
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe use of energy looping between Tm3+ and Er3+ ions to obtain an intense upconversion under the 1208 nm radiation and its use in temperature sensing†
Tomasz
Grzyb
 *a,
Inocencio R.
Martín
b and
Radian
Popescu
c
*a,
Inocencio R.
Martín
b and
Radian
Popescu
c
aDepartment of Rare Earths, Faculty of Chemistry, Adam Mickiewicz University, Poznań, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: tgrzyb@amu.edu.pl
bDepartamento de Fisica, Universidad de La Laguna, Instituto de Materiales y Nanotecnología, 38200 San Cristóbal de La Laguna, Santa Cruz de Tenerife, Spain
cLaboratory for Electron Microscopy, Karlsruhe Institute of Technology, Engesserstrasse 7, 76131 Karlsruhe, Germany
First published on 22nd December 2023
Abstract
The upconversion phenomenon allows for the emission of nanoparticles (NPs) under excitation with near-infrared (NIR) light. Such property is demanded in biology and medicine to detect or treat diseases such as tumours. The transparency of biological systems for NIR light is limited to three spectral ranges, called biological windows. However, the most frequently used excitation laser to obtain upconversion is out of these ranges, with a wavelength of around 975 nm. In this article, we show an alternative – Tm3+/Er3+-doped NPs that can convert 1208 nm excitation radiation, which is in the range of the 2nd biological window, to visible light within the 1st biological window. The spectroscopic properties of the core@shell NaYF4:Tm3+@NaYF4 and NaYF4:Er3+,Tm3+@NaYF4 NPs revealed a complex mechanism responsible for the observed upconversion. To explain emission in the studied NPs, we propose an energy looping mechanism: a sequence of ground state absorption, energy transfers and cross-relaxation (CR) processes between Tm3+ ions. Next, the excited Tm3+ ions transfer the absorbed energy to Er3+ ions, which results in green, red and NIR emission at 526, 546, 660, 698, 802 and 982 nm. The ratio between these bands is temperature-dependent and can be used in remote optical thermometers with high relative temperature sensitivity, up to 2.37%/°C at 57 °C. The excitation and emission properties of the studied NPs fall within 1st and 2nd biological windows, making them promising candidates for studies in biological systems.
Introduction
The photon upconversion process in nanoparticles (NPs) is one of the most intensively investigated phenomena in recent years. The conversion of low energetic photons, usually from the near-infrared range (NIR), into photons with higher energy can be applied in medicine for photodynamic therapy,1,2 in the detection of cancer markers,3–5 in bioimaging,6 or drug delivery.7 Recently, remote temperature sensing is one of the most important areas where the upconversion process finds applications.8–10 Monitoring temperature is essential for understanding biological processes and diagnosing or treating various diseases.11The diversity of applications arises from the distinctive properties of upconversion (UC). NPs showing this process are usually inorganic structures with high chemical and thermal stability. The nature of the UC process in the NPs gives such important features as stability of emission, lack of photobleaching and significant shift between excitation and emission wavelengths.12 The lack of autofluorescence when UC is applied is also important for biological applications. These features result from the properties of lanthanide ions (Ln3+), whose electronic structure, low sensitivity to the local environment and multiple long-living energy states allow for the conversion of NIR photons to higher energetic ones.
UC is possible due to the sequential absorption of multiple photons in a single Ln3+ ion or energy transfer (ET) between two different ions, e.g. Yb3+ and Er3+.13 There are many examples of efficient UC systems with multicolour emissions, various types of Ln3+ ions and their concentrations, and based on different host compounds forming NPs.14–18 Among various types of upconverting NPs, the most popular are those doped with Yb3+ ions as sensitisers for NIR light with a wavelength around 975 nm and co-doped with Er3+, Tm3+ or Ho3+ used as emitters.1,3,6,7,19,20 It is caused by the high effectivity of this system due to the intense absorption of Yb3+ ions and the possibility of energy migration between these ions in the structure of NPs.21,22 Yb3+ ions have a unique electronic structure and only one excited state (2F5/2), limiting the possible quenching processes and energy loss.22
UC based on the properties of the Yb3+/Ln3+ system has its consequences, such as high efficiency23 or the possibility of NIR conversion up to ultraviolet,4,24 but also some drawbacks, e.g. low penetration of the 975 nm wavelength into tissue and high absorption of water in this range causing heating of the biological medium.25–27 The laser wavelength used for excitation of UCNPs doped with Yb3+ ions falls between the 1st and 2nd biological windows, where tissue shows lower transparency for radiation.26–28 Better for medical applications are NPs excitable within biological windows: 1st (650 to 950 nm), 2nd (1100 to 1350 nm) or 3rd (1600 to 1870 nm). Also optimal is the emission of such NPs within these windows, most favourably within the 1st optical window, as many popular optical detectors work within this range.
There are only a few types of Ln3+ ions capable of absorption within the ranges mentioned above, and one of the most promising are Tm3+ ions as they absorb radiation at around 1208 nm, and their emission occurs within the 1st biological window. However, not too many examples of Tm3+-based UC in NPs exist.29–33 Other possibilities are Nd3+ ions used for 808 nm excitation,13,28 Ho3+ ions capable of absorption of 1156 nm,34,35 and Er3+ ions, which show emission under 1490–1580 nm excitation, which is on the edge of the 3rd biological window.27,35,36
UC based on the ions mentioned above can also be efficient and intense.32,37 Such processes as energy looping, photon avalanche (PA) or self-sensitisation can significantly enhance the UC in NPs without Yb3+ in their structure.32,38–40 These processes are based on a cycle or loop of GSA, ESA and/or energy transfers followed by CR, leading eventually to a substantial population of the reservoir level and, therefore, to intense UC emission.38,41
The UC-based optical nanothermometers excitable via 1208 nm radiation have not been reported yet. The most frequently used NIR excitation wavelength for nanothermometry is 975 nm,9,42,43 whereas for biological applications, 808 nm.8 The first excitation wavelength falls outside the biological windows, and the obtained emission is usually based on the properties of Er3+ ions and their emission at around 520 and 545 nm. The application of such thermometers in biology and medicine is therefore limited due to the absorption of green light by, e.g. red blood cells and lowered transparency of water in the 950–1050 nm range.26,27 The conception of using 1208 nm excitation radiation and emission bands in the red to NIR range is new and promising for applications in biology, where lowering interactions of biological environment with excitation beam is of high importance.11
This article reports the spectroscopic properties of Tm3+ and Er3+/Tm3+ doped core@shell NPs based on the NaYF4 host compound and their emission under 1208 nm laser excitation. We selected core@shell structures to obtain better emission intensity, a common practice for upconverting NPs studies.44 We also discuss the mechanism of the observed UC process. The literature reports show that Tm3+ under 1208 nm laser radiation undergo ground state absorption (GSA) and excited state absorption (ESA) to populate 3F3 and 3H4 excited states.29,30,40 Also, ET between Tm3+ ions can be involved in their excitation.29,40 These few reports do not explain all of the processes taking place in Tm3+ and Er3+/Tm3+-doped NPs studied by us, e.g. why number of photons necessary to populate the emitting levels highly exceeds the theoretical values and how exactly Er3+ are being excited. The UC under 1208 nm by using Tm3+ ions seems to be still not sufficiently studied and understood. Additionally, we present here a proof of concept of how the properties of Tm3+-sensitized NPs can be used to detect temperature changes.
