
Borohydride and halide dual-substituted lithium argyrodites†
Ji-Hoon
Han
ab,
Do Kyung
Kim
c,
Young Joo
Lee
 cd,
Young-Su
Lee
*a,
Kyung-Woo
Yi
b and
Young Whan
Cho
*a
cd,
Young-Su
Lee
*a,
Kyung-Woo
Yi
b and
Young Whan
Cho
*a
aEnergy Materials Research Center, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: oze@kist.re.kr
bDepartment of Materials Science and Engineering, Seoul National University, Seoul, 08826, Republic of Korea
cWestern Seoul Center, Korea Basic Science Institute, Seoul 03759, Republic of Korea
dDepartment of chemistry, Chung-Ang University, Seoul 06974, Republic of Korea
First published on 6th November 2023
Abstract
Solid electrolyte is a crucial component of all-solid-state batteries, with sulphide solid electrolytes such as lithium argyrodite being closest to commercialization due to their high ionic conductivity and formability. In this study, borohydride/halide dual-substituted argyrodite-type electrolytes, Li7−α−βPS6−α−β(BH4)αXβ (X = Cl, Br, I; α + β ≤ 1.8), have been synthesized using a two-step ball-milling method without post-annealing. Among the various compositions, Li5.35PS4.35(BH4)1.15Cl0.5 exhibits the highest ionic conductivity of 16.4 mS cm−1 at 25 °C when cold-pressed, which further improves to 26.1 mS cm−1 after low temperature sintering. The enhanced conductivity can be attributed to the increased number of Li vacancies resulting from increased BH4 and halide occupancy and site disorder. Li symmetric cells with Li5.35PS4.35(BH4)1.15Cl0.5 demonstrate stable Li plating and stripping cycling for over 2,000 hours at 1 mA cm−2, along with a high critical current density of 2.1 mA cm−2. An all-solid-state battery prepared using Li5.35PS4.35(BH4)1.15Cl0.5 as the electrolyte and pure Li as the anode exhibits an initial coulombic efficiency of 86.4%. Although these electrolytes have limited thermal stability, it shows a wide compositional range while maintaining high ionic conductivity.
New conceptsIn this study, we have achieved one of the highest ionic conductivities among argyrodite-type electrolytes reported so far by simultaneously substituting halide and BH4 for sulphur via room temperature mechanochemical reaction without a post-heating process. XRD and solid-state NMR spectroscopy suggest that this high ionic conductivity is due to disordered occupancy of BH4 and halide at the 4a and 4d sites in the argyrodite structure. In previous studies, the amount of substitution of BH4 was very small or only BH4 was substituted alone, but this is the first case in which high conductivity was obtained by simultaneously substituting BH4 and halide, with the total amount of substitution up to 1.8. These electrolytes exhibit a very wide compositional range and high tolerance to second phase impurities while maintaining the high ionic conductivity. It also shows good compatibility with Li metal from 0 to 5 V. The high critical current density resulting from the high ionic conductivity, along with the wide composition range and good electrochemical stability, makes these electrolytes very promising for application in all-solid-state lithium rechargeable batteries. |
Introduction
Over the past few decades, research on lithium-ion battery technology has been active due to the environmental crisis caused by global warming. However, the risk of fire and explosion for lithium-ion batteries, which rely on flammable liquid electrolytes, has necessitated the development of solid-state batteries. All-solid-state batteries (ASSBs), in which combustible and volatile liquid electrolytes are replaced with solid electrolytes, are recognized as a good alternative due to their advantages in terms of safety, high energy density, and compact size.1 Furthermore, the solid electrolyte acts as a separator and may prevent cell degradation and short-circuit problems caused by lithium-dendrite growth during charging and discharging.2 Sulphide-type solid electrolytes are particularly attractive for use in ASSBs because of their excellent mechanical properties and easy processability in different forms (glass, glass-ceramic, and crystalline) at low temperatures.3 Various sulphide electrolytes, including the Li-argyrodite Li6PS5X (X = Cl, Br, I), have room-temperature conductivities ranging from 1 to 10 mS cm−1. These conductivities are comparable to, or even surpass, those of commercial organic liquid electrolytes, making sulphide electrolytes promising candidates for practical solid electrolytes.4–7 Li6PS5X has a cubic structure (space group F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m), with halide X− ions and non-bridging S2− ions occupying the 4a and 4d Wyckoff sites.8 The S2−/X− (X = Cl, Br, I) disorder over the 4a and 4d sites significantly affects the ionic conductivity of argyrodites.4,9 In addition, the mixed halide occupancy on these sites by Cl− and Br− produces even higher conductivity (up to 24 mS cm−1, sintered) than mono-substitution by either Cl− or Br−.10 However, Li6PS5I does not exhibit S2−/I− disorder due to the significant ionic-radius difference between S2− and I− and thus exhibits much lower conductivity than Cl−- or Br−-substituted argyrodites.11
3m), with halide X− ions and non-bridging S2− ions occupying the 4a and 4d Wyckoff sites.8 The S2−/X− (X = Cl, Br, I) disorder over the 4a and 4d sites significantly affects the ionic conductivity of argyrodites.4,9 In addition, the mixed halide occupancy on these sites by Cl− and Br− produces even higher conductivity (up to 24 mS cm−1, sintered) than mono-substitution by either Cl− or Br−.10 However, Li6PS5I does not exhibit S2−/I− disorder due to the significant ionic-radius difference between S2− and I− and thus exhibits much lower conductivity than Cl−- or Br−-substituted argyrodites.11
Attempts to substitute S2− with BH4− have been rarely reported and their ionic conductivity was rather low.12–15 Yamauchi et al.14 first reported a glass solid electrolyte with a composition of (100 − x)(0.75Li2S–0.25P2S5)·xLiBH4. In their subsequent studies, they increased the LiBH4 content and obtained a conductivity of 1.8 mS cm−1 at 25 °C.15 The structure was identified as an argyrodite with the chemical formula Li7−xPS6−xXx, where X is BH4−. Dao et al. synthesized an argyrodite in which BH4− anions partially replaced Cl− or I− and obtained a conductivity of 0.4 and 0.76 mS cm−1, respectively.13,16 Wang et al. reported on BH4−-substituted Li6PS5Cl electrolytes with a low conductivity of 0.12 mS cm−1 at room temperature.12 They heated the sample to 550 °C, and it is likely that BH4− did not substitute for X− but decomposed. Furthermore, they ball milled all the precursors together. LiBH4 and P2S5 were reported17 to react with each other and our thermodynamic calculations have indicated that LiBH4 and P2S5 react to form compounds such as H2S, Li2S and B2S3. In fact, when we ball-milled LiBH4 and P2S5 together, the pressure inside the milling container abruptly increased due to the generation of very toxic H2S gas, and a rapid exothermic reaction was also observed.
Sun et al. recently synthesized Li5.91PS4.91(BH4)1.09 and reported a conductivity of 4.8 mS cm−1.18 Very recently, we have synthesized electrolytes by ball milling LiBH4 and Li3PS4 and obtained electrolytes with a high ionic conductivity of up to 11 mS cm−1 at 25 °C.19 We also demonstrated that the changes in conductivity can be attributed to local structural variations caused by adopting different ball milling conditions. Inappropriate ball milling conditions can lead to the formation of non-stoichiometric thiophosphate units such as P2S74− and P2S64−, which negatively impacts the ionic conductivity.19
In the present study, we have synthesized mixed pseudohalide/halide-substituted Li-argyrodite-type electrolytes with a composition of Li7−α−βPS6−α−β(BH4)αXβ (X = Cl, Br, I; 0 ≤ α + β ≤ 1.8) using a two-step ball-milling method without post-annealing. The as-synthesized electrolytes exhibit very high ionic conductivity up to 16.4 mS cm−1 at room temperature. To explain the reason for the high conductivity, the site occupancies of the anionic species were analysed using Rietveld refinement of the XRD data. Solid-state magic angle spinning (MAS) NMR spectroscopy has also been used to analyse the motion of Li ions and the local environments at each crystallographic site. The Li5.35PS4.35(BH4)1.15Cl0.5 electrolyte shows promising electrochemical properties, including high initial coulombic efficiency (ICE), high critical current density (CCD), and good anodic and cathodic stability.
Results and discussion
Synthesis and structural analysis by XRD
As LiBH4 vigorously reacts with P2S5 during milling, Li2S and P2S5 were milled in the first step to form β-Li3PS4, and then LiBH4 and LiX (X = Cl, Br, I) were added in the second step. As shown in a previous study,19 milling with Li2S and P2S5 produces β-Li3PS4, as confirmed by the XRD results in Fig. 1a. Fig. 1a also presents XRD patterns of Li3PS4 + 2LiBH4 and Li3PS4 + 2LiBH4 + 0.5LiX (X = Cl, Br, I). Previous studies have indicated that multiple anionic species in Li-deficient halogen-rich argyrodite diversify the Li chemical environments in terms of the coordination number and shape of the coordination shell.10,20 Therefore, our objective was to incorporate one or two types of X (X = Cl, Br, I), in addition to BH4−, into an argyrodite structure. However, an unknown phase appeared when only LiBH4 was added to β-Li3PS4 or when Br− and I− were added instead of Cl−. Because of this unknown second phase, the conductivity was lower than that of the sample with Cl− (see Tables S1–S5, ESI† and the Correlation between chemical composition and ionic conductivity section for details). For this reason, the present study focused mainly on Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5). If the molar ratio of LiX (X = Cl, Br, I) to Li3PS4 was higher than 0.5, unreacted free lithium halide was detected in the XRD pattern. Therefore, the amount of Cl− was fixed to 0.5 mol. Ionic conductivity data for electrolytes with Br− and/or I− substitutions are included in the ESI† (see Tables S1–S5 for details). In general, the ionic conductivity increased in the following order; I− < Br− < Cl−, when both the LiBH4/Li3PS4 and LiX/Li3PS4 mole ratios are fixed.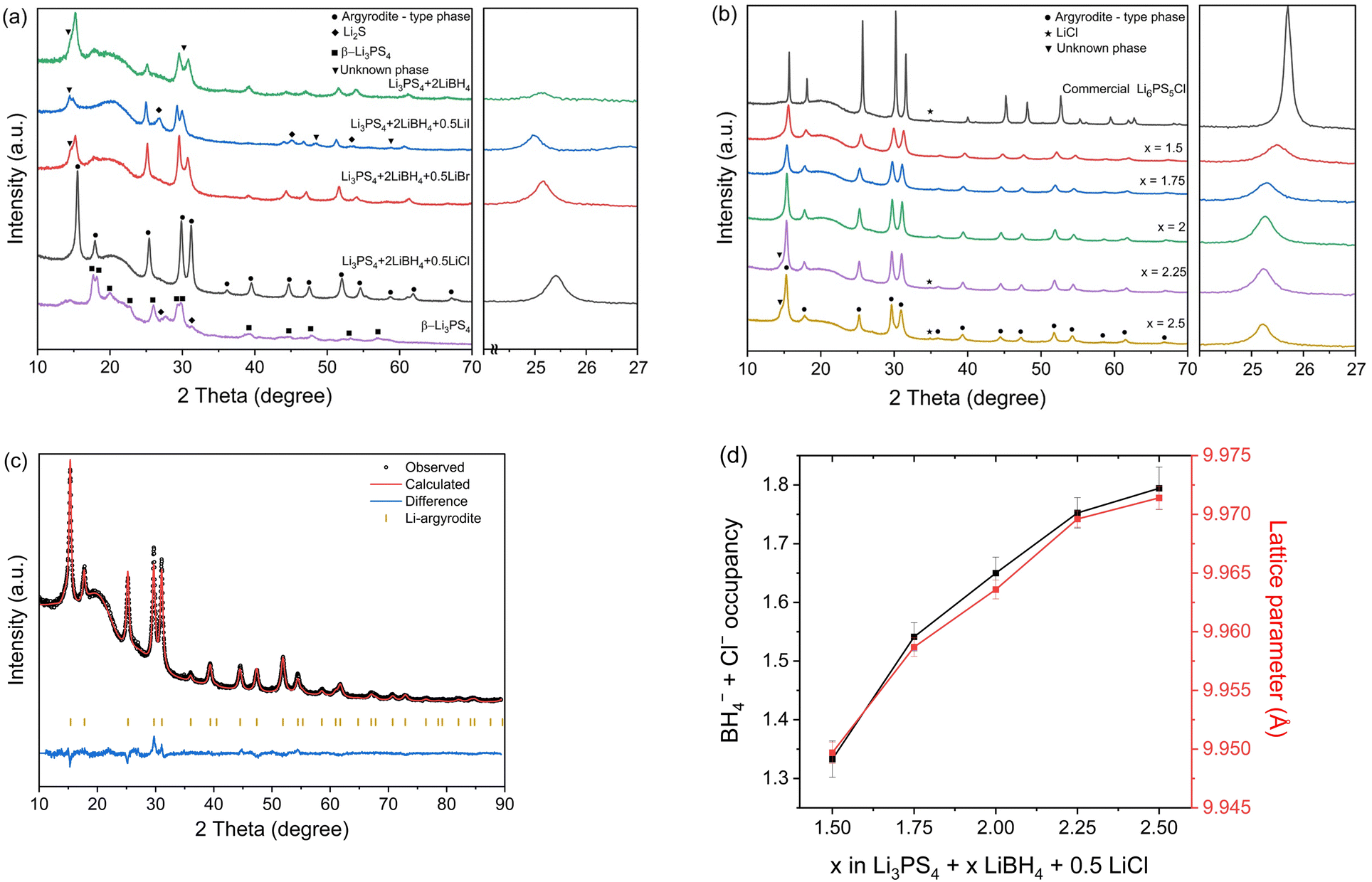 | ||
| Fig. 1 (a) XRD patterns of β-Li3PS4, Li3PS4 + 2LiBH4 + 0.5LiX (X = Cl, Br, I), and Li3PS4 + 2LiBH4, measured at room temperature. When X is a large anion, the peaks are shifted to low angles. (b) XRD patterns of commercial Li6PS5Cl and as-synthesized samples with the composition of Li3PS4+ xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5). (c) Representative Rietveld refinement with XRD data for the as-synthesized Li5.35PS4.35(BH4)1.15Cl0.5. (d) BH4− + Cl− occupancy at 4a and 4d sites and lattice parameter of the argyrodite structure as a function of x in Li3PS4 + xLiBH4 + 0.5LiCl. | ||
Fig. 1b presents XRD patterns of commercial Li6PS5Cl and Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5). Excess LiBH4 is required because LiBH4 is partially decomposed during ball milling. As the content of LiBH4 in the sample increased, the main peak position shifted to a lower angle. This is because the size of the BH4− anion is much larger than that of the Cl− or S2− anion and the lattice parameter increases with an increasing amount of BH4−. Table S6 (ESI†) displays the sizes of the halides and BH4− anions and the volume per formula unit for their respective Li compounds.21 The phase appearing in the XRD pattern was determined to be an argyrodite-type phase, and the diffraction peak shifted to a lower angle with increasing LiBH4, indicating that BH4− occupied 4a and/or 4d sites in the argyrodite structure.15,18,19
The lattice parameters and occupancy of each atomic site of the identified argyrodite-type phase were obtained by Rietveld refinement.22 All the parameters were based on the data of Li6PS5Br, which has similar lattice parameters,23,24 and we introduced a few constraints to extract the occupancies of each ionic species. First, for Li, because of the poor quality of the XRD data, it was not possible to refine the commonly known lithium sites such as T5, T2 in 48h and T5a in 24g.4,23,24 Therefore, all the lithium was assumed to be located at the T5 site for 48h. The results are presented in Table 1 and Tables S8–S11, S13–S17 in the ESI.† The T5 site for 48h, as shown in Table 1, and the corresponding position presented in the ESI† were reported based on the neutron diffraction data.23,24
| Atom | Site | x | y | z | Occupancy |
|---|---|---|---|---|---|
| Space group, F3m; lattice parameter, a = 9.9636(8) Å; Rwp = 3.70%, Rexp = 2.00%, Rp = 2.76%. |
|||||
| Li | 48h (T5) | 0.3139 | 0.0219 | 0.6861 | 0.446(19) |
| P | 4b | 0 | 0 | 0.5 | 1 |
| S | 16e | 0.1189(2) | −0.1189(2) | 0.6189(2) | 1 |
| S | 4d | 0.25 | 0.25 | 0.75 | 0.215(14) |
| B | 4d | 0.25 | 0.25 | 0.75 | 0.535(14) |
| Cl | 4d | 0.25 | 0.25 | 0.75 | 0.25 |
| S | 4a | 0 | 0 | 0 | 0.136(13) |
| B | 4a | 0 | 0 | 0 | 0.614(13) |
| Cl | 4a | 0 | 0 | 0 | 0.25 |
| H | 4d | 0.3135 | 0.1865 | 0.8135 | 0.267(7) |
| H | 4d | 0.3135 | 0.3135 | 0.8135 | 0.267(7) |
| H | 4a | 0.0635 | −0.0635 | 0.0635 | 0.307(6) |
| H | 4a | 0.0635 | 0.0635 | 0.0635 | 0.307(6) |
Again, the quality of the XRD data do not allow us to independently fit the occupancies of the three different anionic species at the 4a and 4d sites. Moreover, the atomic scattering factors of S2− and Cl− are similar. Therefore, we assumed that all 0.5 Cl− occupies 4a and 4d sites evenly (see the ESI† for the details on the assumption).
Applying the aforementioned constraints, we performed the Rietveld refinement and the key features are summarized in Fig. 1c and d. The BH4− occupancy at the 4a and 4d sites and the lattice parameter of the argyrodite phase increase as the amount of LiBH4 increases. The sample with the highest ionic conductivity (x = 2) has BH4− occupancies at the 4a and 4d sites of 0.61 and 0.54, respectively (Table 1). This result indicates that BH4− and Cl− substituted 1.15 (α) and 0.5 (β) of the two non-bridging S2−, respectively. Therefore, the chemical composition of the argyrodite-type phase can be given as Li5.35PS4.35(BH4)1.15Cl0.5.
Structure characterization by solid-state NMR
Solid-state NMR spectroscopy was used to characterize the structures and probe the local environments surrounding Li, P, and B at each crystallographic site. As shown in Fig. 2a, the 7Li MAS NMR spectra of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) exhibit a single main resonance at 0.2–0.3 ppm and very weak signals at 2.3 and −1.1 ppm, which can be attributed to the Li2S and LiCl impurities. These impurity signals are very weak (less than 3% of the total intensity), and no other signals can be assigned to the starting materials or other phases, verifying the formation of a single phase of the argyrodite-type structure. Compared to the structurally analogous Li6PS5Cl and Li7PS6, the 7Li NMR peaks of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) appear at lower frequency with a much larger shift for x = 2.5 than for x = 1.5 and 2.0. A gradual shift toward lower frequency with increasing Cl substitution has been reported and is attributed to the decreased electrostatic interaction between Li ions and the framework resulting from the lower ionic charge of Cl− than S2−.25 The 7Li NMR signal of the samples is extremely narrow, which can result from either high crystallinity or fast dynamics. Because the crystallinity of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) observed by XRD is significantly lower than that of Li6PS5Cl (Fig. 1), the narrow 7Li linewidth indicates that fast Li-ion motion occurs in Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) and that the ionic mobility is in the order of x = 2.0 ≥ x = 1.5 > x = 2.5, as deduced from the linewidth.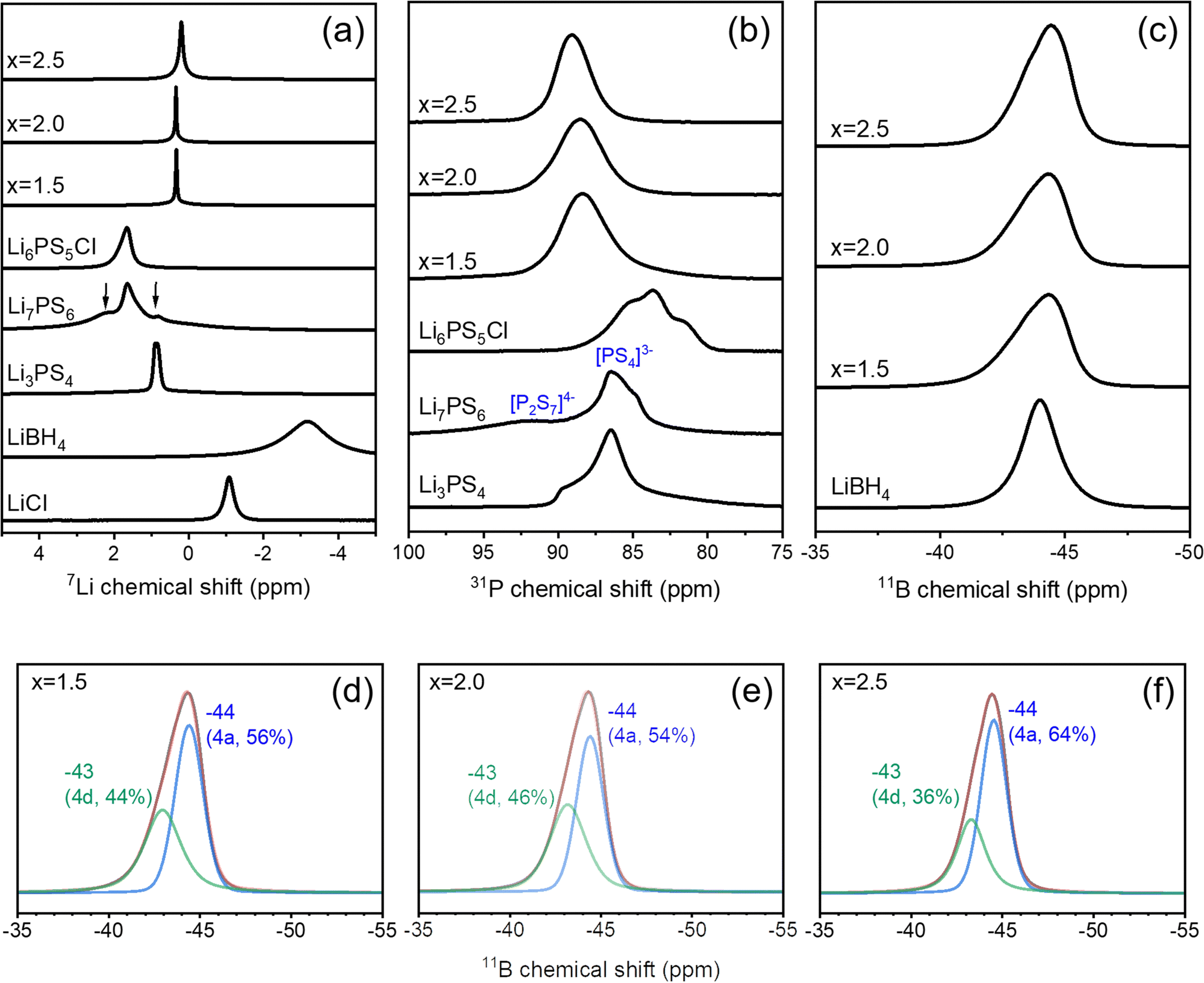 | ||
| Fig. 2 (a)–(c) 7Li, 31P and 11B MAS NMR spectra of argyrodite-type Li3PS4+ xLiBH4 + 0.5LiCl with x = 1.5, 2.0, and 2.5. Spectra for the structural analogous phases of Li6PS5Cl, Li7PS6 and Li3PS4 together with the starting materials LiBH4 and LiCl for the synthesis are plotted for comparison. In the 7Li MAS NMR spectrum of Li7PS6, the signals corresponding to Li2S and Li3PS4 are indicated with arrows. (d)–(f) Deconvolution results of the 11B MAS NMR spectra of Li3PS4+ xLiBH4 + 0.5LiCl with x = 1.5, 2.0, and 2.5. The relative intensity ratio for each peak is denoted in parentheses. | ||
In most halide-substituted argyrodites, the Wyckoff 16e site is solely occupied by S2− forming PS43− tetrahedra, whereas the 4a and 4d sites are occupied by S2− and the halide. It has been reported that ordering, S2− at the 4d site and I− at the 4a site, whereas disordering is favoured for Cl−/Br− substitution.8,26 Interestingly, disordering of anions on 4a/4d sites has been suggested to invoke high conductivity in halide-substituted argyrodites.26 As anion mixing on 4a/4d sites results in variation of the coordination environment of P and 31P MAS NMR is sensitive to the local structure, valuable information about anion disorder can be obtained using 31P MAS NMR. Deiseroth et al. reported that a single sharp 31P signal was observed for ordered I−-substituted argyrodites, whereas broad overlapping signals were detected for disordered Cl−- and Br−-substituted argyrodites.8 Feng et al.6 demonstrated that multiple discrete signals were seen for Li6−xPS5−xCl1+x with varying intensity ratio as a function of Cl− substitution content and they assigned each signal to the local P structure surrounded by (S2−)3(Cl−)1, (S2−)2(Cl−)2, (S2−)1(Cl−)3, and (Cl−)4 in their 2nd coordination shell. 31P MAS NMR spectra of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) display a broad Gaussian-like signal at higher frequency than that of the structural analogues Li6PS5Cl and Li7PS6 (Fig. 2b). The unresolved broad signals suggest that S2−, Cl−, and BH4− are randomly distributed over 4a/4d sites and that PS43− tetrahedra are located at highly disordered local environments, which is consistent with the XRD refinement results (Table 1).
The 11B MAS NMR spectra of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) show a broad dominant signal at a similar frequency to that of LiBH4, indicating that boron is indeed incorporated into the argyrodite structure as BH4. Very weak signals are also present at −2 to 15 ppm and −30 to −20 ppm, which can be assigned to the borates and unknown impurity with no bonded hydrogen, respectively. The amounts of these impurities are negligibly small. This assignment was confirmed by a 11B{1H} cross-polarization experiment (see Fig. S3, ESI†). In contrast to the highly symmetric 11B NMR signal of LiBH4, which is consistent with the highly symmetric environment of BH4− tetrahedra, the 11B NMR signals of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) are asymmetric. There are two possible explanations for the asymmetric line shape. First, the BH4− tetrahedra at 4a/4d sites in argyrodite structures could be distorted, and the effect of the non-zero quadrupolar interaction leads to the line broadening. As 11B (I = 3/2) are quadrupolar nuclei, the characteristic line shape of the central transition influenced by the second-order quadrupolar interaction will appear if boron resides in non-symmetric environments. However, simulation with various quadrupolar coupling constants and asymmetry parameters could not reproduce our spectral pattern. Second, multiple signals resulting from BH4− at different Wyckoff sites could overlap, leading to an asymmetric line shape. This explanation is more plausible considering the Rietveld refinement results, which indicate the distribution of BH4− at both 4a and 4d sites. The 11B NMR signals of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) could be deconvoluted into two components at −43 and −44 ppm with different intensity ratios among samples (Fig. 2d–f). In particular, for the sample with x = 2.0, the intensity ratio of the signals at −43 ppm vs. −44 ppm is 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :54, which is consistent with the occupancy results from the Rietveld refinement (4d site:4a site = 47:53, Table 1). Thus, we tentatively assign the 11B NMR signal at −43 and −44 ppm to BH4− residing at 4d and 4a sites, respectively. Our results are not consistent with previous reports, where two clearly resolved signals for substituted halides were observed, i.e., one sharp and one broad signal. For Cl-substituted argyrodite, a very sharp and a broad 35Cl NMR signal were assigned to 4d and 4a sites, respectively.6 However, Deiseroth et al. reported that a broad 35Cl signal was seen for Cl-substituted argyrodite, whereas a very narrow and a broad 79Br NMR signal were observed for Br-substituted argyrodite.8 These two signals have been assigned to ordered and disordered domains of the material. It is likely that boron in BH4 is dominantly affected by the 1st coordination environment, which is symmetric tetrahedra, and the effect from the 2nd coordination environment is not significant. Thus, boron at 4a and 4d sites gives rise to 11B NMR signals at a similar frequency.
:54, which is consistent with the occupancy results from the Rietveld refinement (4d site:4a site = 47:53, Table 1). Thus, we tentatively assign the 11B NMR signal at −43 and −44 ppm to BH4− residing at 4d and 4a sites, respectively. Our results are not consistent with previous reports, where two clearly resolved signals for substituted halides were observed, i.e., one sharp and one broad signal. For Cl-substituted argyrodite, a very sharp and a broad 35Cl NMR signal were assigned to 4d and 4a sites, respectively.6 However, Deiseroth et al. reported that a broad 35Cl signal was seen for Cl-substituted argyrodite, whereas a very narrow and a broad 79Br NMR signal were observed for Br-substituted argyrodite.8 These two signals have been assigned to ordered and disordered domains of the material. It is likely that boron in BH4 is dominantly affected by the 1st coordination environment, which is symmetric tetrahedra, and the effect from the 2nd coordination environment is not significant. Thus, boron at 4a and 4d sites gives rise to 11B NMR signals at a similar frequency.
Correlation between chemical composition and ionic conductivity
Electrochemical impedance spectroscopy (EIS) was used to measure the ionic conductivity of the cold-pressed Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5). Nyquist plots of the samples shown in Fig. 3a exhibit high ionic conductivities between 10.1 and 16.4 mS cm−1 at room temperature. In particular, the sample with x = 2.0 exhibits a conductivity of 16.4 mS cm−1, which is the highest conductivity, to the best of our knowledge, among argyrodite-type electrolytes reported so far. The EIS spectra were also measured at various temperatures, and the results of Li3PS4 + 2LiBH4 + 0.5LiCl (argyrodite composition of Li5.35PS4.35(BH4)1.15Cl0.5) are shown in Fig. 3b as a representative example. As expected, temperature and resistance are inversely proportional, and very low resistances were measured at high temperatures. All the samples of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) and commercial halide-containing argyrodite Li6PS5Cl show that the temperature dependence of conductivity (σ) can be well described by the Arrhenius equation (Fig. 3c). The activation energy Ea for the Li ion conduction was obtained from the fitting to the following equation: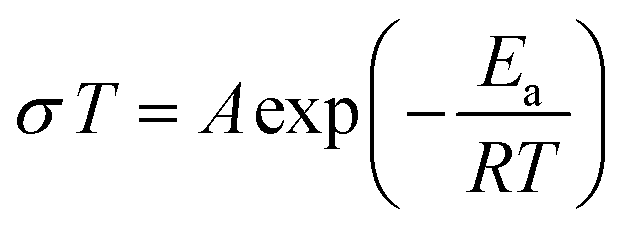 | (1) |
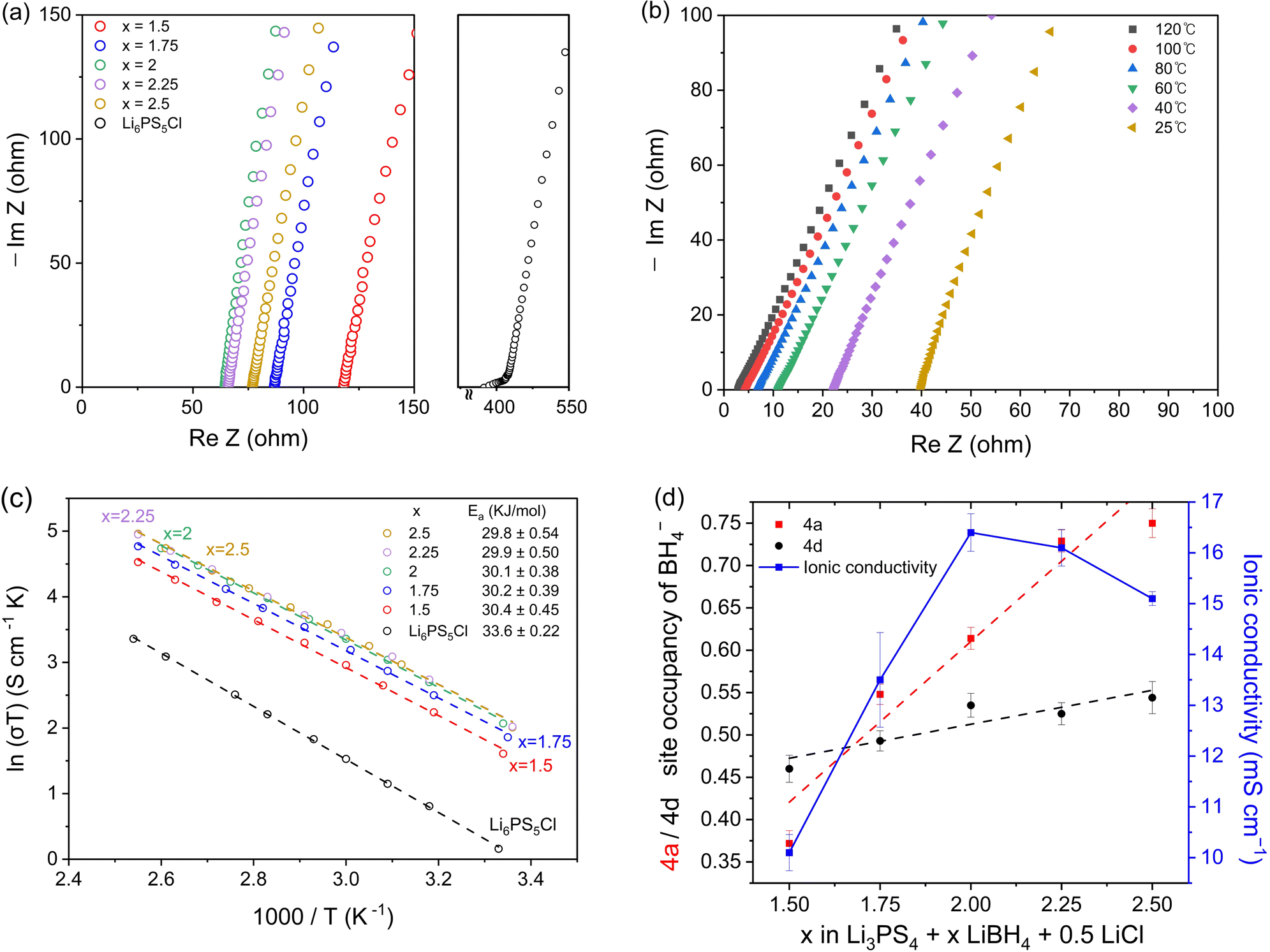 | ||
| Fig. 3 (a) Nyquist plots of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) and commercial Li6PS5Cl. (b) EIS spectra of Li5.35PS4.35(BH4)1.15Cl0.5 at various temperatures. (c) Arrhenius plots and the activation energy of Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5) and commercial Li6PS5Cl. (d) BH4− occupancy of 4a and 4d sites for each electrolyte and its ionic conductivity. The dashed lines are the trend lines of each site. | ||
As the amount of substituted BH4− increases, the conductivity reaches a maximum when x = 2 and starts to decrease when x becomes higher than 2. There are three possible explanations for this behaviour. First, as mentioned earlier, the XRD and solid-state NMR results show that free LiCl was observed when x is higher than 2.25. The same was observed when the molar ratio of LiX (X = Cl, Br, I) to Li3PS4 was 0.75 or higher. As LiX (X = Cl, Br, I) have very low ionic conductivity, their existence would certainly lower the conductivity. However, only a small amount of LiCl remains for Li3PS4 + 2.25LiBH4 + 0.5LiCl (1.4 wt%) and Li3PS4 + 2.5LiBH4 + 0.5LiCl (2.2 wt%). Therefore, the drop in conductivity due to the remaining LiCl is likely to be small.
Second, the decrease in conductivity can be related to the difference in distribution between the 4a and 4d sites of the substituted anion. For Li6PS5Cl and Li6PS5I, it is well known that the difference in conductivity is mainly due to the disorderly distribution of Cl− over the 4a and 4d sites, whereas I− only occupy the 4a site.26–28 According to the Rietveld refinement results in Fig. 3d, as the amount of LiBH4 increases, more BH4− occupy the 4a site, which may cause a drop in conductivity. This result is consistent with the NMR spectra as shown in Fig. 2e and f. Therefore, it is suggested that the difference in distribution of BH4− between the 4a and 4d sites is also responsible for the decrease in conductivity despite the increase in the total amount of substitution.
The third explanation for the decrease in conductivity is that an unknown phase begins to appear in samples containing higher than 2.25 mol of LiBH4. Fig. 1d shows that the amount of total substitution exceeds 1.7 when x is higher than 2.25. As reported in previous studies,29–31 when the total amount of halide substitution exceeds 1.7, the ionic conductivity decreases due to the existence of a secondary phase. In addition, the estimated composition of Li5.35PS4.35(BH4)1.15Cl0.5 indicates that four moles of S2− in β-Li3PS4 is not sufficient to provide the necessary amount of S2−. In fact, the estimated compositions from the Rietveld refinement deviate from the Li3PS4–LiX line in the tentative ternary phase diagram, as illustrated in Fig. S4 (ESI†). Therefore, the second phase has inevitably to form and the existence of this unknown phase is thought to be one of the main causes of the drop in ionic conductivity. Because this unknown phase could not be analysed by XRD or NMR, further study is needed to identify it.
We also compared the conductivity of Li3PS4 + 2LiBH4 (without LiCl) with that of Li3PS4 + 1.5LiBH4 + 0.5LiCl with a similar amount of BH4− + Cl− substitution. For Li3PS4 + 2LiBH4, the BH4− occupancy at the 4a and 4d sites is 0.79 and 0.62, respectively (Table S18, ESI†), giving the composition of Li5.59PS4.59(BH4)1.41. For Li3PS4+ 1.5LiBH4 + 0.5LiCl, the BH4− occupancy of the 4a and 4d sites is 0.37 and 0.46, respectively (Table S8, ESI†), giving the composition of Li5.67PS4.67(BH4)0.83Cl0.5. Although the total amount of substitution is slightly less, the conductivity of Li3PS4+ 1.5LiBH4 + 0.5LiCl is higher (10.1 mS cm−1) than that of Li3PS4 + 2LiBH4 (8.4 mS cm−1). Upon appropriate synthesis, the argyrodite-type electrolytes containing both BH4− and halide anions tend to have higher conductivity than those solely substituted with only one type of X (X = Cl, Br, I, BH4).
In general, Li-argyrodite shows an increasing trend of ionic conductivity with a higher number of substituted halides (see Table S19, ESI†). Nevertheless, the conductivity of cold-pressed Li-argyrodite is generally limited to a maximum of around 9 mS cm−1, even with the maximum halide substitution.6,25,30,32–34 In the case of BH4-substituted argyrodite, when cold-pressed, it exhibits an ionic conductivity of up to 11 mS cm−1 at room temperature. When a halide is simultaneously substituted with BH4−, it reaches a maximum conductivity of 16.4 mS cm−1. In addition, Li5.3PS4.3ClBr0.7 has been reported to have a maximum ionic conductivity of 24 mS cm−1 after high temperature sintering.10 In the present study, Li5.35PS4.35(BH4)1.15Cl0.5 has a higher conductivity of 26.1 mS cm−1 after warm pressing (see Fig. S7, ESI†). Several theoretical studies18,19,35,36 have been conducted to elucidate the possible mechanisms behind the enhanced ionic conductivity of BH4-substituted argyrodite. Fang et al.35 demonstrated through molecular dynamics (MD) simulations and density functional theory (DFT) calculations that BH4− substituted argyrodite can exhibit up to 177 mS cm−1 of ionic conductivity at room temperature. This is possible due to the paddle-wheel effect induced by the rotation of BH4− clusters, promoting the motion of Li ions. It is reported that this paddle-wheel effect also exists in other electrolytes containing PS43− anions.36 Moreover, a recent study has shown that the high ionic conductivity in Na3OBH4, which has an antiperovskite structure with BH4− substitution, is also attributed to the rotation of BH4− clusters.37 Very recently, we have also reported that,19 through DFT calculations, BH4− anions in the argyrodite structure at 4a and 4d sites could exhibit a higher degree of disorder, leading to increased ionic conductivity and that the interaction between Li+ and S2−/PS43− could be weakened, resulting in a more uniform Li+ distribution.
However, Sun et al.18 recently presented results from ab initio molecular dynamics (AIMD) simulations, suggesting that the paddle-wheel effect arising from the BH4− cluster is not correlated with the motion of Li ions in the argyrodite structure. The high conductivity observed in BH4−-substituted argyrodite is attributed to the weak interaction between Li+ and BH4−, resulting in a weaker anchoring effect.18 In summary, while there is strong consensus regarding the positive effect of BH4− substitution on ionic conductivity, a unified explanation of why BH4− substitution enhances conductivity to a greater extent than simple halide substitution has not yet been established.
Thermal stability
To investigate the thermal stability in dry air, differential scanning calorimetry (DSC) was performed, and the results are presented in Fig. S5 (ESI†). The samples did not show any noticeable endothermic or exothermic reaction until the temperature reached 150 °C; however, they began decomposing above 160 °C, indicating that all the samples are thermally stable up to 160 °C in dry air. The intensity of the exothermic reaction increased by increasing the contents of LiBH4. Interestingly, the intensities of the exothermic peaks of our dual substituted electrolytes were much smaller than that of commercial Li6PS5Cl. When DSC was performed under an argon atmosphere, similar exothermic peaks were observed above 160 °C, indicating that the samples simply decompose thermally under any atmosphere as long as it is dry.To investigate the thermal stability of our samples at much higher temperatures, two annealing conditions were applied for Li5.35PS4.35(BH4)1.15Cl0.5: annealing at 200 °C for 4 h followed by 300 °C for 1 h and annealing at 300 °C for 12 h followed by 550 °C for 2 h. The XRD and EIS results are presented in Fig. S6 (ESI†). When the sample was annealed at 300 °C for 1 h, the total resistance increased by about 15 times, and the conductivity was measured to be 1.2 mS cm−1 at room temperature. The XRD data show a notable shift of the peaks towards higher angles, due to a contraction in the lattice parameter of the argyrodite phase in the absence of BH4−. The Rietveld refinement results of the XRD data support this explanation, as the occupancy of BH4− converged toward zero (Tables S21 and S23, ESI†). For the sample annealed at 550 °C for 2 h, its colour turned black, and the conductivity could not be measured. In the XRD pattern, only the peaks corresponding to Li2S and LiCl were detected, indicating that the argyrodite structure was completely destroyed. As mentioned before, LiBH4 decomposes at high temperatures, which may be the reason why the conductivities of some heat-treated samples at high temperatures were quite low.12,16 These results suggest that post-annealing at high temperatures is not suitable for the argyrodites containing BH4−.
As sintering can significantly enhance the ionic conductivity mainly due to the reduction in grain-boundary resistance,38–40 we performed warm pressing for the pellets with the composition Li3PS4 + xLiBH4 + 0.5LiCl (1.5 ≤ x ≤ 2.5). As argyrodite containing BH4− decomposes above 160 °C, we pressed the pellets at 120 °C for 2 h. After cooling to the room temperature, the ionic conductivity was measured again and significantly increased up to 26.1 mS cm−1 (Fig. S7, ESI†). To determine whether compositional or chemical changes occurred during this heat treatment, the conductivity was measured again by cold pressing after finely grinding the warm pressed pellets with a mortar and pestle inside a glove box, and the conductivity returned to the original value before warm pressing. Based on these results, we believe that the main cause of significant increase in ionic conductivity of the warm pressed electrolyte is mainly due to the reduction of grain boundary resistance through the annealing effect, but not from the change in chemistry after thermal treatment.
Electrochemical properties
To measure the anodic electrochemical stability of the electrolyte, cyclic voltammetry (CV) was performed for Li5.35PS4.35(BH4)1.15Cl0.5. Three cycles were performed at 1 mV s−1. As shown in Fig. 4a, a cathode peak was observed at −0.10 V (vs. Li+/Li) due to the deposition of Li (Li+ + e−→ Li). In addition, an anode peak was detected at +0.11 V (vs. Li+/Li), representing the dissolution of Li (Li → Li+ + e−). Up to 5.0 V (vs. Li+/Li), no additional reactions were observed, indicating good electrochemical stability. A Li symmetric cell was also tested to evaluate compatibility with Li metal during extended cycling. At current densities of 0.1 mA cm−2 and 1 mA cm−2, each deposition and dissolution steps lasted for 1 hour, for a total of 100 hours. Throughout this duration, no significant change in overpotential was observed (see Fig. S8, ESI†). Subsequently, a symmetric cell test was conducted at a relatively high current density of 1 mA cm−2 for 2000 hours. As shown in Fig. 4b, after 2000 hours, the overpotential for Li deposition and dissolution slightly changed. This underscores the excellent compatibility of the synthesized electrolyte with Li metal, demonstrating the absence of discernible side reactions or cell failure due to dendrite formation even after long cycling.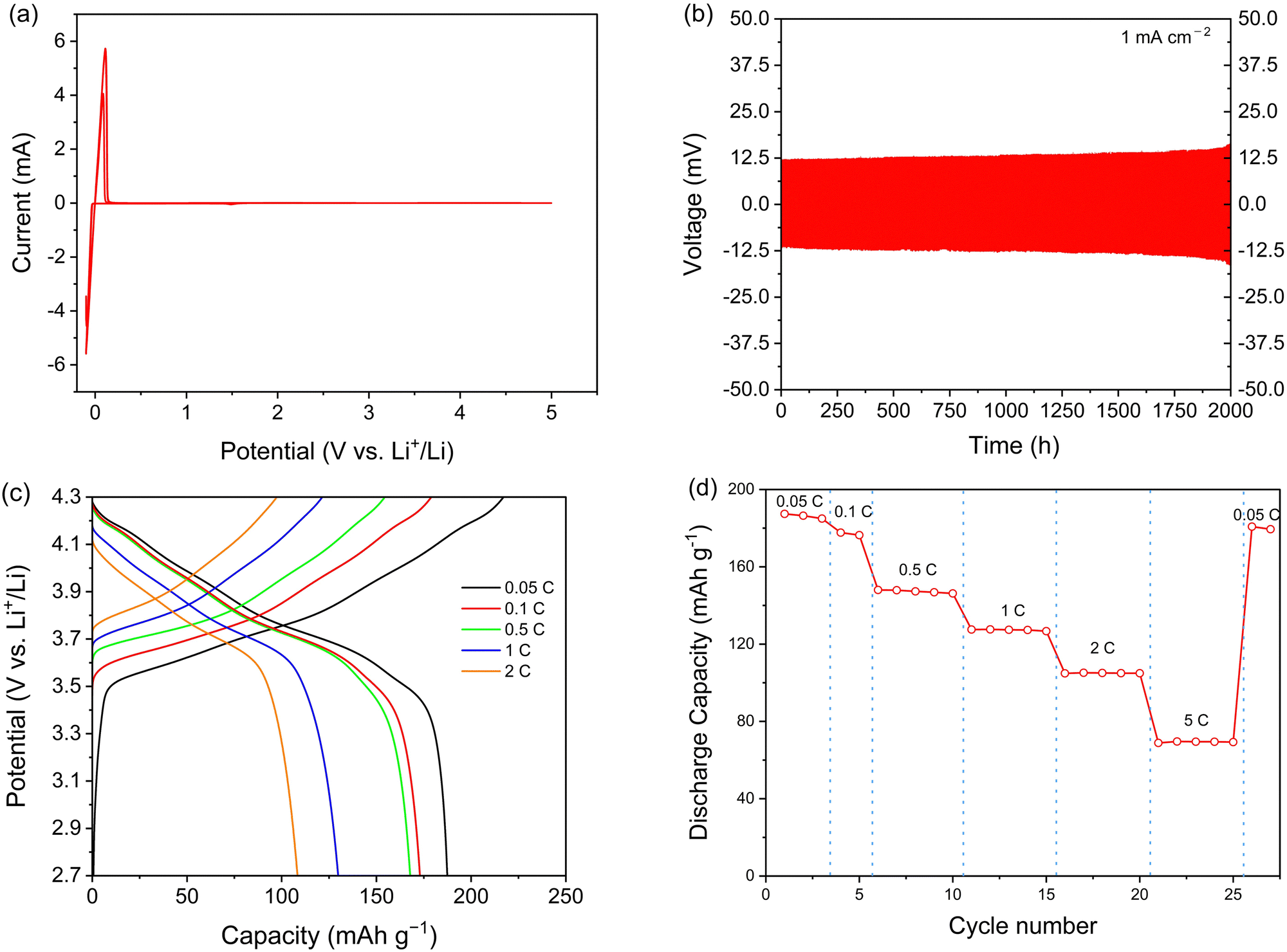 | ||
| Fig. 4 (a) CV curves of the Li/solid electrolyte (Li5.35PS4.35(BH4)1.15Cl0.5)/SS (SUS440C) cell. (b) Voltage profiles of Li metal symmetric cell (Li metal/solid electrolyte (Li5.35PS4.35(BH4)1.15Cl0.5)/Li metal). (c) Voltage profile of the ASSB composed of Li5.35PS4.35(BH4)1.15Cl0.5 solid electrolyte (cathode mixture/solid electrolyte/Li metal). (d) Rate performance of the ASSB composed of Li5.35PS4.35(BH4)1.15Cl0.5 solid electrolyte (cathode mixture/solid electrolyte/Li metal). | ||
Critical current density (CCD) was measured to evaluate the dendrite suppression capability of synthesized electrolyte. CCD represents the maximum current density that a lithium battery can endure through cycling without cell failure or shorting caused by lithium dendrite growth. Electrolytes with an argyrodite structure are generally known to have relatively low (0.1–1 mA cm−2, see Table S24, ESI†) CCD values.41–50 CCD is influenced by various factors, such as temperature, thickness, and stack pressure, leading to variations in its value depending on the measurement conditions.51,52 There are two different CCD testing methods: the time-constant mode, in which the cycling period is kept constant while gradually increasing the areal capacity, and the capacity-constant mode, in which areal capacity is fixed while gradually decreasing the cycling period.53 In this study, both methods were employed for more accurate measurements.
Fig. S9a (ESI†) presents the CCD profile of cold-pressed Li5.35PS4.35(BH4)1.15Cl0.5 measured in the time-constant mode, obtained by setting the fabrication pressure at 356 MPa and the stack pressure of 7 N m using a torque wrench. The sudden voltage drop observed at a current density of 2.1 mA cm−2 indicates a short circuit due to dendrite growth within the solid electrolyte, confirming the high CCD value. On the other hand, the CCD value measured in the capacity constant mode under the same conditions recorded 2.5 mA cm−2, as shown in Fig. S9b (ESI†). It is well-established that materials with elevated ionic conductivity tend to achieve a uniform current density distribution, thereby enhancing dendrite suppression capabilities.51 Consequently, the higher CCD at 2.1/2.5 mA cm−2 aligns with the high ionic conductivity of the electrolyte.
An all-solid-state cell was constructed using Li5.35PS4.35(BH4)1.15Cl0.5 as the electrolyte, a lithium-metal foil anode, and a LiNbO3-coated LiNi0.8Co0.1Mn0.1O2 cathode. The main aim was to investigate the stability between the electrolyte and the cathode. CV measurements were conducted by varying the cut-off voltage from 4.3 up to 4.7 V (vs. Li+/Li), while scanning between 2.6 and a maximum 4.7 V (vs. Li+/Li) at a scanning rate of 0.1 mV s−1. As shown in Fig. S10a (ESI†), all cyclic voltammograms exhibit three distinct pairs of anodic and cathodic peaks. These reversible peaks appear at 3.73, 4.00, and 4.21 V, as well as at 3.71, 3.97, and 4.15 V. These peaks correspond to the phase transitions of NCM 811, a well-known cathode material with transitions from hexagonal to monoclinic (H1 to M), monoclinic to hexagonal (M to H2), and hexagonal to hexagonal (H2 to H3) phases.54–56 Up to a cut-off voltage of 4.5 V, the CV curve demonstrates only slight variations, suggesting that the electrolyte does not induce irreversible reactions or decomposition with the cathode material within this range. However, beyond 4.6 V, undesirable side reactions begin to appear, leading to a shift in the oxidation peak towards higher potentials over a wider voltage range. This indicates a potential promotion of solid electrolyte decomposition, especially at voltages beyond 4.6 V. To check whether these results are primarily due to increased voltage or just a result of cycling, CV measurements were repeated for 10 cycles at a scanning rate of 0.1 mV s−1, with a fixed cut-off voltage of 4.3 V (Fig. S10b, ESI†). Despite undergoing 10 cycles up to 4.3 V, notable changes were limited to a slight increase in overpotential. This result underscores the electrochemical stability of the electrolyte on side reactions at elevated voltages against the LiNbO3-coated LiNi0.8Co0.1Mn0.1O2 cathode.
It is well known that LiBH4 exhibits good chemical compatibility with lithium metal.57 As we confirm that no significant change was observed in the CV measurement, Li metal was used as the anode. The cell was cycled at different C-rates between 2.7 and 4.3 V (versus Li+/Li) at 30 °C. The capacity was calculated based on the mass loading of the cathode active materials. Fig. 4c presents the voltage profiles at various C-rates. At 0.05C, the initial charge and discharge capacities were 217.1 and 187.4 mA h g−1, respectively, and the initial coulombic efficiency was 86.4%. The irreversible capacity is believed to result from contact loss in the charge/discharge cycles and the formation of an interfacial layer between the electrolyte and electrodes. This phenomenon is well-known and has been observed in many ASSB studies.58,59Fig. 4d illustrates the rate performance of the cell. It performed quite well even at 5C (69.5 mA h g−1), indicating that the solid electrolyte functions stably at high C-rates. After 25 cycles, with increasing C-rates up to 5C, the discharge capacity at 0.05C was recovered up to 179.5 mA h g−1, with a capacity retention of 95.8%. This result indicates that the electrolyte performs well even at a high C-rate without decomposition. The cycle performance with the Li–In anode at 0.5C is shown in Fig. S14 (ESI†). The charge and discharge capacity were well maintained up to 100 cycles. The discharge capacity after 100 cycles at 0.5C was 121.46 mA h g−1, 72.6% of the initial discharge capacity.
Conclusion
We synthesized new solid electrolytes by dual substitution of BH4− and halide for non-bridging S2− in a lithium argyrodite structure using a ball-milling method without high-temperature annealing. The as-synthesized electrolytes exhibited a maximum conductivity of 16.4 mS cm−1 at room temperature and 26.1 mS cm−1 after warm pressing at 120 °C. Based on the XRD and solid-state MAS NMR analysis, we confirmed that BH4− was substituted for non-bridging S2− at both 4a and 4d sites and up to 1.8 out of 2 non-bridging S2− in the argyrodite structure were substituted, with the maximum conductivity observed at around 1.7. The ASSB cell using Li5.35PS4.35(BH4)1.15Cl0.5 as an electrolyte and a LiNbO3-coated LiNi0.8Co0.1Mn0.1O2 cathode exhibited an initial discharge capacity of 187.4 mA h g−1 and the ICE was 86.4% at 0.05C. After the 25th cycle under a varying C-rate of up to 5C, the cell maintained 95.8% of the initial discharge capacity. It also had a higher critical current density and electrochemical stability against the lithium metal anode and a LiNbO3-coated NCM cathode. Furthermore, it shows a quite wide compositional range and high tolerance for second phase impurities while maintaining high ionic conductivity. The exact mechanism of how BH4− substitution increases the ionic conductivity needs to be further investigated by neutron powder diffraction to collect quantitative data on Li+ distribution among the well-known three available Li+ sites (T5, T2, and T5a) in the argyrodite structure.Author contributions
Ji-Hoon Han contributed to the data curation, conceptualisation, formal analysis, investigation, validation, visualisation, and writing – original draft. Dokyung Kim contributed to the data curation. Young Joo Lee contributed to the data curation, formal analysis, and writing – original draft (especially for the NMR part). Young-Su Lee contributed to the formal analysis and writing – review & editing. Kyung-Woo Yi contributed to the supervision. Young Whan Cho contributed to the conceptualisation, formal analysis and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Korea Institute of Science and Technology (Grant no. 2E32573), the National Research Foundation grant (No. 2023R1A2C1004094), the Hyundai motor company and the Korea Basic Science Institute grant (C310200 and C210200), funded by the Ministry of Science and ICT of Korea.References
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- H. Huo, Y. Chen, R. Li, N. Zhao, J. Luo, J. G. Pereira da Silva, R. Mücke, P. Kaghazchi, X. Guo and X. Sun, Energy Environ. Sci., 2020, 13, 127–134 RSC.
- J. Wu, S. Liu, F. Han, X. Yao and C. Wang, Adv. Mater., 2021, 33, e2000751 CrossRef.
- M. A. Kraft, S. P. Culver, M. Calderon, F. Bocher, T. Krauskopf, A. Senyshyn, C. Dietrich, A. Zevalkink, J. Janek and W. G. Zeier, J. Am. Chem. Soc., 2017, 139, 10909–10918 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS.
- X. Feng, P.-H. Chien, Y. Wang, S. Patel, P. Wang, H. Liu, M. Immediato-Scuotto and Y.-Y. Hu, Energy Storage Mater., 2020, 30, 67–73 CrossRef.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- H. J. Deiseroth, S. T. Kong, H. Eckert, J. Vannahme, C. Reiner, T. Zaiss and M. Schlosser, Angew. Chem., Int. Ed., 2008, 47, 755–758 CrossRef CAS.
- L. Zhou, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, J. Am. Chem. Soc., 2019, 141, 19002–19013 CrossRef CAS.
- S. V. Patel, S. Banerjee, H. Liu, P. Wang, P.-H. Chien, X. Feng, J. Liu, S. P. Ong and Y.-Y. Hu, Chem. Mater., 2021, 33, 1435–1443 CrossRef CAS.
- I. Hanghofer, M. Brinek, S. L. Eisbacher, B. Bitschnau, M. Volck, V. Hennige, I. Hanzu, D. Rettenwander and H. M. R. Wilkening, Phys. Chem. Chem. Phys., 2019, 21, 8489–8507 RSC.
- H. Wang, L. Gao, Z. Lu, Y. Tang, D. Ye, G. Zhao, H. Zhao and J. Zhang, ACS Appl. Energy Mater., 2021, 4, 12079–12083 CrossRef CAS.
- A. H. Dao, P. Lopez-Aranguren, J. Zhang, F. Cuevas and M. Latroche, Materials, 2020, 13(18), 4028 CrossRef CAS.
- A. Yamauchi, A. Sakuda, A. Hayashi and M. Tatsumisago, J. Power Sources, 2013, 244, 707–710 CrossRef CAS.
- A. Sakuda, A. Yamauchi, S. Yubuchi, N. Kitamura, Y. Idemoto, A. Hayashi and M. Tatsumisago, ACS Omega, 2018, 3, 5453–5458 CrossRef CAS PubMed.
- A. Ha Dao, P. López-Aranguren, R. Černý, O. Guiader, J. Zhang, F. Cuevas, M. Latroche and C. Jordy, Solid State Ionics, 2019, 339, 114987 CrossRef CAS.
- A. El Kharbachi, J. Wind, A. Ruud, A. B. Hogset, M. M. Nygard, J. Zhang, M. H. Sorby, S. Kim, F. Cuevas, S. I. Orimo, M. Fichtner, M. Latroche, H. Fjellvag and B. C. Hauback, Phys. Chem. Chem. Phys., 2020, 22, 13872–13879 RSC.
- Y. Sun, B. Ouyang, Y. Wang, Y. Zhang, S. Sun, Z. Cai, V. Lacivita, Y. Guo and G. Ceder, Matter, 2022, 5, 4379–4395 CrossRef CAS.
- Y. J. Jang, H. Seo, Y. S. Lee, S. Kang, W. Cho, Y. W. Cho and J. H. Kim, Adv. Sci., 2023, 10, e2204942 CrossRef PubMed.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef.
- M. C. Simoes, K. J. Hughes, D. B. Ingham, L. Ma and M. Pourkashanian, Inorg. Chem., 2017, 56, 7566–7573 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- A. Gautam, M. Sadowski, M. Ghidiu, N. Minafra, A. Senyshyn, K. Albe and W. G. Zeier, Adv. Energy Mater., 2020, 11, 2003369 CrossRef.
- N. Minafra, M. A. Kraft, T. Bernges, C. Li, R. Schlem, B. J. Morgan and W. G. Zeier, Inorg. Chem., 2020, 59, 11009–11019 CrossRef CAS PubMed.
- P. Adeli, J. D. Bazak, K. H. Park, I. Kochetkov, A. Huq, G. R. Goward and L. F. Nazar, Angew. Chem., Int. Ed., 2019, 58, 8681–8686 CrossRef CAS.
- P. R. Rayavarapu, N. Sharma, V. K. Peterson and S. Adams, J. Solid State Electrochem., 2011, 16, 1807–1813 CrossRef.
- N. J. J. de Klerk, I. Rosłoń and M. Wagemaker, Chem. Mater., 2016, 28, 7955–7963 CrossRef CAS.
- Y. Liu, H. Peng, H. Su, Y. Zhong, X. Wang, X. Xia, C. Gu and J. Tu, Adv. Mater., 2022, 34, e2107346 CrossRef.
- L. Peng, C. Yu, S. Cheng and J. Xie, Batteries Supercaps, 2023, 6, e202200553 CrossRef CAS.
- W. D. Jung, J. S. Kim, S. Choi, S. Kim, M. Jeon, H. G. Jung, K. Y. Chung, J. H. Lee, B. K. Kim, J. H. Lee and H. Kim, Nano Lett., 2020, 20, 2303–2309 CrossRef CAS PubMed.
- W. D. Jung, J. S. Kim, Y. J. Kim, H. Jeong, D. Han, K. W. Nam, D. Ahn, D. H. Kwon, H. G. Jung, J. H. Lee and H. Kim, Adv. Funct. Mater., 2022, 33, 2211185 CrossRef.
- L. Peng, C. Yu, Z. Zhang, H. Ren, J. Zhang, Z. He, M. Yu, L. Zhang, S. Cheng and J. Xie, Chem. Eng. J., 2022, 430, 132896 CrossRef CAS.
- A. Gautam, M. Ghidiu, E. Suard, M. A. Kraft and W. G. Zeier, ACS Appl. Energy Mater., 2021, 4, 7309–7315 CrossRef CAS.
- S. Wang, A. Gautam, X. Wu, S. Li, X. Zhang, H. He, Y. Lin, Y. Shen and C.-W. Nan, Adv. Energy Sustainability Res., 2023, 2200197 CrossRef.
- H. Fang and P. Jena, Nat. Commun., 2022, 13, 2078 CrossRef CAS PubMed.
- J. G. Smith and D. J. Siegel, Nat. Commun., 2020, 11, 1483 CrossRef CAS PubMed.
- Y. Sun, Y. Wang, X. Liang, Y. Xia, L. Peng, H. Jia, H. Li, L. Bai, J. Feng, H. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 5640–5644 CrossRef CAS.
- M. Cronau, M. Szabo and B. Roling, Mater. Adv., 2021, 2, 7842–7845 RSC.
- Y. Liu, J. Liu, Q. Sun, D. Wang, K. R. Adair, J. Liang, C. Zhang, L. Zhang, S. Lu, H. Huang, X. Song and X. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 27890–27896 CrossRef CAS.
- X. Huang, Y. Lu, Z. Song, K. Rui, Q. Wang, T. Xiu, M. E. Badding and Z. Wen, Energy Storage Mater., 2019, 22, 207–217 CrossRef.
- G. Liu, W. Weng, Z. Zhang, L. Wu, J. Yang and X. Yao, Nano Lett., 2020, 20, 6660–6665 CrossRef CAS PubMed.
- F. Han, J. Yue, X. Zhu and C. Wang, Adv. Energy Mater., 2018, 8, 1703644 CrossRef.
- Y. Subramanian, R. Rajagopal, S. Kang and K.-S. Ryu, J. Alloys Compd., 2022, 925, 166596 CrossRef CAS.
- Z. Zhang, L. Zhang, Y. Liu, X. Yan, B. Xu and L.-M. Wang, J. Alloys Compd., 2020, 812, 152103 CrossRef CAS.
- Y. Subramanian, R. Rajagopal and K.-S. Ryu, J. Alloys Compd., 2023, 940, 168867 CrossRef CAS.
- J. A. Lewis, C. Lee, Y. Liu, S. Y. Han, D. Prakash, E. J. Klein, H. W. Lee and M. T. McDowell, ACS Appl. Mater. Interfaces, 2022, 14, 4051–4060 CrossRef CAS.
- J. Su, M. Pasta, Z. Ning, X. Gao, P. G. Bruce and C. R. M. Grovenor, Energy Environ. Sci., 2022, 15, 3805–3814 RSC.
- Z. Zhang, L. Zhang, X. Yan, H. Wang, Y. Liu, C. Yu, X. Cao, L. van Eijck and B. Wen, J. Power Sources, 2019, 410–411, 162–170 CrossRef CAS.
- C. Wei, C. Yu, R. Wang, L. Peng, S. Chen, X. Miao, S. Cheng and J. Xie, J. Power Sources, 2023, 559, 232659 CrossRef CAS.
- D. Zeng, J. Yao, L. Zhang, R. Xu, S. Wang, X. Yan, C. Yu and L. Wang, Nat. Commun., 2022, 13, 1909 CrossRef CAS PubMed.
- S. Sarkar and V. Thangadurai, ACS Energy Lett., 2022, 7, 1492–1527 CrossRef CAS.
- X. Bai, Y. Duan, W. Zhuang, R. Yang and J. Wang, J. Mater. Chem. A, 2020, 8, 25663–25686 RSC.
- Y. Ruan, Y. Lu, Y. Li, C. Zheng, J. Su, J. Jin, T. Xiu, Z. Song, M. E. Badding and Z. Wen, Adv. Funct. Mater., 2020, 31, 2007815 CrossRef.
- W. Li, J. N. Reimers and J. R. Dahn, Solid State Ionics, 1993, 67, 123–130 CrossRef CAS.
- T. Ohzuku, A. Ueda and M. Nagayama, J. Electrochem. Soc., 1993, 140, 1862–1870 CrossRef CAS.
- H. Arai, S. Okada, H. Ohtsuka, M. Ichimura and J. Yamaki, Solid State Ionics, 1995, 80, 261–269 CrossRef CAS.
- K. Takahashi, K. Hattori, T. Yamazaki, K. Takada, M. Matsuo, S. Orimo, H. Maekawa and H. Takamura, J. Power Sources, 2013, 226, 61–64 CrossRef CAS.
- W. Zhang, D. A. Weber, H. Weigand, T. Arlt, I. Manke, D. Schroder, R. Koerver, T. Leichtweiss, P. Hartmann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 17835–17845 CrossRef CAS.
- Y. Han, S. H. Jung, H. Kwak, S. Jun, H. H. Kwak, J. H. Lee, S. T. Hong and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2100126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01450a |
| This journal is © The Royal Society of Chemistry 2024 |