
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVerticillins: fungal epipolythiodioxopiperazine alkaloids with chemotherapeutic potential
Herma C.
Pierre
a,
Chiraz Soumia M.
Amrine
ab,
Michael G.
Doyle
a,
Amrita
Salvi
c,
Huzefa A.
Raja
 a,
Jonathan R.
Chekan
a,
Andrew C.
Huntsman
d,
James R.
Fuchs
d,
Kebin
Liu
ef,
Joanna E.
Burdette
c,
Cedric J.
Pearce
g and
Nicholas H.
Oberlies
*a
a,
Jonathan R.
Chekan
a,
Andrew C.
Huntsman
d,
James R.
Fuchs
d,
Kebin
Liu
ef,
Joanna E.
Burdette
c,
Cedric J.
Pearce
g and
Nicholas H.
Oberlies
*a
aDepartment of Chemistry and Biochemistry, University of North Carolina at Greensboro, P.O. Box 26170, Greensboro, North Carolina 27402, USA. E-mail: nicholas_oberlies@uncg.edu
bDepartment of Physical and Earth Sciences. Arkansas Tech University, 1701 N. Boulder Ave., Russellville, Arkansas 72801, USA
cDepartment of Pharmaceutical Sciences, University of Illinois at Chicago, 900 S. Ashland Ave (M/C 870), Chicago, Illinois 60607, USA
dDivision of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, Ohio State University, 500 W. 12th Ave., Columbus, Ohio 43210, USA
eDepartment of Biochemistry and Molecular Biology and the Georgia Cancer Center, Medical College of Georgia, Augusta, GA 30912, USA
fCharlie Norwood Veterans Affairs Medical Center, Augusta, GA 30904, USA
gMycosynthetix, Inc., Hillsborough, NC 27278, USA
First published on 17th April 2024
Abstract
Covering: 1970 through June of 2023
Verticillins are epipolythiodioxopiperazine (ETP) alkaloids, many of which possess potent, nanomolar-level cytotoxicity against a variety of cancer cell lines. Over the last decade, their in vivo activity and mode of action have been explored in detail. Notably, recent studies have indicated that these compounds may be selective inhibitors of histone methyltransferases (HMTases) that alter the epigenome and modify targets that play a crucial role in apoptosis, altering immune cell recognition, and generating reactive oxygen species. Verticillin A (1) was the first of 27 analogues reported from fungal cultures since 1970. Subsequent genome sequencing identified the biosynthetic gene cluster responsible for producing verticillins, allowing a putative pathway to be proposed. Further, molecular sequencing played a pivotal role in clarifying the taxonomic characterization of verticillin-producing fungi, suggesting that most producing strains belong to the genus Clonostachys (i.e., Bionectria), Bionectriaceae. Recent studies have explored the total synthesis of these molecules and the generation of analogues via both semisynthetic and precursor-directed biosynthetic approaches. In addition, nanoparticles have been used to deliver these molecules, which, like many natural products, possess challenging solubility profiles. This review summarizes over 50 years of chemical and biological research on this class of fungal metabolites and offers insights and suggestions on future opportunities to push these compounds into pre-clinical and clinical development.
1. Introduction
The structural diversity of fungal metabolites, often coupled with potent biological activity, makes them an attractive source for drug discovery.1–4 The discovery and development of penicillin, perhaps the world's most well-known fungal metabolite,5 was honored with the Nobel Prize in Physiology or Medicine in 1945.6 As such, it is probably not surprising that a great deal of research in this vein, particularly during the antibiotics revolution,7,8 focused on fungi as a source of anti-infective agents.9,10 More recently, researchers have shown growing interest in investigating fungal metabolites for anticancer purposes,11 although to date, no FDA-approved anticancer drugs have been derived from fungi.12 The fungal kingdom is hyper diverse, and various sources estimate 1.5 to 2.5 to 5.1 million species.13–15 The exact number may never be known, and regardless, only about 155![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00015 are taxonomically described in the literature. Of these, it is likely that even fewer have been studied for bioactive secondary metabolites,16 making fungi an under-investigated reservoir of pharmaceutical leads.11,12,17
00015 are taxonomically described in the literature. Of these, it is likely that even fewer have been studied for bioactive secondary metabolites,16 making fungi an under-investigated reservoir of pharmaceutical leads.11,12,17
With over 15 years of research on cytotoxic fungal metabolites,18,19 representing the evaluation of several thousand fungi and the isolation and elucidation of >700 fungal metabolites, the verticillins represent an area of emphasis for our team, including scaled production,20 analogue development,21,22 drug delivery,23,24 and in vitro and in vivo evaluation against a range of cancer models.22–30 While several reviews have been published on specific aspects of the epipolythiodioxopiperazine (ETP) alkaloids,31–36 as a structural class, none of these have focused on the verticillins. Given that research on these compounds now spans over 50 years, a comprehensive summary was timely, particularly with the growing number of publications on members of this class over the last decade.20–30,37–46
2. Structure, biosynthesis, and fungal origin of verticillins
Epipolythiodioxopiperazine (ETPs) alkaloids are a class of fungal secondary metabolites31,47 characterized by the presence of a polysulfur bridge on the dioxopiperazine moiety. These can be subdivided into 14 groups according to structural characteristics,33 and of those, only three are dimeric, specifically verticillins, chaetocins, and leptosins (Fig. 1). The main difference between these is that chaetocins have methyl alcohol groups on both R1 and R2, while leptosins contain at least one isopropyl side chain. For verticillins, the substituents at R1 and R2 vary, ranging from CH3, CH3CHOH, CH3CH2, CH2OH, and/or acetyl, and their substitution can be either identical or non-identical, leading to both symmetric and asymmetric dimers. Admittedly, an argument could be made that verticillins, chaetocins, and leptosins could be lumped into a single grouping. However, for the purposes of this review, we kept them separate, thus focusing on those fungal metabolites that we felt were most closely related structurally to verticillin A (1). Chemotaxonomic studies support this, as the fungi producing these three compound classes are distinct, allowing compound classification to be correlated with their fungal origin. Verticillins are mostly biosynthesized by fungi in the genera Clonostachys, Gliocladium, Penicillium, and Verticillium (Table 1); however, the taxonomic identity of these may need reexamination (as detailed in Sections 3 and 4). Alternatively, chaetocins and leptosins are biosynthesized by Chaetomium sp. and Leptosphaeria sp., respectively.33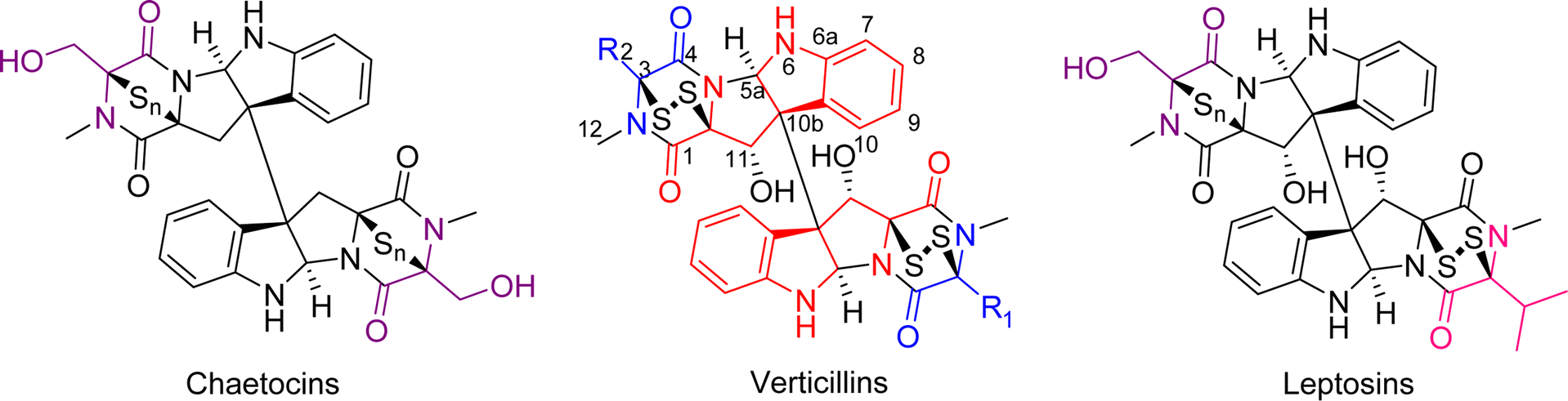 | ||
| Fig. 1 The basic structure of dimeric epipolythiodioxopiperazine (ETP) alkaloids, illustrating their biosynthetic origins. The color coding is used to illustrate the building blocks of these molecules. In red is tryptophan (Trp), common to all three dimeric structural classes. For verticillins, the “blue” substituents dictate the appendages at C-3 and C-3′. Unlike chaetocins and leptosins, the constituents at these positions vary and are typically derived from alanine (Ala), threonine (Thr), and in some rare cases, serine (Ser). The N-methylation, common to all three classes, likely derives from S-adenosylmethionine. For chaetocins and leptosins, biosynthesis often uses Ser (purple) and/or Val (green). Moreover, in these two classes, the sulfur bridge is often greater than two atoms (n = 2–4), whereas in the verticillins, the bridge is typically, but not always, a disulfide.33 | ||
| Compound | Year | Molecular formula | Molecular weight (Da) | Fungus |
|---|---|---|---|---|
| a Recent studies have suggested that the taxonomic identity of this fungus may be different from Verticillium.63,64 b The name of this fungus has been changed to Clonostachys rosea.64,65 c Based on the current rules of fungal nomenclature,66,67 it is more appropriate to refer to fungi in the genus Bionectria as Clonostachys.64,68 | ||||
| Verticillin A (1)49,51 | 1970 | C30H28N6O6S4 | 696.83 | Verticillium sp.a |
| Verticillin B (2)53 | 1973 | C30H28N6O7S4 | 712.83 | Verticillium sp.a |
| Verticillin C (3)53 | 1973 | C30H28N6O7S5 | 744.89 | Verticillium sp.a |
| Sch 52900 (4)54 | 1995 | C31H30N6O7S4 | 726.86 | Gliocladium sp. |
| Sch 52901 (5)54 | 1995 | C31H30N6O6S4 | 710.86 | Gliocladium sp. |
| Verticillin D (6)50 | 1999 | C32H32N6O8S4 | 756.88 | Gliocladium catenulatum |
| Verticillin E (7)50 | 1999 | C32H28N6O8S4 | 752.85 | Gliocladium catenulatum |
| Verticillin F (8)50 | 1999 | C32H30N6O8S4 | 754.87 | Gliocladium catenulatum |
| 11′-Deoxyverticillin A (9)55 | 1999 | C30H28N6O5S4 | 680.83 | Penicillium sp. |
| 11,11′-Dideoxyverticillin A (10)55 | 1999 | C30H28N6O4S4 | 664.83 | Penicillium sp. |
| Gliocladin A (11)56 | 2004 | C24H24N4O3S2 | 480.60 | Gliocladium sp. |
| Gliocladin B (12)56 | 2004 | C24H24N4O2S2 | 464.60 | Gliocladium sp. |
| Gliocladin C (13)56 | 2004 | C22H16N4O3 | 384.40 | Gliocladium sp. |
| Gliocladine A (14)57 | 2005 | C30H28N6O6S5 | 728.89 | Gliocladium roseum |
| Gliocladine B (15)57 | 2005 | C30H28N6O6S6 | 760.95 | Gliocladium roseum |
| Gliocladine C (16)57 | 2005 | C23H20N4O3S2 | 464.56 | Gliocladium roseum |
| Gliocladine D (17)57 | 2005 | C23H20N4O3S3 | 496.62 | Gliocladium roseum |
| Gliocladine E (18)57 | 2005 | C23H20N4O3S4 | 528.68 | Gliocladium roseum |
| Bionectin A (19)58 | 2006 | C22H18N4O3S2 | 450.53 | Bionectria byssicola |
| Bionectin B (20)58 | 2006 | C24H22N4O4S2 | 494.58 | Bionectria byssicola |
| Bionectin C (21)58 | 2006 | C24H24N4O3S2 | 480.60 | Bionectria byssicola |
| Glioclatine (22)59 | 2006 | C23H20N4O2S2 | 448.56 | Gliocladium roseum |
| Verticillin G (23)60 | 2007 | C30H28N6O7S4 | 712.83 | Bionectria byssicola |
| Gliocladicillin A (24)61 | 2009 | C31H30N6O5S4 | 694.86 | Gliocladium sp. |
| Gliocladicillin B (25)61 | 2009 | C31H30N6O4S4 | 678.86 | Gliocladium sp. |
| Gliocladicillin C (26)62 | 2009 | C32H32N6O7S4 | 740.88 | Gliocladium sp. |
| Verticillin H (27)29 | 2012 | C32H32N6O6S4 | 724.88 | Bionectria sp.c |
The pairs of five membered rings in verticillins are cis-fused, and the hydroxy groups at the 11 and 11′ positions add stability to the molecule by forming a hydrogen-bonding network. Indeed, Liu et al.48 reported the crystal structure of verticillin A (1), demonstrating that the O–H⋯O interactions between molecules facilitated the packing of crystals. Most verticillins are ether and methanol insoluble and precipitate as an off-white/yellow powder. The semisynthetic acetate analogues, with the location unspecified in verticillin A acetate49 or a triacetate analogue of verticillin D,50 are reported to have increased aqueous solubility. The structural diversity observed with the dimeric ETP alkaloids stems from differences in the biosynthetic gene clusters used to create these groups. While tryptophan (Trp) is a key unit in all verticillins, alanine (Ala), serine (Ser), or threonine (Thr) are believed to be the other building blocks used in the non-ribosomal peptide synthases (NRPS) for the biosynthesis of these fungal metabolites (see Section 5).33,34
3. Discovery
The first verticillin was reported in 1970 by Minato and colleagues49 of the Shionogi Research Laboratory from the fungus Verticillium sp. (Table 1), which was isolated from the basidiocarp (i.e., fruiting body) of a mushroom identified as Coltricia cinnamomea. This implies that the fungus was mycoparasitic or fungicolous (i.e., growing on another fungus). A pale yellow, ether-insoluble substance was obtained after a series of extractions followed by purification via crystallization from pyridine/acetone. This compound was analyzed using IR, MS, and 1H-NMR, but its structure could not be elucidated completely. During this initial report, the compound was confirmed to contain both disulfide-bridged dioxopiperazine and di-indolyl moieties, and the trivial name verticillin A (1; Fig. 3) was ascribed to this compound of molecular formula C30H28N6O6S4.49 A year later, the structure and absolute configuration of verticillin A (1) were proposed from chemical and physicochemical experiments (particularly CD),51 along with comparisons to the structurally related chaetocin (Fig. 1), which was characterized previously by chemical methods and X-ray crystallography.52 Members of the Shionogi research team continued working on metabolites from this fungus, further refining the absolute configuration of verticillin A and reporting both verticillin B (2) and verticillin C (3) in 1973 (Fig. 2).53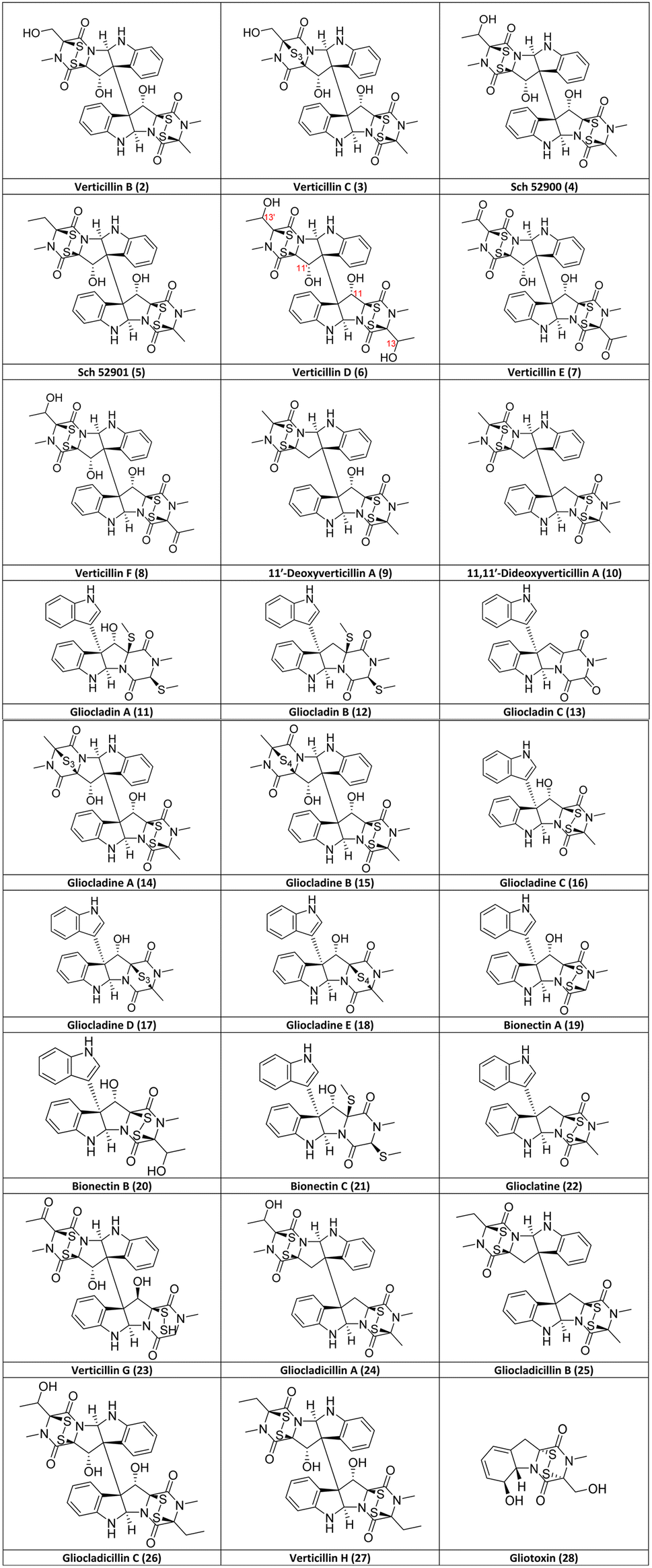 | ||
| Fig. 2 Structures of verticillin analogues (2–27) discovered after A (1) as of June 2023 listed in chronological order and the biosynthetically related ETP alkaloid, gliotoxin (28). Note, while gliocladin A (11)56 and bionectin C (21)58 appear to have the same structure, they were reported in two separate publications with slightly different NMR data. | ||
No other analogues were reported for >20 years until Sch 52900 (4) and Sch 52901 (5) were disclosed by Schering-Plough (Fig. 2);54 while these trivial names may be confusing, they were based on the first three letters of the institute and a sequential serial number, a common practice at that time. Compounds 4 and 5 were isolated from a Gliocladium sp., and their structures were characterized by spectral methods and comparisons to the data for 1, including the use of UV, IR, MS, and 1H- and 13C-NMR data.54 In a separate study about sclerotium survival in soil, reported in 1999,50 a related species, G. catenulatum,64 was investigated after noticing that damage was caused by this mycoparasite to its host. To do so, this fungus was isolated from Aspergillus flavus sclerotia, which had been buried by the research group for two years in a cornfield,50 and after culturing and extracting, three new verticillins [verticillin D (6), verticillin E (7) and verticillin F (8)] were identified. In the same year, another team, who focused on exploring marine sources for fungal metabolites, isolated a Penicillium sp. from Caribbean green algae, Avrainvillea longicaulis. The cytotoxic extract was purified to yield 11′-deoxyverticillin A (9) and 11,11′-dideoxyverticillin A (10), which were both characterized by spectral methods.55 It could be argued that 10 is an outlier in the verticillin group, since it differs from verticillin A (1) by the absence of the hydroxy groups, technically classifying it to both verticillin and chaetocin groups (Fig. 1).33 However, due to the total synthesis of 10 (see Section 6), we consider it a key member of the verticillin structural class.