Experimental
Chemicals
All reagents were used as follows, without further purification. Commercial rare earth oxides Er2O3 (99.99%), Tm2O3 (99.99%), Y2O3 (99.99%), from Stanford Materials (United States), acetic acid (≥99%), ammonium fluoride (99%, ACS Reagent), and n-hexane (≥99%) from Honeywell (Poland), 1-octadecene (ODE, 90%) and oleic acid (OA, 90%) from Alfa Aesar (Germany), sodium oleate (82%) from Sigma-Aldrich (Poland), ethanol (99.8%) from Avantor (Poland), aqueous solutions of acetic acid (40% and 80%) were prepared by dissolution in de-ionised water of acetic acid (≥99%), and nitrogen (99.99%) from Linde (Poland) were used as received.Synthesis of core@shell NPs
To synthesise NPs, we applied the procedure described by Homann et al.23 The core and core@shell NPs were obtained in the high-boiling point solvents, i.e. OA and ODE and from the rare earth acetate salts. Ammonium fluoride was a source of fluorine ions during the synthesis.Synthesis of α-NaYF4 and α-NaYF4:Ln3+ NPs
Initially, the OA and 1-ODE (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture was put into a three-neck flask connected to a reflux condenser and outgassed using a Schlenk line at 100 °C under low pressure. After 1 h, the mixture was cooled to room temperature, and the selected powder rare earth acetates (see ESI for the synthesis procedure†) were added. The solution of acetates was heated to 100 °C under low pressure, and the mixture was kept at this temperature for 1 h. In the next step, the powder of sodium oleate was put into the solution at 100 °C. Then, the solution was outgassed for 20 min until the dissolution of sodium oleate and the powder of ammonium fluoride was added to the mixture. The solution was again outgassed in three cycles and heated to 200 °C under the nitrogen flow. After heating for 1 h, the synthesis mixture was cooled to room temperature and centrifuged. The collected supernatant was applied to the isolation of fluoride NPs. The addition of ethanol to the supernatant led to the precipitation of NPs. The colloidal solution of NPs was centrifuged, and then the NPs were dissolved in n-hexane (1 mmol NPs:5 mL of n-hexane). The NPs were precipitated again after the addition of ethanol and centrifuged. The following materials were obtained: α-NaYF4; α-NaYF4:2%Tm3+; α-NaYF4:2%Er3+,2%Tm3+; α-NaYF4:2%Er3+,5%Tm3+ and α-NaYF4:5%Er3+,2%Tm3+. The obtained materials were applied to the synthesis of β and core@shell NPs.
:1) mixture was put into a three-neck flask connected to a reflux condenser and outgassed using a Schlenk line at 100 °C under low pressure. After 1 h, the mixture was cooled to room temperature, and the selected powder rare earth acetates (see ESI for the synthesis procedure†) were added. The solution of acetates was heated to 100 °C under low pressure, and the mixture was kept at this temperature for 1 h. In the next step, the powder of sodium oleate was put into the solution at 100 °C. Then, the solution was outgassed for 20 min until the dissolution of sodium oleate and the powder of ammonium fluoride was added to the mixture. The solution was again outgassed in three cycles and heated to 200 °C under the nitrogen flow. After heating for 1 h, the synthesis mixture was cooled to room temperature and centrifuged. The collected supernatant was applied to the isolation of fluoride NPs. The addition of ethanol to the supernatant led to the precipitation of NPs. The colloidal solution of NPs was centrifuged, and then the NPs were dissolved in n-hexane (1 mmol NPs:5 mL of n-hexane). The NPs were precipitated again after the addition of ethanol and centrifuged. The following materials were obtained: α-NaYF4; α-NaYF4:2%Tm3+; α-NaYF4:2%Er3+,2%Tm3+; α-NaYF4:2%Er3+,5%Tm3+ and α-NaYF4:5%Er3+,2%Tm3+. The obtained materials were applied to the synthesis of β and core@shell NPs.
Synthesis of core β-NaYF4:Ln3+ NPs
The core β-NaYF4:Ln3+ NPs were obtained by recrystallising the α-core precursor at 300 °C. The purity of the α-core precursor before synthesis was analysed using thermogravimetric analysis (TGA). In the beginning, the α-core precursor was dissolved in the mixture of organic solutions (1 mmol of the α-core precursor was dissolved in 3.72 mL of ODE and 3.72 mL of OA). This mixture was outgassed at 100 °C for 2 h under low pressure. In the next step, the mixture was heated to 300 °C under the flow of nitrogen, and after 55 min, the synthesis mixture was cooled to room temperature. The NPs from the solution were precipitated by adding ethanol and centrifugation. In the next step, the NPs were dissolved again in n-hexane, precipitated one more time after adding ethanol and centrifuged. The following materials were prepared: β-NaYF4:2%Tm3+; β-NaYF4:5%Er3+,2%Tm3+; β-NaYF4:2%Er3+,2%Tm3+; and β-NaYF4:2%Er3+,5%Tm3+.Synthesis of β-NaYF4:Ln3+@β-NaYF4 core@shell NPs
The β-NaYF4:Ln3+ and α-NaYF4 NPs were mixed in a 1:7 ratio and dissolved in the ODE/OA solution, keeping the ratio: 1 mmol of NPs:2 mL of ODE:2 mL of OA. In the beginning, precursors were outgassed at 100 °C for 2 h under low pressure and heated to 300 °C under nitrogen flow. After 135 min, the mixture was cooled to room temperature. The NPs from the solution were precipitated by the addition of ethanol and centrifuged, followed by the dissolution of NPs in n-hexane. The NPs were precipitated again after the addition of ethanol and centrifuged. The following materials were obtained: β-NaYF4:2%Tm3+@β-NaYF4; β-NaYF4:5%Er3+,2%Tm3+@β-NaYF4; β-NaYF4:2%Er3+,2%Tm3+@β-NaYF4; and β-NaYF4:2%Er3+,5%Tm3+@β-NaYF4.
Characterisation of materials
The materials were characterised using selected analytical techniques. TGA was carried out in the flow of pure nitrogen using a Thermogravimetric Analyzer TGA 4000 (PerkinElmer) in the temperature range of 30–600 °C with a temperature ramp of 10 °C min−1.X-ray diffraction (XRD) measurements were performed for fresh powder samples using a Bruker AXS D8 Advance diffractometer equipment with Cu Kα radiation (λ = 0.154 nm), with a step size of 0.05° in the angle range (2θ = 10–80°). The XRD results were used to estimate the size of NPs using the Scherrer formula. The diffraction patterns were compared with those reported in the Inorganic Crystal Structure Database (ICSD).
Before measurements using transmission electron microscopy (TEM), the samples in the form of colloid solution of fluorides in n-hexane were deposited on a copper grid covered with a holey carbon film. TEM images of samples were carried out using a JEOL 1400 electron microscope or Hitachi HT7700 electron microscope operating at 80–120 kV, and the size of particles was estimated using ImageJ™ software.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) combined with energy-dispersive X-ray spectroscopy (EDXS) was used to investigate the chemical composition of core@shell NPs. The experiments were performed on an FEI Osiris ChemiSTEM microscope at 200 keV electron energy, which is equipped with a Bruker Quantax system (XFlash detector) for EDXS. EDXS spectra are quantified with the FEI software package “TEM imaging and analysis” (TIA) version 4.7 SP3. Using TIA, element concentrations were calculated based on a refined Kramers’ law model, which includes corrections for detector absorption and background subtraction. EDXS spectra obtained while scanning a rectangular area inside an NPs ensemble were used to determine their average chemical composition. The concentration profiles of different chemical elements within a single nanoparticle were determined from EDXS spectra measured along a line scan that passes through its centre.
EDXS line profiles were recorded by applying a drift-correction routine via cross-correlation of several images, which yields a local precision better than 1 nm. The drift-corrected EDXS line profiles were taken with a probe diameter of 0.5 nm and a distance of about 1 nm between two measuring points. The quantification of Na-, F-, Y-, Tm- and Er-content from EDXS line or area scans was performed by using the Na-K, Y-L, Tm-L and Er-L series, as well as the F-Kα line.
The crystal structure of selected NPs was investigated by high-resolution transmission electron microscopy (HR-TEM) performed on an aberration-corrected FEI Titan3 80–300 microscope at 300 kV acceleration tension. HR-TEM images were evaluated by calculating the two-dimensional Fourier transform (FT), which yields information on the crystal structure (lattice parameters and crystal symmetry) of single NPs. The analysis was performed by comparing the experimental FT and the calculated diffraction patterns with Miller indices, where the latter were obtained by using the Jems software. The zero-order beam (ZB) is indicated by using a white circle.
Photoluminescence properties were studied at room temperature using a PIXIS:256E Digital CCD Camera equipped with an SP-2156 Imaging Spectrograph (Princeton Instruments) and QuantaMaster™ 40 spectrophotometer (Photon Technology International), equipped with a H10330C-75 photomultiplier (Hamamatsu). A continuous multiwavelength, 2 W diode laser (from CNI) was used as the excitation source at 1208 nm, coupled to a 200 μm optical fibre and collimator (from Thorlabs). For the luminescence risetimes measurements the laser wave was triggered by arbitrary function generator GW Instek AFG-2005. All spectra were corrected for the spectral response of the equipment. A 10A-PPS power meter (Ophir Photonics) determined the beam size and laser powers.
Temperature-dependent emissions were measured by exciting samples with a 1208 nm laser placed in a tubular electric furnace (Gero RES-E 230/3), where the temperature of the sample was controlled via a type K thermocouple in contact with it. A 10 ns pulsed optical parametric oscillator OPO (EKSPLA/NT342/3/UVE) was used as a laser source. Emissions from the oven were focused on the entrance slit of a spectrograph (Andor SR-303i-A) equipped with a cooled CCD camera (Andor Newton). All spectra were corrected from the spectral response of the equipment.
Results and discussion
Structure and morphology
The obtained XRD patterns of the products (Fig. S1†) show the presence of the crystal structure typical for cubic phase (ICSD no. 77099) for samples obtained at a lower temperature (200 °C). At higher temperature (300 °C), cubic NPs transformed to the hexagonal crystal phase (ICSD no. 51916). Also, core@shells crystallised as hexagonal NPs (Fig. 1a).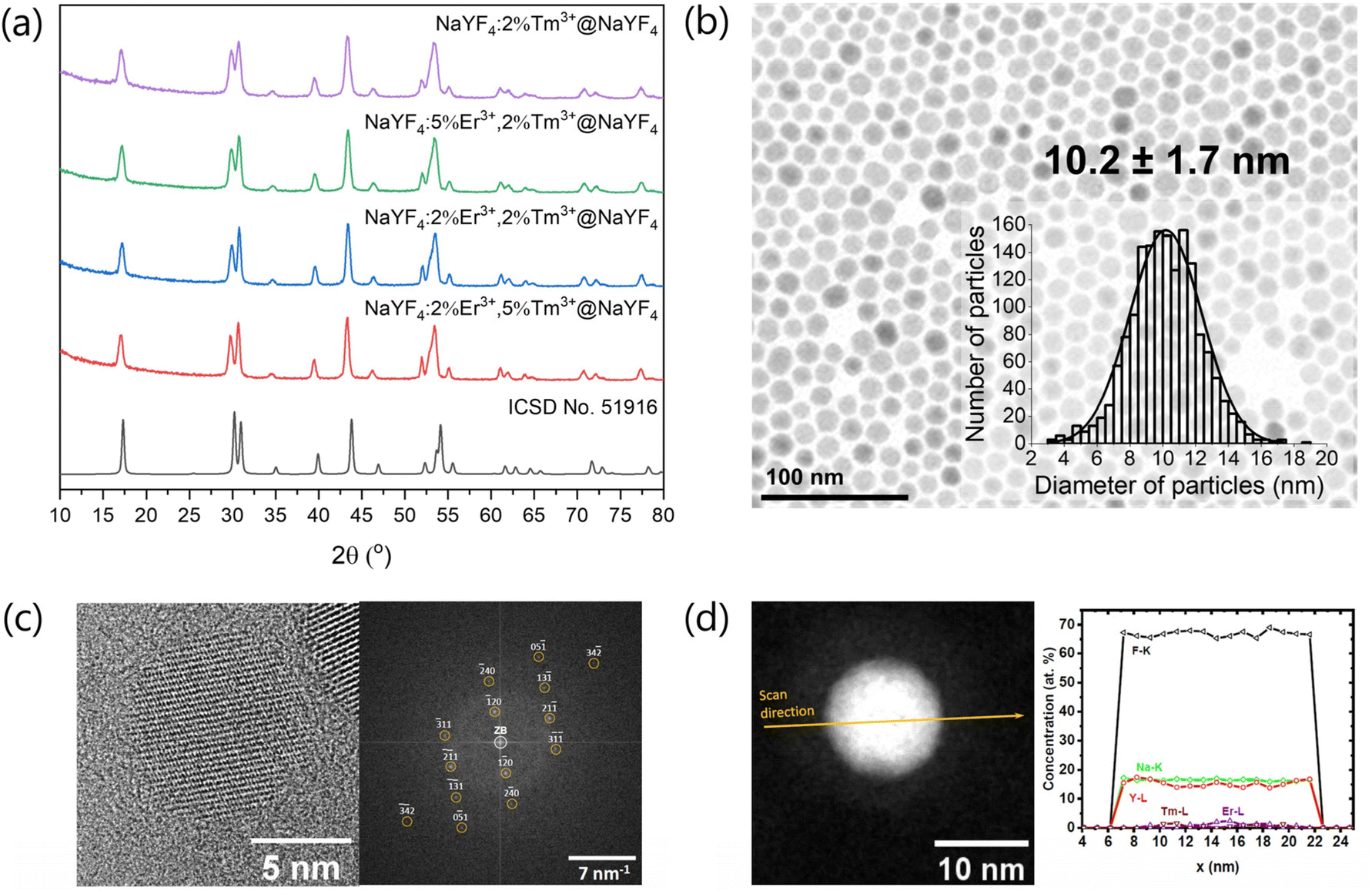 | ||
Fig. 1 (a) XRD patterns performed for core@shell nanomaterials, (b) TEM image and particle size distribution of NaYF4:5%Er3+,2%Tm3+@NaYF4 sample, (c) left panel: HR-TEM image of the NaYF4:5%Er3+,2%Tm3+@NaYF4 nanoparticle, and right panel: its FT pattern, which agrees with the calculated diffraction pattern of bulk hexagonal NaYF4 (space group P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) , No. 174) in the [215]-zone axis (orange circles); (d) left panel: HAADF STEM image with the EDXS line scan direction, which was applied to calculate a chemical composition of a NaYF4:5%Er3+,2%Tm3+@NaYF4 nanoparticle; right panel: concentration profiles determined after the quantification of the EDXS line scan recorded from the core@shell NP on the left panel. , No. 174) in the [215]-zone axis (orange circles); (d) left panel: HAADF STEM image with the EDXS line scan direction, which was applied to calculate a chemical composition of a NaYF4:5%Er3+,2%Tm3+@NaYF4 nanoparticle; right panel: concentration profiles determined after the quantification of the EDXS line scan recorded from the core@shell NP on the left panel. | ||
