
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of ketoclomazone and its analogues as inhibitors of 1-deoxy-D-xylulose 5-phosphate synthases and other thiamine diphosphate (ThDP)-dependent enzymes†
Alex H. Y.
Chan‡
 a,
Terence C. S.
Ho‡
a,
Imam
Fathoni
b,
Rawia
Hamid
cd,
Anna K. H.
Hirsch
cd,
Kevin J.
Saliba
b and
Finian J.
Leeper
*a
a,
Terence C. S.
Ho‡
a,
Imam
Fathoni
b,
Rawia
Hamid
cd,
Anna K. H.
Hirsch
cd,
Kevin J.
Saliba
b and
Finian J.
Leeper
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: fjl1@cam.ac.uk
bResearch School of Biology, The Australian National University, Canberra, ACT 2601, Australia
cHelmholtz Institute for Pharmaceutical Research Saarland (HIPS) – Helmholtz Centre for Infection Research (HZI), Campus Building E8.1, 66123 Saarbrücken, Germany
dDepartment of Pharmacy, Saarland University, Campus Building E8.1, 66123 Saarbrücken, Germany
First published on 2nd April 2024
Abstract
Most pathogenic bacteria, apicomplexan parasites and plants rely on the methylerythritol phosphate (MEP) pathway to obtain precursors of isoprenoids. 1-Deoxy-D-xylulose 5-phosphate synthase (DXPS), a thiamine diphosphate (ThDP)-dependent enzyme, catalyses the first and rate-limiting step of the MEP pathway. Due to its absence in humans, DXPS is considered as an attractive target for the development of anti-infectious agents and herbicides. Ketoclomazone is one of the earliest reported inhibitors of DXPS and antibacterial and herbicidal activities have been documented. This study investigated the activity of ketoclomazone on DXPS from various species, as well as the broader ThDP-dependent enzyme family. To gain further insights into the inhibition, we have prepared analogues of ketoclomazone and evaluated their activity in biochemical and computational studies. Our findings support the potential of ketoclomazone as a selective antibacterial agent.
Introduction
Isoprenoids, comprising a vast family of natural products, are key metabolic components of all organisms.1 They are derived from the five-carbon isoprenoid precursors isopentenyl diphosphate (IDP) and dimethylallyl diphosphate (DMADP), which are biosynthesised in nature via two pathways, namely the 2-methylerythritol phosphate (MEP) and the mevalonate pathways.2–5 The precursors of IDP and DMADP are pyruvate and D-glyceraldehyde 3-phosphate (GAP) in the MEP pathway (Fig. 1a), but solely acetyl coenzyme A in the mevalonate pathway. While mammals exclusively use the mevalonate pathway for isoprenoid biosynthesis, most eubacteria, chloroplast-containing plants and apicomplexan parasites (including Plasmodium falciparum) rely on the MEP pathway.3–6 In the first and rate-limiting step of the MEP pathway, pyruvate and GAP are converted into 1-deoxy-D-xylulose 5-phosphate (DXP) and CO2 by DXP synthase (DXPS), a thiamine diphosphate (ThDP)-dependent enzyme.5–22 This makes DXPS an attractive target for the development of anti-infectious agents and herbicides.5,6 Thus, there have been many studies of DXPS7–22 and its inhibition.23–33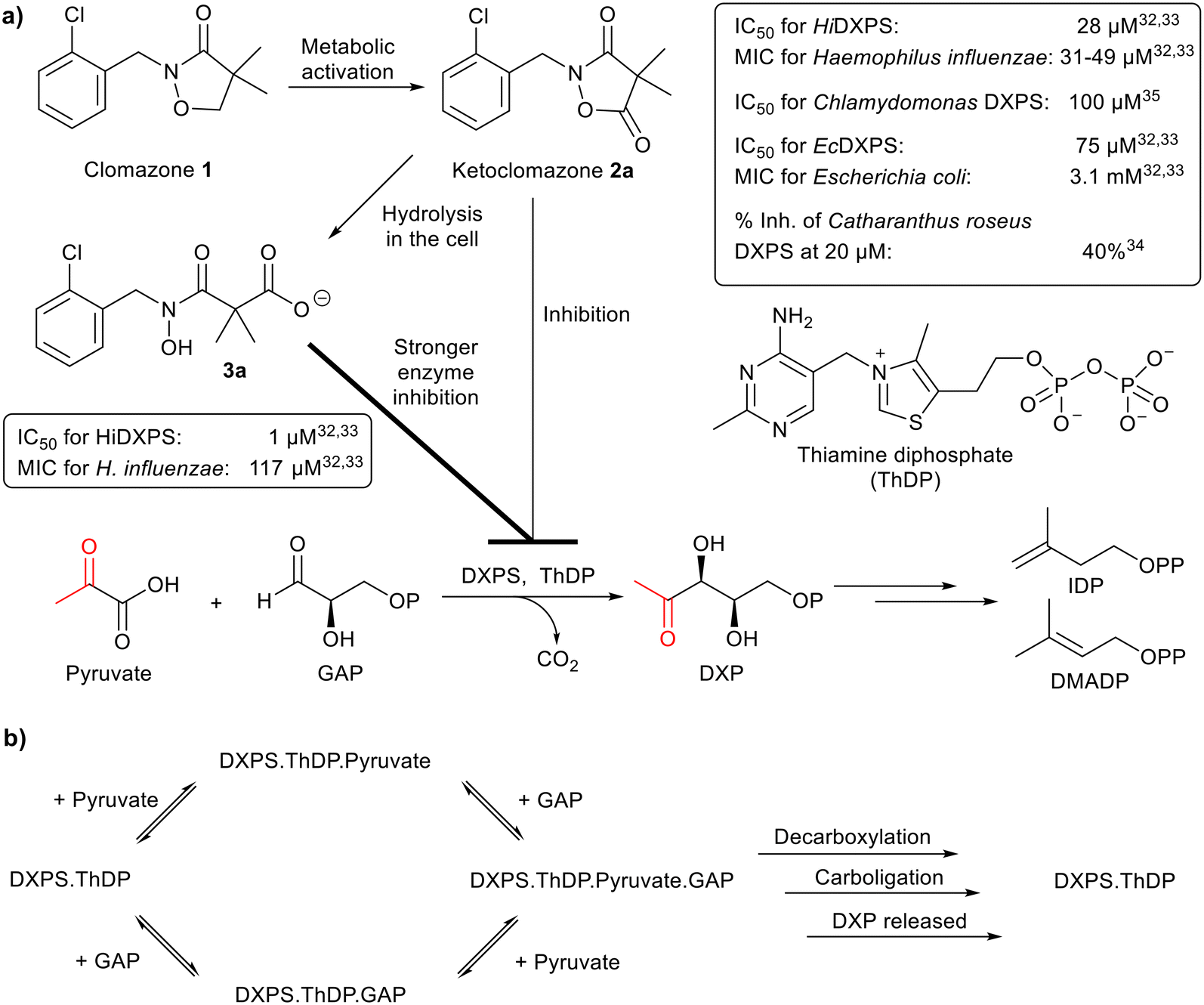 | ||
| Fig. 1 a) Biochemical reaction and inhibition of DXP synthase. DXPS, catalysing the first step of the MEP pathway, is the target of 2a and 3a; their biological activities are summarised in the boxes. MIC, minimum inhibitory concentration. b) Random sequential mechanism of DXPS. | ||
Clomazone (1), a soil-applied herbicide, is effective against grass and broadleaf weeds in many crops.34 Ketoclomazone (2a), a metabolite of 1, is one of the earliest reported DXPS inhibitors.32–34 Clomazone treatment causes bleaching of plant seedlings but to be active, it must be oxidised into 2a. By inhibiting DXPS in the MEP pathway, 2a suppresses isoprenoid biosynthesis in plastids and leads to impaired chloroplast development and pigment loss.5,6,32–34
Further studies showed that 2a and its ring-opened form, carboxylate 3a, suppressed the growth of two pathogenic bacteria, Escherichia coli and Haemophilus influenzae, due to inhibition of DXPS (data summarised in Fig. 1a).32,33 In general, 3a is a stronger inhibitor of DXPS but 2a is more potent in cell-based assays.5,6,32–35 Presumably this is because 2a is more hydrophobic and diffuses better through the membrane and, once inside the cell, 2a is then hydrolysed to 3a for stronger inhibition of DXPS.5 Kinetic studies revealed that the inhibitory action of 2a on HiDXPS and EcDXPS is uncompetitive with respect to pyruvate and mixed type (close to non-competitive) with respect to GAP.32,33 The mode of inhibition by 3a has yet to be determined.
In this study, we studied the activities of 2a and 3a against a range of ThDP-dependent enzymes, DXPS enzymes from four species and four other ThDP-dependent enzymes, namely pyruvate dehydrogenase E1-subunit (PDH E1), pyruvate decarboxylase (PDC), pyruvate oxidase (PO) and 2-oxoglutarate dehydrogenase E1-subunit (OGDH E1). Both 2a and 3a inhibited not only EcDXPS (consistent with the earlier findings32,33) but also PDH E1, and we determined their modes of inhibition. They showed little or no inhibition of the DXPS enzymes from the other bacteria or of the other ThDP-dependent enzymes. To further understand their inhibition, we prepared analogues 2b–i and 3b–i and evaluated them as inhibitors of EcDXPS and PDH E1 in biochemical and computational studies. These results may provide useful insights to facilitate further development of selective inhibitors of DXPS as anti-infectious agents and herbicides.
Results and discussion
Chemical synthesis of compounds 2 and 3 and analogues
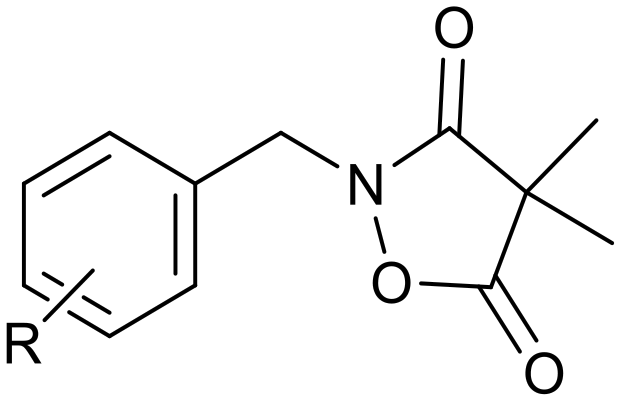
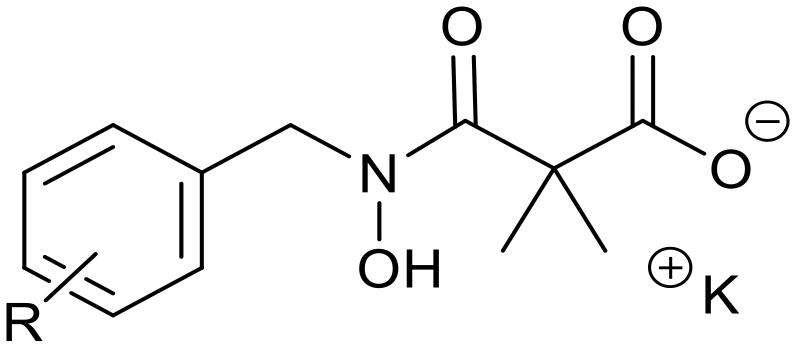
Ketoclomazone 2a and its ring-opened form 3a were synthesised by reported methods.33 As shown in Scheme 1, o-chlorobenzaldehyde was condensed with NH2OH to give oxime 4a, which was reduced with NaBH3CN under acidic conditions to give hydroxylamine 5a. Coupling with dimethylmalonyl dichloride yielded ketoclomazone 2a and hydrolysis under basic conditions afforded the ring-opened form 3a. | ||
| Scheme 1 Chemical synthesis of 2a and 3a and analogues. Identity of the R-groups: a: o-Cl, b: H, c: o-F, d: o-Br, e: p-Cl, f: p-F, g: p-Br, h: o-Me and i: p-tBu. | ||
A decade ago, the Ohkanda group prepared three analogues of 3a as potential antibacterial agents by modifying the acyclic side chain.33 However, all three analogues showed greatly reduced inhibition of DXPS and suppression of the growth of H. influenzae.33 Thus, in this study, we made analogues of 2a and 3a by modifying the aromatic ring. We chose to change the existing o-Cl of 2a and 3a to alternative substituents and we also tested some para-substituted analogues as this is probably the most exposed position, susceptible to metabolic oxidation. Eight analogues of each (2b–i and 3b–i) were synthesised by the synthetic route in Scheme 1; among these, 2c and 2d were described in a patent in 1981.36
Identifying inhibitory activities of compounds 2 and 3 on EcDXPS and porcine PDH E1
Inhibition by 2a and 3a was evaluated on DXPS enzymes from four different species, namely, Klebsiella pneumoniae, Pseudomonas aeruginosa, Deinococcus radiodurans and E. coli. Both were inhibitors of EcDXPS (87–92% inhibition at 1 mM), consistent with earlier reports, but they had low activity on KpDXPS, PaDXPS and DrDXPS (<16% inhibition at 1 mM§) (Table S1†). The selectivity of 2a and 3a as inhibitors of DXPS over other ThDP-dependent enzymes has not yet been reported, so they were also tested against four other ThDP-dependent enzymes: they showed inhibition of porcine PDH E1 (52–71% inhibition at 100 μM) but lacked activity on PDC, PO and OGDH E1 (<5% inhibition at 250 μM).Although apicomplexan parasites (in addition to most bacteria and plants) rely on the MEP pathway for isoprenoid biosynthesis,3–6 only antibacterial and herbicidal activities of ketoclomazone 2a and its ring-opened form 3a have been reported. PfDXPS was not available, so the antiplasmodial activities of 2a and 3a were assessed by determining their effects on in vitro proliferation of the 3D7 strain of P. falciparum. Infected red blood cells were treated with 2a and 3a, and parasite proliferation was measured by performing SYBR-Safe assay, which correlates fluorescence intensity to parasite DNA.37–39 Unfortunately, both compounds showed minimal antiplasmodial activities even at high-micromolar levels (Fig. S1†). Given that fosmidomycin, which targets the second enzyme in the MEP pathway, has potent antimalarial activity,40 this negative result led us to suspect that, as with KpDXPS, PaDXPS and DrDXPS, 2a and 3a may not be good inhibitors of PfDXPS, despite assumptions to the contrary.41
Kinetic studies of EcDXPS inhibition
The Kuzuyama group studied the inhibition of EcDXPS by ketoclomazone 2a in 2010.32 In their assays, conducted in Tris buffer, DXPS seemed to follow a ping-pong bi–bi mechanism (as is normal for ThDP-dependent enzymes42) in which pyruvate reacts with the ThDP-bound DXPS and is decarboxylated and then GAP binds and reacts with the resultant intermediate to yield the DXP product.32KM values of pyruvate and GAP were 48 and 370 μM, respectively. 2a was found to be uncompetitive with respect to pyruvate (with KpyruvateI = 75 μM) and mixed-type with respect to GAP (with KGAPI = 220–460 μM).32 More recent studies, using different buffers, demonstrated that both pyruvate and GAP can bind to the free enzyme (DXPS·ThDP in Fig. 1b), arguing against a strictly ordered mechanism, and a random sequential mechanism has been proposed for EcDXPS8,25 and several other bacterial and protozoan DXPS enzymes, including P. falciparum12 and D. radiodurans.16–18 A buffer-optimisation study conducted by the Freel Meyers group showed that KGAPM values increased with the Tris concentration and they attributed this to the reactivity of the aldehyde group of GAP towards the amino group of Tris.8 Conducting EcDXPS assays in HEPES buffer, they found improved reactivity with KpyruvateM = 49 μM and KGAPM = 24 μM.8 In our work, using the same buffer, we obtained very similar KM values for pyruvate and GAP of 50 and 25 μM, respectively (Fig. S2†). With the more recent findings on the mechanism and the improved assay conditions, we aimed to re-examine the interactions between ketoclomazone (2a) and EcDXPS and extend the kinetic studies to inhibition by 3a and by analogues of both 2a and 3a.Inhibition of EcDXPS by 2a and 3a in HEPES buffer was evaluated in kinetic studies. As Fig. 2 shows, inhibition by 2a is uncompetitive with respect to pyruvate and mixed (approximately non-competitive) with respect to GAP, with KpyruvateI measured to be 84 μM at high [GAP] (250 μM = 10 KM). Our findings on 2a were mostly consistent with those from the Kuzuyama group,32 except our KGAPI at high [pyruvate] (1 mM = 20 KM) was ca. 6 μM compared to their 220–460 μM. This much lower KGAPI value is presumably due to the different assay conditions used (HEPES buffer and higher [pyruvate]) and is consistent with the much lower KGAPM in this buffer. The inhibition by 3a was found to be uncompetitive with respect to both pyruvate and GAP, with KI values of 5 and 9 μM, respectively (Fig. 2). The shift in inhibition modality with respect to GAP from non-competitive (2a) to uncompetitive (3a), though unexpected, is reasonable given their structural differences.
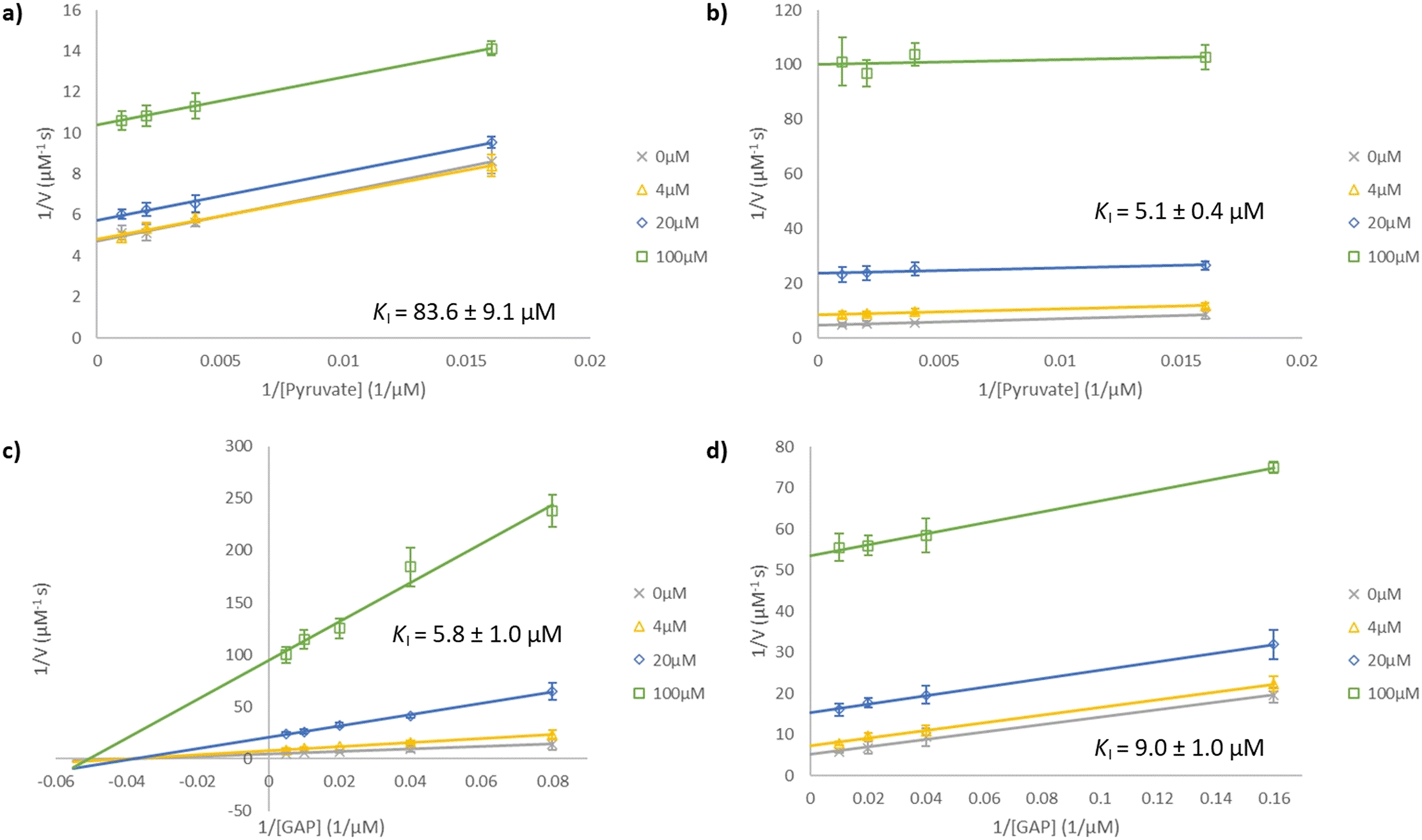 | ||
| Fig. 2 Lineweaver–Burk plots for EcDXPS inhibition. Compounds were assayed at concentrations of 0 μM (×), 4 μM (△), 20 μM (◇) and 100 μM (□). Inhibition by (a) 2a and (b) 3a at a fixed [GAP] of 250 μM (10 KM) under varying [pyruvate] of 62.5, 250, 500 and 1000 μM. Inhibition by (c) 2a and (d) 3a at a fixed [pyruvate] of 1 mM (20 KM) under varying [GAP] of 12.5, 25, 50, 100 and 200 μM and 6.25, 25, 50 and 100 μM respectively. The lines in plot (a), (b) and (d) are essentially parallel, indicating uncompetitive inhibition, while the lines in plot (c) intersect approximately on the x-axis (mixed or non-competitive inhibition). | ||
We then studied how the substitution pattern of the aromatic scaffold affects the inhibition of EcDXPS. The percentage inhibition was measured for compounds at 5 μM under different concentrations of pyruvate and GAP: pyruvate was fixed at either 62.5 μM (= 1.25 KM, shown as [P]) or 1 mM (= 20 KM, shown as [P]) while GAP was fixed at either 50 μM (= 2 KM, shown as [G]) or 250 μM (= 10 KM, shown as [G]) (Table 1). The inhibitory activities of analogues 2b–f increased (from 17–27% to 37–48%) with [pyruvate] but did not change with [GAP], consistent with the mode of inhibition by 2a, i.e. uncompetitive with respect to pyruvate and non-competitive with respect to GAP. The inhibitory activities of analogues 3b–d increased with both [pyruvate] and [GAP], consistent with the modality of inhibition by 3a, i.e. uncompetitive with respect to both pyruvate and GAP. Based on these findings, we speculate that the inhibitory mechanism of ketoclomazone 2a (and 2b–f) is as follows (Fig. 1b): prior to the entry of pyruvate, the inhibitors barely bind to either the DXPS·ThDP free enzyme or the DXPS·ThDP·GAP complex. When pyruvate binds it induces a conformational change in an unidentified inhibitor site (likely to be different from the substrate-binding sites), 2 can then engage the enzyme (with comparable inhibition of both the DXPS·ThDP·pyruvate and the DXPS·ThDP·pyruvate·GAP complexes). In the case of 3a (and 3b–d), it can only engage the enzyme when both substrates are bound as it is uncompetitive towards both, and it exhibits the strongest inhibition when both substrates are bound.
|
|
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Inhibitiona (%) | Compound | Inhibitiona (%) | ||||||
| [P], [G] | [P], [G] | [P], [G] | [P], [G] | [P], [G] | [P], [G] | [P], [G] | [P], [G] | ||
| a Data are the means of measurements in three technical replicates. Percentage inhibition values were determined for compounds at 5 μM with [pyruvate] = 62.5 μM ([P]) or 1 mM ([P]) and [GAP] = 50 μM ([G]) or 250 μM ([G]). | |||||||||
| 2a (o-Cl) | 24 ± 3 | 48 ± 2 | 21 ± 3 | 47 ± 3 | 3a (o-Cl) | 26 ± 2 | 55 ± 5 | 48 ± 3 | 65 ± 5 |
| 2b (H) | 20 ± 3 | 37 ± 3 | 21 ± 2 | 39 ± 2 | 3b (H) | 26 ± 3 | 46 ± 5 | 38 ± 3 | 53 ± 3 |
| 2c (o-F) | 22 ± 2 | 48 ± 3 | 22 ± 3 | 44 ± 4 | 3c (o-F) | 19 ± 2 | 52 ± 3 | 30 ± 2 | 55 ± 2 |
| 2d (o-Br) | 27 ± 2 | 40 ± 4 | 27 ± 3 | 39 ± 4 | 3d (o-Br) | 30 ± 2 | 48 ± 3 | 37 ± 3 | 51 ± 4 |
| 2e (p-Cl) | 19 ± 2 | 42 ± 3 | 20 ± 4 | 42 ± 2 | 3e (p-Cl) | 14 ± 3 | 49 ± 2 | 24 ± 3 | 23 ± 2 |
| 2f (p-F) | 17 ± 3 | 37 ± 3 | 18 ± 2 | 40 ± 4 | 3f (p-F) | 17 ± 3 | 41 ± 3 | 28 ± 3 | 24 ± 2 |
| 2g (p-Br) | 8 ± 2 | 43 ± 4 | 19 ± 3 | 17 ± 4 | 3g (p-Br) | 14 ± 2 | 44 ± 2 | 24 ± 3 | 21 ± 4 |
| 2h (o-Me) | 12 ± 2 | 37 ± 3 | 21 ± 3 | 22 ± 2 | 3h (o-Me) | 17 ± 3 | 39 ± 4 | 23 ± 2 | 21 ± 2 |
| 2i (p-tBu) | 7 ± 2 | 39 ± 3 | 15 ± 2 | 15 ± 3 | 3i (p-tBu) | 8 ± 3 | 39 ± 4 | 18 ± 2 | 16 ± 3 |
Analogues 2g–i and 3e–i showed the weakest inhibition (7–17%) under low level of both substrates, improved inhibition (15–28%) under high [GAP], and the strongest inhibition (37–49%) under high [pyruvate] but low [GAP]; such a change in the inhibition profile may have a steric origin as they are all larger than 2a and 3a. As a result, we did not explore any larger substituents in the ortho- or para-positions. Although none of the para-substituted analogues had improved inhibition relative to the unsubstituted 2a, it is worth noting that 2e and 2f (p-Cl and p-F) have similar activities to 2a. So, if metabolic oxidation at the para-position does prove to be a problem, these substituents (in addition to the o-Cl) may well provide a solution. While the binding location of 2a and 3a is unknown, with no available crystal structures of them bound to EcDXPS, we believe that their binding sites are likely to be distinct due to their different inhibition modalities. As these inhibitor sites are highly sensitive to structural modifications on ligands and they are probably not in the active site, this may help to explain why 2a and 3a bind poorly to several other DXPS enzymes.
Kinetic studies on porcine PDH E1 inhibition
In the preliminary screening on multiple ThDP-dependent enzymes (Table S1†), inhibition of porcine PDH E1 by 2a and 3a was discovered. To gain insights into their binding mode, computational docking was performed: both were predicted to occupy the coenzyme-binding site (Fig. 3). Their competitive relationship with ThDP was confirmed experimentally as the observed potency decreased with increasing [ThDP] (Table 2). With the IC50 values of their dose-dependent inhibition determined (Fig. S3†), the affinities of 2a and 3a were found to be comparable to and 2.5 times weaker than that of ThDP, respectively; as the KM value for ThDP is 50 nM,31 this puts their KI values in the nanomolar range (KI of 2a = 44 nM and KI of 3a = 127 nM). | ||
| Fig. 3 (a) Binding mode of ThDP in PDH E1 showing the V-shaped conformation between the aminopyrimidine and the thiazolium ring. The thiazolium ring is in a relatively hydrophobic region, which facilitates formation of the catalytic ylide. Predicted binding modes of 2a (b) and 3a (c) overlayed with ThDP (blue wires) as in view (a). Molecules were generated using Mercury and dockings were executed using GOLD docking programme with human PDH (PDB: 6CFO) as the target; the reported modelling procedures42 have been adopted in this study. | ||
|
|
|
||||||
|---|---|---|---|---|---|---|---|
| Compounds | Inhibitiona (%) | Compounds | Inhibitiona (%) | ||||
[Compound]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[ThDP] :[ThDP] |
[Compound]:[ThDP] |
||||||
| 4:1 |
1:1 |
1:4 |
4:1 |
1:1 |
1:4 |
||
|
a Data are the means of measurements in three technical replicates. Percentage inhibition values were determined for compounds at 100 μM with [ThDP] = 25 μM (for 4:1); at 100 μM with [ThDP] = 100 μM (for 1:1); at 25 μM with [ThDP] = 100 μM (for 1:4).
|
|||||||
| 2a (o-Cl) | 75 ± 5 | 53 ± 3 | 25 ± 4 | 3a (o-Cl) | 55 ± 3 | 32 ± 3 | 8 ± 3 |
| 2b (H) | 78 ± 4 | 54 ± 2 | 29 ± 2 | 3b (H) | 59 ± 3 | 31 ± 2 | 9 ± 2 |
| 2c (o-F) | 74 ± 4 | 51 ± 3 | 22 ± 2 | 3c (o-F) | 55 ± 4 | 29 ± 4 | <5 |
| 2d (o-Br) | 39 ± 6 | 18 ± 4 | <5 | 3d (o-Br) | 17 ± 3 | <5 | <5 |
| 2e (p-Cl) | 79 ± 4 | 57 ± 3 | 19 ± 4 | 3e (p-Cl) | 58 ± 2 | 37 ± 3 | <5 |
| 2f (p-F) | 73 ± 4 | 44 ± 3 | 13 ± 3 | 3f (p-F) | 55 ± 4 | 23 ± 4 | <5 |
| 2g (p-Br) | 37 ± 5 | 14 ± 4 | <5 | 3g (p-Br) | 13 ± 4 | <5 | <5 |
| 2h (o-Me) | 37 ± 4 | 16 ± 3 | <5 | 3h (o-Me) | 14 ± 3 | <5 | <5 |
| 2i (p-tBu) | <5 | <5 | <5 | 3i (p-tBu) | <5 | <5 | <5 |


The analogues were also tested, to probe the structure–activity relationship (SAR). The ring-opened 3a–h (13–59%) were consistently weaker inhibitors than the cyclic 2a–h (37–79%) at 100 μM with [ThDP] = 25 μM (Table 2). This supports a binding mode in which the non-aromatic ring of our inhibitors occupies the central hydrophobic region (Fig. 3). The charged ring-opened series would suffer a greater desolvation penalty than the cyclic series upon leaving the aqueous environment and binding in this hydrophobic region.31,43–46 If the highly polar moiety of the ring-opened series were able to interact with the Mg2+ ion in the diphosphate pocket, they would be expected to bind better than the cyclic series, so we suggest that the side chain is not long enough to allow any effective interaction with the Mg2+ ion.
Regarding the substitution pattern on the aromatic ring of our inhibitors, the position and identity of the halogen atom (F or Cl) does not seem to affect affinity (compared to the unsubstituted 2b and 3b), but the bromo compounds were weaker binders. For alkyl substituents, having a Me- on the ortho-position reduced affinity while introducing a tBu group on the para-position abolished binding, presumably for steric reasons. These trends were consistent in both series (Table 2). Our docking models suggested that this para-substituent of our inhibitors occupies the same space as the methyl group on the aminopyrimidine ring of ThDP when bound to the enzyme (Fig. 3), and this is supported by the low activity of 2i and 3i: our earlier work44 on developing PDH-selective inhibitors showed that substituents larger than a methyl group at this para-position led to poor binding. So the predicted binding modes in this study were in line with our understanding of PDH E1's ThDP-binding site.
Further evaluation – cytotoxicity and membrane-permeability
Regardless of their precise binding modes, 2a and 3a were found to be potent inhibitors of PDH E1. Ligand efficiency (LE), measuring the binding energy to its target (in kcal mol−1) per heavy atom of the ligand is a widely applied metric in medicinal chemistry;47 drug-discovery efforts often aim to develop clinical candidates with LE > 0.3.47,48 As the LEs of 2a and 3a are 0.61 (binding energy = 10.4 kcal mol−1; heavy atom count = 17) and 0.54 (binding energy = 9.8 kcal mol−1; heavy atom count = 18), respectively, both are highly efficient inhibitors of PDH E1.Given that the PDH complex provides a link between glycolysis and mitochondrial metabolism for energy production,49 concern was raised over possible cytotoxicity due to PDH inhibition. To test this, human foreskin fibroblast (HFF) cells were subjected to compounds 2a and 3a, and the compounds were only weakly cytotoxic, even at concentrations 100 times greater than their KI values (Fig. S4†). Although one possible explanation would be that 2a and 3a are weak inhibitors of human PDH E1 despite their potent inhibition on porcine PDH E1, it is generally accepted that the latter is a good model for the former because the sequence of the two enzymes are >95% identical and the residues that differ are located away from the active site.44 Another possible explanation would be due to hydrolysis of 2a into 3a.25 Extracellular hydrolysis would lead to reduced cell entry of inhibitors as 3a is almost membrane-impermeable (fraction absorbed = 6% in parallel artificial membrane permeability assay, PAMPA,50 as shown in Table S2†). Intracellular hydrolysis would lead to reduced entry into the mitochondria, which is where the PDH complex is active.49 And in either case inhibition of PDH would be lowered as 3a is a less potent inhibitor than 2a.
Given the inhibitory activity on EcDXPS and the lack of cytotoxicity on HFF cells, the potential of ketoclomazone 2a as an antibacterial agent against susceptible bacteria is enhanced. Previous studies had already established its micromolar activities on suppressing bacterial cell growth,32,33 in addition to its herbicidal action. This study provides additional insight into its antibacterial potential. The cyclic form 2a probably serves as a prodrug that facilitates membrane permeability (fraction absorbed = 47% in PAMPA, Table S2†); once inside the cell, it gets hydrolysed to its ring-opened form 3a, which has several important biological consequences: a) this charged form may be trapped within the cells, thus raising the intracellular inhibitor level; b) 3a is a weaker inhibitor of PDH than its parent 2a, thus likely to be less cytotoxic to human cells; and c) although 2a and 3a are both potent inhibitors of DXPS, the shift in inhibition modality with respect to GAP from non-competitive (2a) to uncompetitive (3a) makes the latter a better anti-infectious agent. This is because when an enzyme in a metabolic pathway is inhibited, the upstream metabolic processes continue to make new substrates and this will lead to a build-up of the substrates for that enzyme. As the substrate concentration continues to rise, it is likely to eventually exceed the KM value and approach saturating conditions. This will lead to a relief of inhibition by competitive inhibitors but an increase in the affinity of uncompetitive inhibitors; in the latter case, further suppression of the metabolic pathway could eventually be catastrophic to cells.51 Our biological data may help put these into context. Equimolar concentrations of 2a and ThDP leads to 53% inhibition of PDH E1 activity; if 2a gets hydrolysed to 3a and the cell increases the ThDP content by four-fold, the percentage inhibition would drop to 8% (Table 2). By contrast, the percentage inhibition of DXPS is 24% for 2a at 5 μM with [pyruvate] and [GAP] close to their respective KM values; when hydrolysis occurs and, if the substrates accumulate upon DXPS inhibition to the higher levels used in Table 1, the percentage inhibition would rise to 65%.
Conclusions
Clomazone 1 is a soil-applied herbicide, and ketoclomazone 2a is believed to be the active species. It has also been showed that 2a can be an antibacterial agent and that 2a and 3a probably suppress bacterial growth through inhibition of DXPS.32–34 This study revisited compounds 2a and 3a as inhibitors of a range of DXPS enzymes and also evaluated their activities on the broader ThDP-dependent enzyme family. Among the four tested DXPS enzymes, 2a and 3a only potently inhibited EcDXPS. The kinetics of inhibition of 2a was shown to be uncompetitive with respect to pyruvate and non-competitive with respect to GAP, while that of 3a was found to be uncompetitive with respect to both substrates. Screened on four other ThDP-dependent enzymes, 2a and 3a were identified as ThDP-competitive inhibitors of porcine PDH E1. Despite this, however, 2a and 3a showed little cytotoxicity towards human cells, probably because of hydrolysis of 2a to 3a, which is relatively impermeable towards membranes. Our findings and those of others suggest that the more hydrophobic 2a acts as a prodrug for membrane passage and gets hydrolysed to 3a in cells.5,6,32,33 The differences in inhibitory activities and modalities on the DXPS and PDH enzymes between 2a and 3a, established in this study, support the potential of ketoclomazone 2a as a selective antibacterial agent. This study expands our understanding on ketoclomazone as an inhibitor of the ThDP-dependent enzyme family and may aid work on the development of DXPS inhibitors.Author contributions
AHYC performed the chemical synthesis. TCSH performed the computational studies. IF performed the cell-based assays. AHYC, TCSH and RH performed the enzyme assays. AHYC and TCSH wrote the first draft. FJL supervised the work from AHYC and TCSH. KJS supervised the work from IF. AKHH supervised the work from RH. All authors edited a draft of the manuscript and approved the final version.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the support from K. M. Medhealth (AHYC and TCSH), Research Training Program scholarship from the Australian government (IF), the Canberra Branch of the Australian Red Cross Lifeblood for the provision of red blood cells, members of the van Dooren Lab (Australian National University) for HFF cells, the Schlumberger Foundation Faculty for the Future Fellowship (RH) and the Helmholtz Association Initiative and Networking Fund and the European Research Council, ERC starting grant 757913 (AKHH).References
- S. D. Tetali, Terpenes and isoprenoids: a wealth of compounds for global use, Planta, 2019, 249, 1–8, DOI:10.1007/s00425-018-3056-x.
- K. Bloch, Sterol molecule: structure, biosynthesis, and function, Steroids, 1992, 57, 378–383, DOI:10.1016/0039-128x(92)90081-j.
- T. Kuzuyama and H. Seto, Diversity of the biosynthesis of the isoprene units, Nat. Prod. Rep., 2003, 20, 171–183, 10.1039/B109860H.
- S. Heuston, M. Begley, C. G. M. Gahan and C. Hill, Isoprenoid biosynthesis in bacterial pathogens, Microbiology, 2012, 158, 1389–1401, DOI:10.1099/mic.0.051599-0.
- A. A. DeColli, M. L. Johnston and C. L. Freel Meyers, Recent Insights into Mechanism and Structure of MEP Pathway Enzymes and Implications for Inhibition Strategies, in Comprehensive Natural Products III, ed. H.-W. Liu and T. P. Begley, Elsevier, 2020, vol. 4, pp. 287–322 Search PubMed.
- D. Bartee and C. L. Freel Meyers, Toward Understanding the Chemistry and Biology of 1-Deoxy-d-xylulose 5-Phosphate (DXP) Synthase: A Unique Antimicrobial Target at the Heart of Bacterial Metabolism, Acc. Chem. Res., 2018, 51, 2546–2555, DOI:10.1021/acs.accounts.8b00321.
- L. A. Brammer and C. Freel Meyers, Revealing Substrate Promiscuity of 1-Deoxy-D-xylulose 5-Phosphate Synthase, Org. Lett., 2009, 11, 4748–4751, DOI:10.1021/ol901961q.
- L. A. Brammer, J. M. Smith, H. Wade and C. Freel Meyers, 1-Deoxy-D-xylulose 5-Phosphate Synthase Catalyzes a Novel Random Sequential Mechanism, J. Biol. Chem., 2011, 286, 36522–36531, DOI:10.1074/jbc.M111.259747.
- H. Patel, N. S. Nemeria, L. A. Brammer, C. L. Freel Meyers and F. Jordan, Observation of Thiamin-Bound Intermediates and Microscopic Rate Constants for Their Interconversion on 1-Deoxy-d-xylulose 5-Phosphate Synthase: 600-Fold Rate Acceleration of Pyruvate Decarboxylation by D-Glyceraldehyde-3-phosphate, J. Am. Chem. Soc., 2012, 134, 18374–18379, DOI:10.1021/ja307315u.
- S. Handa, D. Ramamoorthy, T. J. Spradling, W. C. Guida, J. H. Adams, K. G. Bendinskas and D. J. Merkler, Production of recombinant 1-deoxy-d-xylulose-5-phosphate synthase from Plasmodium vivax in Escherichia coli, FEBS Open Bio, 2013, 3, 124–129, DOI:10.1016/j.fob.2013.01.007.
- D. Ramamoorthy, S. Handa, D. J. Merkler and W. C. Guida, Plasmodium vivax 1-deoxy-D-xylulose-5-phosphate synthase: Homology Modeling, Domain Swapping, and Virtual Screening, J. Data Min. Genomics Proteomics, 2014, S1, 003, DOI:10.4172/2153-0602.S1-003.
- M. R. Battistini, C. Shoji, S. Handa, L. Breydo and D. J. Merkler, Mechanistic binding insights for 1-deoxy-D-xylulose-5-phosphate synthase, the enzyme catalyzing the first reaction of isoprenoid biosynthesis in the malaria-causing protists, Plasmodium falciparum and Plasmodium vivax, Protein Expression Purif., 2016, 120, 16–27, DOI:10.1016/j.pep.2015.12.003.
- J. K. White, S. Handa, S. L. Vankayala, D. J. Merkler and H. L. Woodcock, Thiamin Diphosphate Activation in 1-Deoxy-d-xylulose 5-Phosphate Synthase: Insights into the Mechanism and Underlying Intermolecular Interactions, J. Phys. Chem. B, 2016, 120, 9922–9934, DOI:10.1021/acs.jpcb.6b07248.
- D. Bartee and C. L. Freel Meyers, Targeting the Unique Mechanism of Bacterial 1-Deoxy-d-xylulose-5-phosphate Synthase, Biochemistry, 2018, 57, 4349–4356, DOI:10.1021/acs.biochem.8b00548.
- A. A. DeColli, N. S. Nemeria, A. Majumdar, G. J. Gerfen, F. Jordan and C. L. Freel Meyers, Oxidative decarboxylation of pyruvate by 1-deoxy-d-xyulose 5-phosphate synthase, a central metabolic enzyme in bacteria, J. Biol. Chem., 2018, 293, 10857–10869, DOI:10.1074/jbc.ra118.001980.
- S. Handa, D. R. Dempsey, D. Ramamoorthy, N. Cook, W. C. Guida, T. J. Spradling, J. K. White, H. L. Woodcock and D. J. Merkler, Mechanistic Studies of 1-Deoxy-D-Xylulose-5-Phosphate Synthase from Deinococcus radiodurans, Biochem. Mol. Biol. J., 2018, 4(1), 2, DOI:10.21767/2471-8084.100051.
- P. Y.-T. Chen, A. A. DeColli, C. L. Freel Meyers and C. L. Drennan, X-ray crystallography-based structural elucidation of enzyme-bound intermediates along the 1-deoxy-d-xylulose 5-phosphate synthase reaction coordinate, J. Biol. Chem., 2019, 294, 12405–12414, DOI:10.1074/jbc.ra119.009321.
- A. A. DeColli, X. Zhang, K. L. Heflin, F. Jordan and C. L. Freel Meyers, Active Site Histidines Link Conformational Dynamics with Catalysis on Anti-Infective Target 1-Deoxy-d-xylulose 5-Phosphate Synthase, Biochemistry, 2019, 58, 4970–4982, DOI:10.1021/acs.biochem.9b00878.
- R. M. Gierse, E. R. Reddem, A. Alhayek, D. Baitinger, Z. Hamid, H. Jakobi, B. Laber, G. Lange, A. K. H. Hirsch and M. R. Groves, Identification of a 1-deoxy-D-xylulose-5-phosphate synthase (DXS) mutant with improved crystallographic properties, Biochem. Biophys. Res. Commun., 2021, 539, 42–47, DOI:10.1016/j.bbrc.2020.12.069.
- M. L. Johnston and C. L. Freel Meyers, Revealing Donor Substrate-Dependent Mechanistic Control on DXPS, an Enzyme in Bacterial Central Metabolism, Biochemistry, 2021, 60, 929–939, DOI:10.1021/acs.biochem.1c00019.
- R. M. Gierse, R. Oerlemans, E. R. Reddem, V. O. Gawriljuk, A. Alhayek, D. Baitinger, H. Jakobi, B. Laber, G. Lange, A. K. H. Hirsch and M. R. Groves, First crystal structures of 1-deoxy-D-xylulose 5-phosphate synthase (DXPS) from Mycobacterium tuberculosis indicate a distinct mechanism of intermediate stabilization, Sci. Rep., 2022, 12, 7221, DOI:10.1038/s41598-022-11205-9.
- R. Hamid, S. Adam, A. Lacour, L. M. Gomez, J. Köhnke and A. K. H. Hirsch, 1-deoxy-D-xylulose-5-phosphate synthase from Pseudomonas aeruginosa and Klebsiella pneumoniae reveals conformational changes upon cofactor binding, J. Biol. Chem., 2023, 299, 105152, DOI:10.1016/j.jbc.2023.105152.
- J. M. Smith, R. J. Vierling and C. Freel Meyers, Selective inhibition of E. coli 1-deoxy-d-xylulose-5-phosphate synthase by acetylphosphonates, Med. Chem. Commun., 2012, 3, 65–67, 10.1039/C1MD00233C.
- F. Morris, R. Vierling, L. Boucher, J. Bosch and C. L. Freel Meyers, DXP synthase-catalyzed C-N bond formation: Nitroso substrate specificity studies guide selective inhibitor design, ChemBioChem, 2013, 14, 1309–1315, DOI:10.1002/cbic.201300187.
- S. Sanders, R. J. Vierling, D. Bartee, A. A. DeColli, M. J. Harrison, J. L. Aklinski, A. T. Koppisch and C. L. Freel Meyers, Challenges and Hallmarks of Establishing Alkylacetylphosphonates as Probes of Bacterial 1-Deoxy-d-xylulose 5-Phosphate Synthase, ACS Infect. Dis., 2017, 3, 467–478, DOI:10.1021/acsinfecdis.6b00168.
- S. Sanders, D. Bartee, M. J. Harrison, P. D. Phillips, A. T. Koppisch and C. L. Freel Meyers, Growth medium-dependent antimicrobial activity of early stage MEP pathway inhibitors, PLoS One, 2018, 13, e0197638, DOI:10.1371/journal.pone.0197638.
- D. Bartee, S. Sanders, P. D. Phillips, M. J. Harrison, A. T. Koppisch and C. L. Freel Meyers, Enamide Prodrugs of Acetyl Phosphonate Deoxy-d-xylulose-5-phosphate Synthase Inhibitors as Potent Antibacterial Agents, ACS Infect. Dis., 2019, 5, 406–417, DOI:10.1021/acsinfecdis.8b00307.
- R. P. Jumde, M. Guardigni, R. M. Gierse, A. Alhayek, D. Zhu, Z. Hamid, S. Johannsen, W. A. M. Elgaher, P. J. Neusens, C. Nehls, J. Haupenthal, N. Reiling and A. K. H. Hirsch, Hit-optimization using target-directed dynamic combinatorial chemistry: development of inhibitors of the anti-infective target 1-deoxy-d-xylulose-5-phosphate synthase, Chem. Sci., 2021, 12, 7775–7785, 10.1039/D1SC00330E.
- D. Zhu, S. Johannsen, T. Masini, C. Simonin, J. Haupenthal, B. Illarionov, A. Andreas, M. Awale, R. M. Gierse, T. van der Laan, R. van der Vlag, R. Nasti, M. Poizat, E. Buhler, N. Reiling, R. Müller, M. Fischer, J.-L. Reymond and A. K. H. Hirsch, Discovery of novel drug-like antitubercular hits targeting the MEP pathway enzyme DXPS by strategic application of ligand-based virtual screening, Chem. Sci., 2022, 13, 10686–10698, 10.1039/D2SC02371G.
- S. Johannsen, R. M. Gierse, A. Olshanova, E. Smerznak, C. Laggner, L. Eschweiler, Z. Adeli, R. Hamid, A. Alhayek, N. Reiling, J. Haupenthal and A. K. H. Hirsch, Not Every Hit-Identification Technique Works on 1-Deoxy-d-Xylulose 5-Phosphate Synthase (DXPS): Making the Most of a Virtual Screening Campaign, ChemMedChem, 2023, 18, e202200590, DOI:10.1002/cmdc.202200590.
- A. H. Y. Chan, T. C. S. Ho, R. Irfan, R. A. A. Hamid, E. S. Rudge, A. Iqbal, A. Turner, A. K. H. Hirsch and F. J. Leeper, Design of thiamine analogues for inhibition of thiamine diphosphate (ThDP)-dependent enzymes: Systematic investigation through Scaffold-Hopping and C2-Functionalisation, Bioorg. Chem., 2023, 138, 106602, DOI:10.1016/j.bioorg.2023.106602.
- Y. Matsue, H. Mizuno, T. Tomita, T. Asami, M. Nishiyama and T. Kuzuyama, The herbicide ketoclomazone inhibits 1-deoxy-D-xylulose 5-phosphate synthase in the 2-C-methyl-D-erythritol 4-phosphate pathway and shows antibacterial activity against Haemophilus influenzae, J. Antibiot., 2010, 63, 583–588, DOI:10.1038/ja.2010.100.
- D. Hayashi, N. Kato, T. Kuzuyama, Y. Sato and J. Ohkanda, Antimicrobial N-(2-chlorobenzyl)-substituted hydroxamate is an inhibitor of 1-deoxy-d-xylulose 5-phosphate synthase, Chem. Commun., 2013, 49, 5535–5537, 10.1039/C3CC40758F.
- Y. Ferhatoglu and M. Barrett, Studies of clomazone mode of action, Pestic. Biochem. Physiol., 2006, 85, 7–14, DOI:10.1016/j.pestbp.2005.10.002.
- C. Mueller, J. Schwender, J. Zeidler and H. K. Lichtenthaler, Properties and inhibition of the first two enzymes of the non-mevalonate pathway of isoprenoid biosynthesis, Biochem. Soc. Trans., 2000, 28, 792–793, DOI:10.1042/bst0280792.
- M. J. Konz, Herbicidal Isoxazolidine-3,5-diones, US Pat., US4302238, 1981 Search PubMed.
- X. W. A. Chan, C. Wrenger, K. Stahl, B. Bergmann, M. Winterberg, I. B. Müller and K. J. Saliba, Chemical and genetic validation of thiamine utilization as an antimalarial drug target, Nat. Commun., 2013, 4, 2060, DOI:10.1038/ncomms3060.
- A. H. Y. Chan, I. Fathoni, T. Ho, K. J. Saliba and F. J. Leeper, Thiamine analogues as inhibitors of pyruvate dehydrogenase and discovery of a thiamine analogue with non-thiamine related antiplasmodial activity, RSC Med. Chem., 2022, 13, 817–821, 10.1039/D2MD00085G.
- A. H. Y. Chan, T. C. S. Ho, I. Fathoni, R. Pope, K. J. Saliba and F. J. Leeper, Inhibition of Thiamine Diphosphate-Dependent Enzymes by Triazole-Based Thiamine Analogues, ACS Med. Chem. Lett., 2023, 14, 621–628, DOI:10.1021/acsmedchemlett.3c00047.
- H. Jomaa, J. Wiesner, S. Sanderbrand, B. Altincicek, C. Weidemeyer, M. Hintz, I. Türbachova, M. Eberl, J. Zeidler, H. K. Lichtenthaler, D. Soldati and E. Beck, Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs, Science, 1999, 285, 1573–1576, DOI:10.1126/science.285.5433.1573.
- K. Gupta and G. S. Saggu, Isoprenoid Biosynthesis Pathway Enzymes as Promising Drug Candidates Against Malaria Parasites: An In-Silico Investigation, Preprints.org, DOI:10.20944/preprints202312.0032.v1, This is a preprint and has not been peer-reviewed.
- R. A. W. Frank, F. J. Leeper and B. F. Luisi, Structure, mechanism and catalytic duality of thiamine-dependent enzymes, Cell. Mol. Life Sci., 2007, 64, 892–905, DOI:10.1007/s00018-007-6423-5.
- A. H. Y. Chan, T. C. S. Ho, K. Agyei-Owusu and F. J. Leeper, Synthesis of pyrrothiamine, a novel thiamine analogue, and evaluation of derivatives as potent and selective inhibitors of pyruvate dehydrogenase, Org. Biomol. Chem., 2022, 20, 8855–8858, 10.1039/D2OB01819E.
- A. H. Y. Chan, T. C. S. Ho, D. R. Parle and F. J. Leeper, Furan-based inhibitors of pyruvate dehydrogenase: SAR study, biochemical evaluation and computational analysis, Org. Biomol. Chem., 2023, 21, 1755–1763, 10.1039/D2OB02272A.
- A. H. Y. Chan, T. C. S. Ho and F. J. Leeper, Open-chain thiamine analogues as potent inhibitors of thiamine pyrophosphate (TPP)-dependent enzymes, Org. Biomol. Chem., 2023, 21, 6531–6536, 10.1039/D3OB00884C.
- A. H. Y. Chan, T. C. S. Ho and F. J. Leeper, Thiamine analogues featuring amino-oxetanes as potent and selective inhibitors of pyruvate dehydrogenase, Bioorg. Med. Chem. Lett., 2023, 129571, DOI:10.1016/j.bmcl.2023.129571.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, The role of ligand efficiency metrics in drug discovery, Nat. Rev. Drug Discovery, 2014, 13, 105–121, DOI:10.1038/nrd4163.
- N. A. Meanwell, Improving Drug Candidates by Design: A Focus on Physicochemical Properties As a Means of Improving Compound Disposition and Safety, Chem. Res. Toxicol., 2011, 24, 1420–1456, DOI:10.1021/tx200211v.
- O. H. Wieland, The mammalian pyruvate dehydrogenase complex: Structure and regulation, Rev. Physiol., Biochem. Pharmacol., 1983, 96, 123–170, DOI:10.1007/BFb0031008.
- A. Avdeef, The rise of PAMPA, Expert Opin. Drug Metab. Toxicol., 2005, 1, 325–342, DOI:10.1517/17425255.1.2.325.
- R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, Wiley, 1st edn, 2013, DOI:10.1002/9781118540398.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00083h |
| ‡ Equal contribution. |
| § KpDXPS was assayed at 10 mM of inhibitor and showed 64% inhibition by 2a but calculation suggests this would equate to <16% inhibition at 1 mM. |
| This journal is © The Royal Society of Chemistry 2024 |