DOI:
10.1039/D4NJ00824C
(Paper)
New J. Chem., 2024,
48, 8038-8054
Novel fused pyran derivatives induce apoptosis and target cell cycle progression in anticancer efficacy against multiple cell lines†
Received
20th February 2024
, Accepted 16th March 2024
First published on 19th March 2024
Abstract
Nitrogen-based heterocycles such as pyrazole, imidazole, 1,2,4-triazole, benzimidazole, and benzotriazole substituted fused pyran derivatives (6a–e, 8a–e, 10a–e, 12a–e, & 14a–e) have been synthesized and tested for their in vitro anticancer efficacies against MCF7, A549, and HCT116 cancer cell lines. Among the compounds, 6e, 14b, and 8c were identified as the most potent against MCF7, A549, and HCT116, with IC50 values of 12.46 ± 2.72 μM, 0.23 ± 0.12 μM, and 7.58 ± 1.01 μM, respectively. Further studies demonstrated that these compounds can change cellular and nuclear morphology and inhibit colony formation in the tested cancer cells. They also remarkably block/inhibit the cell cycle progression of cancer cells at various phases. DNA damage analysis and apoptosis studies revealed that these compounds have the potential to induce DNA double-strand breaks and apoptosis. In silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the potent compounds were assessed, revealing that all the compounds exhibited favorable pharmacokinetic and toxicological properties. The potent compounds identified from this study can be considered as a lead for further drug design and development.
1. Introduction
Cancer, a multifaceted and relentless disease, continues to pose a significant global health challenge, affecting millions of lives worldwide.1–3 With its diverse range of manifestations and inherent heterogeneity, cancer remains a major focus of scientific investigation, necessitating a comprehensive understanding of its underlying mechanisms and the development of effective therapeutic strategies.1–4 One of the hallmarks of cancer is uncontrolled cell growth, driven by alterations in crucial cellular pathways that regulate proliferation and apoptosis. Genetic mutations disrupt the delicate balance between cell division and cell death, leading to the uncontrolled proliferation of abnormal cells.5–7
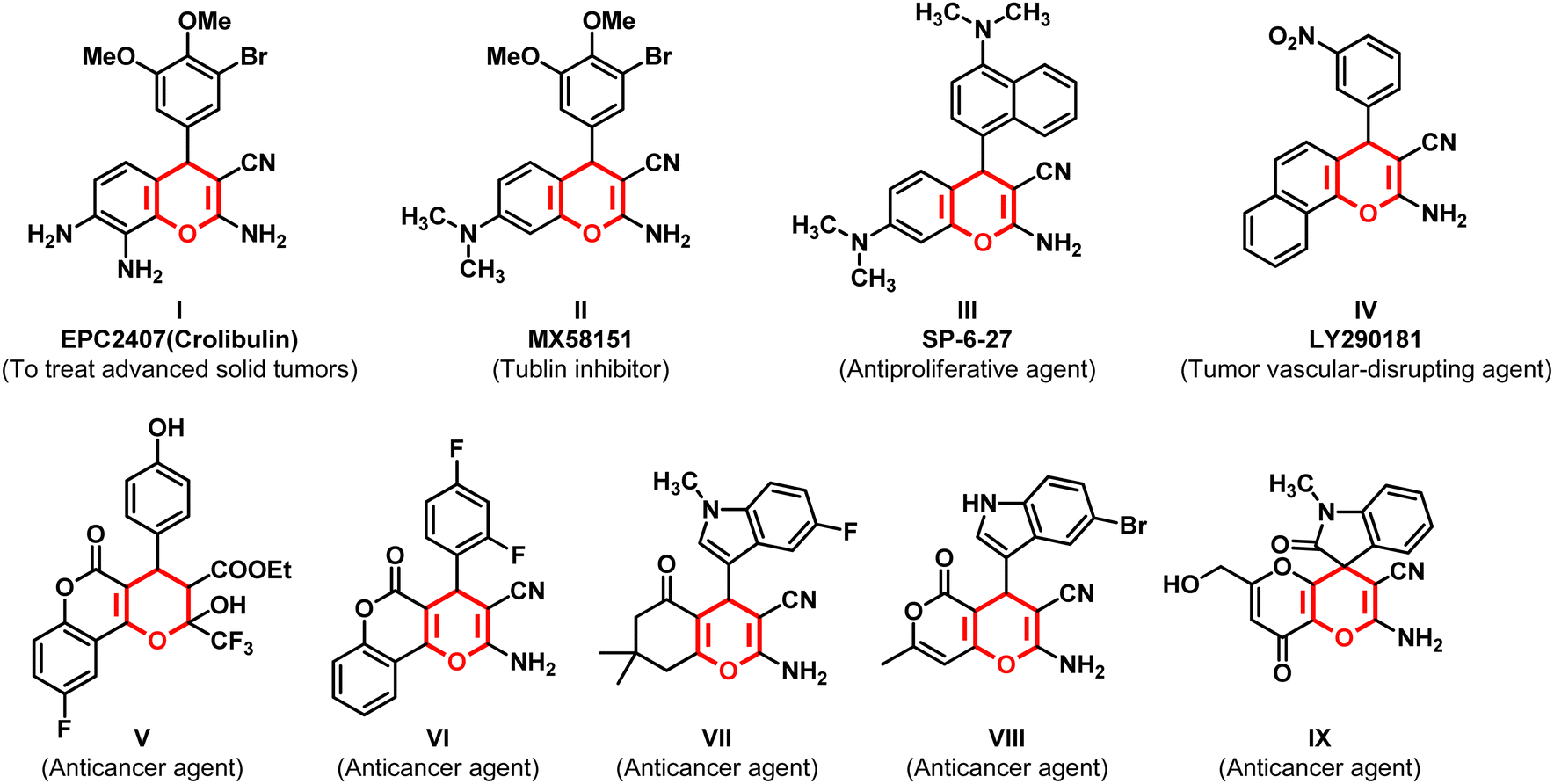
Fused pyran derivatives have emerged as promising candidates in cancer therapeutics due to their ability to target the cellular pathways, resulting in the inhibition of cancer cell growth, induction of apoptosis, and disruption of essential cellular processes that are crucial for tumor progression.8–20 These moieties have also been reported to exhibit a wide array of pharmacological activities that include antimicrobial,21–27 antitubercular,21 antioxidant,24,28 analgesic,25 antileishmanial,29 antiplatelet,30 formyl peptide receptor 1 (FPR1) antagonist,31 and anticonvulsant activities.32 In addition, they were also identified as potent selective estrogen receptor modulators (SERMs),33 inhibitors of enzymes such as acetylcholinesterase,28 monoamine oxidase,34,35 topoisomerase 2,36 c-Src kinase,37 xanthine oxidase,38 and aldehyde reductase 2 (ALR2).39 Structures and pharmacological applications of a few fused pyran derivatives are summarized in Fig. 1. Of these compounds, the chromene analog EPC2407 (Crolibulin, I) is the most promising vascular disrupting agent and apoptosis inducer for treating advanced solid tumors.10,14,40,41 In phase I clinical trials in patients with advanced thoracic and abdominal tumors, cell swelling and decreased tumor perfusion monitored by functional MRI imaging modalities 2–3 days post-treatment with crolibulin were confirmed.41 It is now under phase I/II clinical trials in combination with cisplatin for anaplastic thyroid cancer (ATC).14,40 Similarly, MX58151 (II) was reported as a caspase activator and tubulin inhibitor,15,16 and SP-6-27 (III) displayed the highest potency towards glioma, melanoma, and prostate cancer cell lines, and is known for its high antiproliferative activity.14 Another chromene analog LY290181 (IV) has been identified as a potent tumor vascular-disrupting agent and acts as an inhibitor for mitosis and microtubules.18,42,43 Likewise, fluorine-containing pyrano-chromenes (V)20 and (VI),19 indole-substituted tetrahydro-chromene (VII),14 and indole-tethered pyrano-pyrans (VIII)44 and (IX)45 have been reported as promising anticancer agents. Despite the progress in the development of chemotherapeutic agents, several new target-specific and improved anticancer agents with minimum/no side effects are essential to tackle drug resistance.46–48
 |
| | Fig. 1 Structures of potential chemotherapeutic pyran derivatives available in the clinic (I–IV) and several reported pyran derivatives as anticancer agents (V–IX). | |