TEM measurements (Fig. 1, S2 and S3†) revealed that all β-cores precipitated as small NPs with average 8–14 nm sizes. The shell deposition on their surface increased the size of NPs to around 10–22 nm depending on the size of source core NPs. TEM images in Fig. S2 and S3† reveal that the synthesis procedure successfully generated homogeneous NPs. The composition of NPs influenced their shape. In the case of β-core doped only with Tm3+ ions (NaYF4:2%Tm3+), NPs are hexagonal rounded. The NaYF4:2%Tm3+@NaYF4 core@shell NPs also had a hexagonal structure (Fig. 1b). The addition of a higher concentration of Er3+ ions to the structure of NaYF4 led to the change in the shape of NPs to more elongated, rod-like (Fig. S2†). It is caused by the anisotropic growth of NPs in the presence of Er3+ ions, which have different sizes than Y3+ ions.45 HR-TEM and FT presented in Fig. 1c also confirm the hexagonal structure of prepared NPs similar to pure NaYF4 without significant changes after doping with Er3+ and Tm3+ ions.
The differences in NPs sizes may result from the nature of the synthesis method, which is quite sensitive to changes in conditions and has limited repeatability, mainly affecting small NPs. As evident from the comparison of Fig. S2 and S3,† the final NPs sizes were significantly influenced by the coating with shell applied to the obtained cores. Controlling this process is particularly challenging, and size variations may result from fluctuations in the synthesis conditions.
To investigate the average chemical composition of NaYF4:2%Tm3+,5%Er3+@NaYF4 NPs, we used EDXS spectra obtained by scanning a rectangular area, which includes inside an ensemble of NPs. Fig. 1d shows the distribution profiles of Na, F, Y, Tm and Er elements in the single nanoparticle determined from the EDXS line scan, which was recorded along a line that passes through the object's centre (arrow in Fig. 1d, left panel). The quantification of EDXS line scans performed using the X-ray characteristic lines of the corresponding elements, i.e. lines of the Na-K, Y-L, Tm-L and Er-L series, as well as of the F-K line, from the core@shell NPs gave information about the chemical composition. The chemical composition of NaYF4: 2%Tm3+,5%Er3+@NaYF4 was calculated from the EDXS line scan shown in Fig. 1d, and the obtained results Na16.0±0.5Y16.0±1.3Er1.5±0.5Tm0.3±0.1F66.2±1.4 correspond with the assumed composition.
Spectroscopic properties
NPs doped with Tm3+ can absorb 1208 nm NIR light due to the 3H6 → 3H5 electronic transition. Fig. S4† demonstrates the absorption spectra of NaYF4:2%Tm3+,5%Er3+@NaYF4 NPs, where between 1150 and 1250 nm, a broad and intense absorption band is visible. This fact makes it possible to use 1208 nm to excite Tm3+ ions to higher energy levels due to the sequential absorption of several NIR photons.29 The effect of this process is the UC of Tm3+ ions at 698 and 802 nm. Fig. 2a shows the emission of NaYF4:2%Tm3+@NaYF4 NPs (red line). The main emission occurs at 802 nm due to the 3H4 → 3H6 transition. Under 1208 nm excitation, some of the Tm3+ ions are excited even up to the 1G4 excited state, but emission connected with 1G4 → 3H6 transition is relatively weak (see Fig. 2b). It means a very low probability of the 1G4 state population. NaYF4:2%Tm3+@NaYF4 NPs also show down-shifted emission in the NIR range with the maximum at 1625 nm connected with 3F4 → 3H6 transition (Fig. 2c). | ||
| Fig. 2 (a) emission spectra of the NPs obtained under 1208 nm laser excitation, (b) emission in the blue region of spectra indicating transitions from the higher excited states of Er3+ and Tm3+ ions and (c) emission in the NIR range of the sample containing the highest concentration of Er3+ ions showing down-shifted emission of NPs under 1208 m laser excitation. | ||
The emission properties change dramatically with co-doping with Er3+ ions. Under 1208 nm excitation, all of the NaYF4:Er3+,Tm3+@NaYF4 NPs show red emission with a maximum at 660 nm, related to the 4F9/2 → 4I15/2 transition. Tm3+ ions transfer the absorbed energy to Er3+ ions, which results in their population up to the 2H9/2 excited state (see Fig. 2b). Emission of Er3+ ions under 1208 nm also occurs at around 526 and 546 nm due to the 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions and at 982 nm as the result of the 4I11/2 → 4I15/2 transition. Second, the most intense emission band has its maximum at around 802 nm, which is a result of the 3H4 → 3H6 transition of Tm3+ ions. However, it is also possible that in the case of Er3+ and Tm3+ doped NPs, this band also corresponds to the 4I9/2 → 4I15/2 transition of Er3+ ions in some part. Interesting is the enhancement of Tm3+ ions UC at 475 nm and new emission band at around 455 nm when Er3+ ions are co-dopants (Fig. 2b). It means that Er3+ take part in the population of higher excited states of Tm3+ ions.46 Down-shifted emission of NaYF4:Er3+,Tm3+@NaYF4 NPs is visible in Fig. 2c as two broad bands with the maximum at around 1525 nm, which is the result of 4I13/2 → 4I15/2 transition of Er3+ ions and at 1625 nm which is connected with 3F4 → 3H6 transition of Tm3+ ions.
