
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combined hydrothermal and mechanochemical control of structural modifications of zirconium dioxide for catalytic applications†
V.
Sydorchuk
a,
S.
Levytska
b,
O.
Kiziun
b,
L.
Vasylechko
c,
K.
Simkovicova
 a,
S.
Valtera
a,
B. E.
Billinghurts
d,
S.
Vajda
a and
J. E.
Olszowka
*a
a,
S.
Valtera
a,
B. E.
Billinghurts
d,
S.
Vajda
a and
J. E.
Olszowka
*a
aDepartment of Nanocatalysis, J. Heyrovský Institute of Physical Chemistry v.v.i., Czech Academy of Sciences, Dolejškova 2155/3, 18223 Prague, Czech Republic. E-mail: joanna.olszowka@jh-inst.cas.cz; Tel: +420 266053535
bInstitute for Sorption and Problems of Endoecology of the NAS of Ukraine, 13 Naumova St., Kyiv 03164, Ukraine
cSemiconductor Electronics Department, Lviv Polytechnic National University, 12 Bandera Str., 79013 Lviv, Ukraine
dCanadian Light Source Far-Infrared Beamline, 44 Innovation Blvd, Saskatoon, SK S7N 2V3, Canada
First published on 11th December 2024
Abstract
The influence of hydrothermal treatment (HTT) and subsequent mechanochemical treatment (MChT, milling) on the porous and crystalline structure of precipitated zirconium dioxide was studied. It has been established that HTT at 300 °C promotes the transformation of amorphous ZrO2 into a pure monoclinic phase, as well as the formation of a uniform mesoporous structure which has higher thermal stability. Soft dry milling (300 rpm, 0.5–1 h) of hydrothermally modified monoclinic ZrO2 causes the introduction of defects into its structure without a noticeable change in the phase composition. The presence of defects is confirmed directly using UV-vis spectra and indirectly by the manifestation of photocatalytic activity of milled samples under visible irradiation. Importantly, it is found that after calcination of milled samples at 500 °C a high fraction of defects remains preserved which opens up the potential of using zirconium dioxide modified in this way as a catalyst or catalytic support with added specific properties offered by defects.
1. Introduction
Zirconium dioxide (ZrO2) is a versatile material thanks to the tunability of its physicochemical characteristics. ZrO2 has been receiving increasing attention both as a heterogeneous catalyst and catalyst support since it has adjustable phase composition, redox and acid–base properties, and defects, notably oxygen vacancies, which affect its own catalytic performance and/or of the catalytic moieties dispersed on it.1–5 In particular, ZrO2 has been used in a wide spectrum of industrially relevant processes such as water–gas shift, selective reduction of NO2, or biodiesel production. ZrO2 combined with active metals or metal oxides dispersed on its surface is used in hydrogenation6,7 or ketonization7 reactions, as well as in CO2 and CH4 conversion1,8via CO2 methanation or dry methane reforming (DMR).9 Its role is still not fully understood, but theory calculations indicate that oxygen vacancies significantly lower the adsorption energy of CO2 and promote its dissociation10,11 with the added benefit of suppressing coke formation.12,13One of the most important properties is the phase composition of ZrO2 along its porous vs. crystalline structure, both highly stable in a wide range of applied temperatures. Coking resistance and hydrothermal stability are other major challenges for catalytic materials.1,4,9 Bulk zirconia is hardly reducible, as demonstrated by the high oxygen vacancy formation energies observed by EPR;14 however, reducibility can be altered by lowering ZrO2 particle size. Under-coordination and nanostructuring can strongly affect the properties of ZrO2 making non reducible bulk zirconia reducible at the nanoscale, impacting its role as an oxide support as well.1,4,9 Thus, tailored synthesis protocols offer ways of tuning the structure, morphology and chemical nature of ZrO2 for catalytic applications.12,15–21
The influence of ZrO2 precipitation conditions on its porosity and specific surface area value is relatively well described in the literature.17,22–24 However, the disadvantages of precipitated ZrO2 are the significant content of micropores and an underdeveloped mesoporous structure as well as an amorphous state.25,26 Also, the rapid sintering of micropores and a dramatic decrease in the specific surface area with increasing temperature have been observed.27–30 Post-synthetic modification is necessary to transform amorphous ZrO2 into a crystalline state. Calcination in air, which is most often used for this purpose, results in the formation of an unstable tetragonal phase at low temperature. The complete transformation of tetragonal ZrO2 into a monoclinic phase does not occur even at 500 °C31,32 and is also accompanied by a significant decrease in the specific surface area.33 It appears that it is more advantageous to use hydrothermal treatment (HTT) as a post-synthetic modification of precipitated ZrO2 by either processing the wet gel or the dried xerogel.34 Hydrothermal processes are distinguished as a technique for the synthesis and modification of the structure of porous oxides and nanomaterials31–34 and excel as simple and cost-effective alternative methods.35–44 A specific feature of wet gels HTT is the possibility of obtaining materials that simultaneously have high specific surface area and accessible pore size as was shown for zirconium oxide.26,45 Such modified materials are moreover resistant to sintering,46 as a co-precipitated composition, e.g., CeO2–ZrO2.47,48
Ball milling is considered another environmentally friendly and effective method used for the preparation of oxide materials, including catalysts46,49–51 with an added important feature that nanostructured milled oxides contain various defects (e.g., oxygen vacancies). The latter can act as active sites in different catalytic processes,52–55e.g., in test photocatalytic reactions of safranin degradation.27 It should be noted that studies on ZrO2 milling are not very numerous, do not have a systematic character and are not focused on obtaining catalysts.56–60 As a rule, they are devoted to the milling of monoclinic ZrO2 with its transformation into unstable cubic and tetragonal phases. The effect of milling of ZrO2 on its porous and surface structure has not been studied, to the best of our knowledge.
The purpose of this work is to fill the gap in the synthesis of ZrO2 structures with controllable and thermostable porosity, crystallinity, specific surface area, surface chemistry, redox properties and defects by a unique combination of hydrothermal and mechanochemical routes and explore the properties of this new class of materials.
2. Experimental
2.1 Precipitation
An 11.5 M ammonium hydroxide solution (28%, Sigma-Aldrich, analytical grade) was gradually added to the 0.35 M solution of zirconium(IV) oxynitrate dihydrate (99%, Sigma-Aldrich, analytical grade) under intensive stirring (500 rpm) while monitoring the pH value. During this process, a white gelatinous precipitate was formed. The pH values at the end of the precipitation were chosen to be at the edges of the interval favorable for the formation of the monoclinic phase, 9.5 and 7.0, respectively. The resulting gel was aged in the mother solution for 24 hours at room temperature. Afterward, it was separated by decantation and then washed on a filter to the neutral pH value with distilled water and compacted to a water content of approximately 85% resulting in a wet gel. Part of the obtained wet gel was dried for 48 hours at room temperature to form a dried xerogel.2.2 Modification procedures
The obtained ZrO2 in the form of a wet gel and a dried xerogel was subsequently subjected to hydrothermal treatment (HTT) at 260–320 °C and equilibrium vapor pressure for 5–7 hours. HTT was carried out in Teflon-lined steel autoclaves with a volume of 45 mL. In both cases, the wet gel and dried xerogel were loaded in a quartz tube that was placed in an autoclave. 15 mL of water was added to the bottom of the autoclave in the processing of the dried xerogel so that HTT was carried out in the vapor phase.34 Hydrothermally treated samples were dried at 100 °C for 6 hours. The difference between the HTT option proposed in this paper and the previously described treatments40–45,47–49 can be seen in Scheme 1 presented below. | ||
| Scheme 1 Preparation of ZrO2 using procedures (a) previously described and (b) used in this work. | ||
In the first case (Scheme 1a), HTT of the initial reaction mixture is used, resulting in the formation of powders. Their particles can be porous but in general, they are loosely dispersed materials.61 In the second case (Scheme 1b), washed wet gels or the dried xerogel can be subjected to HTT, the products of which are consolidated or porous materials.61
Hydrothermally treated dried xerogels were subjected to mechanochemical treatment (MChT) in air (dry milling) at 300–500 rpm for 30–60 min. MChT was performed using a planetary ball mill Pulverisette-7, premium line (Fritsch Gmbh) with a vessel of zirconium dioxide. 134 Zirconium dioxide balls with a diameter of 5 mm and a total mass of 91.5 g were used as working bodies. The use of a zirconia ball-milling assembly was chosen to reduce the introduction of contaminants into the product.62 The ratio of the mass of the balls to the mass of the sample (ball-to-powder, BPR) was 10, 13, and 25. The prepared samples, their designations and synthesis conditions are listed in Table 1.
| Sample designation | Method of preparation |
|---|---|
| N1-9.5 | Initial pH 9.5 |
| N2-9.5_HX | HTT xerogel 260 °C 5 h |
| N3-9.5_HXM_300 | HTT xerogel 260 °C + MChT 300 rpm 0.5h BPR 10 |
| N4-9.5_HXM_450 | HTT xerogel 260 °C + MChT 450 rpm 0.5h BPR 10 |
| N5-9.5_HX | HTT xerogel 320 °C 5 h |
| N6-9.5_HXM_300 | HTT xerogel 320 °C 5 h + MChT 300 rpm 0.5h BPR 10 |
| N7-9.5_HG | HTT gel 260 °C 5 h |
| N8-9.5_HGM_300 | HTT gel 260 °C 5 h + MChT 300 rpm 0.5h BPR 10 |
| N9-7 | Initial pH 7 |
| N10-7_HX | HTT xerogel 260 °C 5 h |
| N11-7_HXM_300_1h_10 | HTT xerogel 260 °C 5 h + MChT 300 rpm 1h BPR 10 |
| N12-7_HXM_300_0.5h_13 | HTT xerogel 260 °C 5 h + MChT 300 rpm 0.5 h BPR 13 |
| N13-7_HXM_300_1h_13 | HTT xerogel 260 °C 5 h + MChT 300 rpm 1h BPR 13 |
| N14-7_T | Initial pH 7 +TT 500 °C |
| N15-7_HG | HTT gel 300 °C 5 h |
| N16-7_HG_T | HTT gel 300 °C 5 h + TT 500 °C |
| N17-7_H7hG | HTT gel 300 °C 7 h |
| N18-7_H7hG_T | HTT gel 300 °C 7 h + TT 500 °C |
| N19-7_H7hGM_500_0.5_25 | HTT gel 300 °C 7 h + MChT 500 rpm 0.5h BPR 25 |
| N20-7_H7hGM_500_0.5_25_T | HTT gel 300 °C 7 h + MChT 500 rpm 0.5h BPR 25 + TT 500 °C |
| N21-7_ H7hGM_400_0.5_10 | HTT gel 300 °C 7h + MChT 400 rpm 0.5 h BPR 10 |
2.3 Physicochemical characterization
The porous structure of ZrO2 samples was characterized using the adsorption–structural method. The isotherms of nitrogen adsorption–desorption were obtained using an automatic gas adsorption analyzer ASAP 2405N (Micromeritics Instrument Corp.) after outgassing the samples at 150 °C for 20 h. The specific surface area S, volume of mesopores Vme, and volume of micropores Vmi were calculated from these isotherms using the Brunauer–Emmett–Teller (BET), Barrett–Joyner–Halenda (BJH), and t-methods, respectively. Sorption pore volume V was determined at a relative pressure of nitrogen p/p0 close to 1. The curves of pore size distribution (PSD) were then plotted using the desorption branches of isotherms.X-ray phase analysis and structural and microstructural characterization of the samples were performed by using a DRON-3M diffractometer (CuKα radiation, λ = 0.154 nm), full-profile Rietveld refinement was performed by using WinCSD software63 to obtain lattice parameters of the monoclinic ZrO2 structure, and microstructural parameters of the samples (average grain size D and microstrains 〈ε〉) were derived by full profile Rietveld refinement. The Garvie–Nicholson method allows determination of the molar content of the monoclinic phase Xm according to the following equation:64
| Xm = [Im(111) + Im(−111)]/[Im(111) + Im(−111) + It(101)], |
The volumetric fractions of the monoclinic phase Vm were calculated using the so-called Toraya method based on only some X-ray diffraction (XRD) peak intensity values:56,57
| Vm = 1.311Xm/(1 + 0.311Xm) |
Accordingly, the content of the tetragonal phase Xt(Vt) was calculated as follows:
| Xt(Vt) = 1 − Xm(Vm) |
The size of the crystallites (Dt and Dm) was calculated according to Scherrer's formula using the most intense reflexes of each phase.
The curves of differential thermal analysis (DTA) and thermogravimetry (TG) were recorded using the Derivatograph-C apparatus (F. Paulik, J. Paulik, L. Erdey) in the temperature range 20–1000 °C at a heating rate of 10 °C min−1. The initial mass of the sample was about 200 mg.
The UV-visible diffuse reflectance spectra were recorded with a UV-vis/DRS Lambda 35 spectrometer (PerkinElmer Instruments) in the range of 200–600 nm. MgO was the reference sample. The absorption edge λ was determined based on the spectra plotted in the Kubelka–Munk equation coordination. The bandgap Eg was determined using a Tauc plot.58
The morphology of the ZrO2 powders was characterized with a Hitachi S4800 scanning electron microscope (SEM) equipped with a Nanotrace electron diffraction EDX detector (Thermo Electron).
To study the lattice vibrations of ZrO2, diffuse reflectance measurements were conducted in the Far-Infrared (Far-IR) region (30–600 cm−1), at the Canadian Light Source (CLS) far-infrared beamline using a Bruker IFS125HR spectrometer equipped with a T222 Mylar beamsplitter. The infrared beam was directed out of the sample compartment into a Pike Instruments diffuse IR accessory and then to a QMC superconducting niobium Transition-Edge Sensor (TES) bolometer. A synchrotron was used as a source because of its high flux of photons below 200 cm−1. The ZrO2 samples were packed into ceramic sample cups and placed into the environmental chamber of the DiffusIR accessory. The chamber with an internal volume of 63 cm3 was equipped with an Si window. Argon gas was continuously flowing through the chamber at a rate of 200 mL min−1, regulated by a mass flow controller (Brooks SLA5850). All spectra were collected with 4 cm−1 resolution and averaged over 5 sets of measurements each averaging over 2048 scans. Data processing was done using the OPUS software package to obtain the reflectance spectra, which were produced by dividing the ZrO2 spectra by the spectra of polyethylene powder. The spectra of polyethylene powder were collected directly before and after the collection of ZrO2 spectra and using the same conditions and served as a background.
Selected samples with a pure monoclinic phase were tested in the photocatalytic degradation of safranin T (ST), a dye which is commonly used as a model compound due to its high stability.65 Along with the UV-vis spectra, the reactivity is considered an indirect measure for assessing the formation of defects in the structure of the zirconium dioxide at different stages of its preparation (hydrothermal treatment, milling, and post-calcination). Testing of pure monoclinic ZrO2 samples under visible irradiation was carried out in an aqueous medium using 1 × 10−5 mol dm−3 solution of ST59 with a Philips LED cool daylight (100 W) lamp as an irradiation source. A Shimadzu spectrophotometer UV-2450 was used for the analysis of changes in the concentration of dye during the photocatalytic degradation (band at 520 nm). The optimal dose of catalysts, 1 g dm−3, was used as it was established in other studies.59 Prior to irradiation, the ZrO2 dispersed in the ST-containing solution was stirred until an adsorption–desorption equilibrium was established for 1 h; conditions were applied as those in the testing of other oxide photocatalysts.27,65
3. Results and discussion
3.1 Effect of pH during precipitation
 | ||
| Fig. 1 XRD patterns for ZrO2, precipitated at pH 9.5, after HTT and post-milling at 300 and 450 rpm (T – tetragonal phase, peak from plane [101]; M – monoclinic phase, peaks from planes [111] and [−111]). | ||
Simultaneously, dry milling of the ZrO2 samples, which had been hydrothermally processed as xerogels, gave a very promising result in the formation of the monoclinic phase. For the series of samples with a pH of 9.5 the initial material HTT promotes the formation of crystalline ZrO2 in all cases, which is represented by a mixture of tetragonal and monoclinic phases; however, small content of the amorphous material cannot be excluded. Significant transformation of the tetragonal phase into the monoclinic one (Fig. 1) reaching 93% was achieved already with soft and short ball milling of the xerogel in sample N3-9.5_HXM_300. An increase in the intensity of milling (sample N4-9.5_HXM_450) does not promote further transformation (Table 2). A similar effect of reflexes' intensification can be observed for the gel and its milled counterpart (ESI materials, Fig. S1 and Table S1,† samples N7-9.5_HG and N8-9.5_HGM_300). At the same time, increasing the temperature of the HTT to 320 °C does not give an additional effect (ESI materials, Table S1,† samples N5-9.5_HX and N6-9.5_HXM_300).
It should be noted that in earlier studies, the milling of the tetragonal phase was scarcely studied.60,66,67 It was suggested that this transformation (tetragonal into monoclinic) becomes possible in the presence of water vapor.68,69 Indeed, hydrothermal samples, which are subjected to subsequent milling, contain a lot of adsorbed water, as is shown below based on thermogravimetric measurements. Also, there is a certain critical crystallite size (∼46 nm) above which the transformation of the tetragonal phase into the monoclinic phase may occur.70 However, this mechanism refers to the transformation that occurs during calcination and it is hardly applicable for milling. In this work, the effect is rather attributed to the presence of water and ammonia impurities, which is further discussed in Subsection 3.5.
The size of the crystallites was calculated according to Scherrer's formula using the half-width (FWHM) of the most intense peaks of both phases (Dt and Dm). As can be seen, the crystallites of both monoclinic and tetragonal ZrO2 are reduced after milling (Table 2).
To gather further information about changes in the ZrO2 structure after milling, FIR spectra were acquired for samples N2-9.5_HX, N3-9.5_HXM_300, and N4-9.5_HXM_450, which have different contents of monoclinic phase and defects. Generally, in a crystalline structure, molecules are arranged in a repeating, three-dimensional pattern, leading to well-defined and sharp peaks in the IR spectrum. Commercial ZrO2 was used as a reference material. In general, spectra confirm the results of XRD analysis in terms of the tetragonal and monoclinic mixture of phases. Also, the signal in the lattice vibration range which is characteristic of the monoclinic phase becomes more pronounced with milling (Fig. 2).
 | ||
| Fig. 2 Far IR spectra of samples N2-9.5_HX, N3-9.5_HXM_300, N4-9.5_HXM_450 and commercial ZrO2 (reference). The spectra were obtained at 20 °C with an Si window, and polyethylene was used as a background. Marked ★ are the bands of monoclinic ZrO2 (ref. 71) and ▼ denote bands of tetragonal ZrO2,72 and ♦ bands of tetragonal ZrO2.71 | ||
The spectrum of the reference (commercial monoclinic ZrO2) shows all 9 peaks characteristic of the ZrO2 structure observed by Maczka et al.,73 El Boutaybi et al.74 and previously by Hirata et al.75 with doublets at 494 cm−1 and 524 cm−1. The doublets at 452 and 444 cm−1 and the shoulder at 375 cm−1 are less defined for commercial ZrO2 compared to the ones presented in the cited work73 where highly crystalline ZrO2, calcined at 1200 °C, was studied. The band at 348 cm−1 is the most prominent feature of monoclinic ZrO2. The spectrum of the reference sample also contains intense bands at 411, 262 and 230 cm−1.
For the selected synthesized samples with different tetragonal/monoclinic phase ratios, starting with N2-9.5_HX (the as-prepared hydrothermal material), the features of the spectrum are difficult to distinguish. One possible cause of the loss of spectral details may be a strong scattering on this sample with fine crystallite size. According to the XRD results, the crystal structure of sample N2-9.5_HX is characterized as a mixture of tetragonal and monoclinic phases, reaching about 62% of the former and the rest of the latter (Table 2). The spectrum of this hydrothermal sample is poorly resolved, possibly due to the presence of an amorphous component, as well as adsorbed water and surface hydroxyl groups. Also, the more random distribution of molecular vibrations, leading to a less sharp and often flat IR spectrum,76 may indicate the lack of a well-defined lattice.
With the milling of N2-9.5_HX, the effect of tetragonal into monoclinic transformation is visible already after treatment at 300 rpm for 30 min (N3-9.5_HXM_300) and even more intense bands appear for sample N4-9.5_HXM_450 (appearance of features originating from translations and vibration of the ZrO2 molecules). The spectra of these milled samples, containing about 90% of the monoclinic phase, are similar to each other and the bands characteristic for monoclinic ZrO2 are present at 406, 347, 262, and 230 cm−1. The shape of the spectra is strongly reminiscent of that of the commercial sample, suggesting that milling of the N2-9.5_HX hydrothermal sample leads to a more ordered crystal structure with a monoclinic phase.
For further investigations, pH 7 was chosen for ZrO2 precipitation since its lowering should promote the formation of monoclinic ZrO2,21,31,32,34 albeit with a lower specific surface area.17,23,25,31 HTT of the xerogel, precipitated at pH 7 and 260 °C (N10-7_HX) results in the formation of a sample with a monoclinic phase content reaching 96% but the diffractogram of this sample contains very broad peaks (Fig. 3). This indicates that an imperfect crystal structure with a crystallite size of about 4.2 nm is formed. Subsequent milling of this hydrothermal sample at 300 rpm (N11-7_HXM_300_1h_10, N12-7_HXM_300_0.5h_13, and N13-7_HXM_300_1h_13) practically does not change the diffraction patterns of the resulting samples (Fig. 3). Finally, the elevation of HTT to 300 °C (N15-7_HG) allows for obtaining a pure monoclinic phase with an improved structure and crystallite size of about 12.2 nm.
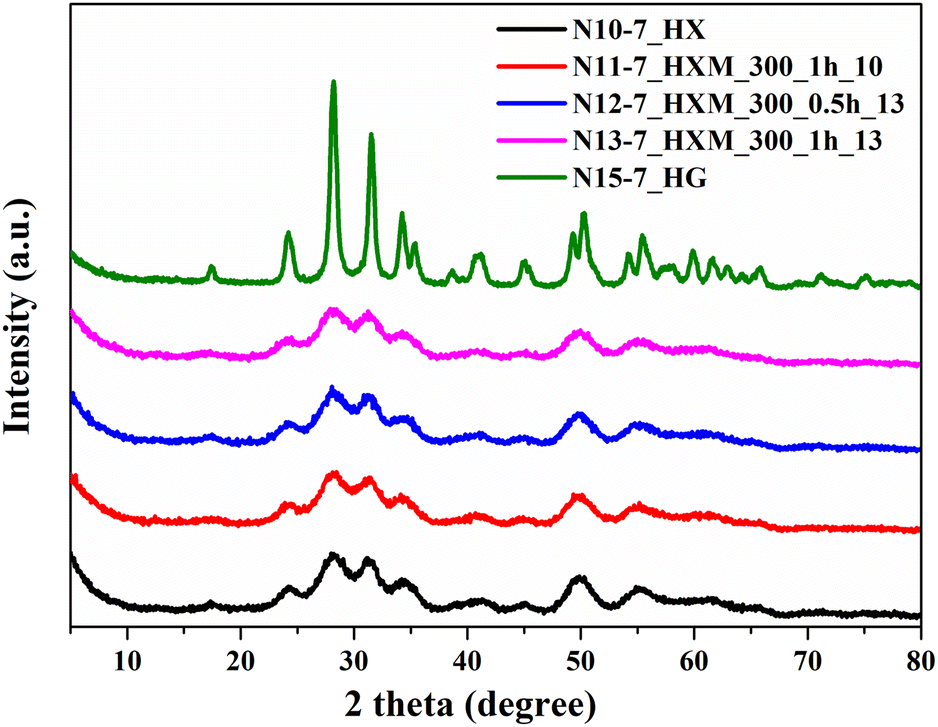 | ||
| Fig. 3 XRD patterns for ZrO2, precipitated at pH 7, after HTT additional MChT under different conditions. | ||
The negligible presence of an amorphous component in the hydrothermal samples can be confirmed by comparing the results of thermogravimetric analysis (DTA-TG) for the initial sample and the sample after HTT at 300 °C (Fig. S2a and b†). It is known that crystallization of the amorphous precipitated ZrO2 is accompanied by an intense exo-effect on the DTA curve at 420–450 °C.26 Indeed, the acquired DTA curve of the initial sample (sample N9-7) contains a sharp exo-effect at 436 °C which corresponds to the crystallization of amorphous ZrO2 (Fig. S2a†). In comparison, this effect is absent on the DTA curve recorded for the sample after HTT at 300 °C (N15-7_HG, Fig. S2b†).
To completely transform the tetragonal phase into a monoclinic phase, HTT at 300 °C was increased to 7 hours (N17-7_H7hG). The diffraction pattern of sample N17-7_H7hG contains only peaks attributed to the monoclinic phase (Fig. 4). The milling of this hydrothermal sample at 400 rpm and BPR = 10 does not cause a change in the phase composition and the diffraction pattern of the milled sample N21-7_H7hGM_400_0.5_10 shows only reflexes of monoclinic ZrO2. Besides, the slight background in its diffraction pattern and calculations indicate the absence of a noticeable amount of amorphous phase. Also interestingly, even harsh milling (500 rpm, BPR 25) of this sample does not lead to broadening of the peaks in the diffraction pattern (N19-7_H7hGM_500_0.5_25 in Fig. 4). This indicates that the crystallite size does not change for this series of samples. The diffractogram shows a low-intensity peak at 2Θ = 30.18°, indicating the presence of a cubic rather than a tetragonal phase. This can be concluded based on the absence of the peak at 43.2° and two peaks in the range of 73–75° by which tetragonal and cubic phases can be distinguished.80 Also, the monoclinic phase can be transformed into a cubic phase during milling, as reported in earlier studies.60,62,66,72,81
 | ||
| Fig. 4 XRD patterns for ZrO2, precipitated at pH 7, after HTT, additional milling and calcination. | ||
It is also important to compare the thermal behavior of the X-ray amorphous initial sample (N9-7) with the samples after HTT and milling. It can be seen that the initial sample after calcination at 500 °C (N14-7_T) contains a predominant monoclinic phase and about 12% of a tetragonal phase (Fig. 5). Calcination of the hydrothermally treated sample (N15_7_HG) at 500 °C results in conservation of the pure monoclinic phase (N16-7_HG_T). The phase composition of the milled sample does not change after its post-calcination at 500 °C. Moreover, the position of the main peak of the monoclinic phase also does not change in all these diffractograms and the values of FWHM for this peak are within 0.55–0.57°.
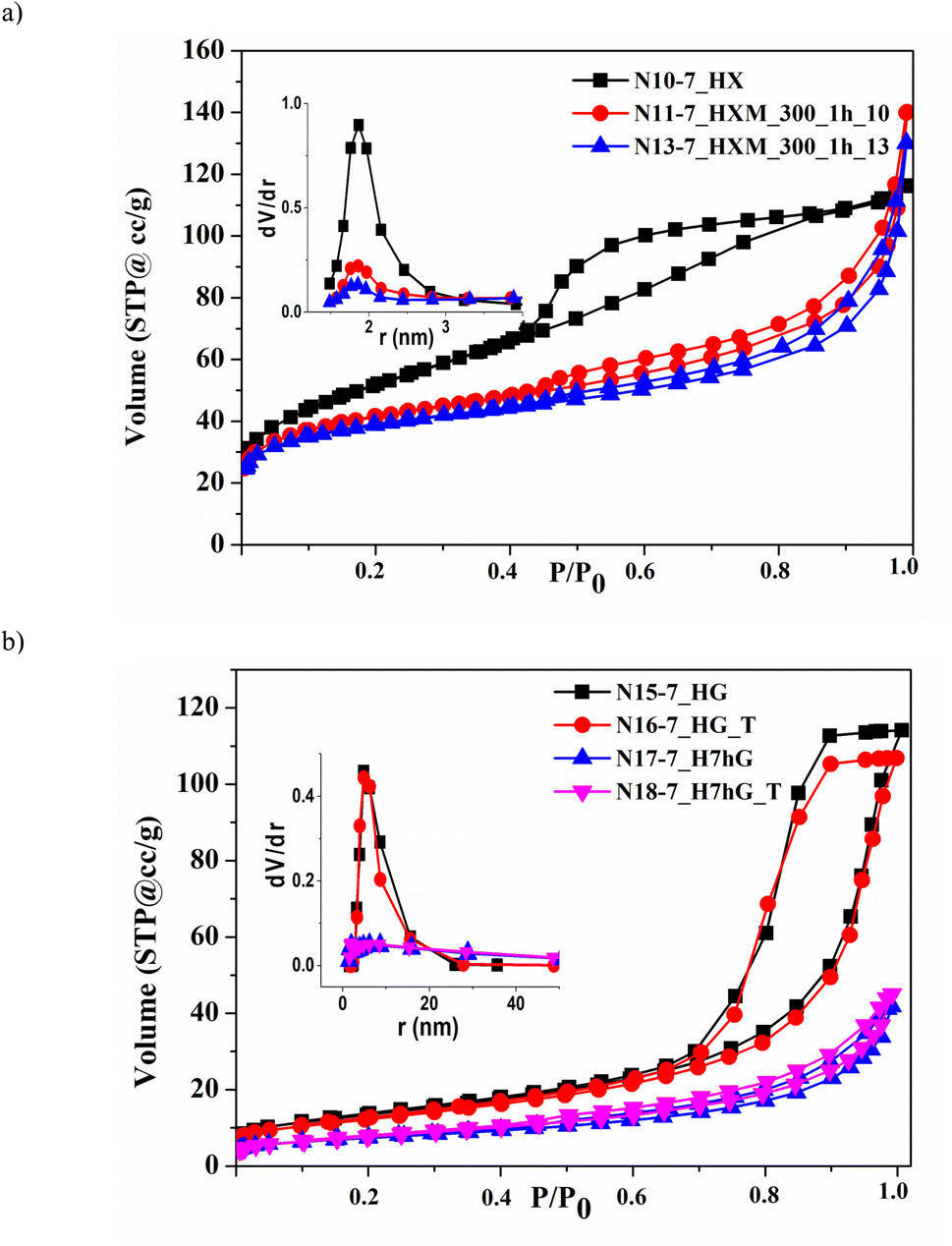 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms and PSD curves (insets) for samples of ZrO2, precipitated at pH 7, after HTT of the xerogel (a) and gel (b). | ||
Increasing the time of HTT from 5 to 7 h (N17-7_H7hG) allowed a material to be obtained that contains only the monoclinic phase. Therefore, for a more precise analysis of the crystal structure of the pure phase samples presented in Fig. 4, lattice parameters and microstructural parameters (average grain size D and microstrains 〈ε〉) were derived by full profile Rietveld refinement, Table 3. It should be noted that the lattice parameters of the sample after HTT at 300 °C are very close to the tabulated data for monoclinic ZrO2 (JCPDS, no. 65-1025).71
| Samples | Lattice parameters | Microstructural parameters | |
|---|---|---|---|
| N17-7_H7hG | a = 5.1484(7) Å | b = 5.2096(8) Å | 〈ε〉 = 0.00208 |
| c = 5.3228(8) Å | β = 99.437(7) o | D = 26.9 nm | |
| V = 140.83(6) Å3 | |||
| N18-7_H7hG_T | a = 5.1488(6) Å | b = 5.1969(8) Å | 〈ε〉 = 0.00228 |
| c = 5.3193(7) Å | β = 99.418(6) o | D = 26.5 nm | |
| V = 140.41(6) Å3 | |||
| N19-7_H7hGM_500_0.5_25 | a = 5.143(2) Å | b = 5.203(2) Å | 〈ε〉 = 0.00318 |
| c = 5.315(2) Å | β = 99.03(1) o | D = 32.7 nm | |
| V = 140.5(2) Å3 | |||
| N20-7_H7hGM_500_0.5_25_T | a = 5.146(1) Å | b = 5.197(2) Å | 〈ε〉 = 0.00325 |
| c = 5.319(2) Å | β = 99.122(9) o | D = 30.1 nm | |
| V = 140.5(1) Å3 | |||
A comparison of the results presented in Table 3 shows that the most detectable decrease in all lattice parameters, monoclinic angle β, and unit cell volume V (by 0.1–0.4%) is observed after milling (samples N19-7_H7hGM_500_0.5_25 and N20-7_H7hGM_500_0.5_25_T). Also, an increase in the concentration of the microstrain value 〈ε〉 by 52% occurs, similarly as reported in the work62 in which 〈ε〉 is approximately 0.00555 after milling commercial monoclinic ZrO2 for 30 min at 500 rpm using a stainless-steel assembly. This indicates the introduction of defects into the crystal structure, which takes place during milling. It is also important that post-calcination of the milled sample (N19-7_H7hGM_500_0.5_25) at 500 °C (as in ref. 60) – sample N20-7_H7hGM_500_0.5_25_T, leads to minimal changes in lattice parameters (less than 0.1%), the microstrain values, and average grain size D. The results provide evidence that the introduced defects are quite stable at this temperature.
3.2 Porous structure of the samples prepared at pH 9.5 and 7
It is known that zirconium dioxide, precipitated from aqueous solutions, is a micro-mesoporous material.26,27 The parameters of the porous structure for all prepared samples are presented in Table 4. It should be noted that only the as-precipitated samples (N1-9.5 and N9-7) contain micropores, the volume of which is given in brackets in Table 4: 0.04 and 0.03 cm3 g−1, respectively. This is 40–50% of the total pore volume (column 3 of Table 4).| Samples | S, m2 g−1 | V, cm3 g−1 | d me, nm |
|---|---|---|---|
| N1-9.5 | 166 | 0.06(0.04) | 3.5 |
| N2-9.5_HX | 61 | 0.20 | 7.9 |
| N3-9.5_HXM_300 | 66 | 0.16 | 6.5 |
| N4-9.5_HXM_450 | 67 | 0.15 | 3.9 |
| N5-9.5_HX | 66 | 0.23 | 9.5 |
| N6-9.5_HXM_300 | 74 | 0.19 | 6.5 |
| N7-9.5_HG | 87 | 0.175 | 6.6 |
| N8-9.5_HGM_300 | 100 | 0.165 | 3.7 |
| N9-7 | 130 | 0.07(0.03) | 2.8 |
| N10-7_HX | 184 | 0.18 | 3.7 |
| N11-7_HXM_300_1h_10 | 149 | 0.215 | 3.7 |
| N12-7_HXM_300_0.5h_13 | 144 | 0.205 | 3.7 |
| N13-7_HXM_300_1h_13 | 139 | 0.20 | 3.7 |
| N14-7_T | 27 | 0.07 | 6.5 |
| N15-7_HG | 60 | 0.17 | 9.7 |
| N16-7_HG_T | 61 | 0.24 | 9.7 |
| N17-7_H7hG | 48 | 0.18 | 9.7 |
| N18-7_H7hG_T | 44 | 0.17 | 9.7 |
| N19-7_H7hGM_500_0.5_25 | 28 | 0.07 | 3.8; 13.0 |
| N20-7_H7hGM_500_0.5_25_T | 28 | 0.07 | 3.8; 13.0 |
HTT of both as-precipitated samples promotes the formation of a developed and uniform mesoporous structure: the volume of mesopores increases to 0.17–0.23 cm3 g−1 and their size is 7.9–9.7 nm, as can be seen for samples N2-9.5_HX, N5-9.5_HX, N7-9.5_HG, N15-7_HG, and N17-7_H7hG (Table 4). For all modified samples, the total pore volume V coincides with the volume of mesopores Vme. This coincidence, as well as the calculations of microporosity using the t-method, indicates the absence of micropores in all modified samples.
One can see that the isotherms recorded for hydrothermal samples of both series contain pronounced capillary-condensation hysteresis which is characteristic of mesoporous materials. This is related to the samples precipitated at pH 9.5 (Fig. S3†). Similar isotherms were obtained for samples precipitated at pH 7 (Fig. 5) which have larger sizes of mesopores – 9.7 nm (Table 4, N15-7_HG and N17-7_H7hG).
The specific surface area of the sample as-precipitated at pH 9.5 (N1-9.5) is higher than that of the as-precipitated sample at pH 7 (N9-7) which is consistent with the results of earlier studies.17,23,25 Its HTT leads to a reduction in specific surface area: all hydrothermal samples modified at 260–320 °C have a specific surface area within 60–87 m2 g−1 (N2-9.5_HX, N5-9.5_HX, and N7-9.5_HG). In contrast, the HTT of the sample, precipitated at pH 7, in the form of a dried xerogel at 260 °C (N10-7_HX), causes an increase in specific surface area to 184 m2 g−1. This is obviously due to the fact that a fine crystalline structure is formed under these conditions, as was shown above according to the XRD data. As a result, the coalescence of these small crystallites leads to the formation of a finely porous structure with a much smaller size of mesopores – 3.7 nm – and, accordingly, a higher specific surface area. On the other hand, an increase in HTT temperature to 300 °C leads to the reduction of specific surface area to 60 m2 g−1 (N15-7_HG).
Milling at low and medium intensity (300–450 rpm, BPR 10–13) causes partial destruction and transformation of the porous structure of all hydrothermally modified samples due to changes in particle morphology and pore shape (N3-9.5_HXM_300, N4-9.5_HXM_450, N6-9.5_HXM_300, N8-9.5_HGM_300, N11-7_HXM_300_1h_10, N12-7_HXM_300_0.5h_13, and N13-7_HXM_300_1h_13). For samples precipitated at pH 9.5, the specific surface area increases slightly after milling and the volume of mesopores, on the contrary, decreases (Table 4, N3-9.5_HXM_300, N4-9.5_HXM_450, N6-9.5_HXM_300, and N8-9.5_HGM_300). Milling of the hydrothermal sample with high specific surface area (precipitation at pH 7 after HTT of the xerogel at 260 °C) is accompanied by its reduction (samples N11-7_HXM_300_1h_10, N12-7_HXM_300_0.5h_13, and N13-7_HXM_300_1h_13). The volume of mesopores for this sample also slightly decreases after milling. But the diameter of the mesopores dme remains unchanged. The change in the shape of nitrogen adsorption–desorption isotherms for milled samples of both series confirms this (Fig. S3† and 5). This can also be seen from the pore volume distribution (PSD) curves by size shown in the inset of Fig. 5a.
Increasing the intensity of milling up to 500 rpm at BPR 25 results in more significant destruction of porosity and a corresponding decrease in specific surface area, volume, and size of mesopores (N19-7_H7hGM_500_0.5_25, Table 4 and Fig. 5b). The PSD curve for this sample contains a second diffuse maximum centered at around 13 nm, which is visible in Fig. S4.† Mesopores with a size greater than 5 nm make a significant contribution to the total pore volume of the milled samples N11-7_HXM_300_1h_10 and N13-7_HXM_300_1h_13, while for the hydrothermal sample N10-7_HX this contribution is minimal. Consequently, the bi-porous structure is formed under milling. The presence of pores with a larger size – 5–50 nm – should contribute to the acceleration of the diffusion of reagents and products in catalytic processes.
It should be noted that the results obtained – multidirectional changes in porosity parameters – coincide with the general trends that are observed during dry milling of porous oxides with different values of specific surface area.50,76,82
As mentioned above, the porous structure of hydrothermal samples has higher thermal stability, i.e. they are sintered to a lesser extent with increasing temperature. This is mainly because they have a larger pore size and a more uniform pore structure. Thus, if the specific surface area of the as-precipitated sample N9-7 after post-calcination at 500 °C decreases almost 5 times and is 27 m2 g−1 (N14-7_T), then it does not change for hydrothermal sample N15-7_HG and has a value of 61 m2 g−1 (N16-7_HG_T). Moreover, the volume of mesopores in this hydrothermal sample is significantly higher after post-calcination. A comparison of samples N19-7_H7hGM_500_0.5_25 and N20-7_H7hGM_500_0.5_25_T also indicates that the sample, successively modified by HTT and milling, does not change the porous structure after post-calcination. The described results are well illustrated in Fig. 5b, which shows corresponding isotherms and PSD curves. As can be seen, the pairs of isotherms and PSD curves for samples before and after post-calcination practically coincide.
The monoclinic ZrO2 phase and the developed uniform mesoporous structure are optimal from the point of view of their application in catalysis. In fact, mesoporous catalysts are the most demanded for many processes because they provide a balance between a good diffusion rate of reactants and a sufficiently high accessible surface, as well as resistance to sintering and coking through the confinement effect.12,15–21
3.3 Morphology
Scanning electron microscopy was used to shed light on the material's morphology undergoing various treatments. Examples of micrographs of hydrothermal, milled and post-calcined samples were acquired for pure phase samples obtained at pH 7 and are shown in Fig. 6. The most pronounced effect can be observed for milling of the sample N10-7_HX (Fig. 6a and b), which results in the diminution of the grains and formation of a loose layer of smaller particles on the top of the bigger ones (N11-7_HXM_300_1h_10, Fig. 6c and d). The following calcination leads to the sintering of those smaller particles yet does not influence the overall size of the grains (N20-7_H7hGM_500_0.5_125_T, Fig. 6e and f). | ||
| Fig. 6 The effect of different treatments on the morphology of ZrO2 precipitated at pH 7, samples N10-7_HX: hydrothermal treatment (a and b), milling (c and d), and post-calcination (e and f). | ||
SEM images of the selected pure monoclinic ZrO2 samples precipitated at pH 7 (Fig. 7a and b) indicate that milling affects their morphology much more than calcination. On the micrometer scale, the diminution of the grains is well visible, when comparing the hydrothermally treated gel (N17-7_H7hG) and its milled counterpart (N19-7_H7hGM_500_0.5_25), see Fig. 7a and e. On the other hand, the changes observable on the nanometer scale show that an initial fine structure of the material is being crushed into agglomerates when milled at 500 rpm (Fig. 7b and f). The post-calcination does not seem to affect the material grains, see the comparison of samples N17-7_H7hG with N18-7_H7hG_T (Fig. 7a and c) and N19-7_H7hGM_500_0.5_25 with N20-7_H7hGM_500_0.5_25_T (Fig. 7e and g).
 | ||
| Fig. 7 The effect on the morphology of the hydrothermal sample N17-7_H7hG (a and b), and its subsequent calcination (c and d), milling (e and f), and post-calcination (g and h). | ||
3.4 Optical properties and photocatalytic activity
In general, zirconium dioxide is a semiconductor with a band gap of 5.0–5.8 eV.36,43,83 Its spectra contain the main band in the UV region, namely at 220–250 nm, which is attributed to the O2− → Zr4+ transition. UV-vis spectra of the samples obtained at pH 7 gathered in Fig. 8 show that a well-crystallized sample after HTT of a gel at 300 °C (N17-7_H7hG) has a band with a maximum of around 227 nm and an absorption edge at 241 nm (Fig. 8a). The absorption edge is determined as the point of the intersection of the tangent with the abscissa axis (solid black line). This corresponds to band gap Eg = 5.10 eV (Tauc plot in Fig. 8b, solid black line).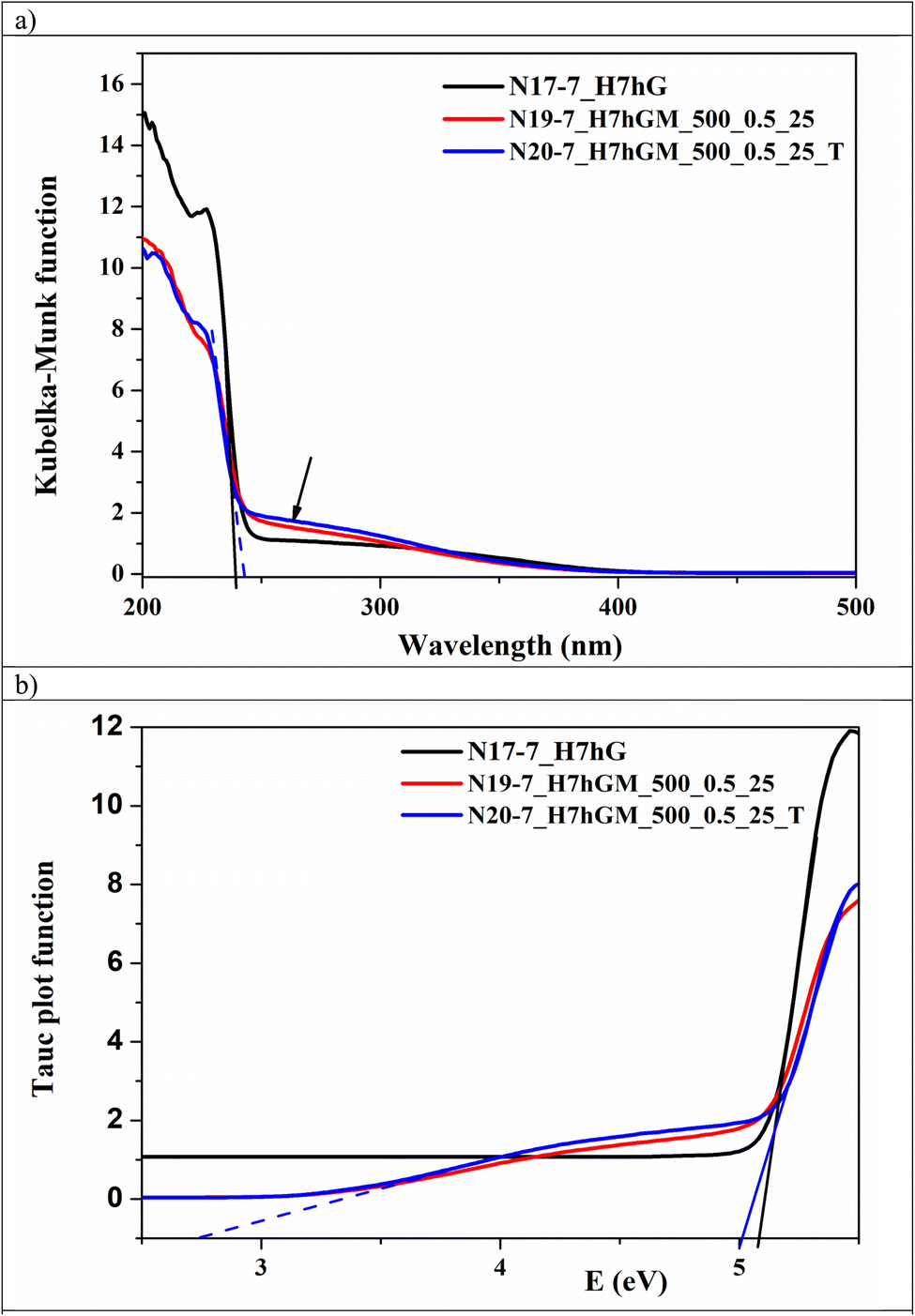 | ||
| Fig. 8 UV-vis spectra (a) and Tauc plots (b) for several ZrO2 samples, precipitated at pH 7. | ||
Milling of this sample at 500 rpm (N19-7_H7hGM_500_0.5_25) and post-calcination of the latter (N20-7_H7hGM_500_0.5_25_T) results in a shift of the absorption edge toward 247 nm (blue dashed line in Fig. 8a) which corresponds to band gap Eg of about 5.02 eV (blue solid line in Fig. 8b). It is noteworthy that these two spectra contain a so-called Urbach tail (marked with an arrow in Fig. 8a), extending to the visible region and characterizing the set of defect states of milled ZrO2.84 As can be seen from Fig. 8a, this is not observed for the hydrothermal sample N17-7_H7hG. The presence of an Urbach tail may be caused by the formation of oxygen vacancies in the ZrO2 structure, which is typical for the dry milling of oxides.85,86 As a result of the extended absorption range, the second band gap at about 2.80 eV is recorded on the Tauc plot for milled and post-calcined samples (Fig. 8b, blue dashed line) which is observed for oxygen vacancy-rich ZrO2.83,87,88 It is important that post-calcination of this milled sample at 500 °C (N20-7_H7hGM_500_0.5_25_T) practically does not lead to a change in the spectra, see Fig. 8a and b. This is consistent with the thermal stability of the crystal structure of milled samples as described above. As can be seen, the second band gap is absent on the Tauc plot for the sample before milling – for sample N17-7_H7hG (Fig. 8b).
Due to its wide band gap, zirconium dioxide is photocatalytically active only under the influence of UV radiation. However, as previously reported in several studies, the introduction of oxygen vacancies into the structure of ZrO2 promotes its acquisition of photocatalytic activity during the decomposition of organic pollutants in visible light.26,87,89 Testing the selected samples with the pure monoclinic phase and high specific surface area under visible radiation was carried out in the process of degradation of a stable model dye molecule – safranin T. The initial sample precipitated at pH 7, as well as the same after HTT at 260 and 300 °C, was predicted to be inactive according to our previous experiments,27 while milling of hydrothermal sample N10-7_HX at 300 rpm and hydrothermal sample N17-7_H7hG at 500 rpm (N11-7_HXM_300_1h_10 and N19-7_H7hGM_500_0.5_25, respectively) activates them as photocatalysts in this process. An example of change in the spectra of an aqueous solution of safranin T vs. time of irradiation for a sample N19-7H7hGM_500_0.5_25 is shown in Fig. S5.†
As can be seen, the intensity of the characteristic band at 520 nm steadily decreases with increasing irradiation time. In parallel, a decrease in the intensity of the bands in the region of 240–280 nm is also observed, which indicates the splitting of the chromophore rings in the safranin molecule.65 As a result, the decolorization of the dye solution reaches 75% for the most active sample N11-7_HXM_300_1h_10 during the applied time window. In addition, the reduction of total organic carbon (TOC) in the solution as a result of the photocatalytic process reached 62%. The degree of TOC reduction is a very important additional signature, in addition to the decolorization of the solution, since it indicates the degree of mineralization of the dye, i.e. the transformation of the organic pollutant into less harmful inorganic substances,65 potentially offering the applications of these new materials in the remedy of notoriously stable water pollutants.
The resulting temporal changes in the optical density of the band at 520 nm were approximated using a pseudo-first-order kinetic equation. The plotted kinetic curves are shown in Fig. S6.† The results fit well with linear fitting (R2 = 0.96–0.99). As mentioned above, the sample subjected to HTT alone was inactive in this process. Milled samples (N11-7_HXM_300_1h_10, N19-7_H7hGM_500_0.5_25), as well as the post-calcined sample (N20-7_H7hGM_500_0.5_25_T), demonstrate fairly high activity. This can be explained by the fact of speculative formation of oxygen vacancies in the ZrO2 structure, which is beneficial for photocatalytic activity, as previously reported.87,89 The observed difference in the activity of tested samples may be caused by their different physicochemical characteristics.59,90,91 Thus, a slightly higher value of the rate constant of safranin degradation kd for a sample milled at 300 rpm (N11-7_HXM_300_1h_10) compared to that milled at 500 rpm (N19-7_H7hGM_500_0.5_25) – reaching 4.6 × 10−5 and 4.2 × 10−5 s−1, respectively – may be associated with a higher specific surface area of the first sample (Table 4). Simultaneously, the lower kd value (3.2 × 10−5 s−1) for the post-calcined sample (N20-7_H7hGM_500_0.5_25_T), despite the same values of the specific surface area and band gap, may be due to dehydroxylation of the surface during calcination. This leads to a decrease in the content of active sites and adsorption of safranin on the surface of the post-calcined sample (N20-7_H7hGM_500_0.5_25_T) compared to the as-milled one (N19-7_H7hGM_500_0.5_25). Since the degree of the dye's photocatalytic breakdown is directly correlated with safranin adsorption, the post-calcined sample's activity is lowered.26,59,90
3.5 Post-treatment and mechanochemical aspects of the preparation
The set of obtained results demonstrates the dependence of the ZrO2 structure (in a broad sense – porosity, crystallinity, and surface design) on the conditions of its precipitation and post-synthetic modification. It is also important that changes in the main structural characteristics due to modification are interrelated. As-precipitated at pH 9.5 and 7 ZrO2 samples (N1-9.5 and N9-7) are mostly X-ray amorphous. But the first of them has a higher specific surface area due to the higher content of micropores. The following HTT contributes to the elimination of microporosity and the development of mesoporosity in both precipitated samples, which is a general tendency of hydrothermal processes.27,35 Differences in the change of the specific surface area may be caused by features of the course of crystallization of both samples under hydrothermal conditions. As mentioned above, crystallization processes during HTT of these samples proceed in different ways. At 260 °C, a mixture of tetragonal and monoclinic phases with a predominance of the first is formed for the sample precipitated at pH 9.5 (N1-9.5), while the HTT product of the sample, precipitated at pH 7 (N9-7), is a pure monoclinic phase (compare Fig. 1 and 3). This difference can be explained in terms of the influence of impurity ions on phase transformation in ZrO2.92 Not only transition metal cations but also alkali metal and ammonium cations have a stabilizing effect on the preservation of the tetragonal phase at low temperatures.93 A sample, precipitated at pH 9.5 (N1-9.5), contains NH4+ cations in ion-exchanged positions which are not completely removed by washing. Due to this, the tetragonal phase is stabilized after HTT at 260 °C (N2-9.5_HX). According to the results of the work,94 the removal of volatile impurities leads to the appearance of shear stresses in the ZrO2 lattice during the desorption of stabilizing species, which can lead to a tetragonal transformation into a monoclinic one. During post-milling of the hydrothermal sample N2-9.5_HX, removal of ammonium impurities is possible due to mechanical action and local temperature increase which takes place under these conditions.52,53 This may be another reason (along with the influence of water) for the rapid transformation of the tetragonal phase into a monoclinic phase.The sample precipitated at pH 7 (N9-7), i.e. near the point of zero charge for ZrO2, absorbs fewer ions, that can stabilize the tetragonal phase, and is more easily washed away from them. Therefore, an almost pure fine-crystalline monoclinic phase is formed already at 260 °C during post-HTT of this sample. The latter factor is the reason for the high specific surface area of this hydrothermally modified sample (Table 4, sample N10-7_HX).
Increasing the HTT temperature and time promotes the complete transformation of the tetragonal phase into a monoclinic phase for both as-precipitated samples, regardless of the conditions of their precipitation. This is also fully consistent with the patterns of hydrothermal transformations in oxides.35 As a result, a temperature of 300 °C and a duration of 5–7 hours are optimal HTT conditions, if several important requirements are taken into account, as confirmed in this work. They are as follows: (i) a well-crystallized monoclinic phase without a tetragonal phase admixture; (ii) a uniformly developed porous structure; (iii) stability of the phase composition and porous structure with increasing temperature of post-calcination. Relatively soft dry milling applied for this series of samples allowed their properties to be influenced in terms of surface structure as shown with the XRD and UV-vis results. Its advantage lies in providing a controlled introduction of defects into the structure while maintaining the monoclinic phase with minimal change in the porosity, which proves mechanochemistry to be a very useful tool for obtaining highly active catalytic materials. By governing the milling intensity one can achieve a spectrum of materials with tuned properties. It was reported in ref. 87 and 88 that oxygen vacancies are responsible for the photocatalytic activity of defected ZrO2. The results obtained on the post-calcined milled samples indicate the stability of the monoclinic phase composition, conserved porous structure, and its defects. This finding is consistent with the conclusion of the authors expressed in studies83,95 about the stabilization effect of the defects on monoclinic ZrO2. Thus, we hypothesize the formation of defects, most likely oxygen vacancies, based on the observed acquisition of photocatalytic activity of ZrO2 with vacancies introduced by milling, and these defects can contribute to the stabilization of the monoclinic phase. The presence of oxygen vacancies is indirectly confirmed by the UV-vis spectra (Fig. 8a) and preservation of the photocatalytic activity of these post-calcined samples (Fig. S5†). Based on this, it can be assumed that thermostable ZrO2, modified in this way, will be promising for application in other catalytic processes, for example in CO2 and CH4 high-temperature conversion.
4. Conclusions
The sequential use of hydrothermal treatment at 260–300 °C and milling at 300–450 rpm as post-synthetic modification strategies makes it possible to vary the physicochemical characteristics of precipitated zirconium dioxide and its following catalytic activity. For both ZrO2 samples – precipitated at pH 9.5 and 7 – elevation of HTT temperature to 300 °C leads to the complete transformation of a mixture of tetragonal/monoclinic phases into pure monoclinic ZrO2 with varying degrees of crystallinity. The milling of these hydrothermally modified samples introduces defects into their structure, as indicated by the results obtained using XRD and UV-vis spectroscopy. Post-milling of low and medium intensity (300–450 rpm, BPR 10–13) allows ZrO2 to be obtained with a sufficiently high specific surface area, volume, and pore size, with defects stable also after post-calcination. More intensive milling (500 rpm, BPR 25) causes significant destruction of the porous structure and partial amorphization of ZrO2. As a result, milled samples acquire photocatalytic activity under visible irradiation.Based on the obtained results, new routes for the synthesis of a thermally stable mesoporous monoclinic ZrO2 support containing stable defects introduced by milling are proposed. This can provide higher material resistance not only against the sintering of the porous structure and indirectly deposited metal particles, but also the reduction of coking during catalytic processes at high temperatures. For in situ characterization under real catalytic conditions, ZrO2 samples with pure phases would facilitate the interpretation of the results and distinguishing particular phase activity. Therefore, the proposed combined synthesis route makes it possible to use the advantages of both hydrothermal and mechanochemical methods in order to prepare pure monoclinic zirconium dioxide with optimal characteristics for application as a catalyst support including in high-temperature catalytic processes.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, V. Sydorchuk and J. E. Olszowka; sample preparation, S. Levytska; milling experiments, O. Kiziun; XRD studies, L. Vasylechko; data acquisition, B. Billinghurst, K. Simkovicova, S. Valtera, and J. E. Olszowka; data curation, K. Simkovicova; visualization, K. Simkovicova and S. Valtera; writing – draft preparation, J. E. Olszowka, V. Sydorchuk, S. Valtera, K. Simkovicova, B. Billinghurst, and S. Vajda. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author V. Sydorchuk acknowledges support from the Risk Fellowships of the Czech Academy of Sciences. The authors J. E. Olszowka, S. Valtera, K. Simkovicova, and S. Vajda acknowledge support from the European Union under Horizon Europe (project 101079142). The funders had no role in the preparation of the article. Part of the research described in this paper was performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.References
- W. Li, et al., ZrO2 support imparts superior activity and stability of Co catalysts for CO2 methanation, Appl. Catal., B, 2018, 220, 397–408 CrossRef CAS.
- T. Yamaguchi, Application of ZrO2 as a Catalyst and a Catalyst Support, Catal. Today, 1994, 20(2), 199–218 CrossRef CAS.
- N. Scotti, et al., Copper-Zirconia Catalysts: Powerful Multifunctional Catalytic Tools to Approach Sustainable Processes, Catalysts, 2020, 10(2), 168 CrossRef CAS.
- A. Ibrahim, et al., Dry Reforming of Methane Using Ni Catalyst Supported on ZrO2: The Effect of Different Sources of Zirconia, Catalysts, 2021, 11(7), 827 CrossRef CAS.
- I. Fatimah, et al., ZrO2-based catalysts for biodiesel production: A review, Inorg. Chem. Commun., 2022, 143, 109808 CrossRef CAS.
- Z. Li, et al., Role of the Cu-ZrO2 interface in the hydrogenation of levulinic acid to γ-valerolactone, J. Energy Chem., 2021, 61, 446–458 CrossRef CAS.
- B. Peng, et al., Manipulating Catalytic Pathways: Deoxygenation of Palmitic Acid on Multifunctional Catalysts, Chem.–Eur. J., 2013, 19(15), 4732–4741 CrossRef CAS PubMed.
- X. Jia, et al., Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- S. Azeem, et al., Dry Reforming of Methane with Mesoporous Ni/ZrO2 Catalyst, Int. J. Chem. Eng., 2022, 2022, 139696 Search PubMed.
- D. M. Ozkan,
et al., A DFT study on the role of oxygen vacancy on m-ZrO2 (
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11) in adsorption and dissociation of CO2, Surf. Sci., 2023, 736, 122336 CrossRef CAS.
11) in adsorption and dissociation of CO2, Surf. Sci., 2023, 736, 122336 CrossRef CAS. - N. H. M. Dostagir, et al., Co Single Atoms in ZrO2with Inherent Oxygen Vacancies for Selective Hydrogenation of CO2to CO, ACS Catal., 2021, 11(15), 9450–9461 CrossRef CAS.
- W. J. Jang, et al., A review on dry reforming of methane in aspect of catalytic properties, Catal. Today, 2019, 324, 15–26 CrossRef CAS.
- Y. W. Budhi, et al., Enhancing the catalytic performance and coke reduction using low-cost Ni-based promoted catalyst for hydrogen production, J. Ind. Eng. Chem., 2023, 128, 487–494 CrossRef CAS.
- C. Gionco, et al., Paramagnetic Defects in Polycrystalline Zirconia: An EPR and DFT Study, Chem. Mater., 2013, 25(11), 2243–2253 CrossRef CAS.
- S. Petrovic, et al., The effect of sintering temperature on mesoporous structure of WO3 doped TiO2 powders, Sci. Sintering, 2018, 50(1), 123–132 CrossRef CAS.
- O. El Samrout, et al., Investigation of new routes for the preparation of mesoporous calcium oxide supported nickel materials used as catalysts for the methane dry reforming reaction, Catal. Sci. Technol., 2020, 10(20), 6910–6922 RSC.
- P. Afanasiev, et al., Control of the textural properties of zirconium oxide, Top. Catal., 1999, 8(3–4), 147–160 CrossRef CAS.
- B. P. Bastakoti, et al., Metal-incorporated mesoporous oxides: Synthesis and applications, J. Hazard. Mater., 2021, 401, 123348 CrossRef CAS.
- X. Gao, et al., Anti-Coking and Anti-Sintering Ni/Al2O3 Catalysts in the Dry Reforming of Methane: Recent Progress and Prospects, Catalysts, 2021, 11(8), 1003 CrossRef CAS.
- Y. Dai, et al., The physical chemistry and materials science behind sinter-resistant catalysts, Chem. Soc. Rev., 2018, 47(12), 4314–4331 RSC.
- A. L. A. Marinho, et al., Embedded Ni nanoparticles in CeZrO2 as stable catalyst for dry reforming of methane, Appl. Catal., B, 2020, 268, 118387 CrossRef CAS.
- C. Normair, P. Goulding and I. McAlpine, Role of anions in the surface area stabilisation of zirconia, Catal. Today, 1994, 20(2), 313–321 CrossRef.
- G. K. Chuah, et al., The influence of preparation conditions on the surface area of zirconia, Appl. Catal., A, 1996, 145(1–2), 267–284 CrossRef CAS.
- G. K. Chuah, S. Jaenicke and B. K. Pong, The preparation of high-surface-area zirconia - II. Influence of precipitating agent and digestion on the morphology and microstructure of hydrous zirconia, J. Catal., 1998, 175(1), 80–92 CrossRef CAS.
- V. Y. Gavrilov, Study of zirconia microporous structure, Kinet. Catal., 2000, 41(5), 715–719 CrossRef CAS.
- K. Kucio, et al., Mechanochemical and microwave treatment of precipitated zirconium dioxide and study of its physical-chemical, thermal and photocatalytic properties, J. Therm. Anal. Calorim., 2022, 147(1), 253–262 CrossRef CAS.
- M. Skovgaard, et al., Nanoporous zirconia microspheres prepared by salt-assisted spray drying, SN Appl. Sci., 2020, 2(5), 784 CrossRef CAS.
- M. Haneda, et al., Synthesis of ordered porous zirconia containing sulfate ions and evaluation of its surface acidic properties, J. Mater. Sci., 2017, 52(10), 5835–5845 CrossRef CAS.
- G. I. Spijksma, et al., Microporous Zirconia–Titania Composite Membranes Derived from Diethanolamine-Modified Precursors, Adv. Mater., 2006, 18(16), 2165–2168 CrossRef CAS.
- N. Mamana, et al., Influence of the synthesis process on the features of Y-stabilized ZrO powders obtained by the sol-gel method, Ceram. Int., 2014, 40(5), 6421–6426 CrossRef CAS.
- Y. Boyjoo, et al., Synthesis and applications of porous non-silica metal oxide submicrospheres, Chem. Soc. Rev., 2016, 45(21), 6013–6047 RSC.
- G. Yang and S.-J. Park, Conventional and microwave hydrothermal synthesis and application of functional materials: A review, Materials, 2019, 12(7), 1177 CrossRef CAS PubMed.
- R. I. Walton, Perovskite Oxides Prepared by Hydrothermal and Solvothermal Synthesis: A Review of Crystallisation, Chemistry, and Compositions, Chemistry, 2020, 26(42), 9041–9069 CrossRef CAS.
- R. Leboda, B. Charmas and V. Sidorchuk, Physicochemical and technological aspects of the hydrothermal modification of complex sorbents and catalysts. Part I. Modification of porous and crystalline structures, Adsorpt. Sci. Technol., 1997, 15(3), 189–214 CrossRef CAS.
- D. K. Qin and H. L. Chen, Transformation of ZrO crystal structure under hydrothermal conditions, Mater. Sci. Technol., 2007, 23(1), 123–126 CrossRef CAS.
- L. Kumari, et al., Controlled Hydrothermal Synthesis of Zirconium Oxide Nanostructures and Their Optical Properties, Cryst. Growth Des., 2009, 9(9), 3874–3880 CrossRef CAS.
- D. Padovini, et al., Facile synthesis and characterization of ZrO 2 nanoparticles prepared by the AOP/hydrothermal route, RSC Adv., 2014, 4(73), 38484–38490 RSC.
- A. Chen, et al., Assembly of monoclinic ZrO 2 nanorods: formation mechanism and crystal phase control, CrystEngComm, 2016, 18(4), 580–587 RSC.
- S. Sagadevan, J. Podder and I. Das, Hydrothermal synthesis of zirconium oxide nanoparticles and its characterization, J. Mater. Sci.:Mater. Electron., 2016, 27(6), 5622–5627 CrossRef CAS.
- S. Ramesh, K. Y. S. Lee and C. Y. Tan, A review on the hydrothermal ageing behaviour of Y-TZP ceramics, Ceram. Int., 2018, 44(17), 20620–20634 CrossRef CAS.
- C. Yang, et al., Modified hydrothermal treatment route for high-yield preparation of nanosized ZrO, Ceram. Int., 2020, 46(12), 19807–19814 CrossRef CAS.
- Z. Shu, X. Jiao and D. Chen, Hydrothermal synthesis and selective photocatalytic properties of tetragonal star-like ZrO 2 nanostructures, CrystEngComm, 2013, 15(21), 4288–4294 RSC.
- K. G. Kanade, et al., Synthesis and characterization of nanocrystallined zirconia by hydrothermal method, Mater. Res. Bull., 2008, 43(3), 723–729 CrossRef CAS.
- Z. Wang, et al., Hydrothermal synthesis and humidity sensing properties of size-controlled Zirconium Oxide (ZrO2) nanorods, J. Colloid Interface Sci., 2013, 396, 9–15 CrossRef CAS PubMed.
- S. Khalameida, et al., Hydrothermal, microwave and mechanochemical modification of amorphous zirconium phosphate structure, J. Therm. Anal. Calorim., 2017, 128(2), 795–806 CrossRef CAS.
- P. Balaz, et al., Hallmarks of mechanochemistry: from nanoparticles to technology, Chem. Soc. Rev., 2013, 42(18), 7571–7637 RSC.
- K. Kucio, et al., Synthesis and modification of Ce-Zr oxide compositions as photocatalysts, Appl. Catal., A, 2020, 603, 117767 CrossRef CAS.
- E. Prates da Costa, et al., Effects of Hydrothermal Treatment on Mesopore Structure and Connectivity in Doped Ceria-Zirconia Mixed Oxides, Langmuir, 2023, 39(1), 177–191 CrossRef CAS PubMed.
- V. Sepelak, S. Begin-Colin and G. Le Caer, Transformations in oxides induced by high-energy ball-milling, Dalton Trans., 2012, 41(39), 11927–11948 RSC.
- B. Szczęśniak, J. Choma and M. Jaroniec, Recent advances in mechanochemical synthesis of mesoporous metal oxides, Mater. Adv., 2021, 2(8), 2510–2523 RSC.
- A. P. Amrute, et al., Mechanochemical Synthesis of Catalytic Materials, Chemistry, 2021, 27(23), 6819–6847 CrossRef CAS PubMed.
- C. Xu, et al., Mechanochemical synthesis of advanced nanomaterials for catalytic applications, Chem. Commun., 2015, 51(31), 6698–6713 RSC.
- R. Amade, et al., Defect formation during high-energy ball milling in TiO and its relation to the photocatalytic activity, J. Photochem. Photobiol., A, 2009, 207(2–3), 231–235 CrossRef CAS.
- G. Rajender and P. K. Giri, Strain induced phase formation, microstructural evolution and bandgap narrowing in strained TiO2 nanocrystals grown by ball milling, J. Alloys Compd., 2016, 676, 591–600 CrossRef CAS.
- F. Huo, et al., Fabrication of NiO/ZrO2 nanocomposites using ball milling-pyrolysis method, Vacuum, 2021, 191, 110370 CrossRef CAS.
- H. Toraya, M. Yoshimura and S. Somiya, Calibration Curve for Quantitative-Analysis of the Monoclinic-Tetragonal ZrO2 System by X-Ray-Diffraction, J. Am. Ceram. Soc., 1984, 67(6), C119–C121 CrossRef CAS.
- M. R. Gauna, et al., Monoclinic - Tetragonal Zirconia Quantification of Commercial Nanopowder Mixtures by Xrd and Dta, Ceram.-Silik., 2015, 59(4), 318–325 CAS.
- P. Makuła, M. Pacia and W. Macyk, How to Correctly Determine the Band Gap Energy of Modified Semiconductor Photocatalysts Based on UV-vis Spectra, ACS Publications, 2018, pp. 6814–6817 Search PubMed.
- V. K. Gupta, et al., Photochemical degradation of the hazardous dye Safranin-T using TiO2 catalyst, J. Colloid Interface Sci., 2007, 309(2), 464–469 CrossRef CAS.
- M. Gateshki, et al., Atomic-scale structure of nanocrystalline ZrO2 prepared by high-energy ball milling, Phys. Rev. B, 2005, 71(22), 224107.1–224107.9 CrossRef.
- J. Rouquerol, et al., Recommendations for the characterization of porous solids (Technical Report), Pure Appl. Chem., 1994, 66(8), 1739–1758 CrossRef CAS.
- G. Štefanić, S. Musić and A. Gajović, Structural and microstructural changes in monoclinic ZrO2 during the ball-milling with stainless steel assembly, Mater. Res. Bull., 2006, 41(4), 764–777 CrossRef.
- L. Akselrud and Y. Grin, WinCSD: software package for crystallographic calculations (Version 4), J. Appl. Crystallogr., 2014, 47(2), 803–805 CrossRef CAS.
- R. C. Garvie, Phase Analysis in Zirconia Systems, J. Am. Ceram. Soc., 1972, 55(6), 303–305 CrossRef CAS.
- B. H. Davis, Effect of pH on crystal phase of ZrO2 precipitated from solution and calcined at 600 °C, J. Am. Ceram. Soc., 1984, 67(8), C168 CrossRef CAS.
- X. Liu, Z. Wang and X. Bian, Monoclinic to cubic phase transformation of ZrO 2 induced by ball milling, J. Mater. Sci., 2004, 39(7), 2585–2587 CrossRef CAS.
- J. D. Lin and J. G. Duh, The use of X-ray line profile analysis in the tetragonal to monoclinic phase transformation of ball milled, as-sintered and thermally aged zirconia powders, J. Mater. Sci., 1997, 32(18), 4901–4908 CrossRef CAS.
- Y. Murase and E. Kato, Phase Transformation of Zirconia by Ball-Milling, J. Am. Ceram. Soc., 1979, 62(9–10), 527 CrossRef CAS.
- S. Xie, E. Iglesia and A. T. Bell, Water-Assisted Tetragonal-to-Monoclinic Phase Transformation of ZrO2 at Low Temperatures, Chem. Mater., 2000, 12(8), 2442–2447 CrossRef CAS.
- T. D. Isfahani, et al., Mechanochemical synthesis of zirconia nanoparticles: Formation mechanism and phase transformation, Int. J. Refract. Met. Hard Mater., 2012, 31, 21–27 CrossRef CAS.
- JCPDS, No. 65-1025. 2024 06. 02. 2024, available from: https://www.icdd.com.
- M. Zakeri, M. R. Rahimipour and B. J. Abbasi, Synthesis of nanostructure tetragonal ZrO by high energy ball milling, Mater. Technol., 2013, 28(4), 181–186 CrossRef CAS.
- M. Maczka, et al., Spectroscopic studies of dynamically compacted monoclinic ZrO2, J. Phys. Chem. Solids, 1999, 60(12), 1909–1914 CrossRef CAS.
- A. El Boutaybi, R. Cervasio, A. Degezelle, T. Maroutian, J.-B. Brubach and V. Demange, et al., Ferroelectric ZrO2 phases from infrared spectroscopy, J. Mater. Chem. C, 2023, 11(32), 10931–10941 RSC.
- T. Hirata, E. Asari and M. Kitajima, Infrared and Raman Spectroscopic Studies of ZrO2 Polymorphs Doped with Y2O3 or CeO2, J. Solid State Chem., 1994, 110(2), 201–207 CrossRef CAS.
- K. Kucio, et al., The effect of mechanochemical, microwave and hydrothermal modification of precipitated TiO on its physical-chemical and photocatalytic properties, J. Alloys Compd., 2021, 862, 158011 CrossRef CAS.
- G. Y. Guo and Y. L. Chen, A nearly pure monoclinic nanocrystalline zirconia, J. Solid State Chem., 2005, 178(5), 1675–1682 CrossRef CAS.
- R. Srinivasan, et al., Zirconium oxide crystal phase: The role of the p H and time to attain the final p H for precipitation of the hydrous oxide, J. Mater. Res., 1988, 3, 787–797 CrossRef CAS.
- R. Srinivasan and B. H. Davis, Influence of Zirconium Salt Precursors on the Crystal-Structures of Zirconia, Catal. Lett., 1992, 14(2), 165–170 CrossRef CAS.
- R. Srinivasan, et al., Identification of Tetragonal and Cubic Structures of Zirconia Using Synchrotron X-Radiation Source, J. Mater. Res., 1991, 6(6), 1287–1292 CrossRef CAS.
- S. Bid and S. K. Pradhan, Preparation and microstructure characterization of ball-milled ZrO powder by the Rietveld method: monoclinic to cubic phase transformation without any additive, J. Appl. Crystallogr., 2002, 35(5), 517–525 CrossRef CAS.
- V. Sydorchuk, et al., Influence of mechanochemical activation in various media on structure of porous and non-porous silicas, Appl. Surf. Sci., 2010, 257(2), 446–450 CrossRef CAS.
- C. Imparato, et al., Unraveling the Charge State of Oxygen Vacancies in ZrO on the Basis of Synergistic Computational and Experimental Evidence, J. Phys. Chem. C, 2019, 123(18), 11581–11590 CrossRef CAS.
- H. Yaghoubi, et al., Toward a Visible Light-Driven Photocatalyst: The Effect of Midgap-States-Induced Energy Gap of Undoped TiO Nanoparticles, ACS Catal., 2015, 5(1), 327–335 CrossRef CAS.
- J. He, et al., Lattice-Refined Transition-Metal Oxides via Ball Milling for Boosted Catalytic Oxidation Performance, ACS Appl. Mater. Interfaces, 2019, 11(40), 36666–36675 CrossRef CAS.
- Y. Sun, et al., Research progress on the formation, detection methods and application in photocatalytic reduction of CO2 of oxygen vacancy, J. CO2 Util., 2023, 67, 102344 CrossRef CAS.
- J. J. Zhang, et al., Oxygen vacancy-rich mesoporous ZrO with remarkably enhanced visible light photocatalytic performance, Sol. Energy Mater. Sol. Cells, 2018, 182, 113–120 CrossRef CAS.
- A. Sinhamahapatra, et al., Oxygen-Deficient Zirconia (ZrO2-x): A New Material for Solar Light Absorption, Sci. Rep., 2016, 6(1), 27218 CrossRef CAS PubMed.
- Y. F. Gao, et al., Reactant effect on visible-light driven photocatalytic performance of sol-gel derived tetragonal ZrO2 nanoparticles, Mater. Res. Bull., 2017, 93, 264–269 CrossRef CAS.
- M. Saeed, et al., Photocatalysis: an effective tool for photodegradation of dyes-a review, Environ. Sci. Pollut. Res. Int., 2022, 29(1), 293–311 CrossRef CAS PubMed.
- Y. Xu, E. Chen and J. Tang, Photocatalytic methane conversion to high-value chemicals, Carbon Future, 2023, 920004 Search PubMed.
- S. Shukla and S. Seal, Mechanisms of room temperature metastable tetragonal phase stabilisation in zirconia, Int. Mater. Rev., 2005, 50(1), 45–64 CrossRef CAS.
- H. Xie, et al., Synthesis of Na-Stabilized Nonporous t-ZrO2 Supports and Pt/t-ZrO2 Catalysts and Application to Water-Gas-Shift Reaction, ACS Catal., 2012, 3(1), 61–73 CrossRef.
- A. P. Bechepeche, et al., Experimental and theoretical aspects of the stabilization of zirconia, J. Mater. Sci., 1999, 34(11), 2751–2756 CrossRef CAS.
- Y. Kanno, Stability of metastable tetragonal ZrO2 in compound powders and nucleation arguments, J. Mater. Sci., 1990, 25(4), 1987–1990 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00094c |
| This journal is © The Royal Society of Chemistry 2025 |