According to the U.S. Food and Drug Administration (FDA) database, nearly 60% of small-molecule drugs are nitrogen-containing heterocycles.49 Incorporation of nitrogen atoms or N-based heterocycles in a pharmacophore such as a fused pyran may not only increase its water solubility but also enhance the binding to a variety of biological targets such as enzymes and receptors.49,50 The structural versatility of fused pyran derivatives enables the synthesis of compounds with tailored properties, optimizing their anticancer efficacy and minimizing off-target effects. By introducing diverse functional groups and modifying specific regions of the fused pyran scaffold, researchers have enhanced their potency and improved their pharmacokinetic profiles, allowing for improved bioavailability and target specificity.14–20 Therefore, we aimed to synthesize a series of novel fused pyran derivatives containing N-based heterocycles to examine their anticancer efficacies against different cancer cell lines, and study their mechanisms of action and in silico ADMET predictions to evaluate their drug-likeness properties.
2. Results and discussion
2.1 Chemical synthesis of pyran/chromene derivatives
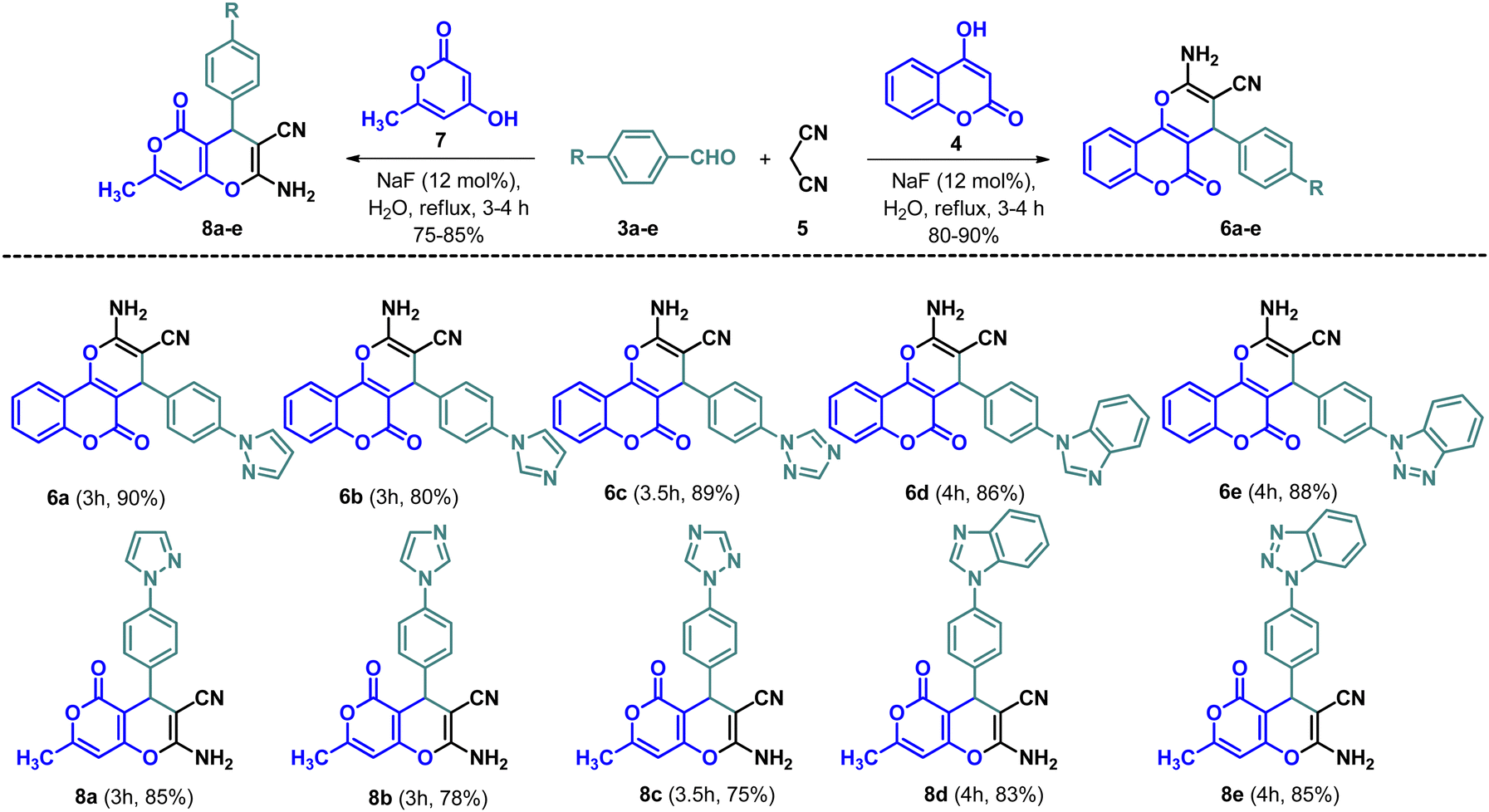
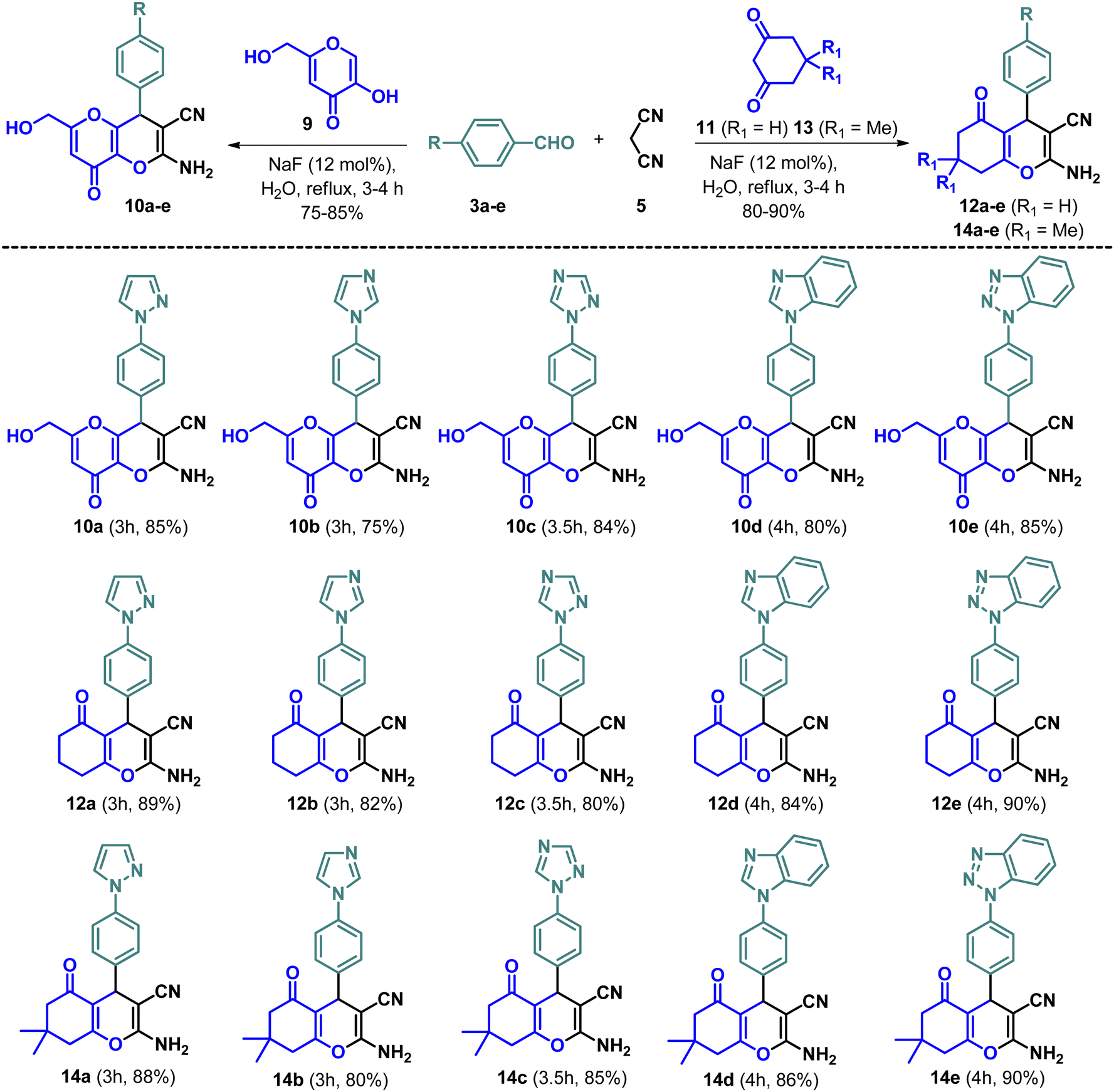
A multicomponent Knoevenagel–Michael reaction of α-naphthol/β-naphthol/4-hydroxycoumarin with aryl/heteroaryl aldehydes and malononitrile in H2O in the presence of sodium fluoride (NaF) catalyst under microwave irradiation resulted in the formation of diverse pyran/chromene derivatives.51 In continuation, we have synthesized a variety of N-based heterocycles substituted fused pyran derivatives using the strategy shown in Schemes 2 and 3. The intermediates, N-based heterocycles substituted aryl aldehydes (3a–e) were obtained by the SNAr reactions of 4-fluorobenzaldehyde (1) with secondary amines such as pyrazole (2a), imidazole (2b), 1,2,4-triazole (2c), benzimidazole (2d), benzotriazole (2e) in the presence of potassium carbonate in DMF at 130 °C (Scheme 1). The desired products (6a–e, 8a–e, 10a–e, 12a–e & 14a–e) were obtained by treating 4-hydroxycoumarin (4), 4-hydroxy-6-methyl-2H-pyran-2-one (7), 5-hydroxy-2-(hydroxymethyl)-4H-pyran-4-one (9), cyclohexane-1,3-dione (11) and 5,5-dimethylcyclohexane-1,3-dione (13) individually with N-based heterocycles substituted benzaldehydes (3a–e) and malononitrile (5) in H2O utilizing 12 mol% of NaF as a catalyst (Schemes 2 and 3). The reaction was carried out in H2O at the reflux temperature. All the compounds were obtained in 75–90% of the yields within 3–4 hours.
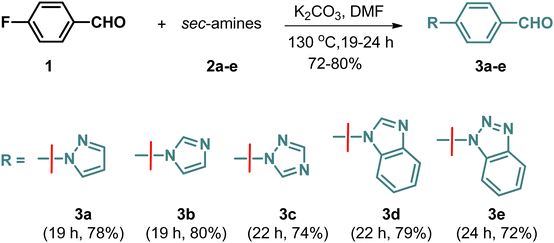 |
| | Scheme 1 Synthesis of N-based heterocycles substituted aryl aldehydes (3a–e). | |
 |
| | Scheme 2 Synthesis of novel fused pyran derivatives (6a–e & 8a–e). | |
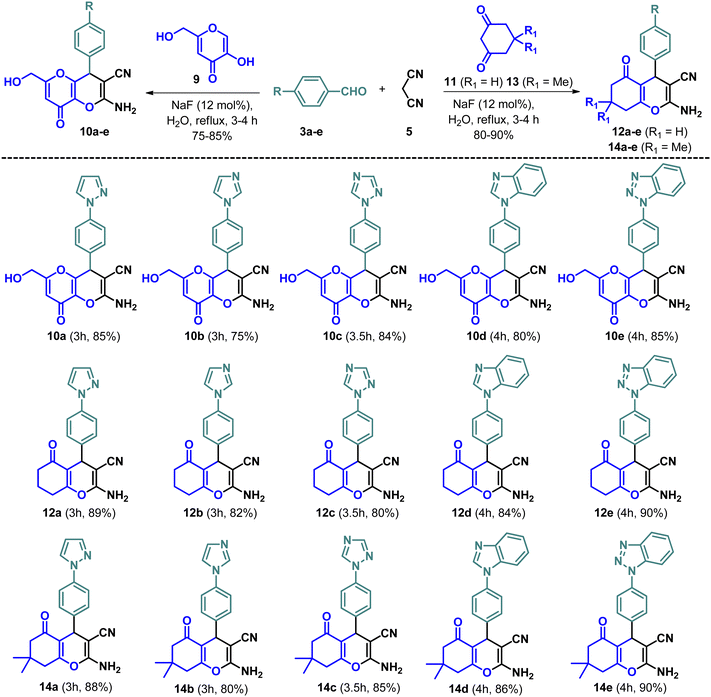 |
| | Scheme 3 Synthesis of novel fused pyran derivatives (10a–e, 12a–e & 14a–e). | |
Structures of all the target molecules were confirmed by spectral studies such as FT-IR, 1H NMR, 13C NMR, and LC-HRMS. From the 1H NMR spectra, the appearance of a singlet peak at 4.30–4.90 ppm confirms the presence of the pyran-C4 proton. Moreover, the presence of a peak at 30–39 ppm in the 13C NMR spectra corresponding to the pyran-C4 carbon provided additional evidence of the formation of the pyran ring. Furthermore, the molecular ion peak from the mass spectra also confirmed the product formation. The purity of all tested compounds was greater than 99.5% determined by LC-HRMS.
2.2 Impact of the fused pyran derivatives on the viability of cancer cells
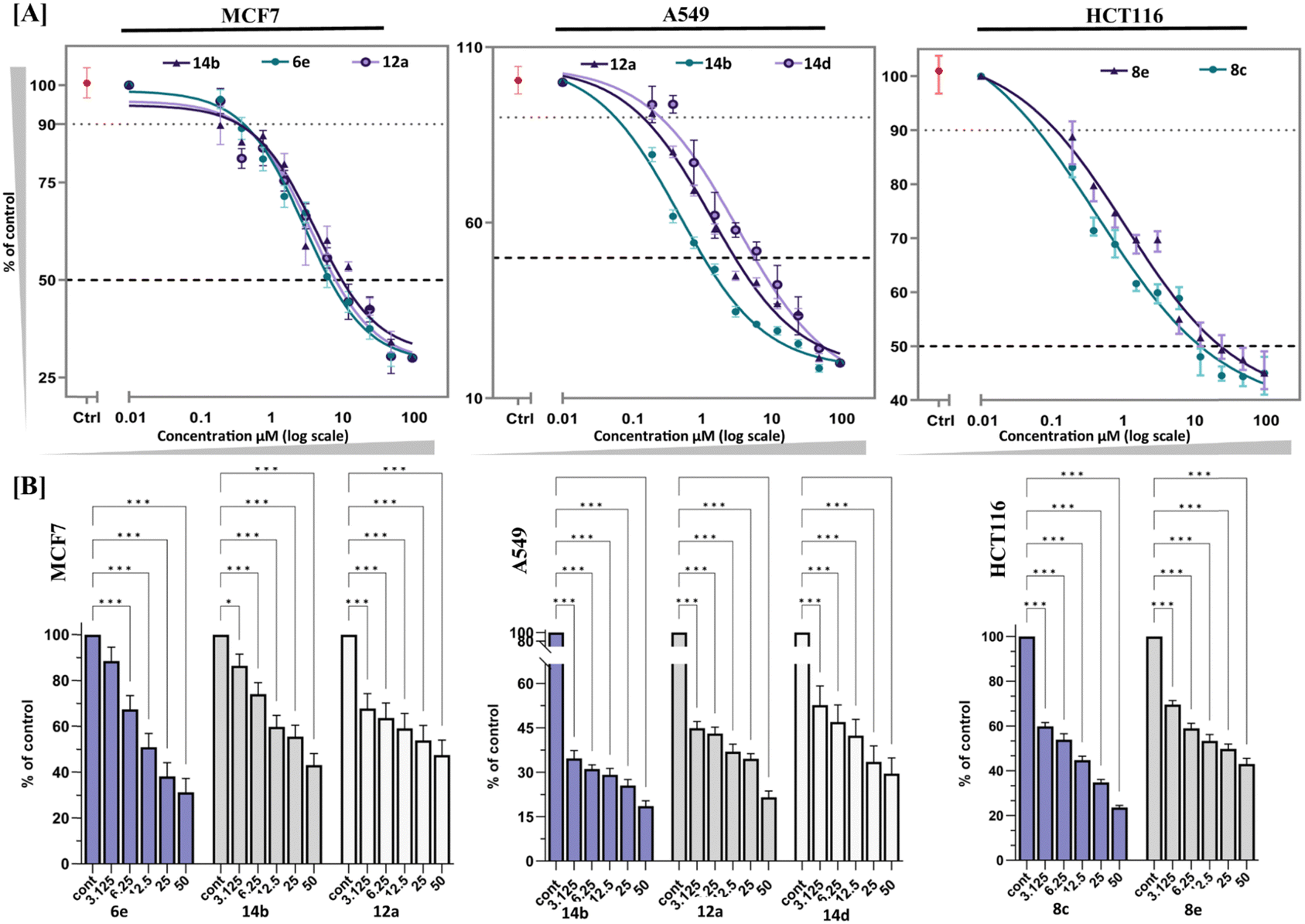
To assess the impact of the synthesized fused pyran derivatives on the viability of cancer cells, an MTT ((3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay was conducted in three different cancer cell lines MCF7 (breast adenocarcinoma), HCT116 (colorectal carcinoma), and A549 (lung adenocarcinoma) and the results were compared with standard drug cisplatin. The results, presented in Table 1, provide valuable insights into the compounds’ half-maximal inhibitory concentration (IC50) values for each tested cell line. Among the compounds tested, compounds 6e, 12a, and 14b exhibited promising results in inhibiting cellular growth in MCF7 cells (Fig. 2). These compounds demonstrated significant inhibitory effects on the proliferation of MCF7 cells, indicating their potential in treating breast adenocarcinoma compared to cisplatin. Similarly, compounds 12a, 12c, 12d, 14b, 14c, and 14d exhibited notable anti-proliferation activity on A549 cells, and compounds 8c and 8e displayed inhibitory effects specifically on HCT116 cells. They may serve as potential candidates for further optimization to treat lung adenocarcinoma and colorectal carcinoma, respectively. It is also noted that some compounds such as 12a and 14b showed cell toxicity in both MCF7 and A549 cells, indicating their broad-spectrum potential against multiple cancer cell lines.