The dependence of UC intensity on laser power densities presented in Fig. 3 shows the complex nature of the studied system. Three to four photons are required to observe the emission of NaYF4:2%Tm3+@NaYF4 NPs at 698 and 802 nm (see ESI for the methodology of calculations†). Above power density of 70 W cm−2, slope values increase from 2.1 to above 3.3. In previous reports, usually, two photons were required to populate the 3F3 and 3H4 excited states of Tm3+ ions under similar excitation wavelength but with lower power densities (<30 W cm−2).29,30
 | ||
| Fig. 3 Upconversion intensity dependencies on laser power density measured for (a) NaYF4:2%Tm3+@NaYF4, (b) NaYF4:5%Er3+,2%Tm3+@NaYF4, (c) NaYF4:2%Er3+,2%Tm3+@NaYF4 and (d) NaYF4:2%Er3+,5%Tm3+@NaYF4 NPs. | ||
Five 1208 nm photons are required to excite Er3+ to the 2H11/2 and 4S3/2 green-emitting levels (see Fig. 3b–d). The slope values determined for emission bands at 526 and 546 nm of NaYF4:Er3+,Tm3+@NaYF4 NPs vary between 4.2 and 4.8 in the 70–130 W cm−2 power density range. The green emission is not even visible below the power density threshold mentioned above. In the case of red emission of Er3+ ions from the 4F9/2 excited state, the slope values vary between 3.9 and 4.4 in the 70–130 W cm−2 power density range. Emission at 982 nm from the 4I11/2 energy level requires fewer photons than from the 4F9/2 excited state as slope values oscillate between 3.2 and 3.6.
Below 70 and above around 130 W cm−2 slopes of presented curves are lower than in the mentioned 70–130 W cm−2 range. Lowering the slope values at high power densities results from saturation effects where intermediate excited states undergo depletion by different nonradiative mechanisms.47 Similar power dependencies were found for Gd2O3 NPs doped with Er3+ ions under 970 nm laser excitation.48 Such anomalous behaviour can be explained by a positive feedback looping upconversion mechanism.41,48 An extreme form of looping is known as photon avalanche.38 Avalanche processes can be seen in many papers,49,50 recently also in core@shell NPs.51 Furthermore, it should also be taken into account that in the case of a typical photon avalanche process, slope values above the critical excitation power density do not represent the number of photons. The high number is associated with the nonlinear effect of photon avalanche. However, in the case of the samples under investigation, we are not dealing with a typical photon avalanche process.
Upconversion mechanism
The mechanism of observed UC is complex and not obvious. Under excitation with 1208 nm laser light, Tm3+ ions undergo GSA to the 3H5, the second excited state. The excitation to higher excited states is possible via the ESA of the second 1208 nm photon.30,40 However, here we postulate different mechanism of excitation of Tm3+ ions. Based on luminescence decays (Fig. 4) and the rate equation model (see ESI Fig. S6†), the 3F3 excited state of Tm3+ is populated mainly by ET1 (Fig. 5) from Tm3+ ions in their 3F4 excited state. This is also consistent with the slope values calculated for the 698 nm emission band at low power densities <70 W cm−2 (Fig. 3a). | ||
| Fig. 4 Luminescence rises and decays measured under laser λex = 1208 nm excitation for (a) NaYF4:2%Tm3+@NaYF4 and (b) NaYF4:5%Er3+,2%Tm3+@NaYF4 NPs. | ||
 | ||
| Fig. 5 Mechanism of up-conversion in (a) NaYF4:Tm3+@NaYF4 NPs and (b) NaYF4:Er3+,Tm3+@NaYF4 NPs, where ET – energy transfer, CR – cross-relaxation process. The Er3+ ions are equivalent; the processes involving them were separated only for the sake of clarity. | ||
Interesting is the origin of emission from the 3H4 excited state of Tm3+ ions at 802 nm, which dominates the emission spectra of NaYF4:2%Tm3+@NaYF4 (Fig. 2a). There are different possibilities to populate the 3H4 level: it can be populated by relaxation from 3F3 excited state or other energy transfer processes from lower levels. However, as evident from Fig. S7 in ESI,† when considering relaxation from the 3F3 level, the temporal evolution of the UC intensity at 802 nm aligns remarkably well with the theoretical pattern.
Once the ions are excited to the 3F3 and 3H4 levels, they can relax through different processes, including CR channels that repopulate the 3F4 and 3H5 levels. From these levels, the upconversion process ET1 could start again. This dynamic can potentially initiate a looping process or an avalanche effect that gains efficiency with higher pumping power. This phenomenon can elucidate the observed slope alterations, as illustrated in the curves of Fig. 3. A change in the rise time can also characterise these processes. Therefore, the rise times of the UC emissions, defined by the Kohlrausch function, have been calculated. As shown in Fig. S5,† about 80 W cm−2, the rise time reaches a maximum that is a characteristic of the avalanche process.49,50 Moreover, this maximum at about 80 W cm−2 coincides with the slope change shown in Fig. 3.
The excitation mechanism of Er3+ ions under 1208 nm laser light is based on the ET process originating from Tm3+ ions. Due to the similar energies between 3H4 of Tm3+ and 4I9/2 state of Er3+ ions, we postulate ET between these two electronic states as the primary population pathway of Er3+ ions (ET2 in Fig. 5). Emission lifetime of Tm3+ ions connected with 3H4 → 3H6 transition is five times shorter when Er3+ ions are present in the structure (see Fig. 4) which confirms our conclusion. Next, Er3+ ions can be excited to the 2H11/2 or 4S3/2 energy states by another ET from Tm3+ ions in their 3F4 excited state (ET3). A higher concentration of Tm3+ ions result in a more intense emission of Er3+ ions. However, the probability of ET3 from the 4I9/2 excited state of Er3+ ions is relatively low as Er3+ ions undergo multiphonon relaxation from the 4I9/2 to the 4I11/2 excited state. Fig. 3b–d shows that up to five photons are necessary to achieve green emitting levels of Er3+ ions. Four of them are consumed to populate Tm3+ to the 3H4 excited state and another one to excite Er3+ to 2H11/2 or 4S3/2 energy states. The role of Tm3+ ions in the population of Er3+ is also visible in Fig. 2b, where the increase of Tm3+ concentration from 2% to 5% allowed for another ET and excitation of Er3+ ions up to the 2H9/2 excited state and their blue emission at around 410 nm.
