
Bromination of organic spacer impacts on the structural arrangement, phase transitions, and optical and electrical properties of a hybrid halide compound:[(CH3)3N(CH2)3Br]2PdBr4†
Mohamed
Saadi
a,
Imen
Dakhlaoui
a,
Fadhel
Hajlaoui
b,
Nidhal
Drissi
c,
Mustapha
Zighrioui
d,
Fethi
Jomni
a,
Nathalie
Audebrand
 e,
Marie
Cordier
e and
Karoui
Karim
*fd
e,
Marie
Cordier
e and
Karoui
Karim
*fd
aUniversité de Tunis El Manar, Laboratoire LMOP, LR99ES17, El Manar, 2092 Tunis, Tunisia
bLaboratoire Physico-chimie de l’Etat Solide, Département de Chimie, Faculté des Sciences de Sfax, Université de Sfax, B.P. 1171, 3000 Sfax, Tunisia
cDepartment of Physics, Faculty of Science, King Khalid University, P. O. Box 9004, Abha 61413, Saudi Arabia
dGREMAN UMR, 7347-CNRS, CEA, INSACVL, Université de Tours, Blois, France
eUniv Rennes, CNRS, INSA Rennes, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France
fLaboratoire des Caractérisations Spectroscopiques et Optique des Matériaux, Faculté des Sciences de Sfax, Université de Sfax, B.P. 1171, 3000 Sfax, Tunisia. E-mail: karouikarim36@yahoo.com; Tel: +00216 25648756
First published on 24th November 2023
Abstract
Understanding the structure and arrangement of organic inorganic hybrid metal halides and their contribution to physical properties remains a challenging topic. In particular, materials involving d8 metal halides incorporating organo-halogen components are still largely unexplored. In this study, we have used precise reagents to design and synthesize a low dimensional semiconductor material, [(CH3)3N(CH2)3Br]2PdBr4. Crystals were prepared through slow evaporation. Through the reactions of (3-bromopropyl)trimethylammonium bromide with PdBr2 in an aqueous HBr solution, we successfully obtained a 0D organic–inorganic hybrid material, [(CH3)3N(CH2)3Br]2PdBr4. This compound exhibits two reversible phase transitions, at 363 K and 393 K, assigned to an order/disorder change and a rare congruent melting. These transitions are confirmed through DSC analysis, temperature dependent Raman studies and electrical measurements. The crystals remained stable up to 520 K. Furthermore, we measured the UV-visible absorption spectrum of the polycrystalline sample to estimate the band gap, which was found to be approximately 2 eV. The halogen atom in the organic spacer has a considerable impact on the structure, high-temperature phase transition, and semiconducting characteristics, as revealed in this work.
1. Introduction
High-temperature phase change materials, which are highly accepted for the design of optoelectronic devices, are desired among many functional materials.1–5 Among the recognized categories of materials, two conventional classes are defined on their stoichiometry, (AmineH)2MX4 and (AmineH)MX3, where M represents a divalent transition metal cation such as Pb2+, Cu2+, Mn2+, Pd2+ and X represents a halogen; M and X atoms form octahedral, tetrahedral or square planar geometries.Aliphatic or aromatic alkylammonium cations, serving as templating agents, are either intercalated between layered metal-halide structures or positioned within cavities surrounded by polyhedral anionic groups, allowing for significant freedom of motion.6,7 Their incorporation into inorganic frameworks can result in substantial polarizability and remarkable structural flexibility, leading to the creation of novel materials with enhanced properties and applications.8–10
However, despite the chemical and structural diversity of hybrid halide compounds, covering the underlying mechanism that governs the high Tc (critical temperature) remains a challenge. As a result, the majority of halogenometallate hybrids used in the crucial systems. Consequently, there is a long way to go in improving the constructive approach to enhance Tc.1 Fortunately, the ability to manipulate fundamental material properties by modulating the molecular structure offers a robust platform for the design of high-temperature phase transition.
For instance, [(CH3)2NH2]PbX3 (X = Cl−, Br−, I−) belong to the ABX3 perovskite family, and exhibit a first-order phase transition. The phase transition of the chloride compound occurs at 320 K, while that of the bromide and iodide compounds occurs at 250 K. These phase transitions are associated with the movement of the organic cations.11,12 Within the crystals, [(CH3)2NH2]+ cations and [PbX6]4− anion chains are linked through hydrogen bond interactions. Both components have the potential to reorient with temperature. These three hybrid compounds are semiconductors with band gap values of 3.5 eV (X: Cl−), 3.0 eV (X: Br−) and 2.59 eV (X: I−).
Meanwhile, the hybrid compound [(C6H14)NH2]2CuBr4, influenced by order–disorder phenomena in [CuBr4]2− anions and n-butyl–ethyl ammonium cations [(C6H14)NH2]+, demonstrates a dielectric anomaly around 194 K. However, the low temperature phase transitions have somewhat limited its applications, which are expected at higher temperatures.13 The approach of halogen substitution, particularly by incorporating fluorine atoms into organic molecules, has been shown to enhance optoelectronic and ferroelectric performances in hybrid halide materials.14–16 By employing the concept of H/F substitution within hybrid halide compounds, scientist can precisely engineer their target materials by modulating the structure and designing high-Tc (Curie temperature) molecular ferroelectrics.17–20 For instance, the notion of introducing electronegative species into organic cations has proven inspiring in the context of compounds like (R)- and (S)-3-(fluoropyrrolidinium)MnCl3.21 The substitution in the (pyrrolidinium)MnCl322 compound induces structural changes that preserve ferroelectric polarization. As anticipated, this H/F substitution results in a successful increase in the Curie temperature Tc of up to 333 K in both compounds representing a remarkable enhancement of 38 K.
Furthermore, the introduction of a single fluorine atom at different positions on the benzene ring structure induces subtle modification in crystal structures and offers significant opportunities for tuning the electrical and optical properties of [2, 3 or 4-fluorobenzylammonium]2PbCl4 hybrid halides.23 For example, the compound [2-fluorobenzylammonium]2PbCl423 clearly demonstrates the impact of positional isomerism on crystal symmetries, ferroelectric performance, and semiconductor characteristics (with an energy gap Eg of 3.62 eV). Notably, this compound exhibits a high phase transition temperature of 448 K. Moreover, the incorporation of NH3+cations such as [X-(CH2)2–NH3]+ (X = Br, Cl, I) into halides MX4 (M = Pb, Cu; X = Cl, Br, I) introduces structural flexibility, making it possible to explore their properties in optoelectronic devices.24–26 These structural features involve a significantly reduced band gap as in hybrid halides: [Cl/Br–(CH2)2–NH3]PbI4 (Eg = 2.2 eV),24 [I–(CH2)2–NH3]PbI4 (Eg = 2.45 eV),24 [C–(CH2)2–NH3]CuCl4 (Eg = 2.17 eV)25 and [Cl–(CH2)2–NH3]CuBr4 (Eg = 2.04 eV).26 These results underscore the impact of hydrogen and halogen bonding at the organic–inorganic interface, which influences the distortions of the MX4 inorganic layers of the structures,27 ultimately resulting in a reduced band gap.24 Similarly, halogen substitutions, such as chlorine or bromine substitution within organic species, affect the thermochromism in layered hybrid halides like [Cl–(CH2)2–NH3]CuCl4 and [Br–(CH2)2–NH3]CuCl4.28 Monitoring the thermochromism through absorption spectroscopy of thin films heated from 323 K to 393 K for these compounds reveals a gradual shift toward the red spectrum, indicating a reduction in the band gap energy.29
More recently, the halogen substitution strategy has been extended to modifying the [(CH3)4N]+ cation by replacing one methyl group, leading to the creation of high-Tc ferroelectric materials such as [(CH3)3NCH2Cl]MCl3 (M = Mn and Cd)30 and [((CH3)3NCH2Cl)FeBr4].31 The remarkable ferroelectric properties of these materials stem from the order–disorder of the (CH3)3NCH2Cl organic cation. These outcomes further demonstrate the potency of organo-halogen cations in conjunction with metal halide anions as powerful tools for tailoring crystal symmetries and influencing physical properties.32
Currently, a novel family of hybrid ferroelectric compounds has emerged through the selective bromination of the alkylammonium salts into hybrid metal–halide materials.33–35 A spherical tetramethylammonium cation has been subject to modification by substituting one of its hydrogen atoms with a CH2Br group. This innovation has led to the creation of innovative hybrid halide compounds, namely [Br(CH2)2N(CH3)3]2CuBr434 and [Br(CH2)2N(CH3)3]2CdBr4,35 in which the (2-bromoethyl)trimethylammonium cation occupies the space enclosed by the [CuBr4]2− or [CdBr4]2− anions. Both compounds undergo high Tc reversible phase transitions around 342 K and 390 K and exhibit properties such as ferroelectricity behavior, and switchable dielectric and semiconducting characteristics.
More recently, the bromination strategy has been effectively applied to synthesize new hybrid halide semiconductors, including compounds such as [Br(CH2)2N(CH3)3]2CoBr436 and [Br(CH2)2N(CH3)3]2MIIBr4 (MII = Cu2+, Zn2+).37 It is noteworthy that the brominated [Br(CH2)3N(CH3)3]2Pd2Cl6 has excellent thermal stability, a high Tc phase transition up to 428 K and a narrow optical band gap of approximately 2.2 eV.38 In the present manuscript, we present and investigate another new hybrid metal halide compound [Br(CH2)3N(CH3)3]2PdBr4 characterized by a remarkable reversible phase transition temperature of 363 K. Herein, we detail the crystal structure, phase transition behavior, Raman spectroscopy results, electric performance and optical properties of this compound.
2. Experimental section
2.1. Materials
Palladium(II) bromide (PdBr2, 99%), hydrobromic acid (HBr, 48%), ethanol, deionized water, and (3-bromopropyl)trimethylammonium bromide (Br(CH2)3N(CH3)3Br, 97%) were used without further purification.2.2. Sample preparation
To synthesize the compound [Br(CH2)3N(CH3)3]2PdBr4, 2 mmol of (3-bromopropyl)trimethylammonium bromide (∼0.522 g) white solid was dissolved in aqueous hydrobromic acid solution (15 ml), and 1 mmol of palladium(II) bromide (∼0.266 g) was added. The mixture was heated with stirring and left in air for 2 days, resulting in the formation of brown, prism shaped crystals. Elemental anal. Calcd (%) for [Br(CH2)3N(CH3)3]2PdBr4: C, 18.29; H, 3.84; N, 3.55. Found: C, 18.01; H, 3.51; N, 3.63.3. Characterization
3.1. Single crystal X-ray diffraction
Single crystal X-ray diffraction data were collected at 150 K using a APEXII Bruker AXS diffractometer equipped with a CDD plated detector. Mo Kα radiation (λ = 0.71073 Å) was employed using a multilayer monochromator. The crystal structure was solved using a dual-space algorithm with the SHELXT program,39 and subsequently refined using full-matrix least-squares methods based on F2 with SHELXL-2018/3,40 which is integrated into the WinGX program.41 Empirical absorption corrections were applied to equivalent reflections based on multiscans using SADABS.42 Hydrogen atoms were placed in their calculated positions and treated as riding on their parent atoms with constrained thermal parameters. All non-H atoms were refined with anisotropic atomic displacement parameters. The MERCURY programs43 were employed to create the graphic representations of a crystal structure. A summary of crystallographic information and the refinement results is provided in Table 1. Details of bond distances and angles are provided in Tables S1 and S2 (ESI†).| Empirical formula | C12H30N2PdBr6 |
| Formula weight (g mol−1) | 788.18 |
| Temperature (K) | 150 |
| Crystal system | Orthorhombic |
| Space group | Pbca |
| a (Å) | 9.0670 (12) |
| b (Å) | 14.456 (2) |
| c (Å) | 17.232 (2) |
| α = β = γ (°) | 90 |
| V (Å3) | 2258.6 (5) |
| Z | 4 |
| λ (MoKα) (Å) | 0.71073 |
| ρ cal (g cm−3) | 2.318 |
| Absorption correction | Multi-scan |
| Transmission factors | T min = 0.055, Tmax = 0.102 |
| μ (mm−1) | 11.43 |
| Crystal size (mm3) | 0.32 × 0.25 × 0.20 |
| Crystal color/shape | Brown/prism |
| hkl range | −11≤ h ≤10; −18 ≤ k ≤ 18; −20 ≤ l ≤ 22 |
| θ range for data collection (°) | 2.818 to 27.499 |
| Refinement method | Full-matrix least-squares on F2 |
| No. of collected reflections | 14501 |
| No. of independent reflections | 2595 |
| Observed reflections/restrains/parameters/refined parameters | 2097/0/100 |
| R int | 0.061 |
| F(000) | 1488 |
| GOF on F2 | 1.06 |
| R indices | R1 = 0.029, wR2 = 0.057 |
| Min/Max (ρ/e Å−3) | 0.64/−1.45 |
| CCDC no. | 2116091 |
3.2. Powder X-ray diffraction
The powder X-ray diffraction (PXRD) data of ground crystals were collected at room temperature on a PANalyticalX’Pert MPD diffractometer with Cu Kα1,2 radiation equipped with an X’Celerator detector in the range of 6°< 2θ < 60° 2θ. The experimental X-ray powder diffraction pattern is in very good agreement with the calculated pattern generated from the single-crystal structure (in Pbca), evidencing that the batch contains the title compound as a unique phase. Indexing of the powder pattern has been performed with the program DICVOL.44 The whole powder pattern fitting (pattern-matching with the Le Bail fit) was done employing the FULLPROF program45 available in the software package WinPLOTR46 (Fig. S1, ESI†).3.3. UV-visible spectral measurements
UV-Vis absorption measurements were conducted at room temperature using a standard UV-visible absorption spectrometer (Shimadzu, UV-3101PC) in a transmission configuration. A pellet with a diameter of approximately 8 mm and a thickness of 0.4 mm was employed. The measurements covered the wavelength range of 200–1200 nm.3.4. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)
TGA data were obtained using a Setaram SETSYS 16/18 instrument in the temperature range of 293 K to 730 K, employing a ramp rate of 5 K min−1 under atmospheric pressure in an air atmosphere.For DSC measurement, a PerkinElmer instrument was employed to heat and cool the sample in an air atmosphere at atmospheric pressure. The heating rate was set to 5 K min−1 with a temperature range of 293–450 K.
