
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDisubstituted thiourea as a suitable sulfur source in the gram-scale synthesis of yellow- and red-emitting CdTeS/CdxZn1−xS core/shell quantum dots
Liudmila
Loghina
 *a,
Jakub
Houdek
a,
Stanislav
Slang
a,
Bozena
Frumarova
a,
Miroslav
Cieslar
c and
Miroslav
Vlcek
ab
*a,
Jakub
Houdek
a,
Stanislav
Slang
a,
Bozena
Frumarova
a,
Miroslav
Cieslar
c and
Miroslav
Vlcek
ab
aCenter of Materials and Nanotechnologies, Faculty of Chemical Technology, University of Pardubice, 53002 Pardubice, Czech Republic. E-mail: liudmila.loghina@upce.cz
bDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, 53210 Pardubice, Czech Republic
cFaculty of Mathematics and Physics, Charles University, 12116 Prague, Czech Republic
First published on 9th May 2024
Abstract
The key parameters of semiconductor quantum dots (QDs) that determine the suitability and efficiency for the design of most optoelectronic devices are the spectral positions of absorbance (ABS) and photoluminescence (PL) maxima, Stokes shift, photoluminescence quantum yield (PL QY) and photoluminescence lifetime (PL LT). All these parameters have been considered in the design of new ternary core CdTeS and core/shell CdTeS/CdxZn1−xS QDs. One-pot synthesis conducted in an organic medium at 160 °C using substituted thioureas as new, highly reactive sulfur sources allowed for the formation of a series of size- and emission-tunable CdTe0.05S0.95 QDs. Gram-scale synthesis of yellow-red emitting CdTe0.06S0.94 and CdTe0.12S0.88 cores was performed through the manipulation of their precursor ratio for the controllable formation of CdTeS/CdxZn1−xS (x = 0.1, 0.2, and 0.3) core/shell QDs. The development of the designed nanomaterials was carried out with a special emphasis on their optical properties, in particular a high PL QY up to 87% and extremely large Stokes shift, reaching ≈280 nm for core/shell QDs. Promisingly, for biolabeling and diagnostics, the synthesized core/shell QDs were transferred into water via surface ligand modification with the expected loss of photoluminescence efficiency. The results indicated that the availability of initial components, high yield of the desired product, stability in the organic phase, and high optical characteristics can scale up the synthesis of the developed nanomaterials from the laboratory level to industrial production.
Introduction
Colloidal chemical approaches comprise continuously advancing current methods and newly emerging methods for the synthesis of various types of nanocrystals. One of the perspective directions of modern nanotechnology is the fabrication of semiconductor nanomaterials called quantum dots (QDs). Owing to their unique size-dependent emission-tunable optical properties, adjustable surface adhesion, and composition flexibility, QDs find many applications in medicine,1 biology,2 and optoelectronics.3,4 The possibility to manipulate the shape, size and content of impurities of QDs opens a significant perspective to improve classical colloid synthesis methods for the fabrication of catalysts,5,6 sensors,7,8 solar cells,9 light-emitting diodes (LEDs)10 and many others.Based on the nature of precursors and reaction media, colloidal synthesis methods can be classified into aqueous and organic syntheses. The main advantage of the aqueous synthesis of QDs is the low cost of the product and minimal environmental impacts. However, the low crystal growth rate limited by the narrow temperature range, as well as the low yield of the nanomaterial, restricts the possibilities of aqueous synthesis. To date, many works on the synthesis of binary CdTe QDs have been conducted, among which most syntheses have been carried out in the aqueous phase with various combinations of capping agents.11–16 Owing to the hydrolysis of thiol ligands, it was possible to synthesize ternary CdTeS QDs in a hydrothermal system.17 At various ligand/cadmium molar ratios, QDs with different PL QYs were obtained, and the best result of PL QY was 68% at a cadmium/MPA (mercaptopropionic acid) ratio of 1.8. With the prospect of further application as detectors of biological information of living organisms, highly photoluminescent NAC-capped CdTeS-alloyed QDs were synthesized as well.18
Homogeneous and heterogeneous CdSxTe1−x alloy nanocrystals were prepared via the pyrolysis of organometallic precursors. Composition-tunable optical properties were achieved through manipulation of the S/Te precursor ratio.19 The fabrication of CdTe0.6S0.4 QD-sensitized TiO2 electrodes was performed for photovoltaic applications.20 The results on core/shell CdTeS/CdZnS QDs have been published recently.21 In this study, the CdTe0.5S0.5 cores were prepared at 280–300 °C using a mixture of octadecylphosphonic acid (ODPA), trioctylphosphine (TOP) and trioctylamine (TOA). The PL quantum efficiency was improved up to 48% by the CdZnS shell overcoating.
It is well known that nanosized metal chalcogenides consist of a stoichiometric core with a surface protective shell in the form of metal–ligand complexes.22,23 Depending on the requirements for the nanomaterial, the existing protective metal–ligand shell can be partially or completely replaced.24 The effect of ligands on optical properties has also been investigated on high-luminescence CdTe/CdS quantum dots.25
In this study, we investigated the possibility of mixing two different chalcogen precursors, which have not previously been mentioned in the literature together. Trialkylphosphine chalcogenides react with a metal carboxylate to form the corresponding metal chalcogenide and trialkylphosphine oxide in a one-step process.26 Substituted thioureas form stable complexes with the metal carboxylate, which begin to decompose at temperatures above 80 °C, releasing the corresponding metal sulfide.27,28 Despite the difference in the mechanisms of parallel reactions, the resulting molecular cadmium sulfide and telluride crystallize together form a sulfide–telluride alloy. One of the selected compositions (CdTe0.05S0.95 QDs) was prepared from various substituted thioureas, with a fixed amount of trioctylphosphine telluride. As expected, the composition and crystal lattice did not change, but the sizes of the nanocrystals and, accordingly, the quantum yield were affected. Using our previously published methodology for applying a shell to an already formed core, core/shell CdTe0.06S0.94/CdxZn1−xS 2 MLs (monolayers) and CdTe0.12S0.88/CdxZn1−xS 2 MLs QDs (x = 0.1, 0.2, and 0.3) were prepared as well. The approbation of the methodology was successful, as confirmed by STEM microscopy and X-ray diffraction analysis. An increase in the quantum yield of up to 87% was also observed. To study the possibility of using the synthesized nanomaterials in medicine and biology, some compositions were subjected to the replacement of the protective ligand in the QD shell with MPA. When replacing the ligand, some change in the optical parameters was detected.
Materials and methods
Characterization
IR spectra in the region of 4000–400 cm−1 (resolution 2 cm−1) were recorded using a Vertex 70V FT-IR spectrometer (Bruker, Germany) with a single-bounce diamond ATR crystal. X-ray diffraction patterns (XRD) were acquired using a PANalytical EMPYREAN powder X-ray diffractometer (ALMELO, Netherlands) with Cu-Kα radiation (λ = 1.5418 Å). Data were obtained across a 2θ range of 20–70° with a step size of 0.05°.The influence of different synthesis conditions on the size and shape of QDs was documented using a scanning electron microscope LYRA 3 (STEM, Tescan, Czech Republic) equipped with an EDX detector AZtec X-Max 20 (Oxford Instruments, UK) at 20 kV accelerating voltage. Energy-dispersive X-ray (EDX/EDS) spectroscopy was performed to analyze the chemical composition of QDs using the same device equipped with a retractable STEM detector (Tescan, Czech Republic) at 30 kV accelerating voltage. To visualize the structure of the QDs with sufficient atomic resolution and contrast, a high-resolution transmission electron microscope (HRTEM, JEOL, Japan) equipped with a Centurio SDDEDS detector was employed. The samples were prepared by dropping a QD solution on a TEM copper grid with a 1 nm-thick graphene oxide film on a lacey carbon membrane.
The optical properties of QDs were measured using a Fluorometer PTI QuantaMaster 400 (Horiba, Germany) to obtain PL data in the spectral range of 300–850 nm at an excitation wavelength (λ) of 300–500 nm and a UV-3600 (Shimadzu, Japan) spectrometer to record UV-vis absorbance spectra in the spectral range of 200–800 nm. The measurements and calculations carried out to determine the PL QY of all the synthesized CdTeS cores and CdTeS/CdxZn1−xS core/shell QDs were repeated 5 times, with the average reported value (error ± 3%).
Nuclear magnetic resonance (NMR) spectra were recorded from solutions of CDCl3 at 295 K using a Bruker Ascend™ 500 spectrometer (equipped with a Z-gradient 5 mm Prodigy cryoprobe) at frequencies of 500.13 MHz for 1H and 125.76 MHz for 13C correspondingly. The solutions were obtained by dissolving approximately 20–40 mg of each compound in 0.6 ml of deuterated solvent. The values of 1H chemical shifts were calibrated to residual signals of CDCl3 (δ (1H) = 7.26 ppm). The values of 13C chemical shifts were referenced to the signals of CDCl3 (δ (13C) = 77.23 ppm). The positive chemical shift values denote shifts to higher frequencies relative to the standards.
Materials
Cadmium oxide (CdO, 99.99%), zinc oxide (ZnO, 99%), oleylamine (OAm, technical grade, 70%), linoleic acid (LA, technical grade, 60–74%), 1-octadecene (ODE, technical grade, 90%), phenyl isothiocyanate (98%), allyl isothiocyanate (95%), benzylamine (BnNH2, 99%), dodecylamine (98%), cyclohexylamine (99%), octylamine (99%), diethylamine (DEA, 99%), carbon disulfide (CS2, 99%), trioctylphosphine (TOP, technical grade, 90%), 2,4,6-trichloro-1,3,5-triazine (TCT, 99%), 3-mercaptopropionic acid (MPA, 98%), tetramethylammonium hydroxide solution (TMAH, 25% in methanol), chloroform-d (CDCl3, 99.8 atom% D) and silica gel (high-purity grade, 60 Å pore size, 230–400 mesh particle size) were purchased from Sigma-Aldrich and used without further purification. Sodium hydroxide (NaOH, p.a.), hydrochloric acid (HCl, 35%), potassium carbonate (K2CO3, p.a.), and solvents were purchased from Fisher Scientific and used without further purification. Syntheses of QDs were carried out using standard Schlenk techniques in an Ar atmosphere.Synthesis of substituted thioureas
Amine (0.2 mol) was added in one portion to a solution of isothiocyanate (0.2 mol) in 150 ml of CH2Cl2 under constant stirring at room temperature. The stirring was continued until complete dissolution. The reaction was monitored by TLC (CH2Cl2). The solvent was distilled off using a rotary evaporator, and the residue was used for the synthesis of QDs without additional purification.
1H NMR (500.13 MHz, CDCl3), ppm: 6.65 (s, 1H, NH), 6.43 (s, 1H, NH), 5.76–5.84 (m, 1H, CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.10–5.20 (m, 2H, CH2–CHCH2), 4.04 (s, 2H, CH2–CHCH2), 3.38 (s, 2H, CH2), 1.47–1.53 (m, 2H, CH2), 1.26–1.33 (m, 2H, CH2), 0.86 (t, 3H, J = 7.52 Hz, CH3). 13C NMR (125.76 MHz, CDCl3), ppm: 181.47 (CS), 133.47, 116.98, 46.78, 44.34, 31.02, 19.94, 13.75.
CH2), 5.10–5.20 (m, 2H, CH2–CHCH2), 4.04 (s, 2H, CH2–CHCH2), 3.38 (s, 2H, CH2), 1.47–1.53 (m, 2H, CH2), 1.26–1.33 (m, 2H, CH2), 0.86 (t, 3H, J = 7.52 Hz, CH3). 13C NMR (125.76 MHz, CDCl3), ppm: 181.47 (CS), 133.47, 116.98, 46.78, 44.34, 31.02, 19.94, 13.75.
1H NMR (500.13 MHz, CDCl3), ppm: 6.16 (br.s, 2H, 2NH), 5.38–5.91 (m, 1H, CHCH2), 5.18–5.27 (dd, 2H, J1 = 17.66 Hz, J2 = 34.87 Hz, CHCH2), 4.08 (br.s, 2H, CH2CH–CH2–NH), 1.54–1.60 (m, 2H), (m, 2H, CH2–CH2–NH), 1.24–1.35 (m, 18H, 9CH2), 0.86 (t, 3H, J = 6.79 Hz, CH3). 13C NMR (125.76 MHz, CDCl3), ppm: 181.75 (CS); 133.58; 117.39; 31.99; 29.73; 29.71; 29.66; 29.62; 29.43; 29.36; 29.04; 27.00; 22.74; 14.21.
1H NMR (500.13 MHz, CDCl3), ppm: 7.27–7.35 (m, 5H, aryl), 6.36 (br.s, 1H, NH), 5.94 (br.s, 1H, NH), 4.60 (br.s, 2H, CH2–NH), 3.87 (br.s, 1H, CH), 1.91–1.95 (m, 2H, CH2), 1.55–1.65 (m, 3H, CH, CH2), 1.27–1.36 (m, 2H, CH2), 1.10–1.17 (m, 3H, CH, CH2). 13C NMR (125.76 MHz, CDCl3), ppm: 180.54 (CS); 128.98; 127.94; 127.62; 32.78; 24.40; 24.64.
1H NMR (500.13 MHz, CDCl3), ppm: 8.35 (br.s, 1H, NH), 7.42 (t, 2H, J = 7.70 Hz, aryl), 7.29 (t, 1H, J = 7.70 Hz, aryl), 7.21–7.23 (d, 2H, J = 7.86 Hz, aryl), 6.06 (br.s, 1H, NH), 3.58–3.62 (m, 2H, CH2–NH), 1.52–1.58 (m, 2H, CH2–CH2–NH), 1.24–1.29 (m, 10H, 5CH2), 0.87 (t, 3H, J = 6.86 Hz, CH3). 13C NMR (125.76 MHz, CDCl3), ppm: 180.34 (CS); 130.26; 127.23; 125.26; 45.60; 31.82; 29.36; 29.02; 26.94; 22.70; 14.19.
1H NMR (500.13 MHz, CDCl3), ppm: 5.99 (br.s, 2H, 2NH), 3.39 (m, 2H, CH2–NH), 1.55–1.61 (m, 4H, 2CH2–CH2–NH), 1.25–1.34 (m, 20H, 10CH2), 0.86 (t, 6H, J = 6.86 Hz, 2CH3). 13C NMR (125.76 MHz, CDCl3), ppm: 181.37 (CS); 31.86; 29.34; 29.28; 27.02; 22.72; 14.17.
1H NMR (500.13 MHz, CDCl3), ppm: 5.33 (br.s, 1H, NH), 3.16–3.67 (m, 6H, –CH2–NH, –N(CH2)2), 1.58–1.63 (m, 2H, CH2–CH2–NH), 1.25–1.35 (m, 18H, 9CH2), 1.21 (t, 6H, J = 7.20 Hz, 2CH3), 0.87 (t, 3H, J = 6.77 Hz, CH3). 13C NMR (125.76 MHz, CDCl3), ppm: 180.23 (CS); 46.29; 45.13; 32.02; 29.73; 29.67; 29.52; 29.47; 29.45; 27.15; 22.79; 14.24; 12.80.
Gram-scale synthesis of CdTeS cores
Cadmium oxide (CdO, 0.021 mol, 2.70 g), linoleic acid (LA, 0.084 mol, 23.56 g), and 1-octadecene (ODE, 80 ml) were mixed in a 250 ml three-necked flask. The resulting mixture was degassed (the air was replaced with argon) at room temperature for 30 minutes, and then heated to 100 °C until complete homogenization, indicating the conversion of cadmium oxide into the corresponding linoleate. The reaction mixture was subsequently naturally cooled to room temperature. Further, TU1 was introduced in one portion, and then a freshly prepared 0.5 M Te–TOP solution was injected. The ratios of the precursors varied depending on the composition of the QDs, maintaining the overall equimolar ratio to cadmium. The temperature of the reaction mixture was increased at a rate of 5 °C min−1 to 160 °C. Starting from 140 °C and until the end of the process, aliquots were taken to assess the growth of nanocrystals. Aliquots of 25 μl were dissolved in CHCl3 (2 ml) and stored at room temperature. From the moment of reaching 160 °C, heating was continued for 30 minutes, after which the reaction mixture was naturally cooled to room temperature. The cold colloidal solution was transferred to an equal volume of chloroform, and then acetone was added in portions until a precipitate began to form. QDs were separated in a centrifuge (Gusto Mini Centrifuge, Fisherbrand, angle rotor with radius 55 mm) by spinning down at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (6160 rcf) for 7 min followed by dissolution in CHCl3 and reprecipitation with acetone. This purification was repeated twice more to separate the solvent and by-products. Before the last purification, the QD solution was separated from the precipitate formed in a small amount to remove unreacted precursors and by-products. Purified QDs were dried under vacuum at room temperature for 2 hours. The nanopowders obtained by this method easily dissolved in low-polarity media without the formation of an insoluble phase, even after long-term storage (2 months). The mass of CdTeS QDs was 3.5–4.43 g.
000 rpm (6160 rcf) for 7 min followed by dissolution in CHCl3 and reprecipitation with acetone. This purification was repeated twice more to separate the solvent and by-products. Before the last purification, the QD solution was separated from the precipitate formed in a small amount to remove unreacted precursors and by-products. Purified QDs were dried under vacuum at room temperature for 2 hours. The nanopowders obtained by this method easily dissolved in low-polarity media without the formation of an insoluble phase, even after long-term storage (2 months). The mass of CdTeS QDs was 3.5–4.43 g.
Synthesis of CdTe0.05S0.95 using different substituted thioureas as sulfur sources
The synthesis of CdTe0.05S0.95 QDs involving five more substituted thioureas was carried out similarly to the above-described method with smaller amounts of starting materials. For 0.007 mol of cadmium oxide, 0.0028 mol of LA and 30 ml of ODE were used. Substituted thioureas (TU2–6) were taken in an amount of 0.00475 mol together with 0.00025 mol Te–TOP. The nanocrystal growth temperature of 160 °C was maintained for 30 minutes for each synthesis. Isolation and purification were conducted in the same way. The mass of CdTe0.05S0.95 QDs was 0.9–1.1 g.Synthesis of core/shell CdTe0.06S0.94/CdxZn1−xS and CdTe0.12S0.88/CdxZn1−xS 2 ML QDs (x = 0.1, 0.2, and 0.3)
Core/shell CdTe0.06S0.94/CdxZn1−xS and CdTe0.12S0.8/CdxZn1−xS QDs (x = 0.1, 0.2, and 0.3) were prepared according to the general method, and the masses and amounts of substances are given below for each nanomaterial. The mixture of cadmium and zinc linoleates required for the application of 2 monolayers (MLs) of the shell was produced separately for each core. For this purpose, CdO, ZnO, LA, and ODE were mixed in a Schlenk flask (100 ml), vacuumed for 30 min at room temperature, and then heated to 100 °C with a periodic exchange of air for Ar until the mixture was homogenized.A freshly prepared solution of cores in a mixture of LA and ODE (1:3) was added to the metal linoleate solution at 100 °C. The temperature of the reaction mixture was increased to 160 °C and the TU1 solution in a mixture of oleylamine (1 ml) and ODE (total volume of 3 ml) was added in portions (0.3 ml/2 min). For individual syntheses, control aliquots were taken to evaluate the growth. After the addition of the TU1 solution was completed, the reaction mixture was stirred at the same temperature for further 12 min (total synthesis time = 30 min). The mixture was naturally cooled to room temperature and further diluted 2-fold with CHCl3. The extraction and purification of core/shell QDs were performed similarly to the purification of the cores. Calculated ratios:
CdTe0.06S0.94/CdxZn1−xS core/shell 2 MLs, x = 0.1:
- CdTe0.06S0.94 QDs (0.305 g + 1 ml LA, 2 ml ODE), CdO (0.000471 mol, 0.054 g), ZnO (0.00375 mol, 0.31 g), LA (0.0167 mol, 4.68 g), ODE (15 ml), TU1 (0.00278 mol, 0.48 g + 1 ml OAm, 1.6 ml ODE).
CdTe0.06S0.94/CdxZn1−xS core/shell 2 MLs, x = 0.2:
- CdTe0.06S0.94 QDs (0.305 g + 1 ml LA, 2 ml ODE), CdO (0.000825 mol, 0.11 g), ZnO (0.0033 mol, 0.27 g), LA (0.0165 mol, 4.63 g), ODE (15 ml), TU1 (0.00275 mol, 0.47 g + 1 ml OAm, 1.6 ml ODE).
CdTe0.06S0.94/CdxZn1−xS core/shell 2 MLs, x = 0.3:
- CdTe0.06S0.94 QDs (0.305 g + 1 ml LA, 2 ml ODE), CdO (0.00122 mol, 0.16 g), ZnO (0.00284 mol, 0.23 g), LA (0.0162 mol, 4.54 g), ODE (15 ml), TU1 (0.00270 mol, 0.465 g + 1 ml OAm, 1.6 ml ODE).
CdTe0.12S0.88/CdxZn1−xS core/shell 2 MLs, x = 0.1:
- CdTe0.12S0.88 QDs (0.269 g + 1 ml LA, 2 ml ODE), CdO (0.00034 mol, 0.04 g), ZnO (0.0031 mol, 0.25 g), LA (0.0136 mol, 3.81 g), ODE (15 ml), TU1 (0.00227 mol, 0.39 g + 1 ml OAm, 1.7 ml ODE).
CdTe0.12S0.88/CdxZn1−xS core/shell 2 MLs, x = 0.2:
- CdTe0.12S0.88 QDs (0.269 g + 1 ml LA, 2 ml ODE), CdO (0.000675 mol, 0.09 g), ZnO (0.0027 mol, 0.22 g), LA (0.0135 mol, 3.79 g), ODE (15 ml), TU1 (0.00225 mol, 0.39 g + 1 ml OAm, 1.7 ml ODE).
Surface ligand exchange of core/shell CdTe0.06S0.94/CdxZn1−xS 2 ML QDs
3-Mercaptopropionic acid (MPA, 0.2 ml) was dissolved in MeOH (15 ml) at room temperature with constant stirring. The mixture was degassed for 15 minutes with replacement by Ar, and then TMAH (25% in methanol) was added in portions of 0.4 ml until pH ≈ 10 was reached. Afterward, it was degassed for another 5 minutes, and then QDs (0.050 g) were added and stirred in Ar until complete dissolution. QDs were precipitated with ethyl acetate and separated by centrifugation (12000 rpm, 10 min). The separated QDs were dried under vacuum at room temperature. The yield was 0.030–0.035 g. The resulting nanopowders were easily soluble in water and methanol.
Results and discussion
Synthesis of the CdTeS cores and core/shell QDs and their precursors
Substituted thioureas have already proven themselves as available and reliable sources of sulfur in the synthesis of chalcogenide QDs.29 An important advantage of these sulfur sources is not only their low cost but also the possibility to synthesize nanocrystals with almost quantitative yields without losses in quality.30 The reaction rate, i.e., the rate of formation of a metal carboxylate complex with substituted thioureas followed by its decomposition to the corresponding sulfide, depends on several factors (environment, temperature, etc.). However, while keeping all other factors constant, the size and shape of QDs were affected by the nature of TU substituents.31 To study this question, the synthesis of substituted thioureas (TU1–6) from the corresponding isothiocyanates and amines was performed (Fig. 1). Some isothiocyanates are commercially available (allyl and phenyl isothiocyanates), and others have been synthesized according to previously published works.32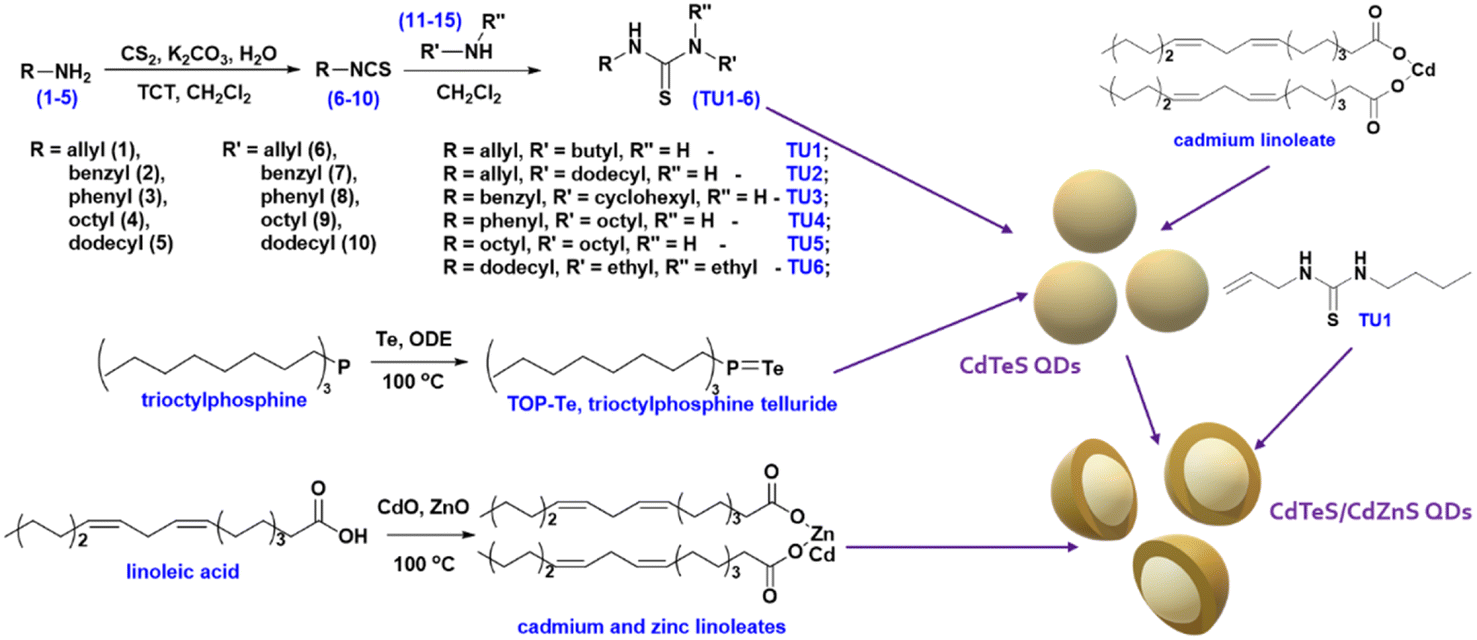 | ||
| Fig. 1 Schematic overview of the CdTeS/CdxZn1−xS core/shell 2 ML QD synthesis. | ||
Classical trioctylphosphine telluride was chosen as the source of tellurium. It was obtained by reacting trioctylphosphine (TOP) with elemental tellurium in an octadecene medium, as the base solvent, in the synthesis of CdTeS and CdTeS/CdxZn1−xS QDs. The interaction was carried out with a 1.5-fold molar excess of TOP.
After conducting preliminary studies, which are not included in this work, it was found that the optimal temperature range for the synthesis of CdTeS QDs is 140–170 °C. The formation of nanocrystals at lower temperatures occurs only partially and randomly, while increasing the synthesis temperature above 170 °C leads to the formation of large amounts of a precipitate consisting of bulk telluride sulfides. Based on the aforementioned parameters, a one-pot synthesis of CdTeS QDs involving substituted thioureas was performed at 160 °C. The selection of substituted thioureas involved in the synthesis of CdTe0.05S0.95 QDs was based on the presence of both aromatic and aliphatic substituents, as well as the ability to dissolve relatively quickly in the selected reaction media. The only one of the selected substituted thioureas TU1, which is in a liquid state in the cold, was chosen for the gram-scale synthesis of CdTeS QDs, as well as for shell formation in the synthesis of core/shell CdTe0.06S0.94/CdxZn1−xS and CdTe0.12S0.88/CdxZn1−xS 2 MLs QDs. During the synthesis of QDs, aliquots were taken for further growth analysis for the measurements of their emission and absorbance spectra. It was found that the participation of various substituted thioureas gives their unique pattern of QD growth. This is due to the rate of their dissolution in the reaction medium, the rate of formation and decomposition of complexes with cadmium linoleate, and, consequently, different levels of saturation of the solution with cadmium sulfides and tellurides. Thus, when the concentration of initial components reaches the critical level for nucleation, the first nuclei begin to form. With a further supply of “building” materials, the growth of crystals reaches a peak and falls according to the LaMer diagram.33
The calculation of the initial components required for the deposition of 2 monolayers (MLs) of the shell was carried out according to the scheme presented earlier.34 Given the known core diameter and desired shell thickness, the weight percentage of materials needed to deposit 2 MLs was calculated. The percentage of the organic phase present in the cores was determined by the decomposition of CdTeS QDs in a 10% HCl solution (ion exchange reaction) followed by extraction, and it was 52.7% for CdTe0.06S0.94 cores and 34.7% for CdTe0.12S0.88 cores. The shell formation was conducted in a 1-octadecene medium at 160 °C, with a portion-wise introduction of the TU1 solution. Further, the colloidal solution was stirred for another 12 minutes and then spontaneously cooled. Isolation, purification, and drying of core/shell CdTe0.06S0.94/CdxZn1−xS and CdTe0.12S0.88/CdxZn1−xS core/shell 2 ML QDs were carried out following the same procedures used for cores. In the perspective of using the resulting nanomaterial for medical purposes, the lipophilic ligand CdTe0.06S0.94/CdxZn1−xS core/shell 2 ML QDs was replaced by a hydrophilic one (MPA). Since thiol ligands have a strong affinity to bind to the nanocrystal surface, the surface energy undergoes some changes, meaning that the expected loss in PL QY occurs when the ligand is replaced.
Morphology and structure of the synthesized QDs
The elemental ratios of the synthesized QDs (Table 1) correspond to the nominal composition with only minor deviations. However, the exact molecular weight of the cores is necessary to calculate the precursor amount required for depositing the shell of a certain thickness. Therefore, in our calculations, we used refined empirical formulas derived from elemental analysis data (CdTe0.06S0.94 and CdTe0.12S0.88). Moreover, carbon and trace amounts of oxygen and nitrogen were found in the EDX spectra, corresponding to metal linoleates and oleylamine, which comprise the protective shell of QDs.| QDs composition | Size (XRD), nm | Size (STEM), nm | a, Å | Elemental compositiona | ||||
|---|---|---|---|---|---|---|---|---|
| Cd | Zn | Te | S | |||||
| a Elemental analysis (EDX) measurements for compositions not listed in the table were hampered by high levels of electroluminescence. | ||||||||
| CdTeS cores | CdTe0.06S0.94 | 3.21 | 3.28 | 5.85 | 58.3 | — | 2.5 | 39.2 |
| CdTe0.12S0.88 | 3.82 | 3.75 | 5.85 | 55.0 | — | 5.3 | 39.7 | |
| CdTe0.05S0.95 QDs synthesized with thioureas TU2–6 | TU2 | 3.41 | 3.78 | 5.81 | 59.6 | — | 1.9 | 38.5 |
| TU3 | 3.32 | 3.46 | 5.85 | |||||
| TU4 | 3.17 | 3.32 | 5.83 | |||||
| TU5 | 3.13 | 3.28 | 5.83 | 60.2 | — | 2.0 | 37.8 | |
| TU6 | 3.70 | 3.71 | 5.84 | |||||
| CdTe0.06S0.94/CdxZn1−xS QDs | x = 0.1 | 4.10 | 4.51 | 5.77 | 37.0 | 20.7 | 0.8 | 41.5 |
| x = 0.2 | 4.15 | 4.68 | 5.78 | |||||
| x = 0.3 | 4.09 | 4.61 | 5.79 | |||||
| CdTe0.12S0.88/CdxZn1−xS QDs | x = 0.1 | 4.80 | 5.04 | 5.79 | 39.0 | 17.6 | 1.1 | 42.3 |
| x = 0.2 | 4.88 | 5.21 | 5.80 | 42.5 | 14.4 | 1.2 | 41.9 | |
Powder X-ray diffraction (XRD) technique was used to determine the crystal structure of the synthesized QDs. The standard diffraction lines of cubic CdTe (ICSD: 620523), CdS (ICSD: 181739), and ZnS (ICSD: 53943) are drawn for comparison. Fig. 2a displays the X-ray diffraction patterns of CdTe0.05S0.95 QDs prepared using various substituted thioureas as sulfur sources.
 | ||
| Fig. 2 (a) XRD patterns of CdTe0.05S0.95 QDs synthesized at 160 °C with thioureas TU1–6. STEM and HRTEM (inset) images of CdTe0.05S0.95 QDs synthesized using substituted thioureas: (b) TU1; (c) TU2; (d) TU3; (e) TU4; (f) TU5; and (g) TU6. | ||
Growing under identical conditions (temperature and concentration of all components), CdTe0.05S0.95 QDs crystallize in the same face-centered cubic phase with the space group F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m. The first of the three diffraction signals at 2θ = 27.3° denotes the dominant (111) orientation of the crystal plane. Its width (FWHM), as well as the FWHM of the other two signals at 2θ = 44.4° and 52.5° corresponding to the (220) and (113) crystal planes, varies from the first to the last sample. These changes indicate different crystallite sizes (Table 1) calculated according to Debye–Scherrer's equation.35
3m. The first of the three diffraction signals at 2θ = 27.3° denotes the dominant (111) orientation of the crystal plane. Its width (FWHM), as well as the FWHM of the other two signals at 2θ = 44.4° and 52.5° corresponding to the (220) and (113) crystal planes, varies from the first to the last sample. These changes indicate different crystallite sizes (Table 1) calculated according to Debye–Scherrer's equation.35
The lattice parameters of core and core/shell QDs were calculated using the standard equation with Miller indices and plane distances.33 The lattice parameters (a) differ slightly between 5.81 and 5.85 for samples prepared using different substituted thioureas. The shift from a = 5.78 for pure CdS to higher values is caused by the presence of Te (for pure CdTe a = 6.48). This shift is in good agreement with Vegard's law for alloys.36 It is observed that thiourea substituents do not affect the overall crystal structure of QDs. It can be assumed that the identity of the crystal lattice of all cores indicates a similar mechanism of crystal formation (growth), and therefore, the possibility of using substituted thioureas in the synthesis of CdTeS cores, both with aliphatic and aromatic substituents.
Moreover, the particle sizes determined from the STEM images (Fig. 2b–g) are listed in Table 1 for comparison. The size data obtained by XRD differ slightly because Debye–Scherrer's equation is designed to calculate ideal spheres, while the synthesized QDs are quasi-spherical. The close size values of quantum dots calculated by both methods and the HRTEM images (Fig. 2b–g inset) together provide consistent confirmation of the single crystallinity of all samples. Fig. 3a displays the diffractograms of the CdTe0.06S0.94/CdxZn1−xS core/shell QDs. When the shell of CdxZn1−xS was deposited, a barely noticeable shift of the main signals toward higher angles was detected. As in the case of cores, CdTe0.06S0.94/CdxZn1−xS core/shell QDs formed a single phase with the cubic zinc blende structure with the space group F3m. The growth of the 2 ML shell composition CdxZn1−xS theoretically increases the total diameter of QDs by 1.256 nm (x = 0.1), 1.272 nm (x = 0.2) and 1.288 nm (x = 0.3).37 The increased size of particles can be seen in all core/shell samples. The conformity of the practical results with the theory is given in Table 1. The lattice parameters of core/shell QDs are lower than those of the core (a = 5.85), and a trend caused by different contents of ZnS (a = 5.40) in the shell was observed (a = 5.77 for x = 0.1; a = 5.78 for x = 0.2; and a = 5.79 for x = 0.3).
 | ||
| Fig. 3 XRD patterns of CdTe0.06S0.94/CdxZn1−xS core/shell QDs (x = 0.1, 0.2, 0.3) (a); STEM and HRTEM (inset) images of CdTe0.06S0.94/CdxZn1−xS core/shell QDs: x = 0.1 (b); x = 0.2 (c); x = 0.3 (d). | ||
The STEM and HRTEM images of the as-prepared CdTe0.06S0.94/CdxZn1−xS core/shell QDs (x = 0.1, 0.2, and 0.3) are provided in Fig. 3b, c and d respectively. For this measurement, a dilute solution (conc. 1 mg ml−1) of QDs in chloroform was sonicated for 10 min and deposited onto a TEM copper grid with a graphene oxide film on an electron transparent membrane. As can be seen, the core/shell QDs were quite monodisperse and had a quasi-spherical shape. Upon overcoating 2 MLs of CdxZn1−xS shell on the surface of the CdTe0.06S0.94 core (Fig. 2b) as well as CdTe0.12S0.88 core (Fig. 4b), epitaxial growth of the shell was observed.
 | ||
| Fig. 4 (a) XRD patterns of the CdTe0.12S0.88 core and CdTe0.12S0.88/CdxZn1−xS core/shell 2 ML QDs (x = 0.1 and 0.2). STEM and HRTEM (inset) images of (b) CdTe0.12S0.88 core, and CdTe0.12S0.88/CdxZn1−xS core/shell 2 ML QDs: (c) x = 0.1 and (d) x = 0.2. | ||
In the case of shell deposition (2 MLs) onto the CdTe0.12S0.88 core, a slight shift (in the order of 0.3°) of the main signals towards larger angles was also observed (Fig. 4a). The increase in the diameters of the obtained CdTe0.06S0.94/CdxZn1−xS core/shell QDs is comparable with the theoretical ones, and the error was 0.05°. The calculated lattice parameters again show the same shift to lower values in the presence of a shell rich in Zn, and the values are summarized in Table 1.
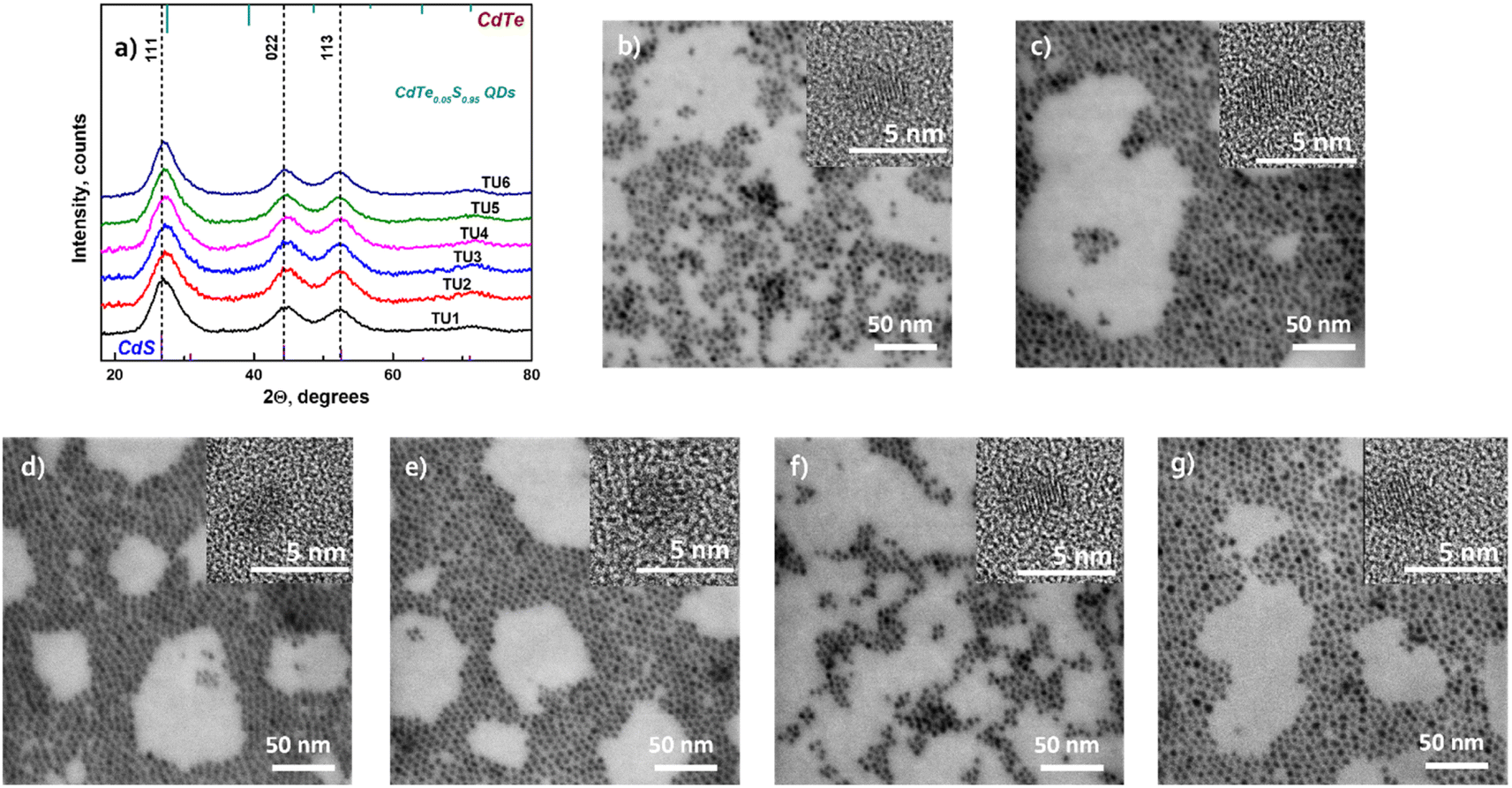
The ATR spectra of two series of CdTe0.06S0.94/CdxZn1−xS and CdTe0.12S0.88/CdxZn1−xS core–shell 2 ML QDs in the far-infrared region are provided in Fig. 5a and b.
 | ||
| Fig. 5 ATR spectra of (a) CdTe0.06S0.94/CdxZn1−xS and (b) CdTe0.12S0.88/CdxZn1−xS core–shell 2 ML QDs in the region of 50–400 cm−1. Decomposition of the ATR spectra of (c) CdTe0.06S0.94 and (d) CdTe0.12S0.88 cores and (e) CdTe0.12S0.88/Cd0.1Zn0.9S and (f) CdTe0.12S0.88/Cd0.2Zn0.8S core–shell 2 ML QDs. | ||
The ATR spectrum of QDs contains a very broad absorbance band between 50 cm−1 and 350 cm−1 with a maximum near 250 cm−1 and a shoulder near 160 cm−1. These frequencies are close to those of CdS and CdTe transverse optical (TO) phonon bands. All the data were scaled by the maximum intensity and fitted with a series of absorbance peaks. In the case of bare CdTe0.06S0.94 (Fig. 5c), the spectra were fitted with 6 bands, a strong band at 249 cm−1, medium bands at 269, 195, 157, and 109 cm−1 and a low-intensity band at 310 cm−1. The dominant band at 249 cm−1 is attributed to CdS-like TO phonon mode and the band at 269 cm−1 should be associated with CdS-like surface optical (SO) phonons. The band at 310 cm−1 should be probably assigned to normally forbidden CdS-like longitudinal (LO) modes. The presence of these three bands can confirm the previously made assumption38,39 that the nature of the confined optical vibrations in nanocrystals has a mixed SO–TO–LO character. In the area of lower wavenumbers, the main band at 157 cm−1 is attributed to the CdTe-like TO mode. The maximum of this band is slightly shifted to higher wavelengths than those of bulk CdTe crystals. The band at 195 cm−1 should be probably assigned to CdTe-like SO modes. This confirms the two-mode type of CdTeS crystal behavior.
The band at 109 cm−1 may consist of contributions of transverse and longitudinal acoustic (TA, LA) modes. These modes are typically IR-inactive in larger nanocrystals; however, they can be activated in QDs in which the crystal symmetry is reduced and thus IR selection rules are broken.38 However, it should be noted that the intensity and the shape of this band, in our case, can be influenced by the background due to the lower sensitivity of the detector and ATR crystals in the region of lower wavenumbers.
With the growing content of CdTe (core CdTe0.12S0.88), the relative intensity of CdTe-like bands increases, and the main band maxima shift towards lower wavenumbers (Fig. 5d). This is due to the subsequent incorporation of the remaining Te atoms into the anion sublattice.
The addition of CdZnS as a shell causes a rise in the intensity in the region between 270 and 360 cm−1. The examples of the fitted spectra of core/shell QDs are shown in Fig. 5e and f. The main band and lower frequency bands can be attributed to the phonon modes of the core. The two bands with higher wavenumbers have a predominant origin in the vibration of the CdZnS shell. In the case of CdTe0.12S0.88/CdxZn1−xS core/shell 2 ML QDs (Fig. 5e), the bands at 287 and 328 cm−1 can be assigned to TO and LO vibrations in the composition of the CdxZn1−xS shell. With the increase in the content of Cd atoms in the shell, the maxima of these bands are shifted to lower wavenumbers (Fig. 5f). This indicates the incorporation of Cd atoms into the lattice positions of Zn. From this, it can be deduced that the core has a two-mode behavior, while the shell behaves as a one-mode system.
Optical properties of the synthesized QDs
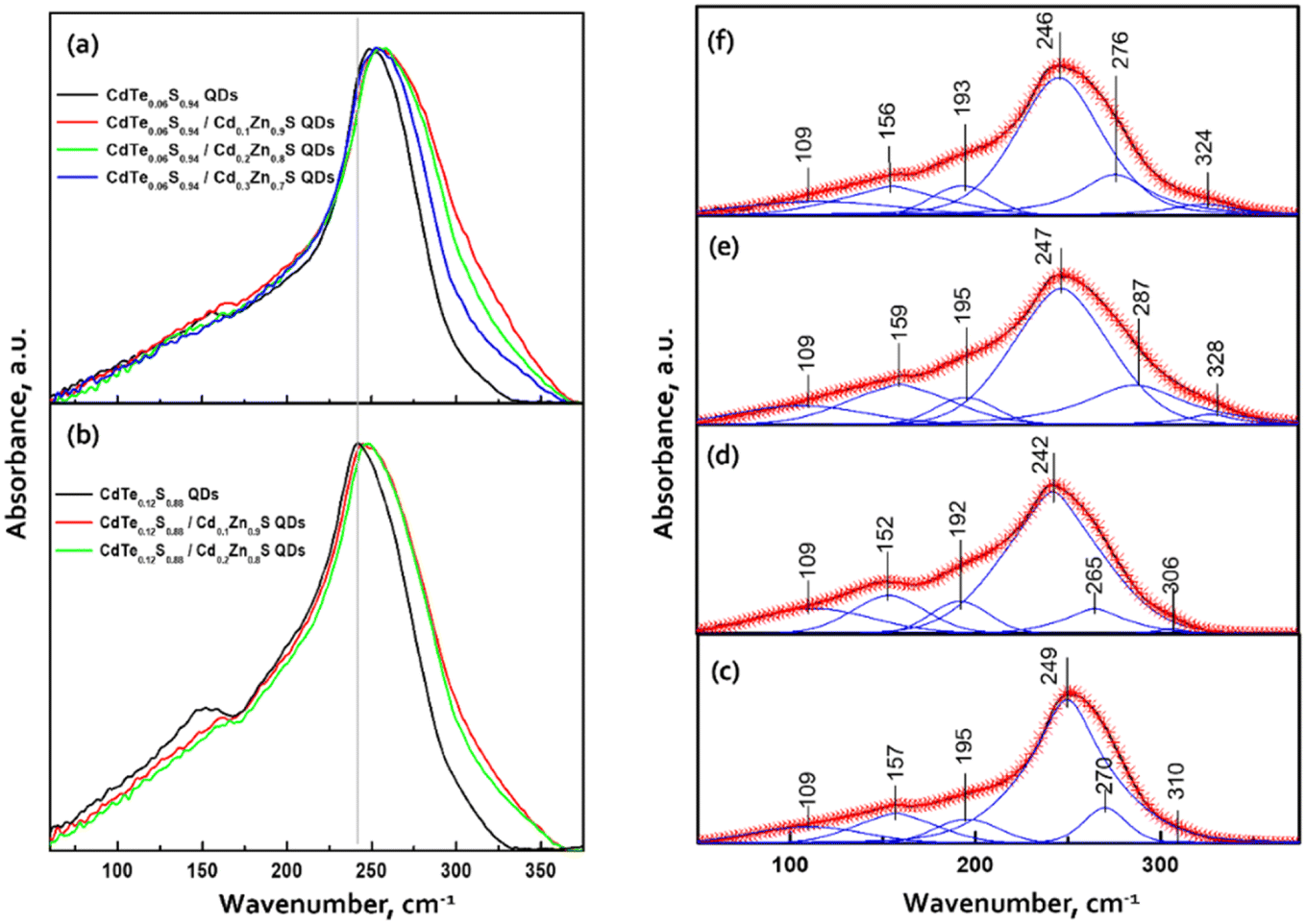
The dominant influence on the optical properties of multicomponent nanocrystals of the same composition is exerted not only by their size and shape, as in the case of binary compositions, but also by the distribution of elements from the center of the crystal to its surface. The level of heterogeneity of a nanocrystal is most influenced by the conditions of its growth; therefore, the process of nanocrystal growth is controlled. However, it is possible to consider all factors experimentally only within the framework of one composition. When replacing one or both thiourea substituents and maintaining other nanocrystal growth conditions, we observed changes primarily in the optical properties. To follow the reaction process and the growth of QDs during synthesis, aliquots were taken from the reaction mixture (see Materials and methods). Temporal evolution of the absorbance (a) and the PL emission spectra (b) of CdTe0.05S0.95 QDs, synthesized using TU4 as the sulfur precursor is presented as an example of the nanocrystal growth (Fig. 6a and b). As follows from the growth picture, the absorbance spectrum of an aliquot taken from the reaction at 120 °C does not have the QD band edge characteristic of CdTe0.05S0.95, and its PL intensity is very weak. With subsequent heating of the reaction to 140 °C, an intense exciton absorbance signal and intense emission appear in the absorbance and PL spectra, respectively. Such a sharp change in the ABS and PL signals indicates spontaneous nucleation and the beginning of the growth of CdTe0.05S0.95 QDs at this temperature. With the increase in temperature to 160 °C and until the end of synthesis, the PL intensity increases and the position of the exciton (ABS) shifts to the red region, which indicates the uniformity in the size of the growing CdTe0.05S0.95 QD. As mentioned above, the elemental composition and crystal lattice of CdTe0.05S0.95 QDs did not undergo noticeable changes when the size varied within the range of 3.28–3.78 nm (Table 1). However, intermediate complexes formed during the interaction of the substituted thiourea with cadmium linoleate have different physical and chemical properties. Consequently, the temperature and rate of decomposition of these complexes with the release of molecular CdS will change when replacing one substituted thiourea with another. Fig. 6c demonstrates the emission spectra of CdTe0.05S0.95 QDs synthesized using TU1–6 as the sulfur precursors. The divergence in the positions of the PL maxima for CdTe0.05S0.95 QDs of the same composition can be explained based on two reasons. First, there is a difference in the sizes of the prepared CdTe0.05S0.95 QDs. According to the well-known phenomenon, within the same QD composition, the position of the PL maximum shifts to the blue region as the sizes decrease; as they increase, a redshift of the PL maxima is correspondingly observed. Second, there is a difference in the rate and temperature of the formation of molecular sulfides during nanocrystal growth. This phenomenon is expressed in the ambiguous distribution of anions from the nucleus to the surface of the nanocrystal. The uneven distribution of chalcogens in the body of a nanocrystal can lead to the formation of a core/shell-like structure, where the CdTe content at the crystal surface will be reduced to zero, and the QD surface itself will be represented by a crystal lattice of the corresponding sulfide. Based on the PL spectra of CdTe0.05S0.95 QDs (Fig. 6d), it is obvious that the dominant reason for the discrepancy between their maxima is the level of heterogeneity of the nanocrystal. | ||
| Fig. 6 Temporal evolution of (a) absorbance and (b) PL emission spectra. (c) PL excitation and PL emission spectra of CdTe0.05S0.95 QDs, synthesized using TU4 as the sulfur precursor. (d) Normalized PL emission spectra, (e) representation of the values of Stokes shift in comparison to ABS (solid lines) and PL emission spectra (dashed lines) and (f) PL decay kinetic curves of CdTe0.05S0.95 QDs, synthesized using TU1–6 as the sulfur precursors. | ||
The relative photoluminescence quantum yield (PL QY) values of the isolated and purified CdTeS cores, core/shell CdTe0.06S0.94/CdxZn1−xS, and CdTe0.12S0.88/CdxZn1−xS 2 ML QDs are summarized in Table 2. The calculation of the PL QY was carried out according to the following formula:
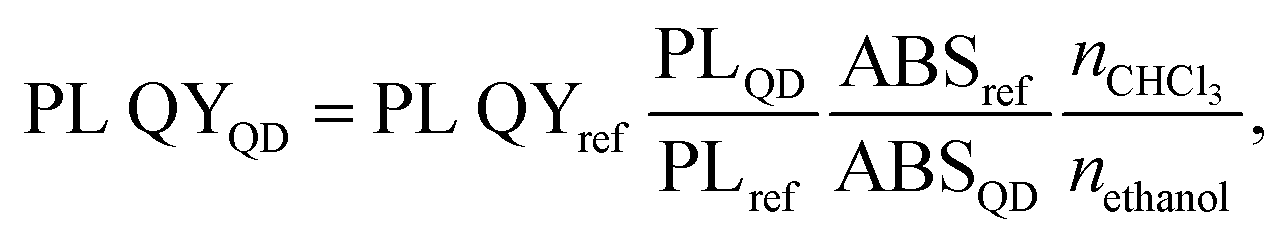 | (1) |
| QDs composition | λ ABS, nm | λ exc, nm | λ em, nm | Stokes shift, nm | PL QY, % | E g, eV | Fitting results of the PL decay curves | |||
|---|---|---|---|---|---|---|---|---|---|---|
| τ 1, ns | τ 2, ns | τ avg, ns | ||||||||
| CdTeS cores | CdTe0.06S0.94 | 395.6 | 424 | 588.0 | 192.4 | 49 | 2.94 | 40.9 | 208.7 | 134.5 |
| CdTe0.12S0.88 | 426.7 | 475 | 678.8 | 252.1 | 19 | 2.69 | 27.2 | 262.3 | 177.6 | |
| CdTe0.05S0.95 QDs synthesized using thioureas TU2–6 | TU2 | 392.9 | 408 | 605.0 | 212.1 | 58 | 2.96 | 39.6 | 214.0 | 154.4 |
| TU3 | 382.4 | 415 | 578.0 | 195.6 | 57 | 3.04 | 42.2 | 200.4 | 140.1 | |
| TU4 | 389.2 | 418 | 568.3 | 179.1 | 52 | 3.03 | 43.8 | 201.3 | 143.4 | |
| TU5 | 395.8 | 425 | 604.1 | 208.3 | 54 | 2.93 | 37.2 | 208.6 | 149.3 | |
| TU6 | 408.4 | 440 | 631.9 | 223.5 | 82 | 2.84 | 37.2 | 231.2 | 182.5 | |
| CdTe0.06S0.94/CdxZn1−xS QDs | x = 0.1 | 412.2 | 443 | 595.4 | 183.2 | 55 | 2.84 | 39.9 | 202.0 | 135.9 |
| x = 0.2 | 413.2 | 458 | 607.9 | 194.7 | 85 | 2.85 | 41.0 | 215.4 | 156.6 | |
| x = 0.3 | 414.1 | 445 | 611.8 | 197.7 | 87 | 2.84 | 36.9 | 215.7 | 156.4 | |
| CdTe0.12S0.88/CdxZn1−xS QDs | x = 0.1 | 413.7 | 440 | 694.3 | 280.6 | 49 | 2.67 | 52.6 | 229.1 | 117.4 |
| x = 0.2 | 412.8 | 435 | 694.3 | 281.5 | 56 | 2.80 | 50.1 | 231.7 | 126.1 | |
| MPA-capped CdTe0.06S0.94/CdxZn1−xS QDs | x = 0.1 | 412.2 | 443 | 638.3 | 226.1 | 42 | 2.82 | |||
| x = 0.2 | 413.2 | 458 | 640.4 | 227.2 | 36 | 2.82 | ||||
| x = 0.3 | 414.1 | 445 | 649.8 | 235.7 | 31 | 2.81 | ||||
The results also revealed that the major advantage of the synthesized CdTe0.05S0.95 QDs lies in their extra-large Stokes shift (varies from 179.1 to 223.5 nm). Due to this, the effect of re-absorbance of intrinsic PL at a high concentration of QDs is eliminated, which significantly expands the possibilities of the CdTe0.05S0.95 QD application. The extremely large values of the Stokes shift of CdTe0.05S0.95 QDs prepared with various substituted thioureas make these nanomaterials attractive for their application as inorganic scintillators. To assess the energy transfer ability, the decay kinetics of PL under pulsed radiation was measured (λexc = 391 nm, FWHM ≤ 1 ns). The PL decay kinetics curves were fitted using the two-exponential function:
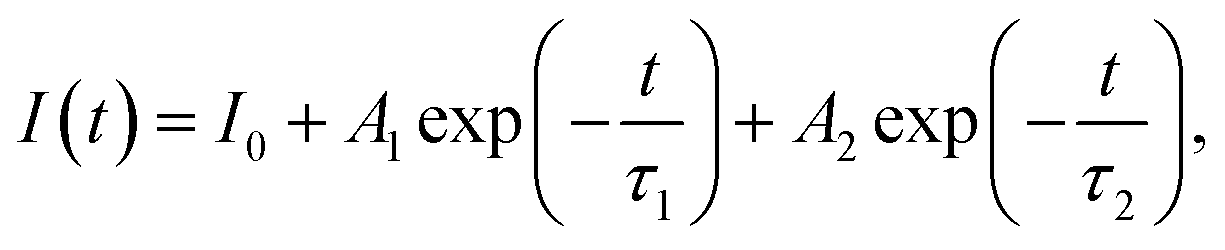 | (2) |
The average lifetime τavg was calculated as follows:
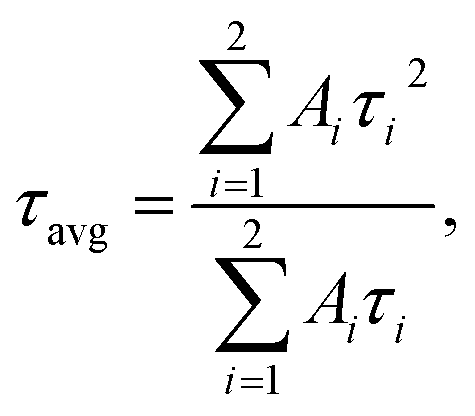 | (3) |
The fitting results are presented in Table 2. The decay kinetics curves of the synthesized CdTe0.05S0.95 QDs consist of two components: a fast one, with a decay time of τ ≈ 40 ns, and a long one, with τ ≈ 200 ns. The main contribution to PL comes from the long component, which, in our opinion, is characteristic of this material. The fast component is formed due to an increase in the non-radiative relaxation paths of excitons. Proof of this is the unquestioning correlation between the average decay time τavg and the PL QY of the synthesized CdTe0.05S0.95 QDs. With the increase in PL QY values, τavg decreases, due to the growth in the contribution of the fast component, that is, the probability of non-radiative relaxation increases. The reason for this PL quenching should be a large number of surface defects in the synthesized CdTe0.05S0.95 QDs. The surface quality of nanomaterials plays a key role in the formation of their optical properties. One of the ways to compensate for surface defects is to increment a shell to the formed core, which is a material with a lower band gap value. To obtain the most pronounced effect of luminescence characteristics, we selected CdTe0.05S0.95 QDs (TU1) with the lowest PL QY (49%) as the core. Fig. 7 illustrates the temporal evolution of the PL (b) and ABS (c) of CdTe0.06S0.94/CdxZn1−xS (x = 0.1, 0.2, and 0.3) core/shell 2 ML QDs.
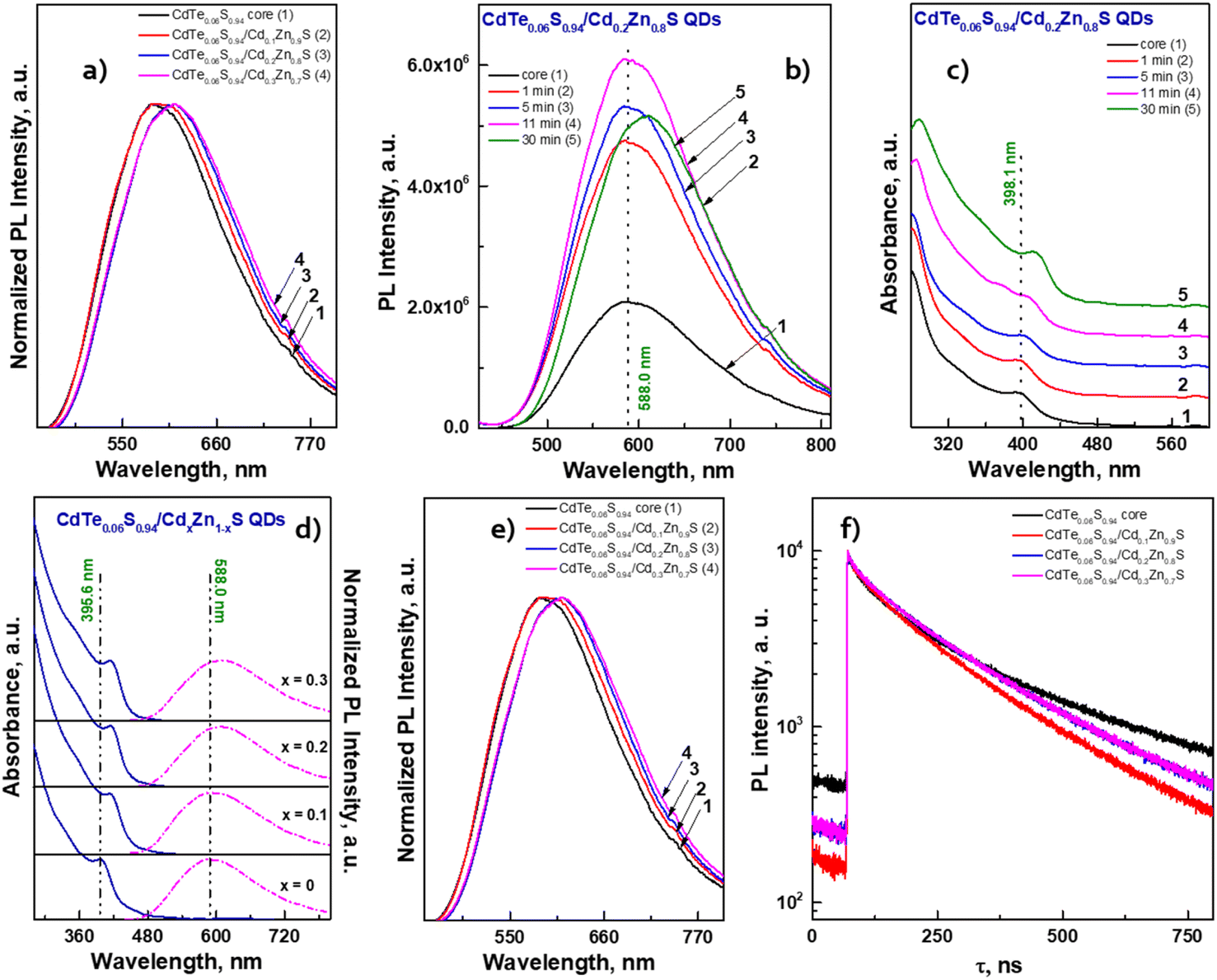 | ||
| Fig. 7 (a) Normalized PL emission spectra of CdTeS QDs prepared at different ratios of chalcogen precursors. Temporal evolution of (b) PL emission spectra and (c) absorbance of core/shell CdTe0.06S0.94/Cd0.2Zn0.8S QDs. (d) Comparison of ABS (solid lines) and PL emission (dashed lines) spectra, (e) normalized PL emission spectra and (f) PL decay kinetic curves of core/shell CdTe0.06S0.94/CdxZn1−xS QDs. | ||
The temporal evolution of the PL spectra (Fig. 7b) demonstrates that after the first minute of shell growth, the PL intensity increased 2 times and continued to increase until the 11th minute. A small drop in intensity at the end of the reaction is probably due to an increase in the concentration of QDs in the reaction mixture, which leads to concentration-induced quenching of luminescence. Moreover, from the absorbance spectra (Fig. 7c), one can see a shift in the exciton absorbance maximum towards longer wavelengths, which indicates an increase in the size of CdTe0.06S0.94/Cd0.2Zn0.8S core/shell QDs. As expected, the largest changes occurred in PL QY values (Table 2). When the increment of a shell with a minimum Cd content, the PL QY value increased by several percent; however, the PL QY value of compositions with a Cd0.3Zn0.7S shell exceeded our expectations and reached the maximum value described in the literature for these compositions (PL QY 87%).
The fitting results of PL decay for CdTe0.06S0.94/CdxZn1−xS (x = 0.1, 0.2, and 0.3) core/shell 2 ML QDs are presented in Table 2. The decay kinetics curves of the synthesized core/shell QDs consist of two components: a fast one, with a decay time of τ = 36.9–41.0 ns, and a long one, with τ = 202.0–215.7 ns. Fast components associated with the recombination of excitons in the cores, as follows, retain their importance during shell growth. However, they contribute more to the average component, and τavg increases with the increasing in the Cd content of the CdxZn1−xS shell material, indicating a decrease in the surface/volume ratio, and also due to the formation of a CdS-rich layer. Such effect has already been reported for CdTe/CdS core/shell nanocrystals.42 With the increase in the tellurium content in the cores (CdTe0.12S0.88), the growth pattern changes dramatically (Fig. 8a).
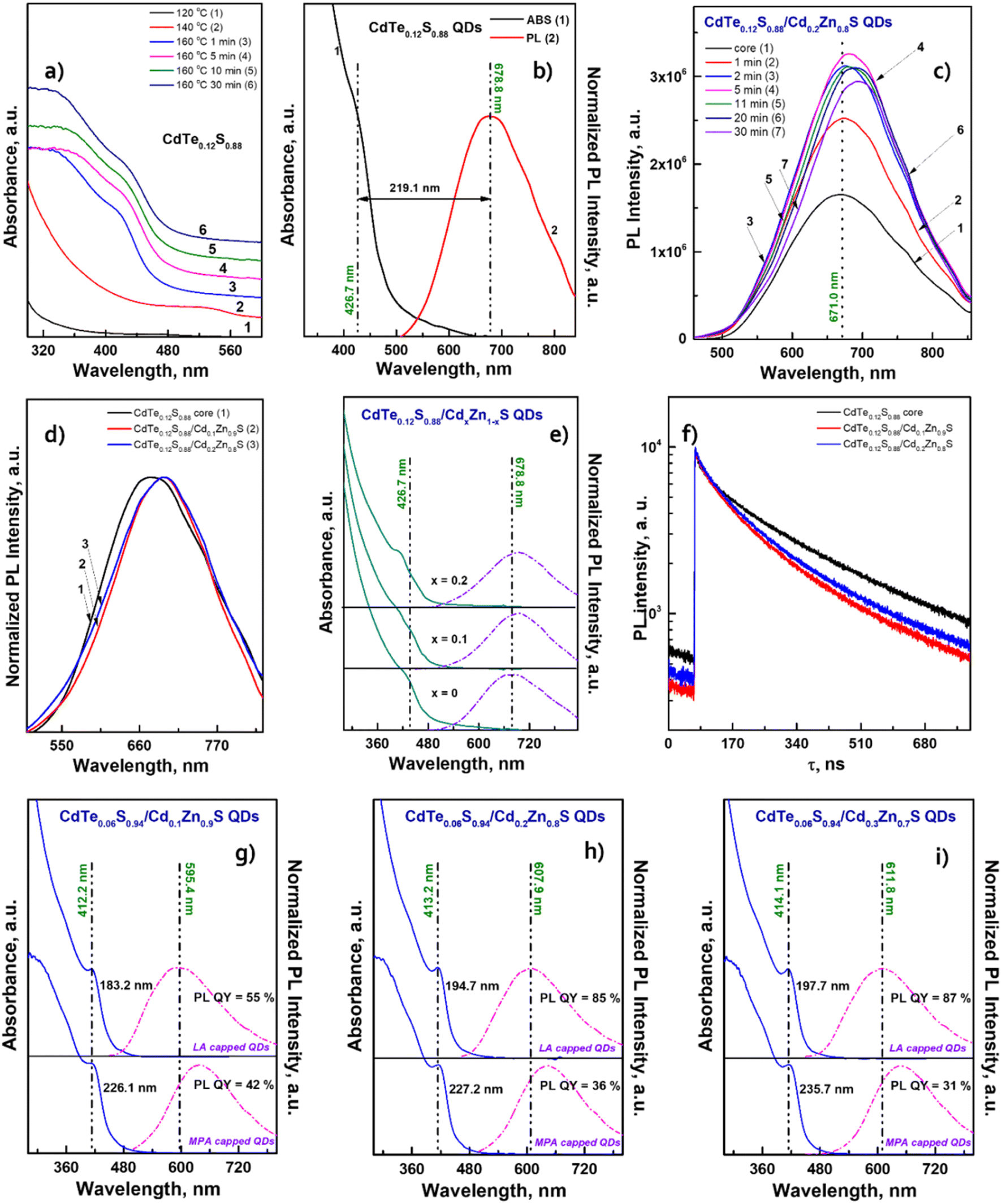 | ||
| Fig. 8 (a) Comparison of ABS (black line) and PL emission spectra (red line) of the core CdTe0.12S0.88. Temporal evolution of (b) PL emission and (c) absorbance spectra of core/shell CdTe0.12S0.88/Cd0.2Zn0.8S 2 MLs QDs. (d) Normalized PL emission spectra, (e) comparison of ABS (solid lines) and PL emission (dashed lines) spectra and (f) PL decay kinetic curves of CdTe0.12S0.88/CdxZn1−xS 2 MLs QDs. Comparison of ABS (solid lines) and PL emission (dashed lines) spectra of CdTe0.06S0.94/CdxZn1−xS LA and MPA capped QDs, where x = 0.1 (g), 0.2 (h) and 0.3 (i). | ||
The exciton band formed even at 140 °C by the first minute of 160 °C gives a significant blue shift of more than 100 nm. It is obvious that in this temperature range, the rate of interaction between cadmium linoleate and Te–TOP, with the formation of molecular CdTe, is significantly higher than the rate of formation of molecular cadmium sulfide. Therefore, the structure of CdTe0.12S0.88 QDs is in a sense a core/shell structure without clear edges between them. It is likely that in the synthesis of CdTe0.06S0.94 cores, this difference in rates can be compensated due to the higher content of the substituted thiourea in the reaction mixture. Thus, the higher level of heterogeneity in CdTe0.12S0.88 QDs leads to an increase in the value of the Stokes shift (Fig. 8b) relative to the composition with a lower tellurium content. The process of shell growth was also studied by measuring the PL spectra (Fig. 8c) of aliquots taken during the synthesis. A change in the position of the PL maxima towards longer wavelengths was observed, as well as, for the core/shell CdTe0.06S0.94/CdxZn1−xS 2 ML QDs, the phenomenon of internal luminescence reabsorbance. Increasing the size of the core/shell QDs resulted in the expected redshift of the PL maxima (Fig. 8d). With a rise in the tellurium content in the cores, the Stokes shift increases significantly, which grows even more during the increment of the CdxZn1−xS shell (Fig. 8e). Such changes indicate partial ion exchange occurring on the surface of the core in the presence of zinc linoleate. Thus, a certain “intermediate layer” is formed, containing all 4 elements, which, in turn, contributes to the exciton energy. The fitting results of PL decay for CdTe0.12S0.88/CdxZn1−xS (x = 0.1 and 0.2) core/shell 2 ML QDs are presented in Table 2. The decay kinetics curves of the synthesized core/shell QDs consist also of two components: a fast one, with a decay time of τ = 50.1–51.6 ns, and a long one, with τ = 229.1–231.7 ns. A correlation is also evident between τavg and PL QY (Table 2), which increased from 19% for cores to 49 and 56% for CdTe0.12S0.88/CdxZn1−xS (x = 0.1 and 0.2) 2 MLs QDs, respectively. This phenomenon is due to the healing of surface defects and a decrease in the probability of non-radiative relaxation.
As the possible application of the prepared nanomaterials, the process of transferring QDs with a hydrophobic surface into aqueous and alcoholic media was investigated. For this purpose, the metal linoleate ligand was replaced with mercaptopropionic acid (see Methods), and its ammonium salt promotes the free dispersion of QDs. This process was carried out on a composition of CdTe0.06S0.94/CdxZn1−xS QDs.
For each of the water-soluble CdTe0.06S0.94/CdxZn1−xS core/shell 2 ML QDs, their optical characteristics were studied. From the comparison of the spectra of ABS and PL (Fig. 8g–i), it was observed that the position of the first exciton for all compositions remains unchanged. However, we observed a redshift of the PL maxima in the order of 30–40 nm. It is well known that each ligand contributes to the overall emission of the nanomaterial, and therefore, it should be noted that the expected drop in PL QY is due to the replacement of the ligand. A selection of the most suitable hydrophilic ligand for these nanomaterials that does not reduce the PL QY values is under development.
Conclusions
Our study is devoted to the development of new routes for the synthesis of highly photoluminescent CdTe0.05S0.95 QDs. The synthesis was carried out by applying an effective combination of two sources of chalcogens, one of which (substituted thioureas) was used for the first time for these compositions. The effect of the thiourea substituent's nature on the structural and optical properties of CdTe0.05S0.95 QDs was demonstrated. The investigation of the structural features of CdTeS cores confirmed the cubic crystal structure and quasi-spherical shape, which remained unchanged when the shell was incremented. The preparation of CdTe0.06S0.94/CdxZn1−xS and CdTe0.12S0.88/CdxZn1−xS core/shell 2 ML QDs was carried out based on our elaborated technique for the shell increment onto already formed cores. The testing was successful and did not cause any technological difficulties. Using EDX and FT-IR spectroscopy methods, the fact of the increment of the shell of the CdxZn1−xS composition was confirmed. According to STEM and HRTEM images, the shell growth occurred epitaxially to the core. The synthesized nanomaterials have promising optical characteristics (PL QY up to 87% and Stokes shift up to 280 nm), which determines their further study, with the prospect of their introduction into industrial production.Author contributions
LL: conceptualization, methodology, data curation, formal analysis, investigation, validation, visualization, writing, and editing. JH: formal analysis, X-ray measurement, validation, visualization, writing, and editing. SS: formal analysis, STEM measurements, review. BF: formal analysis, investigation, FT-IR measurements, review. MC: HRTEM measurements, formal analysis, review. MV: conceptualization, funding acquisition, project administration, resources, supervision, review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors appreciate the financial support from project “High-sensitive and low-density materials based on polymeric nanocomposites” – NANOMAT (No. CZ.02.1.01/0.0/0.0/17_048/0007376), support from the grant LM2023037 from the Ministry of Education, Youth and Sports of the Czech Republic.References
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435, DOI:10.1038/nmat1390.
- L. Pagano, F. Pasquali, S. Majumdar, R. De La Torre-Roche, N. Zuverza-Mena, M. Villani, A. Zappettini, R. E. Marra, S. M. Isch, M. Marmiroli, E. Maestri, O. P. Dhankher, J. C. White and N. Marmiroli, Environ. Sci.: Nano, 2017, 4, 1579, 10.1039/c7en00219j.
- Q. Sun, Y. A. Wang, L. S. Li, D. Wang, T. Zhu, J. Xu, C. Yang and Y. Li, Nat. Photonics, 2007, 1, 717, DOI:10.1038/nphoton.2007.226.
- X. Jin, H. Li, S. Huang, X. Gu, H. Shen, D. Li, X. Zhang, Q. Zhang, F. Li and Q. Li, J. Colloid Interface Sci., 2018, 510, 376, DOI:10.1016/j.jcis.2017.09.080.
- P. Eskandari, F. Kazemi and Z. Zand, J. Photochem. Photobiol., A, 2014, 274, 7, DOI:10.1016/j.jphotochem.2013.09.011.
- H. Zhang, Y. Zhang, Q. He, L. Liu, G. Ding and Z. Jiao, CrystEngComm, 2011, 13, 6650, 10.1039/c1ce05548h.
- C. Liu, Z. Li, T. J. Hajagos, D. Kishpaugh, D. Y. Chen and Q. Pei, ACS Nano, 2017, 11, 6422, DOI:10.1021/acsnano.7b02923.
- I. H. Campbell and B. K. Crone, Adv. Mater., 2006, 18, 77, DOI:10.1002/adma.200501434.
- I. Lokteva, N. Radychev, F. Witt, H. Borchert, J. Parisi and J. Kolny-Olesiak, J. Phys. Chem. C, 2010, 114, 12784, DOI:10.1021/jp103300v.
- X. Liu, L. Wang, Y. Gao, Y. Zeng, F. Liu, H. Shen, L. Manna and H. Li, Nano Lett., 2023, 23, 6689, DOI:10.1021/acs.nanolett.3c01919.
- S. J. Byrne, S. A. Corr, T. Y. Rakovich, Y. K. Gun'ko, Y. P. Rakovich, J. F. Donegan, S. Mitchell and Y. Volkov, J. Mater. Chem., 2006, 16, 2896, 10.1039/B605333E.
- Q. Wang, Y. Kuo, Y. Wang, G. Shin, C. Ruengruglikit and Q. Huang, J. Phys. Chem. B, 2006, 110, 34–16860, DOI:10.1021/jp062279x.
- H. Zhang, L. Wang, H. Xiong, L. Hu, B. Yang and W. Li, Adv. Mater., 2003, 15, 20–1712, DOI:10.1002/adma.200305653.
- Y. Luo, T. Tan, S. Wang, R. Pang, L. Jiang, D. Li, J. Feng, H. Zhang, S. Zhang and C. Li, ACS Appl. Nano Mater., 2022, 5(1), 401, DOI:10.1021/acsanm.1c03246.
- L. Li, H. Qian, N. Fang and J. Ren, J. Lumin., 2006, 116(1–2), 59, DOI:10.1016/j.jlumin.2005.03.001.
- M. Yang, Y. Yan, H. Shi, C. Wang, E. Liu, X. Hu and J. Fan, J. Alloys Compd., 2019, 781, 1021, DOI:10.1016/j.jallcom.2018.12.156.
- W. Mao, J. Guo, W. Yang, C. Wang, J. He and J. Chen, Nanotechnology, 2007, 18(48), 485611, DOI:10.1088/0957-4484/18/48/485611.
- B. Xue, D. Deng, J. Cao, F. Liu, X. Li, W. Akers, S. Achilefu and Y. Q. Gu, Dalton Trans., 2012, 41, 4935, 10.1039/C2DT12436J.
- N. P. Gurusinghe, N. N. Hewa-Kasakarage and M. Zamkov, J. Phys. Chem. C, 2008, 112, 33–12795, DOI:10.1021/jp804045p.
- A. Badawi, K. Easawi, N. Al-Hosiny and S. Abdallah, Mater. Sci. Appl., 2014, 5, 1–27, DOI:10.4236/msa.2014.51004.
- W. Shiquan, L. Chunliang and M. Norio, Adv. Sci., Eng. Med., 2013, 5, 1–23, DOI:10.1166/asem.2013.1229.
- A. Hassinen, I. Moreels, K. De Nolf, P. F. Smet, J. C. Martins and Z. Hens, J. Am. Chem. Soc., 2012, 134(51), 20705, DOI:10.1021/ja308861d.
- N. C. Anderson, M. P. Hendricks, J. J. Choi and J. S. Owen, J. Am. Chem. Soc., 2013, 135, 49–18536, DOI:10.1021/ja4086758.
- G. Mao, Q. Cai, F. Wang, C. Luo, X. Ji and Z. He, Anal. Chem., 2017, 89(21), 11628, DOI:10.1021/acs.analchem.7b03053.
- Y. F. Liu and J. S. Yu, J. Colloid Interface Sci., 2009, 333(2), 690, DOI:10.1016/j.jcis.2009.01.008.
- R. García-Rodríguez, M. P. Hendricks, B. M. Cossairt, H. Liu and J. S. Owen, Conversion Reactions of Cadmium Chalcogenide Nanocrystal Precursors, Chem. Mater., 2013, 25, 1233, DOI:10.1021/cm3035642.
- J. C. Bruce, N. Revaprasadu and K. R. Koch, Cadmium(II) complexes of N,N-diethyl-N’-benzoylthio(seleno)urea as single-source precursors for the preparation of CdS and CdSe nanoparticles, New J. Chem., 2007, 31, 1647, 10.1039/B618254B.
- M. Chylii, L. Loghina, A. Kaderavkova, J. Houdek and M. Vlcek, The Thermal Mode Crucial Influence on the ZnSeS QDs Formation, 2022 IEEE 12th International Conference Nanomaterials: Applications & Properties (NAP), Krakow, Poland, 2022, pp. NSS04-1–NSS04-6, DOI:10.1109/NAP55339.2022.9934209.
- L. Loghina, M. Grinco, A. Iakovleva, S. Slang, K. Palka and M. Vlcek, New J. Chem., 2018, 42, 14779, 10.1039/C8NJ03077D.
- M. P. Hendricks, M. P. Campos, G. T. Cleveland, I. J. Plante and J. S. Owen, A tunable library of substituted thiourea precursors to metal sulfide nanocrystals, Science, 2015, 348(6240), 1226, DOI:10.1126/science.aaa295.
- A. Iakovleva, L. Loghina, Z. Olmrova Zmrhalova, J. Mistrik, P. Svec, S. Slang, K. Palka and M. Vlcek, J. Alloys Compd., 2020, 812, 152159, DOI:10.1016/j.jallcom.2019.152159.
- M. Chylii, L. Loghina, A. Kaderavkova, S. Slang, P. Svec, J. Rodriguez Pereira, B. Frumarova, D. Cizkova, A. Bezrouk and M. Vlcek, Mater. Chem. Phys., 2022, 284, 126060, DOI:10.1016/j.matchemphys.2022.126060.
- T. Sugimoto, J. Colloid Interface Sci., 2007, 309(1), 106, DOI:10.1016/j.jcis.2007.01.036.
- L. Loghina, M. Chylii, A. Kaderavkova, S. Slang, P. Svec, J. Rodriguez Pereira, B. Frumarova, M. Cieslar and M. Vlcek, Nanomaterials, 2021, 11, 2616, DOI:10.3390/nano11102616.
- J. I. Langford and J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102, DOI:10.1107/s0021889878012844.
- L. Vegard, Z. Phys., 1921, 5, 17, DOI:10.1007/BF01349680.
- A. Aharoni, T. Mokari, I. Popov and U. Banin, J. Am. Chem. Soc., 2006, 128(1), 257, DOI:10.1021/ja056326v.
- V. M. Dzhagan, M. Y. Valakh, C. Himcinschi, A. G. Milekhin, D. Solonenko, N. A. Yeryukov, O. E. Raevskaya, O. L. Stroyuk and D. R. T. Zahn, J. Phys. Chem. C, 2014, 118, 33–19492, DOI:10.1021/jp506307q.
- V. M. Dzhagan, Y. M. Azhniuk, A. G. Milekhin and D. R. T. Zahn, J. Phys. D: Appl. Phys., 2018, 51, 503001, DOI:10.1088/1361-6463/aada5c.
- G. A. Reynolds and K. H. Drexhage, Opt. Commun., 1975, 13(3), 222, DOI:10.1016/0030-4018(75)90085-1.
- Y. Shen, R. Tan, M. Y. Gee and A. B. Greytak, ACS Nano, 2015, 9, 3345, DOI:10.1021/acsnano.5b00671.
- Q. Zeng, X. Kong, Y. Sun, Y. Zhang, L. Tu, J. Zhao and H. Zhang, J. Phys. Chem. C, 2008, 112, 23–8587, DOI:10.1021/jp711395f.
| This journal is © The Royal Society of Chemistry 2024 |