Increasing the concentration of Er3+ from 2% to 5% quenched green emission. Er3+ ions undergo CR with Er3+ ions in their 4I15/2 ground state (see Fig. 5b). There are many possible CR pathways between Er3+ ions,52,53 but in the studied system, the CR2 presented in Fig. 5 seems to be the most probable. CR process may also precede the population of Er3+ ions to their 4F9/2 energy level but this population pathway is rather negligible. The excitation of Er3+ ions from 4I11/2 to 4F9/2 excited state occurs via ET from Tm3+ ions. Tm3+ to Er3+ ET is a subject of many reports and explains the shift from green to red upconversion in materials co-doped with Tm3+.54–57 Here, we postulate that the ET3 from the 4I11/2 excited state of Er3+ ions is more probable than from the 4I9/2 state. Red luminescence does not result from multiphonon relaxation from the 4S3/2 excited state.58 Our conclusion also confirms the decrease of the Er3+ emission band at 982 nm with an increased concentration of Tm3+ ions. A higher concentration of Tm3+ ions means depopulation to the 4I11/2 excited state, leading to the excitation of Er3+ to the 4F9/2via ET3. Furthermore, we believe Er3+ ions also participate in energy looping by populating Tm3+ ions to the 3F4 level through ET4. This is also reflected in the prolonged luminescence lifetime from the 3F4 level (1660 nm) in the sample doped with both Er3+ and Tm3+ ions compared to the sample containing only Tm3+ ions (Fig. 4).
Temperature sensing properties
NaYF4:Er3+,Tm3+@NaYF4 NPs are ideal candidates for temperature sensing applications in biological medium. They can be excited within the 2nd biological window, whereas their luminescence is in the 1st biological window. Fig. 6 presents the spectroscopic properties of the NaYF4:2%Er3+,5%Tm3+@NaYF4 NPs measured in the 21–115 °C range. The intensity of emission bands changes with increasing temperature. The dependence of luminescence intensity ratio (LIR) between two luminescence bands on temperature is a perfect tool for remote temperature determination.43,59,60Fig. 6b shows that the 4I11/2 → 4I15/2 transition band of Er3+ ions with a maximum of around 982 nm is the most sensitive to the temperature change (due to the multiphonon relaxation from 4I9/2 state, see Fig. 2), while the 3F3 → 3H6 transition band of Tm3+ ions, with the maximum at 698 nm, shows relatively low sensitivity. These two bands can be used for ratiometric temperature sensing with the best effect. The 982/698 LIR dependence on temperature depicted in Fig. 6c confirms that assumption. Other promising bands for temperature sensing are connected with the 4F9/2 → 4I15/2 transition of Er3+ ions (660 nm) and 3H4 → 3H6 transition of Tm3+ ions (802 nm). Therefore, the 660/698 and 982/802 LIRs are also relatively sensitive to temperature change, but less than in the case of 982/698 LIR. | ||
| Fig. 6 (a) Temperature-dependent upconversion spectra of NaYF4:2%Er3+,5%Tm3+@NaYF4 NPs under λex = 1208 nm excitation, (b) integrated intensity of Tm3+ and Er3+ emission bands in function of temperature, (c) luminescence intensity ratios and (d) relative temperature sensitivities determined from the measured spectra. | ||
The dependencies of measured LIRs on temperature were fitted with Boltzmann-like or cubic functions to calculate relative temperature sensitivities, Sr (see ESI for more details†). The determined Sr values as the dependencies on temperature are presented in Fig. 6d. The 982/698 LIR shows the best sensitivity in the whole temperature range with a maximum value of 2.37%/°C at 57 °C. This value determines that the obtained nanothermometer is very good for temperature-sensing applications. Notably, in the physiological range, 22–50 °C, the obtained nanothermometer offers a relatively high sensitivity between 1.6 and 2.3%/°C. The sensitivity of thermometers close to 3%/°C is recently considered high.61 The relative sensitivities of 660/698 nm and 982/802 nm LIRs are lower than the 982/698 nm LIR but still at a high level, i.e. 1.69%/°C at 77 °C and 1.58%/°C at 67 °C respectively.
The green 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 luminescence bands of Er3+ ions are the most frequently studied for applications in optical thermometry.9,42,43,62 The 2H11/2 and 4S3/2 energy states of Er3+ ions are thermally coupled as their energy difference is around 625 cm−1, considering the recorded emission spectrum (the Boltzmann function fit yields the value of 655 cm−1). Thermalisation processes lead to an increase in the intensity of the higher-energy transition (at 526 nm) and a decrease in the intensity of the lower-energy one (at 546 nm), according to the Boltzmann distribution. In many cases, the relative temperature sensitivity of green-emitting bands is satisfactory and can be used for temperature determination. However, in the case of NaYF4:2%Er3+,5%Tm3+@NaYF4 NPs, the calculated Sr values indicate much lower sensitivity of 526/546 LIR (maximum 1.08%/°C at 22 °C) than those determined for 982/698 and 982/802 or 660/698 LIRs, but similar to those reported in the literature.9,42,63 Additionally, biological materials are usually less transparent for green luminescence, which makes the use of green bands of Er3+ ions less beneficial than those in the red and NIR range.27
Conclusions
In summary, our study shows that Tm3+ ions can be used as sensitisers in upconverting core@shell NPs. Under 1208 nm NIR radiation, NaYF4:Tm3+@NaYF4 and NaYF4:Er3+,Tm3+@NaYF4 NPs showed intense emission. The Tm3+-doped only NPs are able of UC to 698 and 802 nm via the ground state absorption followed by energy transfers and CR between Tm3+ ions. Adding Er3+ as a second co-dopant results in higher luminescence intensity and red luminescence of NPs. The emission from Er3+ ions is possible due to the ET from Tm3+ ions.The studied system revealed the complex mechanism of UC based on energy looping. Such a process generates multiple Tm3+ ions in their 3F4 excited state by a sequence of GSA, ET and CR processes. The proposed mechanism explains high slope values determined from emission intensities’ dependencies on laser powers. The observed UC resulted from 4 photon excitation mechanism in the case of Tm3+ emissions at 698 and 802 nm or 5 photons for Er3+ emissions at 526, 546 and 660 nm.
The Er3+/Tm3+ system is promising for biological applications because of excitation possibility within the 2nd biological window at 1208 nm and emission within the 1st biological window, with major bands at around 660 and 802 nm. The emission at 982 nm is also intense. Additionally, we presented that luminescence of NaYF4:Er3+,Tm3+@NaYF4 NPs is temperature dependent, and the 982/698 nm, 660/698 nm and 982/802 nm band ratios show high relative temperature sensitivities. The emission bands used for temperature-sensing applications must be close to each other, which lowers possible effects and distortions caused by the properties of the biological environment. Our NPs show such property.