3.5 Raman measurements
Raman spectra were recorded in the range of 60 cm−1 to 3200 cm−1, as a function of temperature. Using a RENISHAW InVia Reflex spectrometer in combination with a temperature controller, Linkam THMS600 stage. A He laser with a wavelength of 633 nm and power output of 1 mW served as the excitation source. To focus and collect both the incident and scattered lights, we employed an X50 lens. The scattered light from the sample was dispersed using a grating of 1800 lines per mm. For each temperature point, the spectrum was acquired once the desired temperature was reached, with each acquisition lasting 10 minutes.3.6 Electrical measurements
Electrical measurements were carried out on pellet disks with a thickness of 1 mm, utilizing the SOLARTRON SI 1260 impedance equipment coupled to a dielectric interface. These measurements covered a frequency range of 10−1–106 Hz and temperature intervals between 293 and 413 K, applying an AC voltage of 0.5 V. The pellet was positioned between two copper electrodes.4. Results and discussion
4.1. Structural investigations
Brown prism crystals of [Br(CH2)3N(CH3)3]2PdBr4 were synthesized in high yields (87.5%) through a slow evaporation method. Meanwhile, in our previous work, we have discovered that the transition metal halide anions affiliated with organo-halogen cations affect the crystal symmetry and contribute to the design of new hybrid compounds like [Br(CH2)2N(CH3)3]2MIIBr4 (MII = Co2+, Cu2+, Zn2+).36,37 These materials crystallize in the monoclinic system with a P21/c space group and show some similarity of the structure. With replacement of (2-bromoethyl)trimethylammonium by (3-bromopropyl)trimethylammonium in such a system, the new compound [Br(CH2)3N(CH3)3]2PdBr4 possesses high symmetry. Single-crystal X-ray diffraction analysis revealed that the compound crystallizes in the orthorhombic space group, Pbca, with lattice parameters of a = 9.0670 (12) Å, b = 14.456 (2) Å, c = 17.232 (2) Å and Z = 4. The asymmetric unit consists of one half of a tetrabromopalladate(II) anion and one (3-bromopropyl) trimethylammonium cation (see Fig. 1). | ||
| Fig. 1 The asymmetric unit of [(CH3)3N(CH2)3Br]2PdBr4. Hydrogen bonds are shown using red dotted lines. Symmetry codes: (i) −x + 2, −y, −z. | ||
In the [PdBr4]2− anion, Pd2+ ions adopt a square planar coordination geometry, formed by four bromide ions, with Pd–Br lengths ranging from 2.4358 (4) to 2.4415 (4) Å. The Br–Pd–Br angles vary within the range of 89.805 (15)–90.195 (15)°for the cis configuration and are exactly 180° for the trans configuration (Table S1, ESI†). These values are consistent with those observed in related Pd(II) complexes.47,48 The coordination geometry near the Pd(II) center can be determined by calculating the parameter τ4 [τ4 = 360° − (α + β)/141°], where α and β are the two largest angles in the four-coordinate metal ion species, from 360°, then dividing by 141°.49 For a perfect square-planar geometry, τ = 0, whereas for an ideal tetrahedral geometry, τ = 1 in the title compound. τ4 is zero, indicating a coordination geometry in an ideal square-planar orientation. The packing of the compound reveals an alternating arrangement of two organic cations [Br(CH2)3N(CH3)3]+ and one [PdBr4]2− anions (Fig. S2, ESI†). These entities are alternated forming infinite zigzag chains that run parallel to the c-axis (Fig. 2).
 | ||
| Fig. 2 The packing structure of [(CH3)3N(CH2)3Br]2PdBr4 viewed along the a-axis. Hydrogen bonds are depicted as red dashed lines. | ||
Likewise, [Br(CH2)3N(CH3)3]+ and [PdBr4]2− ions are interconnected by intermolecular hydrogen bonds formed between the –N(CH3)3 and –Br groups creating a supramolecular architecture with C–H⋯Br distances ranging from 3.705 (4) to 3.979 (4) Å. This structure motif bears resemblances to well-studied A2MX4 organically templated metal halides,31,34–36 where “A” represents protonated organo-halogen molecules, “M” denotes a metal (Fe, Cu, Cd, and Co) and “X” is a halogen atom (Cl, Br or I). The shortest Pd⋯Pd distances in [Br(CH2)3N(CH3)3]2PdBr4 are equal to 8.532 Å which is longer than those found in [(R)/(S)-2-methylpiperazinediium]PdCl448 (6.937 Å) and [2,4,6-trimethyl-m-phenyl-enediaminediium]PdBr450 (5.692 Å). These can be explained by the difference in sizes and shapes of the incorporated organic cations, highlighting structural diversity and adjustability. Geometrical characteristics of the organic cation are detailed in Table S1 (ESI†). The Br–C distance is 1.963 (4), C–C and C–N distances range, respectively, from 1.508 (5) to 1.517 (5) Å and from 1.489 (5) to 1.520 (4) Å. Each organic cation through hydrogen atoms is bonded to C atoms in C–H⋯Br hydrogen bonds, either in a monodentate or bidentate manner, leading to an extended supramolecular architecture (see Fig. S3, ESI†). Simultaneously, each [PdBr4]2− anion is surrounded by six [Br(CH2)3N(CH3)3]+ cations connected through C–H⋯Br hydrogen-bonding interactions (Fig. S4, ESI†). The geometric detail of these hydrogen bonds is provided in Table S2 (ESI†). It is important to note that no typical halogen⋯halogen bond interactions under 4 Å were found for terminal Br atoms in the material. Indeed, two weak halogen bonds are observed: the C–Br⋯Br–C bonds of 5.018(2) Å between two organic cations and halogen bonding interaction among cation and anion (C–Br⋯Br–Pd) measure 4.087(2) Å.