Table 1 IC50 values of the fused pyran derivatives against MCF7, A549, and HCT116. Data are shown as mean ± SD, n = 3
| Cell lines (IC50 in μM) |
| Compound |
MCF 7 |
A549 |
HCT116 |
|
6a
|
>50 |
>50 |
>50 |
|
6b
|
>50 |
>50 |
>50 |
|
6c
|
>50 |
>50 |
>50 |
|
6d
|
>50 |
>50 |
>50 |
|
6e
|
12.46 ± 2.72 |
>50 |
>50 |
|
8a
|
>50 |
>50 |
>50 |
|
8b
|
>50 |
>50 |
>50 |
|
8c
|
>50 |
>50 |
7.58 ± 1.01 |
|
8d
|
>50 |
>50 |
>50 |
|
8e
|
>50 |
>50 |
21.43 ± 1.23 |
|
10a
|
>50 |
>50 |
>50 |
|
10b
|
>50 |
>50 |
>50 |
|
10c
|
>50 |
>50 |
>50 |
|
10d
|
>50 |
>50 |
>50 |
|
10e
|
>50 |
>50 |
>50 |
|
12a
|
21.51 ± 4.83 |
2.25 ± 0.67 |
>50 |
|
12b
|
>50 |
>50 |
>50 |
|
12c
|
>50 |
31.12 ± 0.45 |
>50 |
|
12d
|
>50 |
26.58 ± 2.05 |
>50 |
|
12e
|
>50 |
>50 |
>50 |
|
14a
|
>50 |
>50 |
>50 |
|
14b
|
32.14 ± 0.83 |
0.23 ± 0.12 |
>50 |
|
14c
|
>50 |
15.05 ± 0.11 |
>50 |
|
14d
|
>50 |
4.99 ± 3.33 |
>50 |
|
14e
|
>50 |
>50 |
>50 |
| Cisplatin |
30.56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ± 2.54 ± 2.54 |
28.54 ± 1.64 |
22.61 ± 2.03 |
 |
| | Fig. 2 (A) IC50 comparison graph of top selected compounds. (B) Dose-dependent inhibition of the viability of cancer cells under compound treatment at various concentrations (3.125–50 μM). The statistical analysis was conducted by one-way ANOVA and post hoc Dunnett's test. The data represent mean ± SD (n = 3), * P ≤ 0.01, ** P ≤ 0.001, *** P ≤ 0.0001, and **** P ≤ 0.00001. | |
2.3 Effect of the fused pyran derivatives on cancer cell morphology and colony formation
To evaluate the changes caused by the most toxic fused pyran derivatives in cancer cells and nuclear morphology, the morphological analysis using microscopic techniques was performed with the results shown in Fig. 3. After a 48-hour incubation with the tested compounds at their corresponding IC50 concentrations, all compounds induced cytoplasmic morphological alterations (Fig. 3(A)–(C)), suggesting their cytotoxic effect on cellular structures and functions in the cancer cells. Interestingly, it was observed that all compounds, except 14b in MCF7, induced the formation of micronuclei and apoptotic-like nuclear bodies (indicated by red arrows in Fig. 3(A)–(C)). This phenomenon indicates that the compounds have the potential to cause nucleic acid damage and potentially activate the pathways leading to cell death in the tested cell lines. Furthermore, compound 14b exhibited notable variations in its effects across two different cell lines. In MCF7 cells, compound 14b did not induce the formation of apoptotic-like nuclear bodies and micronuclei. However, in the A549 cell line, exposure to 14b resulted in a significant increase in apoptotic nuclear abnormalities. Interestingly, these nuclear changes were accompanied by a distinct phenomenon of cytoplasmic shrinking, suggesting a comprehensive cellular response specific to the A549 cell line upon exposure to compound 14b.
 |
| | Fig. 3 (A)–(C) Morphological and nuclear changes of MCF7, A549, and HCT116 cells after exposure to the selected fused pyran derivatives concerning the non-treated control cells. (D) Representative image of colony formation assay after compound treatment with IC50 and IC90 concentrations. (E) Quantitative analysis of colony formation ability after compound treatment with IC50 and IC90 concentrations. The data represent mean ± SD (n = 3). The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. | |
The colony formation analysis showed a significant decrease in colony formation after treatment with the fused pyran derivatives (Fig. 3(D)). Notably, compound 8c exhibited the highest inhibition rate (70.0 ± 4.96%) against HCT116 cells when treated with its IC50 concentration. Additionally, compounds 6e and 14b showed considerable inhibitions in colony formation with values of 66.4 ± 4.50% and 71.6 ± 2.01% for the MCF7 and A549 cell lines respectively (Fig. 3(E)). These results suggest that these molecules possess the ability to hinder the formation of cell colonies, indicating potential anti-proliferative effects. Specifically, compound 8c displayed remarkable inhibition against HCT116 cells, while compounds 6e and 14b demonstrated notable reductions in colony formation for the MCF7 and A549 cell lines. These results highlight the potential of these compounds as promising candidates for further investigation as new anticancer agents.
2.4 Effect of the fused pyran derivatives on cell cycle
Regulation of cell cycle is a crucial process for maintaining normal cell growth and division. Dysregulation of the cell cycle is one of the hallmarks of cancer, which has emerged as a promising targeting strategy for cancer therapy. We thus evaluated the fused pyran derivatives for their effects on the cell cycle progression in MCF7, A549, and HCT116 cells. As shown in Fig. 4, nearly all tested compounds possess remarkable abilities to arrest the cell cycle progression of the three cancer cells at various phases, including G0/G1, S, and G2/M (Fig. 4). For example, in the A549 cell line (Fig. 4(A)), compound 14b exhibited a strong inhibition of 57.35 ± 3.18% in the G0/G1 phase after 24 h of treatment at its IC50 concentration, suggesting its potential to prevent the cancer cells from entering the DNA synthesis phase and thereby impeding cancer cell proliferation. Similarly, in the MCF7 cell line (Fig. 4(B)), compound 6e showed a robust block in the G0/G1 phase (59.10 ± 0.24%). The G0/G1 phase is critical for regulating cell growth and deciding whether cells will undergo division or remain quiescent. Therefore, the significant inhibition observed with compound 6e suggests its potential to halt the cell division of MCF7 cancer cells. Furthermore, compounds 14b and 12a exhibited inhibition to the S phase of MCF cells, with inhibition rates of 33.60 ± 2.97% and 35.15 ± 7.57%, respectively. This indicated their activity of interfering with DNA replication and halting subsequent cell division. In the HCT116 cell line (Fig. 4(C)), compound 8c showed substantial inhibition to the G2/M phase (60.40 ± 0.99%) after 48 h incubation. The G2/M phase is responsible for ensuring accurate chromosome segregation and cell division. The notable block observed with compound 8c suggests its potential to disrupt these essential processes, thereby inhibiting the growth of HCT116 cancer cells. Additionally, compound 8e exhibited a significant inhibition in the G0/G1 phase (48.15 ± 2.89%) compared to the untreated cells (Fig. 4(C)) after 48 h incubation. These results highlight the potential of fused pyran derivatives as potential agents for disrupting the cell cycle events in cancer cells. However, to provide a more robust assessment, it is essential to further investigate and compare the cell cycle effects of the fused pyran derivatives with those of the drugs employed in clinical settings.
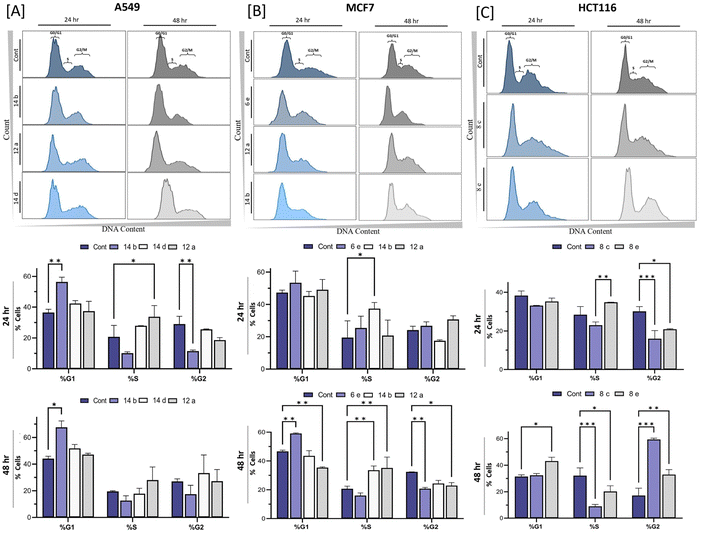 |
| | Fig. 4 (A)–(C) The effects of selected fused pyran derivatives on cell cycle distribution in MCF7, A549, and HCT116 cells and its quantitative analysis after compound treatment for 24 and 48 h at IC50 concentration. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. | |
2.5 Ability of the fused pyran derivatives to induce DNA double-strand breaks (DSBs)
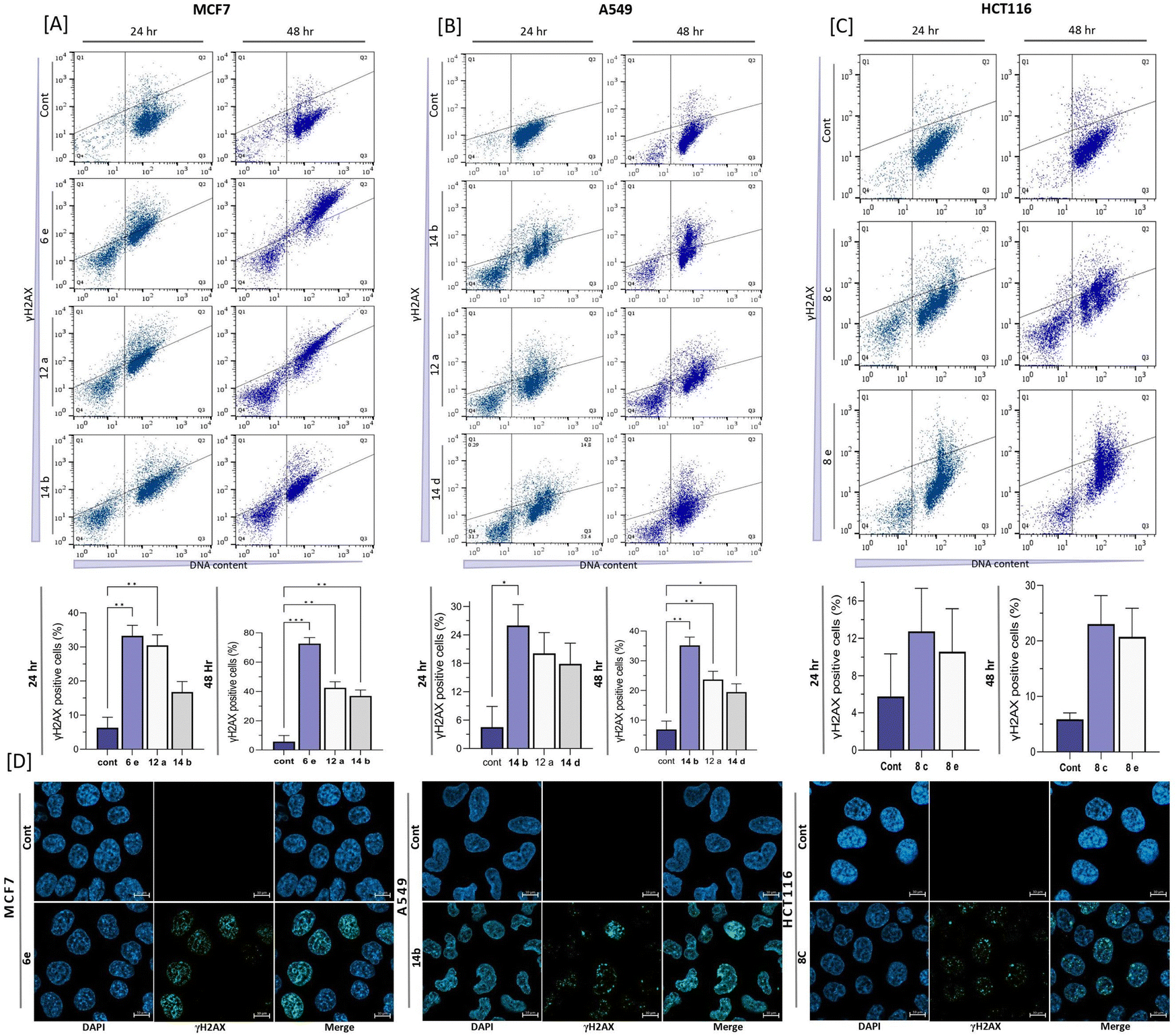
DNA damage analysis conducted by using γH2AX and 7AAD staining has provided significant insights into the capacity of the tested compounds to induce DNA double-strand breaks (DSBs) in A549, MCF7, and HCT116 cells (Fig. 5). In the MCF7 cell line, compound 6e induced a significantly high level of DNA damage of 72.63 ± 2.14% after 48 h incubation. The other two compounds 12a and 14b also exhibited significant DNA damage of 42.53 ± 2.93 and 36.93 ± 3.14%, respectively (Fig. 5(A)) at the 48 h treatment timepoint. Similarly, in the A549 cell line, compound 14b was found to induce a significant amount of DSBs, accounting for 35.17 ± 2.787% of the analyzed cells after 48 h incubation. Compounds 12a and 14d were slightly weaker than 14b, inducing moderate levels of 23.67 ± 1.97% and 19.47 ± 1.87% of DNA damage, respectively (Fig. 5(B)). In the context of the HCT116 cell line, compounds 8c and 8e were observed to induce DNA damage as evidenced by γH2AX detection. Notably, 48 h treatment of compound 8c resulted in 23.05 ± 5.162% of DSBs in the cells, while compound 8e led to 20.75 ± 3.65% γH2AX-positive cells (Fig. 5(C)).
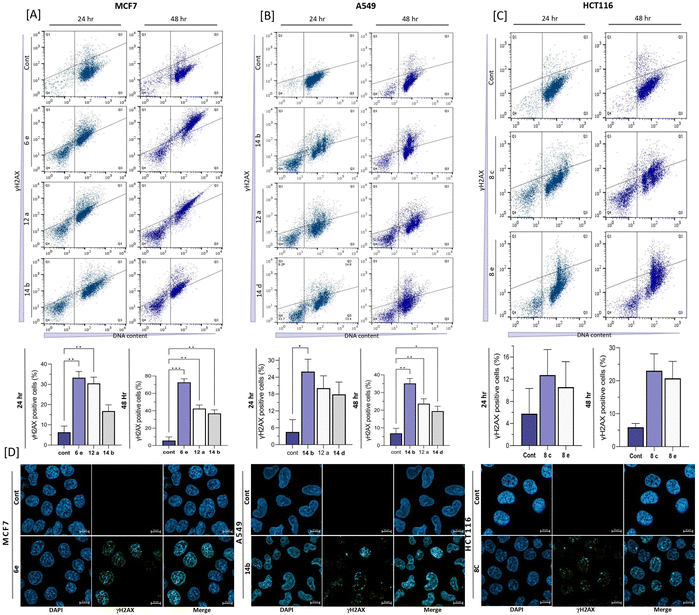 |
| | Fig. 5 Formation of DSBs caused by the fused pyran derivatives on MCF7, A549, and HCT116. (A)–(C) Flow cytometric analysis of γH2AX detection on different cell lines and their quantification after compound treatment for 24 and 48 h at IC50 concentrations. (D) Confocal microscopic images of γH2AX detection in the cells. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. | |
The confocal microscopy analysis provided valuable insights into the presence of significant amounts of γH2AX foci in the cells treated with the tested fused pyran compounds when compared to the untreated cells. Particularly, among the compounds tested, compound 6e exhibited the highest susceptibility for inducing DNA damage in the MCF7 cell line, indicating its promising efficacy as a potential anticancer agent. Similarly, compound 14b showed notable induction of DSBs in the A549 cell line, suggesting its potential therapeutic value against breast and lung cancers. Additionally, compound 8c displayed discernible staining of γH2AX in the HCT116 cell line, further supporting its capacity to induce DNA damage. Identifying the DNA-damaging effects caused by the fused pyran derivatives holds significant implications as they shed light on the potential mechanisms by which the tested compounds can elicit apoptotic pathways and subsequent cell death in cancer cells. By inducing DSBs, the compounds have the ability to disrupt DNA integrity, triggering cellular responses that can ultimately lead to the demise of cancerous cells.
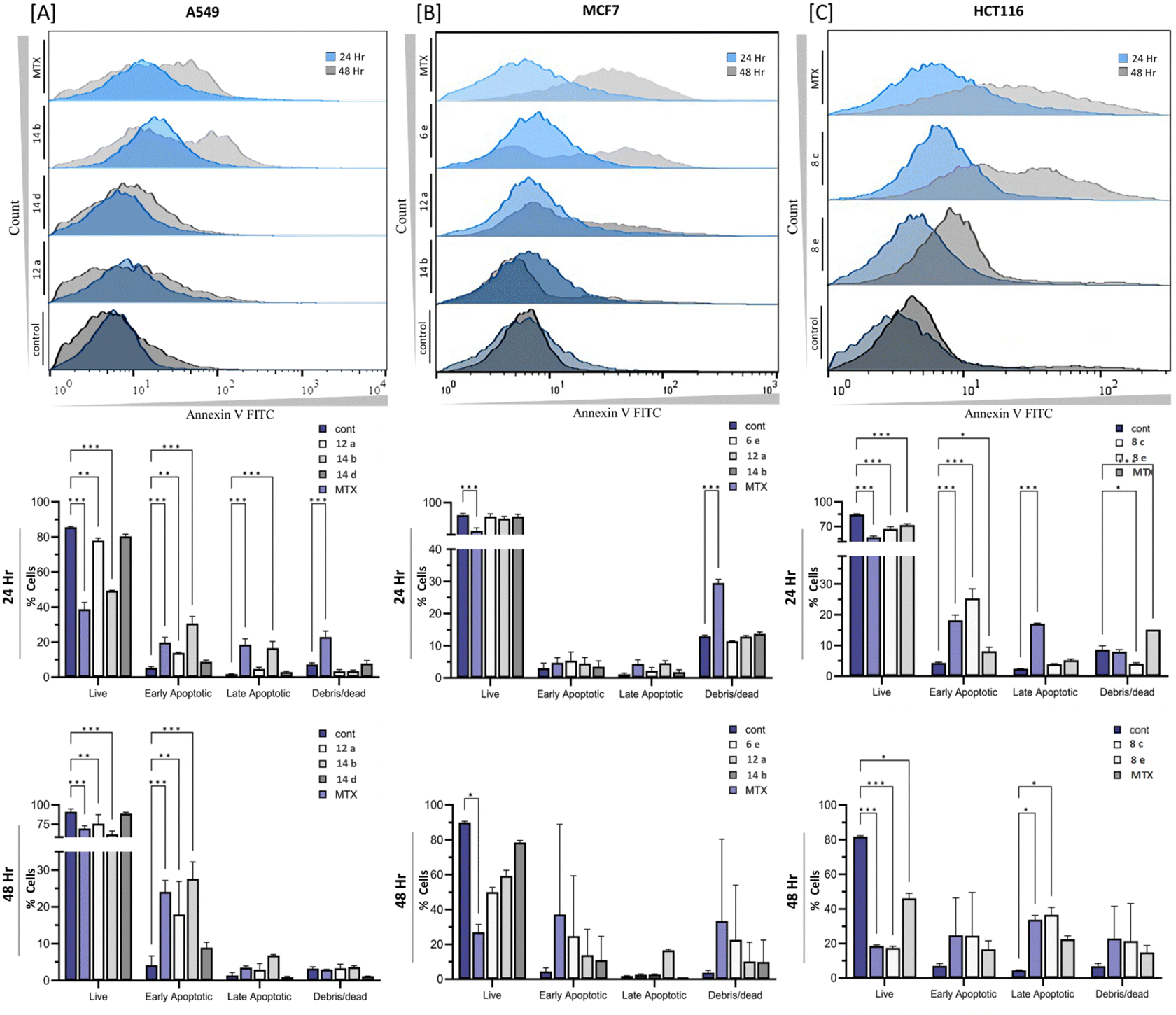
2.6 Ability of the fused pyran derivatives to induce apoptosis
Apoptotic assessment, conducted via annexin V binding and subsequent flow cytometry analysis, aimed to investigate the potential of tested compounds in inducing apoptotic bodies within the cell cultures. Our results indicated that all tested compounds exhibited a time-dependent capacity to induce apoptosis. Specifically, in the A549 cell line, compound 14b displayed a remarkable apoptotic activity, with a substantial increase of apoptotic bodies at 34.48 ± 4.79% and 47.20 ± 0.28% after 24 and 48 h incubation, respectively. Notably, the standard drug Mitoxantrone exhibited a lower percentage of cell death at 27.60 ± 3.56% after 24 h incubation (Fig. 6(A)).
 |
| | Fig. 6 The apoptotic ability of the selected fused pyran derivatives on different cancer cell lines was detected via flow cytometric analysis and its quantification. (A) A549 cell line, (B) MCF7 cell line and (C) HCT116 cell line. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. | |
Furthermore, when evaluating the apoptotic effect of compound 6e on the MCF7 cell line, a significant apoptotic induction ability was observed, resulting in 8.55 ± 3.71% and 57.44 ± 4.16% of apoptotic cells after 24 and 48 h incubation, respectively. In contrast, Mitoxantrone exhibited a lower apoptotic rate of 15.08 ± 2.86% after 24 hours of incubation (Fig. 6(B)). In the HCT116 cell line, compound 8c demonstrated the highest percentage of apoptotic cells, with 11.45 ± 1.14% and 29.20 ± 3.29% observed after 24 and 48 hours of incubation, respectively. Comparatively, Mitoxantrone induced a cell death rate of 35.25 ± 1.90% after 24 h incubation (Fig. 6(C)). These results highlight the apoptotic potential of the tested compounds in different cell lines, with compound 14b showing significant activity in A549 cells, compound 6e demonstrating remarkable effects in MCF7 cells, and compound 8c displaying the most prominent apoptotic induction ability in HCT116 cells. The observed variations in apoptotic responses among the compounds and cell lines merit further investigation, emphasizing the need for comprehensive analysis to elucidate the underlying mechanisms of their action.
2.7 ADMET predictions
Drug-likeness is a qualitative parameter that distinguishes drug candidates from other chemical entities. Approximately 40% of drug failures were allegedly due to their poor pharmacokinetics (ADME) and toxicity properties.52 To become an efficient drug, any promising compound should possess higher activity at minimum concentrations with low toxicity and be stable and active until the desired biological response occurs. Recent developments in combinatorial chemistry and high-throughput screenings (HTS) have significantly increased the capacity of traditional drug discovery programs by early predictions of physicochemical, pharmacokinetic, metabolism, and potential toxicity of a clinical candidate, thereby reducing the late-stage attritions.53
An in silico evaluation encompassing the realms of absorption, distribution, metabolism, excretion, and toxicity (ADMET) was conducted to predict the drug-likeness of potent compounds. We delved into the ADMET attributes of the aforementioned compounds, employing the ADMET Protocol housed within the Discovery Studio software (Accelrys, situated in San Diego, California, USA). These taxations were exclusively predicated upon the molecular structural composition of each constituent. A multitude of parameters were computed, including the two-dimensional Fast Polar Surface Area (2D_FPSA), Atom-based Log P98 (ALogP98), the integrity of the Blood–Brain Barrier (BBB), the presence and influence of Cytochrome P4502D6 (CYP2D6), and the propensity for Hepatotoxicity (HEPATOX).54–56
From Tables 2 and 3, it can be concluded that all the potent compounds displayed appropriate physicochemical properties and obeyed Lipinski's “Rule of Five” and “Veber Rule” and hence, they have drug-likeness and can be considered as probable lead compounds for the development of anticancer drugs. Pharmacokinetic properties like aqueous solubility play a significant role in the drug molecules’ bioavailability; from Table 2, the compounds 8c and 12c have good aqueous solubility levels (level 3). All others have low-level aqueous solubility levels except for ligand 6e, having a very low level (level 2), of solubility (level 1). Table 2 shows that the majority of the compounds have low values for BBB penetration levels (level 3) except for compound 14d, which has a moderate value (level 2), and compounds 6e, 8c, and 8e have undefined BBB penetration levels (level 4). Also, the results indicate that all potent ligands are non-inhibitors concerning CYP2D6 liver, which suggests they are well metabolized in Phase-I and all ligands have good intestinal absorption levels (level 1).
Table 2
In silico physicochemical properties and pharmacokinetic (ADME) predictions of potent compounds
| Comp. no. |
Lipinski's rule-of-five (RO5) |
Veber rule |
ADME |
|
ALogP |
M
W
|
HBD |
HBA |
No. of violations |
PSA_2D |
RB |
Solubility |
BBB |
CYP2D6 |
HIA |
| ≤5 |
≤500 |
≤5 |
≤10 |
≤1 |
≤140 |
≤10 |
|
ALogP = Ghose–Crippen–Viswanadhan octanol–water partition coefficient, MW = molecular weight, HBD = hydrogen bond donar, HBA = hydrogen bond acceptor, HT = hepatotoxic, HIA = human intestinal absorption, NI = non-inhibitor, NT = non-toxic. Solubility level: 0 (extremely low), 1 (very low), 2 (low), 3 (good), 4 (optimal), 5 (too soluble), 5 (molecules with one or more ALogP 98 types). Blood–brain barrier penetration level (BBB): 0 (very high penetrant), 1 (high), 2 (medium), 3 (low), 4 (undefined), 5 (molecules with one or more ALogP calculation). Cytochrome P450 2D6 (CYP2D6) level: 0 (non-inhibitor), 1 (Inhibitor). Hepatotoxic level and prediction: 0 and False (non-toxic), 1 and True (toxic). Human intestinal absorption level (HIA): 0 (good absorption), 1 (moderate), 2 (poor), 3 (very poor). Plasma protein binding (PPB) level and prediction: 0 (binding is <90%), 1 (binding is ≥90%), 2 (binding is ≥95%), and true (binding), false (not-binding). |
|
6e
|
3.98 |
433.42 |
1 |
6 |
0 |
112.51 |
2 |
1 |
4 |
False |
1 |
|
8c
|
1.08 |
347.33 |
1 |
6 |
0 |
112.51 |
2 |
3 |
4 |
False |
0 |
|
8e
|
2.92 |
397.39 |
1 |
6 |
0 |
112.51 |
2 |
2 |
4 |
False |
0 |
|
12a
|
2.08 |
332.36 |
1 |
4 |
0 |
92.32 |
2 |
2 |
3 |
False |
0 |
|
12c
|
1.40 |
333.34 |
1 |
5 |
0 |
103.58 |
2 |
3 |
3 |
False |
0 |
|
12d
|
3.15 |
382.42 |
1 |
4 |
0 |
92.32 |
2 |
2 |
3 |
False |
0 |
|
14b
|
1.98 |
360.41 |
1 |
4 |
0 |
92.32 |
2 |
2 |
3 |
False |
0 |
|
14c
|
1.85 |
361.40 |
1 |
5 |
0 |
103.58 |
2 |
2 |
3 |
False |
0 |
|
14d
|
3.61 |
410.47 |
1 |
4 |
0 |
92.315 |
2 |
2 |
2 |
False |
0 |
Table 3 TOPKAT analysis of potent compounds
| Comp. no. |
NTP rat |
Ames mutagen |
Fathead minnow LC50 |
Rat oral LD50 |
Carcinogenic potency_mouse TD50 |
Daphnia EC50 |
| Male |
Female |
| C = carcinogen, NC = non-carcinogen, NM = non-mutagen, LC50 (g L−1), LD50 (g kg−1 body weight), TD50 (mg kg−1 body weight day−1), EC50 (mg L−1) |
|
6e
|
NC |
C |
NM |
0.000322 |
0.205353 |
3.782 |
0.109699 |
|
8c
|
NC |
NC |
NM |
0.0139834 |
0.188742 |
16.9529 |
2.28863 |
|
8e
|
NC |
NC |
NM |
0.00110458 |
0.172766 |
11.575 |
0.63856 |
|
12a
|
NC |
NC |
NM |
0.0674315 |
0.0890379 |
21.0331 |
1.4037 |
|
12c
|
NC |
NC |
NM |
0.0988727 |
0.0922284 |
15.1738 |
1.26637 |
|
12d
|
NC |
NC |
NM |
0.00806484 |
0.241895 |
10.8802 |
0.40211 |
|
14b
|
NC |
NC |
NM |
0.233863 |
0.201545 |
9.78652 |
4.9743 |
|
14c
|
NC |
NC |
NM |
0.186816 |
0.162781 |
9.84066 |
3.43744 |
|
14d
|
NC |
NC |
NM |
0.0150864 |
0.422685 |
6.98587 |
1.08061 |
Similarly, the toxicological properties of the 2D molecular structures 9 potent ligands were predicted in silico by TOPKAT wizard that utilizes a quantitative structure–toxicity relationship (QSTR) model to assess specific toxicological endpoints such as fathead minnow (LC50), rat oral (LD50), rat (TD50), and Daphnia (EC50) are summarized in Table 3. All potent ligands were depicted to be non-carcinogen and non-mutagen towards NTP rats (male and female) and ames mutagen, except the compound 6e found to be carcinogenic against female NTP rats (Table 3).
3. Conclusion
Twenty-five new fused pyran derivatives containing nitrogen-based heterocycles such as pyrazole, imidazole, 1,2,4-triazole, benzimidazole, and benzotriazole (6a–e, 8a–e, 10a–e, 12a–e & 14a–e) have been synthesized via a three-component reaction involving 4-hydroxycoumarin (4)/4-hydroxy-6-methyl-2H-pyran-2-one (7)/5-hydroxy-2-(hydroxymethyl)-4H-pyran-4-one (9)/cyclohexane-1,3-dione (11)/dimedone (13), N-based heterocycles substituted benzaldehydes (3a–e) and malononitrile (5). All the compounds were screened for their in vitro anticancer potentials against three cancer cell lines MCF7, A549, and HCT116. Among them, compounds 6e, 14b, and 8c were identified as the most potent against MCF7, A549, and HCT116 with IC50 values of 12.46 ± 2.72 μM, 0.23 ± 0.12 μM, and 7.58 ± 1.01 μM, respectively. The top active compounds showed strong activities to induce cytoplasmic morphological alterations and inhibit colony formation in the tested cancer cells. They also remarkably inhibited the cell cycle progression of cancer cells at various phases. DNA damage analysis and apoptosis studies revealed that these compounds have the potential to induce DNA double-strand breaks and apoptosis. The in silico ADMET studies also suggest that these compounds can be considered lead compounds for further development of anticancer drugs. These data together suggest a good potential of the fused pyran derivatives as promising candidates for further investigation as anticancer agents.
4. Experimental section
Chemistry
All the reagents and solvents required for the synthesis were obtained commercially with the highest quality and were used directly without treatment unless otherwise indicated. The melting point of all the compounds was determined using the Stuart-SMP20 capillary melting point apparatus and is uncorrected. IR spectra of the compounds were analyzed by dispersing the compounds in potassium bromide pellets on a JASCO FT-IR 4700 spectrophotometer. 1H NMR and 13C NMR spectra were obtained on a Jeol spectrometer (500 MHz) in CDCl3/DMSO-d6 solvent using TMS as an internal standard. Coupling constant (J) values are expressed in Hertz (Hz) and the chemical shift (δ) values are reported in (ppm). LC-HRMS of all the compounds were recorded on Agilent 6530 ESI-QTOF mass instrument (mass accuracy of 5 ppm) operating in positive ion acquisition mode using reverse phase C18 column and all the compounds were confirmed to be greater than 98% pure. Thin-layer chromatography (TLC) was analyzed using a mixture of (4:1 v/v) hexanes and ethyl acetate. All the compounds were purified through column chromatography using silica gel (Merck 100–200 mesh) as a stationary phase and a mixture and hexanes and ethyl acetate as a mobile phase.
4-(1H-Pyrazol-1-yl)benzaldehyde (3a).
To a solution of 4-fluorobenzaldehyde (248.22 mg, 2 mmol) in dimethylformamide (5 mL) was added 1H-pyrazole (136.15 mg, 2 mmol) and anhydrous potassium carbonate (829.23 mg, 6 mmol). The reaction mixture was heated at 130 °C for 19 h. The reaction mixture was cooled to room temperature and poured into water (50 mL). The resulting mixture was extracted with ethyl acetate (20 mL) three times and the combined organic layer was dried over Na2SO4, concentrated under reduced pressure, and purified by column chromatography using 20% ethyl acetate in hexanes to obtain the pure product. Yield: 78%. Pale yellow solid. M.p.: 83–85 °C. IR (KBr, νmax, cm−1): 3126 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), 1692 (CO), 1604 (CC), 1520 (CN). 1H NMR (500 MHz, CDCl3): δ 9.94 (s, 1H), 7.96 (d, J = 2.5 Hz, 1H), 7.92–7.90 (m, 2H), 7.83–7.81 (m, 2H), 7.71 (d, J = 1.5 Hz, 1H), 6.46 (t, J = 2.2 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 190.898, 144.248, 142.281, 134.064, 131.290, 126.942, 118.774, 108.810. MS (ESI-QTOF) for C10H9N2O [M + H]+ calculated 173.0709, found 173.0712.
C–H), 1692 (CO), 1604 (CC), 1520 (CN). 1H NMR (500 MHz, CDCl3): δ 9.94 (s, 1H), 7.96 (d, J = 2.5 Hz, 1H), 7.92–7.90 (m, 2H), 7.83–7.81 (m, 2H), 7.71 (d, J = 1.5 Hz, 1H), 6.46 (t, J = 2.2 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 190.898, 144.248, 142.281, 134.064, 131.290, 126.942, 118.774, 108.810. MS (ESI-QTOF) for C10H9N2O [M + H]+ calculated 173.0709, found 173.0712.
Compounds 3b–e were synthesized by following the same procedure
4-(1H-Imidazol-1-yl)benzaldehyde (3b).
1H-Imidazole (136.15 mg, 2 mmol) was used as a reagent, and the reaction time was 19 h. Yield: 80%. Pale yellow solid. M.p.: 145–147 °C. IR (KBr, νmax, cm−1): 3112 (C–H), 1689 (CO), 1609 (CC), 1515 (CN). 1H NMR (500 MHz, CDCl3): δ 10.01 (s, 1H), 8.00–7.95 (m, 3H), 7.56–7.54 (m, 2H), 7.33–7.36 (m, 1H), 7.21 (s, 1H). 13C NMR (126 MHz, CDCl3): δ 191.1, 162.6, 142.3, 134.1, 131.3, 127.0, 118.8, 108.9. MS (ESI-QTOF) for C10H9N2O [M + H]+ calculated 173.0709, found 173.0712.
4-(1H-1,2,4-Triazol-1-yl)benzaldehyde (3c).
1H-1,2,4-Triazole (138.13 mg, 2 mmol) was used as a reagent, and the reaction time was 22 h. Yield: 74%. White solid. M.p.: 145–147 °C. IR (KBr, νmax, cm−1): 3099 (C–H), 1693 (CO), 1604 (CC), 1520 (CN). 1H NMR (500 MHz, CDCl3): δ 9.99 (s, 1H), 8.63 (s, 1H), 8.09 (s, 1H), 7.98 (d, J = 8.4 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 190.562, 153.156, 141.177, 141.033, 135.523, 131.405, 119.839. MS (ESI-QTOF) for C9H8N3O [M + H]+ calculated 174.0662, found 174.0658.
4-(1H-Benzo[d]imidazol-1-yl)benzaldehyde (3d).
1H-Benzo[d]imidazole (236.27 mg, 2 mmol) was used as a reagent, and the reaction time was 22 h. Yield: 79%. White solid. M.p.: 108–111 °C. IR (KBr, νmax, cm−1): 3143 (C–H), 1698 (CO), 1604 (CC), 1511 (CN). 1H NMR (500 MHz, CDCl3): δ 10.03 (s, 1H), 8.15 (s, 1H), 8.04 (d, J = 8.6 Hz, 2H), 7.84–7.81 (m, 1H), 7.68–7.65 (m, 2H), 7.56–7.53 (m, 1H), 7.33–7.27 (m, 2H). 13C NMR (126 MHz, CDCl3): δ 190.620, 144.085, 141.724, 141.225, 135.293, 132.912, 131.607, 124.322, 123.717, 123.486, 120.885, 110.442. MS (ESI-QTOF) for C14H11N2O [M + H]+ calculated 223.0866, found 223.0871.
4-(1H-Benzo[d][1,2,3]triazol-1-yl)benzaldehyde (3e).
1H-Benzo[d][1,2,3]triazole (238.25 mg, 2 mmol) was used as a reagent, and the reaction time was 24 h. Yield: 72%. White solid. M.p.: 186–190 °C. IR (KBr, νmax, cm−1): 3148 (C–H), 1693 (CO), 1604 (CC), 1515 (CN). 1H NMR (500 MHz, CDCl3): δ 9.94 (s, 1H), 7.96 (d, J = 2.5 Hz, 1H), 7.93–7.89 (m, 2H), 7.84–7.81 (m, 2H), 7.71 (d, J = 1.5 Hz, 1H), 7.19 (s, 1H), 6.46–6.45 (m, 1H). 13C NMR (126 MHz, CDCl3): δ 190.898, 144.248, 142.281, 134.064, 131.290, 126.942, 118.774, 108.810. MS (ESI-QTOF) for C13H10N3O [M + H]+ calculated 224.0818, found 224.0818.
4-(4-(1H-Pyrazol-1-yl)phenyl)-2-amino-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (6a).
To a solution of 4-(1H-pyrazol-1-yl)benzaldehyde (3a, 172.18 mg, 1 mmol) in water (2 mL) was added 4-hydroxycoumarin (4, 162.14 mg, 1 mmol), malononitrile (5, 66.06 mg, 1 mmol) and sodium fluoride (5 mg, 12 mol%). The reaction mixture was refluxed for 3 h. The mixture was cooled to room temperature and the solid product obtained was filtered, and washed with water (10 mL) three times to remove the NaF. The crude product obtained was purified by column chromatography using 30% ethyl acetate in hexanes to obtain the pure product. Yield: 90%. Pale yellow solid. M.p.: 248–253 °C. IR (νmax, cm−1): 3367, 3255 (–NH2), 3173 (C–H), 2211 (CN), 1710 (CO), 1652 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.45 (d, J = 2.5 Hz, 1H), 7.93–7.91 (m, 1H), 7.77–7.71 (m, 4H), 7.53–7.38 (m, 6H), 6.53–6.52 (m, 1H), 4.53 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 159.589, 157.948, 153.475, 152.179, 141.285, 140.939, 138.770, 132.991, 128.864, 127.750, 124.708, 122.529, 119.217, 118.622, 116.606, 113.307, 107.785, 103.696, 57.690, 36.467. MS (ESI-QTOF) for C22H14N4O3 [M + H]+ calculated 383.1139, found 383.1141.
4-(4-(1H-Imidazol-1-yl)phenyl)-2-amino-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (6b).
Yield: 80%. White solid. M.p.: 210–214 °C. IR (νmax, cm−1): 3460, 3178 (–NH2), 3125 (C–H), 2196 (CN), 1710 (CO), 1671 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.32 (s, 1H), 7.92 (d, J = 7.1 Hz, 1H), 7.7–7.72 (m, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.53–7.44 (m, 6H), 7.16 (s, 1H), 4.56 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 159.608, 157.996, 153.580, 152.198, 142.379, 135.688, 135.266, 133.010, 129.248, 129.133, 124.660, 122.557, 120.945, 119.179, 118.526, 116.549, 112.997, 103.571, 57.584, 36.438. MS (ESI-QTOF) for C22H15N4O3 [M + H]+ calculated 383.1139, found 383.1137.
4-(4-(1H-1,2,4-Triazol-1-yl)phenyl)-2-amino-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (6c).
Yield: 89%. Off-white solid. M.p.: 282–285 °C. IR (νmax, cm−1): 3348, 3241 (–NH2), 3100 (C–H), 2186 (CN), 1715 (CO), 1671 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 9.24 (s, 1H), 8.20 (s, 1H), 7.91–7.90 (m, 1H), 7.78 (d, J = 8.6 Hz, 2H), 7.73–7.69 (m, 1H), 7.51–7.44 (m, 6H), 4.55 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 159.541, 157.929, 153.571, 152.342, 152.189, 142.983, 142.312, 135.679, 132.982, 129.094, 124.660, 122.519, 119.697, 119.083, 116.577, 112.968, 103.466, 57.546, 36.544. MS (ESI-QTOF) for C21H13N5O3 [M + H]+ calculated 384.1091, found 384.1091.
4-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-2-amino-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (6d).
Yield: 86%. Off-white solid. M.p.: 245–248 °C. IR (νmax, cm−1): 3430, 3294 (–NH2), 3153 (C–H), 2201 (CN), 1691 (CO), 1603 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.55 (s, 1H), 7.92 (d, J = 6.7 Hz, 1H), 7.76–7.70 (m, 2H), 7.64–7.46 (m, 9H), 7.31–7.27 (m, 2H), 4.60 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 159.599, 158.101, 153.609, 152.179, 143.617, 143.262, 142.830, 134.834, 133.010, 129.286, 124.698, 123.690, 123.441, 122.500, 122.433, 119.831, 119.179, 116.587, 112.968, 110.703, 103.677, 57.574, 36.553. MS (ESI-QTOF) for C26H16N4O3 [M + H]+ calculated 433.1295, found 433.1292.
4-(4-(1H-Benzo[d][1,2,3]triazol-1-yl)phenyl)-2-amino-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (6e).
Yield: 88%. Yellow solid. M.p.: 220–225 °C. IR (νmax, cm−1): 3387, 3251 (–NH2), 3073 (C–H), 2196 (CN), 1715 (CO), 1667 (CC), 1214 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.60 (s, 1H), 8.50 (d, J = 7.6 Hz, 1H), 8.23–8.16 (m, 3H), 8.00–7.88 (m, 3H), 7.69–7.47 (m, 6H), 4.58 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 160.424, 160.136, 158.552, 154.204, 152.764, 145.421, 144.970, 139.067, 133.595, 132.876, 131.772, 129.823, 129.084, 128.201, 125.254, 123.075, 121.424, 121.012, 118.948, 118.948, 117.172, 113.505, 103.945, 57.948, 37.205. MS (ESI-QTOF) for C25H15N5O3 [M + H]+ calculated 434.1248, found 434.1248.
4-(4-(1H-Pyrazol-1-yl)phenyl)-2-amino-7-methyl-5-oxo-4,5-dihydropyrano[4,3-b]pyran-3-carbonitrile (8a).
Yield: 85%. Mustard yellow solid. M.p.: 232–238 °C. IR (νmax, cm−1): 3465, 3300 (–NH2), 3129 (C–H), 2186 (CN), 1725 (CO), 1671 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.41 (s, 1H), 7.76–7.72 (m, 3H), 7.31 (d, J = 8.2 Hz, 2H), 7.16 (s, 2H), 6.52 (s, 1H), 6.27 (s, 1H), 4.36 (s, 1H), 2.23 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.006, 161.365, 158.197, 158.053, 141.544, 140.843, 138.635, 128.806, 128.566, 127.875, 127.578, 119.284, 118.603, 107.718, 100.442, 98.004, 57.622, 35.766, 19.295. MS (ESI-QTOF) for C26H16N4O3 [M + H]+ calculated 347.1139, found 347.1143.
4-(4-(1H-Imidazol-1-yl)phenyl)-2-amino-7-methyl-5-oxo-4,5-dihydropyrano[4,3-b]pyran-3-carbonitrile (8b).
Yield: 78%. Light brown solid. M.p.: 213–217 °C. IR (νmax, cm−1): 3367, 3125 (–NH2), 3086 (C–H), 2196 (CN), 1695 (CO), 1622 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.25 (s, 1H), 7.72 (t, J = 1.1 Hz, 1H), 7.58 (d, J = 8.4 Hz, 2H), 7.34-7.32 (m, 2H), 7.26 (s, 2H), 7.12 (s, 1H), 6.30 (s, 1H), 4.37 (s, 1H), 2.23 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.083, 161.355, 158.255, 158.101, 142.427, 135.717, 128.969, 128.864, 120.763, 119.265, 118.161, 100.356, 98.023, 96.305, 57.498, 35.747, 19.228. MS (ESI-QTOF) for C19H14N4O3 [M + H]+ calculated 347.1139, found 347.1139.
4-(4-(1H-1,2,4-Triazol-1-yl)phenyl)-2-amino-7-methyl-5-oxo-4,5-dihydropyrano[4,3-b]pyran-3-carbonitrile (8c).
Yield: 75%. Off-white solid. M.p.: 260–265 °C. IR (νmax, cm−1): 3363, 3144 (–NH2), 3105 (C–H), 2192 (CN), 1706 (CO), 1671 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 9.25 (s, 1H), 8.22 (s, 1H), 7.79 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 7.26 (s, 2H), 6.30 (s, 1H), 4.40 (s, 1H), 2.24 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.102, 161.346, 158.284, 158.063, 152.294, 143.271, 142.331, 135.612, 128.960, 119.697, 119.217, 100.231, 98.004, 57.440, 35.833, 19.304. MS (ESI-QTOF) for C18H13N5O3 [M + H]+ calculated 348.1091, found 348.1095.
4-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-2-amino-7-methyl-5-oxo-4,5-dihydropyrano[4,3-b]pyran-3-carbonitrile (8d).
Yield: 83%. Dark yellow solid. M.p.: 255–258 °C. IR (νmax, cm−1): 3353, 3309 (–NH2), 3110 (C–H), 2206 (CN), 1715 (CO), 1667 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.59 (s, 1H), 7.79–7.77 (m, 1H), 7.67–7.63 (m, 3H), 7.46 (d, J = 8.4 Hz, 2H), 7.33–7.31 (m, 4H), 6.33 (s, 1H), 4.44 (s, 1H), 2.25 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.141, 161.403, 158.341, 158.226, 143.751, 143.444, 143.099, 134.777,132.962, 129.219, 123.681, 122.509, 119.860, 119.323, 110.770, 100.442, 97.975, 57.478, 35.929, 19.295. MS (ESI-QTOF) for C23H16N4O3 [M + H]+ calculated 397.1295, found 397.1299.
4-(4-(1H-Benzo[d][1,2,3]triazol-1-yl)phenyl)-2-amino-7-methyl-5-oxo-4,5-dihydropyrano[4,3-b]pyran-3-carbonitrile (8e).
Yield: 85%. Dark brown solid. M.p.: 223–226 °C. IR (νmax, cm−1): 3455, 3319 (–NH2), 3129 (C–H), 2192 (CN), 1710 (CO), 1671 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.45 (d, J = 2.5 Hz, 1H), 7.77–7.73 (m, 4H), 7.31 (d, J = 8.6 Hz, 2H), 7.25 (s, 2H), 6.53 (t, J = 2.1 Hz, 1H), 6.30 (s, 1H), 4.35 (s, 1H), 2.23 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.016, 161.365, 158.197, 158.053, 141.544, 140.843, 138.635, 128.797, 128.566, 127.885, 127.587, 119.284, 118.612, 107.718,100.442, 97.946, 57.613, 35.766, 19.285. MS (ESI-QTOF) for C22H15N5O3 [M + H]+ calculated 398.1248, found 398.1241.
4-(4-(1H-Pyrazol-1-yl)phenyl)-2-amino-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (10a).
Yield: 85%. Dark brown solid. M.p.: 223–227 °C. IR (νmax, cm−1): 3411 (OH) 3304, 3197 (–NH2), 3129 (C–H), 2181 (CN), 1652 (CO), 1613 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.50 (d, J = 1.9 Hz, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.75 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.28 (s, 2H), 6.55 (s, 1H), 6.35 (s, 1H), 5.69 (t, J = 5.5 Hz, 1H), 4.89 (s, 1H), 4.24–4.12 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 169.601, 168.257, 159.224, 148.743, 141.054, 139.250, 138.635, 136.399, 129.037, 128.950, 127.952, 127.674, 119.246, 118.929, 111.433, 107.967, 107.900, 59.091, 55.491. MS (ESI-QTOF) for C19H14N4O4 [M + H]+ calculated 363.1088, found 363.1087.
4-(4-(1H-Imidazol-1-yl)phenyl)-2-amino-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (10b).
Yield: 75%. Grey solid. M.p.: 248–252 °C. IR (νmax, cm−1): 3411 (OH) 3304, 3197 (–NH2), 3129 (C–H), 2181 (CN), 1652 (CO), 1613 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.26 (s, 1H), 7.75 (s, 1H), 7.67 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.29 (s, 2H), 7.11 (s, 1H), 6.35 (s, 1H), 5.70 (t, J = 6.1 Hz, 1H), 4.90 (s, 1H), 4.24–4.12 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 169.581, 168.247, 159.263, 148.628, 139.451, 136.456, 135.324, 129.651, 129.238, 120.964, 119.227, 118.094, 118.017, 118.0, 111.394, 59.101, 55.415. MS (ESI-QTOF) for C19H14N4O4 [M + H]+ calculated 363.1088, found 363.1090.
4-(4-(1H-1,2,4-triazol-1-yl)phenyl)-2-amino-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (10c).
Yield: 84%. Yellowish-white solid. M.p.: 290–294 °C. IR (νmax, cm−1): 3363 (OH) 3319, 3164 (–NH2), 3095 (C–H), 2206 (CN), 1667 (CO), 1608 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 9.30 (s, 1H), 8.25 (s, 1H), 7.89 (d, J = 8.6 Hz, 2H), 7.50 (d, J = 8.6 Hz, 2H), 7.30 (s, 2H), 6.35 (s, 1H), 5.69 (t, J = 6.2 Hz, 1H), 4.94 (s, 1H), 4.24–4.12 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 169.610, 168.276, 159.253, 152.640, 148.512, 142.436, 140.373, 136.485, 136.284, 129.248, 120.052, 119.208, 111.394, 59.101, 55.357. MS (ESI-QTOF) for C18H13N5O4 [M + H]+ calculated 364.1040, found 364.1039.
4-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-2-amino-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (10d).
Yield: 80%. Dark brown solid. M.p.: 210–213 °C. IR (νmax, cm−1): 3416 (OH) 3333, 3178 (–NH2), 3119 (C–H), 2181 (CN), 1657 (CO), 1583 (CC), 1224 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.60 (s, 1H), 7.79–7.73 (m, 3H), 7.68–7.66 (m, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.34–7.30 (m, 4H), 6.37 (s, 1H), 5.73 (t, J = 6.0 Hz, 1H), 4.98 (s, 1H), 4.28–4.17 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 170.138, 168.861, 159.953, 149.222, 144.356, 143.837, 140.775, 137.070, 136.081, 133.423, 129.958, 124.601, 124.054, 123.066, 120.503, 119.821, 112.018, 111.317, 59.676, 55.923. MS (ESI-QTOF) for C23H16N4O4 [M + H]+ calculated 413.1244, found 413.1252.
4-(4-(1H-Benzo[d][1,2,3]triazol-1-yl)phenyl)-2-amino-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (10e).
Yield: 85%. Dark brown solid. M.p.: 268–270 °C. IR (νmax, cm−1): 3382 (OH) 3294, 3178 (–NH2), 3071 (C–H), 2196 (CN), 1628 (CO), 1583 (CC), 1214 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.35 (d, J = 8.6 Hz, 2H), 8.05–8.04 (m, 2H), 7.61 (d, J = 8.8 Hz, 2H), 7.53 (q, J = 3.2 Hz, 2H), 7.34 (s, 2H), 6.36 (s, 1H), 5.69 (t, J = 6.1 Hz, 1H), 5.01 (s, 1H), 4.25–4.13 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 170.138, 168.861, 159.886, 148.963, 145.027, 142.599, 139.604, 137.070, 132.885, 131.781, 129.986, 128.335, 121.405, 119.725, 118.957, 118.794, 111.998, 59.628, 55.769. MS (ESI-QTOF) for C22H15N5O4 [M + H]+ calculated 414.1197, found 414.1195.
4-(4-(1H-Pyrazol-1-yl)phenyl)-2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (12a).
Yield: 89%. Off-white solid. M.p.: 207–210 °C. IR (νmax, cm−1): 3363, 3304 (–NH2), 3110 (C–H), 2196 (CN), 1671 (CO), 1594 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.39 (s, 1H), 7.69 (d, J = 7.6 Hz, 3H), 7.24 (d, J = 8.4 Hz, 2H), 7.02 (s, 2H), 6.48 (s, 1H), 4.21 (s, 1H), 2.63–2.57 (m, 2H), 2.32–2.20 (m, 2H), 1.96–1.86 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 196.448, 165.079, 158.994, 143.348, 141.361, 138.865, 128.796, 128.230, 120.263, 119.121, 114.072, 108.207, 58.418, 36.860, 35.554, 27.031, 20.340. MS (ESI-QTOF) for C19H16N4O2 [M + H]+ calculated 333.1346, found 333.1351.
4-(4-(1H-Imidazol-1-yl)phenyl)-2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (12b).
Yield: 82%. Yellow solid. M.p.: 209–211 °C. IR (νmax, cm−1): 3391, 3324 (–NH2), 3105 (C–H), 2192 (CN), 1681 (CO), 1642 (CC), 1205 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.39 (s, 1H), 7.69 (d, J = 8.4 Hz, 3H), 7.23 (d, J = 8.4 Hz, 2H), 7.01 (s, 2H), 6.48 (s, 1H), 4.21 (s, 1H), 2.60–2.53 (m, 2H), 2.30–2.19 (m, 2H), 1.95–1.85 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 195.920, 164.705, 158.409, 143.934, 135.218, 128.902, 128.537, 120.887, 119.678, 119.361, 118.516, 113.391, 57.843, 36.304, 35.046, 27.914, 26.494. MS (ESI-QTOF) for C19H16N4O2 [M + H]+ calculated 333.1346, found 333.1351.
4-(4-(1H-1,2,4-Triazol-1-yl)phenyl)-2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (12c).
Yield: 80%. Off-white solid. M.p: 231–233 °C. IR (νmax, cm−1): 3402, 3275 (–NH2), 3134 (C–H), 2186 (CN), 1676 (CO), 1608 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 9.23 (s, 1H), 8.22 (s, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 7.08 (s, 2H), 4.28 (s, 1H), 2.68–2.58 (m, 2H), 2.36–2.23 (m, 2H), 2.00–1.86 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 195.920, 164.686, 158.447, 152.448, 152.217, 144.548, 142.302, 135.295, 128.537, 119.649, 113.343, 57.699, 36.304, 35.152, 26.503, 19.775. MS (ESI-QTOF) for C18H15N5O2 [M + H]+ calculated 334.1299, found 334.1302.
4-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (12d).
Yield: 84%. Yellowish-white solid. M.p.: 259–261 °C. IR (νmax, cm−1): 3391, 3300 (–NH2), 3086 (C–H), 2181 (CN), 1691 (CO), 1652 (CC), 1200 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.57 (s, 1H), 7.79–7.76 (m, 1H), 7.64–7.61 (m, 3H), 7.42 (d, J = 8.4 Hz, 2H), 7.33–7.29 (m, 2H), 7.11 (s, 2H), 4.33 (s, 1H), 2.70–2.61 (m, 2H), 2.37–2.28 (m, 2H), 2.03–1.89 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 195.968, 164.811, 158.601, 144.375, 143.607, 143.444, 143.195, 134.383, 132.991, 128.739, 123.690, 123.594, 119.889, 119.755, 113.496, 110.732, 57.766, 36.323, 35.113, 26.503, 19.784. MS (ESI-QTOF) for C23H18N4O2 [M + H]+ calculated 383.1503, found 383.1502.
4-(4-(1H-Benzo[d][1,2,3]triazol-1-yl)phenyl)-2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (12e).
Yield: 90%. Off-white solid. M.p.: 255–257 °C. IR (νmax, cm−1): 3445, 3387 (–NH2), 3076 (C–H), 2202 (CN), 1700 (CO), 1642 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.66 (s, 1H), 8.55 (d, J = 8.4 Hz, 1H), 8.25–8.21 (m, 2H), 8.07–8.03 (m, 2H), 7.58–7.51 (m, 2H), 7.46 (d, J = 8.6 Hz, 1H), 7.13 (s, 1H), 4.34 (s, 1H), 2.70–2.60 (m, 2H), 2.38–2.26 (m, 2H), 2.02–1.92 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 196.496, 165.310, 159.070, 146.755, 144.951, 138.683, 129.276, 128.143, 120.916, 120.186, 118.716, 113.832, 58.073, 36.841, 35.852, 27.069, 20.312. MS (ESI-QTOF) for C22H17N5O2 [M + H]+ calculated 384.1455, found 384.1456.
4-(4-(1H-Pyrazol-1-yl)phenyl)-2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (14a).
Yield: 88%. White solid. M.p.: 271–273 °C. IR (νmax, cm−1): 3372, 3309 (–NH2), 3178 (C–H), 2181 (CN), 1691 (CO), 1652 (CC), 1214 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.44 (s, 1H), 7.75–7.72 (m, 3H), 7.26 (d, J = 8.2 Hz, 2H), 7.06 (s, 2H), 6.52 (s, 1H), 4.24 (s, 1H), 2.53 (s, 2H), 2.27 (d, J = 16.0 Hz, 1H), 2.12 (d, J = 16.0 Hz, 1H), 1.04 (s, 3H), 0.96 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 195.709, 162.526, 158.485, 142.734, 140.891, 140.766, 138.319, 128.413, 128.144, 127.808, 127.530, 119.668, 118.584, 112.498, 107.651, 57.949, 49.963, 35.113, 31.811, 28.366, 26.791. MS (ESI-QTOF) for C21H20N4O2 [M + H]+ calculated 361.1659, found 361.1664.
4-(4-(1H-Imidazol-1-yl)phenyl)-2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (14b).
Yield: 80%. Pale yellow solid. M.p.: 245–247 °C. IR (νmax, cm−1): 3440, 3129 (–NH2), 2950 (C–H), 2177 (CN), 1681 (CO), 1598 (CC), 1209 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.18 (s, 1H), 7.67 (s, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.09 (s, 1H), 6.98 (s, 2H), 4.26 (s, 1H), 2.53 (s, 2H), 2.26 (d, J = 16.0 Hz, 1H), 2.13 (d, J = 16.0 Hz, 1H), 1.05 (s, 3H), 0.98 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 195.488, 162.536, 158.418, 143.377, 135.343, 129.363, 128.537, 128.288, 120.494, 119.342, 117.979, 112.335, 57.987, 49.963, 35.056, 31.648, 28.183, 26.811. MS (ESI-QTOF) for C21H20N4O2 [M + H]+ calculated 361.1659, found 361.1663.
4-(4-(1H-1,2,4-Triazol-1-yl)phenyl)-2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (14c).
Yield: 85%. White solid. M.p.: 254–256 °C. IR (νmax, cm−1): 3440, 3265 (–NH2), 3105 (C–H), 2181 (CN), 1681 (CO), 1594 (CC), 1224 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 9.23 (s, 1H), 8.22 (s, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.6 Hz, 2H), 7.09 (s, 2H), 4.27 (s, 1H), 2.54 (s, 2H), 2.27 (d, J = 16.0 Hz, 1H), 2.12 (d, J = 16.0 Hz, 1H), 1.05 (s, 3H), 0.97 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 195.719, 162.670, 158.476, 152.438, 152.217, 144.481, 142.283, 135.189, 128.547, 119.601, 112.296, 57.786, 49.943, 35.229, 31.821, 28.346, 26.839. MS (ESI-QTOF) for C20H19N5O2 [M + H]+ calculated 361.1612, found 362.1613.
4-(4-(1H-Benzo[d]imidazol-1-yl)phenyl)-2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (14d).
Yield: 86%. Off-white solid. M.p.: 249–251 °C. IR (νmax, cm−1): 3353, 3290 (–NH2), 3086 (C–H), 2196 (CN), 1691 (CO), 1613 (CC), 1214 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.56 (s, 1H), 7.77 (d, J = 7.2 Hz, 1H), 7.63–7.56 (m, 3H), 7.41–7.29 (m, 4H), 7.11 (s, 2H), 4.32 (s, 1H), 2.56 (s, 2H), 2.29 (d, J = 16.0 Hz, 1H), 2.17 (d, J = 16.0 Hz, 1H), 1.06 (s, 3H), 1.01 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 195.805, 162.834, 158.591, 144.289, 143.751, 143.195, 134.441, 132.982, 128.806, 128.720, 123.575, 119.937, 119.707, 112.402, 110.722, 57.910, 49.991, 35.257, 31.859, 28.298, 27.031. MS (ESI-QTOF) for C25H22N4O2 [M + H]+ calculated 411.1816, found 411.1818.
4-(4-(1H-benzo[d][1,2,3]triazol-1-yl)phenyl)-2-amino-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (14e).
Yield: 90%. Off-white solid. M.p.: 266–268 °C. IR (νmax, cm−1): 3387, 3304 (–NH2), 3168 (C–H), 2181 (CN), 1681 (CO), 1642 (CC), 1205 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.19 (d, J = 7.1 Hz, 2H), 7.97 (s, 2H), 7.46–7.38 (m, 4H), 7.08 (s, 2H), 4.27 (s, 1H), 2.49 (s, 2H), 2.22 (d, J = 16.0 Hz, 1H), 2.08 (d, J = 16.0 Hz, 1H), 0.99 (s, 3H), 0.92 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 195.738, 162.718, 158.562, 146.161, 144.394, 138.136, 128.710, 127.645, 120.503, 120.254, 119.591, 118.353, 118.219, 117.988, 112.277, 57.613, 49.924, 35.392, 31.898, 28.289, 26.868. MS (ESI-QTOF) for C24H21N5O2 [M + H]+ calculated 412.1768, found 412.1771.
Cell culturing and maintenance
There are 3 different cancer cell lines were used in this study named MCF 7 (Breast Adenocarcinoma, obtained from ATCC, HTB-22) A549 (Lung Carcinoma, obtained from ATCC, CRM-CCL-185), and HCT116 (Colorectal Carcinoma, obtained from ATCC, CCL-247EMT). The HCT116 was cultured in McCoy's 5A medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics (penicillin 62.6 μg ml−1 and streptomycin 40 μg ml−1). The MCF7 and A549 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics (penicillin 62.6 μg mL−1 and streptomycin 40 μg mL−1). All the culturing experiments were performed with 5% CO2 under controlled humidity.
Determination of cell viability
Cell viability was evaluated using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma-Aldrich) assay. To perform this assay, cells were seeded into 96-well culture plates and allowed to adhere overnight to ensure proper attachment. Subsequently, the cells were exposed to various concentrations of the synthesized compounds for a duration of 72 h. DMSO (Merck) was employed as a control group. After the incubation period, each well was treated with an MTT solution at a final concentration of 0.4 mg mL−1 and incubated for an additional 2–3 hours at 37 °C. Following the completion of incubation, the culture medium was replaced with 100 μl of DMSO. The absorbance of the formazan product was measured at 450 nm using an ASYS UVM340 microplate reader. The IC50 value for each compound was determined using GraphPad Prism 9 software. The IC50 values were estimated based on the dose–response curves obtained from survival as a function of dose. The data were derived from three independent experiments and averaged to ensure the robustness and reliability of the results.
Morphological evaluation
Cell lines HCT116, A549, and MCF7 were cultured in appropriate culture plates and allowed to adhere overnight. Following this, the cells were exposed to the experimental compounds for 24 hours. After the incubation period, the cells were stained using Hoechst 33542 (Sigma-Aldrich) and subsequently observed under a fluorescence microscope (Olympus BX53). Additionally, cellular morphology was assessed using a modified Giemsa Stain solution under a brightfield microscope.
Colony formation assay
In this study, we employed well-established cell lines, HCT-116, MCF7, and A549, each seeded at a density of 170, 140, and 120 cells per well, respectively, in 12-well plates. Before treatment, the cells were allowed to attach and subsequently subjected to various concentrations of test compounds for 24 hours. Following this incubation period, the culture medium was replaced, and the cells were maintained in culture for an additional 8 days. Upon completion of the 8-day culture period, the cells were subjected to a series of preparatory steps. Firstly, thorough washing with PBS was performed to remove any residual compounds. Next, the cells were fixed using 99.5% methanol for 20 minutes, ensuring cellular preservation. Subsequently, a 0.5% crystal violet solution was used to stain the cells for a further 20 minutes. The stained plates were air-dried to facilitate the visualization of colonies. To quantitatively assess the effects of the compounds on cell viability, the visible colonies were meticulously counted using a colony counter. The percentage of viable cells was then calculated by comparing the results to the control.
Cell cycle analysis
Cell lines HCT116, A549, and MCF7 were cultured in appropriate culture plates and allowed to adhere overnight. Subsequently, the cells were exposed to the test compounds for 24 and 48 hours. After the incubation, the cells were fixed using ice-cold ethanol (75%) and stored at a temperature of −20 °C overnight. The cells were then subjected to two washes with PBS and centrifuged. After centrifugation, the cells were stained with a solution containing propidium iodide (PI) at a concentration of 20 μg μL−1 and RNase A at a concentration of 50 μg μL−1 in PBS. This staining process was carried out for 30 minutes at room temperature. Subsequently, the cell cycle distribution was analyzed using a Guava easyCyte 8 flow cytometer (Merck Millipore) in conjunction with FlowJo v10 software.
Evaluation of DNA DSBs induction
The cell lines HCT116, A549, and MCF7 were cultured and seeded onto tissue culture plates, allowing them to adhere overnight. The following day, the cells were exposed to the experimental compounds for durations of 24 and 48 h. Mitoxantrone (1 μM) was utilized as a reference compound. Subsequently, the cells were harvested through trypsinization, fixed in 75% ethanol, and stored at −20 °C until further analysis. To prepare the cells for analysis, they were rehydrated with PBS while placed on ice for 5 minutes. Following this, the cells were permeabilized with a solution consisting of 0.2% Triton X-100 in PBS and incubated at room temperature for 15 minutes. For labeling purposes, the cells were treated with Alexa Fluor 488-conjugated mouse anti-p-γH2AX (Ser139) antibody (#613406, BioLegend, USA) at a dilution of 1:200, and incubated at 37 °C for 1.5 hours. Afterwards, the cells were washed with PBS and stained with propidium iodide (20 μg μL−1) and RNase (50 μg μL−1) from Thermo Fisher Scientific for 20 minutes. Analysis of the labeled cells was analyzed using a Guava EasyCyte 8 cell sorter (Merck Millipore, USA) and FlowJo v10 software (BD Life Sciences, USA).
Immunofluorescence
The cell lines HCT116, A549, and MCF7 were cultured and seeded onto tissue culture plates, allowing them to adhere overnight. On the subsequent day, the cells were exposed to the tested compounds at their IC90 concentration for the specified duration. After exposure, the cells were washed with PBS and fixed with 4% paraformaldehyde (PFA) in PBS for 15 minutes at room temperature. Subsequently, the cells were permeabilized with 0.25% Triton X-100 in PBS for 15 minutes. To minimize non-specific binding, the cells were then blocked with 3% bovine serum albumin (BSA) in PBS for 1 hour at RT. The cells were incubated with Alexa Fluor 488-conjugated mouse anti-H2AX (pS139) antibody (#560445, diluted 1:200) from BD Pharmingen for 1.5 h at 37 °C in a humidified chamber. Following antibody incubation, the cells were washed three times for 10 minutes each with PBS-T. For nuclear staining, the cells were treated with 0.25 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI) for 15 minutes and then mounted onto slides using a mounting medium consisting of PBS-glycerol (90%) containing 2.5% (w/v) 4-diazobicyclo-(2,2,2-octane) (DABCO). The coverslips were washed twice with PBS-T and stained again with 0.25 μg ml−1 DAPI. Finally, images were acquired using an LSM 800 inverted laser scanning confocal microscope (Carl Zeiss, Dresden, Germany) equipped with an Airyscan detector and a ×63 1.4 NA Plan Apochromat objective (Carl Zeiss).
Apoptosis detection
To assess the proapoptotic activity of the compounds, cellular experiments were conducted following standard protocols. Initially, cells were seeded onto suitable Petri dishes and allowed to attach overnight. Subsequently, the cells were treated with the compounds at their respective IC50 concentrations for 24 and 48 hours. To establish a baseline, DMSO at a concentration of 1% (Merck) and Mitoxantrone at 1 μM (Sigma-Aldrich) were utilized as reference samples. After the treatment period, the cells were harvested and subjected to staining with FITC-Annexin V (Thermo Fisher, V13242) as per the manufacturer's instructions. To discriminate between viable and non-viable cells, propidium iodide (Thermo Fisher) staining was subsequently performed. The stained cells were then analyzed using a Guava easyCyte 8 cell sorter (Merck Millipore). The acquired data were further processed and analyzed using FlowJo v10 software.
Statistical analyses
The data are presented as mean ± standard error of the mean. Statistical differences between control and treated groups were assessed using a one-way and two-way analysis of variance (ANOVA) followed by Tukey and Dunnett post-hoc test using GraphPad Prism 9 software. FACS data were quantified by FlowJo software.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
FK and ACG thank the Ministry of Education (MoE) for providing the fellowships. MC is thankful to the DST-INSPIRE (IF190417) for the fellowship. JB sincerely acknowledges the DST-SERB for a research grant (file name: EEQ/2020/000303) and the Director, National Institute of Technology Calicut for providing research facilities. NMT is grateful to DST-SERB for providing fellowship from the project (EEQ/2020/000303). All the authors are thankful to CMC, and NIT Calicut for the characterization data.
References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249, DOI:10.3322/caac.21660.
- J. Ferlay, M. Colombet, I. Soerjomataram, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Int. J. Cancer, 2021, 149, 778–789, DOI:10.1002/ijc.33588.
- C. de Martel, D. Georges, F. Bray, J. Ferlay and G. M. Clifford, Lancet Global Health, 2020, 8, e180–e190, DOI:10.1016/S2214-109X(19)30488-7.
- J. Fares, M. Y. Fares, H. H. Khachfe, H. A. Salhab and Y. Fares, Signal Transduction Targeted Ther., 2020, 5, 28, DOI:10.1038/s41392-020-0134-x.
- American Cancer Society. Learn About Cancer. https://www.cancer.org/cancer.html.
- American Society of Clinical Oncology. Stages of Cancer. https://www.cancer.net/navigating-cancer-care/diagnosing-cancer/stages-cancer.
- National Cancer Institute. What is Cancer?https:// https://www.cancer.gov/about-cancer/understanding/what-is-cancer.
-
G. Feuer, in Progress in Medicinal Chemistry, ed. G. P. Ellis and G. B. West, North-Holland Publishing Company, New York, 1st edn, 1974, vol. 10, p.85 Search PubMed.
-
F. M. Dean, Naturally Occurring Oxygen Ring Compounds, Butterworth-Heinemann, London, 1963, pp. 176–220 Search PubMed.
- S. A. Patil, R. Patil, L. M. Pfeffer and D. D. Miller, Future Med. Chem., 2013, 5(14), 1647–1660, DOI:10.4155/fmc.13.126.
- V. Raj and J. Lee, Front. Chem., 2020, 8, 1–23, DOI:10.3389/fchem.2020.00623.
- M. K. Katiyar, G. K. Dhakad, Shivani, S. Arora, S. Bhagat, T. Arora and R. Kumar, J. Mol. Struct., 2022, 1263, 133012, DOI:10.1016/j.molstruc.2022.133012.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, Polycyclic Aromat. Compd., 2022, 42, 5893–5937, DOI:10.1080/10406638.2021.1962923.
- M. S. Malik, H. Ather, S. M. Asif Ansari, A. Siddiqua, Q. M. S. Jamal, A. H. Alharbi, M. M. Al-Rooqi, R. S. Jassas, E. M. Hussein, Z. Moussa, R. J. Obaid and S. A. Ahmed, Pharmaceuticals, 2023, 16, 1–13, DOI:10.3390/ph16030333.
- W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, C. Crogan-Grundy, D. Labreque, M. Bubenick, G. Attardo, R. Denis, S. Lamothe, H. Gourdeau, B. Tseng, S. Kasibhatla and X. C. Sui, J. Med. Chem., 2008, 51, 417–423, DOI:10.1021/jm7010657.
- W. Kemnitzer, S. Kasibhatla, S. Jiang, H. Zhang, J. Zhao, S. Jia, L. Xu, C. Crogan-Grundy, R. Denis, N. Barriault, L. Vaillancourt, S. Charron, J. Dodd, G. Attardo, D. Labrecque, S. Lamothe, H. Gourdeau, B. Tseng, J. Drewe and S. X. Cai, Bioorg. Med. Chem. Lett., 2005, 15, 4745–4751, DOI:10.1016/j.bmcl.2005.07.066.
- J. M. Doshi, D. Tian and C. Xing, J. Med. Chem., 2006, 49, 7731–7739, DOI:10.1021/jm060968r.
- D. A. N. L. Wood, D. Panda, T. R. Wiernicki, L. Wilson, M. A. N. N. Jordan, J. A. I. P. A. L. Singh and D. L. W. Indiana, Mol. Pharmacol., 1997, 52, 437–444, DOI:10.1124/mol.52.3.437.
- A. M. El-Agrody, A. M. Fouda, M. A. Assiri, A. Mora, T. E. Ali, M. M. Alam and M. Y. Alfaifi, Med. Chem. Res., 2020, 29, 617–629, DOI:10.1007/s00044-019-02494-3.
- N. R. Emmadi, K. Atmakur, G. K. Chityal, S. Pombala and J. B. Nanubolu, Bioorg. Med. Chem. Lett., 2012, 22, 7261–7264, DOI:10.1016/j.bmcl.2012.09.018.
- N. R. Kamdar, D. D. Haveliwala, P. T. Mistry and S. K. Patel, Med. Chem. Res., 2011, 20, 854–864, DOI:10.1007/s00044-010-9399-x.
- G. Singh, A. Sharma, H. Kaur and M. P. S. Ishar, Chem. Biol. Drug Des., 2016, 87, 213–223, DOI:10.1111/cbdd.12653.
- C. Bingi, E. Narender Reddy, M. Chennapuram, Y. Poornachandra, C. G. Kumar, N. Jagadeesh Babu and K. Atmakur, Bioorg. Med. Chem. Lett., 2015, 25, 1915–1919, DOI:10.1016/j.bmcl.2015.03.034.
- N. D. Vala, H. H. Jardosh and M. P. Patel, Chin. Chem. Lett., 2016, 27, 168–172, DOI:10.1016/j.cclet.2015.09.020.
- E. Rajanarendar, M. Nagi Reddy, S. Rama Krishna, K. Rama Murthy, Y. N. Reddy and M. V. Rajam, Eur. J. Med. Chem., 2012, 55, 273–283, DOI:10.1016/j.ejmech.2012.07.029.
- N. C. Lazzara, R. J. Rosano, P. P. Vagadia, M. T. Giovine, M. W. Bezpalko, N. A. Piro, W. S. Kassel, W. J. Boyko, D. L. Zubris, K. K. Schrader, D. E. Wedge, S. O. Duke and R. M. Giuliano, J. Org. Chem., 2019, 84, 666–678, DOI:10.1021/acs.joc.8b02490.
- J. H. Roireau, R. J. Rosano, N. C. Lazzara, T. Chen, J. Bajsa-Hirschel, K. K. Schrader, S. O. Duke, D. Wykoff and R. M. Giuliano, J. Agric. Food Chem., 2020, 68, 9906–9916, DOI:10.1021/acs.jafc.0c02564.
- Y. Dgachi, O. M. Bautista-Aguilera, M. Benchekroun, H. Martin, A. Bonet, D. Knez, J. Godyń, B. Malawska, S. Gobec, M. Chioua, J. Janockova, O. Soukup, F. Chabchoub, J. Marco-Contelles and L. Ismaili, Molecules, 2016, 21, 1–15, DOI:10.3390/molecules21050634.
- A. Foroumadi, S. Emami, M. Sorkhi, M. Nakhjiri, Z. Nazarian, S. Heydari, S. K. Ardestani, F. Poorrajab and A. Shafiee, Chem. Biol. Drug Des., 2010, 75, 590–596, DOI:10.1111/j.1747-0285.2010.00959.x.
- K. S. Lee, L. Y. Khil, S. H. Chae, D. Kim, B. H. Lee, G. S. Hwang, C. H. Moon, T. S. Chang and C. K. Moon, Life Sci., 2006, 78, 1091–1097, DOI:10.1016/j.lfs.2005.06.017.
- I. A. Schepetkin, L. N. Kirpotina, A. I. Khlebnikov, N. Cheng, R. D. Ye and M. T. Quinn, Biochem. Pharmacol., 2014, 92, 627–641, DOI:10.1016/j.bcp.2014.09.027.
- V. T. Angelova, Y. Voynikov, P. Andreeva-Gateva, S. Surcheva, N. Vassilev, T. Pencheva and J. Tchekalarova, Med. Chem. Res., 2014, 26, 1884–1896, DOI:10.1007/s00044-017-1902-1.
- N. Jain, R. M. Kanojia, J. Xu, G. Jian-Zhong, E. Pacia, M. T. Lai, F. Du, A. Musto, G. Allan, D. W. Hahn, S. Lundeen and Z. Sui, J. Med. Chem., 2006, 49, 3056–3059, DOI:10.1021/jm060353u.
- K. Takao, H. Yahagi, Y. Uesawa and Y. Sugita, Bioorg. Chem., 2018, 77, 436–442, DOI:10.1016/j.bioorg.2018.01.036.
- K. Takao, S. U, H. Kamauchi and Y. Sugita, Bioorg. Chem., 2019, 87, 594–600, DOI:10.1016/j.bioorg.2019.03.042.
- A. P. Sarkate, V. S. Dofe, S. V. Tiwari, D. K. Lokwani, K. S. Karnik, D. D. Kamble, M. H. S. H. Ansari, S. Dodamani, S. S. Jalalpure, J. N. Sangshetti, R. Azad, P. V. L. S. Burra and S. V. Bhandari, Bioorg. Med. Chem. Lett., 2021, 40, 127916, DOI:10.1016/j.bmcl.2021.127916.
- H. E. A. Ahmed, M. A. A. El-Nassag, A. H. Hassan, H. M. Mohamed, A. H. Halawa, R. M. Okasha, S. Ihmaid, S. M. Abd El-Gilil, E. S. A. E. H. Khattab, A. M. Fouda, A. M. El-Agrody, A. Aljuhani and T. H. Afifi, J. Mol. Struct., 2019, 1186, 212–223, DOI:10.1016/j.molstruc.2019.03.012.
- R. Kaur, F. Naaz, S. Sharma, S. Mehndiratta, M. K. Gupta, P. M. S. Bedi and K. Nepali, Med. Chem. Res., 2015, 24, 3334–3349, DOI:10.1007/s00044-015-1382-0.
- G. Gopinath, V. Sankeshi, S. Perugu, M. D. Alaparthi, S. Bandaru, V. K. Pasala, P. R. Chittineni, G. L. D. Krupadanam and S. R. Sagurthi, Eur. J. Med. Chem., 2016, 124, 750–762, DOI:10.1016/j.ejmech.2016.08.070.
- Z. Zhang, C. Wang, L. Ma, X. Jiang, C. Wu, Y. Wang, Y. Jiang, W. Zheng, Y. Yang, Y. Ma and J. Yang, Biochem. Biophys. Res. Commun., 2019, 511, 381–386, DOI:10.1016/j.bbrc.2019.02.064.
- A. M. A. Lorza, H. Ravi, R. C. Philip, J. P. Galons, T. P. Trouard, N. A. Parra, D. D. Von Hoff, W. L. Read, R. Tibes, R. L. Korn and N. Raghunand, Sci. Rep., 2020, 10, 1–12, DOI:10.1038/s41598-020-71246-w.
- D. Panda, J. P. Singh and L. Wilson, J. Biol. Chem., 1997, 272, 7681–7687, DOI:10.1074/jbc.272.12.7681.
- F. Schmitt, M. Gold, M. Rothemund, I. Andronache, B. Biersack, R. Schobert and T. Mueller, Eur. J. Med. Chem., 2019, 163, 160–168, DOI:10.1016/j.ejmech.2018.11.055.
- D. Rajguru, B. S. Keshwal and S. Jain, Med. Chem. Res., 2013, 22, 5934–5939, DOI:10.1007/s00044-013-0586-4.
- K. Parthasarathy, C. Praveen, C. Balachandran, P. Senthil Kumar, S. Ignacimuthu and P. T. Perumal, Bioorg. Med. Chem. Lett., 2013, 23, 2708–2713, DOI:10.1016/j.bmcl.2013.02.086.
- L. Braconi, E. Teodori, C. Riganti, M. Coronnello, A. Nocentini, G. Bartolucci, M. Pallecchi, M. Contino, D. Manetti, M. N. Romanelli, C. T. Supuran and S. Dei, J. Med. Chem., 2022, 65, 14655–14672, DOI:10.1021/acs.jmedchem.2c01175.
- S. Wang, S. Q. Wang, Q. Teng, L. Yang, Z. Lei, X. Yuan, J. Huo, X. Chen, M. Wang, B. Yu, Z. Chen and H. Liu, J. Med. Chem., 2020, 63, 15979–15996, DOI:10.1021/acs.jmedchem.0c01741.
- D. J. Hong, S. H. Jung, J. Kim, D. Jung, Y. G. Ahn, K. H. Suh and K. H. Min, J. Enzyme Inhib. Med. Chem., 2020, 35, 227–234, DOI:10.1080/14756366.2019.1693555.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311, 10.1039/d0ra09198g.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909, DOI:10.3390/molecules25081909.
- K. Fabitha, C. G. Arya, M. Chandrakanth and J. Banothu, Res. Chem. Intermed., 2023, 49, 997–1014, DOI:10.1007/s11164-022-04929-w.
- H. van de Waterbeemd and E. Gifford, Nat. Rev. Drug Discovery, 2003, 2, 192–204, DOI:10.1038/nrd1032.
- M. J. Waring, J. Arrowsmith, A. R. Leach, P. D. Leeson, S. Mandrell, R. M. Owen, G. Pairaudeau, W. D. Pennie, S. D. Pickett, J. Wang, O. Wallace and A. Weir, Nat. Rev. Drug Discovery, 2015, 14, 475–486, DOI:10.1038/nrd4609.
- Y. Sivamani, D. Shanmugarajan, T. Durai Ananda Kumar, S. Faizan, B. Channappa, N. L. Naishima and B. R. Prashantha Kumar, Comput. Biol. Chem., 2021, 95, 107600, DOI:10.1016/j.compbiolchem.2021.107600.
- N. Kang, S. Y. Heo, S. H. Cha, G. Ahn and S. J. Heo, Mar. Drugs, 2022, 20, 1–14, DOI:10.3390/md20060399.
- M. Szumilak, W. Lewgowd and A. Stańczak, Acta Pol. Pharm., 2016, 73, 1191–1200 CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Natalia
Maciejewska
b,
Natalia
Maciejewska