The presented article proves that the UC phenomenon is possible in other than Yb3+-based systems, the most frequently used. However, the obtained results indicate that the Er3+/Tm3+ system still requires further research, mainly to optimise its emission and fully understand the mechanism of UC at different concentrations of dopants. It is also essential to test the proposed NPs as nanothermometers in real biological systems where the potential of excitation within 2nd biological system can be fully exploited.
Author contributions
T. G.: conceptualisation, data curation, formal analysis, funding acquisition, investigation, methodology, visualisation, writing – original draft, writing – review & editing. I. R. M.: conceptualisation, data curation, methodology supervision writing – original draft, writing – review & editing, formal analysis. R. P.: formal analysis, methodology, resources, visualisation, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Science Centre, Poland, provided funding for this research under grant no. 2016/22/E/ST5/00016. We want to express our sincere gratitude to Dr Piotr Kamiński for his help with core/shell nanoparticles synthesis and Dr Artur Tymiński for measurements of the power dependencies.References
- M. R. Hamblin, Dalton Trans., 2018, 47, 8571–8580 RSC.
- K. Y. Pham, L. C. Wang, C. C. Hsieh, Y. P. Hsu, L. C. Chang, W. P. Su, Y. H. Chien and C. S. Yeh, J. Mater. Chem. B, 2021, 9, 694–709 RSC.
- A. Hlaváček, Z. Farka, M. J. Mickert, U. Kostiv, J. C. Brandmeier, D. Horák, P. Skládal, F. Foret and H. H. Gorris, Nat. Protoc., 2022, 17, 1028–1072 CrossRef PubMed.
- A. Drozdowski, N. Jurga, D. Przybylska, J. C. Brandmeier, Z. Farka, H. H. Gorris and T. Grzyb, J. Colloid Interface Sci., 2023, 649, 49–57 CrossRef CAS PubMed.
- J. C. Brandmeier, N. Jurga, T. Grzyb, A. Hlaváček, R. Obořilová, P. Skládal, Z. Farka and H. H. Gorris, Anal. Chem., 2023, 95, 4753–4759 CrossRef CAS PubMed.
- H. Li, M. Tan, X. Wang, F. Li, Y. Zhang, L. L. Zhao, C. Yang and G. Chen, J. Am. Chem. Soc., 2020, 142, 2023–2030 CrossRef CAS PubMed.
- D. Yang, P. Ma, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2015, 44, 1416–1448 RSC.
- M. Suta, Ž. Antić, V. Đorđević, S. Kuzman, M. D. Dramićanin and A. Meijerink, Nanomaterials, 2020, 10, 543 CrossRef CAS PubMed.
- A. M. Kaczmarek, M. Suta, H. Rijckaert, A. Abalymov, I. Van Driessche, A. G. Skirtach, A. Meijerink and P. Van Der Voort, Adv. Funct. Mater., 2020, 30, 2003101 CrossRef CAS.
- T. P. van Swieten, D. Yu, T. Yu, S. J. W. Vonk, M. Suta, Q. Zhang, A. Meijerink and F. T. Rabouw, Adv. Opt. Mater., 2021, 9, 3–9 Search PubMed.
- M. Quintanilla, M. Henriksen-Lacey, C. Renero-Lecuna and L. M. Liz-Marzán, Chem. Soc. Rev., 2022, 51, 4223–4242 RSC.
- S. Wu, G. Han, D. J. Milliron, S. Aloni, V. Altoe, D. V. Talapin, B. E. Cohen and P. J. Schuck, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1–5 CrossRef PubMed.
- D. Przybylska, T. Grzyb, A. Erdman, K. Olejnik and A. Szczeszak, Sci. Rep., 2022, 12, 19388 CrossRef CAS PubMed.
- T. Grzyb, P. Kamiński, D. Przybylska, A. Tymiński, F. Sanz-Rodríguez and P. Haro Gonzalez, Nanoscale, 2021, 13, 7322–7333 RSC.
- A. Tymiński, E. Śmiechowicz, I. R. Martín and T. Grzyb, ACS Appl. Nano Mater., 2020, 3, 6541–6551 CrossRef.
- X. Zheng, R. K. Kankala, C. G. Liu, S. Bin Wang, A. Z. Chen and Y. Zhang, Coord. Chem. Rev., 2021, 438, 213870 CrossRef CAS.
- X. Cheng, D. Tu, W. Zheng and X. Chen, Chem. Commun., 2020, 56, 15118–15132 RSC.
- G. Chen, H. Ågren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- N. Jurga, D. Przybylska, P. Kamiński, A. Tymiński, B. F. Grześkowiak and T. Grzyb, J. Colloid Interface Sci., 2022, 606, 1421–1434 CrossRef CAS PubMed.
- N. Jurga, D. Przybylska, P. Kamiński and T. Grzyb, Sci. Rep., 2021, 11, 18846 CrossRef CAS PubMed.
- B. Zhou, L. Yan, J. Huang, X. Liu, L. Tao and Q. Zhang, Nat. Photonics, 2020, 14, 760–766 CrossRef CAS.
- I. Sokólska, W. Ryba-Romanowski, S. Gołab and T. Lukasiewicz, Appl. Phys. B, 1997, 65, 495–498 CrossRef.
- C. Homann, L. Krukewitt, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and M. Haase, Angew. Chem., Int. Ed., 2018, 57, 8765–8769 CrossRef CAS PubMed.
- T. Cheng, R. Marin, A. Skripka and F. Vetrone, J. Am. Chem. Soc., 2018, 140, 12890–12899 CrossRef CAS PubMed.
- Y. Fan and F. Zhang, Adv. Opt. Mater., 2019, 7, 1801417 CrossRef.
- L. A. Sordillo, Y. Pu, S. Pratavieira, Y. Budansky and R. R. Alfano, J. Biomed. Opt., 2014, 19, 056004 CrossRef PubMed.
- N. Jurga, S. Ryszczyńska and T. Grzyb, Spectrochim. Acta, Part A, 2023, 303, 123220 CrossRef CAS PubMed.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- W. Shao, G. Chen, T. Y. Ohulchanskyy, A. Kuzmin, J. Damasco, H. Qiu, C. Yang, H. Ågren and P. N. Prasad, Adv. Opt. Mater., 2015, 3, 575–582 CrossRef CAS.
- H. Zhang, Y. Fan, P. Pei, C. Sun, L. Lu and F. Zhang, Angew. Chem., 2019, 131, 10259–10263 CrossRef.
- A. Tymiński, I. R. Martín and T. Grzyb, Part. Part. Syst. Charact., 2020, 37, 2000068 CrossRef.
