
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the antimicrobial potential of chitosan nanoparticles: synthesis, characterization and impact on Pseudomonas aeruginosa virulence factors†
Dominik
Maršík
 *,
Olga
Maťátková
,
Anna
Kolková
and
Jan
Masák
*,
Olga
Maťátková
,
Anna
Kolková
and
Jan
Masák
Department of Biotechnology, University of Chemistry and Technology, Technická 5, Prague 6, Prague, 166 28, Czechia. E-mail: marsikd@vscht.cz
First published on 22nd April 2024
Abstract
The escalating antibiotic resistance observed in bacteria poses a significant threat to society, with the global prevalence of resistant strains of Pseudomonas aeruginosa on the rise. Addressing this challenge necessitates exploring strategies that would complement existing antimicrobial agents, e.g. by substances mitigating bacterial virulence without eliciting selective pressure for resistance emergence. In this respect, free-form chitosan has demonstrated promising efficacy, prompting our investigation into reinforcing its effects through nanoparticle formulations. Our study focuses on the preparation of chitosan nanoparticles under suitable conditions while emphasizing the challenges associated with stability that can affect biological activity. These challenges are mitigated by introducing quaternized chitosan, which ensures colloidal stability in the culture media. Our approach led to the production of trimethylchitosan nanoparticles with a median size of 103 nm, circularity of 0.967, and a charge of 14.9 ± 3.1 mV, stable within a one-month period in a water stock solution, showing promising attributes for further valorization. Furthermore, the study delves into the antimicrobial activity of trimethylchitosan nanoparticles on Pseudomonas aeruginosa and confirms the benefits of both nanoformulation and modification of chitosan, as our prepared nanoparticles inhibit 50% of the bacterial population at concentration ≥160 mg L−1 within tested strains. Additionally, we identified a concentration of 5 mg L−1 that no longer impedes bacterial growth, allowing reliable verification of the effect of the prepared nanoparticles on Pseudomonas aeruginosa virulence factors, including motility, protease activity, hemolytic activity, rhamnolipids, pyocyanin, and biofilm production. Although trimethylchitosan nanoparticles exhibit promise as an effective antibiofilm agent (reducing biofilm development by 50% at concentrations ranging from 80 to 160 mg L−1) their impact on virulence manifestation is likely not directly associated with quorum sensing. Instead, it can probably be attributed to non-specific interactions with the bacterial surface. This exploration provides valuable insights into the potential of quaternized chitosan nanoparticles in addressing Pseudomonas aeruginosa infections and underscores the multifaceted nature of their antimicrobial effects.
Introduction
Chitosan (CS) is a natural biopolymer consisting of D-glucosamine and N-acetyl-D-glucosamine units connected through β-1,4-glycosidic bonds. It is derived from the naturally occurring chitin polymer through a process of partial N-deacetylation. Chitin is abundantly found in nature, notably in the exoskeletons of crustaceans and insects, and the cell walls of fungi.1 During the N-deacetylation process, secondary amine groups become exposed, exhibiting a pKa value of approximately 6.5. In acidic solutions below this pH, these amine groups become fully protonated, resulting in a positive charge.2CS as a bulk material has received approval in both the EU and USA for dietary and wound dressing applications.3 Currently, a multitude of investigations is underway, exploring CS in the form of nanoparticles (NPs) for the targeted delivery of therapeutic agents, including proteins, vaccines, and nucleic acids.4 With its biocompatibility, biodegradability, and adsorption properties, chitosan serves as an integral component in composite materials, contributing to the acquisition of unique characteristics through diverse combinations.5–8 Furthermore, CS has demonstrated antimicrobial and antibiofilm properties. In the case of G-bacteria, these effects are attributed to the chelation of bivalent ions (Ca2+ and Mg2+) from the bacterial outer membrane and electrostatic interactions with the anionic components of lipopolysaccharides. Next, CS has been shown to disrupt the inner membrane of bacteria. These interactions result in the impairment of cell wall integrity, hindered transport processes, and loss in intracellular materials, and facilitate the entry of CS into the cytosol, where it can interfere with nucleic acid synthesis.9 These properties are particularly intriguing in the context of the escalating problem of antimicrobial resistance.
The unique attributes of nanoparticles, such as their increased surface area-to-volume ratio and surface charge density compared to the bulk material, enable them to interact more effectively with the negatively charged bacterial cell envelopes. This can lead to the formation of an impermeable layer of CS-NPs around the bacterium, thereby preventing transport through the outer membrane of G-bacteria.10 Additionally, the nanoscale dimensions may promote biocompatibility and reduce unwanted interactions with the immune system.11 As a result, we have embarked on an investigation into the antimicrobial effects of CS-NPs using a model microorganism for biofilm formation, Pseudomonas aeruginosa (PA). It is worth noting that since 2017, a carbapenem-resistant strain of PA has been included on the WHO list of microorganisms for which new treatment strategies are urgently needed,12 particularly strategies that do not exert selective pressure for the emergence of resistance.13 Given the evolutionary conservation of the essential negative charge of microbial cell envelopes, it is unlikely that bacteria will develop resistance to CS-NPs.2 Furthermore, there is evidence that CS interferes with the PA quorum sensing (QS) system.14 QS is a bacterial communication mechanism mediated by small, membrane-diffusing signalling molecules that are released into the local environment. QS activation is contingent upon reaching a threshold cell concentration, which subsequently regulates gene expression and shapes specific bacterial phenotypes. These changes are pivotal for the bacteria's ability to thrive in competitive environments, adapt to metabolic demands, and modulate the production of virulence factors, including elastase, exotoxin A, pyocyanin, lipase, pyoverdine, lectins, and, importantly, biofilm formation.15,16 The biofilm formation process relies on the essential involvement of type IV pili and flagella motility.17 Formed biofilm matrices, comprising negatively charged elements such as extracellular polymeric substances (EPS), eDNA, and proteins, represent additional targets for electrostatic interactions with CS-NPs, aided by their nanoscale dimensions penetrating inside.18
In the realm of CS-NP synthesis through the ionic cross-linking methods, the crucial factor is the positive charge inherent to CS. These techniques rely on electrostatic interactions with either negatively charged macromolecules or anionic cross-linking agents.19 Among the various employed techniques, the most widely adopted is the ionic gelation process using trivalent tripolyphosphate anions (TPP).20 This method is advantageous for its ease of execution, absence of undesirable side reactions, utilization of aqueous solutions, non-toxicity of TPP, which makes it acceptable as a food additive by the FDA,21,22 and the absence of high-temperature requirements, making it suitable for encapsulation of thermosensitive active compounds.23 However, despite the relative simplicity of synthesizing CS-NPs, the establishment of appropriate reaction conditions remains a challenging task. This challenge arises from the intricacies associated with various factors such as the initial concentration of reagents,24,25 the ratio of reactants,26 the presence or absence of salts in the reaction medium,27 and the molecular weight and degree of CS deacetylation.28,29 Furthermore, CS-NPs prepared through ionic gelation utilizing TPP often exhibit a broad size distribution and limited colloidal stability.30 This instability poses a challenge, particularly when evaluating the antimicrobial effects of CS-NPs or assessing their cytotoxicity. Of note, standard culture media used for culturing model pathogenic microorganisms and tissue cells, such as LB medium, TSB medium, and DMEM medium, typically maintain a pH range from 6.5 to 7.5. In vitro tests designed for the study of antimicrobial effects and cytotoxicity primarily rely on the inherent buffering capacity of these solutions and often lack a more sophisticated system capable of maintaining a constant pH during cultivation and as a result pH can fluctuate during cultivation due to the influence of metabolites.31 Upon introduction into the culture medium, CS-NPs may undergo deprotonation due to changes in pH, resulting in reduced electrostatic repulsion and decreased affinity between amino groups and TPP. The elevated temperatures typically employed (usually 37 °C) for cultivation of pathogenic microorganisms or tissue cultures further exacerbate the aggregation of CS-NPs.32
To mitigate the challenges posed by deprotonation, quaternization of chitosan's amino groups can be employed. This modification ensures the retention of a positive charge even under neutral and slightly alkaline pH conditions, expanding the solubility range of CS. This increased solubility arises from the substitution of primary amines with alkyl groups, which prevent the formation of hydrogen bonds between the amines and the hydroxylic groups in the CS chain.33 Additionally, nanoparticles prepared from quaternized CS using TPP as a crosslinker exhibit low cytotoxicity, akin to those derived from unmodified CS.1
Results and discussion
Preparation of CS/TMC-NPs for antimicrobial applications
Our objective was to craft CS-NPs possessing characteristics suitable for biomedical applications, including nanoscale size, a narrow size distribution, regular spherical morphology, long-term stability, and stability in culture media to facilitate the evaluation of the antimicrobial efficacy of CS in nanoparticle form against the pathogenic bacterium PA. In this study, two types of chitosan were used, specifically CS representing unmodified chitosan and TMC denoting quaternized chitosan by means of trimethylation.In the initial phase of screening for suitable preparation conditions of CS/TMC-NPs using the ion-crosslinking method, we monitored parameters such as hydrodynamic size, polydispersity index, and ζ-potential. In a study conducted by Sreekumar et al.,28 it was found that the primary factor influencing the mean hydrodynamic size, when transitioning from nano- to micrometer-sized CS-NPs prepared via ionic gelation, was the input concentration of CS. However, the substantial alterations in particle size were observed within a broad input CS concentration range, spanning from 0.10 mg mL−1 to 5.00 mg mL−1. In our narrower range of CS input concentration (ranging from 0.50 to 1.00 mg mL−1), considered as an appropriate range for production of nanosized particles, we observed that changes in the input concentration had a less pronounced effect on the resulting nanoparticle size (Fig. 1). Nevertheless, a slight increase in the hydrodynamic size of the particles was noted with increasing reactant concentrations, particularly in an acetate buffer. This phenomenon aligns with observations made in a study conducted by Liu et al.24 and applies within the appropriate zone of TPP and CS mass ratios. By appropriate zone we mean the point where additional increases in the reaction ratio lead to a significant escalation in the mean hydrodynamic size of particles, as determined by dynamic light scattering (DLS), indicating system aggregation. In the case of CS-NPs, this zone was confined up to a mass reaction ratio of 0.33, with the exception of an input concentration of 0.50 mg mL−1 when diluted acetic acid was used as a solution for CS-NP synthesis. With the further addition of TPP, the aggregate formation becomes apparent from the increase in both hydrodynamic size and PDI (polydispersity index) values. Thus, in our assessment of the parameters suitable for CS-NP preparation, we attach greater significance to the reaction ratio mTPP/mCS than to the input concentration of CS itself. This emphasis on the reaction ratio stems from our hypothesis that, at the verge of aggregation, the maximum conversion of CS into nanoparticles is achieved. Subsequent to the further addition of TPP, the CS-NPs become interconnected through inter-crosslinks between various polymer chains.27
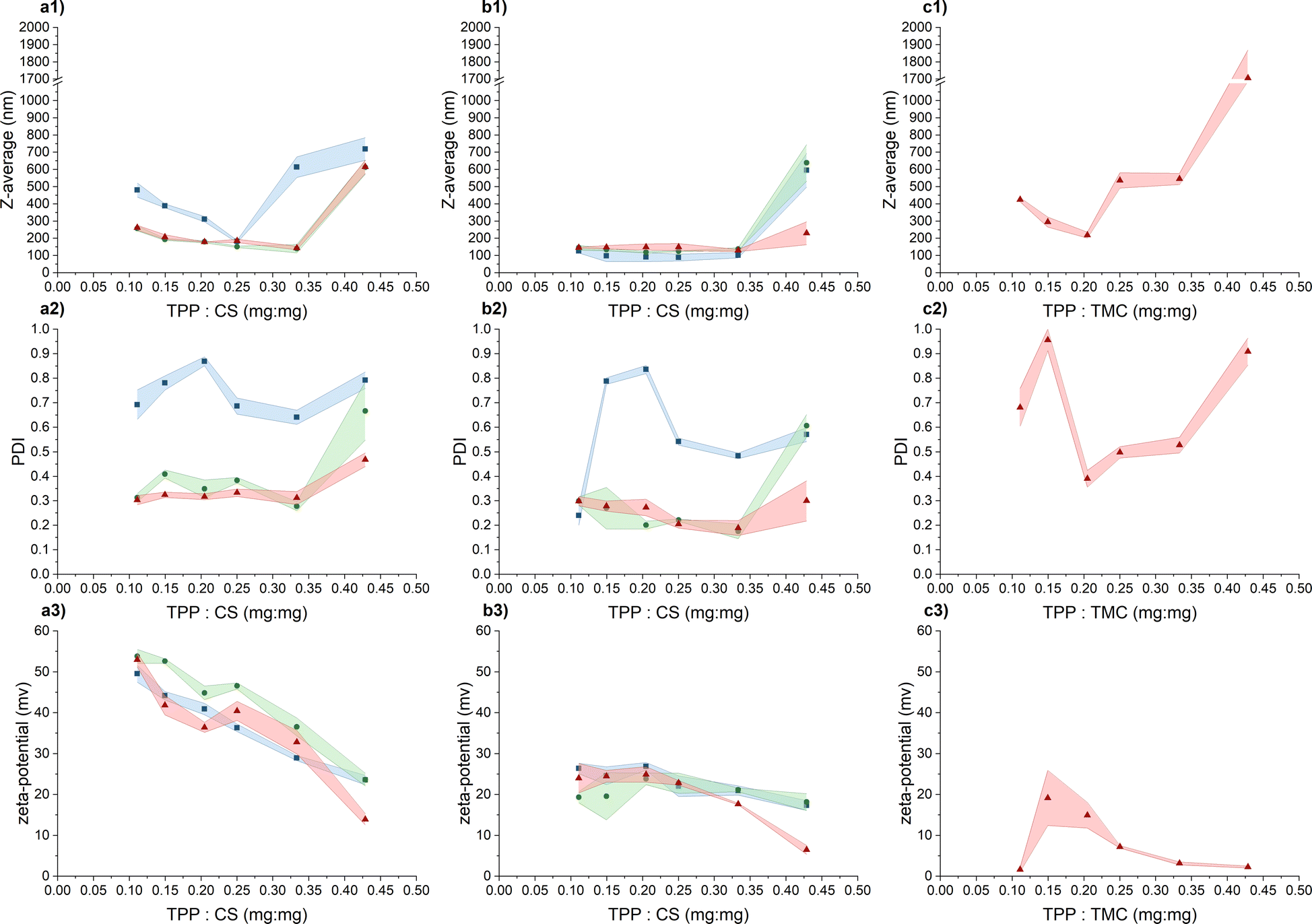 | ||
Fig. 1 Exploring suitable conditions for the CS/TMC-NP preparation by comparing the intensity weighted mean hydrodynamic size (Z-average), polydispersity index (PDI) and zeta-potential values based on the TPP and CS/TMC mass ratio. The appropriate zone of TPP and CS/TMC mass ratio is considered up to the point of significant increase in the mean hydrodynamic size as a result of the increasing concentration of the cross-linking agent (TPP): (a) CS in diluted acetic acid; (b) CS in acetate buffer; (c) TMC in UPW; (1) intensity weighted mean hydrodynamic size; (2) polydispersity index; (3) ζ-potential. Chitosan input concentrations:  0.50 mg mL−1; 0.50 mg mL−1;  0.75 mg mL−1; 0.75 mg mL−1;  1.00 mg mL−1. Coloured areas represent standard deviation values of 3 independent repetitions. 1.00 mg mL−1. Coloured areas represent standard deviation values of 3 independent repetitions. | ||
As previously mentioned, when the input concentration of CS was 0.50 mg mL−1 in an aqueous solution containing acetic acid, the zone of appropriate TPP and CS mass ratio was limited to a value of 0.25. It's important to note that while this shift did not occur in the acetate buffer, the PDI index exhibited a significant increase in comparison to concentrations of 0.75 mg mL−1 and 1.00 mg mL−1. Evidently, at this particular input concentration of CS, particles with a broader size distribution are formed and such high PDI values may also indicate the presence of aggregates. Liu and Gao24 suggest that this phenomenon may be attributed to changes in spatial interaction distances or alterations in the inner structure of the particles, such as increased compactness of the CS molecule chain. The increasing ionic strength of the medium leads to the screening of electrostatic charges within the CS chains by salts, leading to its enhanced compactness and flexibility.34,35 This possibly elucidates why the use of an acetate buffer generally resulted in particles with a smaller hydrodynamic size that in a dilute acetic acid solution. This observation aligns with findings by Jonassen et al.,27 who noted a decrease in particle size after the addition of 0.05 M NaCl at all tested TPP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CS ratios. The general decrease in PDI values following the addition of sodium acetate can be attributed to a combination of the reduction in the intrinsic viscosity of CS and slower kinetics of the CS-NP formation process, a trend observed by Sawtarie et al.36 After the addition of NaCl, the slow kinetics allow for a more thorough mixing of CS and TPP before ionic gelation, resulting in a more uniform rate of CS-NP formation and narrow size distribution.
:CS ratios. The general decrease in PDI values following the addition of sodium acetate can be attributed to a combination of the reduction in the intrinsic viscosity of CS and slower kinetics of the CS-NP formation process, a trend observed by Sawtarie et al.36 After the addition of NaCl, the slow kinetics allow for a more thorough mixing of CS and TPP before ionic gelation, resulting in a more uniform rate of CS-NP formation and narrow size distribution.
When evaluating the ζ-potential values (Fig. 1), the anticipated decrease in the average value with the increasing concentration of negatively charged TPP was confirmed.30 In the acetic acid aqueous solution, the ζ-potential values within the appropriate mass ratio zone range from 32.8 ± 2.9 mV to 53.8 ± 1.7 mV, indicating a high level of colloidal stability. In the acetate buffer, the measured values were lower, ranging from 17.7 ± 0.2 mV to 26.4 ± 1.3 mV, signifying relatively stable to moderately stable colloidal stability.37 Beyond the appropriate mass ratio zone, when TPP is in excess, the surface charge density of the particles decreases to a point where these nanoparticles lose their stability, becoming more prone to mutual interactions and aggregation.30 The lower values recorded in the acetate buffer are likely a result of salt-induced charge screening,24 which also leads to a slight reduction in ζ-potential values compared to the acetic acid aqueous solution.27
Subsequently, transmission electron microscopy (TEM) was employed to assess the morphological characteristics of CS-NPs synthesized in both diluted acetic acid and acetate buffer solutions. Specific samples were chosen based on DLS data at an input CS concentration of 1 mg mL−1 within the mass ratio of 0.33, which remained within the suitable reactant mass ratio zone (Fig. 1). Fig. 2 illustrates the irregular morphologies of CS-NPs produced in acetic acid aqueous solution, alongside the presence of microparticles in the sample. The utilization of acetate buffer as a medium for nanoparticle synthesis led to improved circularity and size distribution of CS-NPs. The median size and circularity were determined to be 30 nm and 0.986, respectively (Fig. 3). Image analysis was not performed for CS-NPs prepared in the acetic acid aqueous solution due to their unsatisfactory morphology and wide size distribution, rendering them unsuitable for consideration as potential antimicrobial agents.
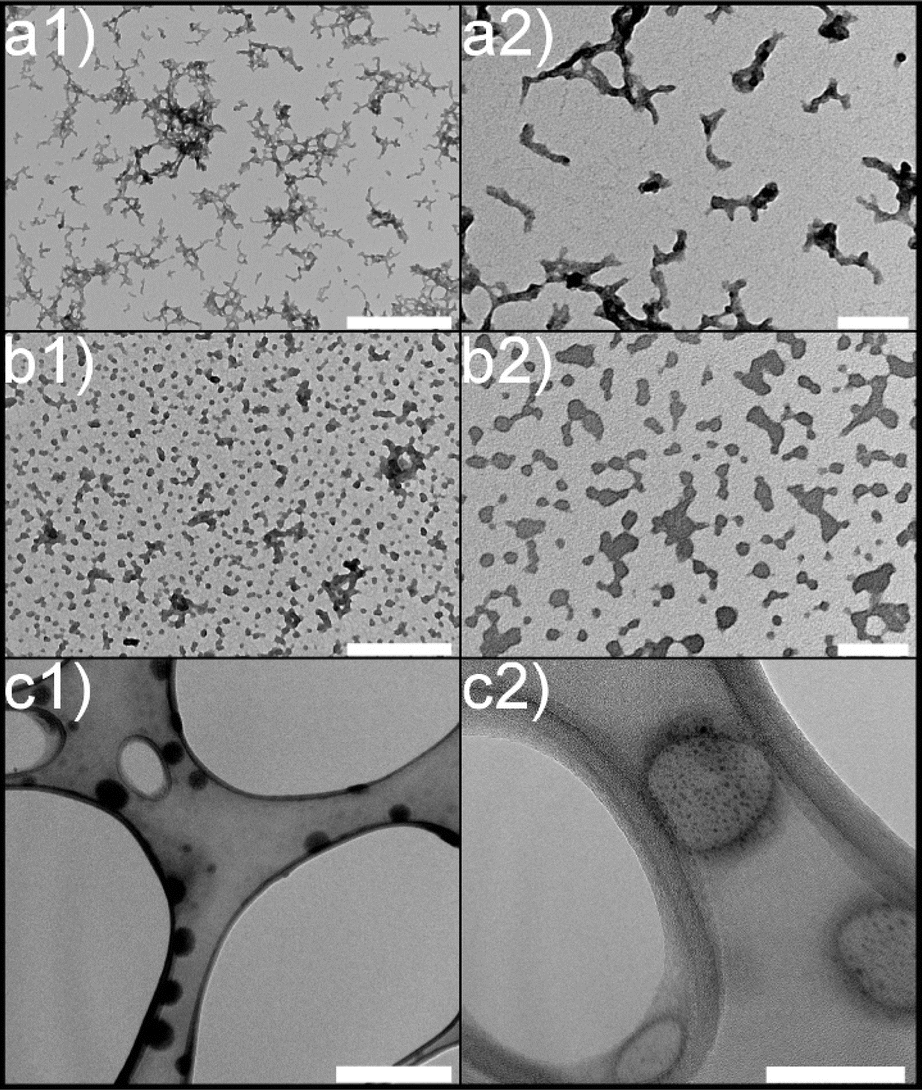 | ||
| Fig. 2 Morphology of chitosan nanoparticles: (a) CS in diluted acetic acid; (b) CS in acetate buffer; (c) TMC in UPW; (1) scale bar represents 500 nm; (2) 100 nm scale. | ||
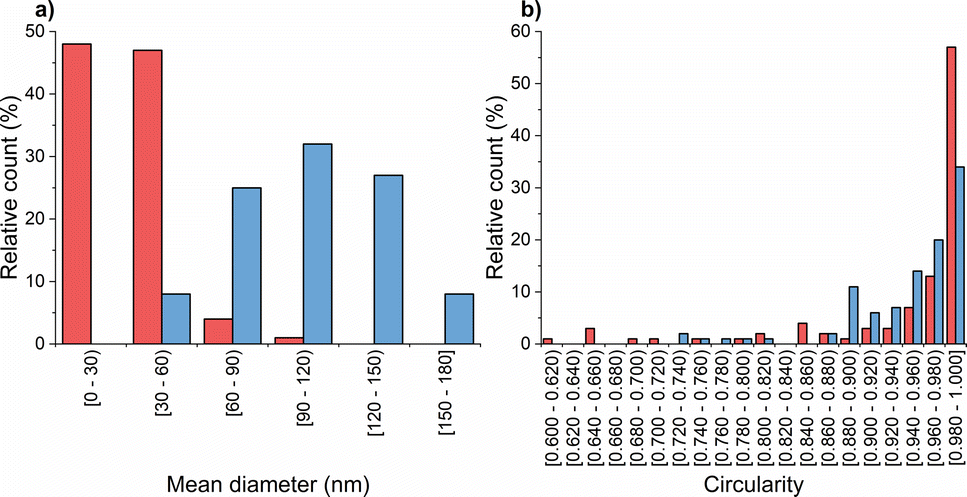 | ||
Fig. 3 Chitosan nanoparticle characteristics: (a) size; (b) circularity of  CS-NPs; CS-NPs;  TMC-NPs. TMC-NPs. | ||
In response to the marginal impact of CS input concentration on NP preparation within our tested range, TMC-NPs were synthesized at a fixed input concentration of 1 mg mL−1. The zone of appropriate reactant mass ratios differed from that observed for CS-NPs, particularly in the case of TMC-NPs, where it was identified at a value of 0.20 (Fig. 1). Both CS and TMC used in our work were LMW chitosans, which under the given conditions exhibit a similar proportion of protonated amines (CS 75–85% deacetylated, TMC > 70% quaternized), consequently providing a similar quantity of available positive charges for interaction with TPP. However, this observed shift in the appropriate ratio might be attributed to steric hindrance introduced by the methyl groups within the TMC structure. This effect aligns with previous observations made by Kiang et al.,38 who explored the impact of chitosan deacetylation levels on the synthesis of CS-NPs using DNA as the negatively charged molecule for ionic crosslinking. The chosen appropriate reaction ratio between TMC and CS closely coincides with those employed by Geçer et al.39 and Sayın et al.40 in their respective studies.
The resulting ζ-potential up to mTPP/mTMC = 0.20 varied from 1.6 ± 0.6 mV to 19.1 ± 6.7 mV (Fig. 1), indicating a range from highly unstable to relatively stable nanoparticles (NPs).37 In general, higher values of the mean hydrodynamic size and polydispersity index (PDI), when compared to the corresponding concentration and reaction mass ratio of CS (Fig. 1), suggest that the prepared TMC-NPs are generally larger than CS-NPs, and the synthesis process is more prone to aggregate formation. Image analysis revealed that TMC-NPs, prepared at a reaction ratio of 0.20, exhibited a median size of 103 nm and a circularity of 0.967 (Fig. 3). In comparison to CS-NPs (prepared in acetate buffer), they displayed a broader size distribution but generally had a narrower circularity distribution. The TMC-NPs prepared in this study exhibit a distinctive morphology and size profile, setting them apart from existing literature on CS-NP preparation (Fig. 2). Our TMC-NPs align with the criteria for biomedical applications41 while adhering to the European Commission's recommended definition of nanomaterials, which classifies nanomaterials as such when 50% or more of their constituent particles fall within the size range of 1–100 nm.42
In the context of TMC-NPs and CS-NPs in an acetate buffer solution, evaluating their stability in culture media is a pivotal step for the subsequent assessment of the antimicrobial capacity of these systems. Luria Bertani (LB) medium serves as a common culture medium for conducting antimicrobial tests against PA.43 After preparing blank samples containing LB media and CS/TMC-NPs at concentrations ranging from 5 mg L−1 to 160 mg L−1, we observed the formation of aggregates within several minutes in the sample containing CS-NPs. In contrast, a stable suspension was formed with TMC-NPs (Fig. S1†). Given the relatively low ζ-potential values of TMC-NPs, we also examined the stability within the stock solution stored at 4 °C (Fig. 4). Over a one-month period, there were no discernible changes in the mean hydrodynamic size or ζ-potential, signifying the absence of time-dependent system destabilization.44 Consequently, we opted to conduct a antimicrobial test with TMC-NPs.
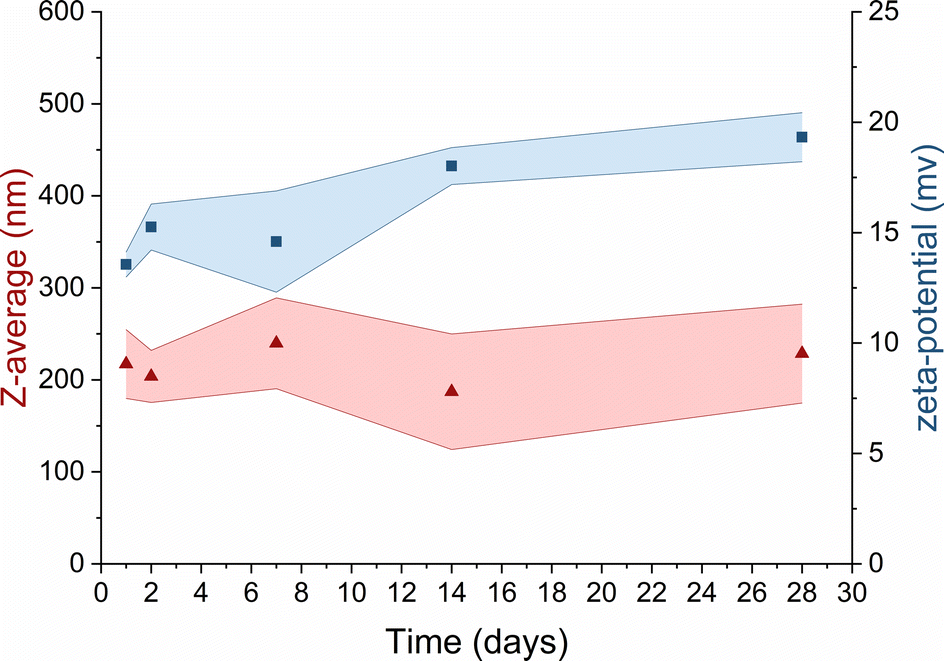 | ||
Fig. 4 Long-term stability of TMC in UPW  mean hydrodynamic size; mean hydrodynamic size;  ζ-potential. Coloured areas represent the standard deviation values of 3 independent repetitions. ζ-potential. Coloured areas represent the standard deviation values of 3 independent repetitions. | ||
Antimicrobial activity of TMC-NPs against PA planktonic cells – MIC and sub-MIC determination
TMC-NPs hold considerable potential as an antimicrobial system, particularly in combination with other antimicrobial agents, thereby presenting viable prospects for utilization in various biomedical fields.45 Furthermore, we investigated their influence on the virulence of PA. To achieve this, it was imperative to identify a sub-MIC concentration, defined as a concentration that does not impede the growth kinetics of PA, as a virulence manifestation is contingent upon cell concentration.46 The sub-MIC concentration of TMC-NPs was consistently identified at 5 mg L−1 across all three tested PA strains, as illustrated in Fig. 5. Beyond this threshold, we observed a progressive enhancement of the antimicrobial effect with increasing TMC-NP concentrations. The MIC50 values were determined to be 160 mg L−1 for strains ATCC 155442 and ATCC BAA-47 (PA01). However, in the case of strain ATCC 10145, the MIC50 value exceeded 160 mg L−1.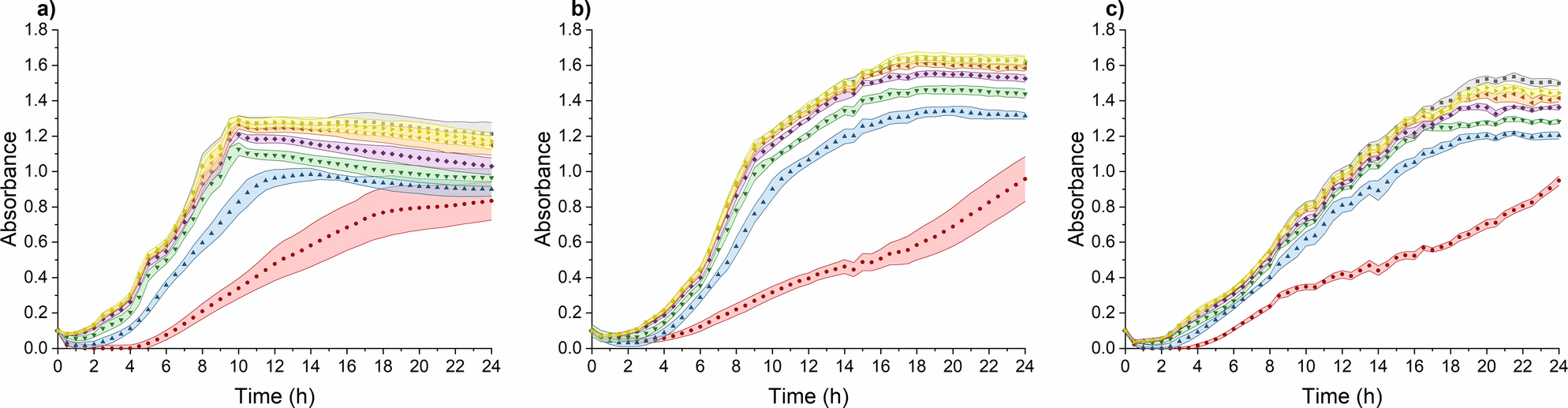 | ||
Fig. 5 Effect of TMC-NPs on PA suspension growth: (a) ATCC 10145; (b) ATCC 15442; (c) ATC BAA-47 (PA01). Concentration (mg L−1) of TMC in TMC-NPs:  0; 0;  5; 5;  10; 10;  20; 20;  40; 40;  80; 80;  160. Coloured areas represent standard deviation values of five parallel and three independent experiments. 160. Coloured areas represent standard deviation values of five parallel and three independent experiments. | ||
In a study by Boudouaia et al.,47 the effectiveness of a CS solution (DDA 95%, LMW) was assessed using evaluation of inhibition zones on agar plates. Interestingly, they observed complete resistance of PA to concentrations up to 0.50% CS solution. In another agar plate experiment, wherein CS (LMW) at a concentration of 1000 mg L−1 was evaluated by measuring the relative inhibition time for visible PA colonies (ATCC 27853) on agar plates, the incubation phase was extended by 5 h to over 85 hours, depending on the N-acetylation degree of CS.48 In a particularly promising study by Tin et al.,49 the antimicrobial effect was quantified as MIC, defined as the lowest CS concentration that prevented visible PA growth. CS (DDA 75–85%, LMW) exhibited impressive results, with an MIC of 32 mg L−1 for four PA strains (ATCC15279, PA01, PT121, and PT149). Conversely, in a study conducted by Liu et al.,50 MIC values for PA (PA01) in response to water-soluble chitosan chloride (91% DDA, LMW) and sulfonated chitosan (86% degree of substitution, LMW) were found to be notably higher, specifically 1000 mg L−1. In addition to sulfonation, a general enhancement in antimicrobial properties was observed after CS methylation. However, with PA (ATCC 43300), the MIC values for TMCNH2/TM with different degrees of substitution ranged from 1024 mg L−1 to ≥8192 mg L−1.51 Similarly, Maisetta et al.52 examined the effect of quaternized chitosan (80% degree of substitution, LMW) against four PA strains (W4, CVC02118, BAL091, and ATCC 27853) and reported MIC values ranging from 2500 to 5000 mg L−1.
The antimicrobial activity of CS and its derivatives, including NPs, is contingent on a complex interplay between intrinsic factors and environmental conditions.48,53 This multifaceted nature is reflected in the variability in reported antimicrobial effects against PA across the existing literature. In our study, we observed a relatively low MIC50 for TMC-NPs, suggesting their effectiveness against PA. This efficacy may be attributed to the maintenance of a stable TMC-NP suspension in LB medium (Fig. S1†). Conversely, the formation of aggregates within unmodified CS-NPs could explain the high concentrations required to inhibit PA in discussed studies, possibly due to the susceptibility of unmodified CS to aggregation after transfer into culture media. This finding aligns with Salis et al.,54 who reported the aggregation of chitosan-modified silica nanoparticles in culture media, leading to reduced uptake by mouse fibroblasts. Additionally, previous studies have demonstrated that methylation of CS enhances interactions with biological membranes, leading to membrane lysis,55 which may contribute to the effectiveness of our TMC-NPs. Furthermore, we hypothesize that the permanent charge conferred by methylation could enable TMC-NPs to penetrate the internal environment of bacterial cells,56 interact with nucleic acids, and thereby enhance antimicrobial efficacy compared to CS-NPs. Indeed, methylation-mediated penetration into the internal cellular environment has been demonstrated in tissue cultures,57 and the size of our 103 nm TMC-NPs indicates suitability for efficient transport through biological membranes.58 However, the currently prevailing understanding of CS action involves primary interactions with bacterial envelopes and their disruption, potentially leading to interactions with internal components.59–61
Antimicrobial activity of TMC-NPs against PA biofilm cells – MBIC determination
CS is recognized in the literature for its antimicrobial properties, particularly as an antibiofilm agent. Often higher concentrations are required to inhibit the planktonic type of growth of PA compared to concentrations sufficient to impede the adhesion and subsequently inhibit biofilm development.62,63 Our study aligns with this trend, revealing MBIC50 values of 80 mg L−1 for ATCC 10145 and ATCC BAA-47 (PA01), and 160 mg L−1 for ATCC 15442 (Fig. 6).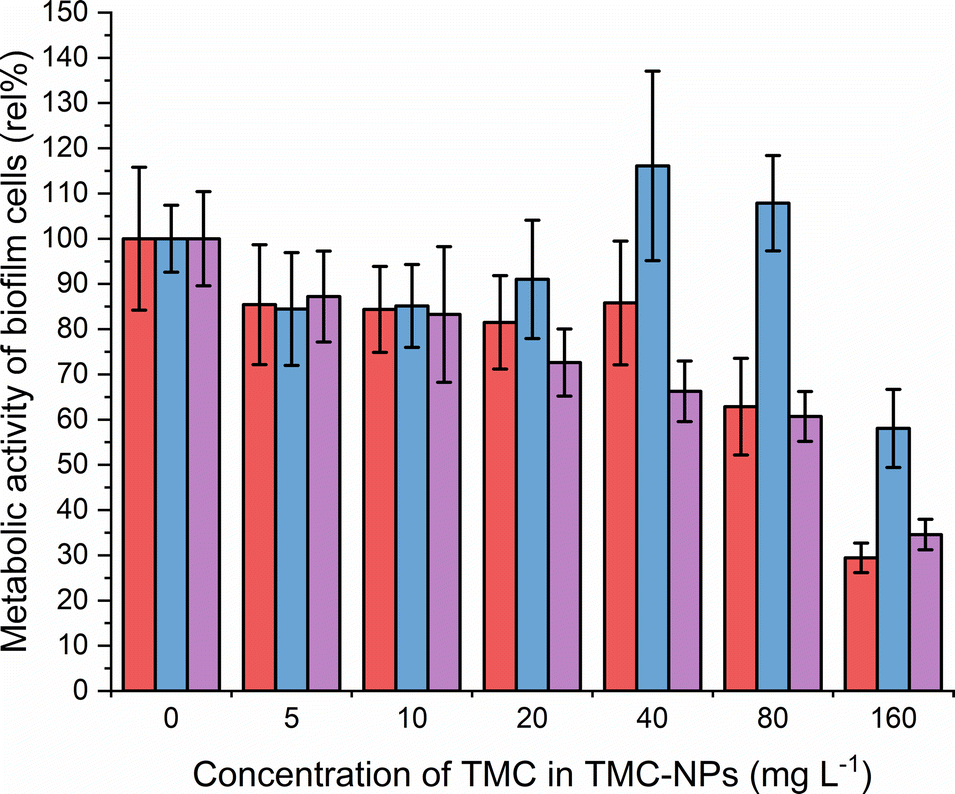 | ||
Fig. 6 Effect of TMC-NPs on adhering PA cells  ATCC 10145, ATCC 10145,  ATCC 15442, and ATCC 15442, and  ATCC BAA-47 (PA01) in comparison to the untreated control. The presented values are a result of eight parallel and three independent repetitions. ATCC BAA-47 (PA01) in comparison to the untreated control. The presented values are a result of eight parallel and three independent repetitions. | ||
The initial adhesion of PA relies on surface virulence factors such as flagella, pili, lipopolysaccharides (LPS), and exopolysaccharides. The negatively charged bacterial cell surface facilitates electrostatic interactions with TMC-NPs, sterically hindering adhesion.64 Intriguingly, a decrease of approximately 15% occurred at a sub-MIC concentration of 5 mg L−1 for all three strains, indicating that the effect is not only related to growth inhibition. In addition to steric hindrance to adhesion, this may be attributed to interference with the quorum sensing system, implicated in biofilm formation, as hypothesised by Piras et al.62 Their study demonstrated that quaternized chitosans inhibited total PA (ATCC 27853) biofilm biomass by 50% in a concentration range of 37–150 mg L−1 in dependence on derivatives tested. Those concentrations were not effective against the planktonic type of growth. Furthermore, interference with quorum sensing was supported by studies revealing downregulation of lasR and rhlR genes after exposure of PA to CS, which govern the PA quorum sensing system.14,52,65,66 In contrast, Maisetta et al.52 observed an increase in PA total biofilm biomass (W4, CVC02118, BAL091, ATCC 27853) in response to the sub-MIC concentration (37 mg L−1) of quaternized chitosan. The benefit of quaternization is evident when comparing the present study with the findings of Liu et al.,50 who observed a decline in the PA01 biofilm metabolic activity by 50% when treated with water-soluble chitosan chloride (91% DDA, LMW) or sulfonated chitosan (86% degree of substitution, LMW) at a concentration of 1000 mg L−1. Additionally, following CS treatment, disruptions in biofilm structural integrity and a reduction in EPS production were observed in their study. Consequently, an alternative action of TMC-NPs at higher concentrations could be electrostatic interactions with biofilm components such as exopolysaccharides or extracellular DNA. The impact of CS-NPs, prepared by CS (LMW) ionotropic gelation, on biofilm eradication was visually confirmed by Rivera Aquayo et al.64 at a concentration of 280 mg L−1. Additionally, the impact of CS in the form of complex nanoparticles has been studied against PA. For instance, NPs synthesized through ionic complexation of CS using alginic acid as a gel core were visually observed to affect mature biofilm at 40 mg L−1.67 Highly effective complex CS-NPs, incorporating the polycationic pyrrole polymer (PPy), were successfully developed in a study by Khan et al.63 The decrease of total biofilm biomass by 50% was determined from 16 mg L−1. However, it is important to note that this effect was not solely attributed to chitosan, as PPy at corresponding concentrations exhibited similar actions against PA adhesion. In conclusion, the TMC-NPs prepared in this study exhibit efficacy at relatively low concentrations, serving as an effective system against PA adhesion and biofilm development.
Effect of TMC-NPs against PA virulence factors
The impact of quaternized chitosan on PA motility was observed, as seen in the strains ATCC 27853 and B910, where swarming motility was reduced by 52% and 40%, respectively, at a concentration of 620 mg L−1.52 In another study involving strain PA KCTC1637, chitosan nanoparticles were found to be more effective against swimming motility, while swarming motility was more inhibited by free chitosan, within a concentration range of 32–256 mg L−1.63 In our study (Fig. 7a), at a concentration of 5 mg L−1 TMC-NPs, the swimming diameter was slightly reduced in all three strains; however, twitching motility was minimally affected. In the case of swarming motility, a reduction was observed in the context of PA01, while in the other two strains, the swarming diameter remained small, even in the untreated control. These results suggest that TMC-NPs likely affect flagellar activity, aligning with the concept that swarming motility is primarily driven by flagella, and type IV pili assist the flagellum during surface spreading.77,78 The reduction in swarming motility as a result of decreased rhamnolipid production after TMC-NP treatment is not supported by our results (see Fig. 7d).
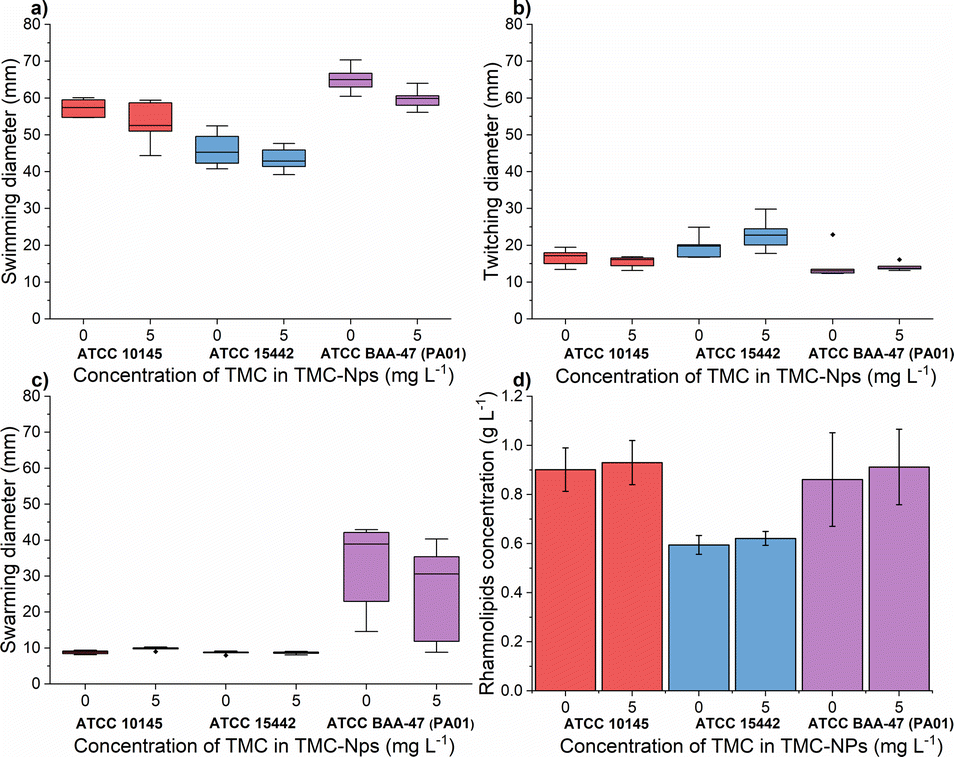 | ||
Fig. 7 Effect of sub-MIC TMC-NPs on PA cells  ATCC 10145, ATCC 10145,  ATCC 15442, and ATCC 15442, and  ATCC BAA-47 (PA01) in comparison to the untreated control, with a focus on (a) swimming motility; (b) twitching motility; (c) swarming motility; and (d) rhamnolipid production. In panels (a–c) dashes indicate the median value. The presented values are a result of duplicate measurements and three independent repetitions. ATCC BAA-47 (PA01) in comparison to the untreated control, with a focus on (a) swimming motility; (b) twitching motility; (c) swarming motility; and (d) rhamnolipid production. In panels (a–c) dashes indicate the median value. The presented values are a result of duplicate measurements and three independent repetitions. | ||
A previous study demonstrated the impact of chitosan extracted from the cell wall of Aspergillus flavus at a concentration of 100 mg L−1 on protease activity, resulting in an almost 60% reduction in the clinical isolate PA.14 Conversely, a slight decrease was observed in PA (KCTC1637) exposed to chitosan in the concentration range of 32–512 mg L−1 (MMW, ≥90% DDA), with no concentration-dependent effect.63 In our investigation, sub-MIC TMC-NPs did not consistently reduce protease activity across the studied strains (Fig. 8a). A minor reduction in total protease activity was observed only in strain ATCC 10145, where LasB elastase activity was slightly stimulated. In strain ATCC 15442, a modest decrease in LasB elastase activity occurred (Fig. 8b) while preserving total protease activity. The correlation between protease activity and quorum sensing (QS) in PA is direct.83 LasB elastase is under the control of the positive regulator LasR, which serves as the main regulator governing the expression of lasB in an otherwise complex regulatory mechanism.84,85 Consequently we can hypothesize that the TMC-NPs prepared in our study likely do not disrupt the direct regulatory pathway of QS-dependent proteases. Minor fluctuations in protease activity may be attributed to interactions with secondary transcription regulators or strain-specific factors.85
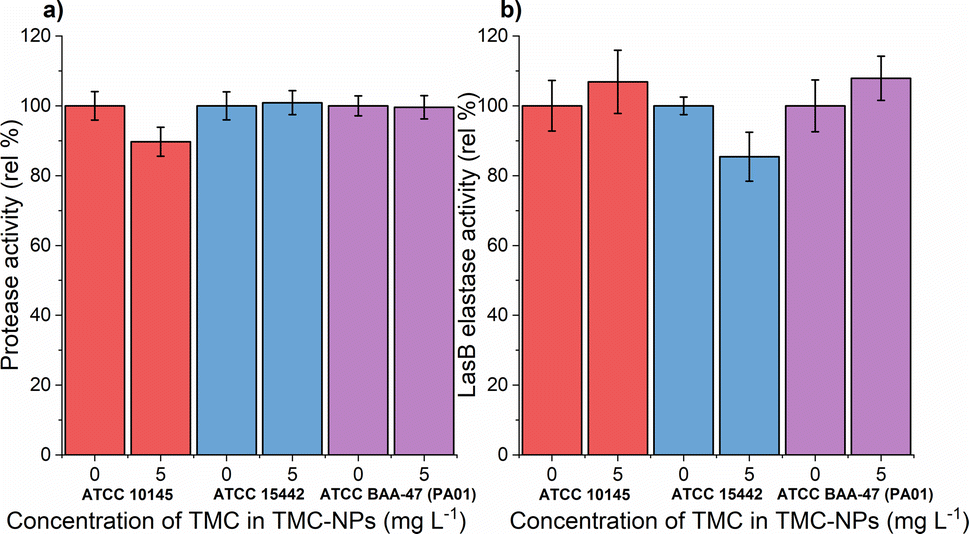 | ||
Fig. 8 Effect of sub-MIC TMC-NPs on PA cells  ATCC 10145, ATCC 10145,  ATCC 15442, and ATCC 15442, and  ATCC BAA-47 (PA01) in comparison to the untreated control, with a focus on (a) protease activity and (b) LasB elastase activity. The presented values are a result of duplicate measurements and three independent repetitions. ATCC BAA-47 (PA01) in comparison to the untreated control, with a focus on (a) protease activity and (b) LasB elastase activity. The presented values are a result of duplicate measurements and three independent repetitions. | ||
The production of pyocyanin, another virulence factor, is associated with tissue damage through the generation of reactive oxygen species.91 Additionally, phenazine compounds present in lung sputum contribute to iron ion reduction (Fe3+ → Fe2+) enhancing the availability of iron and absorption in biofilms.92 Iron is generally a limiting essential element for pathogens during infection since it is sequestered in host organism proteins such as hemoglobin, myoglobin, ferritin, and hemosiderin.93
A comparative study revealed that free-form quaternized chitosan was effective against pyocyanin production in three out of four tested strains at 620 mg L−1, with the fourth strain (CVC02118) exhibiting slight overproduction.52 CS exposure in multiple strains, including PA01, led to decreased pyocyanin levels in a study by Badawy et al.66 Conversely, our study observed a significant increase in pyocyanin production in both tested strains ATCC 10145 and ATCC BAA-47 (PA01) (note: ATCC 155442 is a strain deficient in pyocyanin production) after exposure to sub-MIC TMC-NPs (Fig. 9a). The increased pyocyanin production may be a response to the interaction of TMC-NPs with bacterial surfaces, affecting surface charge and extracellular polymeric substance distance from the substratum surface.94 Attachment of planktonic PA cells to surfaces occurs during the exponential growth phase when cells are phenotypically heterogeneous.95 Pyocyanin's interaction with eDNA has been shown to influence surface properties, promoting intracellular interactions, aggregation, and biofilm development.96 Increased pyocyanin production aligns with the observed reduction in FliA activity, a sigma factor regulating bacterial flagellar gene expression, post TMC-NP exposure. However, a study with a mutated FliA demonstrated increased hemolytic activity in PA,97 which we did not observe (Fig. 9b), probably eliminating pyocyanin overproduction due to flagellar activity disruption.
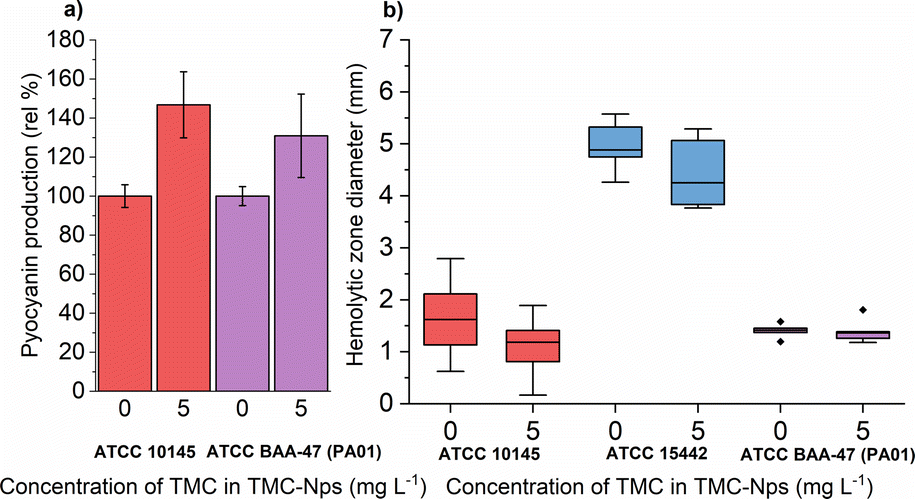 | ||
Fig. 9 Effect of sub-MIC TMC-NPs on PA cells  ATCC 10145, ATCC 10145,  ATCC 15442, and ATCC 15442, and  ATCC BAA-47 (PA01) in comparison to the untreated control, with a focus on (a) pyocyanin production and (b) hemolytic activity. In panel (b) dashes indicate the median value. The presented values are a result of duplicate measurements and three independent repetitions. ATCC BAA-47 (PA01) in comparison to the untreated control, with a focus on (a) pyocyanin production and (b) hemolytic activity. In panel (b) dashes indicate the median value. The presented values are a result of duplicate measurements and three independent repetitions. | ||
Experimental
Preparation of CS/TMC-NPs
For the preparation of chitosan (CS) or trimethylchitosan (TMC) nanoparticles (CS/TMC-NPs) via the ionic gelation method, we employed CS (LMW 50–190 kDa, 75–85% deacetylated) and TMC chloride (LMW, degree of quaternization >70%), both procured from Sigma-Aldrich. All required solutions were prepared using type I ultrapure water (UPW). To create the CS solution of specific concentrations (0.50; 0.75; 1.00 mg mL−1), chitosan was dissolved overnight (RT; 500 rpm) in acetic acid, the mass concentration of which was 1.5 times higher than that of the chitosan, or in 0.05 M acetate buffer (pH 5). The pH of the chitosan solution was adjusted to 5.0 prior to use. The TMC solution at the concentration 1.00 mg mL−1 was prepared through dissolution in UPW. The concentration of TPP crosslinker (purity ≥ 98.0%, Sigma-Aldrich) corresponded to the concentration of CS/TMC used for the reaction and was introduced dropwise into the CS/TMC solution in the required mass ratios mTPP/mCS/TMC (0.43; 0.33; 0.25; 0.20; 0.15; 0.11) under continuous stirring (700 rpm; RT; 30 min). Thus, a solution with a total volume of 10 mL was prepared for subsequent analyses.Characterisation of CS/TMC-NPs
The mean hydrodynamic size (Z-average), polydispersity index (PDI) and zeta potential of CS/TMC-NPs were determined using a Zetasizer Pro instrument (Malvern Panalytical, UK). Data acquired from the measurements were processed with ZS Explorer software v.2.3.0.62 (Malvern Panalytical, UK). In preparation for the analysis, the samples were standardized to a concentration of 0.1 mg mL−1. Subsequently, NPs loaded into folded capillary zeta cell DTS1070 (Malvern Panalytical, UK) were allowed to equilibrate at 25 °C before the measurement were conducted. Measurements were performed in three independent repetitions.The morphological characteristics of CS/TMC-NPs were subjected to investigation through electron microscopy. CS-NPs were scrutinized utilizing a 100 kV JEM-1010 TEM (Jeol, JP). Samples were applied onto copper carbon-coated grids and allowed to adhere for several minutes. Excess solution was carefully eliminated by gently tapping the grid on filtration paper, and the affixed samples were further contrasted with a 1% uranyl acetate solution. Subsequently, the grid was inserted into the TEM column and examined at an acceleration voltage of 80 kV under various magnifications. Images were captured using an Olympus SIS MegaView III CCD camera and processed using Analysis v 2.0 software. For the visualization of TMC-NPs, a TEM (EFTEM Jeol 2200 FS, JEOL, JAPAN) was employed, operating at an electron beam energy of 200 kV. A drop of the sample was air-dried when applied to a copper grid and was examined without the application of staining.
The acquired images underwent manual processing utilizing open-source ImageJ software. For each nanoparticle, measurements of the major axis (2s) and minor axis (2b) were taken, enabling the calculation of the particle's area (A) using eqn (1). This calculated area was then compared with the area determined through a freehand drawing tool. The mean particle diameter was subsequently determined in accordance with eqn (2). Circularity (C) was calculated as the ratio of the particle area (A) to the area of a circle with an equal perimeter (p) (eqn (3)).98 The particle's perimeter (p) was approximated as outlined in eqn (4). This entire measurement process was repeated for 100 nanoparticles in three separate repetitions.
| A = πab | (1) |
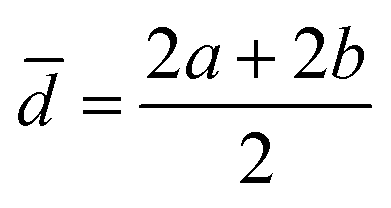 | (2) |
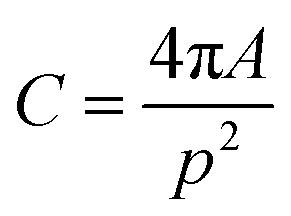 | (3) |
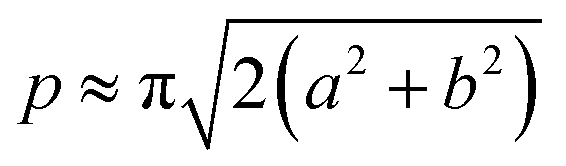 | (4) |
Microorganisms, growth media and conditions
In this work three strains of Pseudomonas aeruginosa were used – ATCC 10145, ATCC 15442, and ATCC BAA-47 (PA01). Before antimicrobial tests Luria–Bertani medium (LB – 10 g L−1 tryptone, 10 g L−1 NaCl, 5 g L−1 yeast extract) was inoculated with colonies of PA, which were stored at 4 °C on LB agar plates. The cultivation process was executed over a duration of 24 h at 37 °C with continuous agitation at 150 rpm using orbital shaking.Antibacterial activity of TMC-NPs against planktonic cells
The assessment of the antibacterial efficacy of TMC-NPs against PA planktonic cells was conducted employing the Bioscreen C microcultivation device (Oy Growth Curves Ab Ltd, Finland) according to Miškovská et al.99 TMC-NPs dispersed in water with a final volume of 70 μl were introduced into a microtiter plate to achieve a final concentration of TMC in TMC-NPs ranging from 5 to 160 mg L−1. Subsequently, 210 μl of LB medium and 30 μl of harvested cells (9000 rcf; RT; 10 min) resuspended in fresh LB medium (OD600 of 0.100 ± 0.010) were added to achieve a total volume of 320 μl. Bacterial growth was monitored for 24 h at 37 °C. Each experiment was performed in five parallel and three independent repetitions from which growth curves were generated, and the minimum concentration required to inhibit 50% of bacterial population (MIC50) was determined.99 Additionally, the sub-minimal inhibitory concentration (sub-MIC) was ascertained, representing a concentration that does not impede bacterial growth analogously to Maisetta et al.52Activity of TMC-NPs against adhering cells
The assessment of the antibiofilm efficacy of TMC-NPs against PA was carried out in 96-well polystyrene microtiter plates (TPP, CH) according to Michailidu et al.100 TMC-NPs dispersed in water with a final volume of 70 μl were introduced into the wells to achieve a final concentration of TMC in TMC-NPs ranging from 5 to 160 mg L−1. Subsequently, 210 μl of harvested cells (9000 rcf; RT; 10 min) resuspended in fresh LB medium (OD600 of 0.800 ± 0.010) were added to attain a total volume of 280 μl. The incubation of the plates was conducted for 24 h at 37 °C with continuous agitation at 150 rpm using orbital shaking. Following incubation, the wells underwent two rounds of washing using an automated microplate washer and dispenser (BioTek 50 TS Washer, USA) with sterile PBS (pH 7.4) to eliminate non-adherent cells. The metabolic activity of adhered cells was evaluated employing the MTT assay, as per the method outlined by Kulišová et al.101 with slight modifications. The washed wells were filled with 60 μl of glucose solution in PBS (57.4 g L−1) and 50 μl of MTT solution in PBS (1.0 g L−1) and incubated for 60 min at 37 °C with continuous shaking at 150 rpm. Following incubation, a solvent solution (pH 4.7) composed of 160 g L−1 SDS, 400 g L−1 DMF, and 20 g L−1 acetic acid, diluted in PBS (pH 7.4), was added to dissolve coloured formazan crystals. The plates were again incubated for 30 min at 37 °C with agitation at 230 rpm, and 100 μl aliquots were analysed at 570 nm using a UV-Vis spectrophotometer, Infinite M200 Pro reader (Tecan, CH). Each experiment was performed in eight parallel and three independent repetitions from which the minimum biofilm inhibiting concentration required to reduce biofilm development by 50% (MBIC50) was determined, in comparison to the nanoparticle-free control, which was assigned to be 100%.Effect of TMC-NPs on virulence of PA
All assessments were conducted at a sub-MIC of TMC-NPs, specifically at 5 mg L−1, a concentration determined not to impact the growth kinetics of PA. This precaution was taken to prevent any inadvertent influence on virulence resulting from a reduction in cell numbers during experiment performance. Each experiment was carried out in duplicate, with three independent repetitions. The treatment protocol for PA cells with TMC-NPs mirrored the approach employed for determining the antibacterial activity of TMC-NPs against planktonic cells, albeit with specific modifications. In the case of agar plate methods, the LB medium, utilized for the resuspension and dilution of PA cells, was substituted with sterile phosphate buffer (pH 7.4). Following a one-hour incubation period (37 °C, 150 rpm) in the presence of TMC-NPs, agar plates were inoculated with 10 μl of the cell suspension. The remaining methods were executed in a 40× larger volume, and the resuspended cells in phosphate buffer (OD600 of 0.100 ± 0.010) were incubated for 24 hours (37 °C, 150 rpm) in the presence of LB media. For the determination of rhamnolipids, the LB medium was replaced with a basic mineral medium (3.4 g L−1 KH2PO4, 4.4 g L−1 K2HPO4, 15.0 g L−1 NaNO3, 1.1 g L−1 KCl, 1.1 g L−1 NaCl, 0.5 g L−1 yeast extract, 20.0 g L−1 sodium citrate, 0.224 g L−1 MgSO4, 0.28 mg L−1 FeSO4·7H2O, 1.45 mg L−1 ZnSO4·7H2O, 1.25 mg L−1 CuSO4·5H2O, 8.40 mg L−1 MnSO4·H2O, 1.20 mg L−1 CaCl2·4H2O). Given that all other methodologies were predicated on the monitoring of extracellular products, the supernatant from free cells (9000 rcf; RT; 10 min) was utilized for subsequent assessments.000 rpm), and 200 μl aliquots were analyzed at 400 nm using a UV-Vis spectrophotometer, Infinite M200 Pro reader (Tecan, CH).
000 rpm, 10 min) were analyzed at 495 nm using a UV-Vis spectrophotometer, Infinite M200 Pro reader (Tecan, CH).
Conclusions
In this study, we investigated the antimicrobial capacity of chitosan nanoparticles (CS-NPs), with a specific emphasis on combatting the biofilm-forming pathogenic bacterium PA. The synthesis of CS-NPs was achieved through ionic gelation utilizing trivalent TPP anions, with subsequent characterization of the resulting nanoparticles regarding size, charge, and stability. While nanoparticles from two different types of chitosan were successfully prepared, namely CS-NPs (median size 30 nm, circularity 0.986) and TMC-NPs (median size 103 nm, circularity 0.967), CS-NPs were excluded from antimicrobial testing. This exclusion was caused by aggregation in culture media which implies a broader usability of quaternized CS TMC-NPs.Our prepared TMC-NPs, exhibiting favourable characteristics for biomedical applications, including nanoscale size, regular spherical morphology, and stability in water and complex culture media, were evaluated for their antimicrobial activity against PA through MIC and sub-MIC determination. Notably, TMC-NPs demonstrated efficacy at a low concentration (MIC50 ≥ 160 mg L−1) compared to existing literature focusing on the antipseudomonal activity of free-form chitosan. However, due to this promoted efficacy the sub-MIC value of TMC-NPs was found at a low concentration (5 mg L−1) and at this concentration we observed a reduction in PA hemolytic activity and motility (swarming, swimming). Importantly, TMC-NPs emerged as a more effective antibiofilm agent (MBIC50 80–160 mg L−1) than disruptors of planktonic growth, suggesting their potential as supportive agents in combating PA biofilm development. Nevertheless, it is imperative to highlight the increased production of pyocyanin as a PA response to the influence of sub-MIC TMC-NPs. We attribute this observation to PA compensating for reduced adhesion efficiency. This statement requires further investigation to elucidate the proposed mechanism.
In summary, our study demonstrates the synthesis conditions for TMC-NPs with properties conducive to biomedical applications. Moreover, it underscores the potential of TMC-NPs as an antipseudomonal agent and demonstrates the beneficial effects of CS methylation and the conversion of TMC into nanoparticles in enhancing the antimicrobial activity of CS, as compared to relevant literature. With the well-established safety profile of TMC, our TMC-NPs could be strategically utilized in combination with other antimicrobial agents to enhance efficacy. Additionally, our comprehensive screening of TMC-NPs against key virulence factors of PA reveals their capacity in combating PA infections. This suggests a possible role for TMC-NPs as adjunctive therapy in infections caused by this pathogen, with their primary benefit likely lying in modulating biofilm formation. Further work should focus on the specific formulation of TMC-NPs and expand screening testing to include additional bacterial strains, thus increasing the prospect of chitosan in antimicrobial applications.
Author contributions
Conceptualization, D. M.; data curation, D. M., A. K.; formal analysis, D. M., A. K.; funding acquisition, D. M., J. M.; investigation, D. M., O. M., J. M.; methodology, D. M., O. M.; project administration, D. M., J. M.; resources, O. M., J. M.; supervision, J. M.; writing – original draft, D. M., writing – review & editing, O. M., J. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grant of Specific university research – grant No. A2_FPBT_2021_029. The authors would like to thanks Pavel Ulbrich and Alena Michalcová for conducting the transmission electron microscopy.Notes and references
- J. Frigaard, J. L. Jensen, H. K. Galtung and M. Hiorth, Front. Pharmacol, 2022, 13, 880377 CrossRef CAS PubMed.
- R. Y. Pelgrift and A. J. Friedman, Adv. Drug Delivery Rev., 2013, 65, 1803–1815 CrossRef CAS PubMed.
- M. A. Mohammed, J. T. M. Syeda, K. M. Wasan and E. K. Wasan, Pharmaceutics, 2017, 9(4), 53 CrossRef PubMed.
- U. Garg, S. Chauhan, U. Nagaich and N. Jain, Adv. Pharm. Bull., 2019, 9, 195–204 CrossRef CAS PubMed.
- S. K. R. Namasivayam, U. K. Pandian, K. Samrat, R. S. Arvind Bharani, A. John, M. Kavisri, S. Kadaikunnan, M. Thiruvengadam and M. Moovendhan, Int. J. Biol. Macromol., 2024, 259, 129264 CrossRef CAS PubMed.
- S. Karthick Raja Namasivayam, V. Pattukumar, K. Samrat, J. A. Kumar, R. S. Arvind Bharani, A. A. Alothman, S. M. Osman, V. A. Tran and M. Rajasimman, Chemosphere, 2022, 308, 135950 CrossRef CAS PubMed.
- S. Karthick Raja Namasivayam, R. S. Arvind Bharani and K. Karunamoorthy, Int. J. Biol. Macromol., 2018, 120, 921–944 CrossRef CAS PubMed.
- S. K. Raja Namasivayam, G. Venkatachalam and R. S. Arvind Bharani, Sustainable Chem. Pharm., 2020, 17, 100300 CrossRef.
- A. Verlee, S. Mincke and C. V. Stevens, Carbohydr. Polym., 2017, 164, 268–283 CrossRef CAS PubMed.
- R. Fattah, F. Fathy, T. A. H. Mohamed and M. S. Elsayed, AIMS Microbiol., 2021, 7, 415–430 CAS.
- R. Ikono, E. Mardliyati, I. T. Agustin, M. M. F. Ulfi, D. Andrianto, U. Hasanah, B. M. Bachtiar, N. Mardianingsih, E. W. Bachtiar and N. N. Maulana, Biomed. Phys. Eng. Express, 2018, 4, 045026 CrossRef.
- J. P. Horcajada, M. Montero, A. Oliver, L. Sorlí, S. Luque, S. Gómez-Zorrilla, N. Benito and S. Grau, Clin. Microbiol. Rev., 2019, 32(4), e00031 CrossRef CAS PubMed.
- P. Piewngam, J. Chiou, P. Chatterjee and M. Otto, Expert Rev. Anti-Infect. Ther., 2020, 18, 499–510 CrossRef CAS PubMed.
- S. N. Muslim, I. Kadmy, A. N. M. Ali, B. K. Salman, M. Ahmad, S. S. Khazaal, N. H. Hussein and S. N. Muslim, Int. J. Biol. Macromol., 2018, 107, 52–58 CrossRef CAS PubMed.
- M. Whiteley, S. P. Diggle and E. P. Greenberg, Nature, 2017, 551, 313–320 CrossRef CAS PubMed.
- G. Tommonaro, Quorum Sensing: Molecular Mechanism and Biotechnological Application, Academic Press, 2019 Search PubMed.
- G. A. O'Toole and R. Kolter, Mol. Microbiol., 1998, 30, 295–304 CrossRef PubMed.
- P. Rivera Aguayo, T. Bruna Larenas, C. Alarcón Godoy, B. Cayupe Rivas, J. González-Casanova, D. Rojas-Gómez and N. Caro Fuentes, Antibiotics, 2020, 9(9), 551 CrossRef PubMed.
- T. A. Ahmed and B. M. Aljaeid, Drug Des., Dev. Ther., 2016, 10, 483–507 CrossRef CAS PubMed.
- P. Calvo, C. Remuñán-López, J. L. Vila-Jato and M. J. Alonso, J. Appl. Polym. Sci., 1997, 63, 125–132 CrossRef CAS.
- K. Songsurang, N. Praphairaksit, K. Siraleartmukul and N. Muangsin, Arch. Pharmacal Res., 2011, 34, 583–592 CrossRef CAS PubMed.
- Y.-H. Lin, K. Sonaje, K. M. Lin, J.-H. Juang, F.-L. Mi, H.-W. Yang and H.-W. Sung, J. Controlled Release, 2008, 132, 141–149 CrossRef CAS PubMed.
- M. C. Di Santo, C. L. D' Antoni, A. P. Domínguez Rubio, A. Alaimo and O. E. Pérez, Biomed. Pharmacother., 2021, 142, 111970 CrossRef CAS PubMed.
- H. Liu and C. Gao, Polym. Adv. Technol., 2009, 20, 613–619 CrossRef CAS.
- S. Vaezifar, S. Razavi, M. A. Golozar, S. Karbasi, M. Morshed and M. Kamali, J. Cluster Sci., 2013, 24, 891–903 CrossRef CAS.
- E. N. Koukaras, S. A. Papadimitriou, D. N. Bikiaris and G. E. Froudakis, Mol. Pharm., 2012, 9, 2856–2862 CrossRef CAS PubMed.
- H. Jonassen, A.-L. Kjøniksen and M. Hiorth, Colloid Polym. Sci., 2012, 290, 919–929 CrossRef CAS.
- S. Sreekumar, F. M. Goycoolea, B. M. Moerschbacher and G. R. Rivera-Rodriguez, Sci. Rep., 2018, 8, 4695 CrossRef PubMed.
- H.-C. Yang and M.-H. Hon, Microchem. J., 2009, 92, 87–91 CrossRef CAS.
- W. Fan, W. Yan, Z. Xu and H. Ni, Colloids Surf., B, 2012, 90, 21–27 CrossRef CAS PubMed.
- R. Sánchez-Clemente, M. I. Igeño, A. G. Población, M. I. Guijo, F. Merchán and R. Blasco, Proceedings, 2018, 2, 1297 Search PubMed.
- K. Ozturk, F. B. Arslan, E. Tavukcuoglu, G. Esendagli and S. Calis, Int. J. Pharm., 2020, 578, 119119 CrossRef CAS PubMed.
- M. Thanou, J. C. Verhoef and H. E. Junginger, Adv. Drug Delivery Rev., 2001, 52, 117–126 CrossRef CAS PubMed.
- J. Cho, M.-C. Heuzey, A. Bégin and P. J. Carreau, J. Food Eng., 2006, 74, 500–515 CrossRef CAS.
- M. L. Tsaih and R. H. Chen, Int. J. Biol. Macromol., 1997, 20, 233–240 CrossRef CAS PubMed.
- N. Sawtarie, Y. Cai and Y. Lapitsky, Colloids Surf., B, 2017, 157, 110–117 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- T. Kiang, J. Wen, H. W. Lim and K. W. Leong, Biomaterials, 2004, 25, 5293–5301 CrossRef CAS PubMed.
- A. Geçer, N. Yıldız, A. Çalımlı and B. Turan, Macromol. Res., 2010, 18, 986–991 CrossRef.
- B. Sayın, S. Somavarapu, X. W. Li, M. Thanou, D. Sesardic, H. O. Alpar and S. Şenel, Int. J. Pharm., 2008, 363, 139–148 CrossRef PubMed.
- B. L. Banik, P. Fattahi and J. L. Brown, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 271–299 Search PubMed.
- H. Rauscher, B. Sokull-Klüttgen and H. Stamm, Nanotoxicology, 2013, 7, 1195–1197 CrossRef PubMed.
- M. Müsken, S. Di Fiore, U. Römling and S. Häussler, Nat. Protoc., 2010, 5, 1460–1469 CrossRef PubMed.
- T. López-León, E. L. S. Carvalho, B. Seijo, J. L. Ortega-Vinuesa and D. Bastos-González, J. Colloid Interface Sci., 2005, 283, 344–351 CrossRef PubMed.
- M. Tré-Hardy, F. Vanderbist, H. Traore and M. J. Devleeschouwer, Int. J. Antimicrob. Agents, 2008, 31, 329–336 CrossRef PubMed.
- J. Lee and L. Zhang, Protein Cell, 2015, 6, 26–41 CrossRef CAS PubMed.
- N. Boudouaia, M. L. Benine, N. Fettal, B. Abbouni and Z. Bengharez, Waste Biomass Valorization, 2023, 15, 1267–1279 CrossRef.
- Y. Hu, Y. Du, J. Yang, Y. Tang, J. Li and X. Wang, Polymer, 2007, 48, 3098–3106 CrossRef CAS.
- S. Tin, K. R. Sakharkar, C. S. Lim and M. K. Sakharkar, Int. J. Biol. Sci., 2009, 5, 153–160 CrossRef CAS PubMed.
- Y. Liu, Y. Jiang, J. Zhu, J. Huang and H. Zhang, Carbohydr. Polym., 2019, 206, 412–419 CrossRef CAS PubMed.
- S. Rathinam, S. Ólafsdóttir, S. Jónsdóttir, M. Á. Hjálmarsdóttir and M. Másson, Int. J. Biol. Macromol., 2020, 160, 548–557 CrossRef CAS PubMed.
- G. Maisetta, A. M. Piras, V. Motta, S. Braccini, D. Mazzantini, F. Chiellini, Y. Zambito, S. Esin and G. Batoni, Microorganisms, 2021, 9(5), 912 CrossRef CAS PubMed.
- N. A. Melake, H. A. Mahmoud and M. T. Al-Semary, Afr. J. Microbiol. Res., 2012, 6, 5387–5398 CAS.
- A. Salis, M. Fanti, L. Medda, V. Nairi, F. Cugia, M. Piludu, V. Sogos and M. Monduzzi, ACS Biomater. Sci. Eng., 2016, 2, 741–751 CrossRef CAS PubMed.
- D. B. Martins, F. D. Nasário, L. C. Silva-Gonçalves, V. A. de Oliveira Tiera, M. Arcisio-Miranda, M. J. Tiera and M. P. dos Santos Cabrera, Carbohydr. Polym., 2018, 181, 1213–1223 CrossRef CAS PubMed.
- F. Chen, Z.-R. Zhang, F. Yuan, X. Qin, M. Wang and Y. Huang, Int. J. Pharm., 2008, 349, 226–233 CrossRef CAS PubMed.
- T. Kean, S. Roth and M. Thanou, J. Controlled Release, 2005, 103, 643–653 CrossRef CAS PubMed.
- J. W. Hickey, J. L. Santos, J. M. Williford and H. Q. Mao, J. Controlled Release, 2015, 219, 536–547 CrossRef CAS PubMed.
- F. Dilnawaz, S. Acharya and A. Kanungo, Polym. Bull., 2024, 81, 1071–1095 CrossRef CAS PubMed.
- M. Chandrasekaran, K. D. Kim and S. C. Chun, Processes, 2020, 8(9), 1173 CrossRef CAS.
- D. Yan, Y. Li, Y. Liu, N. Li, X. Zhang and C. Yan, Molecules, 2021, 26(23), 7136 CrossRef CAS PubMed.
- A. M. Piras, S. Esin, A. Benedetti, G. Maisetta, A. Fabiano, Y. Zambito and G. Batoni, Int. J. Mol. Sci., 2019, 20, 6297 CrossRef CAS PubMed.
- F. Khan, P. Manivasagan, D. T. N. Pham, J. Oh, S.-K. Kim and Y.-M. Kim, Microb. Pathog., 2019, 128, 363–373 CrossRef CAS PubMed.
- P. Rivera Aguayo, T. Bruna Larenas, C. Alarcón Godoy, B. Cayupe Rivas, J. González-Casanova, D. Rojas-Gómez and N. Caro Fuentes, Antibiotics, 2020, 9, 551 CrossRef PubMed.
- D. Rubini, S. F. Banu, P. Subramani, B. N. V. Hari, S. Gowrishankar, S. K. Pandian, A. Wilson and P. Nithyanand, Pathog. Dis., 2019, 77, ftz009 CAS.
- M. S. E. M. Badawy, O. K. M. Riad, F. A. Taher and S. A. Zaki, Int. J. Biol. Macromol., 2020, 149, 1109–1117 CrossRef CAS PubMed.
- R. Thaya, B. Vaseeharan, J. Sivakamavalli, A. Iswarya, M. Govindarajan, N. S. Alharbi, S. Kadaikunnan, M. N. Al-anbr, J. M. Khaled and G. Benelli, Microb. Pathog., 2018, 114, 17–24 CrossRef CAS PubMed.
- D.-G. Ha, S. L. Kuchma and G. A. O'Toole, Pseudomonas Methods and Protocols, 2014, 59–65 Search PubMed.
- M. Bouteiller, C. Dupont, Y. Bourigault, X. Latour, C. Barbey, Y. Konto-Ghiorghi and A. Merieau, Int. J. Mol. Sci., 2021, 22(7), 3337 CrossRef CAS PubMed.
- S. Zhu, M. Schniederberend, D. Zhitnitsky, R. Jain, J. E. Galán, B. I. Kazmierczak and J. Liu, J. Bacteriol., 2019, 201(13), e00117 CrossRef CAS PubMed.
- W. Wu, Y. Jin, F. Bai and S. Jin, in Molecular Medical Microbiology, ed. Y.-W. Tang, M. Sussman, D. Liu, I. Poxton and J. Schwartzman, Academic Press, Boston, 2nd edn, 2015, pp. 753–767, DOI:10.1016/B978-0-12-397169-2.00041-X.
- M. J. Kühn, L. Talà, Y. F. Inclan, R. Patino, X. Pierrat, I. Vos, Z. Al-Mayyah, H. Macmillan, J. Negrete, J. N. Engel and A. Persat, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2101759118 CrossRef PubMed.
- J. Tremblay and E. Déziel, BMC Genomics, 2010, 11, 587 CrossRef PubMed.
- A. Yang, W. S. Tang, T. Si and J. X. Tang, Biophys. J., 2017, 112, 1462–1471 CrossRef CAS PubMed.
- J. Overhage, S. Lewenza, A. K. Marr and R. E. Hancock, J. Bacteriol., 2007, 189, 2164–2169 CrossRef CAS PubMed.
- N. C. Caiazza, R. M. Q. Shanks and G. A. O'Toole, J. Bacteriol., 2005, 187, 7351–7361 CrossRef CAS PubMed.
- T. S. Murray and B. I. Kazmierczak, J. Bacteriol., 2008, 190, 2700–2708 CrossRef CAS PubMed.
- T. Köhler, K. Curty Lasta, F. Barja, C. van Delden and J.-C. Pechère, J. Bacteriol., 2000, 182, 5990–5996 CrossRef PubMed.
- S. Bleves, V. Viarre, R. Salacha, G. P. F. Michel, A. Filloux and R. Voulhoux, Int. J. Med. Microbiol., 2010, 300, 534–543 CrossRef CAS PubMed.
- A. Llanos, P. Achard, J. Bousquet, C. Lozano, M. Zalacain, C. Sable, H. Revillet, M. Murris, M. Mittaine, M. Lemonnier and M. Everett, Sci. Rep., 2023, 13, 14208 CrossRef CAS PubMed.
- M. Mateu-Borrás, L. Zamorano, A. González-Alsina, I. Sánchez-Diener, A. Doménech-Sánchez, A. Oliver and S. Albertí, Front. Cell. Infect. Microbiol., 2022, 11, 816356 CrossRef PubMed.
- F. Bastaert, S. Kheir, V. Saint-Criq, B. Villeret, P. M. Dang, J. El-Benna, J. C. Sirard, R. Voulhoux and J. M. Sallenave, Front. Immunol., 2018, 9, 1675 CrossRef PubMed.
- S. P. Diggle, A. S. Griffin, G. S. Campbell and S. A. West, Nature, 2007, 450, 411–414 CrossRef CAS PubMed.
- Y. Wang, L. Gao, X. Rao, J. Wang, H. Yu, J. Jiang, W. Zhou, J. Wang, Y. Xiao, M. Li, Y. Zhang, K. Zhang, L. Shen and Z. Hua, Sci. Rep., 2018, 8, 13344 CrossRef PubMed.
- M. J. Everett and D. T. Davies, Drug Discovery Today, 2021, 26, 2108–2123 CrossRef CAS PubMed.
- R. M. Berka and M. L. Vasil, J. Bacteriol., 1982, 152, 239–245 CrossRef CAS PubMed.
- L. R. Montes, M. Ibarguren, F. M. Goñi, M. Stonehouse, M. L. Vasil and A. Alonso, Biochim. Biophys. Acta, 2007, 1768, 2365–2372 CrossRef CAS PubMed.
- R. S. Berk, D. Brown, I. Coutinho and D. Meyers, Infect. Immun., 1987, 55, 1728–1730 CrossRef CAS PubMed.
- I. R. Coutinho, R. S. Berk and E. Mammen, Thromb. Res., 1988, 51, 495–505 CrossRef CAS PubMed.
- M. J. Wargo, M. J. Gross, S. Rajamani, J. L. Allard, L. K. Lundblad, G. B. Allen, M. L. Vasil, L. W. Leclair and D. A. Hogan, Am. J. Respir. Crit. Care Med., 2011, 184, 345–354 CrossRef CAS PubMed.
- S. Hall, C. McDermott, S. Anoopkumar-Dukie, A. J. McFarland, A. Forbes, A. V. Perkins, A. K. Davey, R. Chess-Williams, M. J. Kiefel, D. Arora and G. D. Grant, Toxins, 2016, 8(8), 236 CrossRef PubMed.
- P. Cornelis and J. Dingemans, Front. Cell. Infect. Microbiol., 2013, 3, 75 CAS.
- J. Bullen, Rev. Infect. Dis., 1981, 3, 1127–1138 CrossRef CAS PubMed.
- N. P. Boks, W. Norde, H. C. van der Mei and H. J. Busscher, Microbiology, 2008, 154, 3122–3133 CrossRef CAS PubMed.
- S. Yang, X. Cheng, Z. Jin, A. Xia, L. Ni, R. Zhang and F. Jin, Appl. Environ. Microbiol., 2018, 84, e00700–e00718 Search PubMed.
- T. Das, S. K. Kutty, N. Kumar and M. Manefield, PLoS One, 2013, 8, e58299 CrossRef CAS PubMed.
- Y.-L. Lo, C.-L. Chen, L. Shen, Y.-C. Chen, Y.-H. Wang, C.-C. Lee, L.-C. Wang, C.-H. Chuang, R. P. Janapatla, C.-H. Chiu and H.-Y. Chang, Res. Microbiol., 2018, 169, 135–144 CrossRef CAS PubMed.
- S. J. BLOTT and K. PYE, Sedimentology, 2008, 55, 31–63 CrossRef.
- A. Miškovská, M. Rabochová, J. Michailidu, J. Masák, A. Čejková, J. Lorinčík and O. Maťátková, PLoS One, 2022, 17, e0272844 CrossRef PubMed.
- J. Michailidu, O. Maťátková, I. Kolouchová, J. Masák and A. Čejková, Plants, 2022, 11(3), 443 CrossRef CAS PubMed.
- M. Kulišová, O. Maťátková, T. Brányik, J. Zelenka, L. Drábová and I. J. Kolouchová, J. Microbiol. Methods, 2023, 205, 106676 CrossRef PubMed.
- E. K. Saeki, A. Y. Yamada, L. A. de Araujo, L. Anversa, D. d. O. Garcia, R. L. B. de Souza, H. M. Martins, R. K. T. Kobayashi and G. Nakazato, Front. Cell. Infect. Microbiol., 2021, 11, 656984 CrossRef CAS PubMed.
- O. Maťátková, J. Michailidu, R. Ježdík, I. Jarošová Kolouchová, T. Řezanka, V. Jirků and J. Masák, Microorganisms, 2022, 10, 1272 CrossRef PubMed.
- P. Kašparová, E. Vaňková, M. Paldrychová, A. Svobodová, R. Hadravová, I. Jarošová Kolouchová, J. Masák and V. Scholtz, Front. Cell. Infect. Microbiol., 2022, 12, 993029 CrossRef PubMed.
- M. C. Das, P. Sandhu, P. Gupta, P. Rudrapaul, U. C. De, P. Tribedi, Y. Akhter and S. Bhattacharjee, Sci. Rep., 2016, 6, 23347 CrossRef CAS PubMed.
- D. Ziuzina, D. Boehm, S. Patil, P. J. Cullen and P. Bourke, PLoS One, 2015, 10, e0138209 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00064a |
| This journal is © The Royal Society of Chemistry 2024 |