
Interaction of phytate with cyclic polyamines†
Julia
Torres
 *a,
Nicolás
Veiga
a,
Matteo
Savastano
b,
Carlos
Kremer
a and
Antonio
Bianchi
c
*a,
Nicolás
Veiga
a,
Matteo
Savastano
b,
Carlos
Kremer
a and
Antonio
Bianchi
c
aÁrea Química Inorgánica, Departamento Estrella Campos, Facultad de Química, Universidad de la República, Gral. Flores 2124, Montevideo, Uruguay. E-mail: jtorres@fq.edu.uy
bDepartment of Human Sciences and Quality of Life Promotion, University San Raffaele Roma, Via di Val Cannuta 247, 00166 Rome, Italy
cDepartment of Chemistry “Ugo Schiff”, University of Florence, Sesto Fiorentino, Italy
First published on 24th November 2023
Abstract
myo-Inositol hexakisphosphate (InsP6) is a widespread molecule, present in relatively high concentrations in nature, and plays key roles in many metabolic processes. Its complex chemistry arises from the presence of flexible ring bearing six multivalent phosphates that can either be anionic or interact with a myriad of cationic species in solution, including protonated polyamines. Understanding the interaction of this biomolecule with inorganic and organic cations aims to gain a better picture of its chemical and structural behavior in biological systems. Building on our previous work on the interaction of InsP6 with inorganic cations and linear polyamines, here, in this study, we present the corresponding interaction with cyclic polyamines via potentiometry, with the help of computational tools, in order to understand the main determinants of the stability of the formed species and the structural insights that rationalize the interaction. Stable InsP6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polyamine 1:1 species are detected and the strength of the interaction results from an interplay of electrostatic attractions and hydrogen bonding formation. Even though the results show a certain resemblance to the interaction of InsP6 with linear polyamines, the higher rigidity imposed by cyclization increases the selectivity of InsP6 towards polyamines, giving rise to the molecular recognition of hexamine 18N6, due to the spatial geometric fit of the charge distribution of the interacting anionic InsP6 and the protonated polyamine.
:polyamine 1:1 species are detected and the strength of the interaction results from an interplay of electrostatic attractions and hydrogen bonding formation. Even though the results show a certain resemblance to the interaction of InsP6 with linear polyamines, the higher rigidity imposed by cyclization increases the selectivity of InsP6 towards polyamines, giving rise to the molecular recognition of hexamine 18N6, due to the spatial geometric fit of the charge distribution of the interacting anionic InsP6 and the protonated polyamine.
Introduction
myo-Inositol hexakisphosphate (phytate, InsP6) is the most abundant member of biological messengers belonging to the inositol phosphate family. Its intracellular concentration ranges from 10 to 500 μM in living organisms1–4 and it has proved to be of utmost importance in cell functioning,5–9 showing also various pharmacological effects.6,10–14 The detailed biological functions and modes of actions of InsP6 both alone or in the presence of other interacting species still remain a subject of controversy mainly because of its intricate chemistry.8,9The neutral form of InsP6 (H12L) dissociates in aqueous solution to give the corresponding anions, HiL(12−i)−, up to L12− (Fig. 1(a) and (b), showing the two possible conformers) depending on pH values. These anions have shown a very high affinity for metal ions and other positively charged species6 but the precise speciation of InsP6 in any media is still a difficult task, due to the intertwined set of protonation and interaction equilibria that could also lead to the formation of sparingly soluble species.15
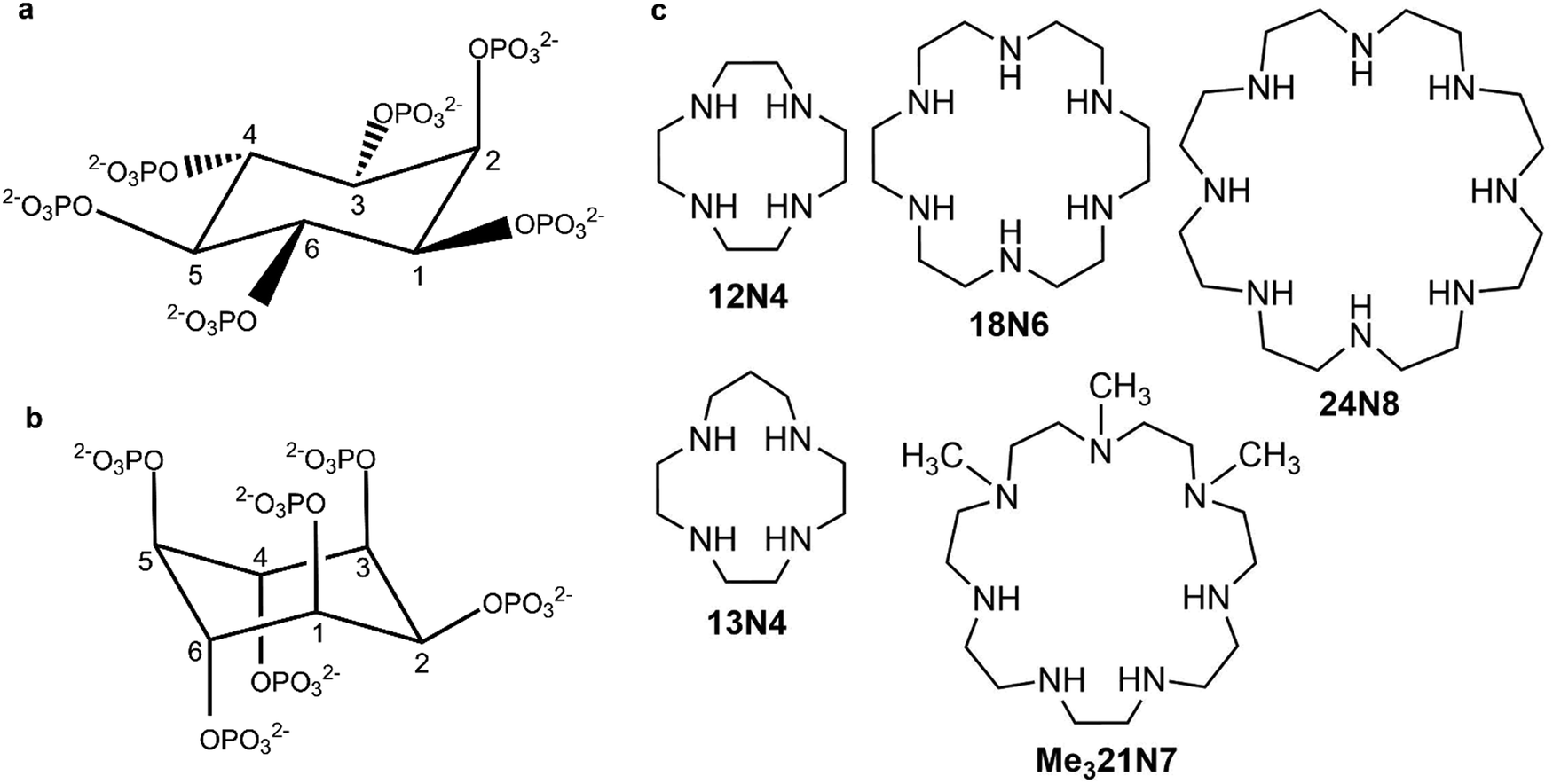 | ||
| Fig. 1 (a) Structure of InsP6 (L12−) for both conformations: (a) 1 axial-5 equatorial (1a5e) and (b) 5 axial-1 equatorial (5a1e). (c) The studied cyclic polyamines. | ||
Among positively charged potentially interacting species, polyamines pose a special challenge for studies in aqueous solutions with anions. They form various protonated species that could strongly interact with the highly charged anions formed by InsP6. Though not totally understood, polyamines have been associated with the observed biological effect of InsP6 under different scenarios.16–18 According to previous reports on linear biogenic polyamines, the most relevant species formed with InsP6 are soluble adducts of 1:1 polyamine:InsP6 stoichiometry in different protonation states.19–21 The formed adducts can be formulated as [(HnA)(HiL)](12−n−i)− where A represents the neutral form of the polyamine, L12− is the fully deprotonated form of InsP6 and the relative protonation is assigned taking into account the comparative basicity of the interacting species HnAn+ and HiL(12−i)−.6 Strong association was evidenced and the adduct stability depends on the absolute charge of the interacting species: the strength of the interaction increases as the protonation state of the polyamine increases and as the protonation degree of InsP6 decreases.19–22 Indeed, it has been proposed that the stability of the adducts can be estimated from the product of the charges of the interacting species.21 Interestingly, apart from charge dependence, the stability also increases with the number of protons bound to InsP6. Thus, logK for the formation of [(HnA)(HiL)](12−n−i)− can be fit to a1ξ + a2i + a3, where ai are constant empirical parameters for all studied polyamines, ξ is the product of the charges of the interacting species and i is the total number of protons bound to InsP6. The influence of parameter i is probably associated with hydrogen bond formation, which gives additional stability to the adduct.21 Indeed, in a previous study, we have shed more light on this matter, measuring the thermodynamic parameters of biogenic polyamine:InsP6 interactions and rationalizing them using computational tools. We found that the general trend determined solely considering the charge of interacting species is not always observed, especially for the less charged species, where the interplay of hydrogen bond formation probably plays a decisive role.19
For the already studied linear polyamines, distribution species show in most cases the predominance of 1:1 polyamine:InsP6 species with the fully protonated form of the corresponding polyamine.19–21 Even though 1:1 polyamine:InsP6 adducts are the predominantly formed species, 2:1 polyamine:InsP6 species can also be formed to a certain extent, especially under excess polyamines and for the lighter polyamines.16,19–21
Cyclic polyamines (see some examples in Fig. 1(c)) can form positively charged species that can interact with InsP6. They are here conventionally labeled by the total number of ring members followed by the number of amine N atoms. These molecules tend to be highly protonated at neutral pH and have proved to be functional receptors of anionic biomolecules, such as nucleotides.23–27 The pre-arranged cyclic topology allows, especially for the large, and thus, more flexible polyaza- and polyaza-crown-macrocycles, the formation of stable adducts based on a multiple-contact interaction.23,25 The stability of the adducts formed with nucleotides depends again principally on the charge of interacting species. Besides, some relevant size effects have been observed: for example, the triprotonated form of 24N6 gives rise to more stable adducts than the triprotonated forms of 15N5, 16N5 or 17N5, probably due to the possibility of more flexible 24N6, allowing it to bend to simultaneously interact with different parts of the anionic nucleotide.26 In line with electrostatic and size effects, the fully protonated form of 32N8 interacts more strongly than the fully protonated form of 24N6 with nucleotides.24,27 It is worth mentioning at this point that unlike what happens with linear polyamines, the higher rigidity imposed by cyclization can decrease the structural diversity of the molecular recognition process, thus leading to a higher selectivity. For example, cyclic polyamines can selectively bind adenosine triphosphate, ATP3− in the presence of either adenosine di or monophosphate (ADP2−, AMP−) at physiological pH.24,26
The recognition pattern of the protonated cyclic polyamines by highly charged InsP6 species is worth comparing to already known behavior of linear polyamines, in order to explore the influence of the macrocyclic effect on the interaction and selectivity, as observed for other polyanions.24,26 Since cyclic polyamines have been considered to mimic the active site of some anion binding proteins, the determination of the main parameters that influence the interaction is especially relevant. However, the interaction of InsP6 with cyclic polyamines in solution has not been systematically studied so far. Very recent reports have concluded, from the large variation in the pH value observed when mixing InsP6 with two different macrocyclic polyamines, that the interaction is strong and dominated by an interplay of electrostatic and hydrogen bond formation.28,29 In this work, we explore the solution chemistry of InsP6 in the presence of various cyclic polyamines (1,4,7,10-tetra-azacyclododecane, 12N4; 1,4,7,10-tetra-azacyclotridecane, 13N4; 1,4,7,10,13,16-hexaazacyclooctadecane, 18N6; 1,4,7-trimethyl-1,4,7,10,13,16,19-heptaazacycloheneicosane, Me321N7; 1,4,7,10,13,16,19,22-octaazacyclotetracosane, 24N8, Fig. 1(c)) via potentiometry at 25.0 °C in 0.15 M NaClO4. The results are discussed with the help of computational tools applied on selected systems in order to shed light on the structural and electronic features of the recognition driving forces.