- C. Lee, E. Z. Xu, Y. Liu, A. Teitelboim, K. Yao, A. Fernandez-Bravo, A. M. Kotulska, S. H. Nam, Y. D. Suh, A. Bednarkiewicz, B. E. Cohen, E. M. Chan and P. J. Schuck, Nature, 2021, 589, 230–235 CrossRef CAS PubMed.
- H. Zhang, Y. Fan, P. Pei, C. Sun, L. Lu and F. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10153–10157 CrossRef CAS PubMed.
- S. Ryszczyńska and T. Grzyb, Methods Appl. Fluoresc., 2022, 10, 024001 CrossRef PubMed.
- X. Cheng, H. Ge, Y. Wei, K. Zhang, W. Su, J. Zhou, L. Yin, Q. Zhan, S. Jing and L. Huang, ACS Nano, 2018, 12, 10992–10999 CrossRef CAS PubMed.
- S. Ryszczyńska, K. Trejgis, Ł. Marciniak and T. Grzyb, ACS Appl. Nano Mater., 2021, 4, 10438–10448 CrossRef.
- G. Chen, T. Y. Ohulchanskyy, A. Kachynski, H. Ågren and P. N. Prasad, ACS Nano, 2011, 5, 4981–4986 CrossRef CAS PubMed.
- E. S. Levy, C. A. Tajon, T. S. Bischof, J. Iafrati, A. Fernandez-Bravo, D. J. Garfield, M. Chamanzar, M. M. Maharbiz, V. S. Sohal, P. J. Schuck, B. E. Cohen and E. M. Chan, ACS Nano, 2016, 10, 8423–8433 CrossRef CAS PubMed.
- L. Yan, B. Zhou, N. Song, X. Liu, J. Huang, T. Wang, L. Tao and Q. Zhang, Nanoscale, 2018, 10, 17949–17957 RSC.
- U. R. Rodríguez-Mendoza and F. Lahoz, J. Lumin., 2016, 179, 40–43 CrossRef.
- S. Sivakumar, F. C. J. M. van Veggel and P. S. May, J. Am. Chem. Soc., 2007, 129, 620–625 CrossRef CAS PubMed.
- F. Paz-Buclatin, F. Rivera-López, O. González, I. R. Martín, L. L. Martin and D. J. Jovanović, Sens. Actuators, A, 2019, 299, 111628 CrossRef CAS.
- D. Jaque and F. Vetrone, Nanoscale, 2012, 4, 4301–4326 RSC.
- M. Quintanilla, E. Hemmer, J. Marques-Hueso, S. Rohani, G. Lucchini, M. Wang, R. R. Zamani, V. Roddatis, A. Speghini, B. S. Richards and F. Vetrone, Nanoscale, 2022, 14, 1492–1504 RSC.
- S. Fischer, N. J. J. Johnson, J. Pichaandi, J. C. Goldschmidt and F. C. J. M. van Veggel, J. Appl. Phys., 2015, 118, 193105 CrossRef.
- H. Zhang, T. Jia, X. Shang, S. Zhang, Z. Sun and J. Qiu, Phys. Chem. Chem. Phys., 2016, 18, 25905–25914 RSC.
- Y. Lei, H. Song, L. Yang, L. Yu, Z. Liu, G. Pan, X. Bai and L. Fan, J. Chem. Phys., 2005, 123, 174710 CrossRef PubMed.
- G. Y. Chen, H. J. Liang, H. C. Liu, G. Somesfalean and Z. G. Zhang, J. Appl. Phys., 2009, 105, 114315 CrossRef.
- I. R. Martín, C. Goutaudier, S. Guy, Y. Guyot, G. Boulon, M. T. Cohen-Adad and M. F. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 7252–7257 CrossRef.
- S. Guy, M. F. Joubert and B. Jacquier, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 8240–8248 CrossRef CAS.
- Y. Liang, Z. Zhu, S. Qiao, X. Guo, R. Pu, H. Tang, H. Liu, H. Dong, T. Peng, L. D. Sun, J. Widengren and Q. Zhan, Nat. Nanotechnol., 2022, 17, 524–530 CrossRef CAS PubMed.
- A. Baride, P. S. May and M. T. Berry, J. Phys. Chem. C, 2020, 124, 2193–2201 CrossRef CAS.
- A. Noculak, A. Podhorodecki, G. Pawlik, M. Banski and J. Misiewicz, Nanoscale, 2015, 13784–13792 RSC.
- T. Grzyb, D. Przybylska, A. Szczeszak, E. Śmiechowicz, P. Kulpiński and I. R. Martín, Carbohydr. Polym., 2022, 294, 119782 CrossRef CAS PubMed.
- E. M. Chan, D. J. Gargas, P. J. Schuck and D. J. Milliron, J. Phys. Chem. B, 2012, 116, 10561–10570 CrossRef CAS PubMed.
- J. F. Suyver, J. Grimm, M. K. Van Veen, D. Biner, K. W. Krämer and H. U. Güdel, J. Lumin., 2006, 117, 1–12 CrossRef CAS.
- Q. Chen, X. Xie, B. Huang, L. Liang, S. Han, Z. Yi, Y. Wang, Y. Li, D. Fan, L. Huang and X. Liu, Angew. Chem., Int. Ed., 2017, 56, 7605–7609 CrossRef CAS PubMed.
- M. T. T. Berry and P. S. S. May, J. Phys. Chem. A, 2015, 119, 9805–9811 CrossRef CAS PubMed.
- P. Cortelletti, A. Skripka, C. Facciotti, M. Pedroni, G. Caputo, N. Pinna, M. Quintanilla, A. Benayas, F. Vetrone and A. Speghini, Nanoscale, 2018, 10, 2568–2576 RSC.
- A. Skripka, A. Benayas, R. Marin, P. Canton, E. Hemmer and F. Vetrone, Nanoscale, 2017, 9, 3079–3085 RSC.
- Y. Shen, J. Lifante, I. Zabala-Gutierrez, M. de la Fuente-Fernández, M. Granado, N. Fernández, J. Rubio-Retama, D. Jaque, R. Marin, E. Ximendes and A. Benayas, Adv. Mater., 2022, 34, 2107764 CrossRef CAS PubMed.
- C. D. S. Brites, E. D. Martínez, R. R. Urbano, C. Rettori and L. D. Carlos, Front. Chem., 2019, 7, 1–10 CrossRef PubMed.
- H. Suo, C. Guo, J. Zheng, B. Zhou, C. Ma, X. Zhao, T. Li, P. Guo and E. M. Goldys, ACS Appl. Mater. Interfaces, 2016, 8, 30312–30319 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures, structure and morphology studies, absorption spectra and luminescence decays, methodology of calculations. See DOI: https://doi.org/10.1039/d3nr04418a |
| This journal is © The Royal Society of Chemistry 2024 |