4.2. Optical properties
UV-vis absorption spectra were obtained to investigate the optical properties of the [(CH3)3N(CH2)3Br]2PdBr4 material. As depicted in Fig. 3, a sharp absorption edge is observed at approximately 370 nm, indicating the direct band gap semiconducting nature of this material. | ||
| Fig. 3 Variation of the absorbance with wavelength and gap energy of [(CH3)3N(CH2)3Br]2PdBr4. | ||
This band gap (Eg) was calculated to be 2 eV using the Tauc equation in conjunction with the Kubelka–Munk relation.51 This band gap value is relatively small compared to that of other materials. For instance, in [(CH3)2NH2]2PdBr4, the band gap Eg = 2.5 eV,52 [(CH3)3N(CH2)2Br]2CoBr4 exhibits an Eg = 3.7 eV36 and [(CH3)3N(CH2)3Br]2Pd2Cl6 has an Eg = 2.2 eV.38
4.3. Thermal analysis
Thermogravimetric analysis (TGA) data display only one main degradation step in the temperature ranging from 293 K to 730 K and reveal that the [(CH3)3N(CH2)3Br]2PdBr4 compound shows good thermal stability, with the onset decomposition temperature reaching 520 K. In the temperature range of 550–700 K, a 67.5% weight loss (theoretical weight loss is 66.22%) is attributed to the decomposition of two organic cations [Br(CH2)3N(CH3)3]+ together with 2Br− anions and corresponds to the formation of PdBr2 (Fig. 4). | ||
| Fig. 4 Differential scanning calorimetric curves on heating and cooling and the TGA curve of [(CH3)3N(CH2)3Br]2PdBr4. | ||
The DSC results measured at heating/cooling rates of 5 K min−1 and 10 K min−1 show two reversible phase transitions at T1 = 363 K/333 K and T2 = 393 K/353 K with the rate of 5 K min−1 and at T1 = 358 K/336 K and T2 = 403 K/355 K with the rate of 10 K min−1 (Fig. 4). The later temperature transition can be attributed to a congruent melting of the organic component, similar to observation in other hybrid halide materials.53,54 The calculated entropy of the first peak aids in determining the type of transition which is equal to 5.64 J mol−1 K−1 and 5.82 J mol−1 K−1 with heating rates of 10 K min−1 and 5 K min−1, respectively. These values exceed 2 J mol−1 K−1 which it is usually considered as an indication of an order–disorder type transition.55 The order of this transition can be determined from the variation of the transition temperature versus the heating/cooling rate (Fig. 5).
 | ||
| Fig. 5 Dependence of temperature hysteresis on the scanning rates 5 °C min−1 and 10 °C min−1 at DSC measurements of [(CH3)3N(CH2)3Br]2PdBr4. | ||
The extrapolation of the heating and cooling curves shows that the linear fits of the heating and cooling transitions had different values at zero rate which indicates that this phase transition is considered as a first order type.56
4.4. Vibrational study and phase transition
We present a comprehensive collection of Raman spectra of the [(CH3)3N(CH2)3Br]2PdBr4 compound covering the frequency range from 60–3200 cm−1 at temperature ranging from 293 K to 433 K. The Raman spectrum at 293 K exhibits four different frequency regions: (a) below 350 cm−1; (b) in the 350–1100 cm−1 frequency range; (c) in the 1200–1600 cm−1 range and (d) in the 2700–3100 cm−1 range (Fig. 6(a)).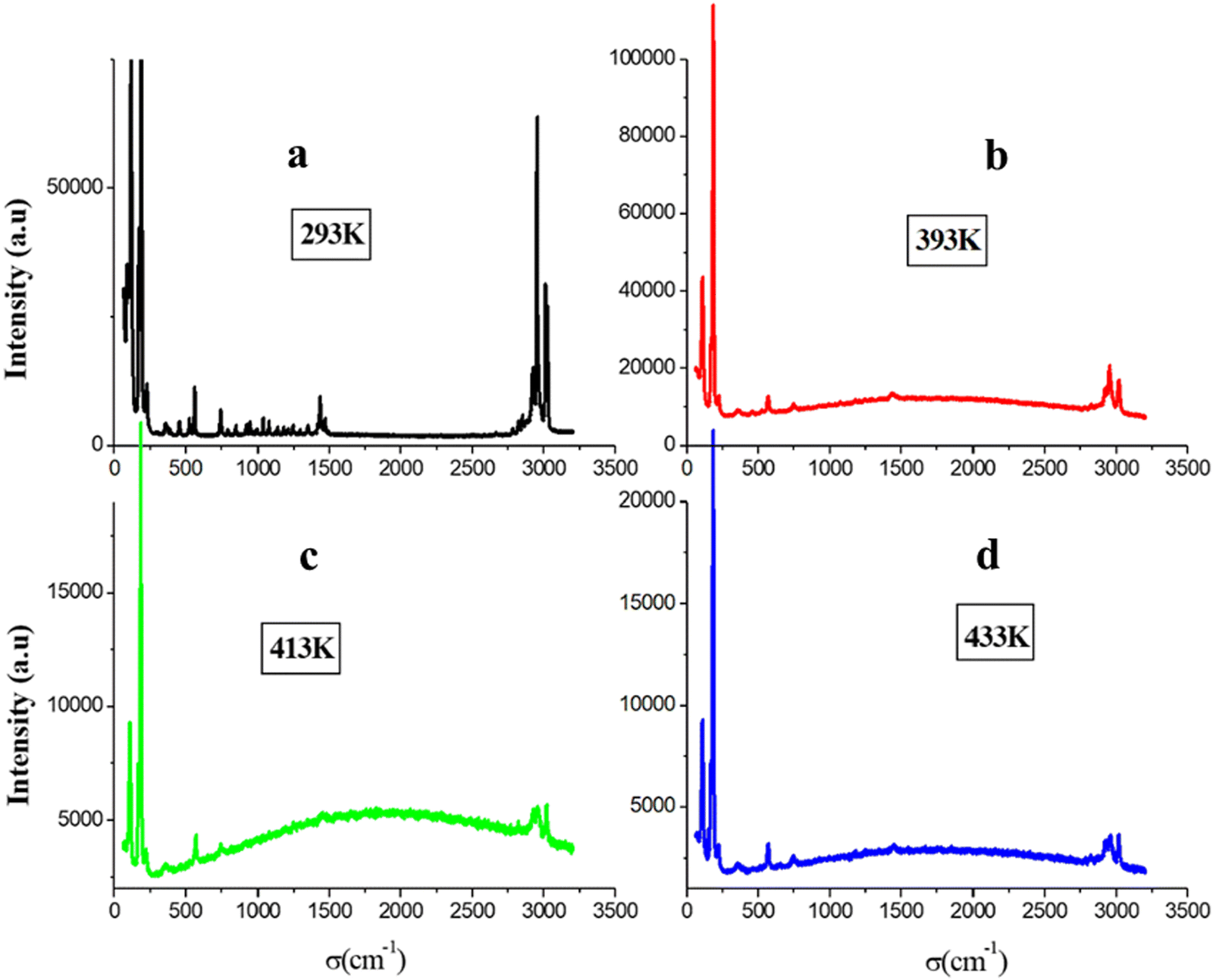 | ||
| Fig. 6 Raman spectra at 293 K (a), 393 K (b), 413 K (c) and 433 K (d) of [(CH3)3N(CH2)3Br]2PdBr4. | ||
In greater detail, the recorded Raman spectrum of the orthorhombic phase reveals vibrational signals at 117, 172, 187 and 226 cm−1, which are attributed to the internal vibration stretching modes of PdBr4.56 The second region encompasses all internal vibration deformation and stretching modes of the C–C, C–Br and N–C bonds.56 The third region features peaks assigned to symmetric and asymmetric deformation involving the molecules CH3 and CH2. The final region at higher wave number corresponds to the symmetric and asymmetric stretching internal vibration modes of C–H bonds.56 The details of the attribution are provided in Table 2.
| σ (cm−1) | Assignements |
|---|---|
| 89 | External modes |
| 117 | v 4 (PdBr4) |
| 172 | v 2 (PdBr4) |
| 187 | v 1 (PdBr4) |
| 226 | v 3 (PdBr4) |
| 359 | δ (CC) |
| 374 | |
| 391 | |
| 455 | |
| 525 | v (C–Br) |
| 562 | |
| 745 | v (N–C) |
| 793 | |
| 850 | |
| 924 | v (C–C) |
| 948 | |
| 968 | |
| 994 | |
| 948 | |
| 968 | |
| 994 | |
| 1036 | |
| 1076 | |
| 1117 | δ as (CH3) and δas (CH2) |
| 1137 | |
| 1178 | |
| 1208 | |
| 1243 | |
| 1294 | |
| 1348 | |
| 1395 | |
| 1416 | |
| 1434 | |
| 1467 | |
| 1491 | |
| 2781 | δ s (C–H) and δas (C–H) |
| 2823 | |
| 2850 | |
| 2923 | |
| 2953 | |
| 2964 | |
| 2972 | |
| 3012 | |
| 3026 |
The presence of all these vibration modes validates the structural analysis and confirms the existence of both organic cation and inorganic anion entities. The additional Raman spectra (Fig. 6(b)–(d)) were recorded at 393 K, 413 K and 433 K, where some peaks associated with the organic cation are absent. This observation indicates the onset of the congruent melting of the organic part at 393 K thereby confirming the second peak observed in the DSC curve. We recorded the two Raman spectra at ambient before heating and after cooling with the Raman spectrum after the fusion to prove the DSC result and the reversibility of the fusion (Fig. S5, ESI†). When these spectra are compared, it is shown that the fusion phase is reversible because the two ambient spectra are the same and distinct from those obtained after the fusion. Furthermore, the breaking of the N–C–H link between the organic cation and the inorganic anion following the congruent melting of the cations part is responsible for the change in the wave number for the inorganic entity and the alteration of the C–H bonds after reaching 393 K (Fig. 7).
 | ||
| Fig. 7 The position of Pd–Br and C–H vibration modes before and after the melting temperature of [(CH3)3N(CH2)3Br]2PdBr4. | ||
The evolution of the Raman spectra as a function of temperature provides valuable insights into changes in molecular bonds, the phase-transition mechanism and the interactions between the organic cations and the inorganic framework. The thermal evolution is depicted in Fig. S6 (ESI†) and the variation in the position of vibration modes is shown in Fig. S7 (ESI†). Near the temperature of 360 K, these spectra show no temperature-dependent changes, proving that this transition is not of a structural character. At 390 K, we notice a sudden change in the position of the Pd–Br modes which confirms the beginning of the fusion of the organic part. The mechanism of the transition at 360 K can be depicted by a multiwell potential energy surface governing the rotator rotational motion of the molecules where, below the phase transition temperature, the rotating component of the CH3 molecule vibrates around the potential minima of one of the wells. Above this temperature (360 K), it possesses enough thermal energy to overcome the rotational energy barrier, leading to a rotational motion as the atoms jump between equivalent lattice sites (referred to as the disorder mechanism) represented at the bottom of each well.57–59 The significant changes at 360 K in the linewidth (FWHM) of CH3 vibration modes during the transition indicate that the cation plays a crucial role in the phase transition (Fig. 8(a) and (b)).
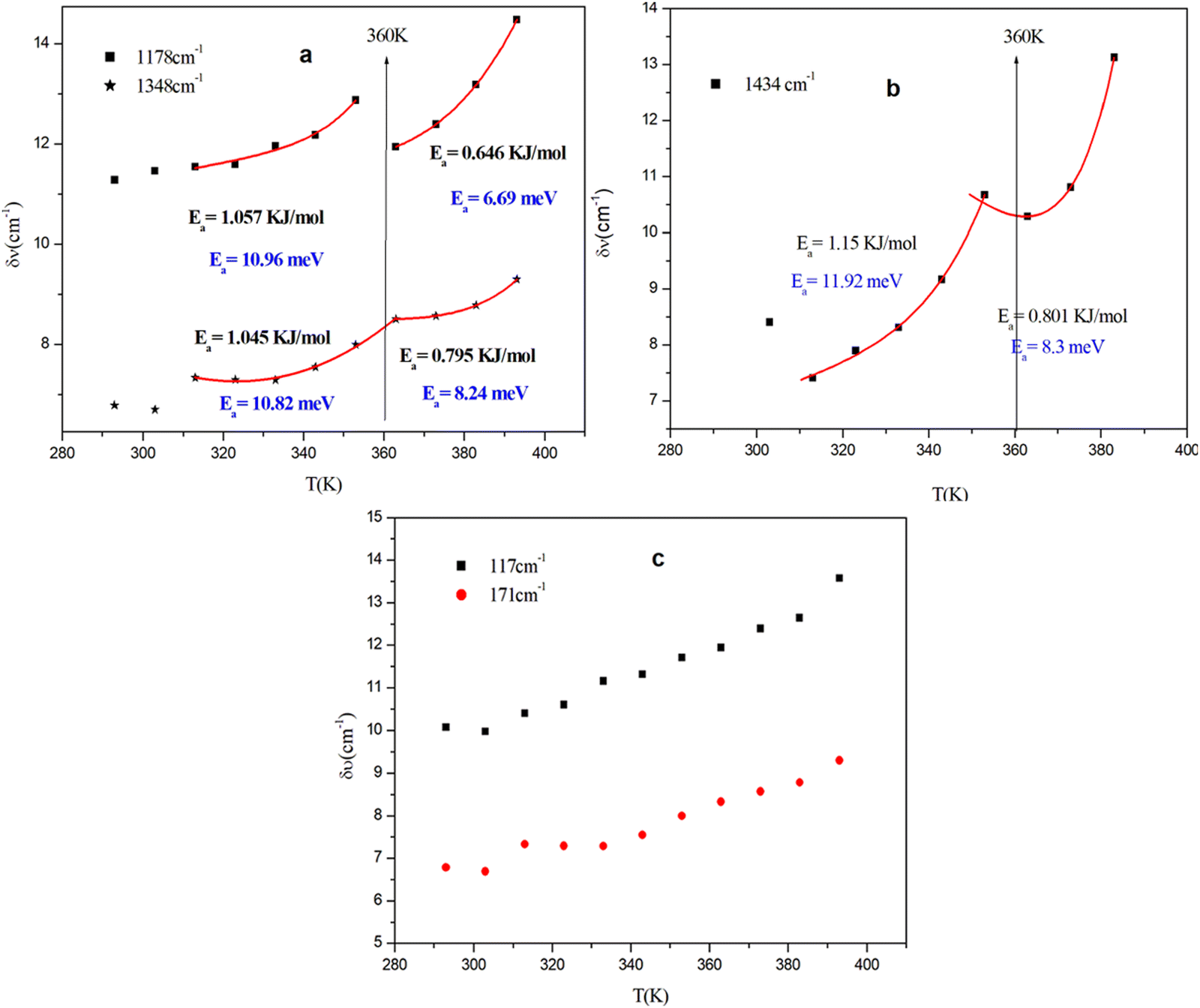 | ||
| Fig. 8 Temperature dependence of the Raman vibrational frequency and FWHM of the CH3 (a) and (b) and Pd–Br (c) Raman modes; solid lines correspond to the fitting results according to eqn (1). | ||
This change is not observed for other vibration modes of the inorganic entity (Fig. 8(c)). This demonstrates that only an intermediate strength of interaction governs the dynamics and phase transitions of the order–disorder type. The potential barrier for the reorientation of the CH3 molecule can be extracted from the broadening of the modes (eqn (1)), as the orientational correlation time represents the mean reorientational time of the atoms jumping from one potential well to another.60,61 The activation energy (Ea) can be calculated using the linewidth for vibrational and reorientation relaxation using eqn (1):
 | (1) |
The activation energies extracted from fitting the FWHM experimental data of several CH3 vibration modes (Fig. 8(a) and (b)) are reported in Table 3.
| T = 393 K | ||
|---|---|---|
| Frequencies (cm−1) | Activation energies: phase I | Activation energies: phase II |
| 1178 | 10.96 meV | 6.69 meV |
| 1348 | 10.82 meV | 8.24 meV |
| 1434 | 11.92 meV | 8.3 meV |
The higher reorientation activation energy of the CH3 vibration modes in the first phase (low temperature) indicates that roto-translational motion is relatively restricted.62 In the second phase (high temperature) the activation energies for most of the vibrational modes are smaller. Consequently, we can conclude that the activation energy significantly decreases in the higher-temperature phase which is consistent with a diminished cation ordering.
4.5. Electrical properties and Nyquist diagrams
An impedance investigation of ionic conductors over a wide frequency range has the advantage of helping to distinguish between electrical resistivity originating from the grain and grain boundary. It provides an accurate representation of the electrical properties of a material, enabling the identification of charge transport processes within both the grains and grain boundaries across a temperature range.62 The Nyquist plots for [Br(CH2)3N(CH3)3]2PdBr4 at different temperatures are shown in Fig. 9(a)–(c).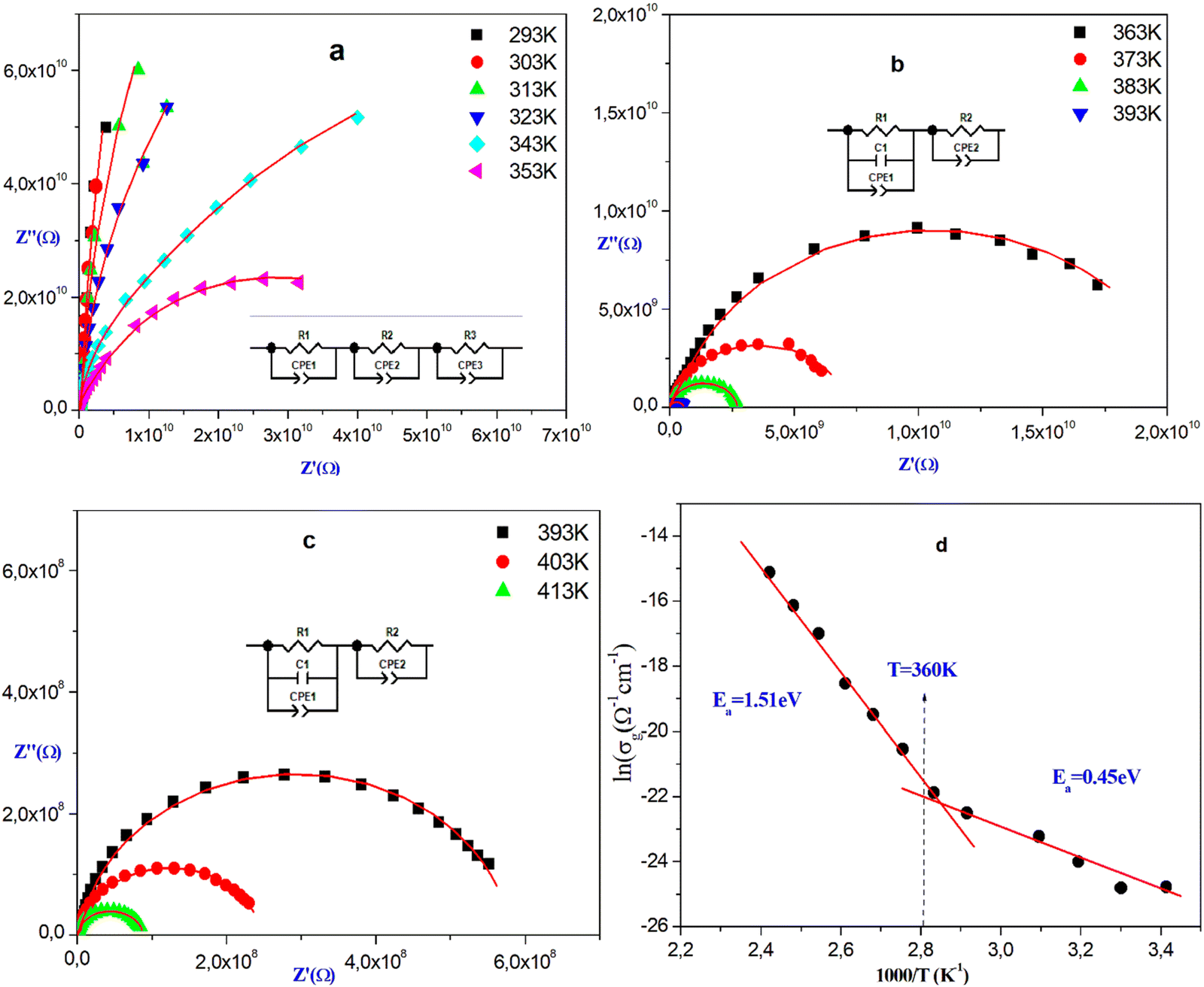 | ||
| Fig. 9 (a)–(c) Nyquist diagrams and equivalent circuit, and (d) variation of the conductivity of grain vs. 1000/T. | ||
The data reveal a semicircle at all temperatures, with their centers shifted toward the real axis indicating a non-Debye type of relaxation. The choosen equivalent circuit shows two semi-circles where the experimental and theoretical curves are superposed. The semicircular arc at higher frequencies corresponds to the grains, and at lower frequencies corresponds to the grain boundaries. The resistance shown in the Nyquist diagrams decreases with increasing temperature, likely due to the increasing conductivity. This confirms that the conduction process is thermally activated, supporting the semiconducting nature of this material.63
At higher temperatures (T > 363 K), all Nyquist diagrams are modeled by the same equivalent circuit consisting of two cells in series (Fig. 9(b) and (c)). The first cell comprises a parallel combination of resistance (Rg), capacitor (C) and a constant phase element (CPE), while the second cell is described by a parallel combination of resistance (Rjg) and a constant phase element (CPE) attributed to the grain and grain boundary effects, respectively. At low temperatures (T ≤ 363 K), the equivalent circuit consists of three cells connected in series, with a pure resistor in parallel with a fractal capacity (R//CPE) (Fig. 9(a)). The good agreement between the calculated lines and the experimental data confirms that the suggested equivalent circuit accurately describes the electrical behavior of this material. Based on the values extracted from the equivalent circuit using Z-view software, the direct conductivities for the different relaxations are determined at each temperature using the following expression:
 | (2) |
Fig. 9(d) shows the grain conductivity vs. 103/T plot of [Br(CH2)3N(CH3)3]2PdBr4 material. It is evident that the increases of the conductivity with rising temperature, suggesting the thermally activated process in this material.64 This behavior follows the Arrhenius law. Notably, there is a change in the slope observed at T = 360 K corresponding to the transition detected by the calorimetric study. This change indicates a shift in the activation energies. The activation energies have been evaluated, EaII = 1.51 eV and EaI = 0.45 eV. These values imply that the charge transport mechanism undergoes a change at this temperature, providing evidence for the order–disorder nature of this transition.