Gliocladin A (11), gliocladin B (12), and gliocladin C (13) were reported in 2004 from a Gliocladium sp. isolated from a marine environment.56 The disulfide bridge, which is a key determinant in most other analogues, has been reduced (with both sulfurs methylated) in 11 and 12, and while the sulfurs are absent in 13, it is the only isolated verticillin analogue with a trioxopiperazine moiety (Fig. 2). One year later, five new verticillin analogues [gliocladine A (14), gliocladine B (15), gliocladine C (16), gliocladine D (17), and gliocladine E (18)] were reported from submerged wood collected from fresh water G. roseum57 (=C. rosea).64 Notably, 14 and 17 have three sulfurs in the bridge across the dioxopiperazine moiety, while 15 and 18 possess four sulfur atoms. Bionectin A (19), bionectin B (20), and bionectin C (21) were isolated from Bionectria byssicola (=Clonostachys byssicola)64 by Zheng et al.58 in the course of a project screening microbial sources for antibacterial leads. It appears that gliocladin A (11)56 and bionectin C (21)58 were published with identical structures; however, since there are slight differences between their reported NMR data, further studies are warranted. Bionectin A (19) and B (20) are structurally related to gliocladine C (16), D (17) and E (18), as all of these are missing a dioxopiperazine ring, making them monomeric ETPs, which are rare compared to the dimers or pseudo-dimers observed with most verticillins. However, we believe that these monomeric analogues still belong to the verticillin group (Fig. 1), since they are observed in fungi that produce the more typical verticillins. In addition, it is believed that all of these compounds are biosynthesized via similar pathways.33,69,70
To close out the list of 27 verticillin analogues, glioclatine (22) was isolated from G. roseum (=C. rosea)64 grown on wheat medium,59 and verticillin G (23) was reported from Bionectra byssicola60 (=Clonostachys byssicola).64 In a study targeting compounds with anticancer activities, Chen et al.61 isolated and characterized the structures of two new dimeric ETPs from a Gliocladium sp., specifically gliocladicillin A (24) and gliocladicillin B (25).61 Gliocladicillin C (26) was isolated by the same research group, but was only published in a Chinese patent.62 Finally, Figueroa et al.29 isolated verticillin H (27), along with six other verticillins (1, 4–5, 9, 24, and 26), after a bioactivity-directed fractionation of extracts of solid phase cultures of Bionectria sp. (=Clonostachys sp.).64 All 27 verticillin analogues reported through June 2023 have been summarized, including both the structures (Fig. 2) and the source organisms (Table 1).
Insights: There are three aspects of the structures of verticillins that are important to clarify. First, their structures are often drawn in two different ways, as shown for verticillin A (1; Fig. 3). It was suggested to us by a colleague71 that the version on the left is more correct, as it defines the absolute configuration of positions 10b and 10b′ in a non-ambiguous manner. Alternatively, the center representation of the molecule has the dash between these two asymmetric centers, and even ChemDraw will warn that this makes the configuration of those positions ambiguous. Minato and colleagues, in their third and final manuscript on the verticillins,53 also drew 1 as shown on the right side of Fig. 3, as they were able to propose its absolute configuration based on CD data. Those assignments were confirmed via X-ray crystallography in 2006,48 establishing the configuration of 1 as 3S, 5aR, 10bS, 11S, 11aS, 3′S, 5a′R, 10b′S, 11′S, 11a′S. For those with a deep interest in the structure of verticillins, we encourage their examination in three dimensions (e.g., see graphical abstract). The elongated bond between 10b and 10b′, while convenient for drawing purposes, does not allow one to see how tightly woven those complex ring systems are. Moreover, in two dimensions, it appears that the disulfide bridges are on the same face of the molecule, but in three dimensions, it is apparent that they are anti to each other.
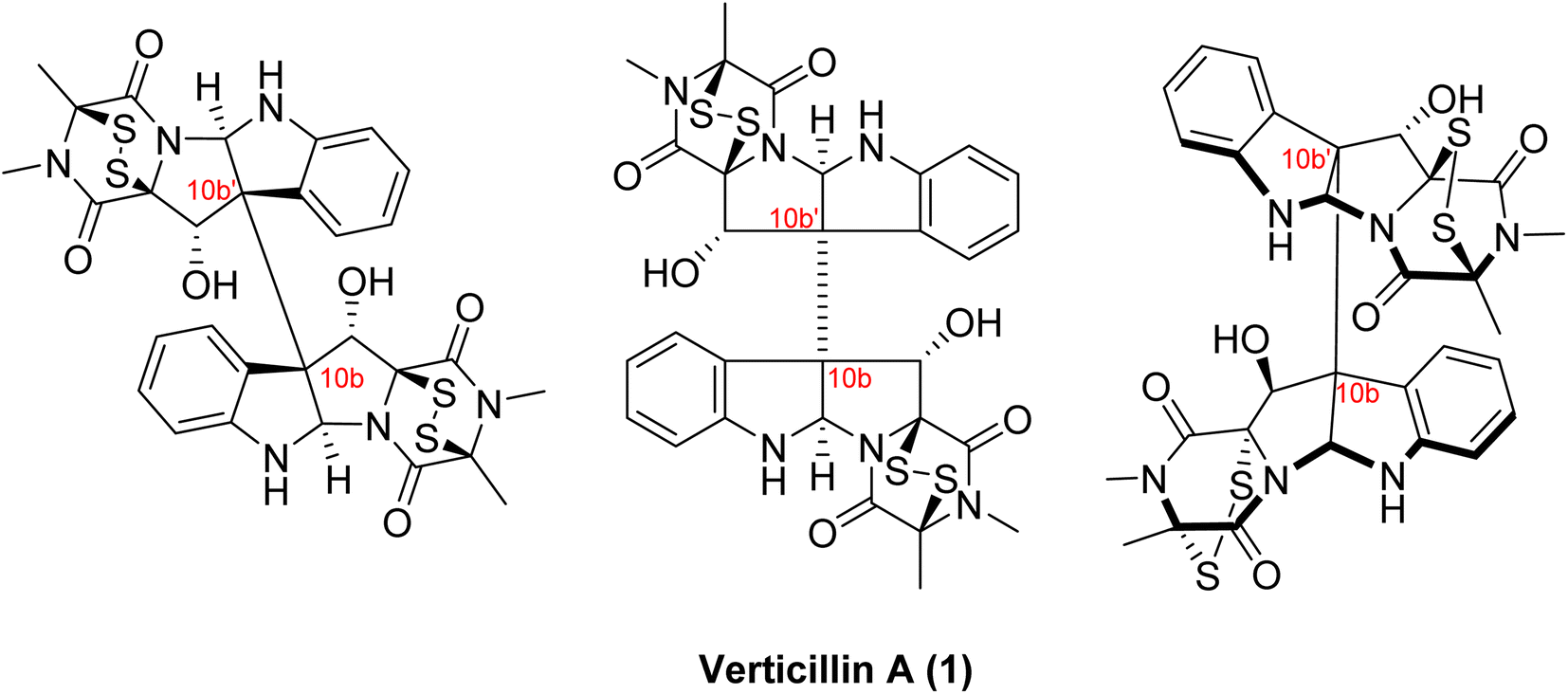 | ||
| Fig. 3 Various structural drawings of verticillin A (1). Verticillin A (1) can be represented several different ways. The representation shown in the middle is seen widely in literature and was how Minato and colleagues first presented the molecule.51 However, the dash line between the two halves of the molecule (i.e., 10b to 10b′) is problematic, as it is connecting two asymmetric centers. Alternatively, the drawing of the molecule on the left displays the same information (i.e. 3S, 5aR, 10bS, 11S, 11aS, 3′S, 5a′R, 10b′S, 11′S, 11a′S) in a non-ambiguous manner. Interestingly, Minato and colleagues subsequently drew 1 as shown on the right,53 where they defined the absolute configuration via circular dichroism; these assignments were confirmed via X-ray crystallography in 2006.48 | ||
Additionally, there is some ambiguity in the literature as to how to number the positions of the atoms throughout the structure of verticillins. For many of the analogues, including 1, the molecule is symmetrical, and thus, the numbering of the top half vs. the bottom half does not matter. However, there are several analogues where there is a difference between those two halves. When the first two asymmetric analogues (2 and 3) were described by Minato and colleagues,53 they did not propose a numbering scheme (likely because they were not assigning NMR spectroscopic data in 1973). However, when Sch 52900 (4) and Sch 52901 (5) were described in 1995,54 those authors assigned the NMR data to distinct positions, and in doing so, they used the prime designation (i.e., 10b′) for the half of the molecule that was different from verticillin A (1). For example, in 5, one half of the molecule has a methyl at the 3-position (i.e., identical to 1), whereas, the other half has an ethyl at the 3′-position. Other scientists have taken a different approach, where the half of the molecule that has the greatest number of atoms is numbered first.22 There are likely pros and cons to both approaches.
Finally, when describing the disulfide-bridged piperazine moiety, the terms dioxopiperazine (or epipolythiodioxopiperazine) and diketopiperazine (or epipolythiodiketopiperazine) seem to be used interchangeably. In fact, in the seminal publications by Minato and colleagues, all of which focused on verticillin A (1), they used the term diketopiperazine49 in their first report and dioxopiperazine51,53 in the latter two. Looking through modern literature, both terms are used interchangeably, although within the biosynthesis community, the term diketopiperazine is probably used more frequently. In discussing this with a colleague who is a journal editor,72 we believe that dioxopiperazine is technically more apropos, as the ‘dioxo’ essentially states that there are two carbonyls attached to the piperazine ring. Obviously, diketo basically implies the same designation, but the confusion is that it also implies ketone carbonyls, whereas the resulting carbonyls are amides. Regardless, both terms are well entrenched in the literature, and it is probably best to simply recognize this fact. Throughout this review, we utilize the terms epipolythiodioxopiperazine or dioxopiperazine.
4. Clarification of fungal taxonomy of strains producing verticillin analogues
It is common practice to ascribe a trivial name to compounds based on the generic or specific epithet of the producing organism. However, this can cause some confusion, especially if the taxonomy of the organism changes or the fungus was misidentified due to morphological similarities with other fungi. Such changes could lead to some confusion in the nomenclature of fungal cultures and the resulting secondary metabolites, especially when looking back over 50 years of research and prior to the use of molecular sequence data (i.e. DNA barcoding).73 For example, the mycoparasitic fungus, Gliocladium roseum, has been transferred (i.e., renamed) to Clonostachys rosea, due to differences in the type species of Gliocladium, G. penicillioides, in morphology, ecology, sexual state, and molecular sequence data.64,65 As such, it is likely that previous reports of the isolation of verticillin analogues from Gliocladium roseum or Gliocladium spp. should really refer to the genus Clonostachys. This was confirmed recently by identifying and characterizing the biosynthetic gene cluster (ver) for biosynthesis of verticillin via genome sequencing, knock out studies, and chemical analysis using an isolate of Clonostachys rogersoniana.39,64As summarized in Table 1, it is apparent that the first three verticillins (i.e., compounds 1–3) were the only ones isolated from Verticillium sp.49,51,53 A 2011 study by Dirk et al.63 investigated the genes that were known to be responsible for the biosynthesis of verticillin-type compounds. Interestingly, their study was performed on V. dahliae, and the authors were unable to detect any genes responsible for the biosynthesis of verticillins. In addition, extraction of a culture of Verticillium sp. also did not yield any verticillins, which led to uncertainties about the ability of this fungal genus to be a producer of these compounds. The authors suggested that the original isolation of verticillin A (1)49 was from a different genus, the identity of which may have been obfuscated; we concur with their explanation. The ability of Clonostachys sp., the fungus which biosynthesizes most of the verticillins, to be a mycoparasite of Verticillium63 suggests the possibility that the original fungal culture reported in the 1970 paper49 was misidentified as Verticillium sp. This is especially true since genera such as Clonostachys, Gliocladium, and Verticillium all have a “verticillate” arrangement of phialides, which means that they are formed in a whorl and have been referred to as Verticillium-like anamorphs (Fig. 4).74 In our laboratory, we have identified several different strains that biosynthesize verticillins, including Clonostachys spp. and C. rogersoniana20 using molecular sequence data.73
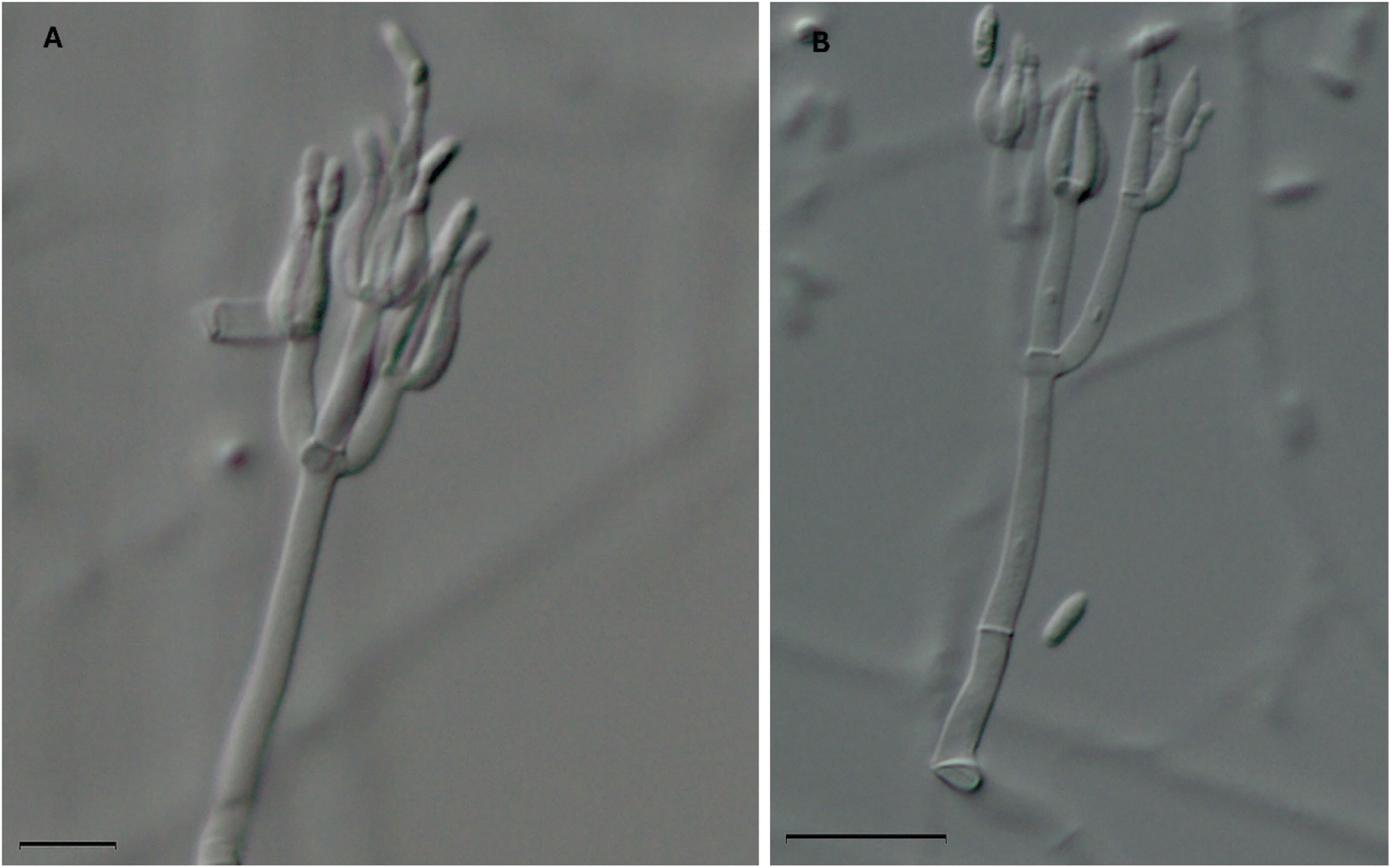 | ||
| Fig. 4 Two microscopic images of the phialides of Clonostachys rogersoniana (strain MSX59553). This fungus biosynthesizes the following verticillins: verticillin A (1), Sch 52900 (4), Sch 52901 (5), verticillin D (6), 11′-deoxyverticillin A (9), gliocladicillin C (26), and verticillin H (27).20 The morphology of the phialides (which have a ‘flask shape’) are characteristic to species of the Clonostachys, Gliocladium, and Verticillium and can cause confusion for non-experts. As such, it is conceivable that fungi used in previous natural products chemistry studies that led to the isolation of some of the analogues shown in Fig. 2 and Table 1, many of which were identified based on morphology, were misidentified. Scale bars = (A) 10 μm and (B) 20 μm. | ||
Finally, there are reports of verticillins being isolated from Bionectria byssicola58 and Bionectria sp.,29 the latter of which was published by some of the authors of this review. The use of the name Bionectria is being phased out due to the adoption of One Fungus = One Name,66,67 in accord with recent rules for pleomorphic fungi in the International Code of Nomenclature for Algae, Fungi, and Plants.75 For pleomorphic names (i.e., where a fungus has a distinct sexual vs. asexual state) in the family Bionectriaceae, Rossman et al.68 proposed the use of the asexual morph (Clonostachys), since it was described first, rather than the sexual morph (Bionectria). Thus, going forward, it is more appropriate to term such fungi as being in the genus Clonostachys (Bionectriaceae).
Insights: The use of trivial names for compounds, which are often derived from the taxonomic nomenclature of the organism, can be problematic and lead to confusion in the literature. As noted by others,76,77 this may be particularly true for microbial products. The various names of the verticillin analogues (Table 1 and Fig. 2) are a glaring illustration of this. While it might be tempting to rename some of these into a consistent format, there is not really a means to do so. Moreover, since most of the pharmacological literature (see Sections 7 and 8) is based on studies of verticillin A (1), it is prudent to keep its trivial name intact.
5. Biosynthesis
The earliest, seminal research on the ETP alkaloids using labeling experiments demonstrated their assembly from amino acids,78,79 and that the formation of the dioxopiperazine (i.e., diketopiperazine) ring occurs during the early stages of the biosynthetic process. In contrast, feeding experiments of sulfur atoms showed incorporation from different sources (e.g. methionine, cysteine or sodium sulfate), suggesting those were incorporated later.80 These results indicate that verticillins, as with other groups of ETP alkaloids, are biosynthesized via a non-ribosomal peptide synthetase (NRPS).34,81The feeding studies were validated with the recent discovery39 of the biosynthetic gene cluster responsible for producing 11′-deoxyverticillin A (9). Previous work identified that an NRPS was responsible for assembling the key dioxopiperazine intermediate in the biosynthesis of chaetocin,82 an ETP structurally related to the verticillins (Fig. 1). Primers based on this NRPS were screened against a genomic fosmid library of the 11′-deoxyverticillin A (9) producer Clonostachys rogersoniana, and a putative biosynthetic gene cluster was identified. This was validated when the NRPS (verP) was knocked out and 11′-deoxyverticillin A (9) production was abolished.39 Closer examination of the nearby genes (Fig. 5A) revealed striking parallels to the biosynthetic gene cluster of gliotoxin (28), an iconic monomeric ETP. The gliotoxin biosynthetic pathway has been thoroughly investigated for many years,70,83–90 making it the prototype for this class of fungal metabolites. Using these results from gliotoxin, it is possible to develop a reasonable hypothesis for the production of the verticillins (Fig. 5B). Importantly, the following proposal is simply based on analogy to gliotoxin and has not been validated. The 11′-deoxyverticillin A (9) pathway is anticipated to begin with VerP, an NRPS that catalyzes the condensation of L-Trp and L-Ala to form the dioxopiperazine skeleton. This is likely followed by bis-hydroxylation catalyzed by the cytochrome P450 (CYP450) monooxygenase VerC. The glutathione-S-transferase VerG can then catalyze the addition of two glutathione moieties, from which the sulfur observed in the verticillins is derived. It is anticipated that VerG catalyzes the nucleophilic attack of the non-enzymatically dehydrated product of the VerC reaction.84 The two glutathiones are proposed to be processed and trimmed by a series of three enzymes: a cyclo-γ-glutamyl-cyclotransferase (VerK), a dipeptidase (VerJ), and a pyridoxal 5′-phosphate (PLP)-dependent C–S bond lyase (VerI).
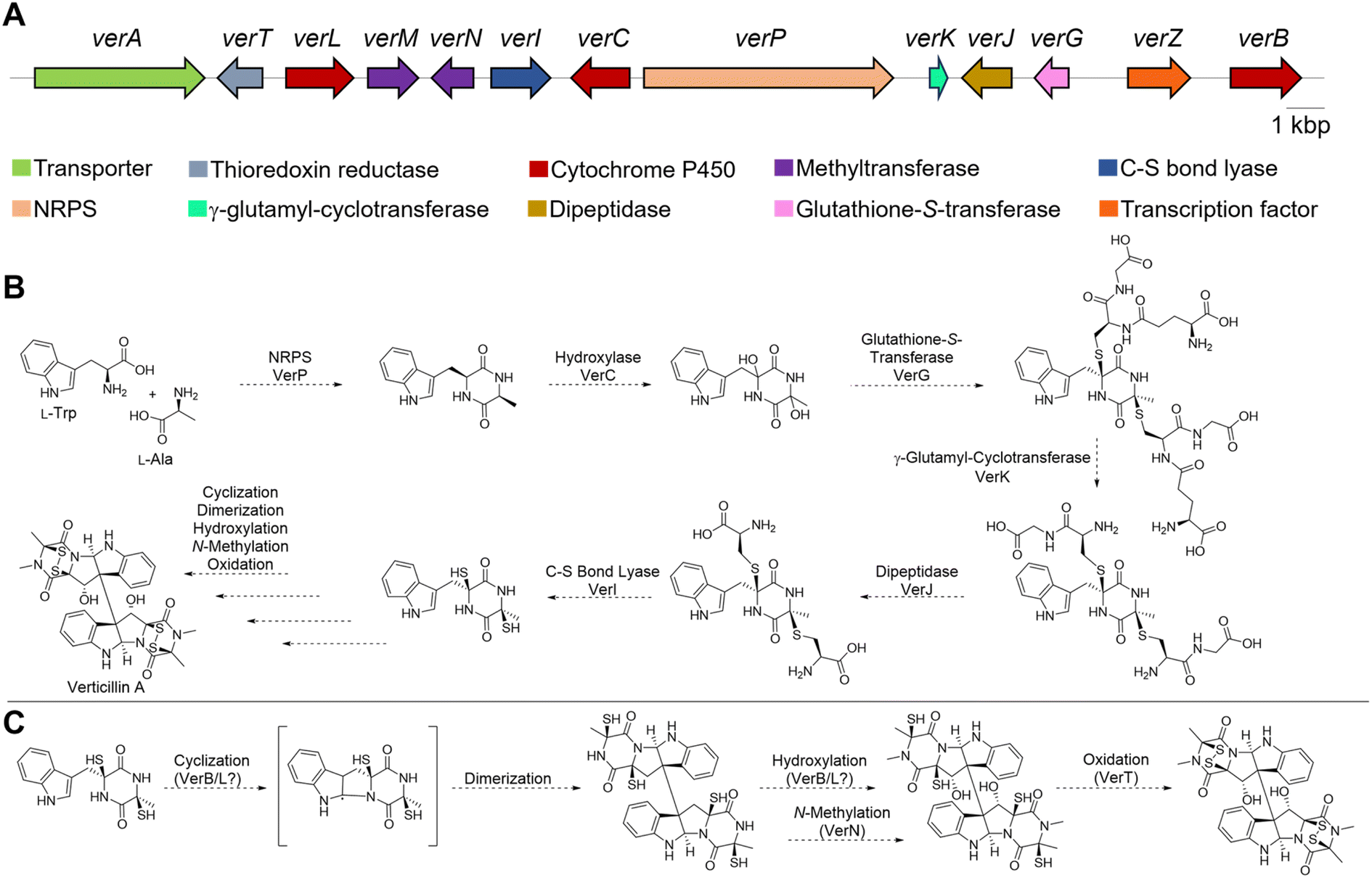 | ||
| Fig. 5 (A) Biosynthetic gene cluster for 11′-deoxyverticillin A (9) from C. rogersoniana. (B) Proposed biosynthetic pathway for verticillin A (1) based on knowledge from gliotoxin. (C) Possible route for the late stage verticillin A (1) biosynthetic steps. | ||
At this point, the obvious similarities to gliotoxin end. Unlike gliotoxin (28), the verticillins are dimeric ETPs and require different biosynthetic steps. In gliotoxin (28), a CYP450 (GliF) was shown to catalyze N-heterocyclization, possibly through an epoxide intermediate, to form a pyrrolidine ring.90 In the verticillin cluster, the two remaining CYP450s in the pathway, VerL and VerB, do not have clear sequence similarity to GliF. Moreover, GliF installs a hydroxy group as part of its cyclization mechanism. Several verticillin family members, such as 11′-deoxyverticillin A (6), lack hydroxy groups at this position. Therefore, a different biosynthetic route is likely. One possibility is found in the dimerization of dioxopiperazines in both bacteria and fungi. It has been shown that in those cases, a CYP450 both cyclizes the indole to form the pyrroloindoline ring and joins two monomers together to form the dimeric scaffold.91–94 If this route is operational in verticillin biosynthesis, one of the two remaining CYP450s, VerB or VerL, could fulfill this role (Fig. 5C). The remaining CYP450 may serve as a monooxygenase and hydroxylate the C11 and/or C11′ positions. An N-methyltransferase, possibly VerN, is also required, which could function on the monomers either prior to or after dimerization. Finally, the free thiols need to be oxidized to the disulfide, either before or after dimerization. VerT is expected to catalyze this reaction, but the timing is not clear.
Enzyme reconstitution has not been completed for the ver cluster; however, thorough knockout studies of the proposed genes support this biosynthetic route.39 Specifically, disruption of verP, verT, verL, verM, verN, verI, verJ, verG, and verB all abolished 11′-deoxyverticillin A (6) production in C. rogersoniana. Knockout of verK lowered production of 11′- deoxyverticillin A (6) significantly, suggesting it is not completely essential. Finally, the disruption of verA lowered the observed levels of 11′-deoxyverticillin A (6), which is consistent with its anticipated role as an ABC transporter to export verticillin from the cell. Recently an ETP-like cluster from the verticillin D (6) producer, C. rosea, was identified that closely matched the ver cluster,95 further supporting this biosynthetic hypothesis.
Insights: Gene knockouts have firmly established the gene cluster responsible for the biosynthesis of 11′-deoxyverticillin A (6) in C. rogersoniana. While in vitro enzyme reconstitution has not been accomplished for any of the proposed enzymes, their similarity to the biosynthetic enzymes of gliotoxin (28) allows for a portion of the biosynthetic route to be proposed. However, the later stages of the biosynthesis, such as the dimerization and hydroxylation, are unclear and will deviate from the gliotoxin pathway. Ultimately, further studies are needed to uncover the dimerization process in ETPs. Additionally, one would predict that a better understanding of verticillin biosynthesis could lead to heterologous expression, a feat that has not been completed to date, both to generate analogues and ameliorate supply from the native host.
6. Synthesis and semisynthesis
The densely functionalized dimeric/pseudo-dimeric bis-pyrroloindoline epipolythiodioxopiperazine scaffolds of the verticillins make them attractive targets for chemical synthesis. From that perspective, however, their C10b–C10b′ vicinal quaternary centers, bicyclic disulfide bridges, and C11/C11′ hydroxy groups pose unique synthetic challenges. Several methods for the preparation of the core ring system of these compounds, the tricyclic pyrroloindoline system also found in numerous other natural products, including the chimonanthines, calycanthidines, and folicanthines, have previously been reported and reviewed.96 Most notably, the elegant work of Overman and coworkers demonstrated the enantioselective construction of the vicinal quaternary centers of the bis-pyrroloindoline system using a cascade Heck cyclization strategy.97,98 Subsequently, Overman also reported the synthesis of (+)-gliocladin C (13), representing the first total synthesis of a member of the verticillin class, by employing a key asymmetric Mukaiyama aldol reaction to establish the stereocenter of the central bis-pyrroloindoline system.99 As mentioned previously, this compound possesses a unique, fused trioxopiperazine ring rather than the more commonly observed dioxopiperazine ring. It also lacks the disulfide bridge and the second dioxopiperazine moiety seen in other members of the verticillin class.The synthesis of the first prototypical verticillin compound was reported in 2009 by Movassaghi.69 His synthesis of (+)-11,11′-dideoxyverticillin A (16), possessing both the disulfide bridge and bis-pyrroloindoline core, set the precedent for C10b-C10b′ bond formation between the vicinal quaternary centers via a biomimetic reductive radical dimerization (Scheme 1). Utilizing a similar strategy, Movassaghi100 and Sodeoka101 separately reported the synthesis of (+)-chaetocin the following year. The installation of the disulfide bridge in each of these syntheses hinged on the generation of a similar oxidized dioxopiperazine intermediate accessed either pre- (Sodeoka) or post- (Movassaghi) reductive radical dimerization. These strategies, along with other methods for the introduction of the disulfide bridges onto dioxopiperazines, have been reviewed elsewhere.102,103
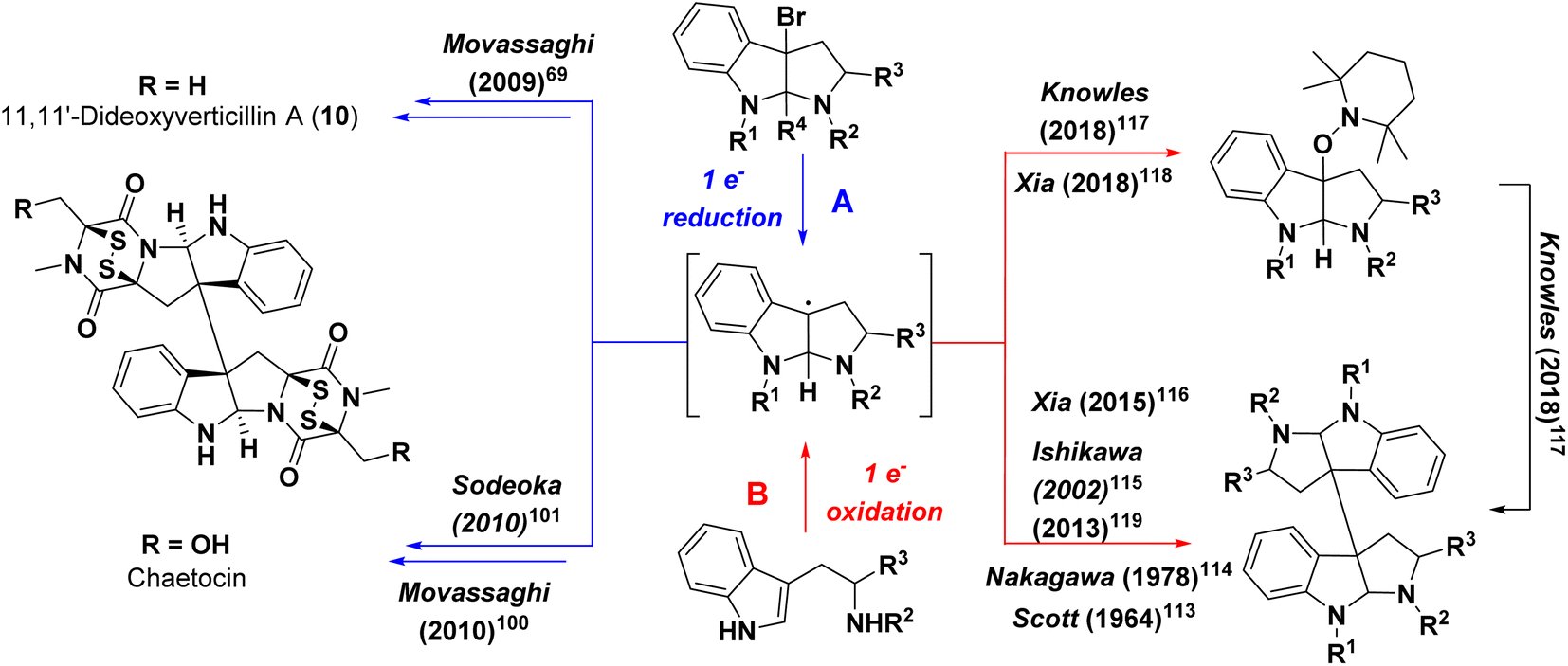 | ||
| Scheme 1 Conceptual strategies for formation of bis-pyrroloindoline containing compounds via (A) reductive or (B) oxidative transformations. | ||
While access to 11,11′-dihydroxy containing bis-pyrroloindoline ETP natural products has yet to be established via chemical synthesis, the Movassaghi lab has reported the synthesis of the monomeric pyrroloindolines (+)-bionectin A (19) and (+)-bionectin C (21) (Scheme 2), which employed an intramolecular Friedel–Crafts reaction to introduce the indole ring at the C3-position of the bis-pyrroloindoline system (step h, Scheme 2). This approach also took advantage of their streamlined access to erythro-β-hydroxy-L-Trp as a precursor.104 Their gram-scale synthesis of this precursor may serve as a stepping stone towards achieving the synthesis of the bis-pyrroloindoline natural products. Additional syntheses of related structures, including (+)-gliocladin B (12) and (+)-gliocladin C (13)105 have also been reported and reviewed103 by Movassaghi. More recently, their lab has extended their synthetic efforts via generation of ETP scaffolds containing appended azide moieties that can be utilized for the synthesis of chemical probes through conjugation reactions.106
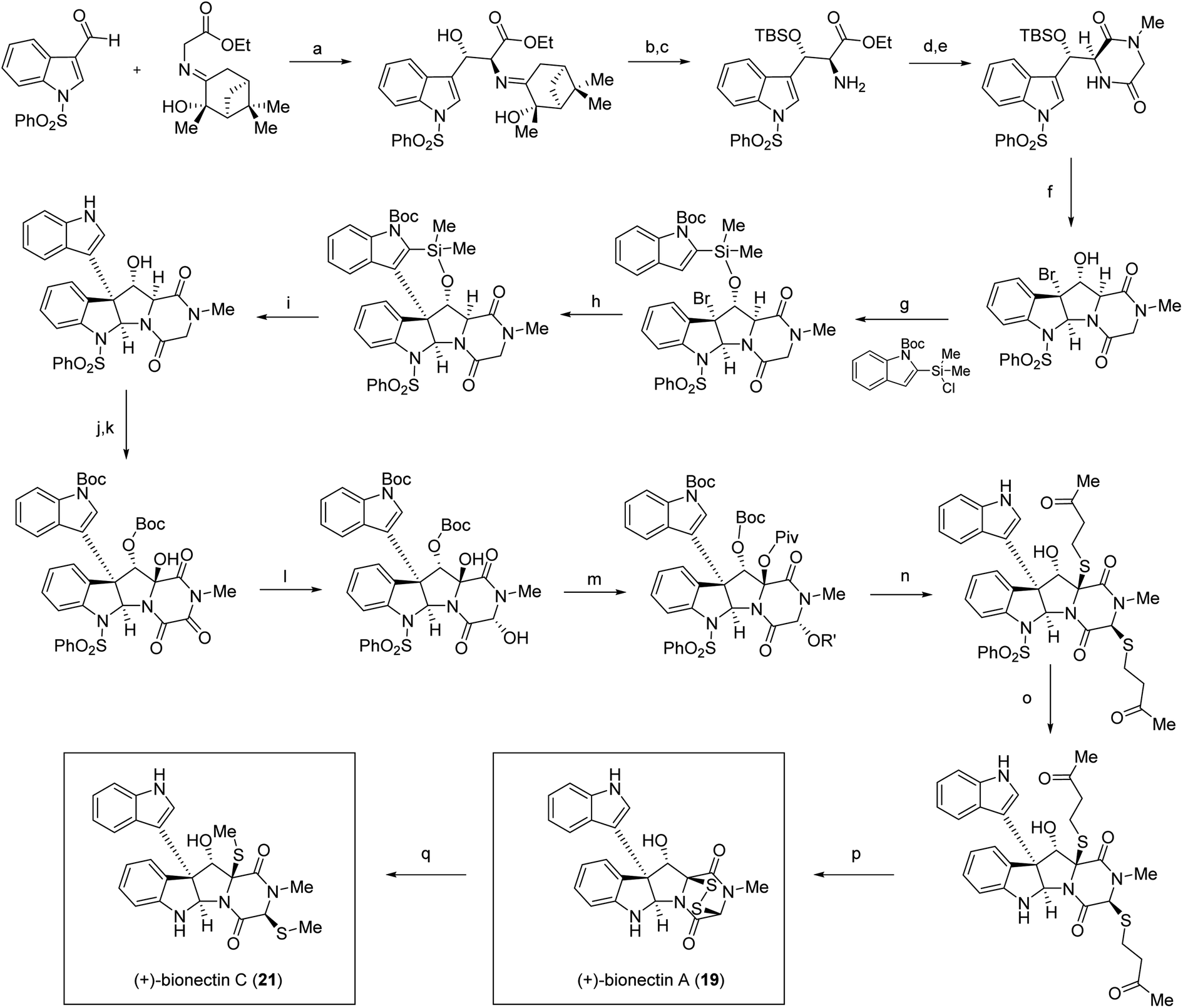 | ||
| Scheme 2 Total synthesis of (+)-bionectins A (19) and C (21) by Coste et al.104 Conditions: (a) TiCl(OEt)3, NEt3, CH2Cl2, 0 °C, 81% (58% desired diastereomer); (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 72%; (c) 2 N HCl, THF, 81%; (d) N-Boc-sarcosine, EDC·HCl, HOBt, CH2Cl2, 23 °C, 98%; (e) TFA, CH2Cl2, 23 °C; AcOH, morpholine, t-BuOH, 80 °C, 97%; (f) Br2, MeCN, 0 °C; anisole, 94%, 9:1 dr; (g) DMAP, THF, 23 °C, 74%; (h) AgBF4, DTBMP, EtNO2, 0 °C, 68%; (i) 6 N HCl, THF, 80 °C, 58%; (j) Boc2O, DMAP, CH2Cl2, 23 °C, 92%; (k) Py2AgMnO4, CH2Cl2, 23 °C, 45%; (l) NaBH4, MeOH, −20 °C, 75%; (m) PivCl, DMAP, CH2Cl2, 23 °C, 83%; (n) 4-mercapto-2-butanone, TFA, MeNO2, 80%, 3:1 dr; (o) 350 nm, 1,4-dimethoxynaphthalene, L-ascorbic acid, sodium L-ascorbate, H2O, MeCN, 25 °C, 56%; (p) pyrrolidine, EtSH, THF, 23 °C; KI3, Py, CH2Cl2, 81%; (q) NaBH4, MeI, Py, MeOH, 0 °C, 97%. | ||
In addition to the total synthesis of members of this class, precursor-directed biosynthesis and semisynthesis experiments have also been explored to generate structural analogues (Fig. 6). This approach is of note for the more complex dimeric members of the class. For example, the Oberlies group has demonstrated that fluorinated derivatives of Sch 52901 (5), verticillin A (1), and verticillin H (27) can be generated at the C9 (and C9′) positions resulting in mono- and di-fluorinated analogues.22 This was accomplished via incorporation of 5-F-DL-Trp during a 28 day fermentation process, producing a ratio of non-, mono-, and di-fluorinated products of approximately 200:20:1. Importantly, in most cases, the fluorinated analogues were equipotent to what was observed with the parent molecules.22
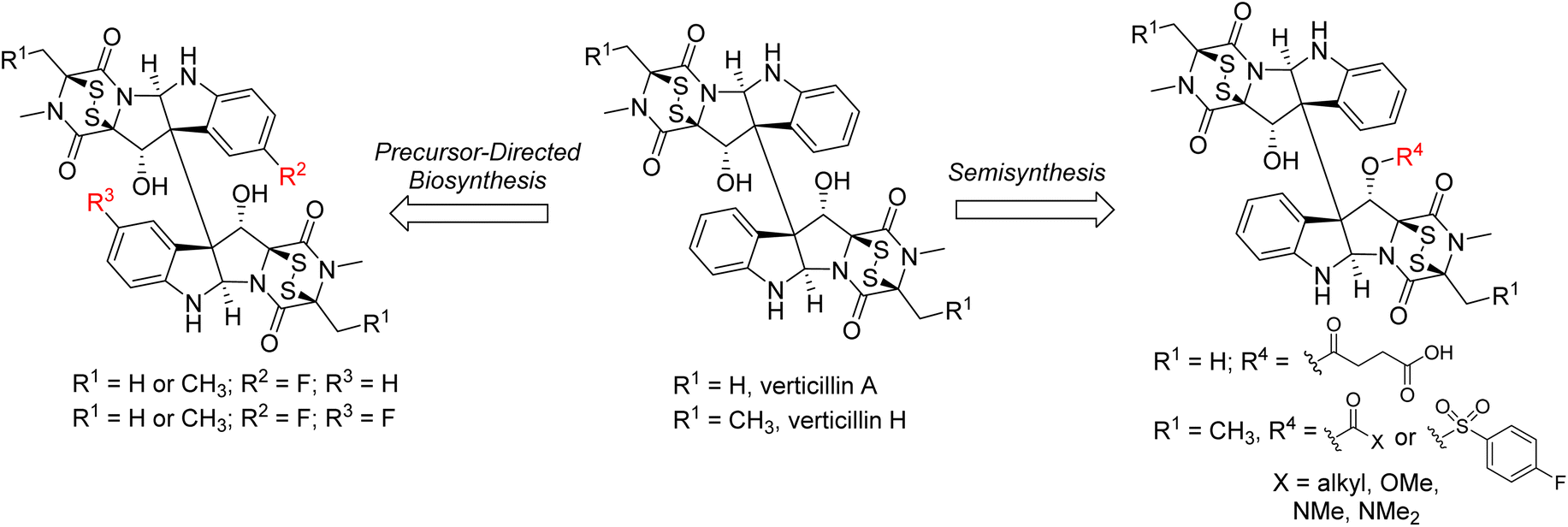
Semisynthesis has largely been limited to the introduction of acetyl groups during the isolation and structural elucidation of the verticillins and related natural products.50,51,53,107–109 In many of these cases, these natural products possessed hydroxy groups at not only the C11/C11′ positions, but also at C13/C13′ (i.e., compound 6 in Fig. 2). Notably, treatment of compounds of this type with excess acetic anhydride resulted in the formation of triacetylated, rather than tetraacetylated products. This result was explained for verticillin D (6)50 as resulting from a “conformational bias” of the C11/C11′ positions that would only permit reaction with one of these “centrally” located alcohol moieties. This bias was confirmed in later semi-synthetic studies by Oberlies and Fuchs,21 who synthesized a series of ester, carbonate, carbamate, and sulfonate analogues of verticillin A (1) and verticillin H (27; Fig. 6). This latter study was unique in that it was both facilitated by a media study to increase verticillin production (i.e., 10 mg of 1 per 10 g oatmeal fungal culture)20 to generate the quantities of the natural products required for this type of work and demonstrated that functionalization with a wide range of groups at the C11 position did not appreciably impact their activity against a variety of cancer cell lines. However, a key point is that high biological activity (with the exception of autophagy110) correlated to the presence of a disulfide bridge across C-3/C-11a, and it was possible to carry out these reactions while keeping that bridge intact. Indeed, removal of the sulfur atoms or reduction of the sulfur bridge diminished/abolished the activity of verticillins.58,111 These observations are critically important, as modifications to the structure likely need to preserve the disulfide bridge.
Insights: The total synthesis of molecules in this class achieved by Overman, Movassaghi, and Sodeoka demonstrate impressive creativity and problem-solving to establish the complex architecture and substitution patterns found in these natural products. These syntheses benefit in large part from methodology developed for the construction of the broader class of tryptamine-derived bis-pyrrolidinoindoline natural products112 like chimonanthine. While they have not yet been specifically utilized for members of the verticillin class, numerous other methods, including oxidative radical coupling strategies (Scheme 1B),113–115 have been developed for the rapid construction of the pyrroloindoline ring systems and installation of the key C10b–C10b′ vicinal quaternary centers from readily available starting materials. More recently, methods have also been described that provide enantioselective access to these tricyclic systems,116–119 including molecules like (+)-WIN 64821 and (−)-ditryptophenaline that also possess fused dioxopiperazine rings, but lack the sulfur bridges found in the verticillins. One of the major synthetic challenges remaining is the synthesis of dimeric verticillin derivatives containing C11 and C11′ hydroxy groups. This means that the total synthesis of verticillin A (1), the prototypical molecule of this class, has not yet been realized. In addition, scalability of the more complex natural products and their analogues for therapeutic development has not yet been demonstrated, although the Movassaghi lab reported the production of several of the monomeric azide probes on 100–200 mg scale.106 In addition to total synthesis, other strategies that promote analogue development and subsequent biological studies, including precursor feeding and semisynthesis, could provide a path toward pharmaceutical development of the verticillins. To do so will require more innovations in the scaled production of verticillins, possibly on the industrial scale. Clearly, there is additional room for compound development in this area, including structure–activity relationship studies and potential drug formulation, so as to advance these compounds into the clinic.
7. Pharmacology part 1: anticancer properties and mechanisms of action of verticillins
The potent biological activities of the verticillins, especially with respect to tumor cell death regulation (i.e., cytotoxicity), has certainly driven the interest in this class of compounds over the decades. More recently, a series of studies has explored their biological activity in more detail. With respect to in-depth mechanistic studies and in vivo tumor efficacy studies, most of the research has focused on verticillin A (1), presenting opportunities for further research on it and other analogues.7.1. Animal models
The very first manuscript on the verticillins included biological evaluation of verticillin A acetate, since, as the authors stated, it was more soluble than verticillin A (1) in water.49 Verticillin A acetate demonstrated cytotoxicity against HeLa cells (ED50 0.2 μg mL−1) and was found to have anti-tumor effects in vivo against Ehrlich ascites tumor, a spontaneous murine mammary adenocarcinoma. For the latter, they found that a dose of 1 mg per kg per day had the best balance of efficacy without toxicity to the animals.49 Interestingly, this dose, determined in 1970, is fairly close to what has been used in many of the subsequent studies (Table 2).| Cancer type | Xenograft | Dosing regimen | Ref. |
|---|---|---|---|
| Ovarian cancer (OVCAR8-RFP) | Intraperitoneal | Intraperitoneal injection twice weekly for 4 weeks | 30 |
| Verticillin A (0.25 mg kg−1 body weight) | |||
| Verticillin D (0.25 mg kg−1 body weight) | |||
| Ovarian cancer (OVCAR8-RFP) | Intraperitoneal | Intraperitoneal injection every 2 days for 14 days | 23 |
| 0.5 mg kg−1 of verticillin A encapsulated nanoparticles (eNP-VA) | |||
| Pancreatic cancer (PANC02-H7, UN-KC-6141) | Orthotopic | Every 2 days for 10 days | 123 |
| 0.5 mg kg−1 body weight | |||
| Soft tissue sarcoma: malignant peripheral nerve sheath tumor (MPNST) (MPNST724) | Sub-cutaneous | Intraperitoneal injection every other day | 27 |
| (1) 0.25 mg kg−1 body weight | |||
| (2) 0.5 mg kg−1 body weight | |||
| Colon carcinoma (SW620-5FU-R) | Sub-cutaneous | Intravenous injection on days 5, 7, 9, 11 and 13 | 28 |
| 1 mg kg−1 body weight | |||
| Colon carcinoma (HepG2, SW620) | Sub-cutaneous | Intravenous injection every 2 days for 14 days | 25 |
| HepG2 tumor: 1 mg kg−1 and 2 mg kg−1 body weight | |||
| SW620 tumor: 0.125 mg kg−1 body weight |
In recent years, target identification studies have shown that verticillin A (1) is a histone methyltransferase (HMTase) inhibitor with selective activity towards G9a, GLP, SUV39H1, SUV39H2, MLL1, and NSD2 methyltransferases (Fig. 7).28,37 The highest selectivity of verticillin A (1) was observed against G9a, SUV39H1 and SUV39H2 methyltransferases with IC50 values of 0.54, 0.57, and 0.48 μM, respectively. Paschall et al.28 discerned the selective role of verticillin A (1) in inhibiting histone H3 lysine 9 (H3K9) trimethylation (H3K9me3) by inhibiting SUV39H1 and SUV39H2 enzyme activity. H3K9me3 was shown to be responsible for Fas transcription silencing, which results in a loss of its expression in human colon carcinoma cells.28,120 Treatment with verticillin A (1) showed a decrease in H3K9me3 deposition levels in the FAS promoter region with an increase of histone H3 lysine 9 acetylation (H3K9ac), which indicates active transcriptional chromatin, thereby enabling Fas expression.28 The ligand of Fas (i.e., Fas-L), present on the surface of cytotoxic T lymphocytes (CTL) when bound to Fas, initiates an immunological reaction that leads to apoptosis.121,122 Modification of H3K9me3 deposition by verticillin A (1) can impact gene expression, and, in certain cancer types, one of the targets is the re-expression of Fas that increases tumor cell sensitivity to CTL FasL-induced cytotoxicity, resulting in tumor growth suppression.28
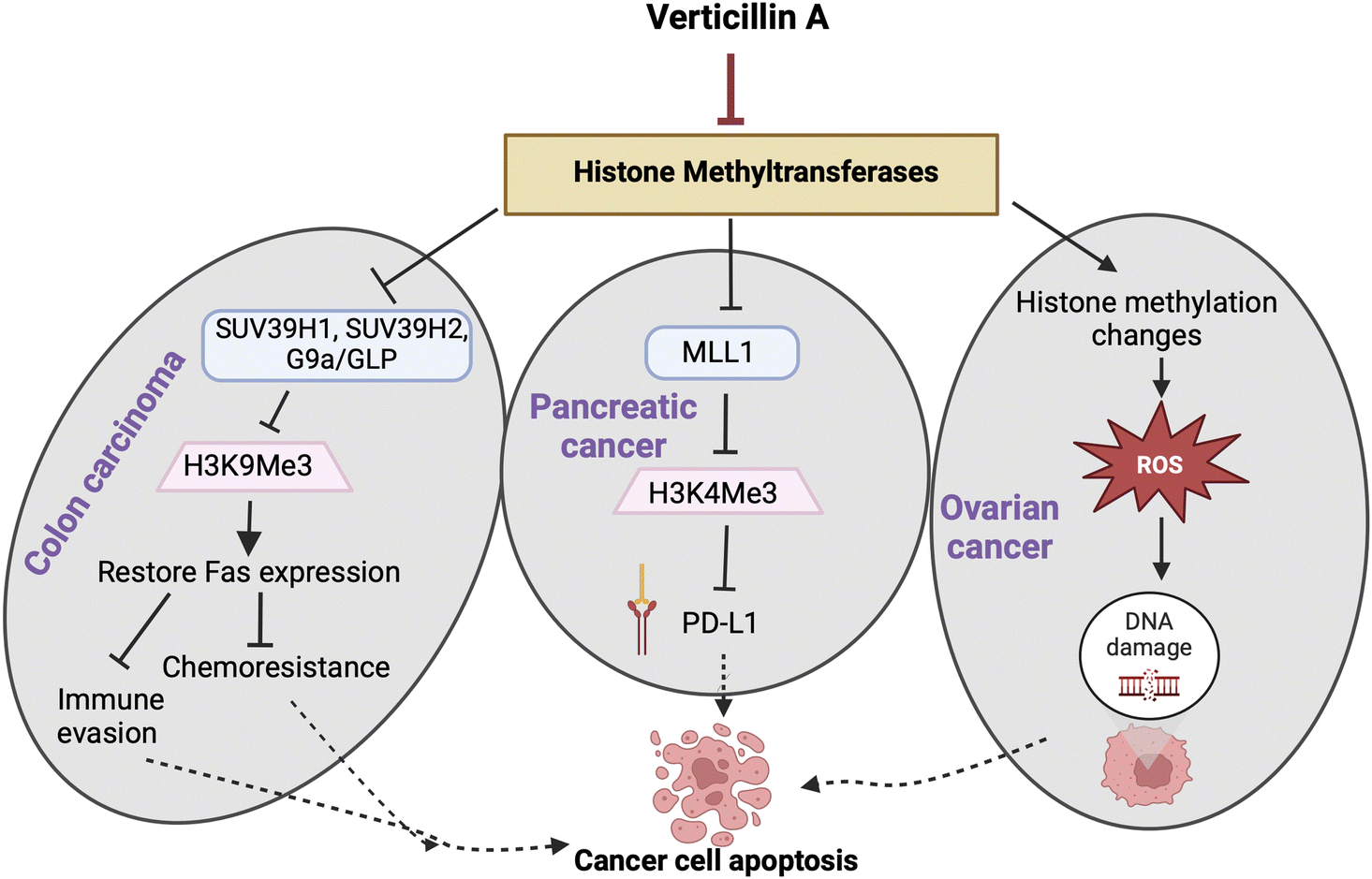 | ||
| Fig. 7 (A) In colon cancer, verticillin A (1) inhibits H3K9me3 levels via selective inhibition of HMTases, specifically G9a, SUV39H1 and SUV39H2, leading to restoration of Fas expression. This, in turn, leads to suppression of immune evasion and overcoming chemoresistance to 5-fluorouracil (5-FU). (B) In pancreatic cancer, verticillin A (1) inhibits MLL1, resulting in a decrease of H3K4me3 levels, consistent with a decrease of expression of PD-L1, and activates the immune response against tumor cells. (C) Verticillin A (1) causes global methylation and acetylation histone modifications in ovarian cancer, which generates oxidative stress mediated DNA damage and apoptosis. | ||
7.2. Verticillin A (1) and colon cancer
Verticillin A (1) showed in vivo efficacy in overcoming metastatic colon carcinoma resistance to 5-fluorouracil (5-FU),28 which is the standard treatment for this type of cancer. The size and weight of tumors decreased significantly when the mice were treated with a combination of verticillin A (1) and 5-FU compared to groups that received individual treatments. Investigating the role of verticillin A (1) in sensitizing the cancer cells to 5-FU revealed that the mechanism of apoptosis induction was facilitated by the combination of FAS expression and 5-FU (Fig. 7). Additionally, an important outcome of this study resulted from analysis of murine liver enzymes, which indicated that verticillin A (1) has minimal liver toxicity at a dose of 1 mg kg−1 body weight when injected every other day over 10 days.28 Interestingly, this is similar to the dose used by Minato and colleagues49 in 1970, but delivering the dose every other day, as performed by Paschall et al.,28 may be important for titrating liver toxicity.7.3. Verticillin A (1) and pancreatic cancer
In pancreatic cancer cell lines PANC02-H7 and UN-KC-6141, H3K4me3 levels were found to be significantly higher at the promoter of CD274, the gene that encodes PD-L1, compared to non-cancerous cells.123 MLL1 was found to be the HMTase responsible for the higher levels of H3K4me3. Silencing the MLL1 gene in both human and mouse pancreatic cancer cells resulted in a decrease of H3K4me3 at the CD274 promoter region and a decrease of PD-L1 expressed by tumor cells, which indicated that it was the HMTase that was key to modifying H3K4me3 in pancreatic tumor cell models (Fig. 7). Since verticillin A (1) is a HMTase inhibitor, it was demonstrated to inhibit MLL1 activity with an IC50 of 0.8 μM.123 Subsequently, verticillin A (1) caused a decrease of H3K4me3 levels, due to MLL1 inhibition, in a dose dependent manner in vitro. In both PANC02-H7 and UN-KC-6141 pancreatic tumors, PD-L1 was found to be expressed in significantly higher levels in vivo; likewise, over 50% of tumor-infiltrating CD8+ cytotoxic T lymphocytes (CTLs) were PD-L1+. Therefore, immunotherapy that mitigates PD/PD-L1 activity should be effective, but in most large clinical trials, pancreatic cancer has a poor response to anti-PD-L1/PD-L1 immunotherapy.124 Interestingly, combined treatment of verticillin A (1) and anti-PD-L1 exhibited the most significant growth suppression as compared to the individual treatments, approaching tumor eradication.123 The mechanism for such an effective treatment was due to verticillin A (1) acting as an HMTase inhibitor of MLL1 and thereby epigenetically repressing PD-L1 expression on the tumor cells. The additive effect of combining the two drugs indicates the important role of verticillin A (1) in increasing the efficacy of the anti-PD-L1 therapy by decreasing the target transcriptionally and also by regulating gene expression in the cancer cell to augment apoptosis.123 Following up on this study, the same group found that verticillin A (1) sensitized pancreatic ductal adenocarcinoma (PDAC) cells to gemcitabine.40 Mechanistically, verticillin A (1) caused epigenetic modification in apoptosis regulating genes, in this case a new set that was different from FAS seen in colon cancer, by altering H3K9me3 and H3K4me3 levels. Chromatin immunoprecipitation (ChIP) revealed that verticillin A (1) caused dysregulation of apoptotic genes by decreasing H3K9me3 levels at the BAK1, BAX, and BCL2L11 promoter regions and H3K4me3 level at BCL2L1, MCL-1, and CFLAR promoter region in PDAC cells.40 These studies collectively demonstrate that combining epigenetic modifiers, such as verticillin A (1), with immune checkpoint blockade immunotherapies can be an effective approach to reduce pancreatic tumor growth.26,1237.4. Verticillins and ovarian cancer
Research conducted by Salvi et al.23 showed that verticillin A (1) has nM-level cytotoxicity against several high-grade serous ovarian cancer (HGSOC) cell lines. This is important because it expanded the tumor types for which verticillins have shown efficacy both in vitro and in vivo. Furthermore, verticillin A (1) induced oxidative stress and DNA damage, causing apoptosis in HGSOC cell lines OVCAR4 and OVCAR8. Consistent with the aforementioned studies, verticillin A (1) caused epigenetic modifications with global changes in histone methylation and acetylation marks, but the change in oxidative stress occurred rapidly (i.e., in a few hours) and likely preceded these gene changes (Fig. 7). When verticillin A (1) was encapsulated in expansile nanoparticles (eNPs),125 its potent cytotoxicity was maintained, the tumor burden was reduced in vivo, DNA damage and apoptosis occurred (which was consistent with the in vitro data), and hepatotoxicity (a side effect of verticillin A (1)) was not observed.23 These studies helped to demonstrate that oxidative stress, along with epigenetic modification, was important for cell death and that encapsulation in an expansile nanoparticle could target the drug after i.p. delivery, both retaining efficacy while reducing liver toxicity.More recently, Kaweesa et al.30 compared the in vitro and in vivo cytotoxicity of verticillin A (1) and verticillin D (6). The authors found that both compounds exhibited cytotoxicity and induced apoptosis in HGSOC cell lines OVCAR4 and OVCAR8 at nanomolar concentrations. The authors performed formulation studies to monitor bioavailability and achieve tolerable drug delivery for future pharmacokinetic studies. Both verticillin A (1) and verticillin D (6) reduced tumor burden in OVCAR8 xenografts at the same dose as was previously tested. Using this optimized formulation, verticillin dosing strategies that are effective in vivo were expanded from expansile nanoparticles to the use of improved formulations that likely increase solubility. Unfortunately, verticillin D (6) exhibited significant liver toxicity in mice,30 and as such, it may require additional formulation development. However, this was the first in vivo study to explore analogues of verticillin A (1), and this seems to be an area where further investigations are warranted.
7.5. Verticillin A (1) and liver cancer and soft tissue sarcoma
Liu et al.25 revealed that verticillin A (1) inhibited HepG2 (human liver carcinoma) tumor cell growth by inducing apoptosis in vitro and inhibited in vivo tumor growth at a dose of 2 mg kg−1 body weight in a murine model. In addition to HepG2, verticillin A (1) showed high potency in vitro against multiple types of cancer cells (Bcap-37, MCF-7, HeLa, SMMC-7721, SPC-A1, and Jurkat) with concentrations in the nanomolar range. It has also been demonstrated that verticillin A (1) is an apoptosis sensitizer for different types of tumors resistant to TRAIL, a potent anticancer agent in vitro and in vivo. This study emphasized the importance of verticillin A (1) as a highly potent anti-cancer agent and a strong adjuvant to overcome drug resistance in cancer treatment.25 It was also the first study to suggest the idea of using verticillin A (1) in a combination therapy, emphasizing that epigenetic reprogramming, which re-expresses or silences targets that possibly drive resistance, provides an opportunity to increase the efficacy of already approved chemotherapies in combinations.In a study on soft tissue sarcoma (STS),27 which is known to be therapeutically challenging due to genetic and histological heterogeneity, verticillin A (1) showed potential in inhibiting malignant peripheral nerve sheath tumor (MPNST) and leiomyosarcoma (LMS) growth by inducing apoptosis, a finding that was consistent with the work of Liu et al.25 noted above. In vitro experiments demonstrated that verticillin A (1) inhibited colony formation in STS cells, while inducing apoptosis. Mice xenografted with MPNST724 revealed tumor growth inhibition and significant reduction of tumor volumes and weights when treated with verticillin A (1) at a dose of 0.25 or 0.50 mg kg−1 of body weight.27
Insights: In addition to exploring the in vivo efficacy of verticillin A (1), all the above studies were mechanistic in their approach and identified verticillin A (1) as an epigenetic modifier in various tumor models (Fig. 7). In general, most studies examine verticillin A (1) at a dose of about 1 mg kg−1, as summarized in Table 2. The narrow therapeutic range of verticillins is an area that warrants further study. For example, the study by Salvi et al.23 showed that a nanoparticle formulation was beneficial, as it resulted in a statistically significant minimization of tumor burden while simultaneously being non-toxic, especially to the liver. Later, Kaweesa et al.30 found that formulation was sufficient to dose verticillin A (1) effectively without toxicity. This suggests that metering the dose of verticillins may provide an avenue to circumvent toxicity concerns, and thus, improve the therapeutic index. In addition, with only one exception,30 all of these in vivo studies were carried out with verticillin A (1). The growing availability of both natural and semisynthetic analogues of the verticillins opens up the opportunity for further in vivo experimentation. Obviously, the ultimate goal is to uncover a verticillin-type molecule that can be formulated effectively, that retains potent anticancer activity, and yet that has minimal toxicity, particularly to the liver.
7.6. Pharmacokinetics and in vitro studies
Pharmacokinetic and bioavailability studies were conducted on verticillin A (1) by Wang and colleagues,24 who administered the drug orally (PO) at a dose of 3 mg kg−1, intraperitoneally (IP) at a dose of 3 mg kg−1, and intravenously (IV) at a dose of 1 mg kg−1. Blood plasma concentrations of verticillin A (1) were then calculated using liquid chromatography-tandem mass spectrometry (LC-MS/MS). The within and between days precision was <9% and the accuracy was between 90 to 105%. IP dosing resulted in the highest systemic exposure, with a maximal concentration (Cmax) of 110 nM and sustained plasma concentrations above 10 nM for 24 h. IV and PO doses achieved 73 nM and 9 nM of Cmax values, respectively. The high levels of verticillin A (1) after IP and IV administration warrant further investigation of these routes for future in vivo studies.24 Importantly, these studies, along with the efficacy data in murine models using concentrations in a similar range, demonstrate the ability to achieve physiologically relevant concentrations of verticillin A (1) that result in reductions in tumor burden.7.7. In vitro cytotoxicity studies of verticillin A (1)
Prior to the more recent mechanistic and animal testing, a number of studies were performed on the in vitro efficacy of verticillins using cell models. In a study by Guan et al.,43 verticillin A (1) was found to be an effective adjuvant of the BH-3 mimetic, ABT-737, against human colon cancer cell lines HT29, LS411N, SW620, DLD1, and RKO. This was accomplished by verticillin A (1) sensitizing colon cancer cells to ABT-737-induced cell death via induction of caspase-mediated apoptosis (specifically, caspase-3 and caspase-9). Further analyses of the protein levels of genes in the mitochondrial-dependent apoptosis pathway showed that verticillin A (1) significantly increased the levels of BIM (BIMEL, BIML, and BIMS), a class of protein that induces apoptosis.126 Although treatment with verticillin A (1) alone led to the upregulation of MCL-1 (an anti-apoptotic protein), its combination with ABT-737 increased the BIMEL/MCL-1 ratio, which in turn sensitized HT29 and LS411N cells to ABT-737. Results from the study also suggested that BIMEL was upregulated via the suppression of the MEK/ERK pathway by verticillin A (1). By blocking the phosphorylation of ERK and BIMEL, verticillin A (1) suppressed the MEK/ERK pathway, resulting in the upregulation of BIMEL. The combination treatment also promoted the translocation and activation of BAX (a pro-apoptotic protein127) that triggered apoptosis.In colon cancer, verticillin A (1) was found to selectively inhibit HMTases leading to a decrease in H3K9me3 levels.28 Building upon this work, metastasis-related genes were investigated in colon cancer cells to identify additional signaling pathways changed by verticillin A (1) mediated epigenetic marks.41,42 After determining the cytotoxicity of 1 against human colon cancer cell lines DLD1 and RKO, as well as the murine colon cancer cell line CT26 (IC50 values of 0.90, 0.31, and 0.18 μM, respectively), the authors found that verticillin A (1) inhibited the migration and invasion ability of these cell lines.41,42 Subsequently, using qRT-PCR, key metastasis-associated genes were identified, including MET, CDH1, PLAU, RHOA, and RHOC. Among these genes, MET and PLAU were significantly downregulated when treated with verticillin A (1). Further results showed that verticillin A (1) selectively decreased C-Met protein levels in DLD1, RKO, and CT26 cells. These findings were substantiated further when it was determined that verticillin A (1) suppressed C-Met at the transcriptional level.41,42 Collectively, these results suggest that C-Met (c-mesenchymal–epithelial transition factor) is a biological target of verticillin A (1) in human colon carcinomas.
Lu et al.45 demonstrated that verticillin A (1) in human gastric (AGS) and cervical cancer (HeLa) cells inhibits migration by targeting C-Met and its downstream FAK/Src (focal adhesion kinase-steroid receptor cofactor) signaling pathways. Specifically, verticillin A (1) represses the expression of C-Met protein and inhibits HGF-induced C-Met phosphorylation, the latter of which results in the suppression of C-Met downstream of FAK/Src signaling. These findings are significant, as the C-Met/FAK/SRc signaling pathway is associated with cancer cell proliferation and invasion.128,129 Importantly, verticillin A (1) displayed cytotoxic activity against AGS and HeLa cells with IC50 values of 69.89 and 319.5 nM at 24 h and 47.59 and 233.9 nM at 48 h, respectively.45
A separate study44 demonstrated that verticillin A (1) decreased trimethylation at H3K9 at the G6pd promoter in tumor-specific 2/20 CTLs (cytotoxic T lymphocytes) co-cultured with mesothelioma AB1 tumor cells (i.e., T-cells exposed to the tumor microenvironment). This, in turn, significantly increased the expression of G6pd in the tumor-specific CTLs. The tumor microenvironment is hostile towards antitumor immune responses and likely responds to T-cells, in particular, by inducing T-cell exhaustion.130,131 Thus, these findings are significant, since the activation of G6pd was found to enhance the acetyl-CoA/H3K9ac pathway to reverse CTL exhaustion and subsequently increase CTL lytic function in tumor cell lysis in vitro and in vivo. These findings suggest that G6pd is a potential biological target in reversing immune suppression in cancer immunotherapy. The changes observed in this study in methylation are consistent with inhibition of HMTase as the target.
In the 1990s, Chu et al. showed that Sch 52900 (4), Sch 52901 (5), and verticillin A (1) inhibited serum-stimulated transcription of human c-fos promoter.54 These verticillin analogues showed potent activity in the fos/lac Z reporter gene assay with in vitro IC50 values of 1.5, 1.8, and 0.5 μM, respectively. Thus, verticillin A (1) exerted antitumor activity by inhibiting the activation of at least two signaling pathways involved in c-fos induction.54 Sch 52900 (4) demonstrated the ability to induce differentiation of 50–69% of HL-60 cells (human promyelocytic cells) at low concentrations (6.8–13.6 nM). Sch 52900 (4) caused induction of the cell cycle inhibitor p21WAF and inhibition of the extracellular signal regulated kinase (ERK) that led to cellular apoptosis and subsequent growth arrest.132 The ultimate result of these verticillins (i.e., compounds 1, 4 and 5) triggering cell death via apoptosis is consistent with the more recent mechanistic data obtained in other cancer cell lines, such as colon, pancreatic, and ovarian.
Several structurally related verticillins have also been studied. The compound 11,11′-dideoxyverticillin A (10) was demonstrated to have inhibitory activity towards epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor-1/fms-like tyrosine kinase-1 (VEGFR-1/FLT-1), and human epidermal growth factor receptor-2 (HER2/ErbB-2), being selectively more potent against EGFR and VEGFR-1 with IC50 values of 0.136 and 1.645 nM, respectively.133 11′-Deoxyverticillin A (9) was shown to induce autophagy in HCT116 human colon carcinoma cells leading to apoptotic cell death.38 Verticillins have also been shown to cause cell cycle G2/M phase arrest in HCT-116 colon cancer cells using in vitro and in vivo experiments.134 Similarly, gliocladicillins A (24) and B (25) are strong anti-proliferative and pro-apoptotic agents and are demonstrated to inhibit proliferation of cancer cell lines HeLa, HepG2, and MCF-7 by cell cycle blockage in the G2/M phase.61 Moreover, 11,11′-dideoxyverticillin A (10) has an anti-angiogenic effect135 and inhibits proliferation of HUVECs (human umbilical vein endothelial cells) with IC50 values of 0.17 and 0.39 μM for VEGF (vascular endothelial growth factor) stimulated cells and serum-stimulated cells, respectively.134
Insights: The cellular target for most of the in vitro studies on verticillins were not clearly defined; however, a common theme is that verticillins induce cytotoxicity and apoptosis in cancer cells. These studies were performed prior to identifying the role of verticillins in modifying histones, and evaluating these studies in light of current knowledge could be informative. For example it would be interesting to pursue whether any of the targets (such as BIM, c-Met, c-fos, etc.) undergo transcription changes via verticillin-mediated epigenetic modifications.
8. Pharmacology part 2: other biological activities
8.1. Nematocidal activity
Verticillin analogues such as gliocladines A (14), B (15), C (16), D (17), and E (18), verticillin A (1), 11′-deoxyverticillin A (9), Sch 52900 (4), and Sch 52901 (5) were tested for their anti-nematocidal effects against Caenorhabditis elegans and Panagrellus redivivus. The dimeric/pseudodimeric verticillins were the most effective in comparison to those lacking an indole moiety (ED50 values of 10–80 vs. 200–250 μg mL−1, respectively). The number of sulfur atoms in the bridge did not influence the potency of the molecules.578.2. Antimicrobial activity
Bionectin A (19) and B (20) were tested for their antibacterial activity against Staphylococcus aureus, methicillin-resistant S. aureus (MRSA), and quinolone-resistant S. aureus, resulting in MIC values of 10–30 μg mL−1. Alternatively, verticillin D (6) and G (23) were slightly more potent with MIC values of 3–10 μg mL−1.58,60 Given the cytotoxic activity of many of the verticillin analogues, it is not surprising that they have not been explored in more depth for antimicrobial activities.9. Summary and conclusions
To date, 27 verticillin analogues (Fig. 2) have been isolated from fungal cultures. The progenitor, verticillin A (1), was reported in 1970 from a Verticillium sp., although the taxonomy of that organism could be incorrect. More recently, Gliocladium sp., Penicillium sp., and Bionectria sp. (now termed Clonostachys sp.20) are reported to be the main producers of verticillins (Table 1). The dioxopiperazine skeleton of verticillins is biosynthesized via NPRSs, using amino acids that are specific for this class of natural products. The biosynthetic proposal was supported by a study of the genome of C. rogersoniana, which identified the gene cluster (ver) and contributed to both a better understanding of the biosynthetic pathway of these molecules and the likely taxonomy of species that make them. The verticillins are characterized by their sulfur bridges, which are essential for their potency. Their potent cytotoxicity has generated interest in the field of cancer drug discovery, since verticillin A (1) has shown promising in vivo activity by modifying the epigenome and sensitizing cancer cells to apoptosis. This interest in the anti-cancer activity led to a series of in vitro and in vivo studies described in this review. The demand for new anticancer drug leads and the potential shown by the verticillins makes them a promising candidate for further development.9.1. Gaps and potential future directions
After examining more than a half century's worth of literature on this fascinating class of fungal metabolites, it is possible to identify opportunities for future study. If these compounds are to be developed further, then it is obvious that reliable supplies are needed. Using a traditional strain and media optimization approach, members of our team are approaching gram-scale production. While that has been via solid phase cultures, it can be envisaged that liquid phase cultures could be desirable for future industrial-scale production. Many new strains of Clonostachys are being discovered,64 indicating a high level of diversity within this genus. Performing chemical analyses on these newly discovered strains may provide valuable information on new analogues. In addition, screening newer strains could shed light on species that could produce higher yields of verticillins. The biosynthetic gene cluster has been identified by at least one research team, and this could facilitate molecular biology approaches to produce verticillins via heterologous expression. Either of those approaches (or some combination thereof) could lead to routes to the multi-gram scale production of verticillins. Alternatively, synthetic approaches have yielded a route to the core structure of verticillins, albeit on an academic scale. Approaches that would lead to scaled production suitable for pharmaceutical development, either synthetically and/or semi-synthetically, would be welcome. Similarly, to develop a strong patent position, non-natural analogues of the verticillins are needed. While this has been explored using both biosynthetic and semi-synthetic approaches, those studies have also largely been driven by academics, and there is room for more innovations. For example, antibody drug conjugates have been developed recently, where natural products (some of which were first discovered many decades ago) have been linked to antibodies, allowing the targeted delivery of potent molecules.136 One can envision how knowledge developed in the synthetic space around verticillins could be applied to this emerging field as well. Like many natural products, their solubility at physiologically relevant concentrations is also a major challenge. Again, analogue development via synthetic and/or semisynthetic techniques could ameliorate this problem. Alternatively, recent studies from the field of material science could yield nanoparticles or thin films that could deliver the verticillins, and given their potent cytotoxicity, targeted delivery could be an added benefit. Finally, from a pharmacological perspective, most of the studies to date have been with an eye toward cancer chemotherapy. If the aforementioned supply issues are solved, giving greater access to these molecules to diverse disciplines, there are other disease states that could be studied, further leveraging the potential of this interesting class of fungal metabolites.10. Author contributions
Writing—review & editing: HCP, AS, HAR, JRC, ACH, JRF, KL, JEB, CJP, NHO; writing—original draft: CSMA, MGD; visualization: HCP, CSMA, AS, HAR, JRC, ACH, JRF, JEB; conceptualization: NHO; funding acquisition: NHO. All authors contributed to the article and approved the final submission.11. Conflicts of interest
The authors declare the following competing financial interest(s): NHO, HAR, and CJP are members of the Scientific Advisory Board of Clue Genetics, Inc. NHO is also a member of the Scientific Advisory Boards of Mycosynthetix, Inc. and Ionic Pharmaceuticals, LLC.12. Acknowledgements
Research support on bioactive compounds from nature has been provided by the National Institutes of Healthvia the National Cancer Institute (P01 CA125066) and the National Center for Complementary and Integrative Health (to HCP; T32 AT008938). ACH was supported in part via an ACS Medicinal Chemistry Division Predoctoral Fellowship.13. Notes and references
- N. P. Keller, G. Turner and J. W. Bennett, Nat. Rev. Microbiol., 2005, 3, 937–947 CrossRef CAS PubMed.
- M. González-Medina, J. R. Owen, T. El-Elimat, C. J. Pearce, N. H. Oberlies, M. Figueroa and J. L. Medina-Franco, Front. Pharmacol., 2017, 8, 180 CrossRef PubMed.
- S. E. Helaly, B. Thongbai and M. Stadler, Nat. Prod. Rep., 2018, 35, 992–1014 RSC.
- A. G. T. Niego, C. Lambert, P. Mortimer, N. Thongklang, S. Rapior, M. Grosse, H. Schrey, E. Charria-Girón, A. Walker, K. D. Hyde and M. Stadler, Fungal Diversity, 2023, 121, 95–137 CrossRef CAS.
- E. Lax, The mold in Dr. Florey's coat: the story of the penicillin miracle, Holt Paperbacks, New York, 2004 Search PubMed.
- The Nobel Prize in Physiology or Medicine, 1945, https://www.nobelprize.org/prizes/medicine/1945/summary/ Search PubMed.
- B. L. Ligon, Semin. Pediatr. Infect. Dis., 2004, 15, 52–57 CrossRef PubMed.
- R. Quinn, Am. J. Public Health, 2013, 103, 426–434 CrossRef PubMed.
- A. L. Demain and E. Martens, J. Antibiot., 2017, 70, 347–360 CrossRef CAS PubMed.
- S. K. Deshmukh, S. A. Verekar and S. V. Bhave, Front. Microbiol., 2015, 5, 1–43 Search PubMed.
- G. Hendrik, M. Ietida, A. Pontius, S. Kehraus, H. Gross and G. König, Phytochem. Rev., 2010, 9, 537–545 CrossRef.
- A. Evidente, A. Kornienko, A. Cimmino, A. Andolfi, F. Lefranc, V. Mathieu and R. Kiss, Nat. Prod. Rep., 2014, 31, 617–627 RSC.
- M. Blackwell, Am. J. Bot., 2011, 98, 426–438 CrossRef PubMed.
- D. L. Hawksworth and R. Lücking, Microbiol. Spectrum, 2017, 5, FUNK-0052-2016 Search PubMed.
- T. Niskanen, R. Lücking, A. Dahlberg, E. Gaya, L. M. Suz, V. Mikryukov, K. Liimatainen, I. Druzhinina, J. R. S. Westrip, G. M. Mueller, K. Martins-Cunha, P. Kirk, L. Tedersoo and A. Antonelli, Annu. Rev. Environ. Resour., 2023, 48, 149–176 CrossRef.
- G. F. Bills and J. B. Gloer, Microbiol. Spectrum, 2016, 4, FUNK-0009-2016 Search PubMed.
- F. Alberti, G. D. Foster and A. M. Bailey, Appl. Microbiol. Biotechnol., 2016, 101, 493–500 CrossRef PubMed.
- L. N. Aldrich, J. E. Burdette, E. Carcache de Blanco, C. C. Coss, A. S. Eustaquio, J. R. Fuchs, A. D. Kinghorn, A. MacFarlane, B. K. Mize, N. H. Oberlies, J. Orjala, C. J. Pearce, M. A. Phelps, L. H. Rakotondraibe, Y. Ren, D. D. Soejarto, B. R. Stockwell, J. C. Yalowich and X. Zhang, J. Nat. Prod., 2022, 85, 702–719 CrossRef CAS PubMed.
- A. D. Kinghorn, E. J. Carcache De Blanco, D. M. Lucas, H. L. Rakotondraibe, J. Orjala, D. D. Soejarto, N. H. Oberlies, C. J. Pearce, M. C. Wani, B. R. Stockwell, J. E. Burdette, S. M. Swanson, J. R. Fuchs, M. A. Phelps, L. Xu, X. Zhang and Y. Y. Shen, Anticancer Res., 2016, 36, 5623–5637 CrossRef CAS PubMed.
- C. S. M. Amrine, H. A. Raja, B. A. Darveaux, C. J. Pearce and N. H. Oberlies, J. Ind. Microbiol. Biotechnol., 2018, 45, 1053–1065 CrossRef CAS PubMed.
- C. S. M. Amrine, A. C. Huntsman, M. G. Doyle, J. E. Burdette, C. J. Pearce, J. R. Fuchs and N. H. Oberlies, ACS Med. Chem. Lett., 2021, 12, 625–630 CrossRef CAS PubMed.
- C. S. M. Amrine, J. L. Long, H. A. Raja, S. J. Kurina, J. E. Burdette, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2019, 82, 3104–3110 CrossRef CAS PubMed.
- A. Salvi, C. S. M. Amrine, J. R. Austin, K. Kilpatrick, A. Russo, D. Lantvit, E. Calderon-Gierszal, Z. Mattes, C. J. Pearce, M. W. Grinstaff, A. H. Colby, N. H. Oberlies and J. E. Burdette, Mol. Cancer Ther., 2020, 19, 89–100 CrossRef CAS PubMed.
- J. Wang, X. Zhu, S. Kolli, H. Wang, C. J. Pearce, N. H. Oberlies and M. A. Phelps, J. Pharm. Biomed. Anal., 2017, 139, 187–192 CrossRef CAS PubMed.
- F. Liu, Q. Liu, D. Yang, W. B. Bollag, K. Robertson, P. Wu and K. Liu, Cancer Res., 2011, 71, 6807–6816 CrossRef CAS PubMed.
- C. Lu and K. Liu, Transl. Cancer Res., 2017, 6, S652–S654 CrossRef CAS PubMed.
- A. Zewdu, G. Lopez, D. Braggio, C. Kenny, D. Constantino, H. K. Bid, K. Batte, O. H. Iwenofu, N. H. Oberlies, C. J. Pearce, A. M. Strohecker, D. Lev and R. E. Pollock, Clin. Exp. Pharmacol., 2016, 6, 221–234 Search PubMed.
- A. V. Paschall, D. Yang, C. Lu, J.-H. Choi, X. Li, F. Liu, M. Figueroa, N. H. Oberlies, C. Pearce, W. B. Bollag, A. Nayak-Kapoor and K. Liu, J. Immunol., 2015, 195, 1868–1882 CrossRef CAS PubMed.
- M. Figueroa, T. N. Graf, S. Ayers, A. F. Adcock, D. J. Kroll, J. Yang, S. M. Swanson, U. Munoz-Acuna, E. J. Carcache de Blanco, R. Agrawal, M. C. Wani, B. A. Darveaux, C. J. Pearce and N. H. Oberlies, J. Antibiot., 2012, 65, 559–564 CrossRef CAS PubMed.
- E. N. Kaweesa, J. M. Bazioli, H. C. Pierre, D. D. Lantvit, S. K. Kulp, K. L. Hill, M. A. Phelps, C. C. Coss, J. R. Fuchs, C. J. Pearce, N. H. Oberlies and J. E. Burdette, Mol. Pharm., 2023, 20, 3049–3059 CrossRef CAS PubMed.
- E. Iwasa, Y. Hamashima and M. Sodeoka, Isr. J. Chem., 2011, 51, 420–433 CrossRef CAS.
- C. S. Jiang and Y. W. Guo, Mini-Rev. Med. Chem., 2011, 11, 728–745 CrossRef CAS PubMed.
- D. M. Gardiner, P. Waring and B. J. Howlett, Microbiology, 2005, 151, 1021–1032 CrossRef CAS PubMed.
- E. M. Fox and B. J. Howlett, Mycol. Res., 2008, 112, 162–169 CrossRef CAS PubMed.
- P. Waring and C. L. L. Chai, Aust. J. Chem., 2015, 68, 178–183 CrossRef CAS.
- L. Wang, Q. Jiang, S. Chen, S. Wang, J. Lu, X. Gao, D. Zhang and X. Jin, Bioorg. Chem., 2023, 137, 106642 CrossRef CAS PubMed.
- F. Liu, P. Wu and K. Liu, US Pat., US20140161785A1, 2014 Search PubMed.
- S. Niu, D. Yuan, X. Jiang and Y. Che, Protein Cell, 2014, 5, 945–949 CrossRef CAS PubMed.
- Y. Wang, P. Hu, Y. Pan, Y. Zhu, X. Liu, Y. Che and G. Liu, Fungal Genet. Biol., 2017, 103, 25–33 CrossRef CAS PubMed.
- C. Lu, D. Yang, M. E. Sabbatini, A. H. Colby, M. W. Grinstaff, N. H. Oberlies, C. Pearce and K. Liu, BMC Cancer, 2018, 18, 149 CrossRef PubMed.
- Q. Liu, X. Zeng, Y. Guan, J. Lu, K. Tu and F. Liu, J. Zhejiang Univ., Sci., B, 2022, 23, 352 CrossRef PubMed.
- Q. Q. Liu, X. L. Zeng, Y. L. Guan, J. X. Lu, K. Tu and F. Y. Liu, J. Zhejiang Univ., Sci., B, 2020, 21, 779–795 CrossRef CAS PubMed.
- Y. Guan, K. Tu, Q. Huang and F. Liu, Biochem. Biophys. Res. Commun., 2021, 567, 22–28 CrossRef CAS PubMed.
- C. Lu, D. Yang, J. D. Klement, Y. L. Colson, N. H. Oberlies, C. J. Pearce, A. H. Colby, M. W. Grinstaff, H. F. Ding, H. Shi and K. Liu, J. Immunother. Cancer, 2022, 10, e003543 CrossRef PubMed.
- J. Lu, X. Li, K. Tu, Y. Guan, K. P. Fung and F. Liu, OncoTargets Ther., 2019, 12, 5823–5833 CrossRef CAS PubMed.
- Z. Guo, T. Hao, Y. Wang, Y. Pan, F. Ren, X. Liu, Y. Che and G. Liu, Microbiology, 2017, 163, 1654–1663 CrossRef CAS PubMed.
- T. W. Jordan and S. J. Cordiner, Trends Pharmacol. Sci., 1987, 8, 144–149 CrossRef CAS.
- F. Liu, S. Wu, Y. Chen, L. Yang and P. Wu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, 974–976 CrossRef.
- K. Katagiri, K. Sato, S. Hayakawa, T. Matsushima and H. Minato, J. Antibiot., 1970, 23, 420–422 CrossRef CAS PubMed.
- B. K. Joshi, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 1999, 62, 730–733 CrossRef CAS PubMed.
- H. Minato, M. Matsumoto and T. Katayama, J. Chem. Soc. D, 1971, 44–45 RSC.
- D. Hauser, H. Weber and H. Sigg, Helv. Chim. Acta, 1970, 53, 1061–1073 CrossRef CAS PubMed.
- H. Minato, M. Matsumoto and T. Katayama, J. Chem. Soc., Perkin Trans. 1, 1973, 17, 1819–1825 RSC.
- M. Chu, I. Truumees, M. L. Rothofsky, M. G. Patel, F. Gentile, P. R. Das and M. S. Puar, J. Antibiot., 1995, 48, 1440–1445 CrossRef CAS PubMed.
- W. B. Son, R. P. Jensen, C. A. Kauffman and W. Fenical, Nat. Prod. Lett., 1999, 13, 213–222 CrossRef.
- U. Yoshihide, Y. Junko and N. Atsushi, Heterocycles, 2004, 63, 1123–1129 CrossRef.
- J. Dong, H. He, Y. Shen and K. Zhang, J. Nat. Prod., 2005, 68, 1510–1513 CrossRef CAS PubMed.
- C. J. Zheng, C. J. Kim, K. S. Bae, Y. H. Kim and W. G. Kim, J. Nat. Prod., 2006, 69, 1816–1819 CrossRef CAS PubMed.
- D. Jin-Yan, Z. Wei, L. Lei, L. Guo-Hong, L. Ya-Jun and Z. Ke-Qing, Chin. Chem. Lett., 2006, 17, 922–924 Search PubMed.
- C. J. Zheng, S. H. Park, H. Koshino, Y. H. Kim and W. G. Kim, J. Antibiot., 2007, 60, 61–64 CrossRef CAS PubMed.
- Y. Chen, H. Guo, Z. Du, X. Z. Liu, Y. Che and X. Ye, Cell Proliferation, 2009, 42, 838–847 CrossRef CAS PubMed.
- X. Liu, S. Liu, Y. C. Ye, S. Yong, H. Guo and Y. Chen, Peop. Rep. China Pat., 2009-10077302-101805699, 2009 Search PubMed.
- S. Dirk, C. Böttcher, J. Lee and D. Scheel, J. Antibiot., 2011, 64, 523–524 CrossRef PubMed.
- L. Zhao, J. Groenewald, M. Hernández-Restrepo, H.-J. Schroers and P. Crous, Stud. Mycol., 2023, 105, 205–266 Search PubMed.
- H.-J. Schroers, G. J. Samuels, K. A. Seifert and W. Gams, Mycologia, 1999, 91, 365–385 CrossRef.
- D. L. Hawksworth, P. W. Crous, S. A. Redhead, D. R. Reynolds, R. A. Samson, K. A. Seifert, J. W. Taylor, M. J. Wingfield, Ö. Abaci, C. Aime, A. Asan, F.-Y. Bai, Z. W. de Beer, D. Begerow, D. Berikten, T. Boekhout, P. K. Buchanan, T. Burgess, W. Buzina, L. Cai, P. F. Cannon, J. L. Crane, U. Damm, H.-M. Daniel, A. D. van Diepeningen, I. Druzhinina, P. S. Dyer, U. Eberhardt, J. W. Fell, J. C. Frisvad, D. M. Geiser, J. Geml, C. Glienke, T. Gräfenhan, J. Z. Groenewald, M. Groenewald, J. de Gruyter, E. Guého-Kellermann, L.-D. Guo, D. S. Hibbett, S.-B. Hong, G. S. de Hoog, J. Houbraken, S. M. Huhndorf, K. D. Hyde, A. Ismail, P. R. Johnston, D. G. Kadaifciler, P. M. Kirk, U. Kõljalg, C. P. Kurtzman, P.-E. Lagneau, C. A. Lévesque, X. Liu, L. Lombard, W. Meyer, A. Miller, D. W. Minter, M. J. Najafzadeh, L. Norvell, S. M. Ozerskaya, R. Öziç, S. R. Pennycook, S. W. Peterson, O. V. Pettersson, W. Quaedvlieg, V. A. Robert, C. Ruibal, J. Schnürer, H.-J. Schroers, R. Shivas, B. Slippers, H. Spierenburg, M. Takashima, E. Taşkın, M. Thines, U. Thrane, A. H. Uztan, M. van Raak, J. Varga, A. Vasco, G. Verkley, S. I. R. Videira, R. P. de Vries, B. S. Weir, N. Yilmaz, A. Yurkov and N. Zhang, IMA Fungus, 2011, 2, 105–112 CrossRef PubMed.
- J. W. Taylor, IMA Fungus, 2011, 2, 113–120 CrossRef PubMed.
- A. Y. Rossman, K. A. Seifert, G. J. Samuels, A. M. Minnis, H.-J. Schroers, L. Lombard, P. W. Crous, K. Põldmaa, P. F. Cannon, R. C. Summerbell, D. M. Geiser, W.-y. Zhuang, Y. Hirooka, C. Herrera, C. Salgado-Salazar and P. Chaverri, IMA Fungus, 2013, 4, 41–51 CrossRef PubMed.
- J. Kim, J. A. Ashenhurst and M. Movassaghi, Science, 2009, 324, 238–241 CrossRef CAS PubMed.
- E. M. Huber, ChemBioChem, 2022, 23, e202200341 CrossRef CAS PubMed.
- D. Ferreira, Personal communication, Department of BioMolecular Sciences, University of Mississippi, University, MS, October 2014 Search PubMed.
- P. J. Proteau, Personal communication, Department of Pharmaceutical Sciences, Oregon State University, Corvallis, OR, January 2023 Search PubMed.
- H. A. Raja, A. N. Miller, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2017, 80, 756–770 CrossRef CAS PubMed.
- K. Seifert, G. Morgan-Jones, W. Gams and B. Kendrick, The Genera of Hyphomycetes, CBS Biodiversity Series no. 9: 1–997, CBS-KNAW Fungal Biodiversity Centre, Utrecht, Netherlands, 2011 Search PubMed.
- N. Turland, J. Wiersema, F. Barrie, W. Greuter, D. Hawksworth, P. Herendeen, S. Knapp, W.-H. Kusber, D.-Z. Li, K. Marhold, T. May, J. McNeill, A. Monro, J. Prado, M. Price and G. Smith, International Code of Nomenclature for algae, fungi, and plants (Shenzhen Code) adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017, Koeltz Botanical Books, 2018 Search PubMed.
- B. Zhou, P. S. Achanta, G. Shetye, S.-N. Chen, H. Lee, Y.-Y. Jin, J. Cheng, M.-J. Lee, J.-W. Suh, S. Cho, S. G. Franzblau, G. F. Pauli and J. B. McAlpine, J. Nat. Prod., 2021, 84, 2644–2663 CrossRef CAS PubMed.
- C. Bailly, Phytochemistry, 2022, 200, 113250 CrossRef CAS PubMed.
- R. J. Suhadolnik and R. G. Chenoweth, J. Am. Chem. Soc., 1958, 80, 4391–4392 CrossRef CAS.
- J. A. Winstead and R. J. Suhadolnik, J. Am. Chem. Soc., 1960, 82, 1644–1647 CrossRef CAS.
- W. Kirby and J. Robins, The biosynthesis of gliotoxin and related epipolythiodioxopiperazines, Academic Press, New York, 1980 Search PubMed.
- H. D. Mootz and M. A. Marahiel, Curr. Opin. Chem. Biol., 1997, 1, 543–551 CrossRef CAS PubMed.
- T. Gerken and C. T. Walsh, ChemBioChem, 2013, 14, 2256–2258 CrossRef CAS PubMed.
- C. J. Balibar and C. T. Walsh, Biochemistry, 2006, 45, 15029–15038 CrossRef CAS PubMed.
- D. H. Scharf, J. D. Dworschak, P. Chankhamjon, K. Scherlach, T. Heinekamp, A. A. Brakhage and C. Hertweck, ACS Chem. Biol., 2018, 13, 2508–2512 CrossRef CAS PubMed.
- D. H. Scharf, N. Remme, A. Habel, P. Chankhamjon, K. Scherlach, T. Heinekamp, P. Hortschansky, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2011, 133, 12322–12325 CrossRef CAS PubMed.
- C. Davis, S. Carberry, M. Schrettl, I. Singh, J. C. Stephens, S. M. Barry, K. Kavanagh, G. L. Challis, D. Brougham and S. Doyle, Chem. Biol., 2011, 18, 542–552 CrossRef CAS PubMed.
- D. H. Scharf, N. Remme, T. Heinekamp, P. Hortschansky, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2010, 132, 10136–10141 CrossRef CAS PubMed.
- D. H. Scharf, P. Chankhamjon, K. Scherlach, T. Heinekamp, K. Willing, A. A. Brakhage and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 11092–11095 CrossRef CAS PubMed.
- D. H. Scharf, A. Habel, T. Heinekamp, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2014, 136, 11674–11679 CrossRef CAS PubMed.
- D. H. Scharf, P. Chankhamjon, K. Scherlach, J. Dworschak, T. Heinekamp, M. Roth, A. A. Brakhage and C. Hertweck, ChemBioChem, 2021, 22, 336–339 CrossRef CAS PubMed.
- T. Saruwatari, F. Yagishita, T. Mino, H. Noguchi, K. Hotta and K. Watanabe, ChemBioChem, 2014, 15, 656–659 CrossRef CAS PubMed.
- J. Liu, X. Xie and S.-M. Li, Chem. Commun., 2020, 56, 11042–11045 RSC.
- C. Sun, Z. Luo, W. Zhang, W. Tian, H. Peng, Z. Lin, Z. Deng, B. Kobe, X. Jia and X. Qu, Nat. Commun., 2020, 11, 6251 CrossRef CAS PubMed.
- V. V. Shende, Y. Khatri, S. A. Newmister, J. N. Sanders, P. Lindovska, F. Yu, T. J. Doyon, J. Kim, K. N. Houk, M. Movassaghi and D. H. Sherman, J. Am. Chem. Soc., 2020, 142, 17413–17424 CrossRef CAS PubMed.
- A. Bahadoor, K. A. Robinson, M. C. Loewen and Z. A. Demissie, BMC Genomics, 2023, 24, 352 CrossRef CAS PubMed.
- J. B. Xu and K. J. Cheng, Molecules, 2015, 20, 6715–6738 CrossRef CAS PubMed.
- L. E. Overman, D. V. Paone and B. A. Stearns, J. Am. Chem. Soc., 1999, 121, 7702–7703 CrossRef CAS.
- A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed.
- L. E. Overman and Y. Shin, Org. Lett., 2007, 9, 339–341 CrossRef CAS PubMed.
- J. Kim and M. Movassaghi, J. Am. Chem. Soc., 2010, 132, 14376–14378 CrossRef CAS PubMed.
- E. Iwasa, Y. Hamashima, S. Fujishiro, E. Higuchi, A. Ito, M. Yoshida and M. Sodeoka, J. Am. Chem. Soc., 2010, 132, 4078–4079 CrossRef CAS PubMed.
- T. R. Welch and R. M. Williams, Nat. Prod. Rep., 2014, 31, 1376–1404 RSC.
- J. Kim and M. Movassaghi, Acc. Chem. Res., 2015, 48, 1159–1171 CrossRef CAS PubMed.
- A. Coste, J. Kim, T. C. Adams and M. Movassaghi, Chem. Sci., 2013, 4, 3191–3197 RSC.
- N. Boyer and M. Movassaghi, Chem. Sci., 2012, 3, 1798–1803 RSC.
- C. R. Olsson, J. N. Payette, J. H. Cheah and M. Movassaghi, J. Org. Chem., 2020, 85, 4648–4662 CrossRef CAS PubMed.
- D. Hauser, H. R. Loosli and P. Niklaus, Helv. Chem. Acta, 1972, 55, 2182–2187 CrossRef CAS PubMed.
- T. Saito, K. Koyama, S. Natori and Y. Iitaka, Tetrahedron Lett., 1985, 26, 4731–4734 CrossRef CAS.
- T. Saito, Y. Suzuki, K. Koyama, S. Natori, Y. Iitaka and T. Kinosita, Chem. Pharm. Bull., 1988, 36, 1942–1956 CrossRef CAS.
- E. Li, B. Hou, Q. Gao, Y. Xu, C. Zhang, X. Liu, X. Jiang and Y. Che, J. Nat. Prod., 2020, 83, 601–609 CrossRef CAS PubMed.
- N. Boyer, K. C. Morrison, J. Kim, P. J. Hergenrother and M. Movassaghi, Chem. Sci., 2013, 4, 1646–1657 RSC.
- Z. Ma, A. Zhou and C. Xia, Nat. Prod. Rep., 2022, 39, 1015–1044 RSC.
- A. I. Scott, F. McCapra and E. S. Hall, J. Am. Chem. Soc., 1964, 86, 302–303 CrossRef CAS.
- T. Hino, S. Kodato, K. Takahashi, H. Yamaguchi and M. Nakagawa, Tetrahedron Lett., 1978, 19, 4913–4916 CrossRef.
- H. Ishikawa, H. Takayama and N. Aimi, Tetrahedron Lett., 2002, 43, 5637–5639 CrossRef CAS.
- M. Ding, K. Liang, R. Pan, H. Zhang and C. Xia, J. Org. Chem., 2015, 80, 10309–10316 CrossRef CAS PubMed.
- E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394–3402 CrossRef CAS PubMed.
- K. Liang, X. Tong, T. Li, B. Shi, H. Wang, P. Yan and C. Xia, J. Org. Chem., 2018, 83, 10948–10958 CrossRef CAS PubMed.
- S. Tadano, Y. Mukaeda and H. Ishikawa, Angew. Chem., Int. Ed., 2013, 52, 7990–7994 CrossRef CAS PubMed.
- C. Lu, J. D. Klement, D. Yang, T. Albers, I. O. Lebedyeva, J. L. Waller and K. Liu, Cancer Lett., 2020, 476, 87–96 CrossRef CAS PubMed.
- J.-S. He, D.-E. Gong and H. L. Ostergaard, J. Immunol., 2010, 184, 555–563 CrossRef CAS PubMed.
- G. Koncz, A. Hancz, K. Chakrabandhu, P. Gogolák, K. Kerekes, É. Rajnavölgyi and A.-O. Hueber, J. Immunol., 2012, 189, 2815–2823 CrossRef CAS PubMed.
- C. Lu, A. V. Paschall, H. Shi, N. Savage, J. L. Waller, M. E. Sabbatini, N. H. Oberlies, C. Pearce and K. Liu, J. Natl. Cancer Inst., 2017, 109, djw283 CrossRef PubMed.
- J. Brahmer, S. Tykodi, L. Chow, W.-J. Hwu, S. Topalian, P. Hwu, C. Drake, L. Camacho, J. Kauh, K. Odunsi, H. Pitot, O. Hamid, S. Bhatia, R. Martins, K. Eaton, S. Chen, T. Salay, S. Alaparthy, J. Grosso, A. Korman, S. Parker, S. Agrawal, S. Goldberg, D. Pardoll, A. Gupta and J. Wigginton, N. Engl. J. Med., 2012, 366, 2455–2465 CrossRef CAS PubMed.
- A. H. Colby, N. H. Oberlies, C. J. Pearce, V. L. M. Herrera, Y. L. Colson and M. W. Grinstaff, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1451 Search PubMed.
- R. V. Sionov, S. A. Vlahopoulos and Z. Granot, Oncotarget, 2015, 6, 23058–23134 CrossRef PubMed.
- R. Carpenter and M. Brady, StatPearls, 2021, NBK555927 Search PubMed.
- S. K. Mitra and D. D. Schlaepfer, Curr. Opin. Cell Biol., 2006, 18, 516–523 CrossRef CAS PubMed.
- A. Y. Hui, J. A. Meens, C. Schick, S. L. Organ, H. Qiao, E. A. Tremblay, E. Schaeffer, S. Uniyal, B. M. Chan and B. E. Elliott, J. Cell. Biochem., 2009, 107, 1168–1181 CrossRef CAS PubMed.
- J. Ma, B. Zheng, S. Goswami, L. Meng, D. Zhang, C. Cao, T. Li, F. Zhu, L. Ma, Z. Zhang, S. Zhang, M. Duan, Q. Chen, Q. Gao and X. Zhang, J. Immunother. Cancer, 2019, 7, 331 CrossRef PubMed.
- P. Gueguen, C. Metoikidou, T. Dupic, M. Lawand, C. Goudot, S. Baulande, S. Lameiras, O. Lantz, N. Girard, A. Seguin-Givelet, M. Lefevre, T. Mora, A. M. Walczak, J. J. Waterfall and S. Amigorena, Sci. Immunol., 2021, 6, eabd5778 CrossRef CAS PubMed.
- G. Erkel, A. Gehrt, T. Anke and O. Sterner, Z. Naturforsch., C: J. Biosci., 2002, 57, 759–767 CrossRef CAS PubMed.
- Y. Zhang, Y. Chen, X. Guo, X. Zhang, W. Zhao, L. Zhong, J. Zhou, Y. Xi, Z. Lin and J. Ding, Anticancer Drugs, 2005, 16, 515–524 CrossRef CAS PubMed.
- Y. Chen, Z. Miao, W. Zhao and J. Ding, FEBS Lett., 2005, 579, 3683–3690 CrossRef CAS PubMed.
- Y. Chen, Y. Zhang, M. Li, W. Zhao, Y. Shi, Z. Miao, X. Zhang, L. Lin and J. Ding, Biochem. Biophys. Res. Commun., 2005, 329, 1334–1342 CrossRef CAS PubMed.
- D. J. Newman, J. Nat. Prod., 2021, 84, 917–931 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |