
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermomechanical properties of confined magnetic nanoparticles in electrospun polyacrylonitrile nanofiber matrix exposed to a magnetic environment: structure, morphology, and stabilization (cyclization)†
Baran
Sarac
 a,
Viktor
Soprunyuk
b,
Gordon
Herwig
c,
Selin
Gümrükçü
d,
Ekrem
Kaplan
de,
Eray
Yüce
f,
Wilfried
Schranz
*b,
Jürgen
Eckert
af,
Luciano F.
Boesel
c and
A. Sezai
Sarac
*g
a,
Viktor
Soprunyuk
b,
Gordon
Herwig
c,
Selin
Gümrükçü
d,
Ekrem
Kaplan
de,
Eray
Yüce
f,
Wilfried
Schranz
*b,
Jürgen
Eckert
af,
Luciano F.
Boesel
c and
A. Sezai
Sarac
*g
aErich Schmid Institute of Materials Science, Austrian Academy of Sciences, 8700, Leoben, Austria
bFaculty of Physics, Physics of Functional Materials, University of Vienna, 1090, Vienna, Austria. E-mail: wilfried.schranz@univie.ac.at
cEmpa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Biomimetic Membranes and Textiles, 9014, St. Gallen, Switzerland
dDepartment of Chemistry, Istanbul Technical University, 34469, Istanbul, Turkiye
eFaculty of Engineering, Doğuş University, 34775, Istanbul, Turkiye
fDepartment of Materials Science, Montanuniversität Leoben, 8700, Leoben, Austria
gPolymer Science & Technology, Istanbul Technical University, 34469, Istanbul, Turkiye. E-mail: sarac@itu.edu.tr
First published on 13th September 2024
Abstract
Electrospun metal oxide–polymer nanofiber composites hold promise for revolutionizing biomedical applications due to their unique combination of electronic and material properties and tailorable functionalities. An investigation into the incorporation of Fe-based nanofillers for optimizing the polyacrylonitrile matrix was conducted, where the systematic and organized arrangement of inorganic components was achieved through non-covalent bonding. These carefully dispersed nanomaterials exhibit the intrinsic electronic characteristics of the polymers and concurrently respond to external magnetic fields. Electrospinning was utilized to fabricate polyacrylonitrile nanofibers blended with Fe2O3 and MnZn ferrite nanoparticles, which were thermomechanically, morphologically, and spectroscopically characterized in detail. With the application of an external magnetic field in the course of dynamic mechanical measurements under tension, the storage modulus of the glass transition Tg of PAN/Fe2O3 rises at the expense of the loss modulus, and a new peak emerges at ∼350 K. For the PAN/MnZn ferrite nanofibers a relatively larger shift in Tg (from ∼367 K to ∼377 K) is observed, emphasizing that in comparison to Fe2O3, Mn2+ ions in particular enhance the material's magnetic response in MnZn Ferrite. The magnetic oxide particles are homogenously dispersed in polyacrylonitrile, corroborated by high-resolution scanning electron microscopy. Both nanopowder additions lead to a slight shift of the peak towards larger angles, related to the shrinkage of the polymer. Produced nanofibers with high mechanical and heating efficiency can optimize the influence of the intracellular environment, magnetic refrigeration systems and sensors/actuators by their magnetic behavior and heat generation.
Introduction
Electrospinning is an important processing method for deriving micro- and nanofibers from polymer solutions in the presence of electrical forces.1 This technique exhibits the unique ability to produce diverse forms of polymeric fibrous assemblies. The remarkable specific surface area and high porosity make electrospun nanomaterials highly attractive to ultrasensitive sensor technologies and increase their importance in other nanotechnological applications. In terms of production, the specific polymer types, that is, electrospinnable polymers, polymer blends, solution, and processing parameters, significantly affect the fiber morphology.2 Adding carefully selected materials to polymer fiber blends can enhance their properties and make them suitable for a wider range of applications, including filtration,3,4 self-cleaning surfaces,5 biotechnology,3 environmental engineering4 and green chemistry.6 In particular, smart textile products play a crucial role in their development, such as medical textiles used in wound dressing, tissue engineering, and drug delivery, as well as protective clothing, touch screen displays, flexible fabric keyboards, energy storage, catalysts, electric double-layer capacitors (EDLC), and sensors7 (Fig. 1). | ||
| Fig. 1 Various application features of electrospun nanofibers. | ||
It has been shown that carbon nanotubes (CNTs) can significantly increase the strength and stiffness of electrospun nanofibers. Studies demonstrated that well-dispersed CNTs can lead to enhanced modulus, strength, and fracture resistance of nanocomposites.8 Our group previously reported the positive influence of CNTs containing polybutadiene–poly(ethylene oxide)9 and styrene–butadiene–styrene10 by increasing the decomposition temperature or promoting the crystallization kinetics. Carbon shells and graphene sheets combined with Fe2O3 nanoparticles have been reported to provide electrical networks that allow fast and efficient electron transport when these composites are used as anode materials in Li-ion batteries.11
Due to the magnetocaloric effect, which describes the influence of a magnetic field on the thermal properties of solids, the presence of iron makes magnetic refrigeration a viable cooling technology. This technology has the potential to compete with traditional gas compression methods used in air conditioning, refrigeration, and gas liquefaction.12
Iron oxide nanoparticles in small sizes exhibit superparamagnetic properties where their properties can be altered by an external magnetic field. Iron oxide nanoparticles provide electrical networks, allowing fast and efficient electron transport when these composites are used as anode materials.13 Specifically, an H atom can bind to the surface of iron oxide (FeO) at unblocked sites on symmetrically equivalent oxygen atoms, forming a hydrogen bond with another symmetrically equivalent O atom, as revealed by scanning tunneling microscopy measurements.13 MnZn ferrites possess almost zero magnetocrystalline anisotropy, high permeability, saturation induction, low power losses and high magnetic induction, which make them suitable candidates for wide range of applications.14 In nanostructure forms, they possess high redox pseudocapacitance, making it highly appropriate for energy storage devices.15 This improvement may stem from several factors, including enhanced magnetic properties, greater surface area for interactions, or alterations in the material's microstructure.16
Fe-based soft magnetic composite polyacrylonitrile (PAN) nanofibers might have improved magnetic characteristics compared to traditional bulk metals with large crystals due to their good thermal and mechanical stability.17 Combinations of the composing units of such nanocomposites with the inclusion of metal-based magnetic nanoparticles (MNPs) in a biocompatible PAN (known as technical textile material) will create an efficient electron transport.18
There are few studies present in the literature for the Fe2O3/PAN systems, but they are mainly focused on the filtration and removal by adsorption of heavy metal ions, i.e., electrospun polyacrylonitrile nanocomposite fibers reinforced with Fe3O4 nanoparticles were studied by ref. 19. The α-Fe2O3/PAN nanofiber mats were prepared via electrospinning followed by a hydrothermal method and used for the adsorption of Pb2+ by ion exchange between the proton of surface hydroxyl groups and Pb2+,20 and mixed-matrix membranes (MMMs), consisting of polyacrylonitrile (PAN) and hematite (α-Fe2O3) nanoparticles (NPs), were produced for the removal of nitrate (NO3−) from aqueous solutions.21 α-Fe2O3/polyacrylonitrile (PAN) hybrid composite adsorbent was also used to eliminate methylene blue (MB) from aqueous solution.22 Functionalized polyacrylonitrile coated with iron oxide nanoparticles was also applied to the nitrate removal from aqueous solution.23
We have recently studied PAN-2-(acryloyloxy)ethyl ferrocenecarboxylate polymer (FcP) blend nanofibers which were electrospun from the solution of N,N′-dimethylformamide and characterized by mechanical, spectroscopic, structural and electrochemical means.24 Frequency-dependent thermomechanical characterization via dynamic mechanical analysis (DMA) reveals pronounced changes in the glass transition, rubbery plateau, and onset of degradation, as well as in their storage and loss modulus values, particularly when small amounts of FcP are present. For wearable sensors made from soft, fabric-like patches on the skin (textile transdermal patches), PAN nanofibers offer several advantages. They improve breathability while effectively delivering drugs and biomolecules to promote cell healing. Additionally, PAN nanofibers have applications in demanding fields like automotive and aerospace, where their electro-active properties create highly sensitive and selective sensors.25 Magnetic nanoparticles have been exploited in intracellular magnetic hyperthermia, novel photothermic treatments, magnetic resonance imaging, magnetic particle imaging, magnetic-guided drug delivery, integrated biosensors and bio-separations.26 By applying various magnetic components for electronic devices, lighter and more compact designs can be generated for current and power transformers, inductors, and current sensors.27
In this study, production and advanced experimental characterizations and interfacial interaction of confined and homogenously dispersed magnetic nanoparticles of iron oxide and manganese zinc ferrite in the PAN matrix as in the nanofiber form are studied in detail. The main focus points are processing novel engineered functional nanocomposites and assessing compositional, and mainly thermomechanical properties under the applied magnetic field of confined magnetic nanoparticles in PAN nanofiber matrix.
Experimental section
Material
Polyacrylonitrile (PAN, average Mw 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), N,N′-dimethylformamide (DMF ≥ 99.8%) were obtained from Sigma Aldrich. Iron oxide nanopowder (γ-Fe2O3, 99.9%, 10 nm average diameter, near-spherical) and MnZn ferrite (MnZnFe2O4, 99.99%, size diameter 28 nm, near-spherical) nanoparticles were obtained from US Research Nanomaterials, Inc. All these chemicals were analytical grade and used as received.
000), N,N′-dimethylformamide (DMF ≥ 99.8%) were obtained from Sigma Aldrich. Iron oxide nanopowder (γ-Fe2O3, 99.9%, 10 nm average diameter, near-spherical) and MnZn ferrite (MnZnFe2O4, 99.99%, size diameter 28 nm, near-spherical) nanoparticles were obtained from US Research Nanomaterials, Inc. All these chemicals were analytical grade and used as received.
Preparation of PAN/metal oxide nanoparticle composite fibers
The electrospinning setup included a grounded collector placed within a syringe, a high voltage direct current (DC) power source capable of producing positive DC voltage up to 50 kV and a syringe pump with a feeding rate ranging from 5.5 μL h−1 to 400 mL h−1. The syringe needle, which had an outside diameter of 0.7 mm, was fitted with a positive electrode. A syringe pump was utilized to regulate the polymer solution's feeding rate, and the solutions were electrospun horizontally onto the aluminum collector. For the production of nanofibers, the polymer mixtures were electrospun at ambient temperature with driving voltages of 10–15 kV. There was a constant distance of around 15 cm between the capillary tip and the collector, and the feed rate was 1 mL h−1. These parameters were chosen based on our previous studies in this subject, which were proven to generate uniform nanofibers with second-phase inclusions.24 Different types of metal oxide nanoparticle contents were always prepared in 100 mL of 10% PAN solution. They were combined continuously and stirred overnight while PAN/metal oxide nanoparticle combinations were dissolved in DMF solution. The precursor solution was prepared by dissolving PAN in DMF solvent containing the MnZn ferrite and iron oxide nanoparticles. The precursor mixtures were prepared to be 5% (w/w, PAN/metal oxide nanoparticles) via mass. The nanoparticles were first dispersed well in a cup horn sonicator to avoid sediment formation, which is a common method in the dispersion of carbon/carbon-based materials. Nanofibers were continuously obtained by electrospinning continuously for 3 hours without sedimentation in polymer nanofiller solution. Nanoparticles were distributed homogeneously without agglomeration during long mixing times using the vibrations imposed by this device. The mixing cup is regularly checked by the lab technician to confirm that there was no nanoparticle sedimentation. After each electrospinning trial, no visual sedimentation was observed in the syringe.Crosslinked membranes were prepared on Al foil under a constant nitrogen flow in a Hönle UVASPOT 400 using an H2 filter over 5 h. Further melted/oxidized samples were prepared either directly during Raman microscopy by irradiation with the integrated 532 nm 75 mW DPSS laser at 10 mW over 0.5 s, or separately (for UV-vis and IR) using a Leister fiber-guided diode laser (940 nm, Novolas Basic AT) including Optic Globo V3.1 on a Gunnar x/y vacuum table (TTC 119 s) set to 4 A, 200 °C, 1 bar, focus of 10.5 and speed of 10 mm s−1 writing parallel lines at 2 mm distance reaching 120–200 °C (according to the integrated pyrometer).
Characterization methods
Dynamic mechanical analysis (DMA) experiments were performed using PerkinElmer DMA-8000 in the temperature range 300 K–800 K with a heating rate of 5 K min−1 and measured in tension mode at different frequencies with the applied sinusoidal force of 0.5, 5 and 15 Hz. The samples were prepared in sheet form for the tension geometry with sizes of about 10 × 7.5 × 0.13 mm3. To account for the effects of a magnetic field on the elastic properties of the sample in the regions of glass transition and crystallization, a static magnetic field of 120 mT (SMF; 120 mT [Bmax]) was applied parallel to the long sample side, which corresponds to the direction of tension. The storage modulus E′ is calculated using E′ = σ0/ε0cos(δ), where σ0 is the stress and ε0 the strain amplitude, and δ is the phase angle between the instantaneous stress and strain at time t. The loss modulus E′′ is calculated using E′′ = σ0/ε0sin(δ), whereas loss tangent (tan δ) is found from the ratio of loss and storage modulus, i.e. E′/E′′. Data was acquired directly from the device software and plotted by OriginLab 2023b. XRD was performed in Bragg–Brentano (θ–2θ) geometry with a Bruker D2Phaser diffractometer with an LYNXEYE-2 detector between 5°–100°. Co Kα radiation of λ = 0.17902 nm using a step size 0.005 was employed. The morphological characteristics and chemical composition analysis of the produced nanofibers were examined by scanning electron microscopy (SEM) and integrated energy-dispersive X-ray (EDX) analysis, respectively. Surface features were investigated on a Hitachi S-4800 SEM at 2 kV acceleration voltage on samples coated with Au/Pd (5 nm) using secondary electron (SE) mode. The same device was used at 20 kV acceleration voltage on C-coated samples (10 nm) for backscattering (BSE) analysis, combined with the subsequent EDX mapping on the same samples with the same acceleration using a ThermoFisher Scientific Axia ChemiSEM LoVac. Samples of electrospun (ES) membranes were prepared by biopsy-punching and gently pressing Al-backed samples on Carbon tape. Analogously, nanopowders were thinly spread on Al-foil and pressed on Carbon tape. Images and maps were then recorded at either 250, 10k or 30k magnification, with 10k images used for average diameter and standard deviation determination on 50 fibers via ImageJ Software.
In contrast, more densely packed nanopowders and flattened ES membranes were prepared in between a glass and cover slide for Raman confocal spectroscopy and microscopy on a WITec (Oxford Instruments) alpha300R using a 532 nm 75 mW DPSS laser. Zeiss EC “Epiplan” 10× and LD “Epiplan-Neofluar” 50× objectives were used for recording Raman spectra over 0.05 s at 20 to 50 accumulations and 10 mW laser power to avoid carbonization. Detailed maps and carbonization spectra were recorded using a Zeiss EC “Epiplan-Neofluar” 100× objective over 0.2 s at 20 accumulations and 10 mW laser power. Finally, similarly compressed samples were prepared for Fourier-transform infrared spectroscopy (FTIR) analysis on a Varian 640-IR by direct pressing with the sample holder. Spread-out UV-vis spectra were recorded on a Varian Cary 4000. Spectra were normalized to the highest peak, usually at 1450 and 2250 cm−1 for IR and Raman, respectively.
Results & discussions
Synthesis and possible interaction mechanism
As detailed in the experimental part, nanocomposites, i.e., PAN nanofibers containing iron oxide and MnZn Ferrite nanoparticles, and pure PAN nanofibers without magnetic nanoparticles, were fabricated by electrospinning technique. C–H functionalization/activation reactions within a range of polymers using Fe2O3 or MFe2O4 (M: metal) containing oxide nanoparticles were reported.28 The Fe–O bonds in the nanoparticles can participate in hydrogen bonding with the C–H bonds adjacent to the cyanide (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) bonds in PAN (Fig. 2), where such non-covalent bonds are interactions between molecules or atoms that do not involve sharing electrons. Their importance lies in their ability to form weak yet specific interactions between molecules, which are crucial for many functions in living systems and materials. This type of interaction can contribute to forming a network between the nanoparticles and PAN, decreasing the fiber dimensions and influencing their strength, elasticity and solubility as discussed in the next sections.
N) bonds in PAN (Fig. 2), where such non-covalent bonds are interactions between molecules or atoms that do not involve sharing electrons. Their importance lies in their ability to form weak yet specific interactions between molecules, which are crucial for many functions in living systems and materials. This type of interaction can contribute to forming a network between the nanoparticles and PAN, decreasing the fiber dimensions and influencing their strength, elasticity and solubility as discussed in the next sections.
 | ||
| Fig. 2 Possible interaction of Fe2O3 with polyacrylonitrile. | ||
Structural characterization
X-ray diffraction can reveal the type of crystalline nanoparticles and the changes in their crystalline structure within an amorphous polymer upon production. Fig. 3 displays the comparison of the PAN, PAN/Fe2O3 and PAN/MnZn ferrite. The diffuse peaks at ∼14.2° and ∼26.0° correspond to PAN.29 The XRD scans of the nanopowders identify that this broad peak corresponds to the PAN as well. Similarly, most of the other peaks of the γ-Fe2O3 and MnZn ferrite nanopowders match the literature findings with Cu Kα radiation.30 For both nanopowder types, there is a slight shift of the peak towards larger angles, i.e., ∼0.3° shift of the (311) peaks. This can be accounted for by the rapid cooling of the electrospun polymers on the Al foil, leading to shrinkage of the polymer. This shrinkage probably imposes compressive stresses on the nanoparticles, leading to a decrease in the lattice sizes. | ||
| Fig. 3 XRD spectra of the PAN, PAN/Fe2O3 and PAN/MnZn ferrite using Co Kα radiation. XRD of the nanoparticles is also provided. | ||
Spectroscopic analysis
FTIR lets us identify the functional groups in polymers and metal oxides (Fig. 4a). The broad peak at ∼3600 cm−1 is associated with the –OH group due to intermolecular hydrogen interactions and water contribution.31 The broadness of the peak might indicate the interaction between the ferrite and cyanoethyl side groups (–CN) of PAN. The peak at ∼2930 cm−1 belongs to the –C–H bonds of PAN.31 The FTIR spectrum of PAN exhibits characteristic peaks at 2238 cm−1 (–CN stretching).32 A vibration peak at 2355 cm−1 is likely due to the interaction of free sites of iron oxide with the PAN ethyl cyano groups. Broad peak between 3000– 3500 cm−1 must be due to the surface –OH groups of the magnetic nanoparticles.31 The peaks confirm the pristine PAN nanofiber at ∼1660 cm−1 and ∼1450 cm−1 attributed to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in the amide structure and CN stretching, respectively.32 The peaks at ∼1070 cm−1, ∼1230 cm−1, ∼1360 cm−1 should be related to the –C–H (non-flat), –C–O and –C–H bonds, respectively.31 Sharp peaks around 780 cm−1 are likely due to the bending vibrations of the –C–CN bonds in the cyanoethyl side groups.31 The change in this peak (I1) was compared for two different powder types by dividing it to the relatively stable PAN peak at 2238 cm−1 (I2). A significant rise in peak ratio is observed by adding MnZn ferrite instead of Fe2O3, referring to the high interaction for this type of blend (Table 1). Hydroxyl groups are chemically or physically interacting with iron atoms on the surface of Fe2O3 or Fe3O4 nanoparticles. In FTIR, the O–H stretching vibration indicates that the hydroxyl groups are involved in hydrogen bonding or coordination with iron atoms (2340 to 2360 cm−1). Hence, a possible interaction between the cyano groups and partial positive charge of Fe+2 through hydroxylation exists.33 Further investigation is required to assess the exact Fe–OH bonding mechanism.
O group in the amide structure and CN stretching, respectively.32 The peaks at ∼1070 cm−1, ∼1230 cm−1, ∼1360 cm−1 should be related to the –C–H (non-flat), –C–O and –C–H bonds, respectively.31 Sharp peaks around 780 cm−1 are likely due to the bending vibrations of the –C–CN bonds in the cyanoethyl side groups.31 The change in this peak (I1) was compared for two different powder types by dividing it to the relatively stable PAN peak at 2238 cm−1 (I2). A significant rise in peak ratio is observed by adding MnZn ferrite instead of Fe2O3, referring to the high interaction for this type of blend (Table 1). Hydroxyl groups are chemically or physically interacting with iron atoms on the surface of Fe2O3 or Fe3O4 nanoparticles. In FTIR, the O–H stretching vibration indicates that the hydroxyl groups are involved in hydrogen bonding or coordination with iron atoms (2340 to 2360 cm−1). Hence, a possible interaction between the cyano groups and partial positive charge of Fe+2 through hydroxylation exists.33 Further investigation is required to assess the exact Fe–OH bonding mechanism.
 | ||
| Fig. 4 (a) FTIR (top), (b) Raman (middle) and (c) UV-vis (bottom) spectra of the different PAN membranes and nanoparticles normalized to the highest peak (1446 and 2245 cm−1 for IR and Raman, respectively). Insert in UV-vis showing a visual representation of membranes before (fibrous) and after (film) laser irradiation – bottom PAN, middle PAN with ferrite (represented as FeO), top PAN with MnZn ferrite (represented as MxO). | ||
| FTIR | |
|---|---|
| Type | I 2 (2255 cm−1)/I1 (2238 cm−1) |
| PAN/MnZn ferrite | 0.72 |
| PAN/Fe2O3 | 0.20 |
| PAN | 0 |
| Raman | |
|---|---|
| Type | I 2 (2915 cm−1)/I1 (2245 cm−1) |
| PAN/MnZn ferrite | 0.52 |
| PAN/Fe2O3 | 0.46 |
| PAN | 0.39 |
Interestingly, an obvious change in the vibrations between 1500 to 1800 cm−1 from pristine over inertly to laser irradiated samples was observed. While pristine PAN membranes seem to exhibit CC vibrations around 1650 cm−1 (ref. 34) potentially because of remaining monomers, these entirely disappear after any irradiation either because of evaporation or polymerization. Generally, pure PAN does not change significantly depending on oxygen. However, particle-containing membranes seem to increase in intensity of CN vibrations under inert conditions, indicating only cyclization without further oxidation, such as in graphene.35 This is followed by a distinct multitude of peaks appearing between 1500 and 1800 cm−1 after laser irradiation without an inert atmosphere, which suggests the formation of graphene oxide. Similarly, such signals appear above 3500 cm−1. It should also be noted that the particle-specific signals between 2300 and 2400 cm−1 disappear in samples irradiated under inert gas, indicating either partial deoxygenation of particles or potential covering of particles with polymer due to induced nanoscopic melting behavior. After laser irradiation, these peaks reappear, indicating either disintegration or phase separation during the macroscopic melting of the membranes into films. Unfortunately, such peaks also appear in “pristine” PAN samples, likely because of cross-contamination during storage or physical handling. Despite this, no transformation into graphene or graphene oxide is observed in the 1500 to 1800 cm−1 region, indicating that superficial particles do not significantly catalyze the carbonization/oxidation processes. Raman spectra of PAN nanofiber and composite nanofiber insights into the chemical bonding and molecular structure of PAN and nanopowder interactions (Fig. 4b). In the Raman spectrum, the dominant band is the nitrile CN stretching band at 2245 cm−1 and the CH stretching band at ∼2925 cm−1. Another characteristic bands of PAN concern the CH2 bending at 1455 cm−1, CH bending at 1350 cm−1, C–OH bonding at 1070 cm−1, and CCH stretching at 830 cm−1.36 The characteristic PAN peaks are suppressed or even disappear after their interaction with nanoparticles since such samples showed significant discoloration and sensitivity to the strong laser light in Raman and hence were recorded in lower resolution. The ratio of the largest two peaks (I1 = 2245 cm−1 and I1 = 2915 cm−1) in each measurement is given in Table 1. Results show that the ratio increases with nanopowders, with MnZn ferrite being higher.
It is well-known that three major stabilization reactions—cyclization of the nitrile groups, dehydrogenation, and oxidation — take place concomitantly during heating PAN over 573–673 K.37 Thermal stabilization is critical in transforming polyacrylonitrile (PAN) fibers into an infusible, stable ladder polymer structure. This process involves the conversion of the cyano (CN) triple bonds in the PAN backbone into aromatic rings with cyano (CN) double bonds.38 The dehydrogenation reaction exhibits a diminished rate when conducted in an inert gas atmosphere.39 Finally, UV-vis spectra were recorded (Fig. 4c) to better understand the more or less visual changes, specifically a slight greying after irradiation under a protected atmosphere and the transparent but stronger colored films obtained after laser irradiation. Since the UV-vis analysis was conducted in transmission mode, no significant change was observed after the irradiation under inert gas to pristine membranes. Presumably, some carbon fibers were formed, which might have protected the voluminous membranes below and, hence, barely changed the bulk of the samples. In contrast, laser welding resulted in the melting of all samples, with all samples significantly shifting their absorption towards UV, which is likely caused by oxidation. The results of optical properties analyzed by UV-vis spectroscopy support that with an increase of nanoparticle content in PAN/Fe2O3 composite fibers, the absorbance as a function of wavelength increases. This absorption was recorded at a similar position (560 nm), confirming the interactions between nanoparticles and the polyacrylonitrile.40
Morphological investigation
High-resolution SEM results showing homogeneous nanofiber formation and nanoparticle and partial cluster distribution (Fig. 5), especially at high magnification, indicate that the particles are well-dispersed and electrospun membranes have a uniform nanofiber morphology. Single nanoparticles throughout both fiber types were observed, with only a slight difference between backscattering and secondary electron SEM. The imaging of the nanopowders is shown in the ESI section (Fig. S1†). Fig. S2† presents environmental scanning electron microscopy (ESEM) images that illustrate the overall morphology of the samples. PAN nanofibers (Fig. 6-top) show a wrinkled texture with a uniform thickness of 970 ± 110 nm. By embedding nanoparticles, the nanofibers exhibit several porous beads (Fig. 6-middle, bottom), which is caused by agglomerates with very high surface area and porosity persisting after spinning, preventing a smooth covering polymer layer. However, the complementary in-depth backscattering image (BSE) reveals that the majority of the particles were homogenously dispersed within the nanofibers (see Fig. 5). Homogeneous fibers and distribution are obtained for all cases, including the PAN/Fe2O3 and MnZn ferrite case, with fiber diameters slightly decreased when particles are incorporated (520 ± 50 nm and 520 ± 90 nm for Fe2O3 and MnZn ferrite, respectively). This is most likely caused by an increase in conductivity in the spinning solution caused by the nanoparticles and hence increased stretching. The impedimetric value of PAN fiber measurement at 1 Hz was measured as |Z|= ∼108–109 Ω (insulator).41 On the other hand, according to our findings at 1 Hz in 0.5 M H2SO4 |Z| of PAN/Fe2O3 and PAN/MnZn Ferrite are ∼44 × 103 Ω and ∼75 × 103 Ω, respectively. The increase in the conductivity of the nanoparticle-containing PAN nanofibers leads to the decrease of the thickness of the PAN fibers from 970 ± 110 nm to 520 ± 50 nm (PAN/Fe2O3), 520 ± 90 nm (PAN/MnZn Ferrite). The viscosity of the solution can vary with metallic oxide particle addition upon electrospinning due to insensitive viscosity measurement conditions in the vicinity of the needle tip as well as the shear-rate dependency of the electrospun polymer.42 | ||
| Fig. 5 HRSEM imaging in BSE (left) and SE (right) modes for three nanofiber types. | ||
 | ||
| Fig. 6 The surface texture of the PAN (top), PAN/Fe2O3 (middle) and PAN/MnZn ferrite (bottom) nanofibers. | ||
Compositional investigation
In order to investigate the distribution of particles and perhaps identify any separation in the MnZn ferrite particles, SEM-energy dispersive X-ray was performed (Fig. 7). The quant map in Fig. 7a depicts the Fe concentration at the local points where nanoparticles are located, confirming the Fe2O3 nanoparticles. Typically, for EDX, the carbon signal is weak and hence unsuitable for spatial or quantitative analyses, especially since carbon coating was used. Analogously, the quant map (Fig. 7b) corroborates the presence and homogenous dispersion of MnZn ferrite nanoparticles within PAN. Here, it needs to be indicated that the signal quality of Zn is low, which is probably due to the very low content, and thus, was not observed in EDX measurement. The fact that Mn and Fe maps overlap indicates no significant separation of the two-particle species, with a rather homogeneous distribution side-by-side. Fig. S3† shows the EDX spectra of the PAN/Fe2O3 and PAN/MnZn Ferrite samples. | ||
| Fig. 7 EDX quant map showing the (a) Fe (PAN/Fe2O3) and (b) Fe + Mn (PAN/MnZn ferrite) accumulation. | ||
Thermomechanical results under magnetic field
To understand the influence of the magnetic Fe2O3 and MnZn–ferrite nanoparticles within the PAN matrix, dynamic mechanical analysis under an external magnetic field (120 mT) was carried out. All samples showed distinct changes in the glass transition (Tg) (around 370 K) and stiffening (around 600 K) regions (Fig. 8). Before the glass transition, there is also a stiffening of the sample as the relaxation processes in the polymer increase, which leads to the reorganization of atoms (chains). The material becomes more homogeneous with fewer vacancies and, accordingly, more rigid. At the same time, we observe an increase in the modulus of elasticity before Tg. The situation is similar to that of samples containing nanoparticles. But when a magnetic field is applied, the magnetic field aligns the dipolar moments of the magnetic nanoparticles. In the vicinity of the glass transition, the material allows for enough deformation under a magnetic field, adding additional stiffness to the material and shifting the glass transition to a high-temperature region. | ||
| Fig. 8 DMA of the (a) PAN sample at three different frequencies without applied external field (0 T), comparison of the PAN/Fe2O3 and PAN/MnZn ferrite samples at (b) 0 T and (c) 120 mT and 0.5 Hz frequency. (Top) Storage modulus, (middle) loss modulus, and (bottom) tanδ. | ||
The pure PAN sample displays a clear Tg and crystallization temperature Tx, which intensify as the frequency rises from 0.5 to 15 Hz (Fig. 8a). The shift in the first glass transition Tg1 is more obvious from the dynamic loss modulus E′′, whereas the second glass transition temperature Tg2 can be observed from the tan δ (ratio of E′′ to dynamic storage modulus E′). The comparison of the PAN with PAN/Fe2O3 and PAN/MnZn ferrite samples at 0 T shows similarities, except that the PAN/Fe2O3 shows a very pronounced Tg2 seen from the tan δ vs. T graph (Fig. 8b). Comparison of all three curves at 0.5 Hz can be found in Fig. S4.† DMA results of the PAN/Fe2O3 and PAN/MnZn Ferrite samples without and with applied magnetic field at three different frequencies are displayed in Fig. S5 and S6,† respectively.
In a magnetic field, the stiffness increases because it aligns the magnetic nanoparticles along the magnetic field lines and creates additional strains (stresses) in the sample (Fig. 8c). For the PAN/Fe2O3 sample, an additional peak Ts shows up at ∼352 K. This means that the presence of Fe2O3 nanoparticles can modify the dynamics of the PAN polymer chains at the interface. This could lead to a distinct relaxation process appearing as a peak before Tg, especially if the magnetic field affects the interfacial interactions. The shift in Tg is more pronounced in the PAN/MnZn ferrite sample because MnZn ferrite exhibits a significant response due to its high magnetic permeability and low coercivity, which strongly interact with the magnetic field, causing notable shift in the glass transition temperature. In contrast, Fe2O3 has weaker magnetic interactions due to lower magnetic permeability and susceptibility, resulting in minimal influence of the magnetic field on its glass transition properties. Furthermore, MnZn ferrites have a more complex spinel structure with mixed cations that promote high permeability and susceptibility. The Mn2+ ions, in particular, enhance the material's magnetic response compared to Fe2+/Fe3+ interactions in γ-Fe2O3, which are less effective at promoting strong magnetic coupling.43 The two peaks in tanδ with two distinct changes in the storage modulus in the glass transition region probably correspond to two relaxation processes. The first relaxation is the glass transition of the PAN matrix. The second one corresponds to the glass transition of the PAN matrix around nanoparticles, where the relaxation processes slow down due to the contact of the PAN matrix with nanoparticles. Also, in the PAN/MnZn ferrite sample, we observe a significant shift of the Tx before the stiffening region (∼600 K, with a shift of 22 K towards larger values) when the magnetic field applied, suggesting that the magnetic field also affects the dynamics of the stiffening process. The change in the condition of the samples containing Fe2O3 and FeMn ferrite before and after DMA testing is given in Fig. S7.†
Fig. 9 compares the activation energies Ea of the three investigated samples with and without external fields. The slope measurement obtained from the Tg1 in E′′ vs. T graph shows a minor increase in the Ea for the PAN/Fe2O3, whereas this change is huge for the PAN/MnZn ferrite after the application of the external field. PAN sample shows very small Ea value measured from the Tg1 in loss modulus graph (Fig. 8a), referring to the samples with magnetic particles have higher stability. The magnetic field enhances the overall activation energy of the samples containing magnetic particles, yielding faster relaxation kinetics.
 | ||
| Fig. 9 Comparison of slopes yielding activation energy of the samples measured from the Tg1 from E′′. | ||
Overall, the results suggest that the magnetic field has a strong influence on the dynamics of the glass transition and the stiffening process. This new finding could have implications for the design and development of new materials with improved properties.
Stabilization (cyclization) of polyacrylonitrile during thermomechanical treatment
The heat treatment process for polyacrylonitrile (PAN)-based carbon fibers critically relies on a chemical transformation known as the cyclization reaction. For example, the cyclization reaction of PAN/Fe2O3 nanofiber is promoted by the catalytic effect and the presence of iron and heat during thermomechanical measurements. Zero-valent iron nanoparticles are reported as nanocatalysts for reduction reactions.44 A key step during this process was the association structures generated by the interaction of hydrogen bonds under heat treatment and the cyclization structures composed of CC and CN bonds.45
The color change observed upon heating is attributed to the formation of polyene structures during the polyacrylonitrile stabilization process. Additionally, it was proposed that the black color arises from the condensed ring structures absorbing all wavelengths within the visible light spectrum.36 This structure is formed by cyclization-induced carbon–nitrogen double bonds (an example provided for PAN/Fe2O3 nanofiber is shown in Fig. 10), known as a ladder structure. During the initial stages of stabilization (low-temperature range) in an air environment, PAN fibers undergo dehydrogenation, a process that introduces double bonds into the polymer backbone. Incorporating double bonds enhances the thermal stability of the PAN chain.46
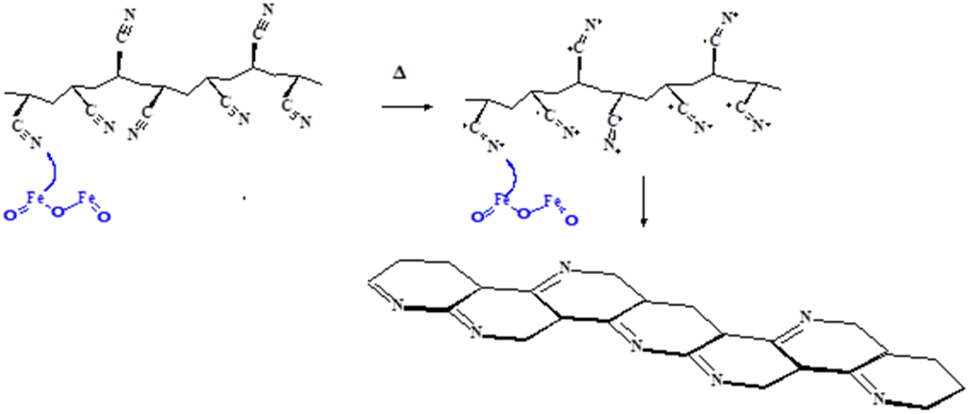 | ||
| Fig. 10 Cyclization of polyacrylonitrile structure by the catalytic action of Fe2O3 present in composite PAN nanofiber. | ||
Post-DMA morphological characterization performed ultra-high resolution SEM shows curvature formation in the nanofibers for the PAN/Fe2O3 sample, most probably related to internal stresses caused by the cyclization and oxidation (Fig. 11a and b). For the PAN/MnZn ferrite (Fig. 11c and d) and PAN (Fig. 11e and f) samples, the catalytic effect is much lower. Thus, the maximum temperature applied in DMA (780 K) cannot initiate the cyclization process completely.
 | ||
| Fig. 11 Post-DMA examination of the (a and b) PAN/Fe2O3, (c and d) PAN/MnZn ferrite, and (e and f) PAN at 20k× and 50k×. | ||
The carbonization process upon heat treatment was corroborated by the confocal Raman microscopy with its intense excitation laser after excess irradiation. The process includes mapping nanofibers via longer-time exposure to a laser compared to recording simple Raman spectra, which eventually carbonizes the particle-loaded nanofibers (Fig. 12). Confocal micrographs of untreated samples are shown in Fig. 12a, and an exemplary close-up comparison before and after carbonization of the PAN/MnZn ferrite is given in Fig. 12b. The patchy structure visible in the Raman intensity maps at different Raman shifts (Fig. 12c) can potentially be explained both by limits in resolution of nanofibrous carbon in combination with agglomeration of carbonized and traces of pristine polymer around or in between particle aggregates. Interestingly, even at the least intense irradiation/mapping conditions (see high noise in PAN spectrum), only D (∼1350 cm−1) and G (∼1600 cm−1) bands of carbon appear after heat treatment of PAN with nanoparticles, with no trace of PAN remaining (Fig. 12d). Additionally, the absence of any further band around 2700 cm−1 and the similar intensity of the D and G bands indicate that graphene oxide is obtained immediately instead of first forming graphene as a stable intermediate. These findings show the effectiveness of transforming intense light irradiation into heat by the nanoparticles used in the carbonization process of PAN. However, due to the complexity of the cyclization process, Raman spectroscopy cannot provide full evidence about the cyclization process by itself, and other methods such as differential scanning calorimetry, thermogravimetric analysis, nuclear magnetic resonance spectroscopy should be utilized to provide a complete conclusion.47
 | ||
| Fig. 12 (a) Confocal imaging of the PAN/Fe2O3 and PAN/MnZn ferrite. (b) Scale-up image of the PAN/MnZn ferrite before and after laser treatment. (c) Raman intensity maps for a single nanofiber at approx. (1) 100 cm−1, (2) 1350 cm−1, (3) 1600 cm−1, (4) 2250 cm−1, (5) 2950 cm−1. Black = no signal, white = highest signal (d) normalized Raman intensity vs. Raman shift graph for three different kinds of nanofibers (enlarged image of the selected spectra range in Fig. 12 middle). | ||
Conclusions
Magnetic nanocatalysts stand out due to their advantageous combination of low toxicity, widespread availability, cost-effectiveness, and ease of catalyst recovery using an environmentally friendly, solvent-free, and rapid magnetic separation method. We present that well-defined Fe2O3 and MnZn ferrite nanoparticle–polyacrylonitrile nanofibers can be obtained by electrospinning, while CN functional groups located at each monomer unit of PAN nanofibers can immobilize magnetic nanoparticles via hydrogen bonding and coordination interactions. Employed advanced characterization techniques, i.e., HRSEM, XRD, spectroscopic and thermomechanical results enlighten the mechanism of interaction between nanoparticle–polymer matrix through the determination of the morphology, interactions, and the crystallinity of the synthesized fibers with the presence of nanoparticles. SEM imaging and EDX mapping indicate NPs are well dispersed in PAN composite nanofibers with minimum bead formation. Fe2O3 and MnZn ferrite nanoparticles embedded in the polymer matrix hinder cross-linking throughout the network and enhance the inter-chain interactions. Compared to Fe2O3, the presence of Mn+2 in MnZn Ferrite nanoparticles leads to the shift of Tg by 10 K towards higher temperatures. On the other hand, for the PAN/Fe2O3, the main peak splits into two peaks under the external magnetic field. The unit lattice of Fe2O3 and PAN nanoparticles shrinks after blending with PAN, confirming the influence of cooling after electrospinning. The emergence of D and G bands of carbonized fibers via long-time laser treatment corroborates the changes in thermomechanical and morphological properties observed during DMA. These features make them the preferred catalysts for accelerating numerous industrially relevant reactions through C–H activation.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
BS: conceptualization, methodology, investigation, visualization, formal analysis, visualization, supervision, writing – original draft. VS: investigation, formal analysis, validation, visualization, writing – review & editing. GH: investigation, formal analysis, validation, visualization, writing – review & editing. SG: investigation, writing – review & editing. EK: investigation, writing – review & editing. EY: investigation, writing – review & editing. WS: supervision, resources, writing – review & editing. JE: supervision, resources, writing – review & editing. LFB: supervision, resources, writing – review & editing. JE: supervision, resources, writing – review & editing. ASS: conceptualization, visualization, formal analysis, writing – original draft.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 13th September 2024, which did not contain changes to the text provided at revision due to an internal processing error. Text, figure ordering and references have been updated, no scientific data has been changed, added, or removed in the updated article.Acknowledgements
We would like to thank for the Short-Term Scientific Mission (STSM) grant (E-COST-GRANT-CA21101-28134500) for ASS between 29/09/2023 -08/10/2023 by the Prof. Dr Wilfried Schranz (Host Institution Faculty of Physics, Physics of Functional Materials, University of Vienna). Empa acknowledges the helpful advice and support of A. Gogos and E. Perret regarding microscopic and spectroscopic investigations. This publication is also based upon work from the COST Action CONTEXT (CA17107), supported by COST (European Cooperation in Science and Technology). Open access funding provided by University of Vienna.Notes and references
- A. S. Sarac, Nanofibers of Conjugated Polymers, Jenny Stanford Publishing, Singapore, 1st edn, 2016 CrossRef; A. Al-Abduljabbar and I. Farooq, Polymers, 2023, 15, 65, DOI:10.3390/polym15010065.
- A. Keirouz, Z. Wang, V. S. Reddy, Z. K. Nagy, P. Vass, M. Buzgo, S. Ramakrishna and N. Radacsi, Adv. Mater. Technol., 2023, 8, 2201723, DOI:10.1002/admt.202201723.
- J. d. A. B. Barbosa, C. A. de França, J. J. d. S. Gouveia, G. V. Gouveia, M. M. da Costa and H. P. de Oliveira, J. Appl. Polym. Sci., 2019, 136, 47479, DOI:10.1002/app.47479.
- S. Ramakrishna, K. Fujihara, W.-E. Teo, T. Yong, Z. Ma and R. Ramaseshan, Mater. Today, 2006, 9, 40, DOI:10.1016/S1369-7021(06)71389-X.
- Q.-L. Huang, Y. Huang, C.-F. Xiao, Y.-W. You and C.-X. Zhang, J. Membr. Sci., 2017, 534, 73, DOI:10.1016/j.memsci.2017.04.015; K. Wang, S. Yu, W. Li, Y. Song, P. Gong, M. Zhang, H. Li, D. Sun, X. Yang and X. Wang, Appl. Surf. Sci., 2022, 595, 153565, DOI:10.1016/j.apsusc.2022.153565.
- W. Han, D. Rao, H. Gao, X. Yang, H. Fan, C. Li, L. Dong and H. Meng, Nano Energy, 2022, 97, 107237, DOI:10.1016/j.nanoen.2022.107237; C. Z. Mosher, P. A. P. Brudnicki, Z. Gong, H. R. Childs, S. W. Lee, R. M. Antrobus, E. C. Fang, T. N. Schiros and H. H. Lu, Biofabrication, 2021, 13, 035049, DOI:10.1088/1758-5090/ac0964; J. Avossa, G. Herwig, C. Toncelli, F. Itel and R. M. Rossi, Green Chem., 2022, 24, 2347, 10.1039/D1GC04252A.
- C. Cleeton, A. Keirouz, X. Chen and N. Radacsi, ACS Biomater. Sci. Eng., 2019, 5, 4183, DOI:10.1021/acsbiomaterials.9b00853; S. Kang, K. Zhao, D.-G. Yu, X. Zheng and C. Huang, Adv. Fiber Mater., 2022, 4, 404, DOI:10.1007/s42765-021-00129-0; M. Li, W. Qiu, Q. Wang, N. Li, L. Liu, X. Wang, J. Yu, X. Li, F. Li and D. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 15911, DOI:10.1021/acsami.1c24131; X. Zhang, R. Lv, L. Chen, R. Sun, Y. Zhang, R. Sheng, T. Du, Y. Li and Y. Qi, ACS Appl. Mater. Interfaces, 2022, 14, 12984, DOI:10.1021/acsami.1c22629; E. Ismar and A. S. Sarac, J. Eng. Fibers Fabr., 2019, 14, 1558925018824890, DOI:10.1177/1558925018824890; D. S. Can, H. Baskan, S. Gumrukcu and A. S. Sarac, J. Nanosci. Nanotechnol., 2019, 19, 3844, DOI:10.1166/jnn.2019.16309; D. Gülercan, İ. Gergin and A. S. Sarac, Fibers Polym., 2018, 19, 2178, DOI:10.1007/s12221-018-8393-7; Z. Guler Gokce, P. Akalın, F. N. Kok and A. S. Sarac, Sens. Actuators, B, 2018, 254, 719, DOI:10.1016/j.snb.2017.07.136.
- P.-C. Ma, N. A. Siddiqui, G. Marom and J.-K. Kim, Composites, Part A, 2010, 41, 1345, DOI:10.1016/j.compositesa.2010.07.003.
- R. Gürbüz, B. Sarac, V. Soprunyuk, A. Rezvan, E. Yüce, W. Schranz, J. Eckert, A. Ozcan and A. S. Sarac, Polym. Adv. Technol., 2022, 33, 3966, DOI:10.1002/pat.5828.
- B. Sarac, R. Gürbüz, M. Micusik, M. Omastova, A. Rezvan, E. Yüce, L. Xi, J. Eckert, A. Ozcan and A. S. Sarac, Mol. Syst. Des. Eng., 2023, 8, 911, 10.1039/D3ME00035D.
- S. Mourdikoudis, A. Kostopoulou and A. P. LaGrow, Adv. Sci., 2021, 8, 2004951, DOI:10.1002/advs.202004951.
- K. A. Gschneidner and V. K. Pecharsky, Mater. Sci. Eng., A, 2000, 287, 301, DOI:10.1016/S0921-5093(00)00788-7.
- M. N. S. Shahrodin, J. Jaafar, A. R. Rahmat, N. Yusof, H. M. Dzarfan Othman and A. M. Rahman, Micro Nanosyst., 2020, 12, 4, DOI:10.2174/1876402911666190716155658.
- P. Thakur, D. Chahar, S. Taneja, N. Bhalla and A. Thakur, Ceram. Int., 2020, 46, 15740, DOI:10.1016/j.ceramint.2020.03.287.
- J.-L. Ortiz-Quiñonez, S. Das and U. Pal, Prog. Mater. Sci., 2022, 130, 100995, DOI:10.1016/j.pmatsci.2022.100995.
- B. H. K. Lopes, R. C. Portes, M. A. d. Amaral Junior, D. E. Florez-Vergara, A. M. Gama, V. A. Silva, S. F. Quirino and M. R. Baldan, J. Mater. Res. Technol., 2020, 9, 2369, DOI:10.1016/j.jmrt.2019.12.068.
- A. Suhasini, K. P. V. Kumar and T. Maiyalagan, Sci. Eng. Compos. Mater., 2018, 25, 189, DOI:10.1515/secm-2015-0531.
- D. Romero-Fierro, M. Bustamante-Torres, F. Bravo-Plascencia, H. Magaña and E. Bucio, Polymers, 2022, 14, 2467, DOI:10.3390/polym14122467; T. Blachowicz, J. Grzybowski and A. Ehrmann, Mater. Today: Proc., 2022, 67, 792, DOI:10.1016/j.matpr.2022.07.362.
- D. Zhang, A. B. Karki, D. Rutman, D. P. Young, A. Wang, D. Cocke, T. H. Ho and Z. Guo, Polymer, 2009, 50, 4189, DOI:10.1016/j.polymer.2009.06.062; D. Zhang, S. Wei and Z. Guo, MRS Online Proc. Libr., 2010, 1240, 1006, DOI:10.1557/PROC-1240-WW10-06.
- J. Chang, J. Wang, J. Qu, Y. Vivian Li, L. Ma, L. Wang, X. Wang and K. Pan, Environ. Sci.: Nano, 2016, 3, 894, 10.1039/C6EN00088F.
- P. Bahmani, A. Maleki, H. Daraei, M. Khamforoush, S. Dehestani Athar and F. Gharibi, J. Mol. Liq., 2018, 271, 557, DOI:10.1016/j.molliq.2018.09.036.
- S. Mohammadreza Miraboutalebi, M. Peydayesh, M. Bagheri and T. Mohammadi, Chem. Eng. Technol., 2020, 43, 1214, DOI:10.1002/ceat.202000140.
- R. Nabizadeh, M. Jahangiri-Rad, M. Yunesian, J. Nouri, F. Moattar and S. Sadjadi, Desalin. Water Treat., 2015, 53, 2636, DOI:10.1080/19443994.2013.867816.
- S. Gumrukcu, V. Soprunyuk, B. Sarac, E. Yüce, J. Eckert and A. S. Sarac, Mol. Syst. Des. Eng., 2021, 6, 476, 10.1039/d1me00008j.
- C. M. Costa, V. F. Cardoso, P. Martins, D. M. Correia, R. Gonçalves, P. Costa, V. Correia, C. Ribeiro, M. M. Fernandes, P. M. Martins and S. Lanceros-Méndez, Chem. Rev., 2023, 123, 11392, DOI:10.1021/acs.chemrev.3c00196; K. Huner, B. Sarac, E. Yüce, A. Rezvan, M. Micusik, M. Omastova, J. Eckert and A. S. Sarac, Mol. Syst. Des. Eng., 2023, 8–394, 10.1039/D2ME00181K.
- R. A. Revia and M. Q. Zhang, Mater. Today, 2016, 19, 157, DOI:10.1016/j.mattod.2015.08.022; S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2019, 138, 302, DOI:10.1016/j.addr.2019.01.005.
- Y. Guo, L. Liu, W. Yin, H. Lu, G. Lei and J. Zhu, Nanomaterials, 2023, 13, 1963, DOI:10.3390/nano13131963; S. J. Salih and W. M. Mahmood, Heliyon, 2023, 9, e16601, DOI:10.1016/j.heliyon.2023.e16601.
- S. Dutta, P. Kumar, S. Yadav, R. D. Sharma, P. Shivaprasad, K. S. Vimaleswaran, A. Srivastava and R. K. Sharma, Catal. Commun., 2023, 175, 106615, DOI:10.1016/j.catcom.2023.106615.
- Y. Zhao, Z. Zhao, M. Wei, X. Jiang, H. Li, J. Gao and L. Hou, Prog. Nat. Sci.: Mater. Int., 2018, 28, 337, DOI:10.1016/j.pnsc.2018.04.013.
- H. A. Alshamsi and B. S. Hussein, Asian J. Chem., 2017, 30, 273, DOI:10.14233/ajchem.2018.20888; D. Arcos, R. Valenzuela, M. Vallet-Regí and M. Vázquez, J. Mater. Res., 1999, 14, 861, DOI:10.1557/JMR.1999.0115; M. Hjiri, N. Zahmouli, K. Khouzami, L. E. Mir, M. S. Aida, K. Moulaee, O. M. Lemine, S. G. Leonardi and G. Neri, Appl. Phys. A, 2020, 126, 788, DOI:10.1007/s00339-020-03829-3.
- E. V. Loginova, I. V. Mikheev, D. S. Volkov and M. A. Proskurnin, Anal. Methods, 2016, 8, 371, 10.1039/C5AY02264A.
- Q. Liu, N. Xu, L. Fan, A. Ding and Q. Dong, Chem. Eng. Sci., 2020, 228, 115993, DOI:10.1016/j.ces.2020.115993.
- G. S. Parkinson, Surf. Sci. Rep., 2016, 71, 272, DOI:10.1016/j.surfrep.2016.02.001.
- I. Karbownik, M. Fiedot, O. Rac, P. Suchorska-Woźniak, T. Rybicki and H. Teterycz, Polymer, 2015, 75, 97, DOI:10.1016/j.polymer.2015.08.015.
- A. Mehmood, N. M. Mubarak, M. Khalid, P. Jagadish, R. Walvekar and E. C. Abdullah, Sci. Rep., 2020, 10, 20106, DOI:10.1038/s41598-020-77139-2.
- G. Konstantopoulos, S. Soulis, D. Dragatogiannis and C. Charitidis, Materials, 2020, 13, 2749, DOI:10.3390/ma13122749.
- Y. Furushima, R. Kumazawa, Y. Yamaguchi, N. Hirota, K. Sawada, M. Nakada and M. Murakami, Polymer, 2021, 226, 123780, DOI:10.1016/j.polymer.2021.123780.
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421, DOI:10.1016/j.polymdegradstab.2007.03.023.
- A. Gupta and I. R. Harrison, Carbon, 1996, 34, 1427, DOI:10.1016/S0008-6223(96)00094-2.
- T. Tański, W. Matysiak and P. Witek, Appl. Eng. Lett., 2017, 2, 54 Search PubMed.
- O. Rac-Rumijowska and H. Teterycz, Materials, 2023, 16, 3085, DOI:10.3390/ma16083085; D. Giray, T. Balkan, B. Dietzel and A. Sezai Sarac, Eur. Polym. J., 2013, 49, 2645, DOI:10.1016/j.eurpolymj.2013.06.012.
- N. Bhardwaj and S. C. Kundu, Biotechnol. Adv., 2010, 28, 325, DOI:10.1016/j.biotechadv.2010.01.004; J. M. Deitzel, J. Kleinmeyer, D. Harris and N. C. Beck Tan, Polymer, 2001, 42, 261–272, DOI:10.1016/S0032-3861(00)00250-0.
- F. Luo, Z. Duan, Y. Zhang and Y. Shang, J. Mater. Sci.: Mater. Electron., 2021, 32, 15633, DOI:10.1007/s10854-021-06112-0.
- Z. H. Farooqi, R. Begum, K. Naseem, W. Wu and A. Irfan, Catal. Rev., 2022, 64, 286, DOI:10.1080/01614940.2020.1807797.
- Z. He, H. Liu, S. Zhang, J. Yang, C. Jiang, M. Ji, J. Yu, M. Wang, C. Zhu and J. Xu, Ind. Eng. Chem. Res., 2020, 59, 9519, DOI:10.1021/acs.iecr.0c00977.
- Y. Xue, J. Liu and J. Liang, Polym. Degrad. Stab., 2013, 98, 219, DOI:10.1016/j.polymdegradstab.2012.10.018; S. Arbab and A. Zeinolebadi, Polym. Degrad. Stab., 2013, 98, 2537, DOI:10.1016/j.polymdegradstab.2013.09.014.
- B. Szepcsik and B. Pukánszky, Thermochim. Acta, 2019, 671, 200, DOI:10.1016/j.tca.2018.12.004.
Footnote |
| † Electronic supplementary information (ESI) available: Additional DMA, SEM and EDX data. See DOI: https://doi.org/10.1039/d4na00631c |
| This journal is © The Royal Society of Chemistry 2024 |