5. Conclusions
In summary, we have successfully synthesized a new material with semiconductor characteristics, featuring a bandgap energy equal to 2 eV. The parent compound [Br(CH2)3N(CH3)3]2PdBr4 crystallizes in an orthorhombic system with the Pbca space group. This material exhibits stability up to 520 K, with a single-phase transition occurring at 363 K. Raman spectroscopy provides confirmation of the order–disorder nature of this transition and the electrical properties further validate its presence through the observed change in the activation energy. We have established an equivalent circuit that describes the material and utilized the fitted parameters to determine grain conductivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through small group Research Project under grand number RGP.1/41/44.References
- S. Han, X. Liu, Y. Liu, Z. Xu, Y. Li, M. Hong, J. Luo and Z. Sun, J. Am. Chem. Soc., 2019, 141, 12470 CrossRef CAS PubMed.
- Q. Pan, Z. B. Liu, H. Y. Zhang, W. Y. Zhang, Y. Y. Tang, Y. M. You, P. F. Li, W. Q. Liao, P. P. Shi, R. W. Ma, R. Y. Wei and R. G. Xiong, Adv. Mater., 2017, 29, 1700831 CrossRef.
- D.-W. Fu, H.-L. Cai, Y. Liu, Q. Ye, W. Zhang, Y. Zhang, X.-Y. Chen, G. Giovannetti, M. Capone, J. Li and R.-G. Xiong, Science, 2013, 339, 425 CrossRef CAS.
- B. Batlogg, R. Cava, A. Jayaraman, R. Van Dover, G. Kourouklis, S. Sunshine, D. Murphy, L. Rupp, H. Chen, A. White, K. T. Short, A. M. Mujsce and E. A. Rietman, Phys. Rev. Lett., 1987, 58, 2333 CrossRef CAS.
- C.-F. Wang, J.-X. Gao, C. Li, C.-S. Yang, J.-B. Xiong and Y.-Z. Tang, Inorg. Chem. Front., 2018, 5, 2413 RSC.
- S. Kataoka, S. Banerjee, A. Kawai, Y. Kamimura, J. Choi, T. Kodaira, K. Sato and A. Endo, J. Am. Chem. Soc., 2015, 137, 4158 CrossRef CAS PubMed.
- D. Guedes-Sobrinho, D. N. Silveira, L. O. de Araujo, J. F. Dalmedico, W. Wenzel, Y. Pramudya, M. J. Piotrowski and C. R. C. Rêgo, Nature, 2023, 13, 4446 CAS.
- M. S. de Holanda, R. F. Moral, P. E. Marchezi, F. C. Marques and A. F. Nogueira, EcoMat., 2021, 3, 12124 CrossRef.
- T. Schmitt, S. Bourelle, N. Tye, G. Soavi, A. D. Bond, S. Feldmann, B. Traore, C. Katan, J. Even, S. E. Dutton and F. Deschler, J. Am. Chem. Soc., 2020, 142, 5060 CrossRef CAS PubMed.
- B. Vargas, G. Rodríguez-López and D. Solis-Ibarra, ACS Energy Lett., 2020, 5, 3591 CrossRef CAS.
- A. García-Fernández, E. J. Juarez-Perez, J. M. Bermúdez-García, A. L. Llamas-Saiz, R. Artiaga, J. J. López-Beceiro, M. A. Señarís-Rodríguez, M. Sánchez-Andújar and S. Castro-García, J. Mater. Chem. C, 2019, 7, 10008 RSC.
- A. García-Fernández, J. M. Bermúdez-García, S. Castro-García, A. L. Llamas-Saiz, R. Artiaga, J. López-Beceiro, S. Hu, W. Ren, A. Stroppa, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, Inorg. Chem., 2017, 56, 4918 CrossRef.
- M. A. Asghar, S. Zhang, T. Khan, Z. Sun, A. Zeb, C. Ji, L. Li, S. Zhao and J. Luo, J. Mater. Chem. C, 2016, 4, 7537 RSC.
- D. B. Mitzi, C. D. Dimitrakopoulos and L. L. Kosbar, Chem. Mater., 2001, 13, 3728 CrossRef CAS.
- N. Song, S.-P. Chen, X.-W. Fan, Y.-H. Tan, W.-J. Wei and Y.-Z. Tang, ACS Omega, 2020, 5, 6773 CrossRef CAS PubMed.
- Z.-Y. Yue, W. Luo, N. Wang, H.-K. Li, Z.-J. Xu, Y. Feng, C. Shi, H.-Y. Ye and L.-P. Miao, CrystEngComm, 2023, 25, 1270 RSC.
- X. Mu, H. Y. Zhang, L. Xu, Y. Y. Xu, H. Peng, Y. Y. Tang and R. G. Xiong, APL Mater., 2021, 9, 051112 CrossRef CAS.
- C. Han, J. A. McNulty, A. J. Bradford, A. M. Z. Slawin, F. D. Morrison, S. L. Lee and P. Lightfoot, Inorg. Chem., 2022, 61, 3230 CrossRef CAS.
- Y.-Y. Tang, Y. Ai, W.-Q. Liao, P.-F. Li, Z.-X. Wang and R.-G. Xiong, Adv. Mater., 2019, 31, 1902163 CrossRef.
- X.-N. Hua, W.-Q. Liao, Y.-Y. Tang, P.-F. Li, P.-P. Shi, D. Zhao and R.-G. Xiong, J. Am. Chem. Soc., 2018, 140, 12296 CrossRef CAS PubMed.
- Y. Ai, X.-G. Chen, P.-P. Shi, Y.-Y. Tang, P.-F. Li, W.-Q. Liao and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 4474 CrossRef CAS PubMed.
- Y. Zhang, W.-Q. Liao, D.-W. Fu, H.-Y. Ye, Z.-N. Chen and R.-G. Xiong, J. Am. Chem. Soc., 2015, 137, 4928 CrossRef CAS.
- P.-P. Shi, S.-Q. Lu, X.-J. Song, X.-G. Chen, W.-Q. Liao, P.-F. Li, Y.-Y. Tang and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 18334 CrossRef CAS PubMed.
- S. Sourisseau, N. Louvain, W. Bi, N. Mercier, D. Rondeau, F. Boucher, J.-Y. Buzaré and C. Legein, Chem. Mater., 2007, 19, 600 CrossRef CAS.
- F. Hajlaoui, N. Audebrand, T. Roisnel and N. Zouari, J. Appl. Organomet. Chem., 2019, 1, 5293 Search PubMed.
- F. Akrout, F. Hajlaoui, K. Karoui, N. Audebrand, T. Roisnel and N. Zouari, J. Solid State Chem., 2020, 287, 121338 CrossRef CAS.
- J. L. Knutson, J. D. Martin and D. B. Mitzi, Inorg. Chem., 2005, 44, 4699 CrossRef CAS PubMed.
- C. Pareja-Rivera and D. Solis-Ibarra, Adv. Opt. Mater., 2021, 9, 2100633 CrossRef CAS.
- G. Marcotrigiano, L. Menabue and G. Pellacani, Inorg. Chem., 1976, 15, 2333 CrossRef CAS.
- Y.-M. You, W.-Q. Liao, D. Zhao, H.-Y. Ye, Y. Zhang, Q. Zhou, X. Niu, J. Wang, P.-F. Li, D.-W. Fu, Z. Wang, S. Gao, K. Yang, J.-M. Liu, J. Li, Y. Yan and R.-G. Xiong, Science, 2017, 357, 306 CrossRef CAS.
- Y. Zhang, X.-J. Song, Z.-X. Zhang, D.-W. Fu and R.-G. Xiong, Matter, 2020, 2, 697 CrossRef.
- C. Han, A. J. Bradford, J. A. McNulty, W. Zhang, P. S. Halasyamani, A. M. Z. Slawin, F. D. Morrison, S. L. Lee and P. Lightfoot, Chem. Mater., 2022, 34, 2458 CrossRef CAS.
- D.-F. Li, X.-H. Deng, Y.-X. Ma, D.-S. Liu, X.-F. Huang, X.-F. Zhao, W.-T. Chen and Y. Sui, J. Phys. Chem. C, 2022, 126, 728 CrossRef CAS.
- Y. Sui, Y.-S. Zhong, J.-J. Wang, Q. Xia, L.-J. Wang and D.-S. Liu, J. Mater. Chem. C, 2019, 7, 14294 RSC.
- Y. Sui, D.-S. Liu, W.-T. Chen, L.-J. Wang, Y.-X. Ma, H.-Q. Lai, Y.-W. Zhou and H.-R. Wen, Chem. – Asian J., 2020, 15, 1 CrossRef.
- I. Dakhlaoui, K. Karoui, F. Hajlaoui, N. Audebrand, T. Roisnel and F. Jomni, J. Mol. Struct., 2021, 1231, 129684 CrossRef CAS.
- K. Karoui, F. Hajlaoui, N. Audebrand, T. Roisnel and A. Ben Rhaiem, J. Alloys Compd., 2020, 844, 156115 CrossRef CAS.
- I. Dakhlaoui, K. Karoui, F. Hajlaoui, M. Zaghrioui, N. Audebrand, M. Dallon and F. Jomnis, New J. Chem., 2023, 47, 8042 RSC.
- G. M. Sheldrick, Acta Cryst., 2015, A71, 3 CrossRef.
- G. M. Sheldrick, SHELXL-2018: Programs for Crystal Structure Refinement, University of Göttingen, Göttingen, 2018 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- G. M. Sheldrick, SADABS version 2.03, Bruker AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- D. Louër and A. Boultif, Some further considerations in powder diffraction pattern indexing with the dichotomy method, Powder Diffr., 2014, 29, S7–S12 CrossRef.
- J. Rodríguez-Carvajal and T. Roisnel, Line Broadening Analysis Using Fullprof: Determination of Microstructural Properties, Mater. Sci. Forum, 2004, 443–444, 123–126 Search PubMed.
- T. Roisnel and J. Rodríguez-Carvajal, WinPLOT: A Windows Tool for Powder Diffraction Pattern Analysis, Mater. Sci. Forum, 2001, 378–381, 118–123 CAS.
- X. Liu, T. J. Huang, L. Zhang, B. Tang, N. Zhang, D. Shi and H. Gong, Chem. – Eur. J., 2018, 24, 4991 CrossRef CAS.
- B. Ben Salah, F. Hajlaoui, K. Karoui, N. Audebrand, T. Roisnel, S. Freslon, N. Zouari and F. Jomni, Mater. Res. Bull., 2023, 164, 112251 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- H. Bouznif, F. Hajlaoui, K. Karoui, N. Audebrand, M. Cordier, T. Roisnel and N. Zouari, J. Solid State Chem., 2022, 311, 123149 CrossRef CAS.
- J. Tauc, Amorphous and liquid semiconductors, Plenum Press, New York, 1974 Search PubMed.
- K. Trabelsi, N. Drissi, F. Hajlaoui, M. Zighrioui, A. Rhaiem, N. Audebrand, T. Roisnel and K. Karim, RSC Adv., 2023, 13, 23348 RSC.
- T. Li, W. A. Dunlap-Shohl, E. W. Reinheimer, P. Le Magueres and D. B. Mitzi, Chem. Sci., 2019, 10, 1168 RSC.
- F. Hleli, N. Mercier, M. Ben Haj Salah, M. Allain, T. Travers, D. Gindre, N. Zouari and C. Botta, J. Mater. Chem. C, 2022, 10, 10284 RSC.
- P. Szklarz, M. Owczarek, G. Bator, T. Lis, K. Gatner and R. Jakubas, J. Mol. Struct., 2009, 929, 48 CrossRef CAS.
- I. Dakhlaoui, K. Karoui and F. Jomni, Appl. Organomet. Chem., 2020, 5545, 1 Search PubMed.
- A. Colin-Molina, D. P. Karothu, M. J. Jellen, R. A. Toscano, M. A. Garcia-Garibay, P. Naumov and B. Rodríguez-Molina, Matter, 2019, 1, 1033 CrossRef.
- H. Chung, D. Dudenko, F. Zhang, G. D’Avino, C. Ruzié, A. Richard, G. Schweicher, J. Cornil, D. Beljonne, Y. Geerts and Y. Diao, Nat. Commun., 2018, 9, 1 CrossRef CAS.
- T. Ivanovska, C. Quarti, G. Grancini, A. Petrozza, F. De Angelis, A. Milani and G. Ruani, ChemSusChem, 2016, 9, 2994 CrossRef CAS.
- C. Carabatos-Nédelec and P. Becker, J. Raman Spectrosc., 1997, 28, 663 CrossRef.
- P. d R. Andrade and S. P. S. Porto, Solid State Commun., 1973, 13, 1249 CrossRef CAS.
- A. Orilukas, A. Dindune, Z. Kanepe, J. Ronis, E. Kazakevicius and A. Kezionis, Solid State Ionics, 2003, 157, 177 CrossRef.
- K. Jawahar, B. Behera and R. N. P. Choudhary, J. Mater. Sci.: Mater. Electron., 2009, 20, 872 CrossRef CAS.
- S. K. Barik, R. N. P. Choudhary and A. K. Singh, Adv. Mater. Lett., 2011, 2, 419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2116091. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj04819e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |