
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt phthalocyanine (CoPc) anchored on Ti3C2 MXene nanosheets for highly efficient selective catalytic oxidation†
Simeng
Zhu
a,
Peng
Liu
b and
Xinlin
Hong
 *a
*a
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, China. E-mail: hongxl@whu.edu.cn
bHubei Province Engineering and Technology Research Center for Fluorinated Pharmaceuticals, School of Pharmaceutical Sciences, Wuhan University, Wuhan, China
First published on 26th April 2024
Abstract
Quinclorac is an important precursor for pharmaceutical, agricultural, and synthetic chemistry. The state-of-the-art synthesis of quinclorac via condensation, chlorination and oxidative hydrolysis often uses homogeneous catalysts and strong acid oxidant agents to promote the catalytic oxidation, which requires huge manpower input for the late-stage purification process and is usually environmentally unfriendly. In this work, we successfully fabricated a stable cobalt phthalocyanine (CoPc) Co-based composite (CoPc/Ti3C2) by anchoring CoPc on the surface of Ti3C2 nanosheets for the selective oxidation of 3,7-dichloro-8-dichloro methyl quinoline (3,7-D-8-DMQ) into quinclorac. More impressively, CoPc/Ti3C2-4.5%–Mn–Br exhibits a high selectivity of 91.8% for the catalytic oxidation of 3,7-D-8-DMQ to quinclorac in acetic acid, with a quinclorac yield of 87.5%, which is approximately 2.46 times higher than that of pristine CoPc–Mn–Br. The obtained heterogeneous catalytic system shows good reusability. Detailed mechanistic investigations reveal that the system works through the free radical mechanism via the formation of Co2+/Co3+ redox cycles. This work provides a new understanding for the stabilization of reaction intermediates and facilitates the design of catalysts for selective catalytic oxidation.
1. Introduction
Selective oxidation is one of the most important transformations in the chemical industry and widely used for producing aldehydes, acid, and several other chemicals.1,2 Compared to strong acid oxidants, oxygen is an inexpensive and green oxidant.3,4 However, directly utilizing clean molecular oxygen as an oxidant is challenging because its ground state is a triplet. According to the Wigner spin selection rules, the direct reaction between the substrate and molecular oxygen is usually spin-forbidden.5 On this basis, activation of ground-state molecular oxygen is a fundamental step in catalytic oxidation processes.6–8 In this regard, it is imperative to develop a novel catalytic strategy to coordinate molecular oxygen and substrates in selective oxidation.The selective oxidation synthesis of quinclorac is an essential laboratory and commercial procedure.9 Industrially, quinclorac is synthesized by the condensation of aniline and glycerol, chlorination and oxidative hydrolysis, processes that require large amounts of strong corrosive or toxic reagents, often in the homogeneous phase, which later require complex separation methods.10–12 To tackle these problems, further exploring the combination of molecular complex catalysts with O2 under green and eco-friendly conditions is one of the key development directions not only for selective oxidation reactions in organic synthesis, but also for the catalytic oxidation synthesis of quinclorac.13,14
Along with the intensive research efforts in developing catalysts with high performance, low cost and environmental friendliness, cobalt-based complex catalysts represented by cobalt phthalocyanine (CoPc) have emerged as a new alternative class of materials.15,16 Transition-metal complexes, for example, CoPc, with well-defined active sites and adjustable structures have been widely used for various catalytic reactions.17–19 However, the major challenge of utilizing homogeneous CoPc for catalysis application lies in the unstable properties, such as uncontrollable aggregation, oxidative self-destruction in the oxidizing solvent, and undesirable reduction of the Pc ligand in the reductive media, which resulted in poor long-term stability.20–22 In addition, homogeneous catalysts suffer from separation and recycling problems. Thus, the heterogenization of CoPc provides a solution to maximize the stability without sacrificing the intrinsic activity and selectivity of homogeneous molecular catalysts.
Two-dimensional (2D) titanium carbide (Ti3C2) nanosheets, as a typical 2D MXene nanomaterial, contain abundant active sites, large surface area and excellent properties. Hence, Ti3C2 MXene could be applied as the solid support for CoPc, increasing its uniform distribution and anchoring impact in Ti3C2, as well as its catalytic capability.23–25 The attempt to produce quinclorac with molecularly dispersed metal phthalocyanine catalysts in the presence of molecular oxygen has never been reported, to the best of our knowledge.26–29
In this work, we report the realization of quinclorac synthesis enabled by anchoring CoPc onto Ti3C2 MXene nanosheets for the design of a new kind of CoPc/Ti3C2–Mn(OAc)2–HBr (CoPc/Ti3C2-4.5%–Mn–Br) system with oxygen as the oxidant. This enables CoPc/Ti3C2-4.5%-Mn–Br to exhibit a high catalytic activity for the production of high value-added quinclorac with a yield of 87.5%, which is even 2.46-fold higher than that of the CoPc–Mn–Br system. By investigating the chemical state of the reaction active species, the favorable effect of Co2+ and Co3+ species leading to the excellent performance is revealed. A catalytic mechanism involving surmised radical generation and the oxidation intermediate species can be proposed based on experimental and theoretic analysis. This strategy provides the exact answer for selective catalytic oxidation and opens a new horizon for efficient organic synthesis conversion owing to its extremely high activity and cost-effective nature.
2. Experimental section
2.1 Synthesis of Ti3C2 MXene nanosheets
Ti3AlC2 powder (1.0 g) was added slowly into 20 mL of 40% HF. The mixture was stirred and heated at 80 °C overnight for selectively exfoliating the Al layers from Ti3AlC2. The as-synthesized product was washed with distilled water by centrifugation until the supernatant reached a pH value of approximately 7. To delaminate the etched MAX powder, the precipitate was stirred vigorously in 20 mL of tetramethylammonium hydroxide (TMAOH) for 24 hours. And the suspension was washed with distilled water several times to completely eliminate the excess TMAOH. The remaining precipitate was further dispersed in fresh distilled water. Then, the solution was ultrasonically treated for 6 h in an ice bath. The resulting material was then treated for the defluorinative functionalization at 300 °C for 4 h under H2 flow. Finally, the resultant products were washed with distilled water followed by vacuum-drying at 60 °C overnight.2.2 Synthesis of CoPc/Ti3C2
Preparation of composite catalyst CoPc/Ti3C2-X using the self-assembly method. CoPc (0.2 g) was dispersed in 10.0 mL of H2O, and then a certain amount of Ti3C2 (containing 3 mg, 6 mg, 9 mg, 12 mg and 15 mg of Ti3C2, respectively) was added and stirred for 1 h. The final product was centrifuged, washed and vacuum-dried at 60 °C overnight. A series of products were prepared and labeled as CoPc/Ti3C2-X, X meant the mass ratio of Ti3C2 to CoPc, which was 1.5%, 3%, 4.5%, 6% and 7.5%, respectively.2.3 Catalytic activity test and characterization
The catalytic experiment and characterization are shown in the ESI.†3. Results and discussion
3.1 Synthesis and characterization of catalysts
As shown in Fig. 1, the CoPc anchored Ti3C2 MXene could be fabricated by a facile self-assembly strategy. Scanning electron microscopy (SEM) measurements of the as-prepared CoPc/Ti3C2 hybrid show that the composite comprises comparatively uniform 2D nanosheets (Fig. 2a and b). High-resolution transmission electron microscopy (HRTEM) images of the composite confirm the high crystallinity of Ti3C2 nanosheets and the amorphous morphology of CoPc molecules (Fig. 2c). As seen in Fig. 2d, the estimated lattice spacing of 0.260 nm can be attributed to the (002) facet of Ti3C2. The presence of rough spots in the HRTEM image of hybrid indicates the anchored CoPc units on Ti3C2, and no remarkable lattice spaces are observed for CoPc, which is attributed to the distribution of amorphous CoPc on the surface of Ti3C2 nanosheets. High angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and corresponding energy dispersive spectroscopy (EDS) elemental analysis have been utilized to observe the distribution of various species in the catalysts. Interestingly, a nanosheet-like composite material is formed according to EDS mapping, which exhibits a relatively homogeneous distribution of Ti, C, Co and N (Fig. 2e–i).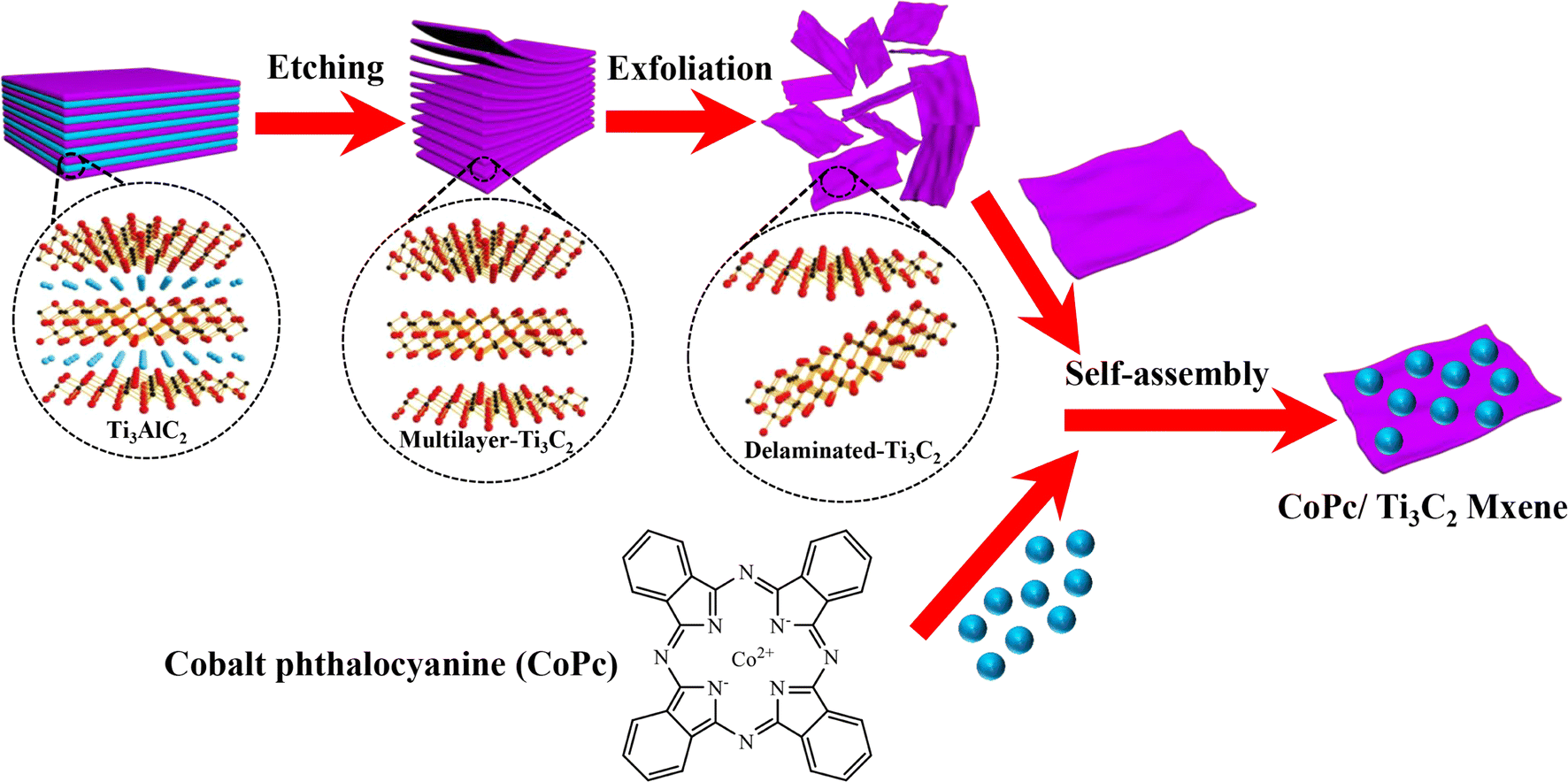 | ||
| Fig. 1 Schematically synthetic procedure of the CoPc/Ti3C2 hybrid. | ||
 | ||
| Fig. 2 SEM images (a and b) and high-resolution TEM (HRTEM) images (c and d) of CoPc/Ti3C2. (e) High-angle annular dark-field (HAADF) image of CoPc/Ti3C2. (f–i) The corresponding EDS elemental mapping images of Ti, C, Co and N elements. | ||
Based on the XRD pattern in Fig. 3a, pristine Ti3C2 nanosheets exhibit three peaks at 8.8°, 27.9°, and 36.5°, corresponding to the (002), (004) and (101) facets of Ti3C2. Moreover, the presence of Ti3C2 and CoPc diffraction signal peaks in the CoPc/Ti3C2 hybrid can be observed, inferring that the combination of CoPc molecules and Ti3C2 does not alter their respective structures. Furthermore, X-ray photoelectron spectroscopy (XPS) was conducted to explore the elementary composition and electronic structure of these catalysts. According to the XPS survey spectra in Fig. 3b, the four elements of Ti, C, Co and N can be evidently observed with the binding energies of Ti 2p, C 1s, Co 2p and N 1s electrons, revealing that the CoPc/Ti3C2 hybrid consists of CoPc and Ti3C2, which is in accordance with the XRD and EDS results. Besides, as for Ti, the peaks centered at 461.6 and 462.5 eV can be assigned to the 2p3/2 and 2p1/2 of Ti, respectively (Fig. 3c). The C 1s spectrum (Fig. 3d) contains three components located at 284.5 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 285.8 eV (C–C) and 288.3 eV (C–Ti), while for Co, two characteristic peaks centered at 782.1 and 797.8 eV of Co 2p3/2 and Co 2p1/2 are observed in Fig. 3e, respectively, suggesting that the valence state of Co is +2. In Fig. 3f, the N 1s peaks centered at 400.5 and 402.1 eV are assigned to the Co–N and aza N in the Pc moieties, respectively. It is interesting to note that the peaks of Ti 2p and C 1s for CoPc/Ti3C2 shift to the higher energy direction compared with pristine Ti3C2, while the binding energies of Co 2p and N 1s peaks of the composite are lower than that of pristine CoPc. Note that the electron binding energy of elements originates from the strong coulombic attraction between the outer electrons of the atom and atomic nucleus. Meanwhile, the changes of element binding energy can directly reflect the changes of electron density. Therefore, these XPS results provide important evidence that CoPc molecules can be anchored on Ti3C2 nanosheets via strong π–π interactions, matching well with the fact that CoPc molecules are uniformly dispersed on Ti3C2 nanosheets.
C), 285.8 eV (C–C) and 288.3 eV (C–Ti), while for Co, two characteristic peaks centered at 782.1 and 797.8 eV of Co 2p3/2 and Co 2p1/2 are observed in Fig. 3e, respectively, suggesting that the valence state of Co is +2. In Fig. 3f, the N 1s peaks centered at 400.5 and 402.1 eV are assigned to the Co–N and aza N in the Pc moieties, respectively. It is interesting to note that the peaks of Ti 2p and C 1s for CoPc/Ti3C2 shift to the higher energy direction compared with pristine Ti3C2, while the binding energies of Co 2p and N 1s peaks of the composite are lower than that of pristine CoPc. Note that the electron binding energy of elements originates from the strong coulombic attraction between the outer electrons of the atom and atomic nucleus. Meanwhile, the changes of element binding energy can directly reflect the changes of electron density. Therefore, these XPS results provide important evidence that CoPc molecules can be anchored on Ti3C2 nanosheets via strong π–π interactions, matching well with the fact that CoPc molecules are uniformly dispersed on Ti3C2 nanosheets.
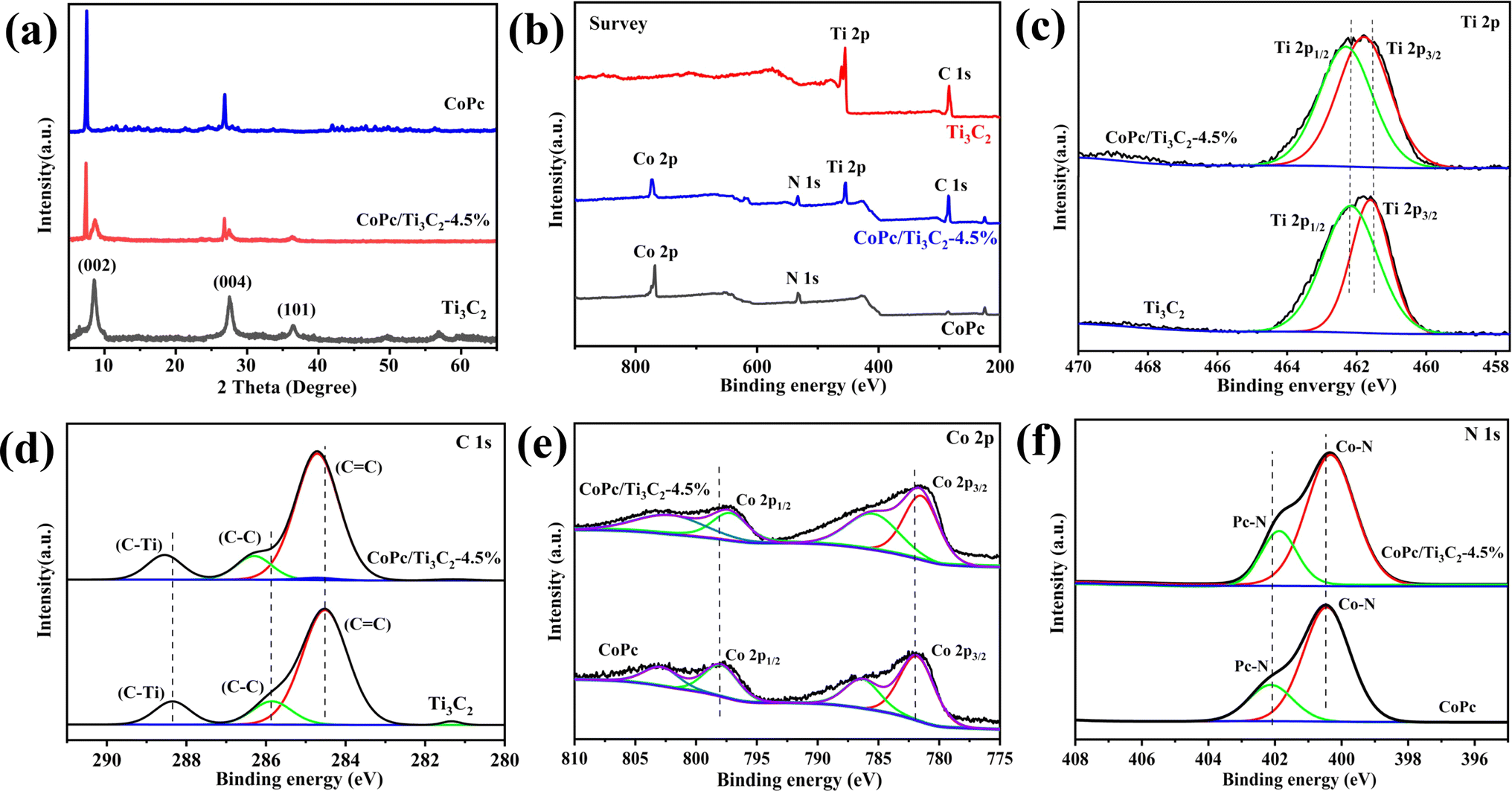 | ||
| Fig. 3 (a) XRD patterns of as-prepared Ti3C2, CoPc and the CoPc/Ti3C2 hybrid. XPS survey spectrum (b), and high-resolution XPS spectra of Ti 2p (c), C 1s (d), Co 2p (e) and N 1s (f) of Ti3C2, CoPc and the CoPc/Ti3C2 hybrid. | ||
Moreover, based on the atomic force microscopy (AFM) images (Fig. 4a and b), the average statistical lateral size of the CoPc/Ti3C2 hybrid is about 1.2 μm, and the thickness of the hybrid is about 61.3 nm, confirming its nanosheet structure. To reveal the crucial role of Co valence, we apply electron paramagnetic resonance (EPR) to acquire related information. Fig. 4c displays a relatively strong signal centered at g = 2.03, which can be attributed to a spin (S = 1/2) Co2+ component in both CoPc and CoPc/Ti3C2, which coincides well with the XPS results. As shown in Fig. 4d, prominent paramagnetic signals located at g = 2.33 are observed, which can be assigned to the Co3+ species. Furthermore, significantly stronger signals shown at the same g value are observed for the CoPc/Ti3C2 hybrid than CoPc, suggesting the most favorable conversion of Co2+ to Co3+ in the CoPc/Ti3C2–Mn–Br catalyst system. This again indicates that the presence of the CoPc/Ti3C2 structure benefits the formation of more Co2+/Co3+ during the reaction.
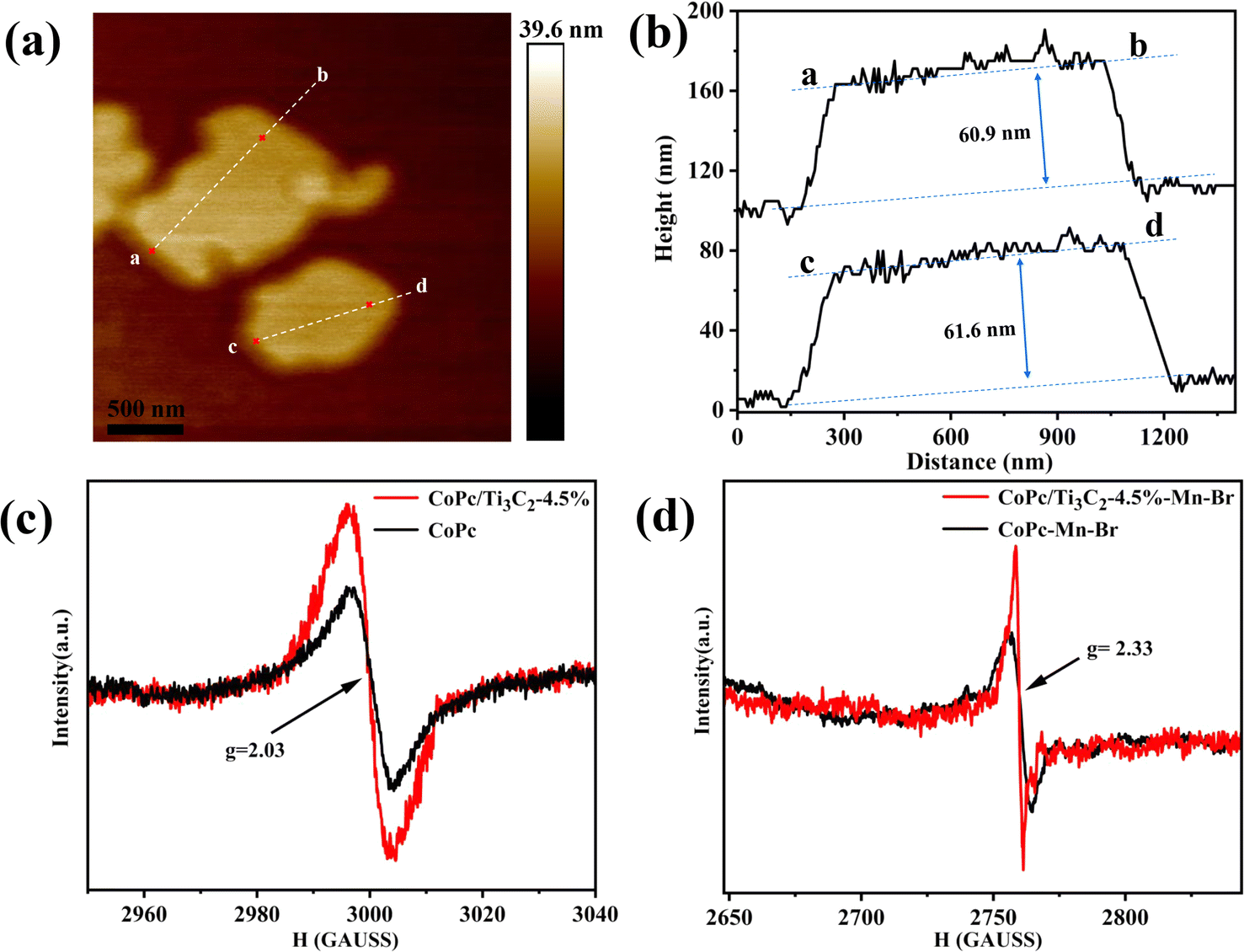 | ||
| Fig. 4 (a) AFM images, and (b) height profiles from AFM analysis of the CoPc/Ti3C2 hybrid. EPR spectra of CoPc and the CoPc/Ti3C2 hybrid (c) without acetic acid as the solvent, (d) with acetic acid as the solvent during the reaction. | ||
Nitrogen (N2) adsorption and desorption isotherms were collected to determine the BET surface area and porous structures of Ti3C2 and the CoPc/Ti3C2 composite. As shown in Fig. 5a, pristine Ti3C2 displays a typical type IV isotherm and H3 hysteresis loop because of capillary condensation steps. The H3 hysteresis loop is also well preserved in the CoPc/Ti3C2 hybrid. The N2 adsorption capacity is improved, implying that the increase of N2 adsorption of the CoPc/Ti3C2 hybrid is attributed to that the CoPc/Ti3C2 composite has a larger pore size and higher BET specific surface area (SBET) than Ti3C2. Besides, the SBET's, pore sizes, and pore volumes of prepared samples are summarized. It can be concluded that the CoPc/Ti3C2 composite has led to a much larger specific surface area of 126.1 m2 g−1, which is higher than that of pristine Ti3C2 (118.5 m2 g−1). The total pore volume (Vtotal) increased after coupling, which suggests that the pores of Ti3C2 are not choked by CoPc (Fig. 5b). The high SBET and Vtotal are expected to create more active sites, facilitate mass transport and promote the utilization of active sites during the reaction.
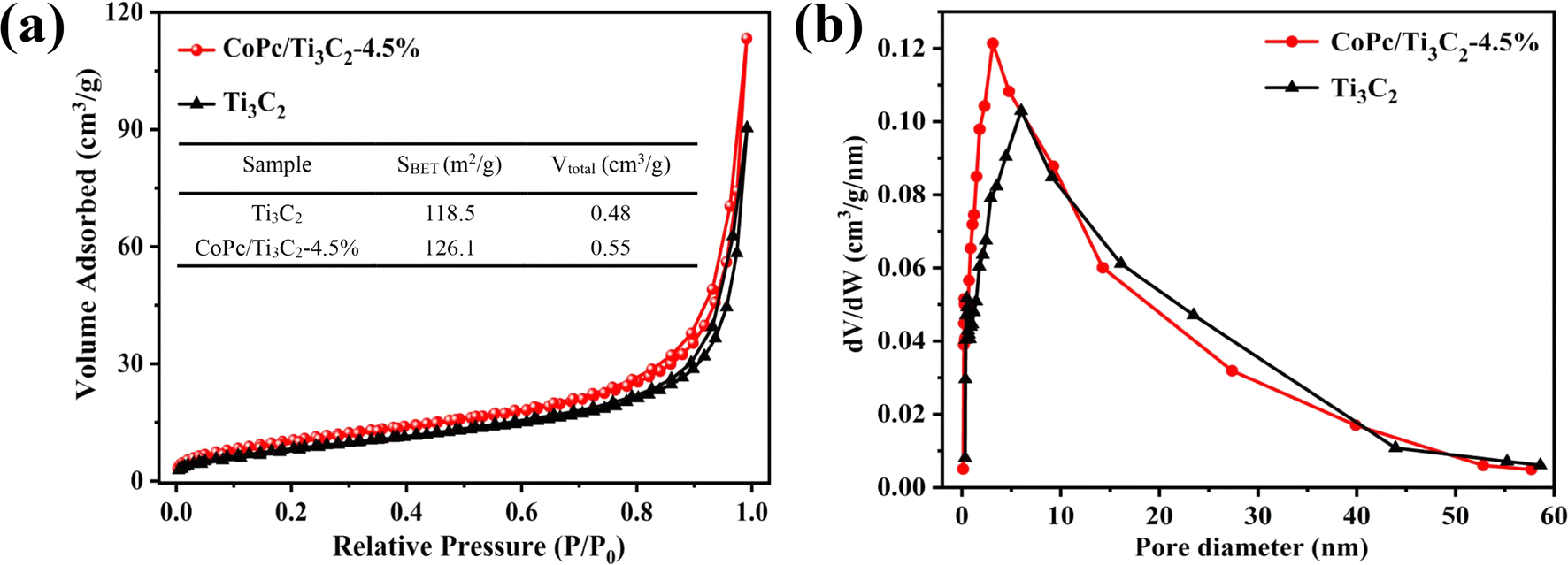 | ||
| Fig. 5 (a) Nitrogen (N2) physisorption isotherms and the inset shows the corresponding BET specific surface area (SBET), total pore volume (Vtotal), and (b) pore size distribution curves of Ti3C2 and the CoPc/Ti3C2 hybrid. | ||
3.2 Selective catalytic oxidation performance of 3,7-dichloro-8-dichloro methyl quinoline to quinclorac
Taking 3,7-dichloro-8-dichloro methyl quinoline (3,7-D-8-DMQ) as the substrate, the catalytic assay of selective oxidation was carried out in acetic acid solution (Fig. 6a). From the experimental results (Fig. 6b), the CoPc–Mn(OAc)2–HBr (CoPc–Mn–Br) catalyst produces less quinclorac with the quinclorac yield at 35.6%. However, the optimized CoPc/Ti3C2-4.5%–Mn–Br catalyst exhibits 3,7-D-8-DMQ conversion at 95.3%, high selectivity to quinclorac at 91.8% with superior quinclorac yield up to 87.5%. CoPc/Ti3C2-4.5%–Mn–Br displays a high catalytic yield for the production of high value-added quinclorac, which is 2.46-fold higher than that of the CoPc–Mn–Br system. These results show that the CoPc/Ti3C2–Mn–Br catalytic system is observably more active and robust compared to the CoPc–Mn–Br under the same conditions. Overall, the catalytic performances of the CoPc/Ti3C2-4.5%–Mn–Br catalyst are much better than that of the Co-based-Mn-Br catalyst (Table S1†). To the best of our knowledge, this is the first successful example of the CoPc/Ti3C2-4.5%–Mn–Br catalyst with excellent catalytic performance in the synthesis of quinclorac. To investigate the origin of the catalytic performance, a series of control experiments were conducted. As shown in Fig. S1,† the quinclorac yield of single CoPc/Ti3C2-4.5% or Mn(OAc)2 or HBr cannot be found under the same conditions. Besides, it is imperative to explore the impact of Mn(OAc)2 and HBr on the quinclorac yield. Significantly, there is no activity without HBr, suggesting that HBr functions as a chain initiator. The introduction of Mn(OAc)2 in the CoPc/Ti3C2-4.5%–Br system can markedly improve the catalytic activity, indicating the cocatalyst role of Mn(OAc)2. For comparison, we performed a series of control experiments without different catalyst components under the same conditions. Moreover, the blank experiments without the presence of the cobalt source show a negligible 3,7-D-8-DMQ conversion, and almost no formation of quinclorac. The acetic acid solvent may have no direct effect on the reaction, and negligible products are obtained without any catalysts. These results confirm that the presence of the cobalt source is responsible for the conversion of 3,7-D-8-DMQ and yield of quinclorac.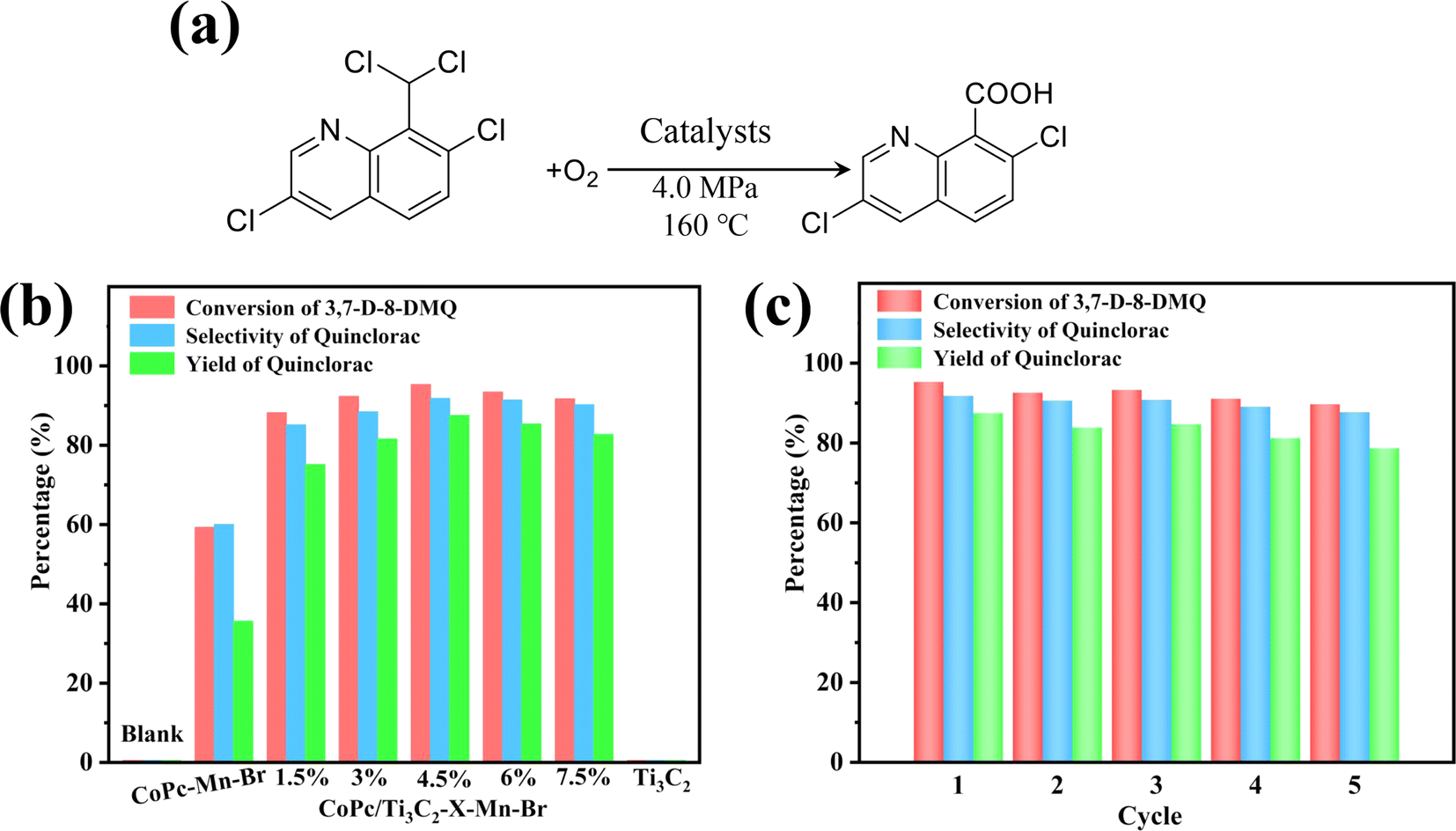 | ||
| Fig. 6 (a) Chemical reactions for selective catalytic oxidation of 3,7-D-8-DMQ to quinclorac. (b) Conversion, selectivity and yield of catalytic oxidation reaction over CoPc/Ti3C2–Mn–Br and CoPc–Mn–Br after 6 h. (c) The stability tests over CoPc/Ti3C2-4.5%–Mn–Br. | ||
The long-term stability of this catalytic system was investigated. The catalytic stability tests of the CoPc/Ti3C2-4.5%–Mn–Br are conducted under the optimum reaction conditions of 160 °C, O2 partial pressure of 4 MPa and reaction time of 6 h for five consecutive cycles (Fig. 6c). After the reaction was completed, the catalysts could be recovered by simple filtration, and used for the next run. The catalytic performance shows very good stability during 30 hours of reaction. After five cycles, there is no observable change in the CoPc/Ti3C2-4.5% composite structure after the cyclic reaction (Fig. S2 and S3†). The as-designed heterogeneous catalyst reveals excellent stability and recyclability in the catalytic application.
3.3 Mechanistic investigations
To further investigate the mechanism, the variation of the metallic valence state was characterized by UV-vis spectra. As shown in Fig. 7a, the broad 560 nm band is a typical peak of an octahedral Co(II)–acetato complex. And the new band appearing at 690 nm is assigned to Co(III) complexes, which demonstrates that there is only Co2+ before the reaction and Co2+ is partly oxidized to Co3+ during the reaction. However, for the CoPc–Mn–Br, the new bands corresponding to Co3+ vanished after 5 h reaction. The Co3+ cations generated previously are quickly reduced to Co2+ by Mn2+, implying that the redox capability of cobaltous cations would change as the reaction intermediates coordinated to the cobaltous cation instead of the acetic acid and bromic anions. As shown in Fig. 7b, the Co3+ peak intensities of CoPc/Ti3C2-4.5%–Mn–Br are much stronger than that of CoPc–Mn–Br, suggesting that the oxidation from Co2+ to Co3+ is promoted in CoPc/Ti3C2-4.5%–Mn–Br, which is beneficial for the production of free radicals.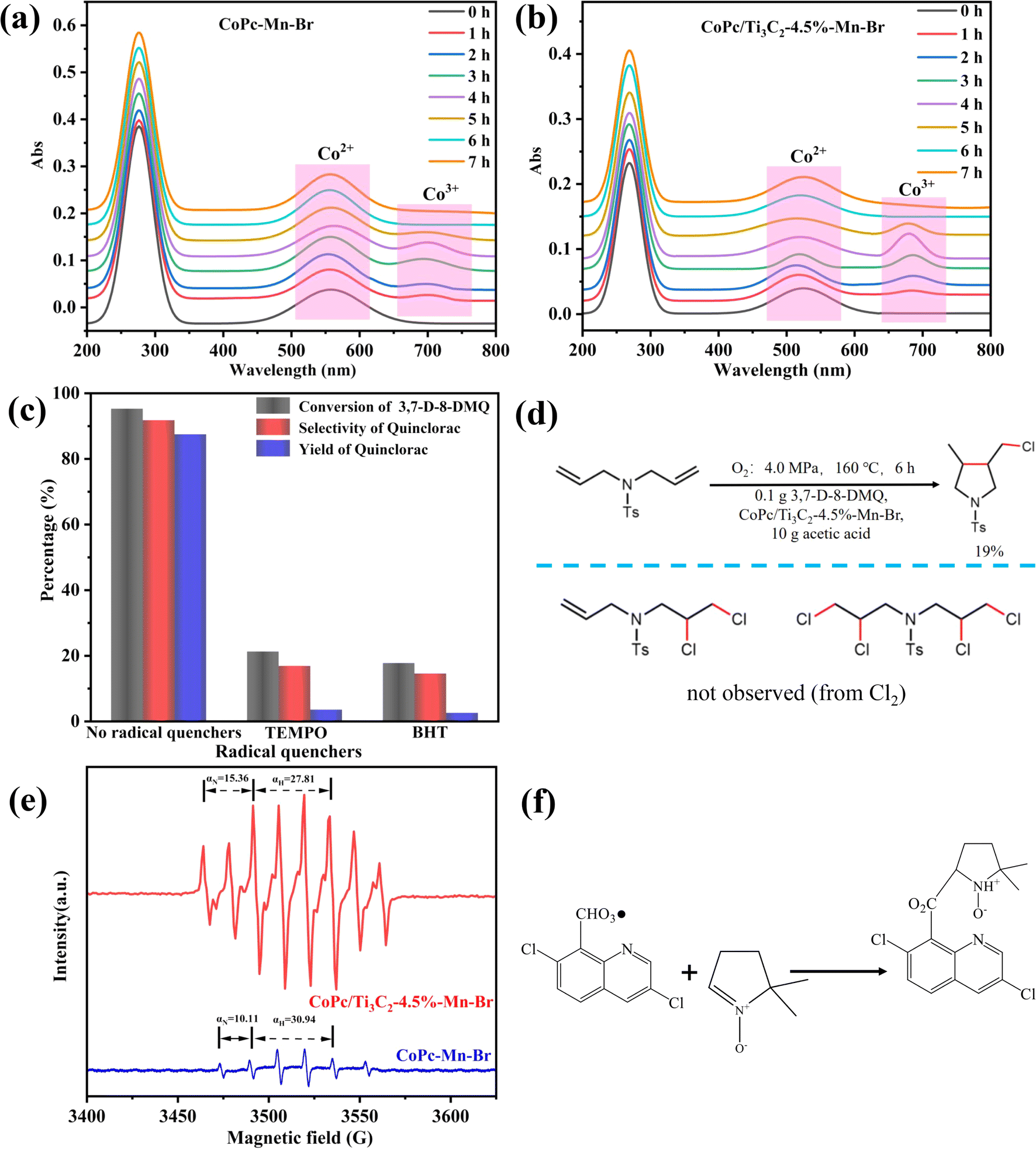 | ||
| Fig. 7 The UV-vis spectra as a function of reaction time: (a) CoPc–Mn–Br and (b) CoPc/Ti3C2-4.5%–Mn–Br. (c) Radical quenching experiments of CoPc/Ti3C2-4.5%–Mn–Br. (d) Cl˙ trapping. (e) EPR spectra of CoPc–Mn–Br and CoPc/Ti3C2-4.5%–Mn–Br. (f) Chemical reactions for intermediate radicals and DMPO. | ||
To gain insight into the transformation of intermediate species in oxidation reaction, a series of radical trapping experiments were conducted. We find that the typical free radical quenchers, 2,2,6,6-tetramethylpiperidoxyl (TEMPO) and 3,5-di-tert-4-butylhydroxytoluene (BHT), significantly suppress the formation of quinclorac (Fig. 7c). These results confirm that the catalytic reaction follows the free radical mechanism. To obtain more details of the chlorine species, a diallyl sulfonamide is submitted to the oxidation reaction of 3,7-D-8-DMQ with a stoichiometric amount of Bu4NCl. As expected, a cyclochlorinated compound is isolated, suggesting the formation of Cl˙ in the reaction (Fig. 7d and S4†). According to former literature, chlorine (Cl2) or Cl− might not be an active intermediate as the characteristic alkenyl dichlorination products are not observed. Thereafter, EPR studies using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as radical scavengers reveal the formation of radicals, as the coupling constants of the signal are consistent with related values of radicals, and the molecular weight is equal to the adduct of DMPO and radicals. As shown in Fig. 7e and f, the special signal with nitrogen hyperfine splitting αN = 15.36 G and hydrogen hyperfine splitting αH = 27.81 G is detected, which can be ascribed to DMPO-3,7-dichloroquinoline-CHO3·. The signal peak intensity of CoPc/Ti3C2-4.5%–Mn–Br is significantly stronger than that of CoPc–Mn–Br. Consequently, with sufficient oxidative driving force from Co2+ to Co3+ for free radical activation and formation, CoPc/Ti3C2-4.5%–Mn–Br possesses more robust capacity to carry out the conversion of 3,7-D-8-DMQ to the key intermediate radicals as compared with CoPc–Mn–Br, which is consistent with previous results.
On the basis of the above experimental results and analyses, a plausible mechanism for the synthesis of quinclorac over the CoPc/Ti3C2-4.5%–Mn–Br catalyst is proposed (Fig. 8). Taken together, these experiments provide support for the surmised radical generation (Br˙ and Cl˙) reaction pathways. Initially, Co2+ and Mn2+ are oxidized to Co3+ and Mn3+. Co3+ and Mn3+ react with Br ions to generate Br˙. Br˙ as the initiator of free radical chain reactions reacts with the substrates, Co2+ and Mn2+ in the presence of oxygen to produce 3,7-dichloro-8-aldehyde quinoline and Cl˙. Then Co3+ and Mn3+ react with 3,7-dichloro-8-aldehyde quinoline to produce the key intermediate species – 3,7-dichloro-8-carbonyl quinoline free radicals. Subsequently, the 3,7-dichloro-8-peroxycarboxylic acid quinoline is generated sequentially under the action of oxygen, Co2+ and Mn2+. 3,7-Dichloro-8-peroxycarboxylic acid quinoline would combine with the generated 3,7-dichloro-8-aldehyde quinoline to form the quinclorac product. More detailed mechanistic studies are still ongoing in our lab, and this encouraging result could enlighten more multifunctional synergistic catalysis strategies for selective oxidation reactions.
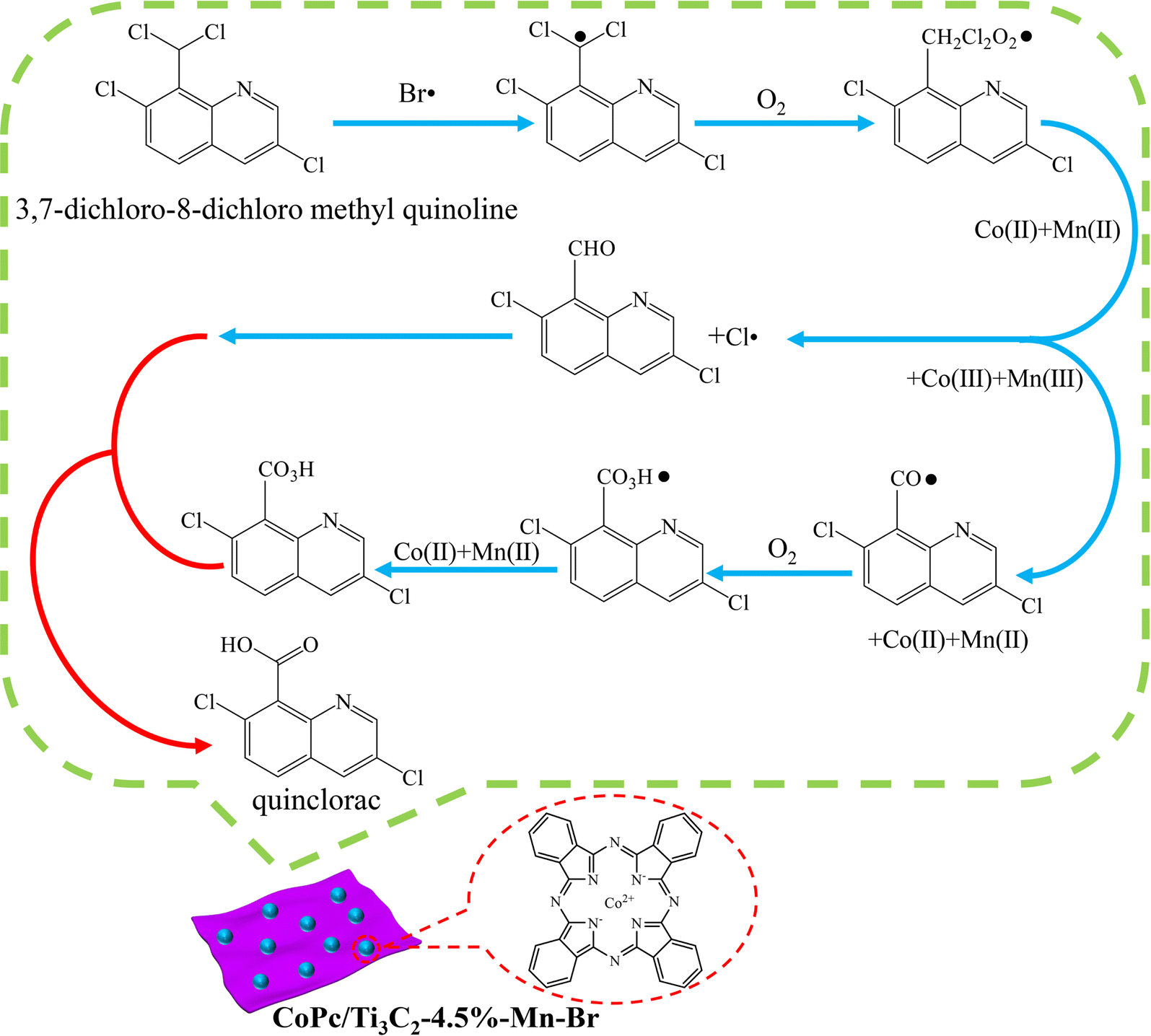 | ||
| Fig. 8 Proposed mechanistic scheme of selective oxidation synthesis of quinclorac. | ||
4. Conclusions
In summary, we have successfully developed a non-noble metal, highly efficient and recyclable CoPc/Ti3C2-4.5%–Mn–Br catalyst system for selective oxidation of 3,7-D-8-DMQ to quinclorac. With this novel catalytic system, quinclorac can be directly obtained from 3,7-D-8-DMQ using O2 as a convenient and clean oxygen source. It is worth noting that the Co oxidation states of the CoPc/Ti3C2 hybrid play a vital role in the catalytic activity and stability during the reaction. Based on the exploration of intermediate radicals, we propose a reasonable catalytic mechanism. The excellent catalytic performance of introducing a CoPc/Ti3C2 heterogeneous structure is attributed to the stronger capacity for stabilizing the accumulated redox cycle of Co2+/Co3+. This study provides a promising strategy for the development of highly effective selective catalytic systems. We believed that this work would advance the excited quinoline chemistry and open more green paths to future oxidation catalysts for large-scale and practical application.Author contributions
Simeng Zhu: investigation, formal analysis, validation and writing – original draft. Peng Liu: writing – review & editing and formal analysis. Xinlin Hong: supervision, project administration and resources.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (grant no. 21872106), and the authors acknowledge financial support from Chibi Research Institute for High-Quality Development.References
- Z. Chen, J. Song, X. Peng, S. Xi, J. Liu, W. Zhou, R. Li, R. Ge, C. Liu, H. Xu, X. Zhao, H. Li, X. Zhou, L. Wang, X. Li, L. Zhong, A. I. Rykov, J. Wang, M. J. Koh and K. P. Loh, Adv. Mater., 2021, 33, 2101382 CrossRef CAS.
- H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed.
- R. Sharma, R. Kumar, I. Kumar and U. Sharma, Eur. J. Org Chem., 2015, 2015, 7519–7528 CrossRef CAS.
- Z. He, H. Liu, Q. Qian, L. Lu, W. Guo, L. Zhang and B. Han, Sci. China: Chem., 2017, 60, 927–933 CrossRef CAS.
- N. J. Pachupate and P. D. Vaidya, J. Environ. Chem. Eng., 2018, 6, 883–889 CrossRef CAS.
- P. L. Arias, J. A. Cecilia, I. Gandarias, J. Iglesias, M. López Granados, R. Mariscal, G. Morales, R. Moreno-Tost and P. Maireles-Torres, Catal. Sci. Technol., 2020, 10, 2721–2757 RSC.
- X. Tian, L. Song, K. Farshadfar, M. Rudolph, F. Rominger, T. Oeser, A. Ariafard and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 59, 471–478 CrossRef PubMed.
- A. K. Bains, V. Singh and D. Adhikari, J. Org. Chem., 2020, 85, 14971–14979 CrossRef CAS PubMed.
- P. E. Hande, M. Mishra, F. Ali, S. Kapoor, A. Datta and S. J. Gharpure, ChemBioChem, 2020, 21, 1492–1498 CrossRef CAS PubMed.
- Z. Zhang, Z. Yuan, D. Tang, Y. Ren, K. Lv and B. Liu, ChemSusChem, 2014, 7, 3496–3504 CrossRef CAS PubMed.
- S. J. Wang, Z. Wang, Y. Tang, J. Chen and L. Zhou, Org. Lett., 2020, 22, 8894–8898 CrossRef CAS PubMed.
- L. Qi, J. Chen, B. Zhang, R. Nie, Z. Qi, T. Kobayashi, Z. Bao, Q. Yang, Q. Ren, Q. Sun, Z. Zhang and W. Huang, ACS Catal., 2020, 10, 5707–5714 CrossRef CAS.
- B. Prabagar, Y. Yang and Z. Shi, Chem. Soc. Rev., 2021, 50, 11249–11269 RSC.
- C. Teja and F. R. N. Khan, Chem.–Asian J., 2020, 15, 4153–4167 CrossRef CAS PubMed.
- N. A. Harry, S. M. Ujwaldev and G. Anilkumar, Org. Biomol. Chem., 2020, 18, 9775–9790 RSC.
- Y. Kodama, S. Ariyasu, M. Karasawa, Y. Aiba and O. Shoji, Catal. Sci. Technol., 2023, 13, 6146–6152 RSC.
- S. G. Makarov, S. Y. Ketkov and D. Wöhrle, Chem. Commun., 2020, 56, 5653–5656 RSC.
- K. Trangwachirachai, I. T. Kao, W. H. Huang, C. L. Chen and Y. C. Lin, Catal. Sci. Technol., 2023, 13, 5248–5258 RSC.
- Q. Huang, M. Zhao, Y. Yang, Y. N. Niu and X. F. Xia, Org. Chem. Front., 2021, 8, 5988–5993 RSC.
- X. Deng, P. Zhao, X. Zhou and L. Bai, Chem. Eng. J., 2021, 405, 126979 CrossRef CAS.
- Z. Hou, L. Dai, Y. Liu, J. Deng, L. Jing, W. Pei, R. Gao, Y. Feng and H. Dai, Appl. Catal., B, 2021, 285, 119844 CrossRef CAS.
- J. Xu, Y. Li, T. Ding and H. Guo, Chem.–Asian J., 2021, 16, 3114–3117 CrossRef CAS PubMed.
- W. An, S. H. Lee, D. Kim, H. Oh, S. Kim, Y. Byun, H. J. Kim, N. K. Mishra and I. S. Kim, J. Org. Chem., 2021, 86, 7579–7587 CrossRef CAS PubMed.
- D. Parmar, A. K. Dhiman, R. Kumar, A. K. Sharma and U. Sharma, J. Org. Chem., 2022, 87, 9069–9087 CrossRef CAS.
- J. Chen, Y. Guo, J. Chen, L. Song and L. Chen, ChemCatChem, 2014, 6, 3174–3181 CrossRef CAS.
- Z. Hao, X. Zhou, Z. Ma, C. Zhang, Z. Han, J. Lin and G. L. Lu, J. Org. Chem., 2022, 87, 12596–12607 CrossRef CAS PubMed.
- N. Zargari, Y. Kim and K. Jung, Green Chem., 2015, 17, 2736–2740 RSC.
- Y. Ding, Y. Huang, W. Sun, F. Li, S. Zhang, S. Fang, M. Wang and X. Hua, Microchem. J., 2022, 183, 107982 CrossRef CAS.
- Y. Ding, T. Guo, Z. Li, B. Zhang, F. E. Kühn, C. Liu, J. Zhang, D. Xu, M. Lei, T. Zhang and C. Li, Angew. Chem., Int. Ed., 2022, 61, e202206284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00123k |
| This journal is © The Royal Society of Chemistry 2024 |