Experimental
Materials
InsP6 solutions were prepared by weighing the dipotassium salt K2H10L·2.5 H2O (Aldrich). Purity and water content of this salt was rechecked by elemental analysis. 12N4, 13N4 and 18N6 were purchased from Merck, while Me321N7 and 23N8 were synthesized according to the reported procedures30,31 Purity was verified by elemental analysis and 1H-NMR spectra.Potentiometric measurements
HCl and NaOH stock solutions were used throughout the potentiometric measurements. They were prepared by diluting concentrated Titripur® (Merck) ampoules and were standardized against Trizma® base (Merck) and potassium biphtalate (Sigma), respectively. All the solutions were prepared with analytical grade water (<18 MΩ cm−1) and were freed of carbon dioxide by Ar bubbling.The protonation constants of the smaller polyamines (12N4 and 13N4) had not been determined previously under the same experimental conditions of this work.25 So, they were measured by four and two potentiometric titrations, respectively (ca. 200 experimental points each), using total concentrations of polyamines in the interval 1–2 mM. The protonation constants of rest of the polyamines (18N6, Me321N7 and 24N8) were taken from previous reports,30,32,33 all of which were obtained under exactly the same experimental conditions of this work (Table S1, ESI†). The protonation constants of InsP6 were also measured in the present work by seven potentiometric titrations in the total concentration interval 0.5–2 mM (ca. 200 experimental points each). Then, the behavior of InsP6 in the presence of the polyamines was analyzed through two to six potentiometric titrations (ca. 200 experimental points each) depending on the complexity of each system, with the total concentration of InsP6 ranging from 0.5 to 1.5 mM and polyamines to InsP6 total molar ratios varying from 0.5:1 to 2:1. The pH interval from 2.0 to 10 was covered.
In each potentiometric experiment, the solutions were poured into a 50 mL titration cell. After thermal equilibrium was reached, hydrogen ion concentrations were determined by successive readings, each performed after a small incremental addition of the stock NaOH solution. The titrant addition and e.m.f. measurements were carried out using an automatic titrator Mettler-Toledo DL50-Graphix. The ionic strength was kept almost constant throughout the titrations by using solutions containing 0.15 M NaClO4 and relatively low initial concentrations of the reactants (the sum of these reactants initial concentrations did not contribute more than 2% to the total ionic strength). Pre-saturated argon (free of CO2) was bubbled through the solutions during the titrations to eliminate the effect of atmospheric carbon dioxide, and the temperature was kept at 25.0 (±0.1) °C. Equilibrium attainment after the addition of each titrant was verified by controlling the deviation of successive e.m.f. readings and performing back titrations. Independent stock solutions were used in some selected titrations to check reproducibility. The cell electrode potential E° and the acidic junction potential were determined from independent titrations of the strong acid with the titrant solution.34 In this way, the pH scale was the free concentration scale. The calibration in the alkaline range was checked by recalculating Kw values for each system. The obtained values (average logKw = −13.70) were always checked to assure that they were in agreement with previously reported data under the same experimental conditions.35 Data were analyzed using the HYPERQUAD program,36 and species distribution diagrams were produced using the HySS program.37 The fit of the values predicted by the model to the experimental data was estimated on the basis of the σ parameter, corresponding to the scaled sum of square differences between the predicted and experimental values. Many other possible stoichiometries were tried for each system, and final models were selected on the basis of the σ parameter, the model confidence level estimator, chi-square, and the internal consistency of data reflected in standard deviations of the formation constants.36
Molecular modelling calculations
The initial geometries of the selected 1:1 and 2:1 adducts were built according to the potentiometric results, starting from the corresponding protonated InsP6 (H4L8− or H6L6−) and polyamine (HnAn+) species. The protonation scheme of InsP6 was taken from our previous work,38 while for the polyamine species, the protons were placed as farther away as possible, taking into account the relative basicity of secondary and tertiary ammine groups. In all cases, the cyclic polyamine molecule was localized in order to maximize the interaction between the ammonium groups and the most negatively charged zones of H4L8− and H6L6− (see the electrostatic potential maps in Fig. S1, ESI†). Those initial geometries were optimized in water without any constraints at the B3LYP/LANL2DZ level of theory,39–41 as implemented in Gaussian 09.42 The effect of the solvent was modelled using an SMD implicit solvation model.43 The use of a continuum instead of a very high number of discrete water molecules allowed us to lower the degrees of freedom of the systems in order to carry out the calculations in a reasonable time. All the DFT-optimized geometries corresponded to minima on the potential energy surface, and the nature of the stationary points was verified through vibrational analysis.
Further studies on structural and electronic features of the InsP6:cyclic polyamine adducts were carried out starting from the optimized structures and using the program Multiwfn (version 3.7).43 A detailed analysis of the electron density was performed employing the noncovalent interaction (NCI) method, allowing to depict the weak interactions with different colors: H-bonds (blue), van der Waals (green) and steric repulsion (red). To do so, the values of the product sign(λ2)ρ (λ2 = second largest eigenvalue of the Hessian matrix of electron density; ρ = electron density) were represented with different colors and mapped on a reduced density gradient (RDG) isosurface (isovalue = 0.35). The location of the conventional intramolecular H-bonds was verified using the NCI method, estimating their energy from the electron density at the bond critical point (ρBCP) through the approach reported by Emamian et al.44 The results were rendered using Discovery Studio Visualizer45 and Gaussview 6.0.46
Statistical analysis
To evaluate the correlation between the stability of the polyamine:InsP6 1:1 adducts and meaningful descriptors such as polyamine nature (linear or cyclic), polyamine chain length and charge of HnAn+ and HiL(12−i)− species, a multiple linear regression model (MLR) was performed using the Real Statistics Resource Pack software (release 7.3.3).47 The effective logK values (response variable) were taken from this work (Table S2, ESI† for cyclic polyamines) or extracted from a previous report by our group (for linear polyamines).19p-Values were employed to assess the statistical significance of the observed associations. The absence of multicollinearity was verified by calculating the variance inflation factor (VIF; all values were lower than 3).48
Results and discussion
Protonation of polyamines and InsP6
The obtained data for the protonation equilibrium constants for polyamines are shown in Table S1 (ESI†). Fig. S2 (ESI†) depicts the final goodness of fit for the selected systems. The de novo measured protonation constants of the polyamines 12N4 and 13N4 are of the same order of previous data measured under similar conditions.49,50 Besides, the expected general trends are verified in the whole set of protonation constants of the cyclic polyamines under study. Indeed, previous results account for a relatively stronger basic behavior in the first half of the polyamine protonation steps and for a weaker basic behavior in the second half.25 So, in comparison to linear polyamines, the cyclic analogues show, in general, stronger first protonation steps but lower last protonation constant values.19–21 This grouping of protonation constants becomes more evident for the smaller polyaza-macrocycles. This fact has been ascribed to the electrostatic repulsion among the positively charged NH2+ groups arranged in the cyclic framework.25 Following the same rationalization, the basicity of polyamines is also influenced by the length of the carbon chain giving rise to a higher basicity as the separation of NH2+ groups is increased.25 This is observed for example in the higher basicity of 13N4 with regard to 12N4.Fig. 2(a)–(e) depict the corresponding species distribution diagrams of cyclic polyamines vs. pH, built from data in Table S1 (ESI†). The predominance of polyamine protonated species is shifted, favoring a higher protonation degree, as the cyclic polyamine becomes larger and bears a higher number of basic groups. Interestingly, the half-protonated polyamine species shows in all cases a wider pH interval predominance, in line with the mentioned grouping of protonation constant values. This is different to what happens to linear polyamines, that can better arrange the increasing charge by separating the NH2+ groups through space, and thus predominantly form the fully protonated species over almost all the acidic intervals.19
