
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ FTIR spectroscopic investigations on rhodium carbonyl complexes in the absence of phosphorus ligands under hydroformylation conditions†
Benedict N.
Leidecker
 a,
Dilver Peña
Fuentes
a,
Chunhong
Wei
a,
Mathias
Sawall
b,
Klaus
Neymeyr
ab,
Robert
Franke
cd,
Armin
Börner
a and
Christoph
Kubis
*a
a,
Dilver Peña
Fuentes
a,
Chunhong
Wei
a,
Mathias
Sawall
b,
Klaus
Neymeyr
ab,
Robert
Franke
cd,
Armin
Börner
a and
Christoph
Kubis
*a
aLeibniz-Institut für Katalyse e. V., Albert-Einstein Str. 29a, 18059 Rostock, Germany. E-mail: christoph.kubis@catalysis.de
bInstitut für Mathematik, Universität Rostock, Ulmenstr. 59, 18057 Rostock, Germany
cEvonik Oxeno GmbH & Co. KG, Paul-Baumann-Str. 1, 45772 Marl, Germany
dLehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, 44780 Bochum, Germany
First published on 2nd September 2024
Abstract
In situ FTIR spectroscopy was combined with the peak group analysis (PGA) for investigations on the composition of ‘unmodified’ rhodium carbonyl complexes in the absence of phosphorus ligands at conditions relevant for alkene hydroformylation. As a precursor complex [Rh(acac)(CO)2] was selected since it is commonly used for rhodium catalyzed hydroformylations. At higher pressures of synthesis gas (CO/H2) and elevated temperatures, [Rh(acac)(CO)2] is decomposed to [Rh4(CO)12] and [Rh6(CO)16] as spectroscopically observable components. [Rh4(CO)12] represents an intermediate that readily reacts towards [Rh6(CO)16] at higher temperatures and lower partial pressures of carbon monoxide. The degradation of [Rh(acac)(CO)2] is hampered significantly at higher concentrations of acetylacetone. The involvement of a hydrido species in the reaction sequence from [Rh(acac)(CO)2] to polynuclear rhodium complexes under H2/CO is in aggreement with the detection of the infrared spectrum of [HRh(CO)4] and the considerable decrease of the decomposition rate in the presence of pure CO and D2/CO. With ethene as the alkene substrate, acyl complexes of the type [CH3CH2C(O)Rh(CO)3(π-C2H4)] and [CH3CH2C(O)Rh(CO)4] have been observed, whereas only the tetra carbonyl complex [(CH3)3CCH2CH2C(O)Rh(CO)4] was detected for neohexene.
1. Introduction
The hydroformylation of short-chain alkenes towards aldehydes catalyzed by rhodium–carbonyl hydride complexes [HxMy(CO)zL] modified with phosphorus-organic ligands is performed on an industrial scale.1–4 Trivalent P-ligands, generally phosphines and phosphites, are of relevance. The degradation of P-ligands due to oxidation and/or hydrolysis leads either to alterations of the active catalyst complexes or to the formation of unmodified rhodium clusters.5–11 In both cases the catalytic performance (activity, chemo-/regioselectivity) is significantly deteriorated. Irreversible rhodium deposition might occur under harsh conditions and depletion of ligands (CO, P-ligands).Time-resolved in situ/operando FTIR-spectroscopy as an online monitoring tool is very powerful to detect changes in the mixture of catalyst complexes or the formation of polynuclear rhodium carbonyl complexes.12–15 Thus, catalyst degradation can be identified in its initial phase and measures can be undertaken to prevent further decomposition.
FTIR-spectroscopy using high-pressure transmission flow-through cells with optical pathlength of ≥400 μm allows the measurement of rhodium carbonyl complexes in the millimolar to submillimolar concentration range.16–18 A chemometric data analysis based on multivariate curve resolution (MCR) techniques performed on acquired spectra series often enables the extraction of pure component spectra and concentration profiles from a multicomponent mixture.19–23 Multiple perturbations of reagents within a multistep dosage experiment are often beneficial to eliminate collinearities between respective components in the spectroscopic data.15,19–21 The infrared spectra of transition metal carbonyl complexes show distinct band patterns related to the molecular structure. The attribution of spectral bands to molecular vibrations can be facilitated by a vibrational mode analysis using DFT methods.11,15–18,20,21 Thus, an interpretation of the spectral patterns of non-isolable complexes and the impact of an isotopic labelling on spectroscopic signatures is readily achieved.
Hydroformylation consists of multiple organometallic elementary steps (Scheme 1). In the catalyst formation step, the 18 VE hydrido Rh(I) complex [HRh(CO)4] 1 is generated from a precursor complex during the treatment under carbon monoxide and hydrogen. Dissociation of a carbonyl ligand provides the 16 VE hydrido complex [HRh(CO)3] 2 which affords the alkene activation. The addition of the alkene substrate generates a π-olefin complex 3 followed by the insertion of the alkene into the Rh–H bond to form a 16 VE alkyl complex 4. The coordination of a CO ligand leads to the corresponding 18 VE alkyl complex 5. Migratory insertion of the alkyl group into the Rh–CO bond gives the 16 VE acyl complex 6 which is in equilibrium with the 18 VE acyl complex 7 after CO coordination. The unsaturated acyl complex 6 might react with hydrogen in an oxidative addition reaction to generate a Rh(III) dihydrido complex (not shown). Reductive elimination releases the aldehyde product and regenerates complex 2, which closes the catalytic cycle. It is also common to regard the aldehyde formation step as hydrogenolysis of the acyl complex 6 as shown in Scheme 1. Regioselectivity, often given as molar fraction of the n-linear aldehyde product or the molar n/iso ratio, is affected by the intrinsic formation of α- and β-alkyl complexes 4 and 4′ (not shown) resulting from Rh–C1 or Rh–C2 bonding, the activity for β-hydride elimination and the partial pressure of carbon monoxide. In addition, the steric bulk caused by P-ligands in hydrido rhodium complexes as catalysts has a tremendous impact on the regioselectivity.3 For unmodified rhodium catalyzed hydroformylation the n-regioselectivity for linear alkenes is generally significantly lower compared to P-ligand modified catalyst systems.
 | ||
| Scheme 1 Multistep catalytic cycle for the unmodified rhodium catalyzed hydroformylation of alkenes based on a Heck–Breslow-type mechanism. | ||
Within the framework of this study, we have investigated several aspects of the P-ligand free rhodium hydroformylation using [Rh(acac)(CO)2] as a precursor complex. The overall aim was to characterize the multicomponent mixture of ‘unmodified’ rhodium carbonyl complexes in the absence of any phosphorus ligand under varying conditions by in situ FTIR spectroscopy. This is of relevance in the case of severe degradation of P-ligands.
In the beginning, a component identification based on a multiple dosage experiment of the precursor complex [Rh(acac)(CO)2] was conducted. [Rh(acac)(CO)2] decomposes under the release of acetylacetone (acacH) towards [Rh4(CO)4] and [Rh6(CO)16] in the presence of synthesis gas (CO/H2) at elevated temperature. The decomposition of [Rh(acac)(CO)2] → [Rh4(CO)4] → [Rh6(CO)16] was also studied stepwise in single experiments. The influence of an excess of acacH on the decomposition of [Rh(acac)(CO)2] was investigated. The detection of the [HRh(CO)4] at lower temperatures based on chemometric data analysis of infrared spectroscopic data is demonstrated. These results are validated by the formation of the corresponding deuteride complex and the cobalt analogues. The generation of acyl rhodium carbonyl complexes in the presence of ethene and neohexene was also investigated.
2. Experimental
2.1 HP-FTIR experiments
The high-pressure (HP) FTIR spectroscopic apparatus consisted of a stainless-steel reactor (either a 25 ml cylinder from Swagelok with a magnetic stirrer bar or a 200 ml batch autoclave from Premex with a gas entrainment impeller) which was connected to a flow-through transmission cell with an optical pathlength of 400 μm using a spacer. CaF2 (Korth Kristalle GmbH, Kiel, Germany) was used as a window material. The liquid reaction solution was circulated via a micro-gear pump (mzr-7255, HNP Mikrosysteme GmbH, Schwerin, Germany). All wetted parts were heated and thermally insulated. Reactants were dosed via a syringe pump (PHD Ultra 4400, Harvard Apparatus GmbH, March-Hugstetten) combined with an 8 or 20 mL syringe made of stainless-steel.A modified Bruker FTIR spectrometer with a MCT-A detector (24 h holding time for the LN2 dewar) was used for the collection of FTIR spectra. The spectral resolution was set to 2 cm−1 and spectroscopic measurements were done at 20 kHz mirror velocity with 10 scans per spectrum (double sided forward–backward).
Further supplementary data and details on the laboratory experiments are given in the ESI.†
2.2 Peak group analysis (PGA)
The peak group analysis (PGA) is a chemometric method based on multivariate curve resolution (MCR) techniques to extract pure component spectra from spectroscopic data of multicomponent mixtures.24–26 PGA, with its focus on vibrational spectra analysis benefits from the typical structure of IR or Raman spectra with their sharp peaks and broad frequency ranges with only low absorption. A characteristic feature of PGA is that it starts by detecting all the more or less isolated peaks in the mixture spectra. For each of these peaks, PGA then attempts to recover a potential pure component spectrum that includes the original peak in its full spectrum. These pure component spectra are built from linear combinations of the right singular vectors, which are first computed by singular value decomposition of the spectral data matrix. If a pure spectrum contains more than one peak, PGA finds this spectrum the same number of times. In a final step, PGA identifies the essentially different pure component spectra. All of these steps are automated in the PGA software, which incorporates powerful numerical optimisation in order to ensure nonnegative spectral profiles that satisfy additional constraints. Once all the pure component spectra have been calculated, the corresponding concentration profiles can be determined using linear regression. The software implementation of the automated PGA algorithm is relatively easy to use. PGA is part of the program package FACPACK which can be downloaded from the website: https://www.math.uni-rostock.de/facpack/.2.3 Vibrational mode analysis by DFT methods
Geometry optimizations and frequency calculations were carried out using the Gaussian 16 (Revision A. 03) program package. All calculations were performed at the B3LYP/def2SVP level in the gas phase. No scaling factor was applied to the calculated frequencies. The assignment of experimental infrared spectra to molecular structures and its vibrational modes can be based on the comparison of spectral patterns, determined by the number and relative intensities of infrared bands.3. Results and discussion
3.1 Component identification by the combination of multiple dosage FTIR experiment and peak group analysis
A multiple dosage experiment with [Rh(acac)(CO)2] as reactant was performed to identify the major components observable during the treatment with synthesis gas at elevated temperature in solution.A solution consisting of diphenyl carbonate as internal FTIR-standard ([DPC] = 2 mM) with n-dodecane as solvent was heated up to 120 °C and pressurized with 2.0 MPa synthesis gas (CO/H2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The precursor complex [Rh(acac)(CO)2] was dosed stepwise with help of a syringe pump covering a concentration range between 0.5–10 mM (Table 1) (Fig. 1). Intervals of 39 min (ID 1–3) and 93–97 min (ID 3–8) were kept between the dosage steps.
:1). The precursor complex [Rh(acac)(CO)2] was dosed stepwise with help of a syringe pump covering a concentration range between 0.5–10 mM (Table 1) (Fig. 1). Intervals of 39 min (ID 1–3) and 93–97 min (ID 3–8) were kept between the dosage steps.
| Dosage ID | [Rh]/mM | Dosage ID | [Rh]/mM |
|---|---|---|---|
| Reaction conditions: ν = 120 °C, p(CO/H2) = 2.0 MPa, solvent: n-dodecane. | |||
| 1 (initial) | 0.5 | 5 (266 min) | 4 |
| 2 (39 min) | 1 | 6 (363 min) | 5 |
| 3 (78 min) | 2 | 7 (458 min) | 7.5 |
| 4 (172 min) | 3 | 8 (551 min) | 10 |
 | ||
| Fig. 1 In situ FTIR-spectra collected during the perturbation experiment monitoring the decomposition of [Rh(acac)(CO)2] under syngas atmosphere. Conditions: [Rh] = 0.5–10 mM, [DPC] = 2 mM, ν = 120 °C, p(CO/H2) = 2.0 MPa, solvent: n-dodecane. | ||
The peak group analysis of the collected IR-spectra series extracted [Rh(acac)(CO)2] (ν(CO) = 1528, 1582, 2012, 2082 cm−1), [Rh4(CO)12] (ν(CO) = 1883, 2042, 2068, 2073 cm−1) and [Rh6(CO)16] (ν(CO) = 1820, 2043, 2073 cm−1) as spectroscopically observable components (Fig. 2(a)). The comparison of the patterns of reconstructed PGA-spectra to calculated vibrational spectra based on DFT calculations confirmed the assignment of the polynuclear complexes to [Rh4(σ-CO)9(μ-CO)3] and [Rh6(σ-CO)12(μ-CO)4] (Fig. 2(b)).27 According to the corresponding absorbance-vs.-time profiles [Rh(acac)(CO)2] is decomposed under the applied conditions without any induction period to [Rh4(CO)12] which further reacts towards [Rh6(CO)16] (Fig. 3(a)). The intermediate [Rh4(CO)12] is not accumulated strongly but seems to react quite readily to [Rh6(CO)16]. While the precursor complex [Rh(acac)(CO)2] is decomposed completely at lower rhodium concentrations (0.5 mM) within ca. 40 min (Fig. 3(b)), the relative degree of the degradation decreased with increasing rhodium concentrations. The ratios between [H2]/[Rh] and [CO]/[Rh] might be relevant for the kinetics of the decay of the precursor complex. Acetylacetone (acacH) was also generated during the process. It shows a broad band feature at 1615 cm−1 corresponding to the enol form. The accumulation of acacH in the reaction solution should also hamper the decomposition of the precatalyst [Rh(acac)(CO)2] at higher concentrations to some extent (vide infra).
 | ||
| Fig. 2 (a) PGA extracted pure component spectra from the in situ FTIR-spectra collected during the perturbation experiment; (b) calculated vibrational spectra based on DFT-methods. | ||
 | ||
| Fig. 3 Extracted profiles of the complexes for the perturbation experiment. (a) Non-scaled absorption-vs.-time profiles for the complete data set; (b) scaled concentration profiles for the first addition of [Rh]0 = 0.5 mM. | ||
At the applied reaction conditions no detection of the hydrido complex [HRh(CO)4] or dinuclear carbonyl complex such as [Rh2(CO)8] was possible. However, it is assumed that they are part of the reaction sequence from [Rh(acac)(CO)2] towards polynuclear carbonyl complexes, even though they might be present only in trace amounts. They were detected spectroscopically by the group of Marc Garland and other researchers during the treatment of [Rh4(CO)12] with synthesis gas at lower temperatures.28–32 Respective in situ FTIR experiments with [Rh(acac)(CO)2] as a precursor have been done within this study (vide infra).
A single control experiment under pure carbon monoxide was performed to verify the significance of hydrogen for the hydrogenolytic conversion of [Rh(acac)(CO)2]. The experiment was carried out in absence of hydrogen using pure carbon monoxide at the following conditions: [Rh] = 1 mM, [DPC] = 2 mM, ν = 120 °C, p(CO) = 1.0 MPa, solvent: n-dodecane. As expected, the reaction was retarded considerably. However, the formation of [Rh6(CO)16] took place even though much more slowly (SI-C, Fig. SI-4, ESI†). A possible explanation could be a release of hydrogen by the dehydrogenation of organic components such as the solvent. This remains a speculation since we did not try to investigate this aspect in any more detail in the framework of this study.
3.2 Decomposition of [Rh(acac)(CO)2] at 100 °C in a single experiment. Impact of acacH concentration
In the next step, the treatment of [Rh(acac)(CO)2] ([Rh] = 1 mM) under 2.0 MPa of synthesis gas at ν = 100 °C was investigated in a single experiment. A solution in n-dodecane was prepared and transferred into to the FTIR-apparatus. Then the system was set to 100 °C and the reaction was started by the addition of synthesis gas (CO/H2).The decomposition of [Rh(acac)(CO)2] started immediately and the formation of [Rh4(CO)12] and [Rh6(CO)16] followed in a consecutive manner. Acetylacetone was released in a stoichiometric manner. The PGA analysis delivered the pure component spectra and concentration profiles were obtained using linear regression in combination with calibration data applied to the last spectra (Fig. 4). After 180 min the degree of decomposition was ca. 83%. The concentration of the [Rh4(CO)12] intermediate was higher for 100 °C in comparison to the dosage experiment at 120 °C, which indicates a strong temperature dependence for the decomposition of [Rh4(CO)12] to [Rh6(CO)16].
 | ||
| Fig. 4 (a) In situ FTIR-spectra series obtained from the treatment of [Rh(acac)(CO)2] with synthesis gas at ν = 100 °C. Conditions: [Rh] = 1 mM, p(CO/H2) = 2.0 MPa, solvent: n-dodecane; (b) extracted PGA-spectra; (c) concentration profiles after scaling based on calibration data. | ||
To investigate whether the concentration of acetylacetone has a noticeably influence on the decomposition of [Rh(acac)(CO)2], a comparative experiment in the presence 10 equivalents of acetylacetone (10 mM) was carried out. The other conditions were not changed. Based on the concentration profiles for the rhodium complexes obtained from the chemometric analysis on the infrared spectroscopic data, the decomposition was hindered, showing a significant decrease in the reaction rate (SI-C, Fig. SI-6, ESI†).
3.3 Impact of the partial pressure of carbon monoxide on the decomposition of [Rh4(CO)12]
From the experimental results discussed so far, the intermediate [Rh4(CO)12] was identified to be quite reactive towards the formation of [Rh6(CO)16]. The rate of this reaction increases with increasing temperature. It is expected that the partial pressure of carbon monoxide should also influence the rate of this reaction step. Comparative FTIR-experiments at ν = 100 °C were conducted starting from [Rh4(CO)12] ([Rh] = 1 mM) which was exposed to a synthesis gas mixture at p(CO/H2) = 2.0 MPa and to argon at p(Ar) = 0.1 MPa in the absence of synthesis gas. While a conversion of ca. 95% under synthesis gas took ca. 160 min, full conversion was reached after ca. 15 min when treated at ambient pressure of argon (Fig. 5). These results confirm a high intrinsic reactivity of [Rh4(CO)12] to [Rh6(CO)16].33 | ||
| Fig. 5 Concentration profiles extracted via PGA and linear regression from in situ FTIR-spectra collected during treatment of [Rh4(CO)12] under (a) synthesis gas p(CO/H2) = 2.0 MPa and (b) argon p(Ar) = 0.1 MPa. Further conditions: [Rh] = 1 mM, ν = 100 °C, solvent: n-dodecane. | ||
3.4 Decomposition of [Rh(acac)(CO)2] at lower temperatures and higher pressures. Detection of [HRh(CO)4]
The mononuclear hydrido rhodium tetracarbonyl complex is known to be very reactive and present only in trace amounts under typical hydroformylation conditions. The group of Marc Garland could detect [HRh(CO)4] during the treatment of [Rh4(CO)12] with synthesis gas based on in situ FTIR spectroscopy combined with the BTEM analysis.28We were interested to identify this component during the reaction of [Rh(acac)(CO)2] with synthesis gas. Under consideration of the results discussed above it was assumed, that the [HRh(CO)4] might be more populated at lower temperatures and higher synthesis gas pressures. Thus, we conducted two in situ FTIR experiments at the following conditions with [Rh(acac)(CO)2] as starting material: (a) [Rh] = 5 mM, p(CO/H2) = 5.0 MPa, ν = 50 °C and (b) [Rh] = 1 mM, p(CO/H2) = 5.0 MPa, ν = 30 °C. For both temperatures the decomposition of [Rh(acac)(CO)2] → [Rh4(CO)4] → [Rh6(CO)16] was observed. The rate for the decomposition of [Rh(acac)(CO)2] and [Rh4(CO)12] significantly decreased leading to the formation of large molar fractions of [Rh4(CO)12] (Fig. 6). This highlights again the strong impact of the temperature on its molar fraction.
 | ||
| Fig. 6 In situ FTIR spectra collected during the treatment of [Rh(acac)(CO)2] under synthesis gas to [Rh4(CO)12] and [Rh6(CO)16] and extracted concentration profiles. (a) and (b) [Rh] = 5 mM, p(CO/H2) = 5.0 MPa, ν = 50 °C, solvent: n-dodecane; (c) and (d) [Rh] = 1 mM, p(CO/H2) = 5.0 MPa, ν = 30 °C, solvent: n-dodecane; (e) and (f) IR-spectra and PGA results from the treatment of [Rh4(CO)12] with p(CO) = 1.0 MP and p(H2) = 4.0 MPa; (g) and (h) p(CO/D2) = 5.0 MPa, ν = 30 °C, solvent: n-dodecane. | ||
However, the peak group analysis (PGA) applied to both data sets did not allow for the detection of [HRh(CO)4]. Then, during the same experiment, the hydrogen partial pressure was increased (p(H2) = 4.0 MPa) and the CO partial pressure decreased (p(CO) = 1.0 MPa). A decomposition of the [Rh4(CO)12] complex took place associated with an alteration in the spectral band at 2043 cm−1 where a shoulder was formed. For the peak group analysis (PGA) a target window from 2025–2035 cm−1 was selected and an infrared spectrum with characteristic bands at ν(CO) = 2039, 2070 cm−1 could be extracted. The spectral pattern and the band positions agree to published data and the calculated spectrum via DFT methods for the mononuclear hydrido rhodium tetracarbonyl [HRh(CO)4]. Since the [HRh(CO)4] complex is of C3v symmetry three vibrations (2A1 + E) are infrared active. The small intense band for the E vibration could not be clearly assigned.
To further confirm this attribution, we conducted an FTIR-experiment for the monitoring of the decomposition of [Rh(acac)(CO)2] using deuterium instead of hydrogen as reactive gas component in the synthesis gas at the following conditions: [Rh] = 1 mM, p(CO/D2) = 5.0 MPa and ν = 30 °C.
In comparison to the reaction with hydrogen, the decomposition of [Rh(acac)(CO)2] in the presence of deuterium took significantly more time (>50 h) to reach full conversion. Interestingly, from the spectroscopic data collected under the 1:1 mixture of CO and D2 a pure component spectrum of the mononuclear deuterido complex [DRh(CO)4] (ν(CO) = 2039, 2061, 2118 cm−1) could be extracted via peak group analysis. The shift of the second A1 band to lower wavenumbers is in accordance with the H/D-Rh–CO trans-orientation in the [H(D)Rh(CO)4] complex. This is also the case for the cobalt analogues [HCo(CO)4] (ν(CO) = 2028, 2050 cm−1) and [DCo(CO)4] (ν(CO) = 2028, 2046 cm−1) which were formed for comparison from [Co2(CO)8], see Fig. 7(c) and SI-C (ESI†) for details of preparation.
 | ||
| Fig. 7 (a) PGA spectra for [HRh(CO)4] and [DRh(CO)4]; (b) calculated infrared spectra for [HRh(CO)4] and [DRh(CO)4] by DFT computations; (c) IR spectra for [HCo(CO)4] and [DCo(CO)4]. | ||
The decrease in reaction rate clearly shows that hydrogen/deuterium is taking part in the formation of [Rh4(CO)12] from [Rh(acac)(CO)2] and the spectroscopic identification of [H(D)Rh(CO)4] suggest the involvement of such a species in this process. This was also concluded from the control experiment under pure carbon monoxide for which a significant lower decomposition rate was observed.
3.5 Population of [Rh2(CO)8] at higher partial pressures of carbon monoxide
Motivated by the successful extraction of the single component spectra of [H(D)Rh(CO)4] via PGA, we attempted the IR-spectroscopic identification of [Rh2(CO)8]. Guided by the works of Marc Garland and other researchers it was to be expected that the dinuclear complex can be populated at lower temperatures and higher partial pressures of CO.29,31,32 Following the objective of this study, we performed an in situ IR experiment starting from [Rh(acac)(CO)2] ([Rh] = 1 mM) as a precursor complex under 5.0 MPa of synthesis gas (CO/H2) at ν = 30 °C. Decomposition took place towards [Rh4(CO)12] as main product complex. We stopped this reaction at ca. 50% conversion. Then, the gas atmosphere was exchanged to pure carbon monoxide p(CO) = 1.0 MPa. With intervals of ca. 1 h, the partial pressure of CO was increased to 3.0 and then 5.0 MPa. Infrared spectra were recorded throughout this process (Fig. 8(a)).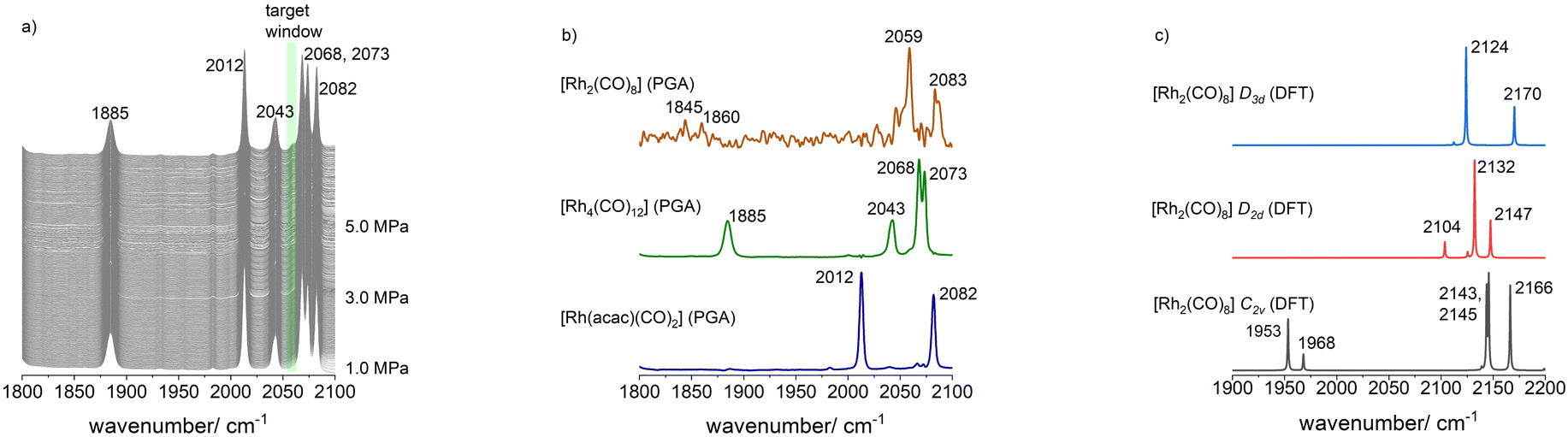 | ||
| Fig. 8 (a) In situ FTIR spectra collected during the CO partial pressure variation (1.0–5.0 MPa) to populate [Rh2(CO)8]. Conditions: [Rh] = 1 mM, ν = 30 °C, solvent: n-dodecane; (b) PGA spectra for [Rh2(CO)8], [Rh4(CO)12] and [Rh(acac)(CO)2] based on the entire IR-spectroscopic data including formation of [Rh4(CO)12] from [Rh(acac)(CO)2]; (c) calculated vibrational spectra for isomeric forms of [Rh2(CO)8] via DFT methods. | ||
In the IR-spectroscopic data an increase of a band feature at around 2060 cm−1 could be noticed with increasing CO pressures. The peak group analysis with a selected target window between 2053–2062 cm−1 gave a spectrum with bands at ν(CO) = 1845, 1860, 2059, 2083 cm−1 (Fig. 8(b)). Even though the signal-to-noise ratio of the obtained spectrum is quite low, the band features are in some accordance with the spectrum of [Rh2(CO)4(μ-CO)2] (C2v). Other possible stereo isomers can be ruled out since other spectral patterns would result (Fig. 8(c)). In addition, [Rh2(CO)4(μ-CO)2] (C2v) is energetically favoured.32 Probably, the dinuclear complex can be more populated at CO pressures >5.0 MPa and temperatures <30 °C.
3.6 Formation of acyl complexes with ethene as alkene of the type [CH3CH2C(O)Rh(CO)4], [CH3CH2C(O)Rh(CO)3(π-C2H4)]
Hydroformylation of ethene yielding propanal, which can be hydrogenated to 1-propanol is of academic and industrial interest.34,35 Ethene (C2H4) as least sterically demanding alkene has a few peculiarities with respect to the formation of unmodified acyl rhodium carbonyl complexes.36–38 Depending on the experimental design, setup and conditions its usage with a P-ligand free rhodium system such as [Rh4(CO)12] and [Rh(acac)(CO)2] allows for the detection of acyl complexes of the type [CH3CH2C(O)Rh(CO)4] and [CH3CH2C(O)Rh(CO)3(π-C2H4)]. There are even reports on the formation of [HRh(CO)3(π-C2H4)].30 We were interested to monitor the treatment of unmodified rhodium precatalysts ([Rh4(CO)12], [Rh(acac)(CO)2]) with a gas mixture of synthesis gas and ethene at the following conditions: [Rh] = 1 mM, p(CO/H2) = 2.0 MPa, p(C2H4) = 1.0 MPa, ν = 30 °C.In the first experiment [Rh4(CO)12] was formed in situ starting from [Rh(acac)(CO)2] under synthesis gas. Thus, acetylacetone is part of the reaction mixture. Starting from the synthesis gas atmosphere, ethene was added and the reaction was monitored by in situ FTIR-spectroscopy.
While the intensities of the vibrational bands for [Rh4(CO)12] (ν(CO) = 1883, 2042, 2068, 2073 cm−1) were decreasing, new bands at ν(CO) = 1695, 2015, 2039, 2089, 2111 cm−1 were increasing (Fig. 9). The results of the peak group analysis of the IR-spectroscopic data are presented in Fig. 10. Besides the pure component spectrum for the starting complex [Rh4(CO)12], two spectra attributable to acyl complexes could be extracted. The spectrum with vibrational bands at ν(CO) = 1965, 2020, 2037, 2065, 2111 cm−1 can be assigned to [CH3CH2C(O)Rh(CO)4] whereas the spectrum with bands at ν(CO) = 1695, 2015, 2039, 2089 cm−1 to [CH3CH2C(O)Rh(CO)3(π-C2H4)].36–38 The molar fraction of the acyl tricarbonyl complex with additional ethene coordination appears to be higher than the acyl tetracarbonyl complex. Under the applied conditions a quasi-stationary plateau was reached after 24 h during which the [Rh4(CO)12] was not completely converted.
 | ||
| Fig. 9 In situ FTIR spectra collected during the treatment of [Rh4(CO)12], preformed from [Rh(acac)(CO)2], with ethene in the presence of synthesis gas. Conditions: [Rh] = 1 mM, p(CO/H2) = 2.0 MPa, p(C2H4) = 1.0 MPa, ν = 30 °C, solvent: n-dodecane. | ||
 | ||
| Fig. 10 Concentration profiles (a) and pure component spectra (b) extracted from a peak group analysis on the in situ FTIR data shown in Fig. 9; (c) calculated spectra of acyl complexes via DFT methods. | ||
A follow-up experiment was conducted with [Rh(acac)(CO)2] as starting material in which synthesis gas and ethene were added together to start the reaction without the preformation to [Rh4(CO)12] under pure synthesis gas. The reaction conditions were otherwise identical: [Rh] = 1 mM, p(CO/H2) = 2.0 MPa, p(C2H4) = 1.0 MPa, ν = 30 °C Within 12 h only a small degree of conversion (ca. 12%) towards a mixture of the acyl complexes ([CH3CH2C(O)Rh(CO)4], [CH3CH2C(O)Rh(CO)3(π-C2H4)]) was noticed (SI-C, ESI†). No, significant amount of [Rh4(CO)12] was detected. Then, the synthesis gas pressure was increased to 4.0 MPa. The formation of the acyl complexes increased steadily during >48 h (SI-C, Fig. SI-8, ESI†). The peak group analysis of the IR-data of the second step gave PGA spectra for [Rh(acac)(CO)2] and an averaged spectrum for an equilibrium mixture of both acyl complexes. The relative mole fraction of the tetra carbonyl complex [CH3CH2C(O)Rh(CO)4] was higher than the tricarbonyl complex [CH3CH2C(O)Rh(CO)3(π-C2H4)] compared to the experiment at 2.0 MPa CO/H2 starting from [Rh4(CO)12]. The reason is obviously the higher partial pressure of carbon monoxide.
3.7 Hydroformylation of neohexene with [Rh(acac)(CO)2] as precatalyst. Formation of [(CH3)3CCH2CH2C(O)Rh(CO)4]
The sterically demanding neohexene (3,3-dimehyl-1-butene) is an interesting model substrate in the class of terminal alkenes because of its incapability for double bond isomerization (Scheme 2). It is often used in in situ spectroscopic and kinetic studies.13,39–44 We performed a hydroformylation experiment with [Rh(acac)(CO)2] as a precatalyst at elevated temperatures to characterize the reaction system by FTIR-spectroscopy. The reaction was conducted at the following conditions: [Rh] = 1 mM, [alkene] = 1 M; p(CO/H2) = 2.0 MPa, ν = 90 °C, solvent: cyclohexane. | ||
| Scheme 2 Hydroformylation of neohexene. | ||
Complete conversion was reached after ca. 180 min with the exclusive formation of aldehyde products (Fig. 11). The n-regioselectivity towards 4,4-dimethyl-pentanal was 0.95. The concentration vs. time data for the product (n + iso) showed a profile typical for saturation kinetics (V = Vsat[S]/(Km + [S])). This means a transition from a 0th order with respect to the substrate concentration [S] at larger initial substrate concentrations towards a 1st order at low substrate concentrations (at higher conversions).13
 | ||
| Fig. 11 Hydroformylation of neohexene. Concentration-time data for the product aldehyde formation (n + iso). Fitted numerical integration/regression data based on the rate equation: V = Vsat[S]/(Km + [S]). Conditions: [Rh] = 1 mM, [alkene] = 1 M, ν = 90 °C, p(Ar) = 0.5 MPa, p(CO/H2) = 2.0 MPa, t = 6 h, solvent: cyclohexane. | ||
Regarding the rhodium carbonyl complexes within the first 15 min, it was observed that [Rh(acac)(CO)2] was partly converted to [Rh4(CO)12] and that subsequently additional bands in the infrared spectra appeared (Fig. 12(a)). By means of the peak group analysis a corresponding pure component spectrum with vibrational bands at ν(CO) = 1969, 2020, 2037, 2110 cm−1 was obtained, which is attributed to the acyl rhodium tetracarbonyl complex [(CH3)3CCH2CH2C(O)Rh(CO)4] (Fig. 13).44,45 The acyl complex was not populated to significant molar fractions with respect to the rhodium mass balance. Neohexene was consumed quite rapidly during the batch reaction under the applied conditions at elevated temperatures. Larger alkene concentrations or [alkene]/[Rh] ratios are needed to populate higher molar fractions of the acyl complex. In course of the reaction [Rh6(CO)16] is generated as dominant rhodium carbonyl complex besides lower amount of [Rh(acac)(CO)2] (Fig. 12(b)).
 | ||
| Fig. 12 In situ FTIR spectra collected during the hydroformylation of neohexene covering the first 15 min (a) and the entire conversion range (b). Conditions: [Rh] = 1 mM, [alkene] = 1 M, ν = 90 °C, p(Ar) = 0.5 MPa, p(CO/H2) = 2.0 MPa, solvent: cyclohexane. | ||
 | ||
| Fig. 13 (a) Extracted pure component IR spectra via PGA based on the spectra collected during the first 15 min of the hydroformylation of neohexene; (b) calculated vibrational spectra based on DFT calculations. | ||
4. Conclusions
Investigations on the composition of rhodium complexes with [Rh(acac)(CO)2] as a precursor complex in the absence of phosphorus organic ligands has been performed using in situ FTIR-spectroscopy under conditions relevant for alkene hydroformylation. Pure component spectra were extracted from infrared spectroscopic data sets using the peak group analysis (PGA). The assignment of the infrared spectra to molecular structures of rhodium carbonyl complexes was facilitated by DFT calculations.The observable decomposition products from [Rh(acac)(CO)2] treated with synthesis gas (CO/H2) are [Rh4(CO)12] and [Rh6(CO)16]. The intermediate [Rh4(CO)12] decomposes rapidly to [Rh6(CO)16] at higher temperatures and lower partial pressures of carbon monoxide. It is assumed that in the entire reaction sequence from [Rh(acac)(CO)2] to [Rh6(CO)16] also the mononuclear hydrido rhodium tetracarbonyl complex [HRh(CO)4] and the dinuclear complex [Rh2(CO)8] are involved. Their pure component spectra can be obtained by PGA from infrared spectroscopic data collected at lower temperatures and adjusted partial pressures of hydrogen and carbon monoxide. These species are obviously present only in trace amounts.
Acetylacetone is formed in stoichiometric amounts during the treatment of [Rh(acac)(CO)2] with synthesis gas. The decomposition rate of [Rh(acac)(CO)2] towards the polynuclear complexes [Rh4(CO)12] and [Rh6(CO)16] is hampered by an excess of acetylacetone.
In the presence of alkenes as substrate and synthesis gas mononuclear acyl rhodium complexes are formed. With neohexene an acyl complex of the type [(CH3)3CCH2CH2C(O)Rh(CO)4] was detected. Using ethene besides the acyl rhodium tetracarbonyl complex also a tricarbonyl complex of the type [CH3CH2C(O)Rh(CO)3(π-C2H4)] with an additional ethene coordination is generated.
The present study has shown that the combination of in situ FTIR spectroscopy and chemometric tools based on MCR techniques is powerful to detect catalyst complexes even at low concentrations. With respect to alkene hydroformylation with rhodium catalysts modified by phosphorus organic ligands (e.g. phosphites, phosphines) this approach could be utilized also in the process monitoring for the detection of critical components such as [Rh4(CO)12] and especially [Rh6(CO)12] which indicates catalyst decomposition due to degradation of the P-ligands.
Data availability
The data supporting the results of this research study is given as figures/tables of spectra and concentration-time-data in the main manuscript and in the (ESI†). Original and processed data available in specific data formats can be provided on request from the corresponding author.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the state of Mecklenburg–Western Pomerania for partly funding this work. Chunhong Wei is appreciative of her funding within the Research Unit FOR5538 of the German Research Foundation (DFG, 501735683).References
-
R. Franke and A. Börner, Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2016 Search PubMed
.
- R. Franke, D. Selent and A. Börner, Applied Hydroformylation. Chem. Rev., 2012, 112, 5675–5732 CAS
-
Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen, C. Claver, Kluwer, Dordrecht, 2002 Search PubMed
-
Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Wiley-VCH, Weinheim, 3rd edn, 2018 Search PubMed
-
P. W. N. M. van Leeuwen and J. C. Chadwick, Homogeneous Catalysts, Wiley-VCH, Weinheim, 2011 Search PubMed
- P. W. N. M. van Leeuwen, Appl. Catal., A, 2001, 212, 61–81 CrossRef CAS
- R. C. How, P. Dingwall, R. T. Hembre, J. A. Ponasik, G. S. Tolleson and M. L. Clarke, Mol. Catal., 2017, 434, 116–122 CrossRef CAS
- K. Köhnke, N. Wessel, J. Esteban, J. Jin, A. J. Vorholt and W. Leitner, Green Chem., 2022, 24, 1951–1972 RSC
- B. Zhang, H. Jiao, D. Michalik, S. Kloß, L. M. Deter, D. Selent, A. Spannenberg, R. Franke and A. Börner, ACS Catal., 2016, 6, 7554–7565 CrossRef CAS
- S. Kloß, D. Selent, A. Spannenberg, R. Franke, A. Börner and M. Sharif, Catalysts, 2019, 9, 1036 CrossRef
- M. Gerlach, F. Jameel, A. Seidel-Morgenstern, M. Stein and C. Hamel, Catal. Sci. Technol., 2023, 13, 1788–1801 RSC
-
Mechanisms in Homogeneous Catalysis, ed. B. Heaton, Wiley-VCH, Weinheim, 2005 Search PubMed
-
D. Selent and D. Heller, In Situ Techniques for Homogeneous Catalysis, in Catalysis: From Principles to Applications, ed. M. Beller, A. Renken, R. A. van Santen, Wiley-VCH, Weinheim, 2012, pp. 465–490 Search PubMed
- O. Diebolt, P. W. N. M. van Leeuwen and P. C. J. Kamer, ACS Catal., 2012, 2, 2357–2370 CrossRef CAS
- M. Garland, C. Li and L. Guo, ACS Catal., 2012, 2, 2327–2334 CrossRef CAS
- C. Kubis, M. König, B. N. Leidecker, D. Selent, H. Schröder, M. Sawall, W. Baumann, A. Spannenberg, A. Brächer, K. Neymeyr, R. Franke and A. Börner, ACS Catal., 2023, 13, 5245–5263 CrossRef CAS
- C. Kubis, D. Selent, M. Sawall, R. Ludwig, K. Neymeyr, W. Baumann, R. Franke and A. Börner, Chem. – Eur. J., 2012, 18, 8780–8794 CrossRef CAS PubMed
- J. M. Dreimann, E. Kohls, H. F. W. Warmeling, M. Stein, L. F. Guo, M. Garland, T. N. Dinh and A. J. Vorholt, ACS Catal., 2019, 9, 4308–4319 CrossRef CAS
-
M. Garland in Mechanisms in Homogeneous Catalysis, ed. B. Heaton, Wiley-VCH, Weinheim, 2005, pp. 151–193 Search PubMed
- M. Garland and C. Li, Top. Catal., 2009, 52, 1334–1341 CrossRef CAS
- M. Garland, Catal. Today, 2010, 155, 266–270 CrossRef CAS
- K. Neymeyr, M. Sawall and D. Hess, J. Chemom., 2010, 24, 67–74 CrossRef CAS
- M. Sawall, A. Börner, C. Kubis, D. Selent, R. Ludwig, K. Neymeyr and J. Chemom, J. Chemom., 2012, 26, 538–548 CrossRef CAS
- M. Sawall, C. Kubis, E. Barsch, D. Selent, A. Börner and K. Neymeyr, J. Iran. Chem. Soc., 2016, 13, 191–205 CrossRef CAS
- H. Schröder, M. Sawall, C. Kubis, A. Jürß, D. Selent, A. Brächer, A. Börner, R. Franke and K. Neymeyr, Chemom. Intell. Lab. Syst., 2017, 163, 55–63 CrossRef
- M. Sawall, C. Kubis, B. Leidecker, L. Prestin, T. Andersons, M. Beesea,J. Hellwig, R. Franke, A. Börner and K. Neymeyr, An Automated Peak Group Analysis for Vibrational Spectra Analysis, Technical Report, University of Rostock, 2024.
- A. D. Allian, Y. Wang, M. Saeys, G. M. Kuramshina and M. Garland, Vib. Spectrosc., 2006, 41, 101–111 CrossRef CAS
- C. Li, E. Widjaja, W. Chew and M. Garland, Angew. Chem., Int. Ed., 2002, 41, 3785–3789 CrossRef
- A. D. Allian and M. Garland, Dalton Trans., 2005, 1957–1965 RSC
- J. L. Vidal and W. E. Walker, Inorg. Chem., 1981, 20, 249–254 CrossRef CAS
- L. A. Hanlan and G. A. Ozin, J. Am. Chem. Soc., 1974, 96, 6324–6329 CrossRef CAS
- X. Feng, C. Xie, Z. Liu, Y. Xie, R. B. King and H. F. Schaefer III, Dalton Trans., 2009, 2599–2608 RSC
- C. Cesari, J.-H. Shon, S. Zacchini and L. A. Berben, Chem. Soc. Rev., 2021, 50, 9503–9539 RSC
- H. Tricas, O. Diebolt and P. W. N. M. van Leeuwen, J. Catal., 2013, 298, 198–205 CrossRef CAS
- O. Diebolt, H. Tricas, Z. Freixa and P. W. N. M. van Leeuwen, ACS Catal., 2013, 3, 128–137 CrossRef CAS
- G. Liu and M. Garland, J. Organomet. Chem., 2000, 613, 124–127 CrossRef CAS
- J. Zhang, M. Poliakoff and M. W. George, Organometallics, 2003, 22, 1612–1618 CrossRef CAS
- C. Li, L. Guo and M. Garland, Organometallics, 2004, 23, 2201–2204 CrossRef CAS
- C. Kubis, R. Ludwig, M. Sawall, K. Neymeyr, A. Börner, K.-D. Wiese, D. Hess, R. Franke and D. Selent, ChemCatChem, 2010, 2, 287–295 CrossRef CAS
- C. Kubis, M. Sawall, A. Block, K. Neymeyr, R. Ludwig, A. Börner and D. Selent, Chem. – Eur. J., 2014, 20, 11921–11931 CrossRef CAS PubMed
- M. Garland and P. Pino, Organometallics, 1991, 10, 1693–1704 CrossRef CAS
- B. Moasser, W. L. Gladfelter and D. C. Roe, Organometallics, 1995, 14, 3832–3838 CrossRef CAS
- C. Li, E. Widjaja and M. Garland, J. Am. Chem. Soc., 2003, 125, 5540–5548 CrossRef CAS PubMed
- C. Li, E. Widjaja and M. Garland, J. Catal., 2003, 213, 126–134 CrossRef CAS
- G. Liu, R. Volken and M. Garland, Organometallics, 1999, 18, 3429–3436 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02288b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |