional electrocatalysts without performing any further modification such as pyrolysis, doping, metal incorporation, etc.
Experimental section
The organic linker L1 (1,1′-bis(2,4-dinitrophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride) and the corresponding triamines [guanidine chloride (GC), 1,2,3-triaminoguanidine chloride (GAC), melamine (MEL) and N,N′-bis(4-aminophenyl) benzene-1,4-diamine (BPDP)] were used as starting materials to synthesize all vCONs via the Zincke reaction. First, a sealed tube was charged with L-1 (0.5 mmol, 0.28 g) and respective triamines (0.5 mmol) in a 10 mL solution mixture of ethanol and water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) under an N2 atmosphere. Then the reaction mixture was degassed through three freeze–pump–thaw cycles and kept in a preheated oven at 120 °C for 72 h. After the completion of the reaction, the precipitate was collected via centrifugation followed by vacuum filtration and washing with polar solvents like dimethylformamide (DMF), dimethylacetamide (DMAc), ethanol (EtOH), boiling water (H2O), methanol (MeOH), dichloromethane (DCM) and acetone to remove the small polymers, oligomers and other impurities. After washing, the desired product was kept in a mixture of chloroform and tetrahydrofuran (1:1) solutions for three days. Finally, the product was collected via vacuum filtration and dried overnight at 120 °C. The details of experimental procedures are available in section S2, ESI.†
:1 ratio) under an N2 atmosphere. Then the reaction mixture was degassed through three freeze–pump–thaw cycles and kept in a preheated oven at 120 °C for 72 h. After the completion of the reaction, the precipitate was collected via centrifugation followed by vacuum filtration and washing with polar solvents like dimethylformamide (DMF), dimethylacetamide (DMAc), ethanol (EtOH), boiling water (H2O), methanol (MeOH), dichloromethane (DCM) and acetone to remove the small polymers, oligomers and other impurities. After washing, the desired product was kept in a mixture of chloroform and tetrahydrofuran (1:1) solutions for three days. Finally, the product was collected via vacuum filtration and dried overnight at 120 °C. The details of experimental procedures are available in section S2, ESI.†
Results and discussion
Synthesis of viologen CONs and their characterization
Viologens or 4,4′-bipyridinium salts exhibit excellent redox behaviour and undergo two consecutive electron reduction reactions from the dicationic phase to form the radical cation and neutral bipyridinylidene.26 Hence, viologens have been recently used as attractive components for the building of adjustable charged covalent organic networks.27 Such cationic organic networks would enable the insertion of greater electrostatic forces for host–guest identification. In addition, the redox-active sites would allow for the creation of a wide range of tunable properties and functionalities.28 These dicationic viologen-based covalent networks also have an inherent opportunity to convert to radical cationic states, which might enhance several properties including catalytic functions.29 The unique nature of such redox-active network structures motivated us to synthesize a new series of viologen-based covalent organic networks (vCONs) and investigate their exclusive electrocatalytic properties. Four unique template-free vCONs (vGC, vGAC, vMEL and vBPDP) were synthesized solvothermally by reacting 0.5 mmol of L1 with 0.5 mmol of the respective triamines (BPDP, MEL, GC and GAC) in the presence of a 4:1 EtOH:H2O solvent mixture at 120 °C for 72 h (Fig. 1, S1 and Section S2, ESI†). Various combinations of solvent mixtures have been used for the synthesis of these vCONs (Table S1a to S1d†). However, we found that using a 4:1 mixture of EtOH and H2O solvents is the ideal option to obtain the pure phase product because the water has the ability to protect the Zincke salt from deterioration.30 The four as- synthesized vCONs were activated by exchanging the 1:1 solvent mixture of dry THF and chloroform for 72 h followed by the treatment of dry DCM for 12 h at 25 °C. After filtration, all the samples were dried and activated at 120 °C under vacuum for 12 h. All these activated pristine vCONs samples (solvent-free) were characterized by FT-IR spectroscopy, PXRD, TGA, solid UV-vis spectroscopy, BET surface area analysis, solid-state cross-polarization magic angle spinning 13C-NMR (CP-MAS) spectroscopy, solid-state EPR spectroscopy, XPS analysis, FESEM, HR-TEM and DLS particle size analysis. The FT-IR spectra (Fig. S2a–d†) of all vCONs showed the absence of the peak at 1550 cm−1 corresponding to the ∼NO2 groups of the 2,4-dinitroaniline unit of linker L1, which supported the formation of the desired product by the Zincke reaction. The absence of ∼NH2 symmetric and asymmetric stretching bands at 3412 and 3437 cm−1 corresponding to the amine group of the starting triamine precursor also confirmed the completion of the polymerization process by the Zincke reaction. The solid-state 13C CP-MAS NMR spectra of all vCONs displayed a wide signal in the 110–170 ppm region, which confirms the presence of aromatic carbons in the polymeric network of the structures of all vCONs (Fig. 3c–f). In the case of vBPDP, carbons marked as ‘a’ (Fig. 3f) exhibited a greater downfield chemical shift (∼157 ppm), owing to their attachment to the nitrogen and conjugation with an aromatic ring. Meanwhile the pyridinium carbons exhibited chemical shifts in the range of 130–160 ppm denoted as e, f, and g. In vGAC, the carbons attached to the amines, denoted as ‘a’ (Fig. 3d), were most de-shielded toward the downfield (∼160 ppm) due to their connection to three nitrogen atoms with a delocalized positive charge. The pyridinium carbons of vGAC exhibited a region of 130–160 ppm, approximately the same as those of vBPDP. On the other hand, for vMEL the carbon denoted as ‘a’ (Fig. 3e) was shifted more towards the downfield due to the presence of nitrogen adjacent to the carbon (approx. ∼170 ppm).In the case of vGC, the carbon denoted as ‘a’ shows a similar type of downfield shifting to the remaining vCONs (Fig. 3c). It should be noted that crystalline viologen-based polymeric networks are rare and most of the reports mentioned that such viologen-based polymeric networks are amorphous in nature.31,32 We used several combinations of solvents to get crystalline products of these vCONs (Table S1a to S1d†). However, our attempts to improve the crystallinity of vGC and vBPDP were unsuccessful every time and they exhibited broad hump peaks, indicating the amorphous nature of both compounds (Fig. S3 and S4†). Interestingly, we found the crystalline nature of vGAC and vMEL by using an EtOH:H2O (4:1) solvent mixture, which was also confirmed by experimental PXRD (Fig. 3a). vGAC displayed crystalline peaks at 2θ = 2.44° (6.44 Å) and small hump intense crystalline peaks at 2θ = 13.10° (6.70 Å) and 16.32° (5.43 Å) in the experimental PXRD profile (Fig. 3a and S5†). The crystal structure of vGAC was solved in the monoclinic P1121 space group and a probable two-dimensional crystal structure was constructed using Materials Studio software after the models were geometrically optimized using a Forcite-based module. After analyzing the crystal data, we found that the vGAC structure is similar to a distorted honeycomb, as both the building unit L1 and GAC structures are non-planar in nature. The bipyridinium rings of viologen moieties are in rotational mode with respect to each other and also with the GAC moiety. As a result, the structure of vGAC becomes a distorted shape. The simulated powder pattern of the AA-stacking structure has good agreement with the experimental powder pattern of vGAC (Fig. S22†). Furthermore, this result was validated through the Pawley refinement with a good profile fitting (Rwp-3.43%). The unit cell parameters of vGAC are as follows: a, 45.0069; b, 37.0058; c, 15.0022; α, 90.00; β, 90.00; γ, 120.00. On the other hand, the vMEL structure exhibits a crystalline peak at 2θ = 2.36° (34.5 Å) and small hump peaks at 13.47° (6.57 Å) and 16.62° (5.33 Å). There are also two low-intensity peaks residing at 8.46° (10.45 Å) and 9.67° (9.13 Å) (Fig. 3b, Fig. S6†). The crystal structure of vMEL was resolved in the monoclinic P21 space group and the structure was arranged as an inter penetrable honeycomb structure in three-dimensional directions. The powder pattern of the AA-stacking model structure has good agreement with the experimental powder pattern and after Pawley refinement, with very good (Fig. S23†) refinement fitting (Rwp-1.24%). The unit cell parameters for vMEL are as follows: a, 33.4728; b, 9.9472; c, 33.4507; α, 90.0000; β, 59.8337; γ, 90.0000.
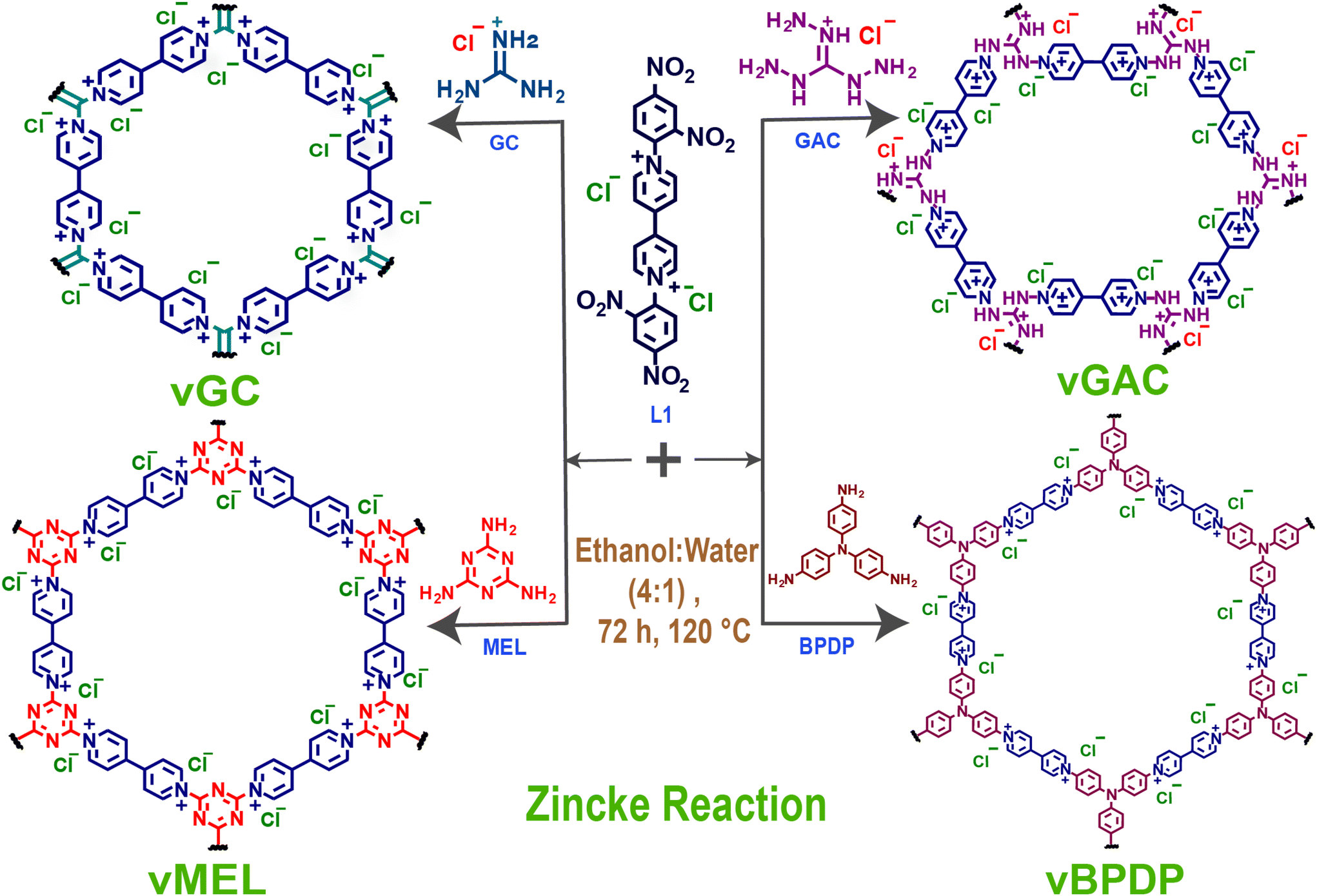 | ||
| Fig. 1 Schematic representation of the synthesis of vCONs (vGC, vGAC, vMEL, and vBPDP) by the Zincke reaction of L1 with the respective triamines (GC, GAC, MEL, and BPDP). | ||
In general, viologen moieties display three distinct intermediates, i.e., dicationic, radical cationic and neutral states. In order to investigate the existence of the radical cation state from the synthesized dicationic state, 30 mg of each pristine and degassed vCON was immersed in an over-saturated aqueous solution of alkaline sodium dithionite under an N2 atmosphere. The mixtures were stirred at 25 °C for 30 minutes and significant colour changes were observed for all the vCONs due to the formation of their radical cationic state (Fig. S7†). In the case of vGC, a colour change was observed from yellow to dark brown, and vGAC changed to blackish green from yellow colour. On the other hand, vMEL and vBPDP transformed from dark juniper to dark brown and from dark blue to black, respectively (Fig. S7†). These color changes in vCONs indicate the formation of a radical cationic state and are further confirmed by solid-state ESR and UV-vis spectroscopy techniques. All the radical cationic states of these vCONs exhibited a prominent paramagnetic peak in the solid state ESR spectra due to the presence of unpaired electrons, whereas the pristine vCONs were silent in the ESR spectra because of their diamagnetic nature in the dicationic state (Fig. 2a–d).33 In general, the viologen radical cations show the g-factor approximately in the range of 2.002–2.004 due to their single unpaired electrons.34 All four vCONs displayed similar g-factor values aligned with theoretical values (Fig. 2a–d).
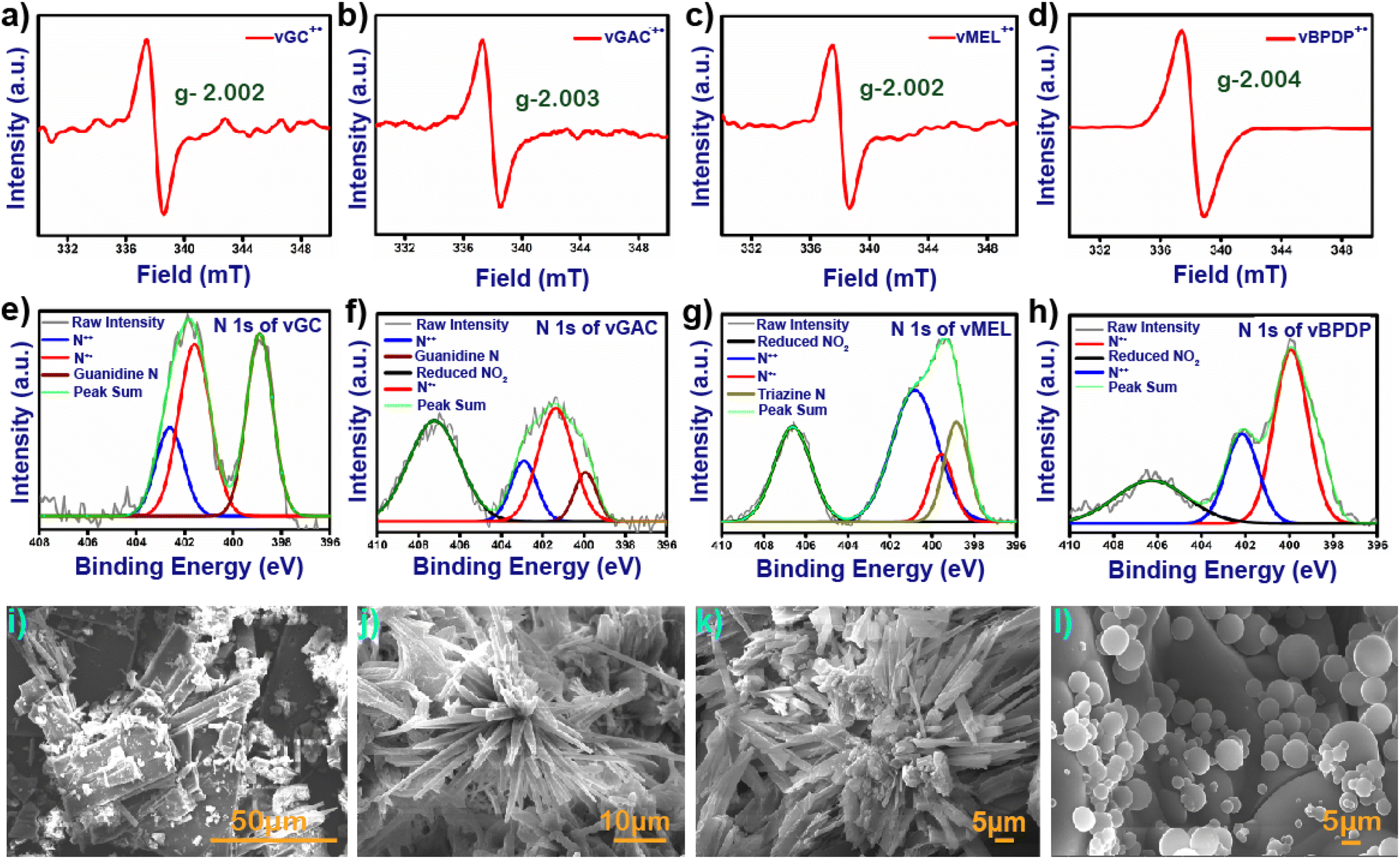 | ||
| Fig. 2 Solid-state EPR spectra of (a) vGC, (b) vGAC, (c) vMEL and (d) vBPDP in the radical cationic state. N 1s XPS spectra of (e) vGC, (f) vGAC, (g) vMEL and (h) vBPDP. FE-SEM micrographs of (i) vGC, (j) vGAC, (k) vMEL and (l) vBPDP. | ||
The g-factors shown by vGC, vGAC, vMEL and vBPDP are 2.002, 2.003, 2.002 and 2.004, respectively, which clearly confirm the formation of radical cationic intermediate states of these four viologen-based covalent organic networks. The solid-state UV/vis spectra of all four alkaline sodium dithionite-treated vCONs exhibit a prominent viologen radical peak centered at approximately between 600 and 900 nm, confirming the formation of the radical cationic state of vCONs (Fig. S8†).
The FE-SEM micrographs of all vCONs demonstrated a high degree of self-aggregated morphological diversity under the same experimental conditions (Fig. 2). The morphology of vGC was similar to that of nanorods (Fig. 2a) with an average length of 320 nm and a width of 142 nm. As shown in Fig. 2b, the arrangement of molecules in vGAC was similar to the arrangement of the petals that is generally observed in flowers. The average diameter of each flower was 122.4 μm, which indicates the micro-flower-like morphology among the structural networks (Fig. S12a–d†). The morphology of vMEL was similar to that of microtubes with an average length 5.5 μm and an average width of 2 μm (Fig. 2c). These microtubes are further self-assembled together and grow in three-dimensional directions. The FESEM image of vBPDP shows a uniformly arranged spherical-like morphology (Fig. 2d). The arrangements of all spheres are very uniform but the diameters of these spheres are different. The average diameter of the bigger spheres is ∼5 μm and other spheres have a diameter of approximately 2 μm. The HR-TEM micrographs of vCONs (Fig. S13†). were found to be similar to the FESEM images of vGC, which displayed a rod-shaped structure, while vGAC and vMEL demonstrated a tubular morphology, as shown in Fig. S13a–c.† In contrast, vBPDP exhibited a spherical morphology (Fig. S13d†). The hydrodynamic diameters of vCONs were measured by the dynamic light scattering (DLS) method, and were found to be 93.83 nm, 82.76 nm, 73.85 nm and 110.30 nm with polydispersities of 1.3, 1, 0.75 and 0.02 for vGA, vGAC, vMEL and vBPDP, respectively. SFESEM-EDX analysis and mapping (Fig. S20†) of all vCONs clearly showcase the existing C, N and C![[l with combining macron]](https://www.rsc.org/images/entities/char_006c_0304.gif) ions homogeneously distributed among the structural moieties (Fig. S15–18†). The analysis showed that the order of weight % of N content is as follows: vMEL (3 8.0%) > vGC (31.6%) > vGAC (30.5%) > vBPDP (16.3%). vMEL has the highest weight % of nitrogen content among the four vCONs. This finding also has a significant impact on the electrochemical properties of vCONs that are discussed later.
ions homogeneously distributed among the structural moieties (Fig. S15–18†). The analysis showed that the order of weight % of N content is as follows: vMEL (3 8.0%) > vGC (31.6%) > vGAC (30.5%) > vBPDP (16.3%). vMEL has the highest weight % of nitrogen content among the four vCONs. This finding also has a significant impact on the electrochemical properties of vCONs that are discussed later.
The porosities of these vCONs were measured by nitrogen gas adsorption isotherms at 77 K. All vCONs exhibited reversible type-II and type-IV N2 adsorption isotherms. The Brunauer–Emmett–Teller (BET) surface areas were 34.20, 33.77, 47.20 and 22.2 m2 g−1 for vGC, vGAC, vMEL and vBPDP, respectively (Fig. S19a–d†). The low surface areas among all the vCONs were due to the existence of charged chloride counter ions inside the pore area. Such a low surface area in the viologen-based covalent networks is aligned with the existing report.35 Our attempt to derive the pore size distributions using non-local density functional theory (NLDFT) was inappropriate due to the presence of several Cl− ions inside the framework architecture. The chemical and solvent stabilities of the vCONs were ratified through their unchanged physical appearance as well as the constant stretching bands in FT-IR spectroscopy. We have tested the water, acid and alkaline stabilities of these four vCONs. 10 mg of each vCON sample was individually immersed in 10 mL of boiling water (100 °C), 6 M aqueous HCl (25 °C), and 1 M aqueous NaOH solution (25 °C) for 72 h. After discarding the acid, alkaline solution and boiling water, the products were washed with ethanol and dried in air. All the treated vCON samples were characterized by FTIR spectroscopy. As shown in Fig. S10,† the boiling water-, acid- and alkaline-treated vGC samples maintain all the peak positions corresponding to their pristine sample, indicating the preservation of the structural integrity of vGC. Similar results have also been observed in the FTIR spectra of 1 M aqueous NaOH, 6 M aqueous HCl and boiling water-treated vGAC, vMEL and vBPDP samples (Fig. S10a–d†). For, vGAC and vMEL, we recorded the PXRD patterns as well for acid-, base- and boiling water-treated samples (Fig. S10a′ and b′†). In all these cases the powder patterns of treated samples were matched with that of the parent sample, which proves that the structural integrity was preserved even after such harsh treatment. These results clearly show that all the vCONs are very stable in acid, alkaline and boiling water. To study in more depth the chemical stability of these vCONS, we checked their stability in different polar solvents and confirmed their stability from the FT-IR spectra. 20 mg of each vCON was immersed in a variety of organic solvents such as DMF, DMSO, NMP, THF, methanol and DCM for 72 h at 25 °C. Then, the solids were washed with water, followed by acetone and dried at 40 °C for 12 h. The FTIR spectral analysis of these solvent-treated samples showed (Fig. S11a and b†) that all the bands are constant in their corresponding pristine samples. These results clearly indicate that the four unique vCONs were exceptionally stable in various polarities of solvents as well.
Thermo-gravimetric analysis of vGC, vGAC, vMEL, and vBPDP showed excellent thermal stability up to decomposition temperatures of 460 °C, 415 °C, 430 °C and 440 °C, respectively (Fig. S9†). During the first heating, slight weight losses were observed due to the evaporation of trapped solvents. At higher temperatures, an increased weight loss was observed, which is consistent with framework deterioration. To elucidate the nature of chemical bonding in the vCONs, we performed an X-ray photoelectron spectroscopy (XPS) analysis. All four vCONs exhibited C1s, N1s, O1s and Cl 2p peaks in the XPS spectra, which gave a clear indication towards the purity of samples (Fig. S21a†). In the case of C1s, we found three different peaks at 284.4, 287.1 and 285.8 eV, indicating the presence of C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N and CN bonds, respectively (Fig. S21b–e†). The N 1s spectra of vCONs could be deconvoluted into four types of peaks with binding energies of 398.5, 400.5, 402.5 and 406.0 eV (Fig. 2e–h). The peak at 398.5 eV was assigned for the triazinic nitrogen for vMEL and the quaternary nitrogen or the guanidine nitrogen for vBPDP, vGAC and vGC respectively. The peak at 402.5 eV is denoted as the dicationic nitrogen (N++) of the viologen moiety for all vCONs. Interestingly, all vCONs showed a prominent peak at 400.5 eV, which indicates the presence of a reduced radical cationic nitrogen state (N˙+) of the viologen moiety. Intriguing aspects of this finding clearly indicate the redox-active nature of the viologen moiety. In the case of the Cl 2p orbital, we found peaks at 197.3 and 198.5 eV assigned to Cl− 2p3/2 and Cl− 2p1/2, respectively (Fig. S21f–i†). For some cases, we fitted peaks at 200.3 and 201.9 eV for the Cl 2p1/2 and Cl 2p3/2 orbitals. Sometimes Cl− ion insertion occurs inside the aromatic ring due to the high temperature, which results in Cl–C bond formation.
C, C–N and CN bonds, respectively (Fig. S21b–e†). The N 1s spectra of vCONs could be deconvoluted into four types of peaks with binding energies of 398.5, 400.5, 402.5 and 406.0 eV (Fig. 2e–h). The peak at 398.5 eV was assigned for the triazinic nitrogen for vMEL and the quaternary nitrogen or the guanidine nitrogen for vBPDP, vGAC and vGC respectively. The peak at 402.5 eV is denoted as the dicationic nitrogen (N++) of the viologen moiety for all vCONs. Interestingly, all vCONs showed a prominent peak at 400.5 eV, which indicates the presence of a reduced radical cationic nitrogen state (N˙+) of the viologen moiety. Intriguing aspects of this finding clearly indicate the redox-active nature of the viologen moiety. In the case of the Cl 2p orbital, we found peaks at 197.3 and 198.5 eV assigned to Cl− 2p3/2 and Cl− 2p1/2, respectively (Fig. S21f–i†). For some cases, we fitted peaks at 200.3 and 201.9 eV for the Cl 2p1/2 and Cl 2p3/2 orbitals. Sometimes Cl− ion insertion occurs inside the aromatic ring due to the high temperature, which results in Cl–C bond formation.
vCONs for the oxygen reduction reaction (ORR)
The excellent redox-active nature, high N content and outstanding acid and base stabilities of all the vCONs motivated us to measure their electrocatalytic properties. At first, we tested these vCONs as potential electro-catalysts for the oxygen reduction reaction (ORR). Our initial step was to conduct cyclic voltammetry (CV) and linear sweep voltammetry (LSV) experiments by using one by one pristine vCONs as active electrocatalysts coated on glassy carbon electrodes in 1 M KOH electrolyte solution. To maintain a homogeneous ionic concentration surrounding the working electrode and to avoid the build-up of O2 bubbles, all measurements were performed on a rotating disc electrode (RDE) revolving at a rate of 1000 rpm. Fig. S24a–d† in the ESI show the CV curves of all electrodes in N2- and O2-saturated KOH electrolytes at a scan rate of 10 mV s−1 with 1000 rpm. The current densities of all the vCONs were found to show a remarkable enhancement in O2-saturated KOH electrolyte. However, negligible current densities were observed of these vCONs in N2-saturated KOH electrolyte. Fig. 4a shows the LSV curves of all four vCONs at 1000 rpm (Fig. S25†). vBPDP showed a high value in the LSV curve, which indicates the highest effective ORR activity of vBPDP compared with the other vCONs. The onset potential (Eo) and the half-wave potential (E½) of vBPDP are 0.80 V and 0.72 V, respectively, with a diffusion-limit current density (jlim) of 6.2 mA cm−2 (Fig. 4a), which is the highest among all the vCONs. vGAC displayed an E½ value of 0.71 V with Eo and jlim values of 0.79 V and 6.11 mA cm−2, respectively. On the other hand, the E½ values of vMEL and vGC are 0.67 V and 0.69 V with jlim values of 5.35 and 5.65 mA cm−2, respectively (Fig. 4a). This indicates that vGC and vMEL were less effective towards the electrocatalytic ORR. From the above results, we conclude that electrocatalytic activity towards the ORR is expected to be ∼vBPDP > vGAC > vGC > vMEL. In addition, we have also measured and compared the ORR activity of commercial Pt/C (state-of-the-art catalyst for the ORR) under the same instrumental conditions. The Eo, E½ and jlim values of Pt/C were found to be 0.87 V, 0.74 V and 7.0 mA cm−2 respectively, which are highly comparable with those of vBPDP. Even though vBPDP exhibits less efficiency than the Pt/C catalyst, however, the observed value of vBPDP is excellent in terms of metal-free bare vCONs as electrocatalysts. As per our knowledge, vBPDP is the exceptionally rare example of a metal-free viologen-based covalent organic network that shows high ORR performance (Table S2†). A possible reason for the high performance of vBPDP is the electron-donating nature and hole-transporting nature of the organic linker BPDP (Fig. 1). Hence, the BPDP linker coupled with the bipyridine unit is very effective towards the electrocatalytic ORR. In order to calculate the number of oxygen transfers (n) per ORR cycle, we analyzed the Koutechy–Levich (K–L) equation. The linear K–L plots for all four vCONs indicate the first-order kinetics for oxygen reduction reactions (Fig. 4b). The electron transfer number (n) for vBPDP was found in the range of 3.8–3.95, which implies the selectivity of vBPDP followed a four-electron pathway for the ORR. It should be noted that the four-electron pathway for the ORR leads to the formation of H2O, which is also the preferred reaction pathway in metal–air batteries. The unique catalyst vBPDP is selective for H2O formation and one of the greatest outcomes as an electrocatalyst for further application. The current–time chronoamperometric measurements of vBPDP display benchmark stability and the operating efficiency exhibits a fairly stable response at higher current densities (Fig. 4c). The aforementioned measurements justified the high efficiency and enhanced robustness of the vBPDP sample toward the ORR activity. We also recorded the electrochemical impedance spectroscopy (EIS) spectra of four vCONs at 1.606 V under ambient conditions. The corresponding Nyquist plots (Fig. 4d and S29†) were fitted to an equivalent circuit composed of two semicircles, one in the region of high frequency and another in the low frequency region, owing to the intrinsic/contact resistance (Ri) and charge transfer resistance (Rct), respectively. The calculated Rct values followed the order: vBPDP (6.8 Ω) < vGAC (7.15 Ω) < vGC (7.39 Ω) < vMEL (7.73 Ω). The lower Rct value of vBPDP signifies an efficient electron transfer pathway inside the structural integrity that facilitates the high ORR activity as well.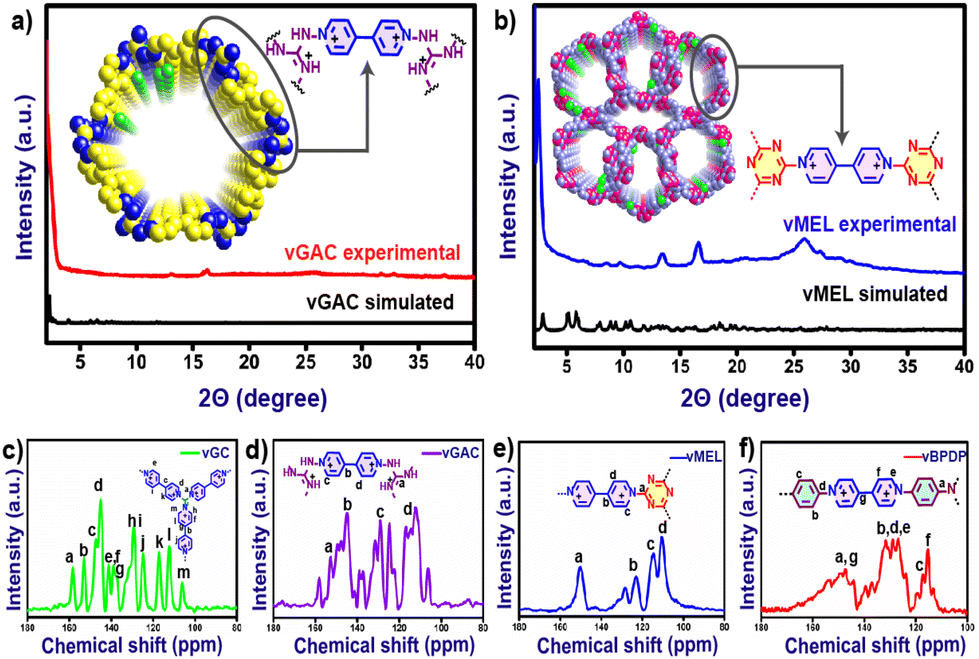 | ||
| Fig. 3 Comparison of experimental PXRD pattern with the simulated powder pattern of (a) vGAC and (b) vMEL. Solid-state 13C CP-MAS NMR spectra of (c) vGC, (d) vGAC, (e) vMEL and (f) vBPDP. | ||
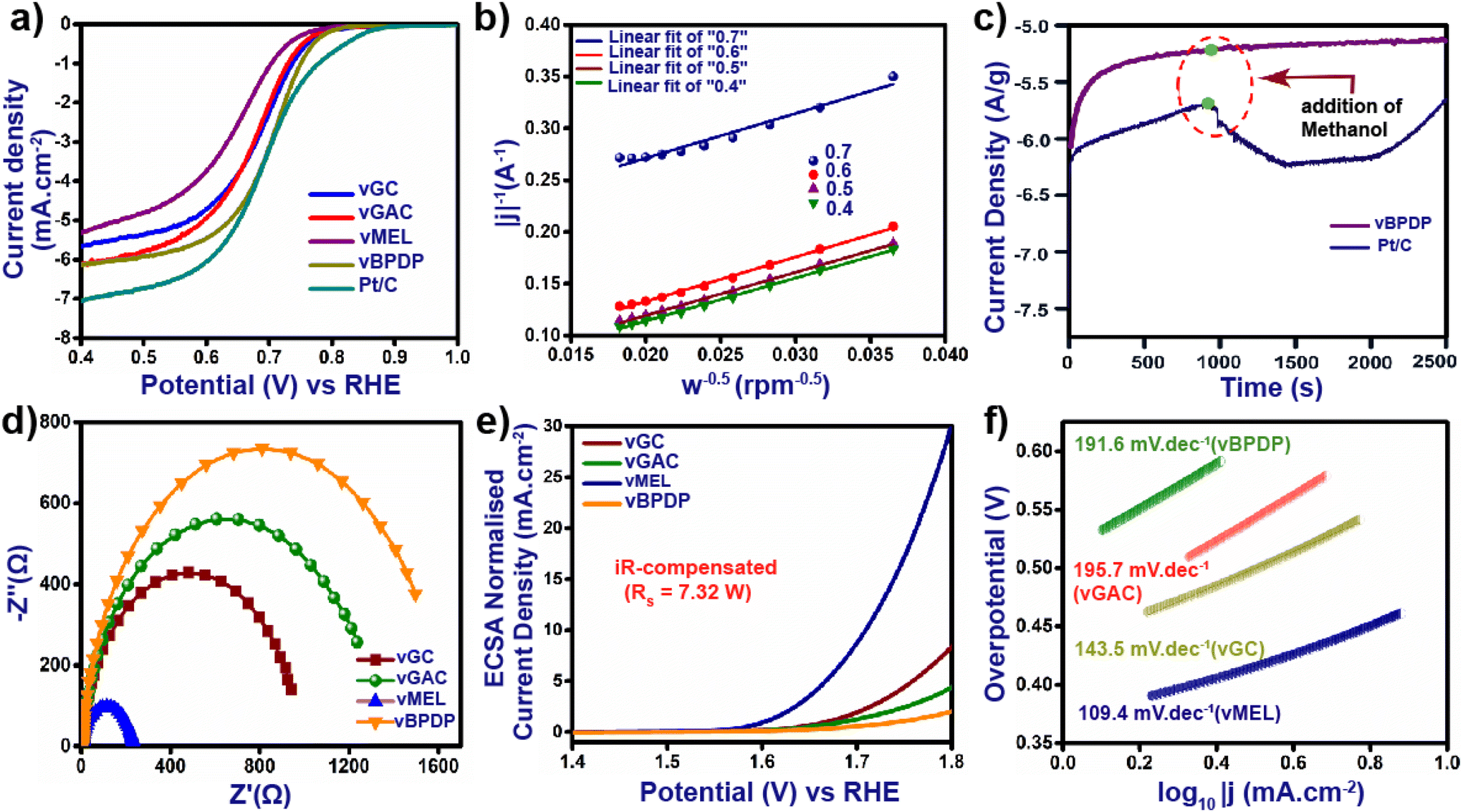 | ||
| Fig. 4 (a) LSV curves of all vCONs, (b) K–L plot of vBPDP, (c) methanol stability of vBPDP, (d) EIS spectra of all vCONs, (e) LSV curves for the OER of all vCONs, and (f) Tafel plots of all the vCONs. | ||
vCONs for the oxygen evolution reaction (OER)
Next, we investigated the electrocatalytic activity of all vCONs toward the oxygen evolution reaction (OER). The experiments were performed by using a standard three-electrode system in O2-saturated 1 M aqueous KOH electrolyte solution. Fig. 4e displays the LSV curve after the iR compensation of all four vCON electrocatalysts. vMEL (Fig. 4e) showed high catalytic efficiency towards the OER at an overpotential (η10) of 320 mV with a current density of 10 mA cm−2 compared to the others. vGC, vGAC and vBPDP exhibited inferior electrocatalytic OER activity with overpotentials of 339, 350 and 370 mV at 10 mA cm−2, respectively (Fig. 4e). To calculate the electrical double-layer capacitance Cdl, we first recorded the cyclic voltammograms (CVs) of all vCONs in the non-faradaic region in 1 M KOH solution at a scan rate of 10–100 mV s−1 and in a potential range of 0.05–0.15 V (Ag/AgCl) (Fig. S26†). The calculated Cdl values (Fig. S28†) for vGC, vGAC, vMEL, and vBPDP were found to be 62, 57.6, 61.6 and 60.4 μF cm−2 respectively. The roughness factors (Rf) of vGC, vGAC, vMEL and vBPDP have also been calculated, which were found to be 22.24, 20.57, 22 and 21.57 respectively. The electrochemically active surface area (ECSA) of the four vCONs samples were determined by using Cdl, and the details of comprehensive calculations are provided in Section S-2, ESI.† The ECSAs of vGC, vGAC, vMEL and vBPDP were found to be 1.55, 1.44, 1.54 and 1.51, respectively (Fig. S27†). The larger ESCAs of vMEL and vGC reveal that their pore areas are more permeable to electrolytes than vBPDP and vGAC. Although vGC and vMEL have similar ECSAs and roughness factors, the overpotential of vMEL is higher than that of vGC. The wide surface area and redox-active cationic structure of these vCONs increase the electrolyte's ability to carry a larger number of reactant species on the electrode surface. To understand the kinetics and mechanism of the vCON-driven OER, Tafel slopes were calculated as shown in Fig. 4f. The Tafel slopes of vGC, vGAC, vMEL, and vBPDP were calculated as 143.5, 195.7, 109.4 and 191.6 mV dec−1, respectively (Fig. 4f). vMEL exhibited the lowest Tafel slope, which also validates that vMEL is a good electrocatalyst towards the OER. IrO2 is the state-of-the-art material known to be the best electrocatalyst for OER performance, and hence we compared the electrocatalytic activity of IrO2 with the activities of these vCONs. IrO2 displayed an overpotential of 250 mV at a current density of 10 mA cm−2. Even though vMEL showed relatively less activity (320 mV at 10 mA cm−2) compared to the benchmark IrO2, it is still the highest among the other metal-free bare organic polymer electrocatalysts (Table S2†). We assume that the strong coupling interactions and electronic structural alteration in vMEL (due to the multiple redox centers and high N content)36 enhance the electrocatalytic OER activity.Proposed mechanism for bifunctional electrocatalysis
The preceding electrochemical experiments established that all vCONs are bifunctional in nature. We propose here the possible mechanism of the bifunctional electrocatalytic properties of these vCONs. As per the literature report, if a structure contains both pyridinic and quaternary nitrogen atoms, then the electron-withdrawing pyridinic nitrogen (p-type) is the active side area for the OER, whereas the electron-donating quaternary nitrogen sites (n-type) are responsible for the ORR.37 Additionally, recent DFT studies have shown that not only the integrated nitrogen atoms but also the adjacent carbon atoms bonded with the respective nitrogen have lower energy barriers for electrocatalysis due to the charge induced by nitrogen atoms thoroughly redistributed inside the π-conjugated system.37,38 We expect that in all vCONs, the adjacent carbons of the pyridinic nitrogen of the viologen unit (p-type) and the quaternary nitrogen of the amine moiety (n-type) also act as the active site zone for the individual OER and ORR, respectively (Fig. 5). The coexistence of both p-type and n-type domains in each of these vCON structures could be the possible reasons for their bifunctional behaviors. In fact, our experimental trend of ORR and OER results in these vCONs supports this prediction. We found that vBPDP showed high ORR activity and vMEL displayed the high performance in the OER. Note that the viologens unit (p-type) is common for all vCONs and hence the contribution of viologen moieties towards the OER will be similar in all vCONs. However, in vBPDP, the linker BPDP contains tertiary amine (quaternary nitrogen) that is well known for its inherent redox-active nature (already discussed above) and that shows high ORR activity. On the flip side, the weak OER activity of vBPDP is only due to the contribution of viologen moieties (p-type). In the case of vMEL, the ORR activity is significantly diminished as n-type domains (the absence of quaternary nitrogen) are inactive in this structure. However, the structure of vMEL contains a large number of pyridinic N domains (p-type) from melamine addition with p-type viologen units, which enhances the OER efficiency. To understand the clear mechanism of the bifunctional electrochemical behavior of these vCONs, detailed computational studies are necessary.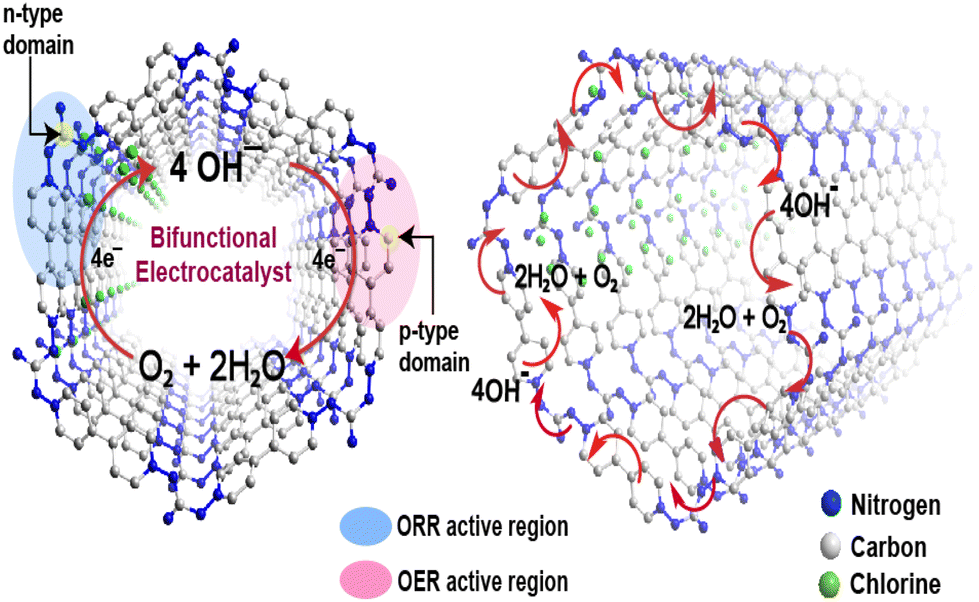 | ||
| Fig. 5 Possible mechanism and active site regions for the bifunctional electrocatalysis. Here we represent the simulated structure of vGAC to understand the active sites and probable mechanistic pathways for the ORR and OER in a single framework. | ||
Conclusion
In conclusion, we successfully synthesized a series of viologen-based covalent organic networks vCONs. They are very stable in various polar solvents, boiled aqueous solutions, and harsh acidic [6 M HCl] and basic [1 M NaOH] solutions. All the vCONs are highly redox active in nature due to the unique architecture made by the viologen moieties as building blocks and they can also undergo a transition from a dicationic to a radical cation state. These pristine vCONs are directly used as excellent bifunctional electrocatalysts toward the ORR and OER (confirmed from the CV curves). vBPDP exhibits high efficiency towards the ORR with a half-wave potential of 0.72 V and a diffusion-limiting current density (jlim) of 6.2 mA cm−2. The K–L plot of vBPDP also supports the high ORR activity achieved by the four-electron pathway with the formation of water as the final product. On the other hand, vMEL showed high OER efficiency with an overpotential of 320 mV at a current density of 10 mA cm−2 and a Tafel slope of 109.4 mV dec−1 in 1 M KOH solution. These four pristine vCONs exhibit inherent bifunctional behaviour without the fabrication of any metals or conductive materials or pyrolysis products. We strongly believe that these types of vCONs are potential efficient bifunctional electrocatalysts towards the ORR and OER, which will be applicable in next-generation energy storage devices.Abbreviation
| vCONs | Viologen-based covalent organic networks |
| GC | Guanidine hydrochloride |
| GAC | 1,2,3-Triaminoguanidine chloride |
| MEL | Melamine |
| BPDP | N,N′-Bis(4-aminophenyl)benzene-1,4-diamine |
Author contributions
Pampa Jhariat: synthesis and characterization of vCONs and writing and editing the manuscript. Arjun Warrier: writing, reviewing and editing the manuscript. Dr Subhadip Das: structural determination of vCONs. Dr Arpan Kumar Nayak and Ananta Sasmal: helped in performing the electrochemical experiments. Shafeeq Sarfudeen and Priyanka Kumari: reviewing and editing the draft. Dr Tamas Panda: supervised the whole project and modified and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PJ, AW, AS, SK, and PK acknowledge VIT Vellore for the teaching and research assistant (TRA) fellowship. TP acknowledge the financial assistance from SERB, the start-up research grant SRG/2020/001108 from the Government of India and VIT Vellore for the seed grant. We acknowledge Prof. Rajeswara Rao M (IIT Dharwad) and Aswani Raj K for giving us the opportunity to use the solid state 13C CP-MAS NMR instrument. We also acknowledge Prof. T. G. Ajith kumar and Ankita Shelke (NCL Pune) for useful structural discussion of SSNMR.References
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- J. Li, A. Alsudairi, Z.-F. Ma, S. Mukerjee and Q. Jia, J. Am. Chem. Soc., 2017, 139, 1384–1387 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- Y. Liu, H. Jiang, Y. Zhu, X. Yang and C. Li, J. Mater. Chem. A, 2016, 4, 1694–1701 RSC.
- H. Wu, T. Yang, Y. Du, L. Shen and G. W. Ho, Adv. Mater., 2018, 30, 1804341 CrossRef PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11758 CrossRef CAS PubMed.
- B.-Q. Li, S.-Y. Zhang, C. Tang, X. Cui and Q. Zhang, Small, 2017, 13, 1700610 CrossRef PubMed.
- N. Ullah, R. Ullah, S. Khan and Y. Xu, Front. Mater. Sci., 2021, 15, 543–552 CrossRef.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867–5871 CrossRef CAS PubMed.
- R. Gao, Q. Dai, F. Du, D. Yan and L. Dai, J. Am. Chem. Soc., 2019, 141, 11658–11666 CrossRef CAS PubMed.
- S. Mondal, B. Mohanty, M. Nurhuda, S. Dalapati, R. Jana, M. Addicoat, A. Datta, B. K. Jena and A. Bhaumik, ACS Catal., 2020, 10, 5623–5630 CrossRef CAS.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- J. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- V. Briega-Martos, A. Ferre-Vilaplana, A. de la Peña, J. L. Segura, F. Zamora, J. M. Feliu and E. Herrero, ACS Catal., 2017, 7, 1015–1024 CrossRef CAS.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- H. Lei, Q. Zhang, Z. Liang, H. Guo, Y. Wang, H. Lv, X. Li, W. Zhang, U.-P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202201104 CrossRef CAS PubMed.
- A. Singh, D. Samanta and T. K. Maji, ChemElectroChem, 2019, 6, 3756–3763 CrossRef CAS.
- X. Lin, P. Peng, J. Guo and Z. Xiang, Chem. Eng. J., 2019, 358, 427–434 CrossRef CAS.
- Q. Xu, J. Qian, D. Luo, G. Liu, Y. Guo and G. Zeng, Adv. Sustainable Syst., 2020, 4, 2000115 CrossRef CAS.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gándara, M. Lusi, D. S. Shankar Rao, S. Vimala, S. Krishna Prasad, J. Raya, D. S. Han, R. Jagannathan, J.-C. Olsen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K.-C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC.
- N. Zeghbib, P. Thelliere, M. Rivard and T. Martens, J. Org. Chem., 2016, 81, 3256–3262 CrossRef CAS PubMed.
- T. Škorjanc, D. Shetty, M. A. Olson and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 6705–6716 CrossRef PubMed.
- P. Samanta, P. Chandra, S. Dutta, A. V. Desai and S. K. Ghosh, Chem. Sci., 2018, 9, 7874–7881 RSC.
- L.-Z. Peng, P. Liu, Q.-Q. Cheng, W.-J. Hu, Y. A. Liu, J.-S. Li, B. Jiang, X.-S. Jia, H. Yang and K. Wen, Chem. Commun., 2018, 54, 4433–4436 RSC.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D'Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J.-C. Olsen, K. Polychronopoulou, C. Platas-Iglesias, F. Ravaux, M. Jouiad and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361–15369 RSC.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- H. B. Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, e1501122 CrossRef PubMed.
- R. Ma, G. Lin, Y. Zhou, Q. Liu, T. Zhang, G. Shan, M. Yang and J. Wang, npj Comput. Mater., 2019, 5, 78 CrossRef.
- S. Ghosh and R. N. Basu, Nanoscale, 2018, 10, 11241–11280 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: All the additional characterization data, graphs and electrochemical measurement results. See DOI: https://doi.org/10.1039/d3nr05277j |
| This journal is © The Royal Society of Chemistry 2024 |