
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting thiol-functionalized benzosiloxaboroles for achieving diverse substitution patterns – synthesis, characterization and biological evaluation of promising antibacterial agents†
Krzysztof
Nowicki
 *a,
Joanna
Krajewska
b,
Tomasz M.
Stępniewski
c,
Monika
Wielechowska
a,
Patrycja
Wińska
a,
Anna
Kaczmarczyk
a,
Julia
Korpowska
a,
Jana
Selent
c,
Paulina H.
Marek-Urban
a,
Krzysztof
Durka
a,
Krzysztof
Woźniak
d,
Agnieszka E.
Laudy
*b and
Sergiusz
Luliński
*a
*a,
Joanna
Krajewska
b,
Tomasz M.
Stępniewski
c,
Monika
Wielechowska
a,
Patrycja
Wińska
a,
Anna
Kaczmarczyk
a,
Julia
Korpowska
a,
Jana
Selent
c,
Paulina H.
Marek-Urban
a,
Krzysztof
Durka
a,
Krzysztof
Woźniak
d,
Agnieszka E.
Laudy
*b and
Sergiusz
Luliński
*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: krzysztof.nowicki2.dokt@pw.edu.pl; sergiusz.lulinski@pw.edu.pl
bDepartment of Pharmaceutical Microbiology and Bioanalysis, Medical University of Warsaw, Banacha 1b, 02-097, Warsaw, Poland. E-mail: alaudy@wp.pl
cGPCR Drug Discovery Lab, Research Programme on Biomedical Informatics (GRIB), Hospital del Mar Medical Research Institute (IMIM) – Department of Medicine and Life Sciences, Pompeu Fabra University (UPF), Carrer del Dr. Aiguader, 88, 08003 Barcelona, Spain
dFaculty of Chemistry, University of Warsaw, Pasteura 1, 00-093 Warsaw, Poland
First published on 20th March 2024
Abstract
Benzosiloxaboroles are an emerging class of medicinal agents possessing promising antimicrobial activity. Herein, the expedient synthesis of two novel thiol-functionalized benzosiloxaboroles 1e and 2e is reported. The presence of the SH group allowed for diverse structural modifications involving the thiol-Michael addition, oxidation, as well as nucleophilic substitution giving rise to a series of 27 new benzosiloxaboroles containing various polar functional groups, e.g., carbonyl, ester, amide, imide, nitrile, sulfonyl and sulfonamide, and pendant heterocyclic rings. The activity of the obtained compounds against selected bacterial and yeast strains, including multidrug-resistant clinical strains, was investigated. Compounds 6, 12, 20 and 22–24 show high activity against Staphylococcus aureus, including both methicillin-sensitive (MSSA) and methicillin-resistant (MRSA) strains, with MIC values in the range of 1.56–12.5 μg mL−1, while their cytotoxicity is relatively low. The in vitro assay performed with 2-(phenylsulfonyl)ethylthio derivative 20 revealed that, in contrast to the majority of known antibacterial oxaboroles, the plausible mechanism of antibacterial action, involving inhibition of the leucyl-tRNA synthetase enzyme, is not responsible for the antibacterial activity. Structural bioinformatic analysis involving molecular dynamics simulations provided a possible explanation for this finding.
Introduction
In the last two decades, strong interest in boron heterocycles was stimulated to a significant extent by the discovery of the biological activity of benzoxaboroles – internal hemiesters of 2-(hydroxymethyl)phenylboronic acid.1–6 They emerged as a novel class of small-molecule therapeutic agents possessing strong antimicrobial,7–11 anti-inflammatory12,13 as well as anti-cancer activity.14–16 Recent intensive efforts resulted in the preparation of over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 benzoxaborole derivatives. So far, two of them have been successfully commercialized,17,18 while several others are in various phases of clinical trials (Fig. 1a).19–22 Recently, our group has given attention to silicon analogues of benzoxaboroles–benzosiloxaboroles.23–28 The introduction of the SiMe2 group to the oxaborole ring in place of the methylene group resulted in increased Lewis acidity and lipophilicity, which may be beneficial for biological activity.23 Moreover, the strategy for the synthesis of these compounds is different, which opens up possibilities for enriching substitution patterns at the boracyclic scaffold.23–27,29 Our preliminary microbiological studies show that simple fluorinated benzosiloxaboroles are active against selected yeast strains,23 whereas more extended systems demonstrate potent antibacterial activity, especially against Gram-positive cocci, including multidrug-resistant clinical strains.26,27 Some derivatives were also found to be effective inhibitors of KPC-2/AmpC β-lactamases responsible for drug resistance in bacteria (Fig. 1b).25
000 benzoxaborole derivatives. So far, two of them have been successfully commercialized,17,18 while several others are in various phases of clinical trials (Fig. 1a).19–22 Recently, our group has given attention to silicon analogues of benzoxaboroles–benzosiloxaboroles.23–28 The introduction of the SiMe2 group to the oxaborole ring in place of the methylene group resulted in increased Lewis acidity and lipophilicity, which may be beneficial for biological activity.23 Moreover, the strategy for the synthesis of these compounds is different, which opens up possibilities for enriching substitution patterns at the boracyclic scaffold.23–27,29 Our preliminary microbiological studies show that simple fluorinated benzosiloxaboroles are active against selected yeast strains,23 whereas more extended systems demonstrate potent antibacterial activity, especially against Gram-positive cocci, including multidrug-resistant clinical strains.26,27 Some derivatives were also found to be effective inhibitors of KPC-2/AmpC β-lactamases responsible for drug resistance in bacteria (Fig. 1b).25
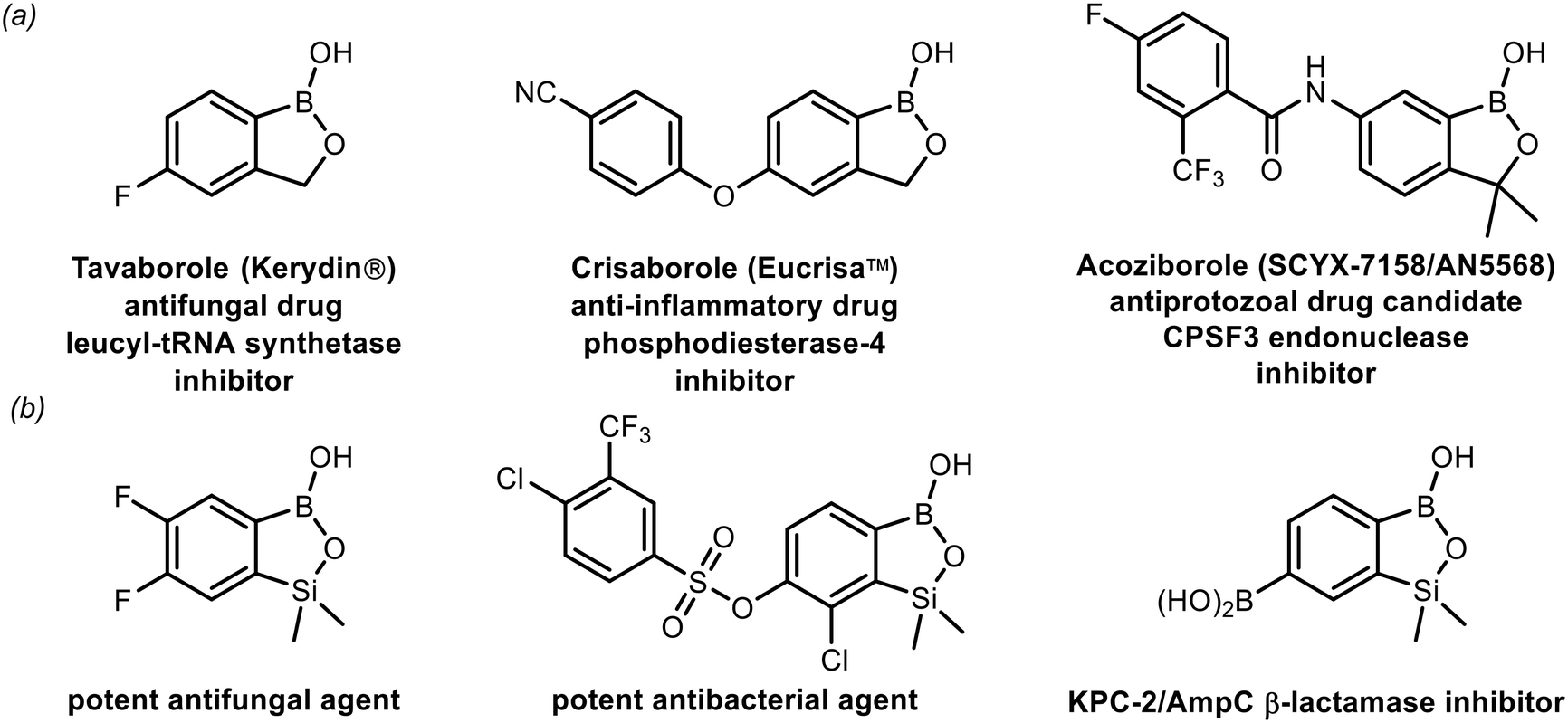 | ||
| Fig. 1 (a) Examples of biologically active benzoxaboroles already introduced into clinical use (both marketed and under clinical trials); (b) examples of benzosiloxaboroles showing antimicrobial activity. | ||
The sulfide group is widely used in medicinal chemistry as a linker and many benzoxaboroles containing this moiety have been already reported.30,31 However, thiol-functionalized benzoxaboroles themselves have not been widely exploited so far. Only two S-functionalized benzoxaboroles were synthesized through direct transformation of a thiol group at the benzoxaborole (Fig. 2). These compounds were investigated as potential anti-Wolbachia agents, however they did not exhibit significant potency.32 Considering the growing potential of benzosiloxaboroles in medicinal chemistry we decided to extend the library of these compounds by utilization of a thiol substituent. The proposed general concept is practical due to the high reactivity of the thiol group, which can be easily converted into other sulfur-based groups, such as thioether, thioester, sulfonamide, etc. Thus, it was successfully validated by synthesis of 27 new functionalized benzosiloxaboroles followed by comprehensive evaluation of antimicrobial activity and cytotoxicity. This work was complemented by a study on the plausible mechanism of action of one of the obtained compounds.
 | ||
| Fig. 2 Pleuromutilin-functionalized benzoxaboroles obtained from respective thiol precursors.32 | ||
Results and discussion
Synthesis
The approach to fluorinated thiol-functionalized benzosiloxaboroles 1e and 2e involved a general four-step protocol (Scheme 1) starting with the preparation of appropriate halogenated thiophenols from inexpensive starting materials. The synthesis of thiophenol 1b was accomplished through deprotonative lithiation of 1-bromo-3,5-difluorobenzene with LDA in THF at −78 °C followed by addition of sulfur and hydrolytic workup. The respective disulfide 1b_D was also formed as a byproduct (ca. 5%). Notably, when the reaction mixture was warmed to room temperature prior to hydrolysis, bis((4-bromo-2,6-difluorophenyl)thio)methane 1b_CH2 was formed to a significant extent (ESI, Scheme S1). The structures of 1b_D and 1b_CH2 were confirmed by single-crystal X-ray diffraction. The extensive formation of 1b_CH2 is intriguing but not fully clear. It was rationalized by the reaction of 1b_D with lithium enolate formed from LDA-induced cleavage of THF according to the mechanism proposed for a similar transformation33 (ESI,† Scheme S2). Thiophenol 2b was synthesized by the reduction of 4-bromo-2-fluorobenzene-1-sulfonyl chloride with PPh3. Compounds 1b and 2b were converted to respective TBDMS thioethers 1c and 2c which were subjected to deprotonation with LDA in THF at −78 °C followed by trapping of corresponding aryllithium intermediates with chlorodimethylsilane.23 In both cases, the reaction occurred regioselectively at the position between fluorine and bromine atoms in agreement with a strong cumulated ortho-acidifying effect of those two halogen substituents.34,35 Finally, the conversion of functionalized arylsilanes 1d and 2d to respective benzosiloxaboroles 1e and 2e was performed as described by us previously.23,25 It involved a Br/Li exchange reaction using t-BuLi in Et2O and subsequent boronation of resultant aryllithiums with B(OMe)3 at very low temperatures (≤95 °C) followed by hydrolysis. The simultaneous Si–H bond cleavage and deprotection of the thiol group was cleanly performed under alkaline conditions. After acidification, 1e and 2e were isolated as white powders. | ||
| Scheme 1 Synthesis of thiol-functionalized fluorinated benzosiloxaboroles 1e and 2e. | ||
The incorporation of various side chains into the structure of benzosiloxaboroles was achieved through the thiol-Michael addition reaction with 1e and 2e as effective S-nucleophiles (Table 1). The thiol-Michael addition is broadly applicable and usually proceeds under mild conditions; moreover, it can be regarded as a “click chemistry” method owing to 100% atom economy.36,37 The syntheses involving selected Michael acceptors proceeded smoothly under relatively mild conditions (temperature range from 0 to 25 °C) and resulted in preparation of 19 S-linked functionalized benzosiloxaboroles with good yields (>70%).
|
|
|||||
|---|---|---|---|---|---|
| Compound | X | R | Michael acceptor | Base | Yield, % |
| 3 | F |

|
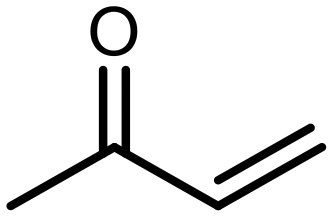
|
— | 90 |
| 4 | H |

|

|
NaHCO3 | 93 |
| 5 | F |

|

|
Et3N | 79 |
| 6 | F |

|

|
K2CO3 | 89 |
| 7 | F |

|
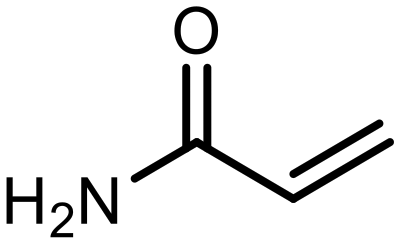
|
K2CO3 | 80 |
| 8 | F |

|

|
K2CO3 | 90 |
| 9 | F |

|

|
K2CO3 | 82 |
| 10 | F |

|

|
K2CO3 | 86 |
| 11 | F |

|

|
K2CO3 | 75 |
| 12 | F |

|

|
K2CO3 | 87 |
| 13 | H | 90 | |||
| 14 | F |

|

|
Et3N | 83 |
| 15 | H | 85 | |||
| 16 | F |

|
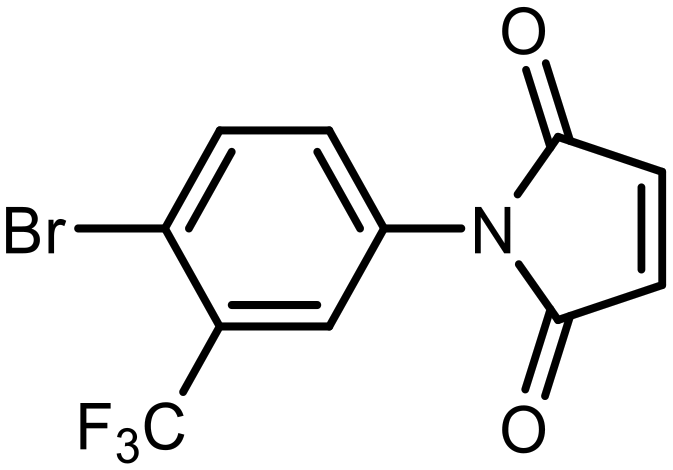
|
Et3N | 79 |
| 17 | H | 85 | |||
| 18 | F |
|

|
K2CO3 | 83 |
| 19 | H | 79 | |||
| 20 | F |

|

|
K2CO3 | 81 |
| 21 | H | 72 | |||

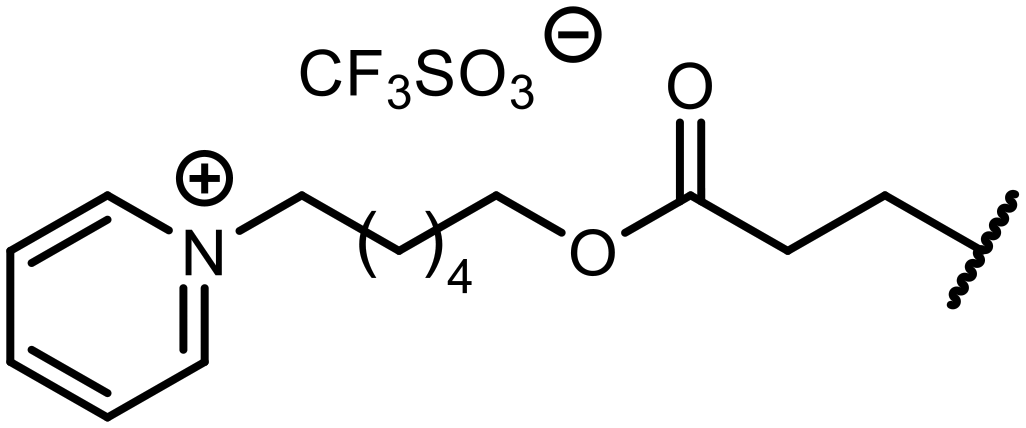
In the case of compound 3, the reaction occurred in water without the need for the use of a base. All other syntheses required the use of a base to generate more effectively active anionic forms of 1e and 2e. The wide representation of Michael acceptors used in the syntheses of benzosiloxaboroles 3–21 includes α,β-unsaturated ketones, esters, nitriles, amides, imides and sulfones. Reactions leading to compounds 3, 4, 6, 7, 12, 13, 20 and 21 utilized readily available substrates. Protocols for the preparation of the Michael acceptors used in the syntheses of 5, 9–11, 14–19 are available in the ESI.† We assumed that the introduction of pendant substituents containing various polar end groups to the benzosiloxaborole may result in specific interactions with targeted biomolecules which would be beneficial for antibacterial activity. Moreover, benzosiloxaboroles 7–12 containing an amide group constitute an interesting group as it seems that bioconjugates of benzosiloxaboroles with amino acids or peptides could be obtained analogously. Benzosiloxaboroles 12–17 possess a pendant succinimide ring where the presence of a nitrogen atom enables further functionalization. Compounds 18 and 19 are specific as they can be regarded as ionic liquids (ILs) which might potentially improve solubility and the drug delivery process.38 Derivatives 20 and 21 feature a pendant phenylsulfonyl moiety and thus show structural similarity to benzosiloxaboroles decorated with arylsulfonate and arylsulfonamide groups which exhibit potent activity against Gram-positive cocci.26,27
Benzosiloxaboroles bearing side chains linked via the thioether group can also be obtained by treatment of 1e or 2e with electrophilic partners comprising reactive C–Hal (Hal = Cl, Br) or P–Cl bonds. Thus, respective products 22–25 were isolated using α-bromoketones, 4-(trifluoromethyl)benzoyl chloride and diethyl chlorophosphate (Scheme 2).
 | ||
| Scheme 2 Synthesis of benzosiloxaboroles 22–25 through treatment of 1e or 2e with electrophiles featuring reactive C–Hal or P–Cl bonds. | ||
Thiols can also be easily oxidized into sulfonyl chlorides using trichloroisocyanuric acid (TCCA).39,40 Accordingly, benzosiloxaborole 26 was prepared by chemoselective oxidation of the SH group in 1e and subsequently converted to sulfonamides 27–29 (Scheme 3). Since sulfonamides are widely exploited as antibacterial drugs,41–44 we reasoned that their combination with the benzosiloxaborole scaffold could result in enhanced antimicrobial potency.
 | ||
| Scheme 3 Oxidation of 1e to the corresponding sulfonyl chloride derivative 26 and subsequent conversion to sulfonamides 27, 28 and 29. | ||
The obtained compounds are white solids that are well soluble in most organic solvents. They were characterized by multinuclear NMR (1H, 13C, 11B, 19F, 31P) spectroscopy and HRMS analyses. The molecular structures of selected benzosiloxaboroles 1e, 6, 11, 12, 14, 15, 20, 22, 24, 26, 27 and 29 were determined by X-ray crystallography (see Fig. S3 in the ESI†) showing that the geometric parameters of benzosiloxaborole cores are similar as in structures reported previously.45
Since acidity is an important parameter in medicinal chemistry, the pKa values of the benzosiloxaborole precursors 1e and 2e, as well as the selected promising compounds 6, 20 and 22–24 were measured (Table 2) by potentiometric titration with aq. 0.1 M NaOH in H2O/MeOH (1:1 v/v). In the case of 1e and 2e possessing an acidic thiol group, both apparent pKa1 and pKa2 values were obtained. Since the acidic properties of the aromatic thiol group46 and siloxaborole23 moiety are comparable, it is likely that pKa1 values represent the formation of an equilibrium mixture of both possible monoanions, i.e., the species with the deprotonated SH group and the boronate anion resulting from the coordination of OH− to the boron atom. Difluoro-substituted benzosiloxaboroles 1e, 6, 20 and 24 are characterized by approximately one unit lower pKa values than their monofluoro counterparts 2e, 22 and 23, which is consistent with previous findings.23 Overall, pKa values of mono- and difluoro derivatives are very similar within each of these groups indicating that the type of pendant sulfur-based moiety plays a minor role. All studied compounds are sufficiently acidic to exist in respective anionic forms under standard physiological conditions (pH = 7.4), which should be beneficial for solubility in biological systems.
:1)
Antimicrobial activity
We have recently proven that various benzosiloxaboroles display high antibacterial and antifungal activities.23,25–27 Thus, in this study, we tested the ability of all obtained compounds to inhibit the growth of selected standard strains of bacteria (6 Gram-positive strains and 11 Gram-negative strains) and yeasts (7 strains). All obtained results are presented in Table 3 and Tables S1 and S2 in the ESI.† Most compounds displayed moderate to weak activity against Gram-positive cocci, with MICs ranging from 12.5 to >400 μg mL−1 (Table 3). However, derivatives 6, 12, 20, and 22–24 were highly active against standard staphylococci, including MRSA, with MIC = 1.56–6.25 μg mL−1. MRSA strains are of great clinical concern. Not only are they resistant to almost all β-lactams (except ceftaroline and ceftobiprole), but they are often resistant to many other antibiotic classes (macrolides, tetracyclines, aminoglycosides, fluoroquinolones), which severely limits therapeutic options.47,48 Consequently, they entered the WHO list of the most dangerous high-priority pathogens, for which the search for new antibiotics is urgently needed.49 Thus, in this study, agents highly active against the standard MRSA strain were subsequently tested against five clinical MRSA strains (Table 4). Compound 23 bearing the benzoylmethylthio functionality displayed the highest potency (MIC = 1.56–3.12 μg mL−1), followed by 2-(phenylsulfonyl)ethylthio- and acetylmethylthio derivatives 20 and 22, respectively (MRSA: MIC = 3.12–6.25 μg mL−1). Their activity was comparable or only 3- to 6-fold lower than our reference agent linezolid, which is an antibiotic indicated in infections caused by multi-drug resistant Gram-positive cocci, including MRSA.48 Interestingly, their MICs for S. aureus were comparable with linezolid breakpoints (according to CLSI, strains are classified as sensitive when linezolid MIC is ≤ 4 μg mL−1 and as resistant when MIC is ≥8 μg mL−1).50 Thus, 20, 22, and 23 are promising anti-MRSA agents comparable with the previously described N-methyl arylsulfonamide benzosiloxaboroles (MRSA: MIC = 3.12–6.25 μg mL−1).27 It should be noted that the structural homologue of 22, namely compound 3, bearing the additional methylene spacer between the sulfur atom and the carbonyl group, is much less active.| Agentc | MIC/[MBC],a μg mL−1 (diameter of inhibition zone, mm) | |||||
|---|---|---|---|---|---|---|
| S. aureus ATCC 6538P | S. aureus ATCC 43300 MRSA | S. epidermidis ATCC 12228 | E. faecalis ATCC 29212 | E. faecium ATCC 6057 | B. subtilis ATCC 6633b | |
| The highest activity against Gram-positive bacteria indicated by the low MIC values (≤ 6.25 μg mL−1) is shown in boldface.(—): the inhibition zone was not observed in the disc-diffusion method. The diameter of the paper discs was 9 mm.a Only the MBC values ≤400 μg mL−1 are presented.b The growth type of B. subtilis in MHB medium prevented reading the MIC values of tested agents.c The MIC and MBC values of the agent were determined up to 200 μg mL−1. In the table, only MBC values ≤200 μg mL−1 are presented. The tested agent dissolved in DMSO precipitated after implementation into MHB medium at a concentration above 200 μg mL−1.d The MIC and MBC values of the agent were determined up to 100 μg mL−1. In the table, only MBC values ≤100 μg mL−1 are presented. The tested agent dissolved in DMSO precipitated after implementation into MHB medium at a concentration above 100 μg mL−1.e The MIC and MBC values of the agent were determined up to 50 μg mL−1. In the table, only MBC values ≤50 μg mL−1 are presented. The tested agent dissolved in DMSO precipitated after implementation into MHB medium at a concentration above 50 μg mL−1.f The Eagle effect was observed during the determination of the MBC value of the same tested agents against S. aureus strains.51,52 The Eagle effect is shown in italic face.g LIN, linezolid, was used as a reference agent active against Gram-positive bacteria. The diameter of a commercial disc containing 0.03 mg of linezolid was 6 mm; the MIC of linezolid was determined according to the CLSI recommendations.53 | ||||||
| 1e | 12.5 [25] (19) | 12.5 [25] (19) | 1.56 [50] (24) | 12.5 [100] (17) | 12.5 [400] (24) | NT (22) |
| 2e | 12.5 [25] (20) | 12.5 [25] (20) | 6.25 [25] (23) | 12.5 (18) | 12.5 (18) | NT (23) |
| 3 | 50 [100] (23) | 50 [200] (19) | 25 [200] (25) | 25 [200] (19) | 12.5 [200] (21) | NT (25) |
| 4 | 12.5 [25/400]f (27) | 12.5 (24) | 25 (22) | 200 (12) | 100 (13) | NT (23) |
| 5 | 400 (15) | 400 (—) | 400 (—) | 400 (—) | 400 (—) | NT (—) |
| 6 | 6.25 [12.5/>400]f (30) | 12.5 (19) | 50 (—) | 400 (—) | 400 (—) | NT (23) |
| 7 | 50 (24) | 100 (—) | 100 (19) | 400 (12) | 200 (12) | NT (15) |
| 8 | 50 (25) | 50 (25) | 100 (19) | 400 (—) | 200 (—) | NT (14) |
| 9 | 25 [400] (17) | 50 [400] (12) | 50 [400] (11) | 200 (13) | 200 (15) | NT (15) |
| 10 | 50 [400] (24) | 50 [400] (25) | 50 [400] (22) | 200 (13) | 100 (15) | NT (19) |
| 11 | 25 [400] (21) | 50 [400] (14) | 25 [400] (13) | 200 (12) | 200 (14) | NT (17) |
| 12 | 6.25 [100] (20) | 12.5 [200] (23) | 6.25 [400] (28) | 25 (20) | 12.5 (23) | NT (27) |
| 13 | 25 [200] (20) | 25 (22) | 25 (24) | 25 (19) | 12.5 (20) | NT (24) |
| 14 | 50 (22) | 50 (24) | 50 (20) | 25 (20) | 50 (20) | NT (19) |
| 15 | 25 (23) | 25 (21) | 12.5 (21) | 12.5 (20) | 25 (22) | NT (19) |
| 16 | 50 [100] (20) | 50 [100] (20) | 50 [100] (18) | 25 [200] (18) | 50 (18) | NT (19) |
| 17 | 25 [100] (19) | 25 [100] (19) | 12.5 [100] (18) | 12.5 [200] (17) | 25 (18) | NT (19) |
| 18 | 25 [100] (19) | 50 [200] (17) | 50 [200] (19) | 200 (—) | 100 (—) | NT (18) |
| 19 | 25 [100] (20) | 25 [100] (18) | 25 [100] (21) | 100 [400] (12) | 100 (12) | NT (19) |
| 20 | 3.12 [12.5/200]f (30) | 6.25 [200] (28) | 25 [400] (25) | 100 (14) | 100 (15) | NT (25) |
| 21 | 12.5 [200] (26) | 12.5 [400] (25) | 25 (22) | 200 (15) | 200 (13) | NT (21) |
| 22 | 1.56 [200] (25) | 6.25 [200] (25) | 6.25 [12.5] (23) | 25 [400] (16) | 25 (12) | NT (24) |
| 23 | 1.56 [3.12/200]f (31) | 3.12 [25/100]f (31) | 12.5 [50] (25) | 50 [200] (17) | 50 (17) | NT (24) |
| 24 | 6.25 [12.5/>400]f (32) | 12.5 [>400] (27) | 100 (20) | >400 (11) | 400 (11) | NT (21) |
| 25 | 12.5 [25] (11) | 12.5 [25] (11) | 3.12 [25] (16) | 12.5 [25] (11) | 12.5 [>50] (12) | NT (15) |
| 26 | 200 [400] (14) | 200 (15) | 200 (13) | 200 (14) | 100 (14) | NT (15) |
| 27 | 400 (—) | >400 (—) | >400 (—) | 400 (—) | 400 (—) | NT (—) |
| 28 | 50 (22) | 100 (16) | 50 [100] (19) | 400 (—) | 400 (—) | NT (22) |
| 29 | 200 [400] (16) | 400 (—) | 400 (—) | > 400 (—) | 400 (—) | NT (12) |
| LIN | 1 [>128] (25) | 2 [>128] (25) | 1 [>128] (26) | 2 [>128] (15) | 2 [>128] (14) | NT (30) |
| Agent | MIC [MBC], μg mL−1 | |||||
|---|---|---|---|---|---|---|
| ATCC 43300 MRSA | NMI 664 K MRSA | NMI 1576 K MRSA | NMI 1712 K MRSA | NMI 1991 K MRSA | NMI 2541 K MRSA | |
| The highest activity against S. aureus strains indicated by the low MIC values (≤6.25 μg mL−1) is shown in boldface.a The Eagle effect was observed when determining the MBC value of the same tested agents against S. aureus strains.51,52 The Eagle effect is shown in italic face.b LIN, linezolid, was used as a reference agent, active against S. aureus strains. The MIC of linezolid was determined according to the CLSI recommendations.53 | ||||||
| 6 | 12.5 [>400] | 12.5 [25/>400]a | 12.5 [25/>400]a | 12.5 [12.5/>400]a | 12.5 [25/>400]a | 12.5 [25/>400]a |
| 12 | 12.5 [200] | 12.5 [200] | 6.25 [100] | 12.5 [200] | 12.5 [100] | 12.5 [200] |
| 20 | 6.25 [200] | 6.25 [12.5/400]a | 6.25 [12.5/400]a | 6.25 [12.5/400]a | 6.25 [12.5/400]a | 6.25 [25/400]a |
| 22 | 6.25 [200] | 6.25 [200] | 6.25 [200] | 6.25 [400] | 3.12 [200] | 3.12 [200] |
| 23 | 3.12 [25/100]a | 3.12 [3.12/200]a | 3.12 [6.25/100]a | 3.12 [25/200]a | 1.56 [3.12/200]a | 1.56 [6.25/200]a |
| 24 | 12.5 [>400] | 6.25 [25/>400]a | 6.25 [12.5/400]a | 12.5 [25/>400]a | 12.5 [12.5/>400]a | 12.5 [12.5/>400]a |
| 25 | 12.5 [25] | 12.5 [25] | 25 [50] | 12.5 [25] | 25 [50] | 12.5 [25] |
| LIN | 2 [>128] | 1 [>128] | 1 [>128] | 1 [>128] | 1 [>128] | 1 [>128] |
The minimal bactericidal concentrations (MBCs) of most tested agents for staphylococci were established at 2–16 × MIC, whereas for enterococci these values were usually above the highest tested concentration (>400 μg mL−1) (Table 3). The lowest MBC values against S. aureus strains were obtained for compounds 1e, 2e and 25 (MBCs 25–50 μg mL−1). Interestingly, for 4, 6, 20, 23, and 24 the Eagle effect (also known as the paradoxical growth)51,52 was observed in the case of S. aureus ATCC 6538P and five MRSA clinical strains, which is in line with previous results obtained for benzosiloxaboroles decorated with arylsulfonate26 and N-methyl arylsulfonamide27 groups. Consequently, two MBC values were determined (Tables 3 and 4). According to CLSI recommendations, MBC is the lowest concentration that kills at least 99.9% of bacteria.54 In our study, the first MBCs were 2- or 4-fold higher than the MICs. However, a progressive increase in the number of surviving bacteria was observed at concentrations beyond it, followed by a subsequent decrease at 100–>400 μg mL−1. If the bacterial population was reduced again to the MBC threshold, a second MBC value was reported.
Furthermore, only agents 12 and 13 displayed weak activity against Gram-negative rods with MICs ranging from 25 to >400 μg mL−1 (ESI,† Table S1). However, considering that Gram-negative bacilli resistance is frequently associated with efflux pumps' activity,55–57 we also determined the MICs in the presence of their inhibitor, i.e., phenylalanine-arginine-β-naphthylamide (PAβN).57–59 It turned out that the activity of agents 12 and 13 against Enterobacterales is affected by the efflux phenomenon, as their MICs are reduced at least 4-fold in the presence of PAβN. Compounds 4, 20, and 21 are also actively extruded from bacterial cells, whereas for other compounds the efflux assay confirmed the lack of any activity.
We have previously reported that some benzosiloxaboroles are potent antifungals, with MICs ranging from 0.78–12.5 μg mL−1.23,26,27 Thus, we investigated the activity of our new derivatives against 7 standard yeasts. The moderate antifungal activity was found for agents 14–17, and 25 with MICs ranging from 12.5 to >200 μg mL−1 for Candida spp. and from 6.25–200 μg mL−1 for Saccharomyces cerevisiae (ESI,† Table S2). Compounds 1e, 3, 4, 18, 19, 21–23 displayed only weak activity with MICs ranging from 100 to >400 μg mL−1 for Candida spp. and 6.25–400 μg mL−1 for S. cerevisiae.
Cytotoxicity studies
To evaluate the cytotoxic effect of the tested compounds, an MTT-based assay was performed. Human normal lung fibroblasts MRC-5 were treated with representative compounds including the most active ones in the concentration range of 6.25–400 μg mL−1 for 48 h. All viability data are summarized in Table S3 (ESI†). Whenever possible, the respective IC50 values were calculated and summarized in Table 5. The representative plots demonstrating sigmoidal dose–response curves for the tested compounds are shown in the ESI† (Fig. S1). Overall, the results indicate rather weak cytotoxicity as for most compounds (except for 16 and 28), IC50 values are above 100 μg mL−1. Moreover, based on available IC50 and MIC (for standard and clinical S. aureus MRSA strains) values, the selectivity index (SI) was calculated for several compounds as the IC50/MIC ratio (Table 5).60 Within the group of most active compounds (6, 12, 20, 22–24), SI values were generally higher than 10, indicating their potential as antibacterial agents with respect to S. aureus MRSA.| Agent | IC50 [μg mL−1] | SI for S. aureus ATCC 43300 MRSA | SI range for S. aureus MRSA clinical strains (NMI) |
|---|---|---|---|
| n/a – not applicable; SI values higher than 10 are shown in boldface. | |||
| 6 | 140.0 | 11.2 | 11.2 |
| 12 | 167.4 | 13.4 | 13.4–26.8 |
| 14 | 133.3 | 2.7 | n/a |
| 15 | 145.0 | 5.8 | n/a |
| 16 | 76.2 | 1.5 | n/a |
| 17 | 139.5 | 5.6 | n/a |
| 20 | 122.0 | 19.5 | 19.5 |
| 22 | >200 | >32.0 | >32.0 |
| 23 | 126.1 | 40.4 | 40.4–80.8 |
| 24 | >400 | >32.0 | >32.0 |
| 25 | 111.7 | 8.9 | 4.5–8.9 |
| 27 | >400 | > | n/a |
| 28 | 74.9 | 0.7 | n/a |
| 29 | >400 | > | n/a |
| LIN | >400 | >200 | >400 |
| Cisplatin | 1.9 | n/a | n/a |
Studies on the mechanism of antibacterial activity
We have undertaken research devoted to determining the most probable mechanism of antibacterial action. Numerous reports indicate that related benzoxaboroles exhibit antimicrobial activity through the oxaborole tRNA trapping (OBORT) mechanism10,61–65 involving inhibition of leucyl-tRNA synthetase (LeuRS). Considering structural similarity to benzoxaboroles we assumed that benzosiloxaboroles are active through this mechanism. Our working hypothesis was supported by the fact that benzosiloxaboroles are rather bacteriostatic than bactericidal,26,27 which is consistent with the specificity of the OBORT mechanism. The enzyme inhibition assay was performed for compound 20 as it features high activity against S. aureus MRSA strains and the respective favorable SI = 19.5 (Table 5). Moreover, due to the presence of the SO2 group in the pendant substituent, it shows structural analogy to previously reported benzosiloxaboroles bearing sulfonate and sulfonamide groups which also exhibit high activity against S. aureus.26,27 We obtained S. aureus MRSA LeuRS in the E. coli expression system. From 400 mL culture, approximately 20 mg of 6× His tagged LeuRS (with a molecular mass of 92.3 kDa) was purified to more than 90% homogeneity, which was confirmed by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Fig. S2, ESI†). Next, we used an in vitro aminoacylation assay to investigate the ability of 20 to inhibit S. aureus MRSA LeuRS activity consistent with affecting the transfer of the 14C-radiolabeled leucine to a total tRNA. We found that 20 did not show the expected activity (residual activity of 92.9% at 25 μM), compared to reference arylsulfonamide-substituted benzoxaborole PT662 known as a potent S. aureus MRSA LeuRS inhibitor10 (residual activity 30.8% at 25 μM).Subsequent bioinformatic analysis provided possible explanations for this disclosure. Compound 20 possesses a high degree of structural similarity to potent antibacterial compound 1-((4-((1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-6-yl)oxy)phenyl)thio)propan-2-one,65AceSPhO_BOB, which has been crystallized in complex with Streptococcus pneumoniae leucyl-tRNA synthetase (Fig. 3a, right). By using the crystallized complex as a reference, we generated a putative S. aureus MRSA LeuRS/(20 bound to AMP) complex through docking. A comparison of binding modes of both compounds (Fig. 3b) revealed that although the oxaborole moiety of 20 occupies a similar space within the protein as for AceSPhO_BOB,65 the pendant groups of both compounds are oriented very differently. In the case of 20, the phenylsulfonyl group forms tight interactions with LeuRS, penetrating the cavity between structured regions of the protein and a loop (Fig. 3c). Clearly, it is not surprising as in 20 the large substituent is attached at the para position with respect to the boron atom, while for AceSPhO_BOB (ref. 65) an analogous moiety is located at the meta position (Fig. 3a). It should be stressed that the loop comprises mostly polar and negatively charged residues (Fig. 3c). Such an environment is highly unfavorable for the primarily hydrophobic pendant group of 20 which would likely aggravate binding of this compound.
 | ||
| Fig. 3 Structural determinants of LeuRS inhibition by oxaborole derivatives; (a) 2D representations of ligands. Since known LeuRS oxaborole inhibitors work by forming complexes with tRNA nucleotides, both compounds have been represented as AMP adducts. The studied compound 20 is depicted on the left and a known potent inhibitor (AceSPhO_BOB (ref. 65)) is depicted on the right; (b) comparison of the binding modes of AMP adducts of both compounds with LeuRS. On the left, the binding mode of compound 20 obtained through docking (using MOE (ref. 66)) into a homology model of the S. aureus MRSA LeuRS, and on the right, the binding mode of AceSPhO_BOB (ref. 65) as seen in the crystal structure [PDB code: 7BZJ]. Residues in proximity of the ligands are colored according to the residue property (white – nonpolar, green – polar, blue – negatively charged, red – positively charged). Labels were added to selected residues adjacent to the ligand for reference; (c) contacts between the pendant group of both compounds and its interaction with a polar LeuRS loop. The loop is colored according to the residue property (the same color code as above). | ||
To further evaluate the stability of the generated complex, we have carried out extensive (5 × 1 μs) unbiased MD simulation of the generated ligand, i.e., (20 bound to AMP)/LeuRS complex. Importantly, as parameters for silicon atoms are not available within the utilized simulation forcefield, this atom in the ligand structure was replaced with a carbon atom (as it shares multiple chemical properties with silicon atoms). Unbiased simulations were performed in order to observe changes in the structure of the complex, which could help rationalize the lack of affinity of studied compounds towards LeuRS. The relative positions of the protein and atoms in the benzosiloxaborole core and the pendant moiety were plotted to monitor the stability of the ligands (Fig. 4a). Intriguingly, we found that in each of the replicates both fragments assume distances characteristic to unbound conformations. A more detailed analysis of one of the unbinding events (Fig. 4b) observed in the unbiased simulations reveals the following sequence of events: in the initial frames the polar loop quickly unbinds (likely due to unfavorable interactions with the ligand). This is followed by destabilization of the pendant group, which later leads to full unbinding and displacement of 20. The observed unbinding event, preceded by the opening of the polar loop, would suggest that 20 does not interact with LeuRS as the largely polar and electronegative environment of the loop cannot accommodate the hydrophobic and electronegative pendant moiety of the studied ligand.
 | ||
| Fig. 4 MD evaluation of interactions between the ligand – AMP adduct of benzosiloxaborole 20 and S. aureus MRSA LeuRS; (a) initial structure of the (AMP adduct of 20)/LeuRS complex subjected to 5 × 1 μs of unbiased MD simulations. The distances between the protein and the benzosiloxaborole core (blue) and the pendant group (yellow) are depicted as lines, and their evolution across the simulations is plotted; (b) evolution of the position of the AMP adduct of 20 across one unbiased simulation run. The loop initially interacting with the ligand is colored based on the residue property (white – nonpolar, green – polar, red – negatively charged). | ||
The lack of activity of 20 towards LeuRS indicates a different mechanism of antibacterial activity. Bioinformatic analysis suggests that this inability might be linked with the placement of its bulky sulfur-linked moiety in the para position with respect to the boron atom. Importantly, this would also suggest that none of the compounds within this series, which have a large hydrophobic/negatively charged pendant group in the para position, would inhibit LeuRS. Although LeuRS remains the most frequently identified molecular target for antimicrobial organoboron compounds, few examples of differently acting boron-based antimicrobial agents are also known. For instance, in the case of bis(indolyl)methylboronic acid derivatives, it was found that their antibacterial activity might be linked with a strong binding affinity to the peptidoglycan layer of the Gram-positive bacteria cell wall.67 On the other hand, some benzoxaboroles were identified as potent inhibitors of other bacterial enzymes, such as NADH dehydrogenase,9 enoyl acyl carrier protein FabI68 or carbonic anhydrases.69 These findings are in line with our disclosure and prove that benzosiloxaboroles, like other boron-based antimicrobials, could utilize another antibacterial mechanism than OBORT as well.
Conclusions
Thiol-functionalized benzosiloxaboroles proved to be very useful precursors for the preparation of a wide variety of novel derivatives. Most of them were obtained by the thiol-Michael addition reaction, which turned out to be a convenient tool for the preparation of many structurally diverse benzosiloxaboroles. Some of them were also synthesized through the nucleophilic substitution reaction of thiolates with appropriate electrophilic partners as well as through chemoselective SH-oxidation and subsequent transformations. We intended to decorate new derivatives with extended substituents containing multiple polar groups as we assumed that such functionalizations may result in a stronger and specific binding to biological targets by means of polar interactions, e.g., hydrogen bonds. Regardless of the substitution pattern at the sulfur atom, the studied benzosiloxaboroles display a comparable acidity (pKa of 4.7–6.4 in H2O/MeOH, 1:1). Compounds 6, 12, 20 and 22–24 show strong bacteriostatic activity, especially against S. aureus, including multidrug-resistant clinical strains (S. aureus MRSA). Overall, moderate activity against Gram-positive bacteria is common. Furthermore, SAR analysis indicates that the antibacterial properties are enhanced for difluorinated derivatives. Following the initial premises, the antibacterial activity of the studied benzosiloxaboroles seemed to be based on the tRNA-targeting OBORT mechanism. However, experimental investigation revealed that the studied agent 20 does not effectively inhibit LeuRS as IC50 > 200 μM. Hence, it can be concluded that benzosiloxaboroles have a different molecular target in a bacterial cell than benzoxaboroles. The lack of affinity to LeuRS was also confirmed by bioinformatic structural analysis. To summarize, the potential of SH-substituted benzosiloxaboroles for the synthesis of a diverse library of structurally extended derivatives was amply demonstrated and represents a significant progress in the field. Selected results of antimicrobial activity screening are promising. Hopefully, the presented findings will be followed by future research, eventually leading to development of new boron-based antibiotics.
Experimental section
General comments
Solvents used for reactions were dried by heating to reflux with sodium/benzophenone and distilled under argon. Starting materials and other reagents including halogenated benzenes, alkyllithiums, diisopropylamine, trimethyl borate, tert-butyl(chloro)dimethylsilane, or chlorodimethylsilane were used as received without further purification. Reactions in which organometallic compounds were used were carried out under an argon atmosphere. Detailed procedures for the synthesis of Michael acceptors used in reactions leading to the formation of compounds 5, 9–11, 14–19 as well as α-bromoketones used in syntheses of 22 and 23 are given in ESI,† section S1. 1H, 13C, 19F and 31P NMR spectra were recorded on an Agilent NMR 400 MHz DDR2 spectrometer. 11B NMR spectra were recorded on a Bruker AVANCE III 300 MHz spectrometer. In the 13C NMR spectra the resonances of boron-bound carbon atoms were not observed in most cases as a result of their broadening by a quadrupolar boron nucleus. 1H and 13C NMR chemical shifts are given relative to TMS using residual solvent resonances. 11B and 19F NMR chemical shifts are given relative to BF3·Et2O and CFCl3, respectively. 31P NMR chemical shifts are given relative to 85% phosphoric acid solution in D2O. CSD deposition numbers 2190658 (for 2e), 2190659 (for 1b_D), 2190660 (for 1b_CH2), 2190661 (for 6), 2190662 (for 11), 2190663 (for 12), 2190664 (for 14), 2190665 (for 15), 2190666 (for 20), 2296881 (for 22), 2190667 (for 24), 2258836 (for 26), 2190668 (for 29), 2190669 (for 27), contain the ESI† crystallographic data for this paper.Synthesis
:1) was added dropwise to a solution of t-BuLi (1.8 M in pentane, 52.0 mL, 0.094 mol) in Et2O (150 mL) at −95 °C. After 30 min of stirring at −95 °C, B(OMe)3 (10.4 mL, 9.73 g, 0.094 mol) was added slowly to the colorless mixture at −95 °C. The resulting white suspension was warmed slowly to ca. 0 °C, quenched with 1 M aq. NaOH (100 mL) and stirred at room temperature until evolution of H2 ceased. The two-phase mixture was concentrated under reduced pressure in order to remove solvents and other volatile organic components. The residual aqueous alkaline solution was transferred to a separatory funnel and washed with hexane (60 mL). Then it was placed in a beaker, cooled in an ice bath and carefully neutralized by an addition of ca. 50 mL 2 M aq. H2SO4 (to reach the pH = 1). The precipitated abundant white solid was filtered and washed several times with distilled water. Then it was recrystallized with heptane (70 mL) and filtered. Finally, the product was dried in vacuo to give 1e as a white powder. Yield 8.80 g (76%). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 7.9 Hz, 1H), 5.82 (s, 1H), 3.71 (s, 1H), 0.49 (s, 6H) ppm. 11B NMR (96 MHz, CDCl3) δ 29.4 ppm. 13C NMR (101 MHz, CDCl3) δ 161.1 (dd, J = 249.5, 3.9 Hz), 160.3 (dd, J = 244.5, 5.3 Hz), 159.1 (d, J = 5.3 Hz), 138.6 (broad), 129.6 (dd, J = 35.5, 2.5 Hz), 114.1 (dd, J = 19.8, 3.2 Hz), 112.1 (dd, J = 25.0, 22.0 Hz), −0.8 (s) ppm. 19F NMR (376 MHz, CDCl3) δ −100.05 (d, J = 8.1 Hz), −106.07 (t, J = 7.9 Hz) ppm. HRMS (ESI, negative ion mode) calcd. for C8H8BF2O2SSi− [M − H]−: 245.0070; found: 245.0077.
:1, v/v). The mixture was stirred for 10 minutes at 50 °C. Then it was concentrated under reduced pressure to remove the organic solvent. The aqueous residue was acidified with 1.5 M aq. H2SO4. Upon addition of the acid, formation of a white solid was observed. The acidic aqueous phase was discarded and the solid residue was washed with water and hexane and vigorously stirred to give the suspension. Then it was filtered and the precipitate was dried in vacuo (ca. 10−3 mbar). The pure product 6 was obtained as a white powder. Yield 254 mg (80%). 1H NMR (400 MHz, DMSO-d6) δ 9.45 (s, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.31 (s, 1H), 6.85 (s, 1H), 3.05 (t, J = 7.2 Hz, 2H), 2.31 (t, J = 7.2 Hz, 2H), 0.40 (s, 6H) ppm. 11B NMR (96 MHz, DMSO-d6) δ 28.9 ppm. 13C NMR (101 MHz, DMSO-d6) δ 172.2, 164.6 (dd, J = 249.6, 3.0 Hz), 164.1 (dd, J = 245.5, 4.3 Hz), 145.3, 130.6 (dd, J = 33.9, 2.7 Hz), 115.0 (dd, J = 21.0, 3.3 Hz), 113.0 (dd, J = 25.3, 22.4 Hz), 35.8, 30.1 (t, J = 3.0 Hz), −0.3 ppm. 19F NMR (376 MHz, DMSO-d6) δ −96.36 (d, J = 6.8 Hz), −102.67 (t, J = 7.5 Hz) ppm. HRMS (ESI, positive ion mode) calcd. for C11H15BF2NO3SSi+ [M + H]+: 318.0598; found: 318.0595.
Determination of acidity constants (pKa values)
The acidity constants of benzosiloxaboroles 1e, 2e, 6, 20 and 22–24 were determined by standard pH titration with 0.05 M aq. NaOH. The measurements were performed using a thermostated (25.0 °C) glass vessel equipped with a magnetic stirrer and a 10 mL burette. Prior to the measurement, the pH glass electrode was calibrated using borax and phosphate buffers. A solution of the analyzed compound (ca. 20–40 mg) in a mixture of MeOH/H2O (v/v 1:1, 30 mL) was titrated until the pH of the solution exceeded the value of 12.0.
Antimicrobial activity
Direct antimicrobial activity was determined against the following standard strains: (1) Gram-positive cocci: methicillin-sensitive Staphylococcus aureus ATCC 6538P (MSSA), methicillin-resistant S. aureus subsp. aureus ATCC 43300 (MRSA), S. epidermidis ATCC 12228, Enterococcus faecium ATCC 6057, E. faecalis ATCC 29212, Bacillus subtilis ATCC 6633; (2) Gram-negative bacteria from Enterobacteriales order: Klebsiella pneumoniae ATCC 13883, Escherichia coli ATCC 25922, Enterobacter cloacae DSM 6234, Proteus mirabilis ATCC 12453, Serratia marcescens ATCC 13880; (3) Gram-negative non-fermentative rods: Acinetobacter baumannii ATCC 19606, Pseudomonas aeruginosa ATCC 27853, Stenotrophomonas maltophilia ATCC 12714, S. maltophilia ATCC 13637, Burkholderia cepacia ATCC 25416, Bordetella bronchiseptica ATCC 4617; (4) yeasts: Candida albicans ATCC 90028, C. krusei ATCC 6258, C. parapsilosis ATCC 22019, C. tropicalis (Castellani) Berkhout ATCC 750, C. tropicalis IBA 171, C. guilliermondii IBA 155, and Saccharomyces cerevisiae ATCC 9763. Moreover, 5 clinical strains of methicillin-resistant S. aureus were also included: NMI 664 K, NMI 1576 K, NMI 1712 K, NMI 1991 K, and NMI 2541 K. All strains were stored at −80 °C. Prior to testing, each bacterial strain was subcultured twice on tryptic soy agar TSA (bioMerieux) medium and yeast strains on Sabouraud dextrose agar (bioMerieux) for 24–48 h at 30 °C to ensure viability. The antimicrobial activity was evaluated as previously described26 by the disc-diffusion method and the MIC determination assays according to the EUCAST (ref. 71 and 72) and CLSI (ref. 53 and 73) recommendations. Assessment of bactericidal (MBC) and fungicidal (MFC) activities was performed according to the CLSI recommendations.54,74 The following reference agents were used: fluconazole (in the case of fungi), linezolid (for Gram-positive bacteria), and nitrofurantoin (for Gram-negative rods). The new benzosiloxaboroles were dissolved in DMSO (Sigma). Depending on the solubility, the MIC and MBC/MFC values were determined up to 50 mg L−1 for agent 23, up to 100 μg mL−1 for agent 2e, up to 200 μg mL−1 for agents 14, 15, 16, and 25, and up to 400 μg mL−1 for the remaining compounds.Considering that the MDR efflux pumps often contribute to Gram-negative rod resistance, the MIC values of studied agents, with or without the pump inhibitor, PAβN (20 μg mL−1) (Sigma), were determined.75 The MIC was determined using Mueller–Hinton II broth medium (MHB) (Becton Dickinson) by the broth microdilution method, according to the CLSI guideliness.53 To minimize the influence of PAβN on the destabilization of bacterial cell covers, the tests were conducted in the presence of 1 mM MgSO4 (Sigma).76 At least a 4-fold reduction in the MIC value after the addition of PAβN was considered significant.56,77
Cytotoxicity studies
MRC-5 pd30 human fibroblasts (ECACC) were cultured in MEME, Minimum Essential Medium Eagle (Merck), supplemented with 10% fetal bovine serum (Merck), 2 mM L-glutamine, antibiotics (100 U mL−1 penicillin, 100 μg mL−1 streptomycin, Merck) and 1% non-essential amino acids (Merck). Cells were grown in 75 cm2 cell culture flasks (Sarstedt), in a humidified atmosphere of CO2/air (5/95%) at 37 °C. MTT-based viability assay was conducted as described previously.26 Optical densities were measured at 570 nm using a BioTek microplate reader. All measurements were carried out in three replicates and the results expressed as a percent of viable cells versus control cells. Cisplatin (0.5–30 μg mL−1) and linezolid (6.25–400 μg mL−1) were used as the positive and negative control, respectively. The selectivity index (SI) was calculated as , in accordance with previously described protocols.60
, in accordance with previously described protocols.60
Studies on the mechanism of antibacterial action
000g for 20 min at 4 °C. The cleared lysate was loaded onto the HisTrap HP 5 ml column mounted on a AKTA Purifier10 FPLC system (GE Healthcare). S. aureus MRSA LeuRS was eluted with imidazole gradient in extraction buffer (50–500 mM). Fractions containing S. aureus MRSA LeuRS were dialyzed overnight against 25 mM HEPES (pH 8.0), 150 mM NaCl, 5 mM 2ME, 0–20% glycerol and stored at −20 °C. The protein concentration in the final solution was 2.86 mg mL−1 (determined by the Bradford method and bovine serum albumin as a standard).78
The pressure and temperature of the system were maintained at 1 bar and 303 K, respectively using the Nose-Hoover thermostat and the Parrinello–Rahman barostat. For non-bonding interactions, we applied a cutoff at 12 Å using the Verlet scheme, and a force switch at 10 Å for vdW interactions. Electrostatic interactions were treated using PME. System parameters were obtained from the CHARMM36m forcefield.84 Parameters of the ligand were extracted from the CGenFF forcefield85,86 using Paramchem.87–89 As the simulated ligand contained a silicon atom, for which parameters are not available in the CGenFF forcefield, it was replaced with a carbon atom (which holds similar chemical properties). Simulations were analyzed using VMD.90
Author contributions
S. L. and A. E. L.: conceptualization of the paper and supervision of the research; S. L.: funding acquisition, S. L. and A. E. L.: design of the experiments; K. N., A. K., and J. Ko.: synthesis and compound characterization, J. Kr.: antimicrobial activity studies; M. W. enzyme inhibition assay; P. W.: cytotoxicity assay; P. H. M.-U., K. D. and K. W.: single-crystal X-ray diffraction; K. N., T. M. S. and J. S.: bioinformatic analysis; K. N., A. E. L., and S. L.: analysis of all data; K. N., J. Kr., T. M. S., A. E. L. and S. L.: writing the original draft. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by National Science Centre (Poland) in the framework of the project UMO-2018/31/B/ST5/00210. Work implemented as a part of Operational Project Knowledge Education Development 2014–2020 co-financed by the European Social Fund (the TRIBIOCHEM interdisciplinary PhD Program for P. H. M.-U.). The computational part of the work has been performed under the Project HPC-EUROPA3 (INFRAIA-2016-1-730897), with the support of the EC Research Innovation Action under the H2020 Program; in particular, the authors gratefully acknowledge the support of Hospital del Mar Medical Research Institute (IMIM), Pompeu Fabra University and the computer resources and technical support provided by Barcelona Supercomputing Center (BSC). The work was supported by the Warsaw University of Technology.References
- K. Torssell, Ark. Kemi, 1957, 10, 507–511 CAS.
- V. V. Zhdankin, P. J. Persichini, L. Zhang, S. Fix and P. Kiprof, Tetrahedron Lett., 1999, 40, 6705–6708 CrossRef CAS.
- C. T. Liu, J. W. Tomsho and S. J. Benkovic, Bioorg. Med. Chem., 2014, 22, 4462–4473 CrossRef CAS PubMed.
- A. Adamczyk-Woźniak, K. M. Borys and A. Sporzyński, Chem. Rev., 2015, 115, 5224–5247 CrossRef PubMed.
- A. Nocentini, C. T. Supuran and J.-Y. Winum, Expert Opin. Ther. Pat., 2018, 28, 493–504 CrossRef CAS PubMed.
- G. R. Mereddy, A. Chakradhar, R. M. Rutkoski and S. C. Jonnalagadda, J. Organomet. Chem., 2018, 865, 12–22 CrossRef CAS.
- S. J. Baker, Y.-K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. R. K. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447–4450 CrossRef CAS PubMed.
- D. Ding, Q. Meng, G. Gao, Y. Zhao, Q. Wang, B. Nare, R. Jacobs, F. Rock, M. R. K. Alley, J. J. Plattner, G. Chen, D. Li and H. Zhou, J. Med. Chem., 2011, 54, 1276–1287 CrossRef CAS PubMed.
- A. Korkegian, T. O'Malley, Y. Xia, Y. Zhou, D. S. Carter, B. Sunde, L. Flint, D. Thompson, T. R. Ioerger, J. Sacchettini, M. R. K. Alley and T. Parish, Tuberculosis, 2018, 108, 96–98 CrossRef CAS PubMed.
- Y. Si, S. Basak, Y. Li, J. Merino, J. N. Iuliano, S. G. Walker and P. J. Tonge, ACS Infect. Dis., 2019, 5, 1231–1238 CrossRef CAS PubMed.
- T. Akama, Y. R. Freund, P. W. Berry, D. S. Carter, E. E. Easom, K. Jarnagin, C. S. Lunde, J. J. Plattner, F. Rock, R. Stefanakis, C. Fischer, C. A. Bulman, K. C. Lim, B. M. Suzuki, N. Tricoche, A. Mansour, U. DiCosty, S. McCall, B. Carson, J. W. McCall, J. McKerrow, M. P. Hübner, S. Specht, A. Hoerauf, S. Lustigman, J. A. Sakanari and R. T. Jacobs, ACS Infect. Dis., 2020, 6, 173–179 CrossRef CAS PubMed.
- T. Akama, S. J. Baker, Y.-K. Zhang, V. Hernandez, H. Zhou, V. Sanders, Y. Freund, R. Kimura, K. R. Maples and J. J. Plattner, Bioorg. Med. Chem. Lett., 2009, 19, 2129–2132 CrossRef CAS PubMed.
- Y. R. Freund, T. Akama, M. R. K. Alley, J. Antunes, C. Dong, K. Jarnagin, R. Kimura, J. A. Nieman, K. R. Maples, J. J. Plattner, F. Rock, R. Sharma, R. Singh, V. Sanders and Y. Zhou, FEBS Lett., 2012, 586, 3410–3414 CrossRef CAS PubMed.
- J. Zhang, F. Yang, Z. Qiao, M. Zhu and H. Zhou, Bioorg. Med. Chem. Lett., 2016, 26, 5797–5801 CrossRef CAS PubMed.
- J. Zhang, J. Zhang, G. Hao, W. Xin, F. Yang, M. Zhu and H. Zhou, J. Med. Chem., 2019, 62, 6765–6784 CrossRef CAS PubMed.
- S. K. Jonnalagadda, K. Wielenberg, C. T. Ronayne, S. Jonnalagadda, P. Kiprof, S. C. Jonnalagadda and V. R. Mereddy, Bioorg. Med. Chem. Lett., 2020, 30, 127259 CrossRef CAS PubMed.
- A. Markham, Drugs, 2014, 74, 1555–1558 CrossRef CAS PubMed.
- FDA approves Eucrisa for eczema, https://www.fda.gov/news-events/pressannouncements/fda-approves-eucrisa-eczema, (accessed 1 June 2023) Search PubMed.
- R. J. Wall, E. Rico, I. Lukac, F. Zuccotto, S. Elg, I. H. Gilbert, Y. Freund, M. R. K. Alley, M. C. Field, S. Wyllie and D. Horn, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9616–9621 CrossRef CAS PubMed.
- Safety and Tolerability Study of Acoziborole in g-HAT Seropositive Subjects (OXA004), https://www.clinicaltrials.gov/ct2/show/NCT05256017, (accessed 9 January 2023) Search PubMed.
- D. Tenero, G. Derimanov, A. Carlton, J. Tonkyn, M. Davies, S. Cozens, S. Gresham, A. Gaudion, A. Puri, M. Muliaditan, J. Rullas-Trincado, A. Mendoza-Losana, A. Skingsley and D. Barros-Aguirre, Antimicrob. Agents Chemother., 2019, 63(8), e00240 CrossRef CAS PubMed.
- Early Bactericidal Activity, Safety & Tolerability of Oral GSK3036656 in Combination With Delamanid or Bedaquiline, Delamanid in Combination With Bedaquiline, or Standard of Care in Male and Female Participants Aged 18 to 65 Years With Pulmonary Tuberculosis, https://clinicaltrials.gov/ct2/show/NCT05382312, (accessed 9 January 2023).
- A. Brzozowska, P. Ćwik, K. Durka, T. Kliś, A. E. Laudy, S. Luliński, J. Serwatowski, S. Tyski, M. Urban and W. Wróblewski, Organometallics, 2015, 34, 2924–2932 CrossRef CAS.
- M. Czub, K. Durka, S. Luliński, J. Łosiewicz, J. Serwatowski, M. Urban and K. Woźniak, Eur. J. Org. Chem., 2017, 2017, 818–826 CrossRef CAS.
- K. Durka, A. E. Laudy, Ł. Charzewski, M. Urban, K. Stępień, S. Tyski, K. A. Krzyśko and S. Luliński, Eur. J. Med. Chem., 2019, 171, 11–24 CrossRef CAS PubMed.
- P. Pacholak, J. Krajewska, P. Wińska, J. Dunikowska, U. Gogowska, J. Mierzejewska, K. Durka, K. Woźniak, A. E. Laudy and S. Luliński, RSC Adv., 2021, 11, 25104–25121 RSC.
- J. Krajewska, K. Nowicki, K. Durka, P. H. Marek-Urban, P. Wińska, T. Stępniewski, K. Woźniak, A. E. Laudy and S. Luliński, RSC Adv., 2022, 12, 23099–23117 RSC.
- K. Nowicki, P. Pacholak and S. Luliński, Molecules, 2021, 26, 5464 CrossRef CAS PubMed.
- K. Durka, M. Urban, M. Czub, M. Dąbrowski, P. Tomaszewski and S. Luliński, Dalton Trans., 2018, 47, 3705–3716 RSC.
- T. Akama, C. Virtucio, C. Dong, R. Kimura, Y.-K. Zhang, J. A. Nieman, R. Sharma, X. Lu, M. Sales, R. Singh, A. Wu, X.-Q. Fan, L. Liu, J. J. Plattner, K. Jarnagin and Y. R. Freund, Bioorg. Med. Chem. Lett., 2013, 23, 1680–1683 CrossRef CAS PubMed.
- Y. Xia, K. Cao, Y. Zhou, M. R. K. Alley, F. Rock, M. Mohan, M. Meewan, S. J. Baker, S. Lux, C. Z. Ding, G. Jia, M. Kully and J. J. Plattner, Bioorg. Med. Chem. Lett., 2011, 21, 2533–2536 CrossRef CAS PubMed.
- R. T. Jacobs, C. S. Lunde, Y. R. Freund, V. Hernandez, X. Li, Y. Xia, D. S. Carter, P. W. Berry, J. Halladay, F. Rock, R. Stefanakis, E. Easom, J. J. Plattner, L. Ford, K. L. Johnston, D. A. N. Cook, R. Clare, A. Cassidy, L. Myhill, H. Tyrer, J. Gamble, A. F. Guimaraes, A. Steven, F. Lenz, A. Ehrens, S. J. Frohberger, M. Koschel, A. Hoerauf, M. P. Hübner, C. W. McNamara, M. A. Bakowski, J. D. Turner, M. J. Taylor and S. A. Ward, J. Med. Chem., 2019, 62, 2521–2540 CrossRef CAS PubMed.
- Q. Chen, G. Yu, X. Wang, Y. Huang, Y. Yan and Y. Huo, Org. Biomol. Chem., 2018, 16, 4086–4089 RSC.
- J. Moyroud, J.-L. Guesnet, B. Bennetau and J. Mortier, Tetrahedron Lett., 1995, 36, 881–884 CrossRef CAS.
- F. Mongin and M. Schlosser, Tetrahedron Lett., 1996, 37, 6551–6554 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- P. Wadhwa, A. Kharbanda and A. Sharma, Asian J. Org. Chem., 2018, 7, 634–661 CrossRef CAS.
- C. Liu, B. Chen, W. Shi, W. Huang and H. Qian, Mol. Pharmaceutics, 2022, 19, 1033–1046 CrossRef CAS PubMed.
- N. Maraš, ChemSpider Synthetic pages, 2013, p. 597 Search PubMed.
- D. K. H. Ho, L. Chan, A. Hooper and P. E. Brennan, Tetrahedron Lett., 2011, 52, 820–823 CrossRef CAS.
- C. Supuran, Molecules, 2017, 22, 1642 CrossRef PubMed.
- F. Carta, A. Scozzafava and C. T. Supuran, Expert Opin. Ther. Pat., 2012, 22, 747–758 CrossRef CAS PubMed.
- S. Kumar Verma, R. Verma, F. Xue, P. Kumar Thakur, Y. R. Girish and K. P. Rakesh, Bioorg. Chem., 2020, 105, 104400 CrossRef CAS PubMed.
- S. S. A. Shah, G. Rivera and M. Ashfaq, Mini-Rev. Med. Chem., 2013, 13, 70–86 CrossRef CAS PubMed.
- K. Durka, A. Zuba, P. H. Marek-Urban, K. Nowicki, J. Drapała, K. Woźniak and S. Luliński, CrystEngComm, 2023, 25, 6329–6342 RSC.
- B. G. Cox, Acids and Bases: Solvent Effects on Acid-Base Strength, Oxford University Press, Oxford, UK, 1st edn, 2013 Search PubMed.
- T. J. Foster, FEMS Microbiol. Rev., 2017, 41, 430–449 CrossRef CAS PubMed.
- Y. Guo, G. Song, M. Sun, J. Wang and Y. Wang, Front. Cell. Infect. Microbiol., 2020, 10, 107 CrossRef PubMed.
- World Health Organization, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, on World Health Organization (WHO), 2017, https://apps.who.int/iris/handle/10665/311820, (accessed 1 June 2023) Search PubMed.
- CLSI, Performance standards for antimicrobial susceptibility testing, CLSI guideline M100-ED33, Clinical and Laboratory Standards Institute, Wayne, PA, USA, 33rd edn, 2023 Search PubMed.
- H. Eagle and A. D. Musselman, J. Exp. Med., 1948, 88, 99–131 CrossRef CAS PubMed.
- A. Prasetyoputri, A. M. Jarrad, M. A. Cooper and M. A. T. Blaskovich, Trends Microbiol., 2019, 27, 339–354 CrossRef CAS PubMed.
- CLSI, Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, Approved Standard, CLSI guideline M07-A9, Clinical and Laboratory Standards Institute, Wayne, PA, USA, 9th edn, 2012 Search PubMed.
- CLSI, Methods for determining bactericidal activity of antimicrobial agents, CLSI guideline M26-A, Clinical and Laboratory Standards Institute, Wayne, PA, USA, 1999 Search PubMed.
- A. E. Laudy, Pol. J. Microbiol., 2018, 67, 129–135 CrossRef PubMed.
- A. E. Laudy, A. Mrowka, J. Krajewska and S. Tyski, PLoS One, 2016, 11, e0147131 CrossRef PubMed.
- O. M. Zając, S. Tyski and A. E. Laudy, Biology, 2022, 11, 1044 CrossRef PubMed.
- H. Nikaido and J.-M. Pagès, FEMS Microbiol. Rev., 2012, 36, 340–363 CrossRef CAS PubMed.
- T. J. Opperman and S. T. Nguyen, Front. Microbiol., 2015, 6, 421 Search PubMed.
- (a) I. Orme, J. Secrist, S. Anathan, C. Kwong, J. Maddry, R. Reynolds, A. Poffenberger, M. Michael, L. Miller, J. Krahenbuh, L. Adams, A. Biswas, S. Franzblau, D. Rouse, D. Winfield and J. Brooks, Antimicrob. Agents Chemother., 2001, 45, 1943–1946 CrossRef CAS PubMed; (b) P. Da Silva, B. Bonifácio, R. Frem, A. Godoy Netto, A. Mauro, A. Ferreira, E. Lopes, M. Raddi, T. Bauab, F. Pavan and M. Chorilli, Molecules, 2015, 20, 22534–22545 CrossRef CAS PubMed; (c) T. S. Ibrahim, E. S. Taher, E. Samir, A. M. Malebari, A. N. Khayyat, M. F. A. Mohamed, R. M. Bokhtia, M. A. AlAwadh, I. A. Seliem, H. Z. Asfour, N. A. Alhakamy, S. S. Panda and A. M. M. Al-Mahmoudy, Molecules, 2020, 25, 3125 CrossRef CAS PubMed.
- V. Hernandez, T. Crépin, A. Palencia, S. Cusack, T. Akama, S. J. Baker, W. Bu, L. Feng, Y. R. Freund, L. Liu, M. Meewan, M. Mohan, W. Mao, F. L. Rock, H. Sexton, A. Sheoran, Y. Zhang, Y.-K. Zhang, Y. Zhou, J. A. Nieman, M. R. Anugula, E. M. Keramane, K. Savariraj, D. S. Reddy, R. Sharma, R. Subedi, R. Singh, A. O'Leary, N. L. Simon, P. L. De Marsh, S. Mushtaq, M. Warner, D. M. Livermore, M. R. K. Alley and J. J. Plattner, Antimicrob. Agents Chemother., 2013, 57, 1394–1403 CrossRef CAS PubMed.
- Q.-H. Hu, R.-J. Liu, Z.-P. Fang, J. Zhang, Y.-Y. Ding, M. Tan, M. Wang, W. Pan, H.-C. Zhou and E.-D. Wang, Sci. Rep., 2013, 3, 2475 CrossRef PubMed.
- A. Palencia, X. Li, W. Bu, W. Choi, C. Z. Ding, E. E. Easom, L. Feng, V. Hernandez, P. Houston, L. Liu, M. Meewan, M. Mohan, F. L. Rock, H. Sexton, S. Zhang, Y. Zhou, B. Wan, Y. Wang, S. G. Franzblau, L. Woolhiser, V. Gruppo, A. J. Lenaerts, T. O'Malley, T. Parish, C. B. Cooper, M. G. Waters, Z. Ma, T. R. Ioerger, J. C. Sacchettini, J. Rullas, I. Angulo-Barturen, E. Pérez-Herrán, A. Mendoza, D. Barros, S. Cusack, J. J. Plattner and M. R. K. Alley, Antimicrob. Agents Chemother., 2016, 60, 6271–6280 CrossRef CAS PubMed.
- X. Li, V. Hernandez, F. L. Rock, W. Choi, Y. S. L. Mak, M. Mohan, W. Mao, Y. Zhou, E. E. Easom, J. J. Plattner, W. Zou, E. Pérez-Herrán, I. Giordano, A. Mendoza-Losana, C. Alemparte, J. Rullas, I. Angulo-Barturen, S. Crouch, F. Ortega, D. Barros and M. R. K. Alley, J. Med. Chem., 2017, 60, 8011–8026 CrossRef CAS PubMed.
- G. Hao, H. Li, F. Yang, D. Dong, Z. Li, Y. Ding, W. Pan, E. Wang, R. Liu and H. Zhou, Bioorg. Med. Chem., 2021, 29, 115871 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), 2020.09 Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022, https://www.chemcomp.com/ Search PubMed.
- S. M. Mandal, R. Pegu, W. F. Porto, O. L. Franco and S. Pratihar, Bioorg. Med. Chem. Lett., 2017, 27, 2135–2138 CrossRef CAS PubMed.
- S. Mandal and T. Parish, Antimicrob. Agents Chemother., 2021, 65, e0262220 CrossRef PubMed.
- A. Bonardi, A. Nocentini, R. Cadoni, S. Del Prete, P. Dumy, C. Capasso, P. Gratteri, C. T. Supuran and J. Y. Winum, ACS Med. Chem. Lett., 2020, 11, 2277–2284 CrossRef CAS PubMed.
- E. Bellale, M. Chaudhari and K. Akamanchi, Synthesis, 2009, 2009, 3211–3213 CrossRef.
- European Committee on Antimicrobial Susceptibility Testing, Method for the determination of broth dilution MIC of antifungal agents for yeasts. Document E.DEF 7.3.2., 2020, https://www.eucast.org/ Search PubMed.
- European Committee on Antimicrobial Susceptibility Testing, EUCAST disk diffusion method for antimicrobial susceptibility testing. Document version 11.0., 2023, https://www.eucast.org/ Search PubMed.
- CLSI, Method for antifungal disk diffusion susceptibility testing of yeasts, Approved Standard, CLSI guideline M44-A2, Clinical and Laboratory Standards Institute, Wayne, PA, USA, 2nd edn, 2009 Search PubMed.
- E. Cantón, J. Pemán, A. Viudes, G. Quindós, M. Gobernado and A. Espinel-Ingroff, Diagn. Microbiol. Infect. Dis., 2003, 45, 203–206 CrossRef PubMed.
- R. Misra, K. D. Morrison, H. J. Cho and T. Khuu, J. Bacteriol., 2015, 197, 2479–2488 CrossRef CAS PubMed.
- R. P. Lamers, J. F. Cavallari and L. L. Burrows, PLoS One, 2013, 8, e60666 CrossRef CAS PubMed.
- A. Laudy, E. Kulińska and S. Tyski, Molecules, 2017, 22, 114 CrossRef PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- Staphylococcus aureus strain MRSA_S24 MRSA_S24_contig005, whole genome shotgun sequence, https://www.ncbi.nlm.nih.gov/nuccore/NZ_LFVO01000005.1, (accessed 1 December 2022) Search PubMed.
- E. Von Dach, S. M. Diene, C. Fankhauser, J. Schrenzel, S. Harbarth and P. François, J. Infect. Dis., 2016, 213, 1370–1379 CrossRef CAS PubMed.
- F. L. Rock, W. Mao, A. Yaremchuk, M. Tukalo, T. Crépin, H. Zhou, Y.-K. Zhang, V. Hernandez, T. Akama, S. J. Baker, J. J. Plattner, L. Shapiro, S. A. Martinis, S. J. Benkovic, S. Cusack and M. R. K. Alley, Science, 2007, 316(4), 1759–1761 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed.
- W. Yu, X. He, K. Vanommeslaeghe and A. D. MacKerell, J. Comput. Chem., 2012, 33, 2451–2468 CrossRef CAS PubMed.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- The CHARMM General Force Field (CGenFF) program, https://cgenff.umaryland.edu Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2190658–2190669, 2258836 and 2296881. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4md00061g |
| This journal is © The Royal Society of Chemistry 2024 |