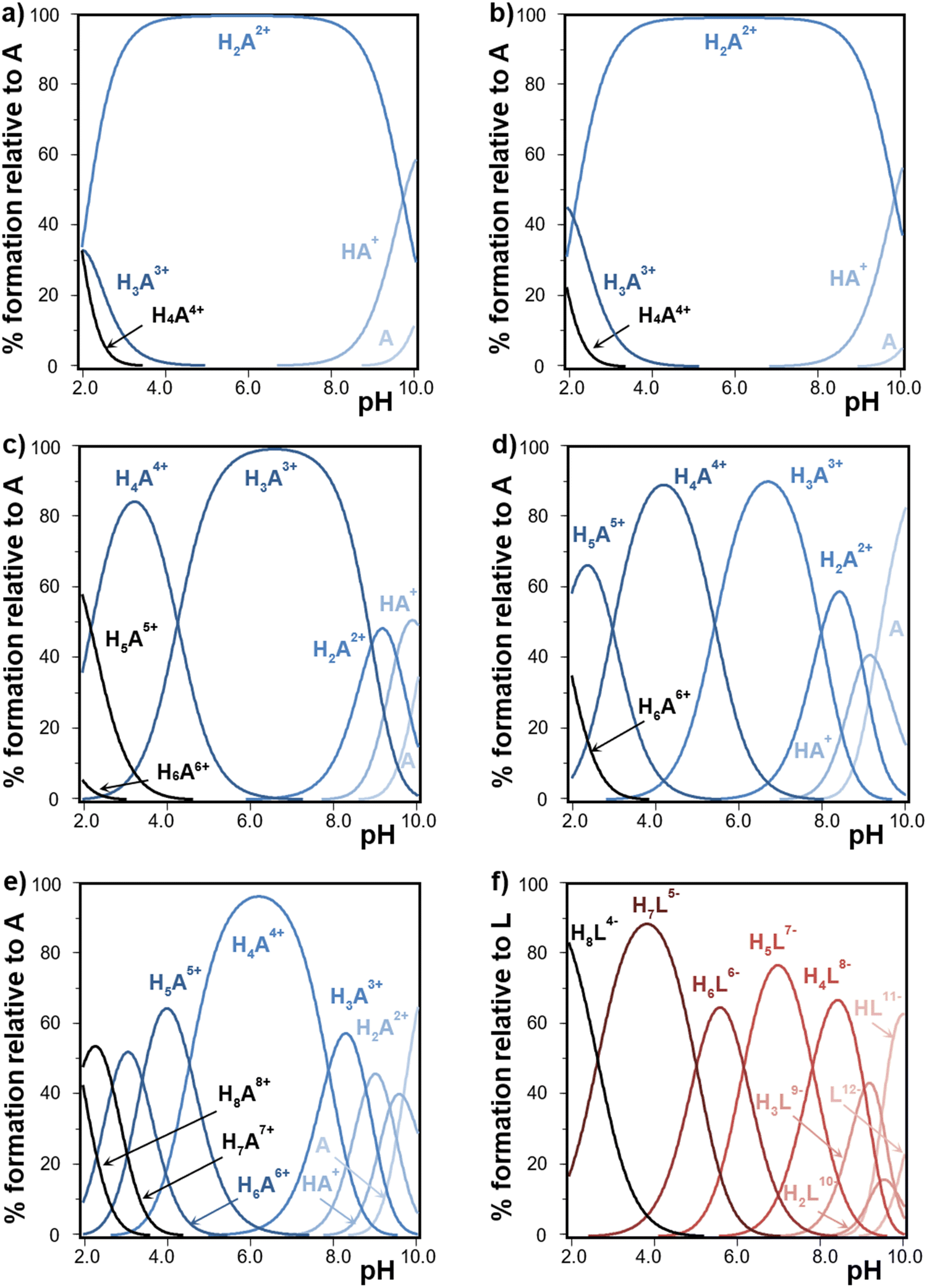 | ||
| Fig. 2 Species distribution diagrams built from data in Table 1 and Table S1 (ESI†), showing the protonation patterns of polyamines (A represents the neutral form) and InsP6 (L12− represents the fully deprotonated form) vs. pH at 25.0 °C in 0.15 M NaClO4. (a) 12N4, (b) 13N4, (c) 18N6, (d) Me321N7, (e) 24N8, and (f) InsP6. | ||
InsP6 protonation constants measured at 25.0 °C in 0.15 M NaClO4 (Table S1, ESI† and Fig. 2(f)) are similar to those previously obtained for the same medium but at 37.0 °C, in line with general protonation stages being almost athermic and favored by dominating entropic contributions.19 The second protonation step corresponds to the equilibrium HL11− + H+ ⇌ H2L10− and it is known to occur along with the conformational transition from the 5a1e conformation of HL11− to the 1a5e conformation of H2L10− (Fig. 1(a) and (b)).38 Even though this process has been determined to be exothermic19 mainly due to the rearrangement of phosphate groups, this is not reflected in any special decrease for that stepwise protonation constant as temperature is decreased, indicating that the decrease in phosphate groups’ repulsion must be compensated by the many influencing factors comprised in this complex process.
Polyamines interaction with InsP6:potentiometric results
The overall equilibrium constants for the association of cyclic polyamines with InsP6 determined potentiometrically are listed in Table 1. Overall stability constant values are associated with the formation reaction equilibria defined by the following general equation:| p A + L12− + r H+ ⇌ [HrApL](12−r)− |
| p q r | 12N4 | 13N4 | 18N6 | Me321N7 | 24N8 |
|---|---|---|---|---|---|
| 1 1 4 | 43.95(6) | 46.39(8) | |||
| 1 1 5 | 53.0(1) | 54.63(9) | |||
| 1 1 6 | 62.23(6) | 63.72(7) | 61.71(8) | 62.39(3) | |
| 1 1 7 | 70.6(1) | 71.55(4) | |||
| 1 1 8 | 79.64(4) | 79.73(4) | |||
| 1 1 9 | 87.40(7) | 81.91(1) | 86.47(5) | ||
| 1 1 10 | 93.87(9) | 92.59(4) | |||
| 1 1 11 | 99.3(1) | 97.22(7) | |||
| 1 1 12 | 103.1(2) | 101.84(3) | |||
| 2 1 9 | 93.0(1) | 94.73(9) | |||
| 2 1 10 | 99.1(2) | 99.4(3) | 102.7(2) | 97.3(2) | 103.1(1) |
| 2 1 11 | 110.7(3) | 111.0(3) | |||
| σ | 0.5 | 2.4 | 2.0 | 1.1 | 0.5 |
For all the systems and under the conditions of this study, the detected adducts have a 1:1 or 2:1 polyamine:InsP6 molar ratio and different protonation states, defining a behavior similar to that already observed for linear polyamines.19–21 The inclusion of species with polyamine:InsP6 2:1 stoichiometry increases in all cases the goodness of fit, since their formation is relevant, especially in the potentiometric experiments set out with a 2:1 polyamine:InsP6 molar ratio. Indeed, even though σ and χ2 do not markedly change, they all systematically decrease when these species are included.
In order to analyze the obtained formation constant values, it is important to take into account the protonation patterns of both the polyamines and InsP6vs. pH (Fig. 2). Since in the studied pH range, both cyclic polyamines and InsP6 can be present in different protonation states, the formed adducts should be better formulated as [(HnA)(HiL)](12−n−i)− where n is the number of protons expected to be associated with the polyamine and i is the number of protons expected to be associated with InsP6. The rationale of this is that protonation can be understood as governed by the different basicity of the interacting species.51–54 The resulting formulation does not necessarily reflect reality, because basicity might change upon binding. Nevertheless, it gives a better comparative understanding of the possible prevailing species in solution vs. pH, indicating the expected relative localization of protons within the adduct. According to this criterion, we calculated the complex stability constants for the 1:1 polyamine:InsP6 adducts shown in Table S2 (ESI†). These data account for a strong electrostatic association between the anionic forms of InsP6 and the protonated polyamines, due to their simultaneous presence in solution of ions with high and opposite charges. In agreement with electrostatic expectations, the strength of interaction varies with the charge of the interacting species: the protonated forms of the cyclic polyamines associate more strongly in solution with more deprotonated (more negatively charged) forms of InsP6 (Table S2, ESI†). This marked electrostatic influence had been also observed for linear polyamines19–21 and in the interaction of cyclic polyamines with nucleotides.25 Indeed, following previous studies,21 calculating logK/ξ values, (ξ is the product of the charges of the interacting species) gives a fairly constant value of 0.24 ± 0.12. The reported value corresponding to linear tetra- to octamines is 0.26 ± 0.09.19 In the case of the cyclic polyamines with ethylene connections, the corresponding values of 12N4 (average logK/ξ = 0.20) and 24N8 (average logK/ξ = 0.21) are also in line with previous values, whereas 18N6 shows much higher values (average logK/ξ = 0.35) especially for the more protonated species, indicating a much stronger interaction than that expected only for the electrostatic attraction (Table S2, ESI†).
According to our results, and also in line with the marked electrostatic character of the interaction, the studied cyclic polyamines need to be at least doubly protonated in order to give rise to the formation of detectable species, a fact that has also been observed previously for the interaction of linear polyamines both with InsP6 and with other negatively charged biomolecules.19–21,55 Also, the interaction of cyclic polyamines with less charged polyanions (AMP−, ADP2−or ATP3−) showed the need of cyclic polyamine triple protonation in order to detect adduct formation.26
Apart from electrostatics, the total chain length of the polyamine also proved to be a key point in the interaction in the case of linear polyamines.19,20 Accordingly, our results show that 13N4 forms more stable adducts than 12N4, following the basicity order, as expected from the higher flexibility originated in the longer carbon chain that favors a larger separation of neighbor NH2+ groups.52 Also, higher values of average logK/ξ are observed for 13N4 compared to 12N4 (Table S2, ESI†). On the other hand, the effect of the increasing number of amine groups among the studied polyamines is more difficult to compare, because not only the number of NH2+ groups but also other highly relevant parameters are concomitantly changed. A general increase in the affinity of InsP6 has been observed as the number of NH2+ groups increases for linear polyamines19 and a similar effect has been reported in the interaction of cyclic polyamines with nucleotides.24,27,49 Our results are partially in line with this, even though 18N6 shows a stronger association even when compared with 24N8. This will be discussed in more depth below. Besides, the methylated Me321N7 shows a much weaker association. This last fact is probably due to the fact that the disposition of methyl groups partially inhibits the interaction, as it also happens in the association of cyclic polyamines with nucleotides.56 In addition to the steric effect, methyl groups can reduce the ability to form hydrogen bonds decreasing the basicity of the amino groups. In turn, protonation is redirected to the secondary amino groups, generating the localization of the ammonium groups. This fact accounts in some cases for methylated polyazacycloalkanes being more selective in the interaction with anions than with the corresponding unmethylated ones.57
Another complex aspect within the obtained results is to elucidate if cyclic polyamines form comparatively less stable adducts with InsP6, with regard to linear polyamines as it happens in the interaction with other anions.26 Again, it is difficult to select data in which the only changing factor is cyclization. The formation constants of the species of identical stoichiometry formed by 24N8 are lower than those of Me2heptaen (a linear octamine).19 The same occurs for 12N4 with regard to spermine (N,N′-bis(3-aminopropyl)-1,4-diaminobutane) or 3,3,3-tet (N,′-bis(3-aminopropyl)-1,3-propanediamine).19 In both cases, the flexibility of the linear polyamine is also expected to be higher due to a longer carbon chain between neighbor NH2+ groups.
Fig. 3 shows selected species distribution diagrams built from data in Table S1 (ESI†) and Table 1. The results show that different adducts are observed to be predominant in the millimolar scale over a wide pH interval. In line with the already discussed stability constant values, for the smaller tetramines represented by 12N4 as shown in Fig. 3(a), the adduct is formed to a relatively lower extent in the pH interval, probably because these +2 charged molecules do not interact so strongly with the highly protonated, thus less negatively charged, InsP6 species. As pH is raised, 2:1 polyamine:InsP6 species [(H2A)2(H5L)]3− and [(H2A)2(H6L)]2− are formed to some extent. Around neutrality, H4L8− and H2A2+ form [(H2A)(H4L)]6− which is a specially relevant adduct in speciation. In line with the determined stability constant values, speciation for 13N4 is very similar but shows even higher predominance of the complex species (not shown). The maximum percentage formed at pH 8 of the species [(H2A)(H4L)]6− for [InsP6]total = [polyamine]total = 1 mM is 42% in the case of 12N4 and raises to 69% for 13N4. On the other hand, for the bigger polyamines 18N6 and 24N8, even though 2:1 polyamine:InsP6 species are detected, they become comparatively less relevant in speciation at equimolar amounts. Indeed, 1:1 species are predominant over a wide pH interval and this is especially true for 18N6 and 24N8 (Fig. 3(b) and (c), respectively). Under the conditions of excess polyamine, the 2:1 polyamine:InsP6 species are favored, especially for concentrated solutions. This strategy led Bowman-James group to succeed in the isolation and X-ray structure determination of compounds of InsP6 and two macrocyclic polyamines: [(H2A)2(H8L)]·12.5H2O·3CH3OH·CH3CN (where A is a macrocyclic polyamine containing six amine and two pyridine groups)28 and [(H3A)2(H6L)]·36.6H2O·CH3OH (where A is a macrocyclic polyamine containing nine amine and three pyridine groups).29
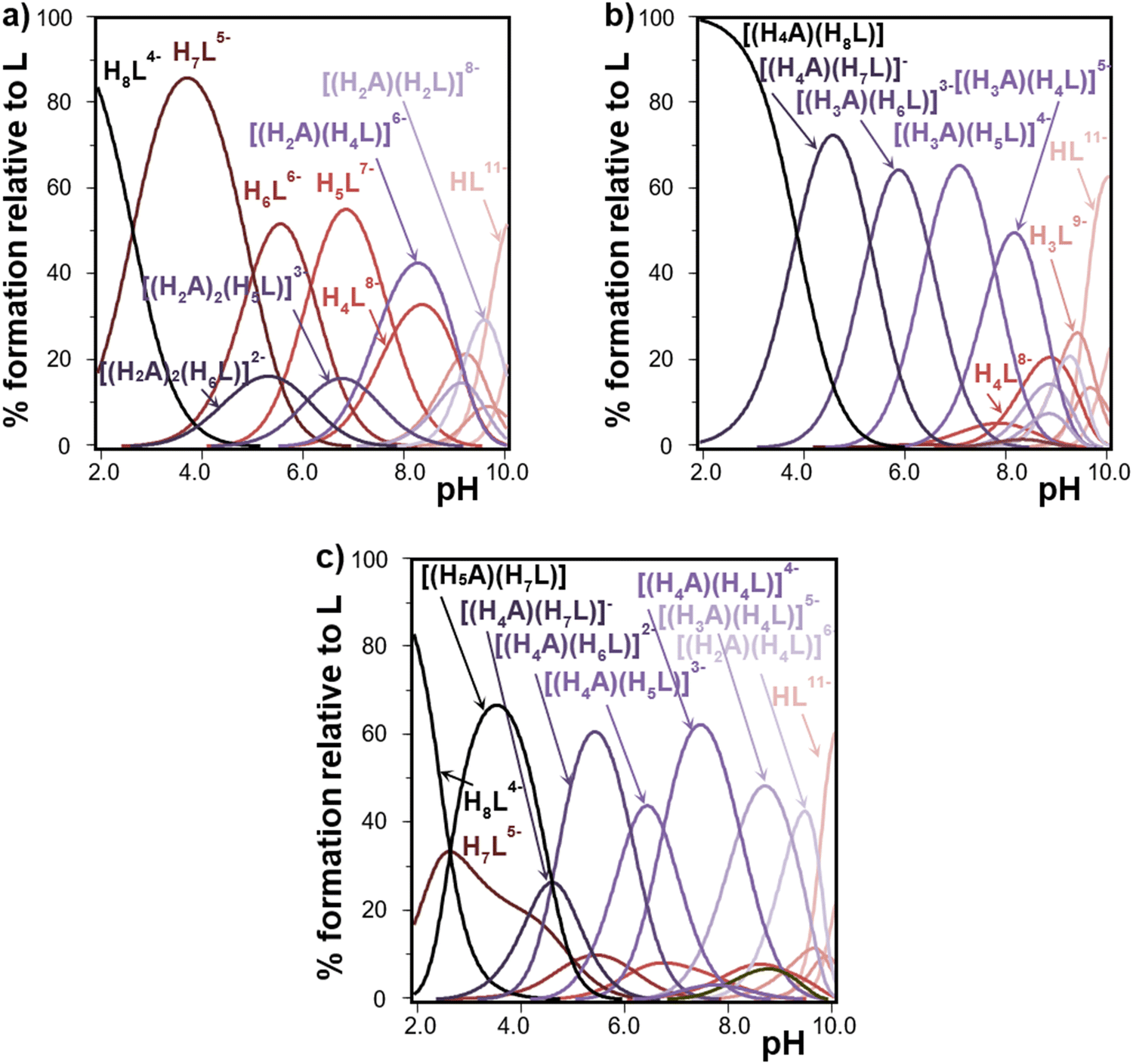 | ||
| Fig. 3 Speciation of InsP6 (L12− represents the fully deprotonated form) in the presence of polyamines (A represents the neutral form) vs. pH calculated from data in Table 1 and Table S1 (ESI†). Conditions: 25.0 °C in 0.15 M NaClO4, [InsP6]total = [polyamine]total = 1 mM. (a) A = 12N4, (b) A =18N6, (c) A = 24N8. Most relevant species are labelled. Code color: red for polyamines, blue for InsP6 and purple for adducts. The color fades as species become less protonated. | ||
In line with stability constant values, Me321N7 does not form adducts in high proportions (the highest predominance is 39% for [(H3A)(H6L)]3−, the only detected species). On the other hand, the interaction proves to be strong for unsubstituted hexa- and octamines, giving rise to highly predominant adducts with many different protonated forms of InsP6. In general, H4L8− and H6L6− strongly interact with the half-protonated polyamines.
Coming back to polyamine interaction with InsP6, a general comparison can be carried out from the plot of unbound InsP6 under the same conditions for different polyamines ([InsP6]total = [polyamine]total = 1 mM). This is shown in Fig. 4(a) for 12N4, 18N6 and 24N8. (Fig. S3, ESI†) shows the same calculation for ([InsP6]total = [polyamine]total = 1 μM). The free InsP6 for 12N4 is higher than that calculated for the bigger polyamines (with more N atoms to be protonated but also with a higher general flexibility to bind InsP6). Interestingly, 18N6 retains a high percentage of InsP6 almost in the whole pH interval, showing even stronger affinity below pH 7.8 than that calculated for 24N8 (24N8 is tetraprotonated at pH below 7.8, whereas 18N6 remains in the triprotonated form up to the acid region, see successive protonation constants in Table S1, (ESI†)).
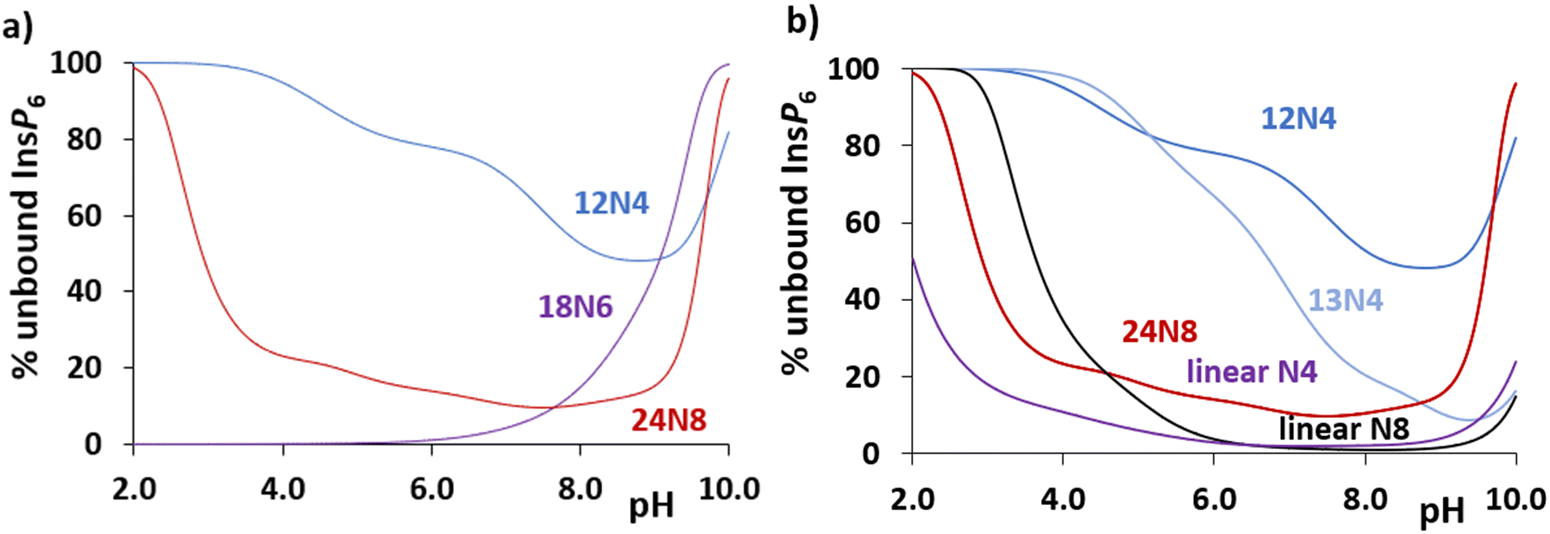 | ||
| Fig. 4 Percentage of unbound InsP6 in the presence of (a) selected cyclic polyamines and (b) selected tetra and octamines. [InsP6]total = [polyamine]total = 1 mM, calculated from data in Tables S1 (ESI†) and Table 2 and previously reported values.19 Selected tetramines are 12N4, 13N4 and the linear 3,3,3-tet (N,N′-bis(3-aminopropyl)-1,3-propanediamine): octamines are 24N8 and 1,22-dimethyl-1,4,7,10,13,16,19,22-octaazadocosane. | ||
Besides, Fig. 4(b) shows the comparative unbound InsP6 proportion left by the cyclic 12N4, 13N4 and 24N8, in comparison with other tetra- and octamines. The flexibility of a longer cyclic carbon chain (13N4 vs. 12N4) or of a linear disposition that allows a higher flexibility and a bigger separation of NH2+ groups through space (linear N4 or N8 vs. the cyclic polyamines) favors the interaction, decreasing the unbound % of InsP6 and also favoring the formation of the adducts from lower pH values.
Polyamine interaction with InsP6: relative stability and structural insights
In order to identify the structural features that modulate the stability of the polyamine:InsP6 adducts, we carried out a multiple linear regression (MLR) analysis, employing the logarithm of the effective formation constants as the response variable (y = logK) and the polyamine nature (linear or cyclic), polyamine chain length and charge of HnAn+ and HiL(12−i)− species as descriptors (x1, x2,…). Both cyclic and linear polyamines were taken into account, extracting the logK data from this work and another previously published by our group.19 All attempts to fit the data to the MLR model were unsuccessful, because 18N6 and Me321N7 display substantially higher and lower logK values, respectively, showing no clear correlation with the descriptors. For Me321N7, a much lower affinity for the InsP6 species is expected. The presence of methyl groups decreases the basicity of the polyamine and probably hampers the phytate–polyamine interaction. On the other hand, in the case of 18N6, a molecular recognition mechanism might be operative, which alters the expected order of affinity. Indeed, when both polyamines are taken out of the analysis, the rest of the data (57 observations) can be fitted to the linear model that explains 54% of the logK variability, showing a standard error of 0.91 log units (see Table 2). Looking at the statistically significant regression coefficients, the influence of each descriptor when the others are kept fixed can be revealed. To aid in this discussion, the structure in the solution of the most relevant 1:1 and 2:1 polyamine:InsP6 adducts was modelled by DFT, allowing to evaluate the number (n) and total energy (EHB) of InsP6-A conventional intra-adduct hydrogen bonds. The results are depicted in Fig. 5–8.
:InsP6 adducts
| Variables | Regression coefficients | p value |
|---|---|---|
| InsP6 charge (HiL(12−i)− species) | +0.48* | 0.0035 |
| Polyamine charge (HnAn+ species) | +0.29 | 0.11 |
| Polyamine chain length | +0.11* | 5.4 × 10−6 |
| Polyamine nature (linear = 1; cyclic = 0) | −0.78* | 0.0099 |
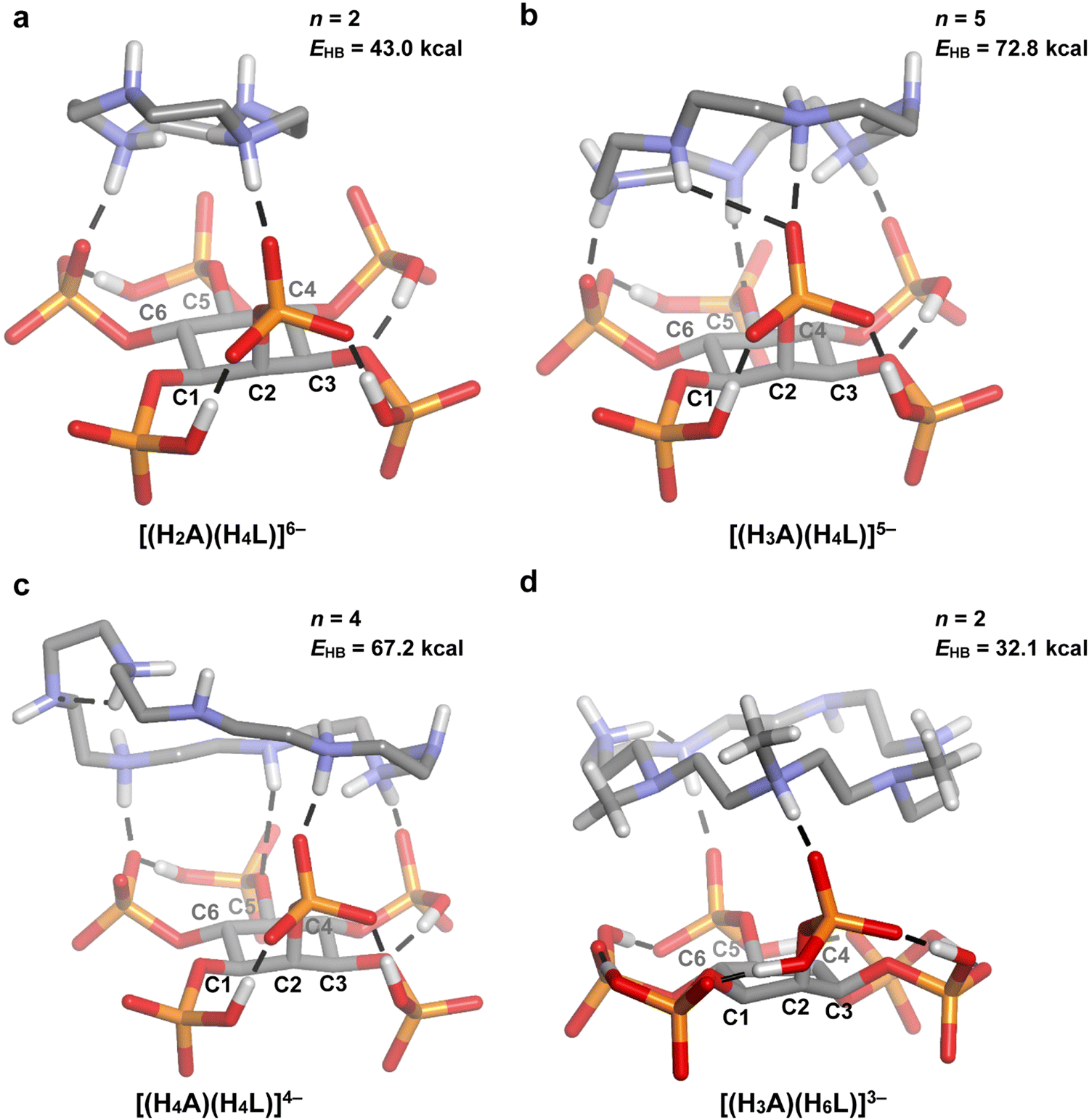 | ||
| Fig. 5 DFT optimized geometries for some of the 1:1 InsP6–cyclic polyamine adducts. A = 12N4 (a), 18N6 (b), 24N8 (c), and Me321N7 (d). The conventional hydrogen bonds are represented as dashed lines and some non-polar hydrogen atoms are omitted for clarity. The number (n) and total energy (EHB) of InsP6-A conventional hydrogen bonds are also shown. Atom color code: C (grey), H (white), O (red), P (orange), N (blue). | ||
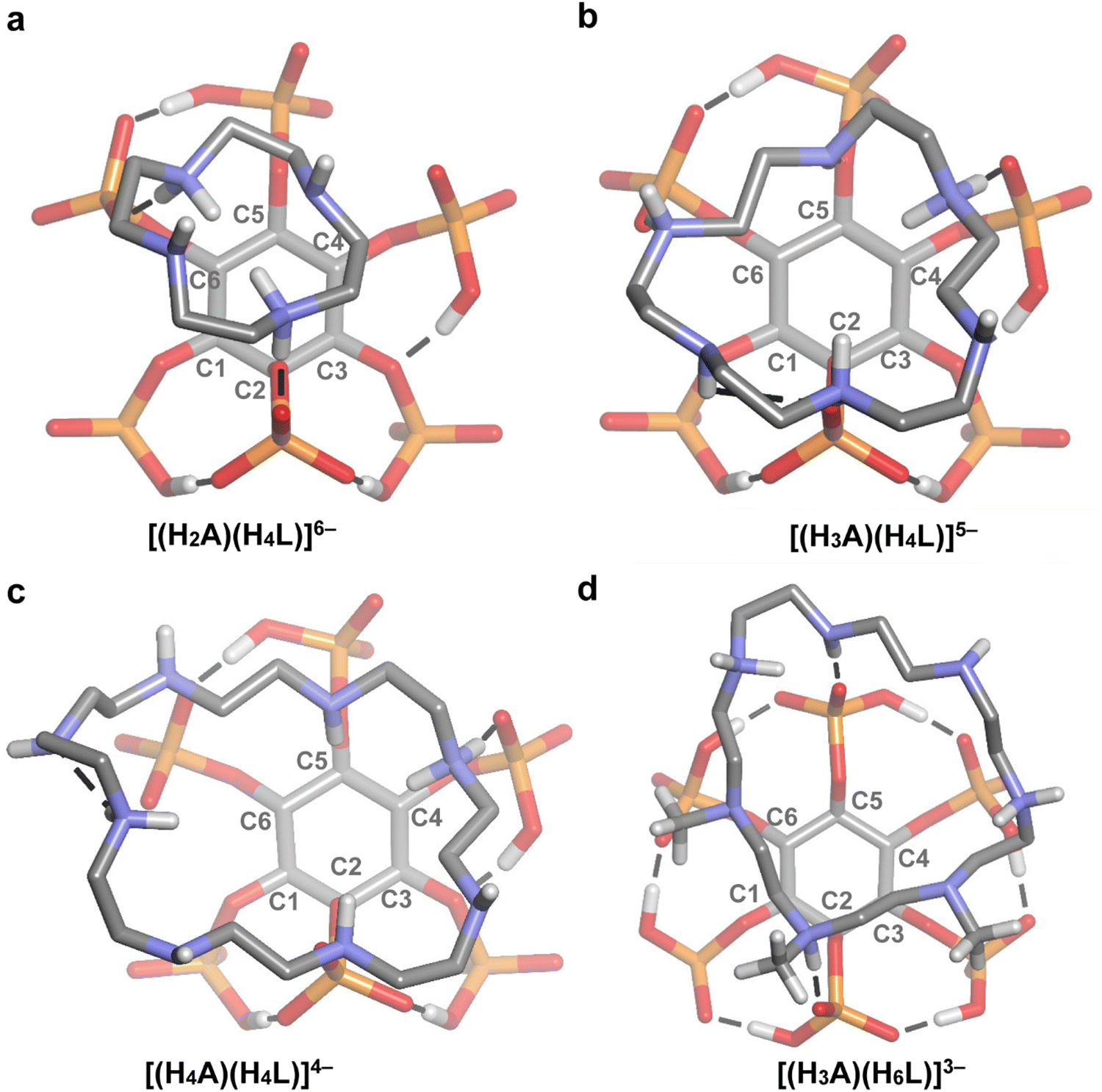 | ||
| Fig. 6 Top view of the DFT-optimized structures for some of the 1:1 InsP6-cyclic polyamine adducts. A = 12N4 (a), 18N6 (b), 24N8 (c), and Me321N7 (d). The conventional hydrogen bonds are represented as dashed lines and some non-polar hydrogen atoms are omitted for clarity. Atom color code: C (grey), H (white), O (red), P (orange), N (blue). | ||
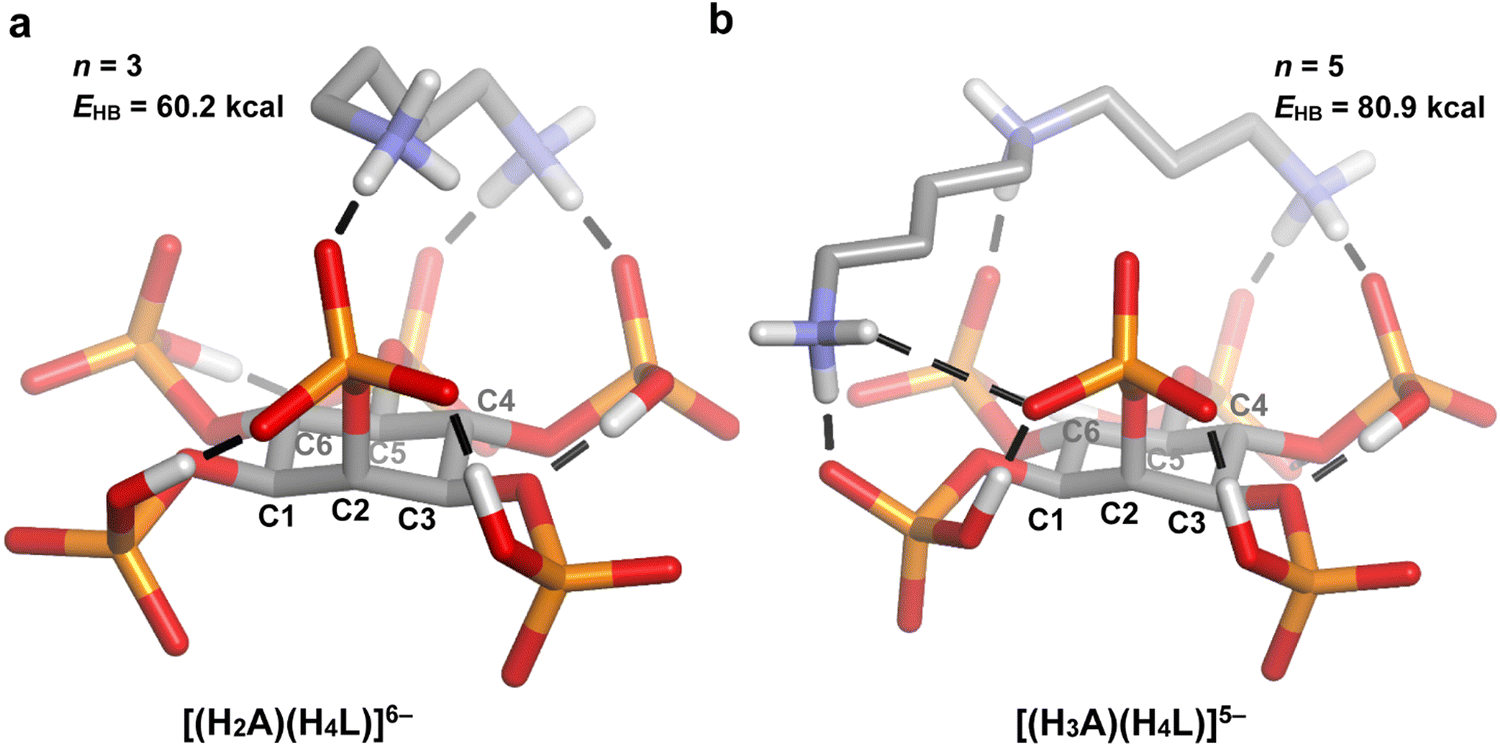 | ||
| Fig. 7 DFT optimized geometries for some of the 1:1 InsP6–linear polyamine adducts. A = putrescine (a) and spermidine (b). The conventional hydrogen bonds are represented as dashed lines and non-polar hydrogen atoms are omitted for clarity. The number (n) and total energy (EHB) of InsP6-A conventional hydrogen bonds are also shown. Atom color code: C (grey), H (white), O (red), P (orange), N (blue). | ||
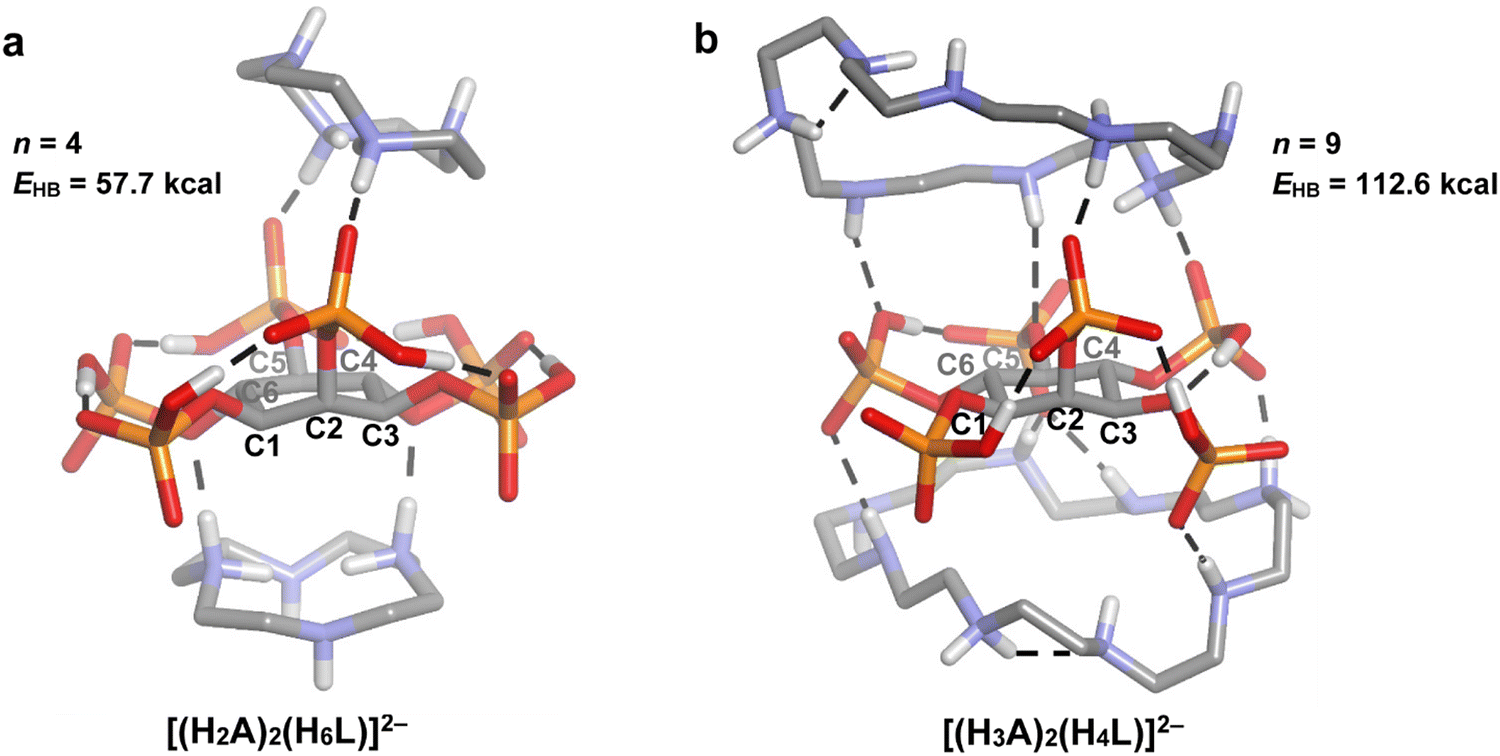 | ||
| Fig. 8 DFT optimized geometries for some of the 2:1 polyamine:InsP6 adducts. A = 12N4 (a) and 24N8 (b). The conventional hydrogen bonds are represented as dashed lines and non-polar hydrogen atoms are omitted for clarity. The number (n) and total energy (EHB) of InsP6-A conventional hydrogen bonds are also shown. Atom color code: C (grey), H (white), O (red), P (orange), N (blue). | ||
Returning to Table 2, the charge of HiL(12−i)− species positively influences the logK values (p = 0.0035). Hence, the higher the charge of the phytate anionic species, the higher the stability of the adducts. Regarding the charge of HnAn+ species, however, there is not enough statistical evidence to draw a general conclusion (p = 0.11), even though for this set of cyclic and linear polyamines, the overall influence on the stability is also positive. These findings are in complete agreement with the above-mentioned electrostatic model of recognition.
Moving on to the polyamine chain length, the MLR model indicates a positive correlation with the 1:1 adducts’ stability (p = 5.4 × 10−6; Table 2). As it was discussed in the previous section, longer polyamines are expected to give rise to higher logK values, since they bear more NH2+ groups and are more flexible, allowing them to establish stronger contacts with the phytate anion. Indeed, according to the n and EHB values estimated by DFT modeling (Fig. 5–7), large cyclic and long linear polyamines (24N8 and spermidine) establish more H-bond contacts with the InsP6 moiety than the shortest members (12N4 and putrescine), exhibiting a higher total H-bond energy. The computational model also suggests that the higher flexibility of the biggest polyamines allows them to adapt to the shape of the HiL(12−i)− species, enabling their ammonium and amine groups to easily approach several phosphate groups at the same time.
According to the regression coefficient listed in Table 2, the cyclic polyamines (p = 0.0099) form adducts with lower stability compared to the linear counterparts, even when the chain length and charge of HiL(12−i)− and HnAn+ species are kept constant. In fact, as it was discussed, the linear structure allows for a better separation between the NH2+ groups, favoring the interaction with the phytate species. In line with this, a comparison between the modeled structures of 24N8 (Fig. 5(c) and (c)) and spermidine (Fig. 7(b)) indicates that the linear polyamines tend to display higher n and EHB values. While spermidine establishes 5 hydrogen bonds (total energy = 80.9 kcal mol−1), 24N8 gives rise to 4 H-bond contacts (total energy = 67.2 kcal mol−1). The computational model indicates that linear polyamines are able to arch over the HiL(12−i)− species, optimizing the hydrogen bonding network with the phosphate groups. The cyclic polyamines, however, display certain rigidity brought about by the ring structure and the proximity of the positively charged NH2+ groups. In this regard, they are less able to match the specific charge distribution of the HiL(12−i)− species, preferring in some cases to establish intramolecular H-bonds, as it is seen for 24N8 in Fig. 5(c) and 6(c). This phenomenon observed for the cyclic polyamines gives rise to less and weaker H-bond contacts between InsP6 and the cyclic polyamines, rendering lower average logK values.
Another interesting aspect of polyamine–phytate speciation is the incipient formation of 2:1 complexes. These adducts are comparatively less abundant than the analogous 1:1 species. To furnish a picture of the structural basis behind this phenomenon, Fig. 8 depicts the DFT optimized geometries for the selected 2:1 polyamine:InsP6 adducts of 12N4 and 24N8. While for the 1:1 adducts, the polyamine molecule faces the phytate syn to the phosphate at C2, in the 2:1 complexes, the second polyamine molecule approaches from the other side of the inositol ring. Comparison of Fig. 5 and 8 shows that when going from the 1:1 to the 2:1 adducts, the charge of the reactant species is reduced by protonation: [(H2A)(H4L)]6− → [(H2A)2(H6L)]2− for 12N4 and [(H4A)(H4L)]4− → [(H3A)2(H4L)]2− for 24N8. Therefore, the electrostatic interaction energy between the polyamine and phytate moieties is lower for the ternary complexes. However, a stronger hydrogen bonding network is also expected to occur, because there are more hydrogen bond donors, partly counteracting this effect. To assess this phenomenon, the total number and energy of H-bonds for the 2:1 adducts of 12N4 and 24N8 (Fig. 8) can be compared with the corresponding values for the 1:1 complexes (Fig. 5(a) and (c)). While the binding of the first polyamine molecule gives rise to hydrogen bond network energies of 43.0 and 67.2 kcal mol−1 for 12N4 and 24N8, respectively, the binding of the second polyamine molecule produces less stabilization, with increments in the EHB of 14.7 and 45.4 kcal mol−1, respectively. Then, it is feasible that during the binding of the second polyamine molecule, the reduction in the electrostatic interaction energy is not compensated by the less marked increment in the H-bond energy. Notwithstanding, the steric hindrance brought about by the simultaneous binding of the two polyamines cannot be ruled out, especially for the bigger cyclic members. This gives a plausible explanation for the much less abundance of the 2:1 adducts in comparison with the 1:1 counterparts and for the fact that species with a higher polyamine:InsP6 ratio were not detected.
Finally, 18N6 and Me321N7 require special analysis. Both molecules had to be left behind during the MLR adjustment, because they are outliers of the general trend, exhibiting substantially higher and lower logK values, respectively. Indeed, when the stability of 1:1 InsP6–polyamine adducts is plotted as a function of chain length, the data for both polyamines are localized far from the adjusted linear model (Fig. 9). To unveil the structural features associated with this phenomenon, we resorted to the DFT models for these cyclic polyamines (Fig. 5 and 6). Of all the complexes tested, 18N6 displays the maximum number of H-bond connections, leading to the highest InsP6–polyamine hydrogen bond network energy (72.8 kcal mol−1). This is possible because its diameter fits the size of the phytate inositol ring (Fig. 6(b)), which, in conjunction with its flexibility, allows to adequately match the binding sites of the InsP6 species. As is seen for 12N4, shorter cyclic polyamines are more rigid and have fewer number of amine/ammonium groups, being not able to establish a high number of strong H-bonds with the phytate moiety (leading to lower stability). On the other hand, the largest cyclic polyamines like 24N8 have larger diameters that even exceed the size of the phytate inositol ring (Fig. 6(c)). Consequently, a part of the ammine/ammonium groups is not able to properly interact with the phosphate groups. In line with this, in the reported InsP6 structure with a dodecamine (containing three ammonium, three amide and three pyridine groups), the pyridine–dicarboxamide groups form a loop that points away from the H6L6− species.28 Because these large polyamines are also very flexible, they are prone to establish intramolecular H-bonds, which in turn causes the decrease of the interaction energy (see Fig. 5(c)). In the case of Me321N7, however, the computational model shows that the methyl substituents have a detrimental impact on the polyamine–phytate interaction (Fig. 5(d) and 6(d)). Indeed, the substitution withdraws three potential H-bond donor/acceptor positions over the polyamine. Besides, the steric repulsion brought about by the –CH3 groups not only hinders the interaction with InsP6 but also provokes a stiffening effect, leading to an almost planar configuration of the polyamine ligand that is not able to adapt to the geometric fit of the protonated groups to the phytate moiety. In this scenario, Me321N7 is only able to set up two H-bond connections with H6L6−, with a total H-bond energy (32.1 kcal mol−1) lower than that for 12N4 (43.0 kcal mol−1). This gives a plausible explanation for the observed weaker interaction with InsP6.
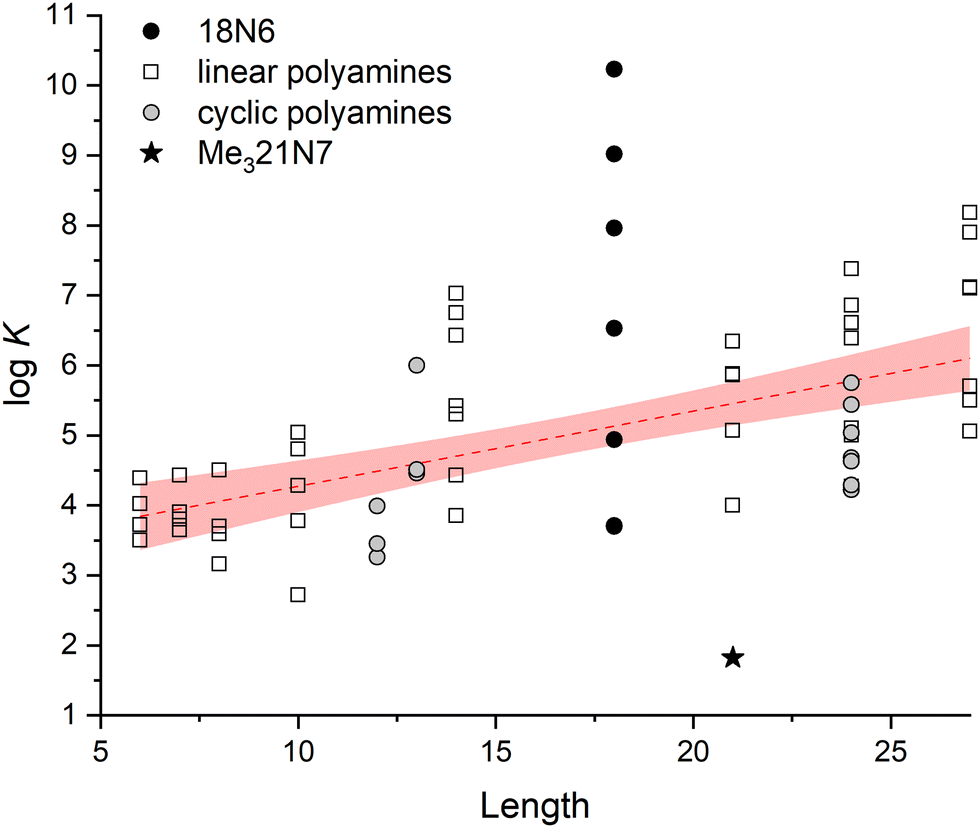 | ||
| Fig. 9 Values of logK for the formation of 1:1 InsP6–polyamine adducts as a function of the length of the polyamine (number of atoms). The linear regression model and its 95% confidence band for the fitting of the data of all polyamines except for 18N6 and Me321N7 are also shown. | ||
Concluding remarks
In this work, we presented the aqueous solution acid–base and complexation results of InsP6 in the presence of cyclic polyamines. We established the thermodynamic and structural parameters of the InsP6–polyamine interaction and calculated the corresponding chemical distribution.The potentiometric study evidenced a relatively strong association between the anionic forms of InsP6 and the protonated species of the cyclic polyamines, especially for the larger ones. The complexes detected have 1:1 and 2:1 polyamine:InsP6 stoichiometries, in different states of protonation, with the former being generally predominant in speciation. The half-protonated forms of polyamines are especially relevant for the interaction with InsP6. Due to the predominantly electrostatic character of the interaction model, the protonated forms of the polyamines associate more strongly in solutions with more deprotonated forms of InsP6. The stability of the complex also increases in general with cyclic polyamine dimension because of the increase in flexibility and in the potential number of hydrogen bonds to be formed.
Multiple linear regression analysis confirmed that the charge of the phytate species increases the stability of the formed adducts. Also, our computational results show that as polyamines get bigger, more H-bond contacts can be formed with the InsP6 moiety and they behave as more flexible cations, thus increasing the strength of interaction. In the case of linear polyamines, this stability enhancement is favored by the capacity of the longer polycations to arch over the anionic species as the total length is increased. However, for the bigger more rigid cyclic polyamines, the size and charge distribution of the polyamine with regard to the possible match should be also taken into account. Thus, 18N6 shows the strongest interaction due to a better dimensional fit compared to the corresponding smaller polyamines. Also, hydrogen bond formation favored in the more protonated species plays a role in enhancing the interaction between the more protonated species of InsP6 and 18N6. Thus, in the simulated equimolar simultaneous presence of 12N4 and 18N6 competing for InsP6, the selectivity for 18N6 is remarkable (Fig. 10a). Besides, this hexamine shows a comparable retention of InsP6 even in the presence of 24N6 having extra ammonium binding groups (Fig. 10(b)). This is indicative of the molecular recognition of InsP6 towards 18N6.
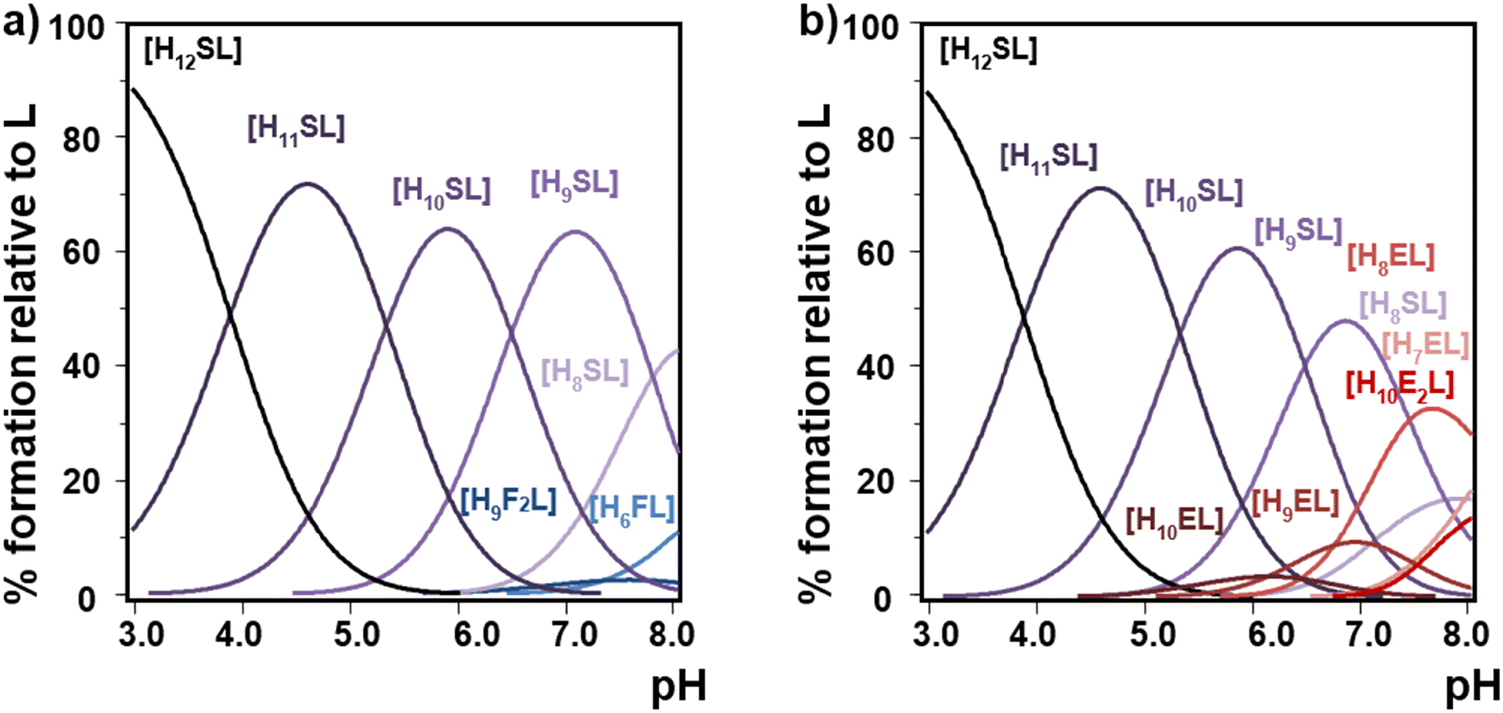 | ||
| Fig. 10 Species distribution diagrams of InsP6 (L12− represents the fully deprotonated form) in the presence of equimolar amounts of 18N6 (S represents the neutral form of this amine containing six amino groups) and (a) 12N4 (F represents the neutral form of this amine containing four amino groups) and (b) 24N8 (E represents the neutral form of this amine containing eight amino groups). Data from Table 1 and Table S1 (ESI†) were used. Conditions: 25.0 °C in 0.15 M NaClO4, [InsP6] = [18N6] = 1 mM; (a) [12N4] = 1 mM, (b) [24N8] = 1 mM. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Comisión Sectorial de Investigación Científica (CSIC, Programa de Apoyo a Grupos) and PEDECIBA, both from Uruguay, for financial support. We also thank Prof. Oscar Ventura and Prof. Kenneth Irving (Facultad de Química, Universidad de la República) for the access to the University computer cluster.References
- A. J. Letcher, M. J. Schell and R. F. Irvine, Biochem. J., 2008, 416, 263–270 CrossRef CAS.
- F. Pisani, T. Livermore, G. Rose, J. R. Chubb, M. Gaspari and A. Saiardi, PLoS One, 2014, 9, e85533 CrossRef PubMed.
- D. Pittet, W. Schlegel, D. P. Lew, A. Monod and G. W. Mayr, J. Biol. Chem., 1989, 264, 18489–18493 CrossRef CAS.
- B. S. Szwergold, R. A. Graham and T. R. Brown, Biochem. Biophys. Res. Commun., 1987, 149, 874–881 CrossRef CAS.
- S. B. Shears, in myo-Inositol Phosphates, Phosphoinositides, and Signal Transduction, ed. B. B. Biswas and S. Biswas, Springer, US, Boston, MA, 1996, vol. 26, pp.187–226 Search PubMed.
- C. Kremer, J. Torres, A. Bianchi, M. Savastano and C. Bazzicalupi, Coord. Chem. Rev., 2020, 419, 213403 CrossRef CAS.
- R. F. Irvine, J. Physiol., 2005, 566, 295–300 CrossRef CAS.
- R. H. Michell, Nat. Rev. Mol. Cell Biol., 2008, 9, 151–161 CrossRef CAS.
- S. B. Shears, Adv. Biol. Regul., 2015, 57, 203–216 CrossRef CAS.
- E. Graf, Free Radicals Biol. Med., 1990, 8, 61–69 CrossRef CAS PubMed.
- W. N. Addison and M. D. McKee, Bone, 2010, 46, 1100–1107 CrossRef CAS PubMed.
- D. Xu, T. Xu, X. Guo, Q. Liu, J. Liu, W. Lv, X. Jing, H. Zhang and J. Wang, New J. Chem., 2017, 41, 5305–5312 RSC.
- N.-H. Liu, M.-J. Dong, H. Liu, X.-L. Lu, D. Tian, J. Zhang, J. Yang, J.-H. Sun, L.-H. Wu, J.-L. Bi and B. Zhang, Nucl. Med. Commun., 2018, 39, 818–824 CrossRef.
- I. Vucenik, J. Nutr. Sci. Vitaminol., 2019, 65, S18–S22 CrossRef.
- N. Veiga, J. Torres, H. Y. Godage, A. M. Riley, S. Domínguez, B. V. L. Potter, A. Díaz and C. Kremer, JBIC, J. Biol. Inorg. Chem., 2009, 14, 1001–1013 CrossRef CAS.
- F. Crea, C. De Stefano, D. Milea and S. Sammartano, Coord. Chem. Rev., 2008, 252, 1108–1120 CrossRef CAS.
- B. I. Naik and S. K. Srivastava, Phytochemistry, 1978, 17, 1885–1887 CrossRef CAS.
- M. Riche and D. L. Garling, Aquacult. Nutr., 2004, 10, 389–400 CrossRef CAS.
- J. Torres, C. Giorgi, N. Veiga, C. Kremer and A. Bianchi, Org. Biomol. Chem., 2015, 13, 7500–7512 RSC.
- A. Odani, R. Jastrzab and L. Lomozik, Metallomics, 2011, 3, 735 CrossRef CAS PubMed.
- C. De Stefano, O. Giuffrè, D. Milea, C. Rigano and S. Sammartano, Chem. Speciation Bioavailability, 2003, 15, 29–36 CrossRef CAS.
- F. Crea, C. De Stefano, N. Porcino and S. Sammartano, Biophys. Chem., 2008, 136, 108–114 CrossRef CAS.
- H. P. Ferguson Johns, E. E. Harrison, K. J. Stingley and M. L. Waters, Chem. – Eur. J., 2021, 27, 6620–6644 CrossRef CAS PubMed.
- B. Dietrich, M. W. Hosseini, J. M. Lehn and R. B. Sessions, J. Am. Chem. Soc., 1981, 103, 1282–1283 CrossRef CAS.
- A. Bianchi, M. Micheloni and P. Paoletti, Coord. Chem. Rev., 1991, 110, 17–113 CrossRef CAS.
- E. Kimura, M. Kodama and T. Yatsunami, J. Am. Chem. Soc., 1982, 104, 3182–3187 CrossRef CAS.
- M. W. Hosseini and J.-M. Lehn, Helv. Chim. Acta, 1987, 70, 1312–1319 CrossRef CAS.
- S. Pramanik, R. M. Steinert, K. R. Mitchell-Koch and K. Bowman-James, Chem. – Eur. J., 2023, e202301764 CrossRef CAS.
- S. Pramanik, V. W. Day and K. Bowman-James, Chem. Commun., 2020, 56, 3269–3272 RSC.
- A. Bencini, A. Bianchi, M. Micheloni, P. Paoletti, E. Garcia-España and M. A. Niño, J. Chem. Soc., Dalton Trans., 1991, 1171–1174 RSC.
- A. Bianchi, S. Mangani, M. Micheloni, V. Nanini, P. Orioli, P. Paoletti and B. Seghi, Inorg. Chem., 1985, 24, 1182–1187 CrossRef CAS.
- A. Andrés, C. Bazzicalupi, A. Bencini, A. Bianchi, V. Fusi, E. Garcia-España, C. Giorgi, N. Nardi, P. Paoletti, J. A. Ramirez and B. Valtancoli, J. Chem. Soc., Perkin Trans. 2, 1994, 2367–2373 RSC.
- A. Bencini, A. Bianchi, P. Dapporto, E. Garcia-Espana, M. Micheloni and P. Paoletti, Inorg. Chem., 1989, 28, 1188–1191 CrossRef CAS.
- P. Gans, Talanta, 2000, 51, 33–37 CrossRef CAS.
- L. D. Petit and K. J. Powell, Stability Constants Database, SC-Database for Windows, IUPAC and Academic Software in cooperation with the Royal Society of Chemistry, 1997 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- N. Veiga, J. Torres, I. Macho, K. Gómez, G. González and C. Kremer, Dalton Trans., 2014, 43, 16238–16251 RSC.
- E. G. Lewars, Computational Chemistry, Springer International Publishing, Cham, 2016 Search PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Ab initio, Gaussian 09, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868–2881 CrossRef CAS PubMed.
- Discovery Studio Visualizer, Accelrys Software Inc., 2009 Search PubMed.
- R. Dennington; T. A. Keith and J. M. Millam, GaussView (version 6.0), Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- C. Zaiontz, Real Statistics Resource Pack 2020.
- G. James, D. Witten, T. Hastie and R. Tibshirani, An Introduction to Statistical Learning, Springer, US, New York, NY, 2021, pp.1–14 Search PubMed.
- M. Kodama and E. Kimura, J. Chem. Soc., Dalton Trans., 1976, 116 RSC.
- M. Kodama and E. Kimura, J. Chem. Soc., Dalton Trans., 1976, 1720 RSC.
- C. Bazzicalupi, A. Bencini, A. Bianchi, M. Cecchi, B. Escuder, V. Fusi, E. Garcia-España, C. Giorgi, S. V. Luis, G. Maccagni, V. Marcelino, P. Paoletti and B. Valtancoli, J. Am. Chem. Soc., 1999, 121, 6807–6815 CrossRef CAS.
- E. García-España, P. Díaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952–2986 CrossRef.
- P. Mateus, N. Bernier and R. Delgado, Coord. Chem. Rev., 2010, 254, 1726–1747 CrossRef CAS.
- A. Bianchi, M. Micheloni and P. Paoletti, Inorg. Chim. Acta, 1988, 151, 269–272 CrossRef CAS.
- L. Lomozik, A. Gasowska, G. Krzysko and R. Bregier-Jarzebowska, Bioinorg. Chem. Appl., 2010, 2010, 1–13 CrossRef.
- F. P. Schmidtchen, Chem. Ber., 1981, 114, 597–607 CrossRef CAS.
- A. Bencini, A. Bianchi, C. Giorgi, P. Paoletti, B. Valtancoli, V. Fusi, E. García-España, J. M. Llinares and J. A. Ramírez, Inorg. Chem., 1996, 35, 1114–1120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj04652d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |