DOI:
10.1039/D4NJ01774A
(Paper)
New J. Chem., 2024,
48, 17947-17952
New approach for predicting crystal densities of energetic materials†‡
Received
17th April 2024
, Accepted 19th September 2024
First published on 27th September 2024
Abstract
Predicting crystal density of energetic materials accurately critically impacts assessment of their detonation potential, with higher density directly improving energetic performance. This new approach will help in density prediction to enable accelerated computational material discovery. By refining Politzer's approach using rigorously validated B3LYP quantum calculations and Multiwfn's precise structural elucidation, our optimized protocol substantially enhances predictive accuracy, marking a significant leap over traditional approaches. This streamlined and efficient framework spearheads high-fidelity virtual screening to transformatively expedite the development of novel, potent energetic candidates. Moreover, this research accentuates the decisive impact of refined computational techniques on elucidating structure–property relationships early-on, paving the way to accelerated development of promising energetic materials. Future work may concentrate on expanding training datasets to further improve model robustness and computational efficiency. Overall, this transformative density prediction workflow delivers a more precise approach to fundamentally advanced energetic material innovation.
The pursuit of novel energetic materials is an intricate process encompassing design, synthesis, characterization, testing and scale-up, amounting to a costly R&D endeavour. Heightened costs stem from the advanced techniques required to safely design powerful yet stable explosives, hindering rapid advancements. Therefore, the scientific community has increasingly embraced computationally efficient quantum mechanics (QM) protocols offering expedited and economic performance estimates to help accelerate development while reducing dangers with a focus on structure–property evaluations. QM methods particularly excel at predicting critical detonation attributes through virtual screening, minimizing risky experiments. Specifically, QM methods can efficiently predict key detonation and propulsion figures-of-merit like density, enthalpy of formation, detonation pressures and velocities that enable preliminary screening.1–3 When applied to density predictions, the computed density of prospective materials strongly informs detonation performance assessments, underpinning shock wave propagation and blast efficiency. Structure–density relationships quantify how intermolecular packing impacts bulk density values, although traditional divisions of molecular weight by molar volume overlook these interactions. Pioneering works derived structure-density links by correlating electrostatic potentials with statistical variance measures.4,5 Extending this foundation through recent sophisticated improvements, approaches by Willer and coworkers,6 Pan-Lee,7 Rice-Byrd8 and Kim and coworkers,9 Keshavarz and coworkers10 and Klapötke and coworkers11 have been introduced. Later, Ghule and coworkers compared the experimental densities with the densities obtained from these approaches12 and found that the Politzer and Rice approaches give calculated values for density which are closer to the experimental densities. However, opportunities for enhanced predictive precision remain. This new approach suggests a refined density prediction model delivering noticeably improved accuracy through targeted reparameterization of Politzer's approach (Fig. 1). By incorporating contemporary density functional theory and multi-scale simulations, our density estimates excel preceding methods in reliability. The optimized protocol pioneers a simulation toolkit to further innovation in high-energy materials discovery.
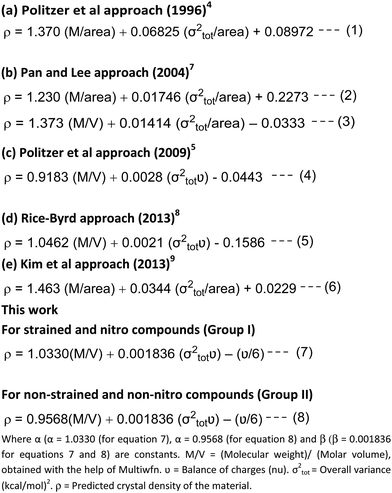 |
| | Fig. 1 Recently developed methods and the present work.4,5,7–9 | |
Inspired by these approaches, we have further extended Politzer's approaches4,5 for predicting crystal density more precisely using the B3LYP/6-311++G(d,p) level of DFT13 selected for its accurate portrayal of intermolecular interactions pivotal for density models (eqn (7) and (8)). Optimization and frequency analysis were pursued using the Gaussian 03 suite to ensure that the obtained structures represent true local minima.14 Subsequently, Multiwfn enabled rigorous quantitative elucidation of electron densities and molecular surfaces to derive electrostatic potentials (ESPs) for density prediction.15
Molecules were strategically selected based on the availability of reliable crystal density characterizations in the literature. Most studied compounds hold distinction in energetic materials, supplemented by recent publications.16–31 For optimal density predictions, these are classified into two groups – strained or nitro-containing compounds (Group I, Schemes 1 and 2), and non-strained aliphatic/aromatic hydrocarbons (Group II, Scheme 3) based on their distinct intermolecular interaction profiles. These bespoke classification schemes account for variances in packing interactions, working to improve predictive accuracy.
 |
| | Scheme 1 Group I (crystal density predicted using eqn (7)): (a) polycyclic strained/nitro compounds. | |
 |
| | Scheme 2 Group I (crystal density predicted using eqn (7)): (b) polynitro compounds. | |
 |
| | Scheme 3 Group II (crystal density predicted using eqn (8)): hydrocarbon and compounds without nitro group. | |
Crystal densities of compounds 1–24 (Schemes 1 and 2) were computed via eqn (7), selected specifically for strained and nitro-containing systems where intermolecular interactions substantially influence density. For non-strained aliphatic/aromatic hydrocarbons (compounds 25–36, Scheme 3), eqn (8) was pursued to account for their distinct packing effects.
For benchmarking, densities were additionally predicted using the preceding eqn (1), (2) and (4)–(6) established in the literature (Fig. 1).4–8 These comparative results are tabulated in Tables 1 and 2 and depicted in Fig. 2–5.
Table 1 Comparison of crystal densities of compounds 1–24 (Group I) determined by our approach and using eqn (1), (2) and (4)–(6)
| Compound |
υ
|
M/Vb |
υ × σtot2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
β × υ × σtot2d |
ρ
|
ρ
(Lit.) |
Δ
|
ρ
(eqn (1)) |
ρ
(eqn (2)) |
ρ
(eqn (4)) |
ρ
(eqn (5)) |
ρ
(eqn (6)) |
|
υ = balance of charges (nu).
M/V = (molecular weight)/(molar volume), obtained with the help of Multiwfn.
Product of σtot2 (overall variance kcal mol−2) and υ (balance of charges).
Product of σtot2 (overall variance kcal mol−2), υ (balance of charges) and β.
ρ = crystal density of the material predicted according to eqn (7).
ρ (Lit.) = experimental crystal density.
Δ = difference between experimental crystal density and crystal density predicted according to eqn (7).
ρ (eqn (1)) = predicted crystal density according to eqn (1).
ρ (eqn (2)) = predicted crystal density according to eqn (2).
ρ (eqn (4)) = predicted crystal density according to eqn (4).
ρ (eqn (5)) = predicted crystal density according to eqn (5).
ρ (eqn (6)) = predicted crystal density according to eqn (6). α = 1.0330 and β = 0.001836 are constants.
|
|
1
|
0.0584 |
1.9601 |
13.9932 |
0.025691625 |
2.0407 |
2.0400 |
0.0007 |
1.9763 |
1.8972 |
1.7948 |
1.9214 |
2.0165 |
|
2
|
0.1758 |
1.8792 |
27.6664 |
0.050795492 |
1.9626 |
1.9850 |
−0.0223 |
1.8020 |
1.7383 |
1.7588 |
1.8655 |
1.8283 |
|
3
|
0.1089 |
1.8821 |
22.0477 |
0.040479687 |
1.9665 |
1.9590 |
0.0075 |
1.9986 |
1.9194 |
1.7457 |
1.8567 |
2.0423 |
|
4
|
0.1716 |
1.7897 |
29.4705 |
0.054107856 |
1.8742 |
1.8130 |
0.0612 |
1.9036 |
1.8350 |
1.6816 |
1.7756 |
1.9416 |
|
5
|
0.0943 |
1.7773 |
20.5503 |
0.037730516 |
1.8579 |
1.8140 |
0.0439 |
1.6467 |
1.5916 |
1.6453 |
1.7439 |
1.6561 |
|
6
|
0.0302 |
2.0004 |
4.8985 |
0.008993664 |
2.0703 |
1.9790 |
0.0913 |
2.0438 |
1.9664 |
1.8063 |
1.9445 |
2.0962 |
|
7
|
0.1143 |
1.2317 |
0.7875 |
0.001445813 |
1.2547 |
1.2900 |
−0.0352 |
1.0827 |
1.1159 |
1.0889 |
1.1316 |
1.0807 |
|
8
|
0.0092 |
2.2766 |
1.3556 |
0.002489028 |
2.3526 |
2.4290 |
−0.0763 |
1.9046 |
1.8306 |
2.0500 |
2.2260 |
1.9381 |
|
9
|
0.1397 |
1.7276 |
25.9716 |
0.047683894 |
1.8090 |
1.8300 |
−0.0209 |
1.5716 |
1.5154 |
1.6148 |
1.7033 |
1.5682 |
|
10
|
0.1273 |
1.7291 |
27.2892 |
0.050103045 |
1.8150 |
1.8100 |
0.0050 |
1.6081 |
1.5483 |
1.6199 |
1.7076 |
1.6073 |
|
11
|
0.1340 |
1.7690 |
33.0870 |
0.060747842 |
1.8657 |
1.9100 |
−0.0442 |
1.7599 |
1.6903 |
1.6728 |
1.7616 |
1.7744 |
|
12
|
0.2085 |
1.6404 |
22.4788 |
0.041271132 |
1.7010 |
1.6500 |
0.0510 |
1.5244 |
1.4946 |
1.5250 |
1.6047 |
1.5368 |
|
13
|
0.1979 |
1.7158 |
36.2283 |
0.066515159 |
1.8059 |
1.7800 |
0.0259 |
1.6192 |
1.5732 |
1.6327 |
1.7125 |
1.6322 |
|
14
|
0.2490 |
1.5677 |
26.7377 |
0.049090527 |
1.6270 |
1.5700 |
0.0570 |
1.4166 |
1.3906 |
1.4701 |
1.5376 |
1.4153 |
|
15
|
0.1372 |
1.7310 |
34.9834 |
0.064229596 |
1.8294 |
1.8560 |
−0.0265 |
1.6215 |
1.5578 |
1.6432 |
1.7258 |
1.6194 |
|
16
|
0.1815 |
1.6904 |
84.0449 |
0.154306495 |
1.8702 |
1.8900 |
−0.0197 |
1.6294 |
1.4726 |
1.7433 |
1.7863 |
1.5468 |
|
17
|
0.2468 |
1.7185 |
63.4310 |
0.116459334 |
1.8505 |
1.8340 |
0.0165 |
1.6113 |
1.5427 |
1.7114 |
1.7725 |
1.6032 |
|
18
|
0.1399 |
1.6509 |
20.1076 |
0.036917535 |
1.7189 |
1.6970 |
0.0219 |
1.5701 |
1.5441 |
1.5280 |
1.6107 |
1.5930 |
|
19
|
0.1820 |
1.7559 |
27.2569 |
0.050043700 |
1.8335 |
1.7670 |
0.0665 |
1.6093 |
1.5629 |
1.6444 |
1.7357 |
1.6205 |
|
20
|
0.0465 |
1.9699 |
6.1808 |
0.011347894 |
2.03850 |
2.0070 |
0.0315 |
1.8477 |
1.7898 |
1.7819 |
1.9152 |
1.8864 |
|
21
|
0.0836 |
1.9774 |
13.9262 |
0.025568522 |
2.0542 |
2.0060 |
0.0482 |
1.7385 |
1.6849 |
1.8105 |
1.9394 |
1.7637 |
|
22
|
0.1792 |
1.8882 |
41.7622 |
0.076675454 |
1.9973 |
1.9990 |
−0.0016 |
1.7522 |
1.6811 |
1.8065 |
1.9045 |
1.7642 |
|
23
|
0.1434 |
1.8509 |
24.1799 |
0.044394443 |
1.9324 |
1.9020 |
0.0304 |
1.7002 |
1.6416 |
1.7230 |
1.8285 |
1.7149 |
|
24
|
0.0446 |
1.8882 |
13.8161 |
0.025366341 |
1.9684 |
1.9400 |
0.0284 |
1.7659 |
1.6807 |
1.7283 |
1.8458 |
1.7677 |
Table 2 Comparison of crystal densities of compounds 25–36 (Group II) determined by our approach and using eqn (1), (2) and (4)–(6)
| Compound |
υ
|
M/Vb |
υ × σtot2c |
β × υ × σtot2d |
ρ
|
ρ (Lit.)f |
Δ
|
ρ
(eqn (1)) |
ρ
(eqn (2)) |
ρ
(eqn (4)) |
ρ
(eqn (5)) |
ρ
(eqn (6)) |
|
υ = balance of charges (nu).
M/V = (molecular weight)/(molar volume), obtained with the help of Multiwfn.
Product of σtot2 (overall variance kcal mol−2) and υ (balance of charges).
Product of σtot2 (overall variance kcal mol−2), υ (balance of charges) and β.
ρ = crystal density of the material predicted according to eqn (8).
ρ (Lit.) = experimental crystal density.
Δ = difference between experimental crystal density and crystal density predicted according to eqn (7).
ρ (eqn (1)) = predicted crystal density according to eqn (1).
ρ (eqn (2)) = predicted crystal density according to eqn (2).
ρ (eqn (4)) = predicted crystal density according to eqn (4).
ρ (eqn (5)) = predicted crystal density according to eqn (5).
ρ (eqn (6)) = predicted crystal density according to eqn (6). α = 0.9568 and β = 0.001836 are constants.
|
|
25
|
0.0554 |
0.6218 |
0.3596 |
0.000660 |
0.5863 |
0.5170 |
0.0693 |
0.4795 |
0.5595 |
0.5277 |
0.4927 |
0.4235 |
|
26
|
0.0210 |
1.9500 |
0.5871 |
0.001078 |
1.8633 |
1.8230 |
0.0403 |
1.6517 |
1.6217 |
1.7480 |
1.8827 |
1.6839 |
|
27
|
0.0935 |
0.7484 |
0.4235 |
0.000778 |
0.7012 |
0.6900 |
0.0072 |
0.5855 |
0.6659 |
0.6441 |
0.6252 |
0.5466 |
|
28
|
0.1047 |
1.2332 |
32.9179 |
0.060437 |
1.2229 |
1.2300 |
−0.0071 |
1.2687 |
1.1096 |
1.1803 |
1.2007 |
1.1272 |
|
29
|
0.2456 |
1.2545 |
44.8608 |
0.082364 |
1.2417 |
1.2660 |
−0.0242 |
1.1445 |
1.0410 |
1.2333 |
1.2480 |
1.0322 |
|
30
|
0.2483 |
1.1979 |
87.9804 |
0.161532 |
1.2662 |
1.2600 |
0.0062 |
1.3910 |
1.0509 |
1.3020 |
1.2794 |
1.1098 |
|
31
|
0.2380 |
1.0846 |
8.9239 |
0.016384 |
1.0144 |
1.0160 |
−0.0015 |
0.9634 |
0.9906 |
0.9766 |
0.9948 |
0.9374 |
|
32
|
0.1358 |
0.9770 |
0.4493 |
0.000825 |
0.9129 |
0.8560 |
0.0569 |
0.8854 |
0.9400 |
0.8541 |
0.8644 |
0.8711 |
|
33
|
0.1911 |
1.7574 |
7.6850 |
0.014110 |
1.6637 |
1.6540 |
0.0097 |
1.5236 |
1.5034 |
1.5910 |
1.6961 |
1.5443 |
|
34
|
0.1966 |
1.3230 |
8.0502 |
0.014780 |
1.2478 |
1.2250 |
0.0228 |
1.1697 |
1.1810 |
1.1931 |
1.2424 |
1.1622 |
|
35
|
0.1929 |
1.2031 |
27.9871 |
0.051384 |
1.1703 |
1.1550 |
0.0153 |
1.1375 |
1.1005 |
1.1388 |
1.1588 |
1.0825 |
|
36
|
0.2494 |
1.3131 |
40.9579 |
0.075199 |
1.2900 |
1.3220 |
−0.0320 |
1.2406 |
1.2017 |
1.2762 |
1.3011 |
1.2002 |
 |
| | Fig. 2 Plots of predicted crystal densities using our approach (eqn (7)) versus literature value calculated using eqn (1), (2) and (4)–(6). | |
 |
| | Fig. 3 Plots of predicted crystal densities using our approach (eqn (8)) versus literature value calculated using eqn (1), (2) and (4)–(6). | |
 |
| | Fig. 4 Difference in crystal densities of compounds 1–24 determined using eqn (1), (2) and (4)–(7) and experimental values. | |
 |
| | Fig. 5 Difference in crystal densities of compounds 25–36 determined using eqn (1), (2), (4)–(6) and (8) and experimental values. | |
A good comparison of the results produced by eqn (1), (2), and (4)–(8) suggests that by refining Politzer's approach using rigorously validated B3LYP quantum calculations and Multiwfn's precise structural elucidation, our protocol substantially enhances predictive accuracy involving the individual contributions of ν/6, marking a significant leap over traditional approaches as listed in Tables 1 and 2, Fig. 2–5 and Tables S15 and S16 (ESI‡).
Conclusions
This study revealed that the predictions of the crystal density of organic compounds could be significantly improved by strategically modifying Politzer's approach. Compounds were categorized into two classes (Group I and Group II) based on the presence/absence of strain and nitro functionalities to optimize model parameters for these distinct chemical spaces. The universal constants α, β, and γ were replaced by 1.0330, 0.001836, and ν/6 specifically for Group I compounds, whereas values of 0.9568, 0.001836, and ν/6 were utilized for Group II systems. Notably, substituting γ with ν/6 to capture intrinsic charge differences between materials delivered superior density predictions over all preceding methods. This work not only enhances predictive accuracy, but pioneers a broadly applicable platform to accelerate the discovery of novel, high-performance energetic candidates.
S. L. investigation, methodology, conceptualization and manuscript writing. H. G. manuscript writing – review and editing. J. M. S. conceptualization, manuscript writing – review and editing, supervision.
Data availability
All data relevant to the work described here are available in the ESI,‡ or from the current literature.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We are grateful for the support from the Fluorine-19 fund.
Notes and references
-
J. P. Agrawal and R. D. Hodgson, Organic chemistry of explosives, John Wiley & Sons, Ltd, Chichester, 2007, p. 243 Search PubMed.
-
(a)
T. M. Klapötke, Chemistry of high-energy materials, De Gruyter, Berlin, 2nd edn, 2012 CrossRef;
(b) S. Lal, H. Gao and J. M. Shreeve, Design and computational insight into two novel CL-20 analogues, BNMTNIW and BNIMTNIW: high performance energetic materials, New J. Chem., 2022, 46, 16693–16701 RSC.
-
(a) H. Gao and J. M. Shreeve, Azole-Based Energetic Salts, Chem. Rev., 2011, 111, 7377–7436 CrossRef CAS;
(b) L. Mallick, S. Lal, S. Reshmi, I. N. N. Namboothiri, A. Chowdhury and N. Kumbhakarna, Theoretical studies on the propulsive and explosive performance of strained polycyclic cage compounds, New J. Chem., 2017, 41, 920–930 RSC.
- J. S. Murray, T. Brinck and P. Politzer, Relationships of molecular surface electrostatic potentials to some macroscopic properties, Chem. Phys., 1996, 204, 289–299 CrossRef CAS.
- P. Politzer, J. Martinez, J. S. Murray, M. C. Concha and A. Toro-Labbe, An electrostatic interaction correction for improved crystal density prediction, Mol. Phys., 2009, 107, 2095–2101 CrossRef CAS.
- R. L. Willer, Calculation of the density and detonation properties of C, H, N, O and F compounds: use in the design and synthesis of new energetic materials, J. Mex. Chem. Soc., 2009, 53, 108–119 CAS.
- J.-F. Pan and Y.-W. Lee, Crystal density prediction for cyclic and cage compounds, Phys. Chem. Chem. Phys., 2004, 6, 471–473 RSC.
- B. M. Rice and E. F. Byrd, Evaluation of electrostatic descriptors for predicting crystalline density, J. Comput. Chem., 2013, 34, 2146–2151 CrossRef CAS PubMed.
- C. K. Kim, S. G. Cho, C. K. Kim, M.-R. Kim and H. W. Lee, Prediction of physicochemical properties of organic molecules using semi-empirical methods, Bull. Korean Chem. Soc., 2013, 34, 1043–1046 CrossRef CAS.
- M. H. Keshavarz, H. Soury, H. Motamedoshariati and A. Dashtizadeh, Improved method for prediction of density of energetic compounds using their molecular structure, Struct. Chem., 2014, 2, 455–466 Search PubMed.
- S. Wahler and T. M. Klapötke, Research output software for energetic materials based on observational modelling 2.1 (RoseBoom2.1©), Mater. Adv., 2022, 3, 7976–7986 RSC.
-
(a) A. Nirwan, A. Devi and V. D. Ghule, Assessment of density prediction methods based on molecular surface electrostatic potential, J. Mol. Model., 2018, 24, 166 CrossRef;
(b) P. Politzer and J. S. Murray, Electrostatic potentials at the nuclei of atoms and molecules, Theor. Chem. Acc., 2021, 140, 7 Search PubMed;
(c) F. A. Bulat, A. T. Labbé, T. Brinck, J. S. Murray and P. Politzer, Quantitative analysis of molecular surfaces: areas, volumes, electrostatic potentials and average local ionization energies, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS.
-
R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. L. Caricato, X. H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision D.01 ed., Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. V. Viswanath, K. J. Venugopal, N. V. S. Rao and A. Venkataraman, An overview on importance, synthetic strategies and studies of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (HNIW), Def. Technol., 2016, 12, 401–418 CrossRef.
- V. T. Ramakrishnan, M. Vedachalam and H. Boyer, 10-Dinitro-2,6,8,12-tetraoxa 4,10-diazatetracyclo[5.5.0.05,9.03,11] dodecane, Heterocycles, 1990, 31, 179–180 Search PubMed.
- J. Zhang, T. Hou, L. Zhang and J. Luo, 2,4,4,6,8,8-Hexanitro-2,6-diazaadamantane: A High-Energy Density Compound with High Stability, Org. Lett., 2018, 20, 7172–7176 CrossRef CAS PubMed.
- T.-J. Hou, H.-W. Ruan, G.-X. Wang and J. Luo, 2,4,4,8,8-Pentanitro-2-Azaadamantane: A High-DensityEnergetic Compound, Eur. J. Org. Chem., 2017, 6957–6960 CrossRef CAS.
- K. A. Lukin, J. Li, P. E. Eaton, N. Kanomata, J. Hain, E. Punzalan and R. Gilardi, Synthesis and chemistry of 1,3,5,7-tetranitrocubane including measurement of its acidity, formation of-nitro anions, and the first preparations of pentanitrocubane and hexanitrocubane, J. Am. Chem. Soc., 1997, 119, 9591–9602 CrossRef CAS.
- K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Cubane: 50 years later, Chem. Rev., 2015, 115, 6719–6745 CrossRef CAS.
- M. Sugiyama, M. Akiyama, Y. Yonezawa1, K. Komaguchi, M. Higashi, K. Nozaki1 and T. Okazoe, Electron in a cube: synthesis and characterization of perfluorocubane as an electron acceptor, Science, 2022, 377, 756–759 CrossRef CAS.
- T. G. Archibald, R. Gilardi, K. Baum and C. George, Synthesis and X-ray crystal sructure of 1,3,3-trinitroazetidine, J. Org. Chem., 1990, 55, 2920–2924 CrossRef CAS.
- S. Lal, R. J. Staples and J. M. Shreeve, FOX-7 derived nitramines: Novel propellants and oxidizers for solid rocket propulsion, Chem. Eng. J., 2023, 468, 143737 CrossRef CAS.
- S. Lal, R. J. Staples and J. M. Shreeve, Design and synthesis of high-performance planar explosives and solid propellants with tetrazole moieties, Org. Lett., 2023, 27, 5100–5104 CrossRef.
- W. Huang, Y. Tang, G. H. Imler, D. A. Parrish and J. M. Shreeve, Nitrogen-Rich tetrazolo[1,5-b] pyridazine: promising building block for advanced energetic materials, J. Am. Chem. Soc., 2020, 142(7), 3652–3657 CrossRef CAS.
- S. Lal, R. J. Staples and J. M. Shreeve, Design and synthesis of phenylene-bridged isoxazole and tetrazole-1-ol based energetic materials of low sensitivity, Dalton Trans., 2023, 52, 3449–3457 RSC.
- Q. Yu, P. Yin, J. Zhang, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, Pushing the limits of oxygen balance in 1,3,4-oxadiazoles, J. Am. Chem. Soc., 2017, 139, 8816–8819 CrossRef CAS PubMed.
- W. Zhang, Y. Yang, Y. Wang, T. Fei, Y. Wang, C. Sun and S. Pang, Challenging the limits of the oxygen balance of a pyrazole ring, Chem. Eng. J., 2023, 451, 138609 CrossRef CAS.
- S. Lal, R. J. Staples and J. M. Shreeve, Energetic performance of trinitromethyl nitrotriazole (TNMNT) and its energetic salts, Chem. Commun., 2023, 59, 11276–11279 RSC.
- S. Lal, R. J. Staples and J. M. Shreeve, Trinitromethyl-triazolone (TNMTO): a highly dense oxidizer, Dalton Trans., 2023, 52, 12341–12346 RSC.
Footnotes |
| † Dedicated to Professor Peter Politzer for his invaluable contribution to theoretical and computational chemistry. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj01774a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Haixiang
Gao
a,
Haixiang
Gao







