
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of imidazo[1,2-b]pyridazine-containing TAK1 kinase inhibitors with excellent activities against multiple myeloma†
Desmond
Akwata
a,
Allison L.
Kempen
 a,
Jones
Lamptey
a,
Neetu
Dayal
a,
Nickolas R.
Brauer
a and
Herman O.
Sintim
*abc
a,
Jones
Lamptey
a,
Neetu
Dayal
a,
Nickolas R.
Brauer
a and
Herman O.
Sintim
*abc
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA. E-mail: hsintim@purdue.edu
bPurdue Institute for Drug Discovery, 720 Clinic Drive, West Lafayette, IN 47907, USA
cPurdue Institute for Cancer Research, 201 S. University St., West Lafayette, IN 47907, USA
First published on 28th November 2023
Abstract
Current treatment options for patients with multiple myeloma (MM) include proteasome inhibitors, anti-CD38 antibodies, and immunomodulatory agents. However, if patients have continued disease progression after administration of these treatments, there are limited options. There is a need for effective targeted therapies of MM. Recent studies have shown that the transforming growth factor-β activated kinase (TAK1) is upregulated and overexpressed in MM. We have discovered that 6-substituted morpholine or piperazine imidazo[1,2-b]pyridazines, with an appropriate aryl substituent at position-3, inhibit TAK1 at nanomolar concentrations. The lead compound, 26, inhibits the enzymatic activity of TAK1 with an IC50 of 55 nM. Under similar conditions, the known TAK1 inhibitor, takinib, inhibits the kinase with an IC50 of 187 nM. Compound 26 and analogs thereof inhibit the growth of multiple myeloma cell lines MPC-11 and H929 with GI50 values as low as 30 nM. These compounds have the potential to be translated into anti-MM therapeutics.
Introduction
Multiple myeloma (MM), a blood cancer, is characterized by dysregulated and proliferative plasma cells, which facilitate bone destruction.1–6 MM cells suppress the differentiation of osteoblasts from bone marrow stroma cells (BMSCs) and enhance osteoclastic formation and activity.5–7 This process creates an imbalance leading to extensive bone damage and enhanced angiogenesis. MM cells have been found to overexpress several soluble inhibitors of osteoblastogenesis, including transforming growth factor-beta (TGF-β), tumor necrosis factor-alpha (TNF-α), interleukin-3 (IL-3), interleukin-7 (IL-7), and activin.7Previous studies showed that the transforming growth factor-β activated kinase (TAK1) is constitutively upregulated and phosphorylated in MM.1,6,8 TAK1 is a serine/threonine kinase that is important for cell growth, differentiation, and apoptosis.8–10 Various extracellular signals trigger TAK1 activation, including cytokines, growth factors, and Toll-like receptor ligands. In the classical TAK1 signaling pathways, TAK1 is activated by receptor-associated proteins, such as TGF-β receptors or interleukin-1 receptors. Activation of these receptors recruits TAB1 and TAB2/3 to TAK1. TAK1 then phosphorylates downstream signaling molecules, including MAP kinases and various transcription factors.11–15 TAK1 is involved in numerous cellular processes, including the regulation of immune responses, inflammation, cell survival, and differentiation.15 Dysregulation of TAK1 has been implicated in various diseases, including cancer, autoimmune disorders, and inflammatory diseases.15 Therefore, TAK1 has emerged as a potential target for therapeutic intervention in these diseases.
Overexpression of TAK1 has been found in many multiple myeloma cell lines as well as patient samples, suggesting that it may play a role in the development and progression of this disease.16–19 Several studies have investigated the role of TAK1 in multiple myeloma. For example, a study by Teramachi et al. has shown that TAK1 is consistently upregulated and phosphorylated in MM cells. They found that inhibition of TAK1 leads to the suppression of NF-κB, p38MAPK, ERK, and STAT3 signaling pathways, which consequently inhibits the expression of key regulators involved in the growth and survival of MM, such as PIM2, MYC, Mcl1, IRF4, and Sp1. They also found that TAK1 inhibition significantly reduces the levels of the angiogenic factor VEGF in MM cells.20 Another study by Harada et al. in 2021 highlighted that the dysregulation of the TAK1-PIM2 pathway is a key factor in promoting tumor growth and bone destruction in MM and that targeting the TAK1 pathway could be a therapeutic strategy for effectively addressing MM and its associated complications.21 Overall, these findings suggest that TAK1 may be a promising therapeutic target for the treatment of multiple myeloma.
A few TAK1 inhibitors, such as OTS964, NG-25, LX-2343, 5Z-7-oxozeaenol, ponatinib, and takinib, have been described in the literature (Fig. 1).22,23 Unfortunately, the reported GI50 values for these compounds against TAK1 overexpressing cancers, such as MM, are in the micromolar range and, as such, it would be difficult to achieve effective concentrations in blood without encountering dose-limiting toxicities.24 Here, we present new and potent inhibitors of TAK1, which inhibit MM cell growth in the nanomolar range.
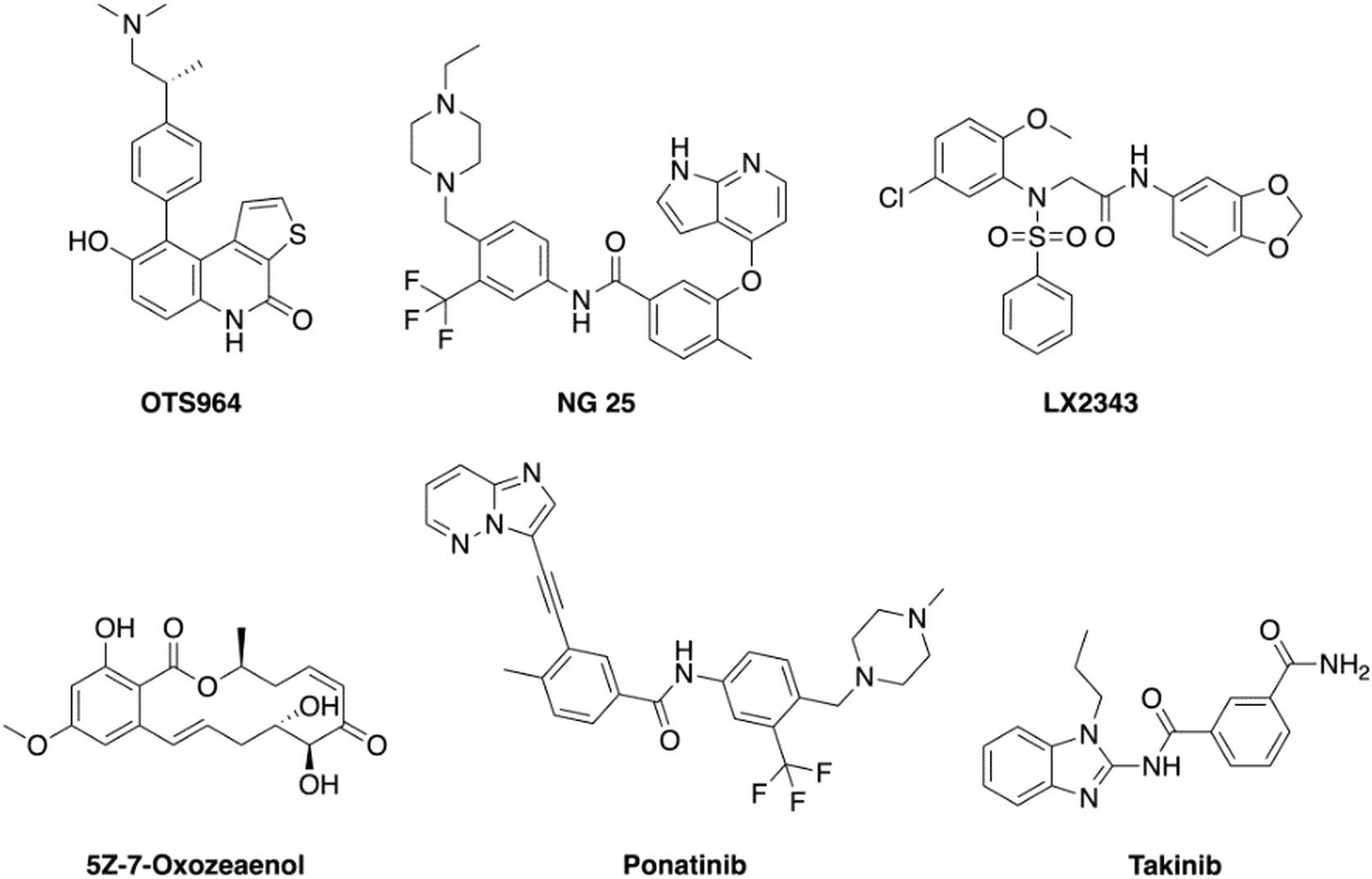 | ||
| Fig. 1 Structures of reported small molecule TAK1 inhibitors. | ||
Results and discussion
The imidazo[1,2-b]pyridazine moiety is a privileged drug moiety that is found in many approved and experimental drugs.24 It has been demonstrated in multiple reports that while the imidazo[1,2-b]pyridazine moiety binds to the hinge region of kinases, substitutions at positions 2, 3, 6, 7, and 8 dictate kinase selectivity and potency.24 During a different project, we discovered that substituting position 6 of imidazo[1,2-b] pyridazine with morpholine or piperazine afforded compounds that displayed enhanced kinase inhibition when compared to analogs that were not substituted at position C6 of the imidazo[1,2-b]pyridazine core. We chose morpholine and piperazine because these moieties are known to enhance the drug-like properties of lead compounds and could also partake in ligand interactions with target proteins.25,26 We designed compounds that fused the morpholine or piperazine imidazo[1,2-b]pyridazine scaffold with indazole (another privileged kinase scaffold), see Fig. 2 for structures. We initially synthesized a small set of such compounds with the imidazo[1,2-b]pyridazine as the main scaffold for a preliminary kinase screening. Compounds within this library were synthesized through a nucleophilic aromatic substitution (SNAr) reaction at the C6 position. The final products were obtained by performing a Suzuki–Miyaura cross-coupling reaction with aromatic boronic acid substrates. This reaction was carried out using potassium carbonate as a base, a palladium catalyst, and a solvent mixture of acetonitrile and water in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 1).
:1 ratio (Scheme 1).
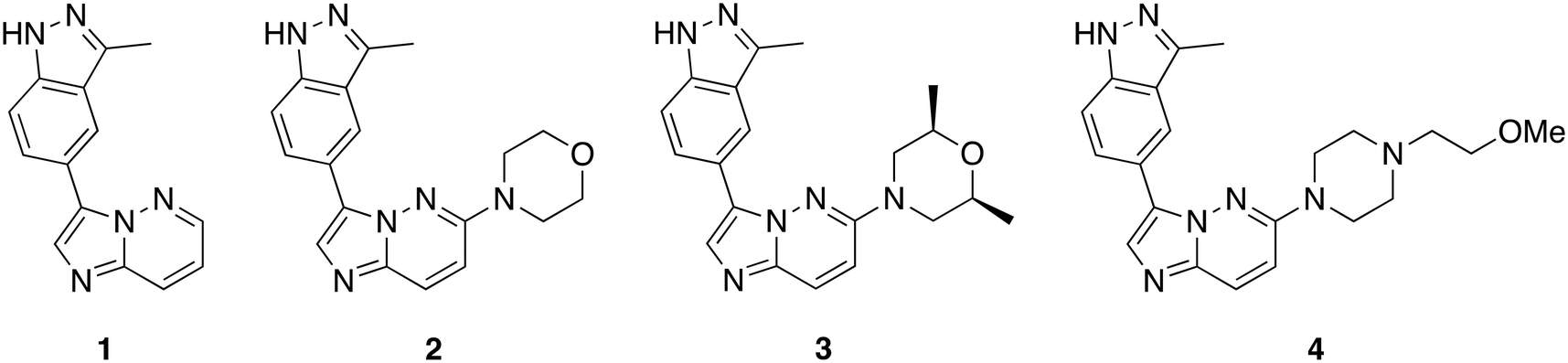 | ||
| Fig. 2 Preliminary compounds synthesized and screened against TAK1. | ||
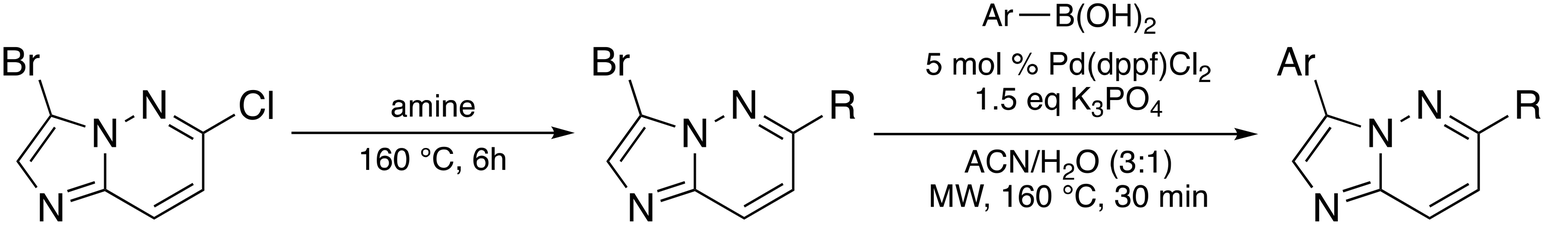 | ||
| Scheme 1 Synthesis of compounds via nucleophilic aromatic substitution and Suzuki–Miyaura cross-coupling reaction. | ||
To evaluate the impact of substitution at C6 of the imidazo[1,2-b]pyridazine core, we synthesized compound 1, allowing us to compare the differences between a substituted and unsubstituted structure at the C6 position. With the initial set of compounds on hand (Fig. 2), we began a kinase screening of the compounds at 100 nM against TAK1 utilizing the ADP-Glo kinase assay (Promega, Madison, WI).27 Our preliminary data suggested that compound 3, containing a cis-dimethylmorpholine moiety, showed the best inhibition of TAK1 (95%) compared to no substituent (1, 26% inhibition), the unsubstituted morpholine moiety (2, 91% inhibition), or the piperidine substituent (4, 46% inhibition). Thus, we decided to move forward with a structure–activity relationship (SAR) study based on compound 3.
To investigate the potential binding mode of our preliminary compounds, we performed molecular docking studies of compound 3 and takinib against TAK1 (Fig. 3). We found that the oxygen in the cis-dimethylmorpholine interacts with the conserved lysine residue Lys-63 in the ATP-binding site of TAK1 (Fig. 3), which is crucial for kinase activity as observed in the kinase inhibition data for 2 and 3. The methyl groups of the morpholine formed favorable hydrophobic interactions with the surrounding residues, Cys-174, Lys-63, and Gly-45. In addition, we identified a hydrogen bonding interaction between Ala-107 and H2 of the imidazo[1,2-b]pyridazine core. We therefore hypothesized that blocking this position with a methyl group at position 2 of the imidazo[1,2-b]pyridazine (as in 28) would sterically hinder this hydrogen bonding and lead to reduced activity, vide infra.
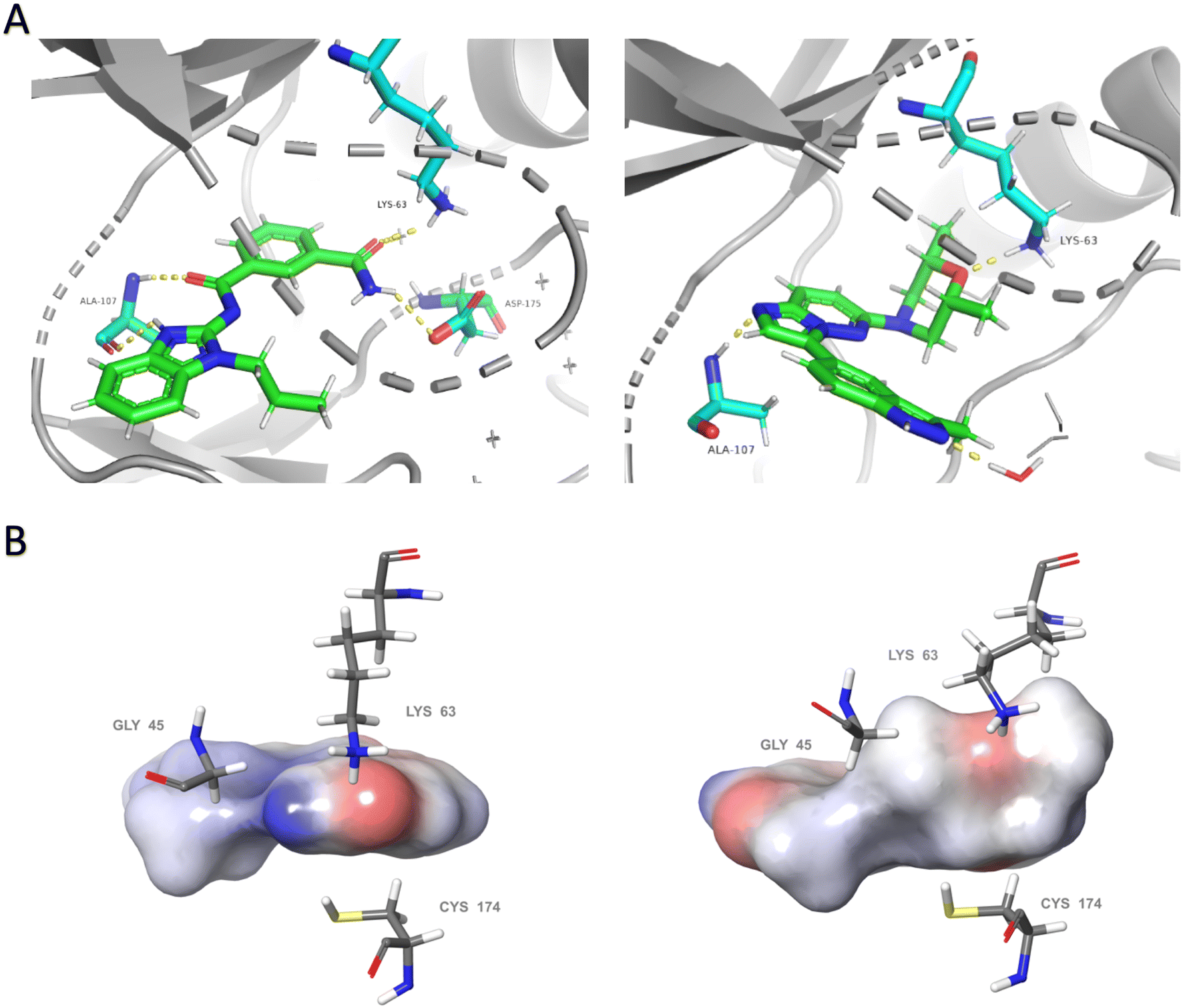 | ||
| Fig. 3 Comparative analysis of binding modes of takinib and docking model of compound 3 within the binding pocket of TAK1 (PDB: 5V5N). (A) Polar interactions (yellow dashes) between takinib (left) and compound 3 (right) and active site of TAK1. Each ligand forms hydrogen bonds with the conserved lysine Lys-63 and with Ala-107 within the hinge region. Takinib makes an additional interaction with DFG residue Asp-175. (B) Surface models of each ligand surrounded by representative hydrophobic residues within the binding pocket. Slight conformational shifts to residues Lys-63, Gly-45, and Cys-174 from the bound structure of takinib to the docked structure of compound 3 indicate a possible induction of entropically favored hydrophobic interactions. Docking was performed using the Induced Fit Docking protocol within Schrodinger. Visualization of (A) was performed in PyMol. Visualization of (B) utilized Maestro. | ||
We synthesized three types of compound 3 analogs, where series A explores the use of phenyl derivatives in place of the indazole, series B examines indazole derivatives, and series C evaluates the effect of modifying the morpholine moiety.
In series A (Fig. 4), we probed the use of substituted phenyls and pyridine rings in place of the indazole as found in compound 3. Specifically, we synthesized compounds with privileged moieties, such as halides, –OCF3, –CN, and –CF3 at the C3 position. These groups are known to enhance the potency of a drug by increasing its binding affinity to the target protein and improve the pharmacokinetic properties of a drug by increasing its lipophilicity, which can enhance its membrane permeability and metabolic stability.28,29 We explored the use of a pyridine ring (17 and 18) as a bioisostere of phenyl (as in 5). The so-called “necessary nitrogen” scanning (for example phenyl to pyridyl) could enhance solubility and/or activity of a lead compound via hydrogen bonding and polar interactions with water and/or active site residues.30 Additionally, we tested substituents such as amides on the phenyl (12 and 13) as their polarity, electron-withdrawing capability, and hydrogen-bonding ability are known to improve the potency and selectivity of a drug by enhancing its binding affinity to the target protein or receptor. Compounds 8 and 15 were made with sulfonyl and sulfonamide groups, which can enhance the metabolic stability of drugs (compared to amides) by resisting enzymatic degradation.31,32
 | ||
| Fig. 4 Series A: analogs of 3 with different 6-membered ring substitutions at position C3. | ||
We went on to explore different indazole derivatives in series B (Fig. 5). In this series, we synthesized analogs with different indazole derivatives attached to the imidazo[1,2-b]pyridazine core scaffold at positions 4, 5, and 7 of the indazole (20, 21, and 22). We also synthesized methylated analogs (23 and 24) at the 1H-position of compounds 21 and 20, respectively. Based on the activity of 3, we synthesized 25 and 26, to investigate how different alkyl groups on the indazole moiety affected kinase activity. 27 substitutes an indolin-2-one as a bioisostere of indazole. The introduction of different indazole derivatives/mimics at the C3 position of the imidazo[1,2-b]pyridazine scaffold was expected to modulate the steric and electronic properties of the molecule, which in turn could affect its interactions with the target protein kinase.
 | ||
| Fig. 5 Series B: analogs of 3 with various indazole derivatives. | ||
The introduction of morpholine at C6 of the imidazo[1,2-b]pyridazine core improved TAK1 kinase inhibition when compared to 1, which has no substitution at C6, and 4, which has a piperazine moiety at C6. For series C, we made analogs of 3 exploring the substitution of different substituted morpholine moieties at the C6 position (Fig. 6). Morpholines in drugs are known to enhance the water solubility and metabolic stability of a drug and improve its bioavailability.33 Morpholines can also be used to enhance the receptor binding affinity of a drug by engaging in additional hydrogen-bonding interactions with the protein target. This can improve the potency and the selectivity of the drug, as well as reduce its off-target effects.33–35 As the cis-dimethylmorpholine analog showed higher inhibition compared to the unsubstituted morpholine (2), we made analogs of 3 exploring the substitution of different substituted morpholines at the C6 position (Fig. 6).
 | ||
| Fig. 6 Series C: analogs of 3 with different heterocyclic substitutions at position C6. | ||
With the library on hand, we screened the compounds against TAK1 using the ADP-Glo kinase assay (Promega, Madison, WI) at a compound concentration of 500 nM (Fig. 7). We identified the best-performing compounds that exhibited over 50% inhibition at 500 nM. Then, we subjected these compounds to further screening at a concentration of 100 nM. We then conducted a final screening of the remaining compounds, which had >50% inhibition at 100 nM, at a concentration of 20 nM (Fig. 7).
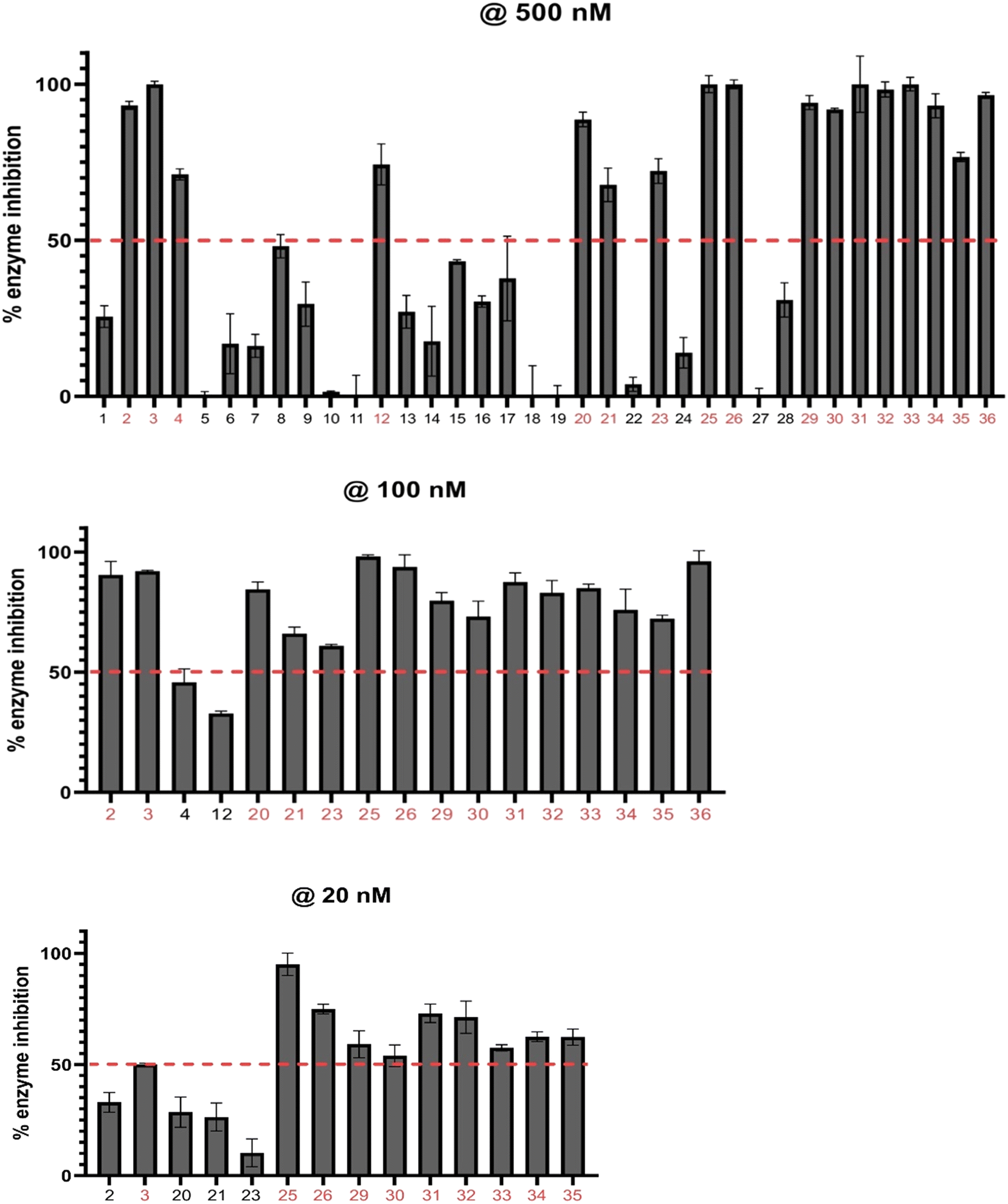 | ||
| Fig. 7 Histograms of TAK1 inhibition at compound concentrations of 500 nM, 100 nM, and 20 nM. Compounds were screened against TAK1 using the ADP-Glo kinase assay (Promega). The dashed red line denotes 50% enzyme inhibition, and compounds performing above the line were assessed at the next concentration. Values reported represent means of triplicates, and error bars represent standard deviation. | ||
To evaluate the significance of kinase inhibition, a cutoff value of 50% was used to classify compounds as having good kinase inhibition or not. At 500 nM screening, 16 compounds out of the 35 demonstrated good kinase inhibition, surpassing the 50% cutoff. Notably, compounds 3, 25, 26, 31, and 33 exhibited exceptional potency, achieving 100% kinase inhibition. Conversely, all the compounds in series A apart from 12 (74%) showed less than 50% kinase inhibition, with 6, 7, 8, 9, 13, 14, 16, and 17 showing moderate kinase inhibition. Compounds 5, 11, 18, and 19 exhibited negligible kinase inhibition. Additionally, compounds 21, 22, 24, 27, and 28 in series B also showed moderate kinase inhibition below the 50% cutoff. These compounds may not effectively target the kinase of interest but may still have some inhibitory potential with further modifications. The compounds demonstrating kinase inhibition above 50% were subjected to a second round of screening at a lower concentration of 100 nM.
Comparing the kinase inhibition at 100 nM to the previous screening at 500 nM, some compounds show consistent inhibition, while others demonstrate variations in their inhibitory activities. Notably, compounds 2, 3, 25, 26, 31, 32, 33, and 35 continue to exhibit significant TAK1 kinase inhibition above the 50% cutoff, indicating their consistent inhibitory potential. Compounds 4 and 12 show a decrease in TAK1 kinase inhibition compared to the previous screening, suggesting a potential concentration-dependent effect. Again, all compounds in series C (29–35) exhibited excellent TAK1 kinase inhibition.
To confirm our in-house kinase assay results, an enzymatic IC50 assay was performed on the top ten compounds, with takinib used as a control, at a contract research service company (Reaction Biology). The results showed that compound 26 was the most potent TAK1 inhibitor, with an IC50 value of 55 nM; under similar experimental conditions, the IC50 for the known TAK1 inhibitor, takinib, was 187 nM. Additionally, six of the ten analogs tested also had a lower IC50 than takinib (Fig. 8).
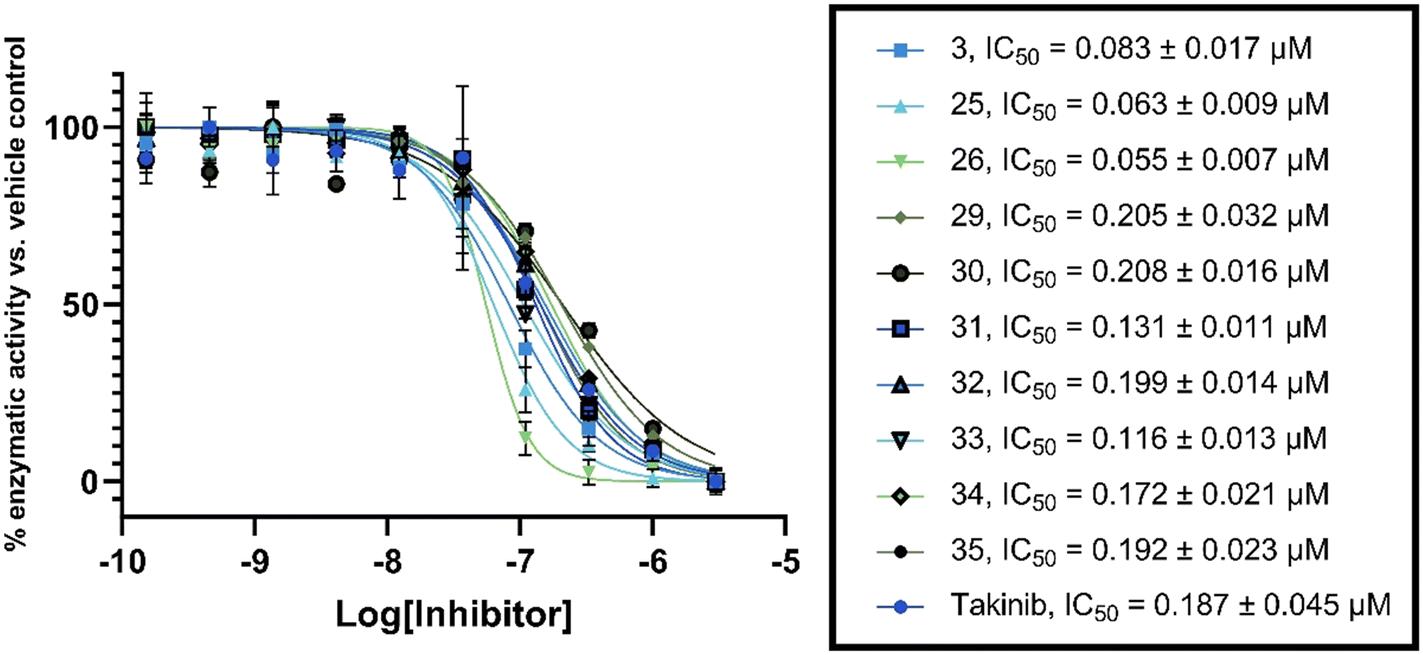 | ||
| Fig. 8 TAK1 inhibition. IC50 values obtained by Reaction Biology. Data were fitted to a non-linear regression equation using GraphPad Prism 9.0 software. Each data point represents the mean and error bars represent the SD of duplicates. | ||
As stated earlier, studies have shown that TAK1 is overexpressed in many multiple myeloma cells. To investigate the performance of these compounds in cell culture, we explored the effect of the library on MM cell growth inhibition (Fig. 9). Compounds were screened at 1 and 0.2 μM against the MPC-11 cell line.
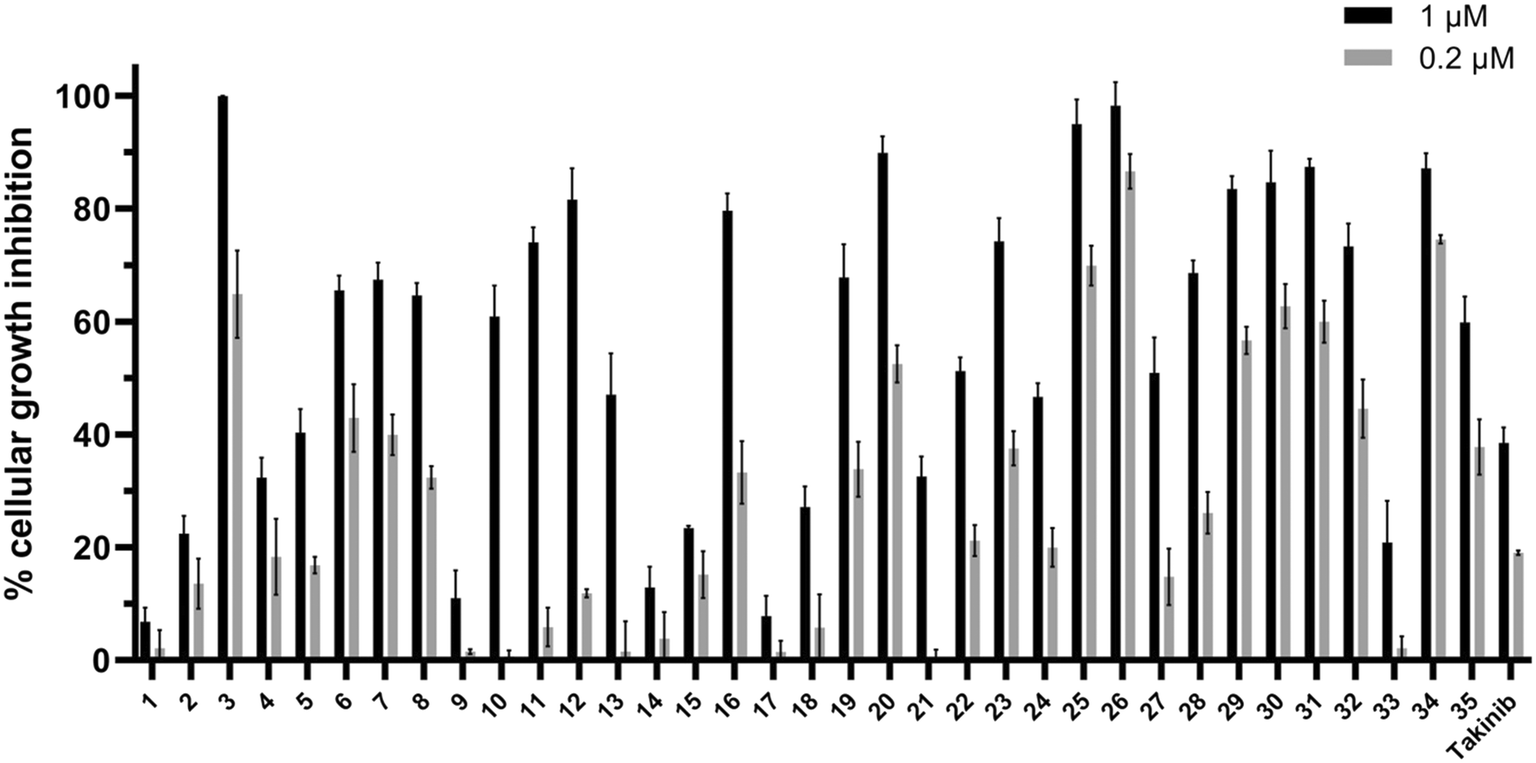 | ||
| Fig. 9 Compounds (1 and 0.2 μM) against MPC-11 cells. Each data point represents the mean and error bars represent the SD of triplicates. | ||
The compounds showed a relative consistency between activity against TAK1 and antiproliferative activity against multiple myeloma cells. All compounds in series C (29–35) showed excellent inhibitory activity against TAK1, and, with the exception of 33, had significant cellular growth inhibition. It is interesting to note that 32 and 33 are enantiomers and show similar TAK1 inhibitions but display different cellular activities. Compound 32 inhibited ∼50% of MPC-11 at 200 nM, while at the same concentration 33 was inactive. We do not have a hypothesis for this discrepancy but appreciate that before a compound can engage with cellular targets it has to get into the cell and differences in permeation and/or compound stability might account for such differences. Nonetheless, because 2-oxa-5-azabicyclo[2.2.1]heptane, found in 32 or 33, is not the best substituent, we did not expend our resources looking into the origin of differences between 32 and 33.
Based on our cell growth inhibition results, we moved forward to collect GI50 values of the top compounds to evaluate their effectiveness (Table 1). We determined the GI50 values of these compounds against two distinct multiple myeloma (MM) cell lines: H929, a human plasma MM cell line, and MPC-11, a murine plasmacytoma cell line. Interestingly, our findings revealed that all the top compounds demonstrated remarkable potency against the H929 cells, with compound 31 exhibiting the most exceptional activity, recording a GI50 value of 0.024 μM (24 nM). Notably, the lead compounds 3 and 26 displayed GI50 values of 0.087 μM (87 nM) and 0.043 μM (43 nM), respectively, indicating their high efficacy against the H929 cells. In comparison, takinib, a known TAK1 inhibitor, exhibited a significantly higher GI50 value of 51 μM (51000 nM), making it over 500 times less effective than the least active compound 3 (GI50 = 0.087 μM).
| Compound | H929 (GI50) | MPC-11 (GI50) |
|---|---|---|
| 3 | 0.087 ± 0.024 μM | 0.072 ± 0.011 μM |
| 25 | 0.030 ± 0.006 μM | 0.094 ± 0.023 μM |
| 26 | 0.043 ± 0.014 μM | 0.041 ± 0.012 μM |
| 29 | 0.071 ± 0.012 μM | 0.155 ± 0.019 μM |
| 30 | 0.045 ± 0.009 μM | 0.101 ± 0.015 μM |
| 31 | 0.024 ± 0.005 μM | 0.126 ± 0.028 μM |
| 34 | 0.033 ± 0.008 μM | 0.131 ± 0.026 μM |
| Takinib | 51 ± 9 μM | 8.5 ± 2.9 μM |
Similarly, the evaluation of these top compounds against the MPC-11 cells demonstrated promising results, with all compounds exhibiting favorable GI50 values ranging from 0.041 μM (26) to 0.155 μM (29). Once again, takinib proved to be substantially less potent with a GI50 value of 8.5 μM against the MPC-11 cells. These findings highlight the outstanding potential of our identified compounds as potent inhibitors against both human and murine MM cell lines. Notably, their efficacy against the H929 cells, particularly compound 31, stands out as a highly promising avenue for further investigation (see ESI† Fig. S1 for dose–response curves).
TAK1 ser412 phosphorylation has been shown to be essential for regulation of downstream pathways by TAK1; the TAK1 S412A mutant could not support the activation of the p38, JNK, and NF-κB pathways.36 Thus we wanted to know if our compounds could inhibit TAK1 phosphorylation at ser412 in murine MM MPC-11 cells (Fig. 10). The results showed that both compounds 3 and 26 were successful in reducing levels of phosphorylated TAK1 (S412) compared to the DMSO control. Treating with 0.5 μM compound decreased phosphorylation levels more significantly than treatment at 0.1 μM. Additionally, takinib did not show a significant difference in phosphorylated TAK1 (S412) levels at 0.1 or 0.5 μM.
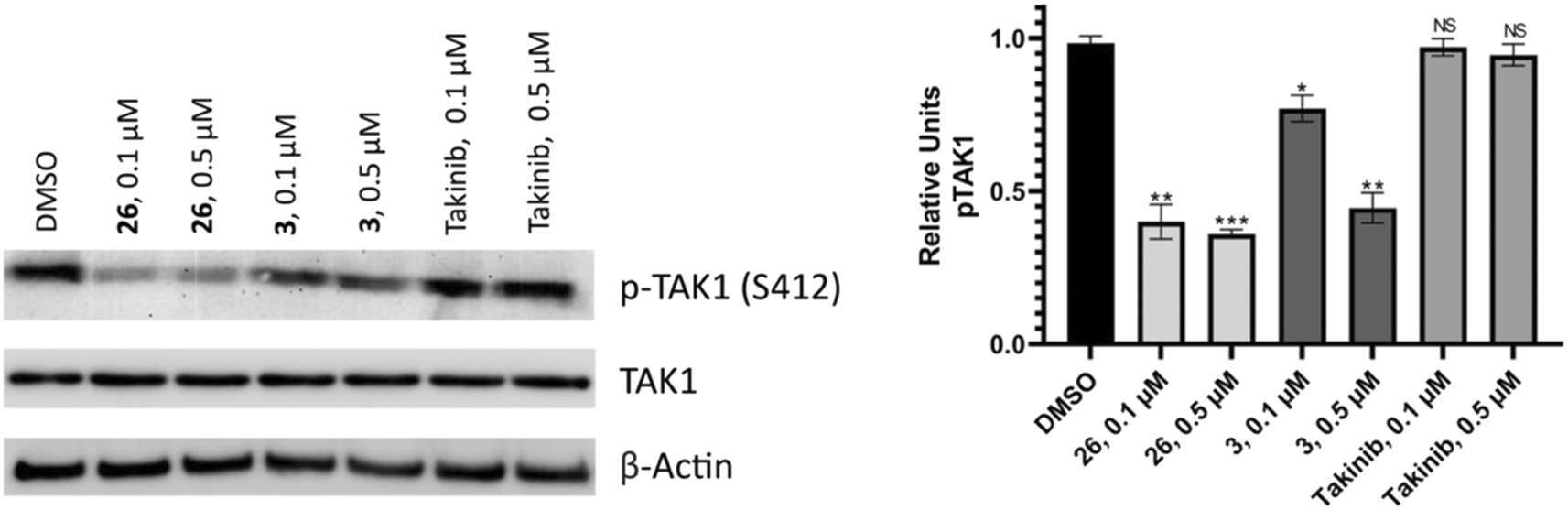 | ||
| Fig. 10 Western blot analysis. MPC-11 cells were treated with compounds 26 or 3 at 0.1 or 0.5 μM or the DMSO control for 48 hours. Bands were quantified relative to the actin loading control. Values are reported as means of duplicates and error bars represent standard deviation. | ||
Since compound 26 was effective in inhibiting TAK1 phosphorylation in cells, it was profiled against some essential kinases to determine potential liabilities, using Eurofins KinomeScan but displayed minimal inhibition of kinase anti-targets, which are essential kinases and/or kinases that are important for immune, kidney or cardiovascular or skin functions, such as KIT, ZAP70, JAK2, RET, KDR (VEGFR2), PDGFR, FGFR, EGFR, BRAF, AKT, AURKA or B (see the ESI,† Table S1). Interestingly, compound 26 showed low affinity for PKAC alpha26 (ESI,† Table S1) and hence the inhibition of p-TAK1 (S412) is intriguing, which requires future investigation.
Conclusion
TAK1 is an important kinase that is involved in many critical processes and has been proposed as a potential target for various disease states, including cancer and inflammatory diseases. While a few TAK1 inhibitors have been reported, to the best of our knowledge, none has been shown to potently inhibit TAK1 phosphorylation or inhibit MM cell growth at low nanomolar concentrations. Here, we show that the strategic substitution of the imidazo[1,2-b]pyridazine core with morpholine and indazole affords excellent TAK1 inhibitors with activities against MM cell lines. We also utilize the so-called “magic methylation” on the morpholine ring to increase TAK1 inhibition.Experimental
General procedure for the synthesis of substituted imidazo[1,2-b] pyridazine substrates
In a sealed tube, 3-bromo-6-chloroimidazo[1,2-b] pyridazine (500 mg) and appropriate amine (4 equiv.) in n-propanol (1 mL) was refluxed at 150 °C overnight. After completion, the reaction was extracted with ethyl acetate and washed with brine. The organic layer was collected, dried over sodium sulfate, and concentrated under reduced pressure. The crude was purified via silica gel column chromatography to yield the desired product.Suzuki cross-coupling synthesis of compound 3 and analogs
A solution of (2S,6R)-4-(3-bromoimidazo[1,2-b] pyridazin-6-yl)-2,6-dimethyl morpholine (0.5 mmol), respective boronic acid substrates, Pd(dppf)2Cl2 (5 mol%), K3PO4 (1.5 equiv.) in degassed acetonitrile (3 mL) and water (1 mL) was stirred at 160 °C in a sealed microwave vial for 30 minutes in a microwave oven. The organic layer was collected after workup with water and ethyl acetate, and the organic layer was washed with brine, followed by concentration using a rotary evaporator and purification by silica gel column chromatography to obtain the final product.3-(3-Methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazine (1)
Pale yellow solid (69 mg, 49%). 1H NMR (500 MHz, methanol-d4) δ 8.58–8.52 (m, 2H), 8.09 (s, 1H), 8.05 (dd, J = 9.2, 1.7 Hz, 1H),7.99 (dd, J = 8.8, 1.6 Hz, 1H), 7.55 (dd, J = 8.7, 0.9 Hz, 1H), 7.25 (dd, J = 9.2, 4.4 Hz, 1H), 2.60 (s, 3H). 13C NMR (126 MHz, methanol-d4) δ 143.5, 142.8, 140.6, 139.7, 130.9, 129.1, 126.2, 124.9, 122.2, 120.3, 118.4, 117.1, 110.0, 10.3. HRMS (ESI†) m/z calcd. for C14H11N5 [M+H]+ 250.1093, found 250.1090.4-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)morpholine (S1)
Brown solid (552 mg, 87%). 1H NMR (500 MHz, methanol-d4) δ 7.72 (d, J = 10.0 Hz, 1H), 7.50 (s, 1H), 7.15 (d, J = 10.0 Hz, 1H), 3.85–3.79 (m, 4H), 3.59–3.53 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 155.7, 136.9, 130.8, 124.9, 110.8, 100.2, 66.1, 45.9. HRMS (ESI†) m/z calcd. for C13H14N6O [M + H]+ 284.1311, found 284.1311.4-(3-(3-Methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (2)
Synthesized using substrate S1. Brown solid (95 mg, 50%). 1H NMR (500 MHz, methanol-d4) δ 8.60–8.56 (m, 1H), 7.93 (dd, J = 8.8, 1.6 Hz, 1H), 7.84 (s, 1H), 7.75 (d, J = 9.9 Hz, 1H), 7.49 (dd, J = 8.8, 0.8 Hz, 1H), 7.09 (d, J = 9.9 Hz, 1H), 3.86–3.78 (m, 4H), 3.57–3.48 (m, 4H), 2.55 (d, J = 2.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 155.4, 142.2, 140.3, 137.3, 130.9, 128.0, 126.7, 125.3, 122.7, 121.0, 117.5, 110.7, 109.9, 66.2, 46.7, 12.1. HRMS (ESI†) m/z calcd. for C18H18N6O [M + H]+ 335.1621, found 335.1621.(2S,6R)-4-(3-Bromoimidazo[1,2-b] pyridazin-6-yl)-2,6-dimethylmorpholine (S2)
Pale yellow solid (640 mg, 95%). 1H NMR (500 MHz, chloroform-d) δ 7.66 (d, J = 9.9 Hz, 1H), 7.52 (s, 1H), 6.80 (d, J = 9.9 Hz, 1H), 4.01–3.89 (m, 2H), 3.81–3.65 (m, 2H), 2.65 (dd, J = 12.8, 10.6 Hz, 2H), 1.28 (d, J = 6.3 Hz, 6H). 13C NMR (125 MHz, Chloroform-d) δ 154.8, 136.9, 132.1, 126.0, 109.4, 100.3, 71.3, 51.3, 18.9. HRMS (ESI†) m/z calcd. for C12H16BrN4O [M + H]+ 311.0507, found 311.0507.(2S,6R)-2,6-Dimethyl-4-(3-(3-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (3)
Synthesized using substrate S2. Off-white solid (142 mg, 42%). 1H NMR (500 MHz, methanol-d4) δ 8.69 (dd, J = 1.6, 0.8 Hz, 1H), 7.86 (dd, J = 8.8, 1.6 Hz, 1H), 7.84 (s, 1H), 7.69 (d, J = 9.8 Hz, 1H), 7.46 (dd, J = 8.8, 0.9 Hz, 1H), 7.06 (d, J = 10.0 Hz, 1H), 3.97–3.90 (m, 2H), 3.73 (dqd, J = 12.4, 6.2, 2.4 Hz, 2H), 2.57–2.51 (m, 2H), 2.51 (s, 3H), 1.22 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.9, 142.5, 140.2, 137.0, 128.8, 128.2, 125.6, 124.8, 122.0, 121.0, 117.2, 109.9, 71.3, 51.4, 17.8, 10.5. HRMS (ESI†) m/z calcd. for C20H22N6O [M + H]+ 363.1928, found 363.1939.3-Bromo-6-(4-(2-methoxyethyl)piperazin-1-yl)imidazo[1,2-b]pyridazine (S3)
Off-white solid (583 mg, 79%). 1H NMR (500 MHz, Methanol-d4) δ 7.66 (d, J = 9.9 Hz, 1H), 7.47 (s, 1H), 7.10 (d, J = 10.0 Hz, 1H), 3.61–3.51 (m, 6H), 3.34 (s, 3H), 2.66–2.58 (m, 6H); 13C NMR (125 MHz, Methanol-d4) δ 155.3, 136.7, 130.6, 124.8, 110.9, 100.1, 69.6, 57.6, 57.2, 52.6, 45.1. HRMS (ESI†) m/z calcd. for C13H19BrN5O [M + H]+ 340.0773, found 340.0770.6-(4-(2-Methoxyethyl)piperazin-1-yl)-3-(3-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazine (4)
Synthesized using substrate S3. Yellow solid (174 mg, 78%). 1H NMR (500 MHz, methanol-d4) δ 8.54 (dd, J = 1.6, 0.8 Hz, 1H), 7.86 (dd, J = 8.8, 1.6 Hz, 1H), 7.78 (s, 1H), 7.64 (d, J = 9.9 Hz, 1H), 7.44 (dd, J = 8.8, 0.9 Hz, 1H), 6.98 (d, J = 9.9 Hz, 1H), 3.55 (t, J = 5.5 Hz, 2H), 3.54–3.48 (m, 4H), 3.34 (s, 3H), 2.65–2.62 (m, 4H), 2.60 (d, J = 5.5 Hz, 2H), 2.50 (s, 3H). 13C NMR (126 MHz, methanol-d4) δ 154.8, 142.5, 140.2, 136.8, 128.8, 128.3, 125.6, 124.7, 122.0, 121.0, 117.4, 110.0, 109.9, 69.5, 57.6, 57.2, 52.6, 45.5, 10.3. HRMS (ESI†) m/z calcd. for C21H25N7O [M + H]+ 392.2199, found 392.2202.(2S,6R)-2,6-Dimethyl-4-(3-phenylimidazo[1,2-b]pyridazin-6-yl)morpholine (5)
Synthesized using substrate S2. Off-white solid (241 mg, 81%). 1H NMR (500 MHz, methanol-d4) δ 8.05 (dd, J = 8.4, 1.2 Hz, 2H), 7.80 (s, 1H), 7.72 (d, J = 9.9 Hz, 1H), 7.44 (dd, J = 8.5, 7.1 Hz, 2H), 7.36–7.29 (m, 1H), 7.11 (d, J = 10.0 Hz, 1H), 4.01–3.94 (m, 2H), 3.72 (dqd, J = 10.6, 6.2, 2.4 Hz, 2H), 2.57 (dd, J = 12.9, 10.6 Hz, 2H), 1.22 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.8, 137.1, 129.4, 128.9, 128.2, 128.2, 127.3, 126.2, 125.0, 110.5, 71.2, 51.4, 17.7. HRMS (ESI†) m/z calcd. for C18H20N4O [M+H]+ 309.1715, found 309.1718.4-(6-((2S,6R)-2,6-Dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)benzonitrile (6)
Synthesized using substrate S2. Off-white solid (246 mg, 77%). 1H NMR (500 MHz, methanol-d4) δ 8.32–8.25 (m, 2H), 7.99 (s, 1H), 7.78 (dd, J = 9.3, 8.4 Hz, 3H), 7.22 (d, J = 10.0 Hz, 1H), 4.05–3.98 (m, 2H), 3.76 (dqd, J = 12.6, 6.2, 2.4 Hz, 2H), 2.63 (dd, J = 12.8, 10.6 Hz, 2H), 1.26 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 155.1, 138.1, 133.5, 132.1, 131.2, 126.2, 126.0, 125.2, 118.4, 111.5, 109.9, 71.3, 51.3, 17.7. HRMS (ESI†) m/z calcd. for C19H19N5O [M+H]+ 334.1668, found 334.1671.(2S,6R)-2,6-Dimethyl-4-(3-(4-(trifluoromethyl)phenyl)imidazo[1,2-b]pyridazin-6-yl)morpholine (7)
Synthesized using substrate S2. Pale yellow solid (248 mg, 68%). 1H NMR (500 MHz, methanol-d4) δ 8.34–8.28 (m, 2H), 7.97 (s, 1H), 7.81 (d, J = 9.9 Hz, 1H), 7.78–7.72 (m, 2H), 7.23 (d, J = 10.0 Hz, 1H), 4.04 (ddd, J = 12.0, 2.3, 1.0 Hz, 2H), 3.78 (dqd, J = 10.5, 6.2, 2.4 Hz, 2H), 2.65 (dd, J = 12.9, 10.6 Hz, 2H), 1.26 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 155.1, 137.8, 132.8, 130.6, 128.8 (d, J = 31.5 Hz),, 126.7, 126.2, 125.2, 125.1, 111.3, 71.3, 51.4, 17.7. HRMS (ESI†) m/z calcd. for C19H19F3N4O [M + H]+ 377.1589, found 377.1590.(2S,6R)-2,6-Dimethyl-4-(3-(3-(methylsulfonyl)phenyl)imidazo[1,2-b]pyridazin-6-yl)morpholine (8)
Synthesized using substrate S2. Pale yellow solid (254 mg, 68%). 1H NMR (500 MHz, methanol-d4) δ 9.25 (t, J = 1.8 Hz, 1H), 8.23 (ddd, J = 7.9, 1.8, 1.0 Hz, 1H), 8.05 (s, 1H), 7.89 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 7.81 (d, J = 10.0 Hz, 1H), 7.71 (t, J = 7.9 Hz, 1H), 7.24 (d, J = 10.0 Hz, 1H), 4.11 (dt, J = 12.1, 2.2 Hz, 2H), 3.78 (dqd, J = 10.5, 6.2, 2.4 Hz, 2H), 3.16 (s, 3H), 2.63 (dd, J = 12.9, 10.6 Hz, 2H), 1.29 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 155.1, 141.2, 137.8, 130.4, 130.4, 130.3, 129.4, 125.9, 125.5, 125.2, 123.9, 110.9, 71.5, 51.2, 43.2, 17.7. HRMS (ESI†) m/z calcd. for C19H22N4O3S [M + H]+ 387.1491, found 387.1493.(2S,6R)-4-(3-(4-Fluorophenyl) imidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (9)
Synthesized using substrate S2. Yellow solid (217 mg, 69%). 1H NMR (500 MHz, methanol-d4) δ 8.10–8.02 (m, 2H), 7.78 (s, 1H), 7.73 (d, J = 9.9 Hz, 1H), 7.22–7.14 (m, 2H), 7.13 (d, J = 9.9 Hz, 1H), 4.01–3.94 (m, 2H), 3.73 (dqd, J = 10.5, 6.2, 2.4 Hz, 2H), 2.58 (dd, J = 12.9, 10.6 Hz, 2H), 1.23 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 161.6 (d, J = 244.9 Hz), 155.0, 137.5, 131.4, 128.4 (d, J = 7.9 Hz), 126.7, 126.5, 126.1, 116.0 (d, J = 21.4 Hz), 110.6, 71.1, 51.5, 19.2. HRMS (ESI†) m/z calcd. for C18H19FN4O [M + H]+ 327.1621, found 327.1623.(2S,6R)-4-(3-(4-Chlorophenyl)imidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (10)
Synthesized using substrate S2. Yellow solid (246 mg, 74%). 1H NMR (500 MHz, methanol-d4) δ 8.08–8.01 (m, 2H), 7.83 (s, 1H), 7.74 (d, J = 9.9 Hz, 1H), 7.48–7.40 (m, 2H), 7.15 (d, J = 9.9 Hz, 1H), 3.98 (ddd, J = 12.1, 2.3, 1.0 Hz, 2H), 3.74 (dqd, J = 10.5, 6.2, 2.4 Hz, 2H), 2.59 (dd, J = 12.9, 10.6 Hz, 2H), 1.24 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.9, 137.4, 132.8, 129.6, 128.3, 127.6, 127.5, 127.0, 125.0, 110.7, 71.3, 51.4, 17.7. HRMS (ESI†) m/z calcd. for C18H19ClN4O [M+H]+ 343.1326, found 343.1331.(2S,6R)-2,6-Dimethyl-4-(3-(4-(trifluoromethoxy)phenyl)imidazo[1,2-b]pyridazin-6-yl)morpholine (11)
Synthesized using substrate S2. Yellow solid (276 mg, 73%). 1H NMR (500 MHz, methanol-d4) δ 8.22–8.15 (m, 2H), 7.87 (s, 1H), 7.77 (d, J = 10.0 Hz, 1H), 7.37 (ddt, J = 7.9, 2.1, 1.0 Hz, 2H), 7.18 (d, J = 9.9 Hz, 1H), 4.01 (ddd, J = 12.1, 2.3, 1.0 Hz, 2H), 3.76 (dqd, J = 10.4, 6.2, 2.4 Hz, 2H), 2.62 (dd, J = 12.9, 10.6 Hz, 2H), 1.24 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 155.0, 148.2, 129.8, 128.1, 127.7, 126.9, 125.1, 121.6, 120.7, 119.6, 110.9, 71.3, 51.4, 17.7. HRMS (ESI†) m/z calcd. for C19H19F3N4O2 [M + H]+ 393.1538, found 393.1539.4-(6-((2S,6R)-2,6-Dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)benzamide (12)
Synthesized using substrate S2. Yellow solid (160 mg, 47%). 1H NMR (500 MHz, methanol-d4) δ 8.24–8.18 (m, 2H), 8.00–7.92 (m, 3H), 7.77 (d, J = 10.0 Hz, 1H), 7.19 (d, J = 10.0 Hz, 1H), 4.06–3.99 (m, 2H), 3.76 (dqd, J = 12.5, 6.2, 2.3 Hz, 2H), 2.62 (dd, J = 12.9, 10.6 Hz, 2H), 1.25 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 170.4, 155.0, 137.7, 132.3, 132.0, 130.4, 127.6, 127.2, 125.7, 125.1, 111.1, 71.3, 51.4, 17.7. HRMS (ESI†) m/z calcd. for C19H21N5O2 [M + H]+ 352.1774, found 352.1777.4-(6-((2S,6R)-2,6-dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)-N-methylbenzamide (13)
Synthesized using substrate S2. Off-white solid (265 mg, 75%). 1H NMR (500 MHz, methanol-d4) δ 8.20 (dt, J = 8.4, 2.3 Hz, 2H), 7.95–7.87 (m, 3H), 7.77 (dq, J = 10.1, 2.0 Hz, 1H), 7.19 (dq, J = 10.0, 2.0 Hz, 1H), 4.05–3.99 (m, 2H), 3.81–3.72 (m, 2H), 2.94 (dt, J = 3.1, 1.6 Hz, 3H), 2.62 (ddd, J = 15.1, 11.6, 3.4 Hz, 2H), 1.25 (d, J = 6.3 Hz, 6H).13C NMR (126 MHz, methanol-d4) δ 168.7, 155.0, 137.7, 132.6, 132.0, 130.4, 127.2, 127.1, 125.7, 125.1, 111.0, 71.3, 51.4, 25.5, 17.7. HRMS (ESI†) m/z calcd. for C20H23N5O2 [M+H]+ 366.1930, found 366.1933.2-(4-(6-((2S,6R)-2,6-Dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)phenyl)acetonitrile (14)
Synthesized using substrate S2. Off-white solid (221 mg, 66%). 1H NMR (500 MHz, methanol-d4) δ 8.07–8.01 (m, 2H), 7.80 (s, 1H), 7.68 (d, J = 9.9 Hz, 1H), 7.42–7.36 (m, 2H), 7.06 (d, J = 10.0 Hz, 1H), 3.91 (d, J = 14.4 Hz, 4H), 3.69 (dqd, J = 10.6, 6.2, 2.4 Hz, 2H), 2.52 (dd, J = 12.8, 10.6 Hz, 2H), 1.21 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 153.3, 135.7, 128.5, 128.1, 127.0, 126.3, 125.8, 124.9, 123.4, 116.6, 109.0, 69.7, 49.8, 20.2, 16.2. HRMS (ESI†) m/z calcd. for C20H21N5O [M + H]+ 348.1824, found 348.1815.4-(6-((2S,6R)-2,6-Dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)benzenesulfonamide (15)
Synthesized using substrate S2. Off-white solid (231 mg, 62%). 1H NMR (500 MHz, DMSO-d6) δ 8.34 (d, J = 8.6 Hz, 2H), 8.12 (s, 1H), 7.96 (d, J = 9.9 Hz, 1H), 7.90 (d, J = 8.6 Hz, 2H), 7.37 (s, 2H), 7.28 (d, J = 10.0 Hz, 1H), 4.08–4.02 (m, 2H), 3.69 (dqd, J = 12.4, 6.1, 2.3 Hz, 2H), 2.57 (dd, J = 12.8, 10.6 Hz, 2H), 1.18 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 155.2, 142.5, 138.3, 132.8, 132.7, 126.8, 126.5, 126.1, 126.0, 111.3, 71.2, 51.5, 19.2. HRMS (ESI†) m/z calcd. for C18H21N5O3S [M + H]+ 388.1443, found 388.1441.5-(6-((2S,6R)-2,6-Dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)-2-fluorobenzonitrile (16)
Synthesized using substrate S2. Pale yellow solid (225 mg, 66%). 1H NMR (500 MHz, Methanol-d4) δ 8.70 (dd, J = 6.2, 2.3 Hz, 1H), 8.30 (ddd, J = 8.9, 5.0, 2.3 Hz, 1H), 7.95 (s, 1H), 7.79 (d, J = 10.0 Hz, 1H), 7.43 (t, J = 9.0 Hz, 1H), 7.22 (d, J = 10.0 Hz, 1H), 4.01 (ddd, J = 12.1, 2.4, 1.1 Hz, 2H), 3.80 (dqd, J = 10.5, 6.2, 2.4 Hz, 2H), 2.65 (dd, J = 12.9, 10.7 Hz, 2H), 1.29 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 161.59 (d, J = 258.0 Hz), 155.0, 137.6, 132.7, 132.6, 130.3, 130.1, 126.6, 125.2, 124.9, 116.5 (d, J = 20.2 Hz), 113.4, 111.1, 100.9 (d, J = 15.7 Hz) 71.3, 71.2, 51.3, 17.7. HRMS (ESI†) m/z calcd. for C19H18FN5O [M + H]+ 352.1574, found 352.1571.(2S,6R)-2,6-Dimethyl-4-(3-(pyridin-4-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (17)
Synthesized using substrate S2. Off-white solid (176 mg, 59%). 1H NMR (500 MHz, methanol-d4) δ 8.56–8.51 (m, 2H), 8.15–8.10 (m, 2H), 8.09 (s, 1H), 7.76 (d, J = 10.0 Hz, 1H), 7.21 (d, J = 10.0 Hz, 1H), 3.99 (ddd, J = 12.1, 2.3, 1.0 Hz, 2H), 3.75 (dqd, J = 10.4, 6.2, 2.4 Hz, 2H), 2.62 (dd, J = 12.8, 10.6 Hz, 2H), 1.26 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 155.2, 148.9, 138.6, 137.3, 131.9, 125.2, 124.8, 119.7, 111.7, 71.2, 51.3, 17.7. HRMS (ESI) m/z calcd. for C17H19N5O [M + H]+ 310.1668, found 310.1668.(2S,6R)-2,6-Dimethyl-4-(3-(pyridin-3-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (18)
Synthesized using substrate S2. Off-white solid (227 mg, 78%). 1H NMR (500 MHz, methanol-d4) δ 9.28 (d, J = 2.4 Hz, 1H), 8.48–8.39 (m, 2H), 7.92 (d, J = 2.0 Hz, 1H), 7.73 (dd, J = 10.0, 2.0 Hz, 1H), 7.49 (ddd, J = 7.7, 5.0, 2.0 Hz, 1H), 7.15 (dd, J = 10.0, 2.0 Hz, 1H), 3.95 (dd, J = 13.2, 2.5 Hz, 2H), 3.77–3.67 (m, 2H), 2.56 (td, J = 11.7, 2.1 Hz, 2H), 1.23 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 155.0, 147.1, 145.9, 137.7, 133.8, 130.1, 126.0, 125.1, 124.6, 123.8, 111.2, 71.2, 51.2, 17.7. HRMS (ESI†) m/z calcd. for C17H19N5O [M + H]+ 310.1668, found 310.1667.3-(6-((2S,6R)-2,6-Dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)benzonitrile (19)
Synthesized using substrate S2. Brown solid (224 mg, 70%). 1H NMR (500 MHz, methanol-d4) δ 8.66–8.61 (m, 1H), 8.23–8.17 (m, 1H), 7.93 (s, 1H), 7.76–7.64 (m, 1H), 7.64–7.51 (m, 2H), 7.15 (dt, J = 10.1, 1.9 Hz, 1H), 4.01–3.93 (m, 2H), 3.75 (dqd, J = 12.5, 6.2, 2.1 Hz, 2H), 2.60 (dd, J = 12.8, 10.5 Hz, 2H), 1.25 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 154.8, 138.1, 132.4, 130.7, 130.4, 130.1, 130.0, 128.7, 126.6, 124.9, 119.2, 112.1, 110.9, 71.2, 71.0, 51.4, 19.1. HRMS (ESI†) m/z calcd. for C19H19N5O [M + H]+ 334.1668, found 334.1667.(2S,6R)-4-(3-(1H-Indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (20)
Synthesized using substrate S2. Pale yellow solid (90 mg, 27%). 1H NMR (500 MHz, methanol-d4) δ 8.57 (dd, J = 1.6, 0.8 Hz, 1H), 8.06 (d, J = 1.0 Hz, 1H), 7.99 (dd, J = 8.8, 1.6 Hz, 1H), 7.82 (s, 1H), 7.75 (d, J = 9.9 Hz, 1H), 7.61 (dt, J = 8.8, 1.0 Hz, 1H), 7.13 (d, J = 10.0 Hz, 1H), 4.02 (ddd, J = 12.1, 2.3, 1.0 Hz, 2H), 3.75 (dqd, J = 12.5, 6.2, 2.4 Hz, 2H), 2.61 (dd, J = 12.9, 10.6 Hz, 2H), 1.24 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.9, 139.5, 136.9, 134.0, 129.1, 128.6, 126.0, 125.0, 123.0, 121.8, 118.5, 110.3, 109.9, 71.2, 51.5, 17.7. HRMS (ESI†) m/z calcd. for C19H20N6O [M+H]+ 349.1772, found 349.1785.(2S,6R)-4-(3-(1H-Indazol-6-yl)imidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (21)
Synthesized using substrate S2. Pale yellow solid (111 mg, 33%). 1H NMR (500 MHz, DMSO-d6) δ 8.50 (d, J = 1.5 Hz, 1H), 8.07 (d, J = 2.5 Hz, 2H), 7.95 (d, J = 9.9 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.72 (dd, J = 8.5, 1.4 Hz, 1H), 7.26 (d, J = 10.0 Hz, 1H), 4.11–4.04 (m, 2H), 3.71 (dqd, J = 12.4, 6.1, 2.2 Hz, 2H), 2.59 (dd, J = 12.8, 10.6 Hz, 2H), 1.20 (d, J = 6.2 Hz, 7H). 13C NMR (126 MHz, DMSO-d6) δ 155.1, 140.6, 137.9, 134.0, 132.1, 127.6, 127.1, 126.8, 122.2, 121.1, 119.7, 110.5, 106.9, 71.2, 51.6, 19.3. HRMS (ESI†) m/z calcd. for C19H20N6O [M + H]+ 349.1772, found 349.1783.(2S,6R)-4-(3-(1H-Indazol-4-yl)imidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (22)
Synthesized using substrate S2. Yellow solid (141 mg, 42%). 1H NMR (500 MHz, methanol-d4) δ 8.17 (s, 1H), 7.90 (s, 1H), 7.81–7.75 (m, 2H), 7.57 (d, J = 8.4 Hz, 1H), 7.47 (dd, J = 8.5, 7.1 Hz, 1H), 7.15 (d, J = 10.0 Hz, 1H), 3.96–3.89 (m, 2H), 3.67 (dqd, J = 12.4, 6.2, 2.2 Hz, 2H), 2.51 (dd, J = 12.9, 10.6 Hz, 2H), 1.16 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.7, 140.6, 137.2, 133.9, 130.8, 127.2, 126.4, 125.0, 121.4, 120.5, 120.0, 110.9, 109.6, 71.3, 51.2, 17.6. HRMS (ESI†) m/z calcd. for C19H20N6O [M + H]+ 349.1772, found 349.1783.(2S,6R)-2,6-Dimethyl-4-(3-(1-methyl-1H-indazol-6-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (23)
Synthesized using substrate S2. Off-white solid (313 mg, 90%). 1H NMR (500 MHz, methanol-d4) δ 8.57 (s, 1H), 7.98 (s, 1H), 7.96 (s, 1H), 7.77–7.71 (m, 2H), 7.68 (d, J = 8.6 Hz, 1H), 7.14 (dt, J = 10.0, 1.7 Hz, 1H), 4.03–3.95 (m, 5H), 3.77 (dtt, J = 12.4, 6.1, 3.6 Hz, 2H), 2.59 (t, J = 11.5 Hz, 2H), 1.24 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.9, 139.8, 137.5, 132.2, 130.1, 127.4, 127.1, 124.8, 122.4, 120.7, 119.0, 110.3, 105.4, 71.2, 51.3, 34.1, 17.8. HRMS (ESI†) m/z calcd. for C20H22N6O [M + H]+ 363.1928, found 363.1932.(2S,6R)-2,6-Dimethyl-4-(3-(1-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (24)
Synthesized using substrate S2. Off-white solid (334 mg, 96%). 1H NMR (500 MHz, methanol-d4) δ 8.50 (dd, J = 1.6, 0.8 Hz, 1H), 8.01–7.95 (m, 2H), 7.80 (s, 1H), 7.72 (d, J = 9.9 Hz, 1H), 7.57 (dt, J = 8.9, 0.9 Hz, 1H), 7.09 (d, J = 9.9 Hz, 1H), 4.07 (s, 3H), 3.97 (ddd, J = 12.0, 2.3, 1.0 Hz, 2H), 3.71 (ddt, J = 12.5, 6.2, 3.1 Hz, 2H), 2.56 (dd, J = 12.8, 10.6 Hz, 2H), 1.22 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.8, 139.1, 136.9, 132.8, 129.1, 128.3, 125.7, 124.9, 123.8, 121.7, 118.5, 110.2, 109.0, 71.2, 51.4, 34.3, 17.7. HRMS (ESI†) m/z calcd. for C20H22N6O [M + H]+ 363.1928, found 363.1934.(2S,6R)-4-(3-(3-Ethyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (25)
Synthesized using substrate S2. Off-white solid (49 mg, 25%). 1H NMR (500 MHz, DMSO-d6) δ 8.85 (dd, J = 1.6, 0.8 Hz, 1H), 8.02 (s, 1H), 7.96–7.87 (m, 2H), 7.56–7.50 (m, 1H), 7.22 (d, J = 9.9 Hz, 1H), 4.10–4.03 (m, 2H), 3.72 (dqd, J = 12.3, 6.0, 2.2 Hz, 2H), 2.96 (q, J = 7.6 Hz, 2H), 2.58 (dd, J = 12.6, 10.5 Hz, 2H), 1.34 (t, J = 7.6 Hz, 3H), 1.18 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 155.2, 147.5, 140.4, 137.5, 131.0, 127.9, 126.6, 125.4, 121.8, 121.1, 116.9, 111.0, 110.1, 71.2, 51.7, 20.3, 19.2, 14.1. HRMS (ESI†) m/z calcd. for C21H14N6O [M + H]+ 377.2089, found 377.2089.(2S,6R)-4-(3-(3-Cyclopropyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (26)
Synthesized using substrate S2. Pale yellow solid (50 mg, 30%). 1H NMR (500 MHz, DMSO-d6) δ 8.83 (d, J = 1.2 Hz, 1H), 8.00 (s, 1H), 7.96–7.86 (m, 2H), 7.54–7.49 (m, 1H), 7.22 (d, J = 10.0 Hz, 1H), 4.10–4.04 (m, 2H), 3.71 (dqd, J = 12.3, 6.1, 2.2 Hz, 2H), 2.58 (dd, J = 12.7, 10.6 Hz, 2H), 2.27 (tt, J = 7.6, 6.1 Hz, 1H), 1.16 (d, J = 6.2 Hz, 6H), 1.03–0.96 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 155.2, 147.3, 140.5, 137.5, 131.0, 127.9, 126.6, 125.6, 122.0, 121.1, 116.9, 111.0, 110.1, 71.2, 51.7, 19.2, 8.5, 7.9. HRMS (ESI†) m/z calcd. for C22H24N6O [M + H]+ 389.2089, found 389.2086.5-(6-((2S,6R)-2,6-Dimethylmorpholino)imidazo[1,2-b]pyridazin-3-yl)indolin-2-one (27)
Synthesized using substrate S2. Off-white solid (76 mg, 45%). 1H NMR (500 MHz, methanol-d4) δ 7.95–7.88 (m, 2H), 7.75–7.68 (m, 2H), 7.09 (d, J = 10.0 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 4.00–3.94 (m, 2H), 3.74 (dqd, J = 12.4, 6.1, 2.3 Hz, 2H), 3.54 (s, 2H), 2.58 (dd, J = 12.8, 10.6 Hz, 2H), 1.23 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 178.4, 154.8, 142.7, 136.9, 128.7, 128.2, 126.0, 126.0, 124.9, 123.1, 122.6, 110.1, 109.2, 71.2, 51.5, 35.7, 17.7. HRMS (ESI†) m/z calcd. for C20H21N5O2 [M + H]+ 364.1774, found 364.1771.(2S,6R)-4-(3-Bromo-2-methylimidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (S4)
Pale yellow solid (425 mg, 82%). 1H NMR (500 MHz, methanol-d4) δ 7.56 (d, J = 9.8 Hz, 1H), 7.04 (d, J = 9.9 Hz, 1H), 4.00 (ddd, J = 12.1, 2.4, 1.1 Hz, 2H), 3.70 (dqd, J = 10.5, 6.2, 2.4 Hz, 2H), 2.51 (dd, J = 12.9, 10.6 Hz, 2H), 2.33 (s, 3H), 1.22 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.9, 139.1, 136.0, 124.0, 110.2, 98.3, 71.3, 51.0, 17.7, 12.2. HRMS (ESI†) m/z calcd. for C13H17BrN4O [M + H]+ 325.2189, found 325.2186.(2S,6R)-2,6-Dimethyl-4-(2-methyl-3-(3-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (28)
Synthesized using substrate S4. Yellow solid (242 mg, 67%). 1H NMR (500 MHz, methanol-d4) δ 8.04 (dd, J = 1.6, 0.9 Hz, 1H), 7.67–7.57 (m, 2H), 7.51 (dd, J = 8.7, 0.9 Hz, 1H), 6.99 (d, J = 9.9 Hz, 1H), 3.89–3.81 (m, 2H), 3.63 (dqd, J = 12.3, 6.1, 2.2 Hz, 2H), 2.52 (s, 3H), 2.50–2.38 (m, 5H), 1.13 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 154.4, 142.3, 140.3, 138.1, 134.8, 127.9, 125.0, 123.9, 122.0, 120.7, 120.5, 109.8, 109.4, 71.3, 71.2, 51.3, 51.2, 17.7, 13.4, 10.3. HRMS (ESI†) m/z calcd. for C21H24N6O [M + H]+ 377.2089, found 377.2087.(1R,4R)-5-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (S5)
Off-white solid (478 mg, 75%). 1H NMR (500 MHz, methanol-d4) δ 7.66 (d, J = 9.8 Hz, 1H), 7.45 (s, 1H), 6.86 (d, J = 9.8 Hz, 1H), 4.90–4.86 (m, 1H), 4.73–4.69 (m, 1H), 3.88 (s, 2H), 3.59 (dd, J = 10.0, 1.6 Hz, 1H), 3.48–3.42 (m, 1H), 2.06–1.96 (m, 2H); 13C NMR (126 MHz, methanol-d4) δ 153.3, 136.7, 130.3, 124.8, 111.0, 99.7, 76.2, 72.7, 57.2, 56.2, 35.9. HRMS (ESI†) m/z calcd. for C11H12BrN4O [M + H]+ 295.0194, found 295.0193.(1R,4R)-5-(3-(3-Methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (29)
Synthesized using substrate S5. Off-white solid (51 mg, 64%). 1H NMR (500 MHz, methanol-d4) δ 8.66 (s, 1H), 7.96 (dd, J = 8.8, 1.6 Hz, 1H), 7.84 (s, 1H), 7.75 (d, J = 9.8 Hz, 1H), 7.52 (d, J = 8.8 Hz, 1H), 6.88 (d, J = 9.8 Hz, 1H), 4.74 (d, J = 2.4 Hz, 1H), 3.98 (d, J = 7.6 Hz, 1H), 3.92 (dd, J = 7.6, 1.5 Hz, 1H), 3.66 (dd, J = 9.8, 1.5 Hz, 1H), 3.50 (d, J = 9.8 Hz, 1H), 2.57 (s, 3H), 2.11–1.94 (m, 2H), 1.27 (s, 1H). 13C NMR (126 MHz, methanol-d4) δ 152.8, 142.6, 128.6, 125.8, 125.0, 122.1, 121.2, 117.6, 110.1, 109.9, 76.3, 72.6, 57.5, 56.5, 47.1, 36.1, 10.3. HRMS (ESI†) m/z calcd. for C19H18N6O [M + H]+ 347.1620, found 347.1624.(1S,4S)-5-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (S6)
Off-white solid (506 mg, 79%). 1H NMR (500 MHz, methanol-d4) δ 7.71–7.63 (m, 1H), 7.48–7.43 (m, 1H), 6.91–6.84 (m, 1H), 4.89 (d, J = 5.3 Hz, 2H), 4.72 (s, 1H), 3.91–3.86 (m, 2H), 3.63–3.56 (m, 1H), 3.49–3.43 (m, 1H), 2.07–1.95 (m, 2H); 13C NMR (126 MHz, methanol-d4) δ 153.3, 136.7, 130.3, 124.8, 111.0, 99.8, 76.2, 72.7, 57.3, 56.2, 35.9. HRMS (ESI†) m/z calcd. for C11H12BrN4O [M + H]+ 295.0194, found 295.0193.(1S,4S)-5-(3-(3-Methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (30)
Synthesized using substrate S6. Off-white solid (65 mg, 93%). 1H NMR (500 MHz, methanol-d4) δ 8.69 (t, J = 1.1 Hz, 1H), 8.00 (dd, J = 8.8, 1.5 Hz, 1H), 7.90–7.69 (m, 2H), 7.54 (d, J = 8.8 Hz, 1H), 6.92 (d, J = 9.7 Hz, 1H), 4.00 (d, J = 7.6 Hz, 1H), 3.94 (dd, J = 7.6, 1.5 Hz, 1H), 3.69 (dd, J = 9.9, 1.5 Hz, 1H), 3.52 (d, J = 9.7 Hz, 1H), 2.59 (s, 3H), 2.13–2.01 (m, 2H), 1.28 (s, 1H). 13C NMR (126 MHz, methanol-d4) δ 152.8, 142.6, 140.4, 131.5, 126.4, 125.8, 125.0, 122.1, 121.2, 117.6, 110.1, 109.9, 108.8, 76.3, 72.6, 57.5, 56.5, 36.1, 10.3, 10.2. HRMS (ESI†) m/z calcd. for C19H18N6O [M + H]+ 347.1620, found 347.1625.4-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)-2,2-dimethylmorpholine (S7)
Off-white solid (571 mg, 85%). 1H NMR (500 MHz, DMSO-d6) δ 7.86 (d, J = 10.0 Hz, 1H), 7.58 (s, 1H), 7.21 (d, J = 9.9 Hz, 1H), 3.77–3.67 (m, 2H), 3.48 (dd, J = 6.2, 4.0 Hz, 2H), 1.19 (s, 6H); 13C NMR (126 MHz, chloroform-d) δ 155.3, 136.9, 132.2, 126.1, 109.4, 100.3, 71.3, 60.4, 55.4, 45.7, 24.6. HRMS (ESI†) m/z calcd. for C12H16BrN4O [M + H]+ 311.0507, found 311.0506.2,2-Dimethyl-4-(3-(3-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (31)
Synthesized using substrate S7. Yellow solid (178 mg, 51%). 1H NMR (500 MHz, methanol-d4) δ 8.68 (s, 1H), 7.95 (dt, J = 8.9, 1.8 Hz, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.79 (dd, J = 9.9, 2.3 Hz, 1H), 7.54 (dd, J = 8.8, 2.4 Hz, 1H), 7.15 (dd, J = 9.9, 2.4 Hz, 1H), 3.90 (q, J = 3.9 Hz, 2H), 3.57 (dt, J = 6.9, 3.0 Hz, 2H), 3.45 (d, J = 2.4 Hz, 2H), 2.59 (d, J = 2.5 Hz, 3H), 1.32 (d, J = 2.4 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 155.4, 142.1, 140.2, 137.3, 130.9, 127.8, 126.6, 125.3, 122.7, 121.1, 117.2, 110.8, 109.9, 71.2, 60.1, 55.3, 46.2, 24.9, 12.1. HRMS (ESI†) m/z calcd. for C20H22N6O [M + H]+ 363.1933, found 363.1931.(R)-4-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)-2-methylmorpholine (S8)
Off-white solid (501 mg, 78%). 1H NMR (500 MHz, DMSO-d6) δ 7.87 (d, J = 9.9 Hz, 1H), 7.59 (s, 1H), 7.22 (d, J = 10.0 Hz, 1H), 4.14–3.84 (m, 3H), 3.71–3.51 (m, 2H), 2.92 (ddd, J = 12.9, 11.9, 3.5 Hz, 1H), 2.70–2.54 (m, 1H), 1.15 (d, J = 6.2 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ 155.6, 137.1, 132.0, 126.5, 111.0, 99.8, 71.3, 65.8, 51.8, 45.4, 19.1. HRMS (ESI†) m/z calcd. for C11H14BrN4O [M + H]+ 297.0351, found 297.0348.(R)-2-Methyl-4-(3-(3-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (32)
Synthesized using substrate S8. Off-white solid (151 mg, 42%). 1H NMR (500 MHz, DMSO-d6) δ 8.77 (dd, J = 1.6, 0.8 Hz, 1H), 8.02 (s, 1H), 7.98–7.87 (m, 2H), 7.52 (dd, J = 8.7, 0.8 Hz, 1H), 7.20 (d, J = 10.0 Hz, 1H), 4.07 (dt, J = 12.5, 2.1 Hz, 1H), 4.03–3.92 (m, 2H), 3.71–3.61 (m, 2H), 3.02–2.93 (m, 1H), 2.67 (dd, J = 12.7, 10.4 Hz, 1H), 2.51 (s, 3H), 1.18 (d, J = 6.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 155.2, 142.1, 140.2, 137.4, 130.9, 127.9, 126.6, 125.3, 122.7, 121.1, 117.2, 110.8, 109.9, 71.2, 66.0, 52.3, 46.1, 19.3, 12.0. HRMS (ESI†) m/z calcd. for C19H20N6O [M + H]+ 349.1777, found 349.1779.(S)-4-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)-2-methylmorpholine (S9)
Off-white solid (449 mg, 69%). 1H NMR (500 MHz, methanol-d4) δ 7.71 (d, J = 10.0 Hz, 1H), 7.50 (s, 1H), 7.17 (d, J = 10.0 Hz, 1H), 4.12–3.96 (m, 3H), 3.76–3.65 (m, 2H), 3.02 (ddd, J = 13.0, 11.9, 3.5 Hz, 1H), 2.69 (dd, J = 12.9, 10.4 Hz, 1H), 1.24 (d, J = 6.3 Hz, 3H); 13C NMR (126 MHz, methanol-d4) δ 155.5, 136.9, 130.7, 124.9, 110.8, 100.2, 71.4, 65.8, 51.6, 45.1, 17.6. HRMS (ESI†) m/z calcd. for C11H14BrN4O [M + H]+ 297.0351, found 297.0348.(S)-2-Methyl-4-(3-(3-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (33)
Synthesized using substrate S9. Off-white solid (146 mg, 41%). 1H NMR (500 MHz, DMSO-d6) δ 8.79–8.75 (m, 1H), 8.01 (s, 1H), 7.97–7.89 (m, 2H), 7.52 (dd, J = 8.8, 0.8 Hz, 1H), 7.19 (d, J = 9.9 Hz, 1H), 4.06 (dt, J = 12.5, 2.1 Hz, 1H), 3.96 (dddd, J = 12.9, 8.4, 3.1, 1.3 Hz, 2H), 3.71–3.61 (m, 2H), 3.02–2.92 (m, 1H), 2.66 (dd, J = 12.7, 10.4 Hz, 1H), 2.51 (s, 3H), 1.18 (d, J = 6.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 155.2, 142.1, 140.2, 137.4, 130.9, 127.9, 126.6, 125.3, 122.7, 121.1, 117.2, 110.8, 109.9, 71.2, 66.0, 52.3, 46.1, 19.3, 12.0. HRMS (ESI†) m/z calcd. for C19H20N6O [M+H]+ 349.1779, found 349.1774.3-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)-8-oxa-3-azabicyclo[3.2.1]octane (S10)
Off-white solid (514 mg, 77%). 1H NMR (500 MHz, DMSO-d6) δ 7.85 (d, J = 10.0 Hz, 1H), 7.58 (s, 1H), 7.13 (d, J = 10.0 Hz, 1H), 4.57–4.37 (m, 2H), 3.78 (d, J = 12.3 Hz, 2H), 3.06 (dd, J = 12.6, 2.6 Hz, 2H), 1.95–1.67 (m, 4H); 13C NMR (126 MHz, DMSO-d6) δ 156.3, 137.1, 131.9, 126.3, 110.4, 99.6, 73.1, 51.3, 28.0. HRMS (ESI†) m/z calcd. for C12H14BrN4O [M + H]+ 309.0351, found 309.0349.3-(3-(3-Methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)-8-oxa-3-azabicyclo[3.2.1]octane (34)
Synthesized using substrate S10. Off-white solid (39 mg, 19%). 1H NMR (500 MHz, methanol-d4) δ 8.65 (dd, J = 1.6, 0.8 Hz, 1H), 7.99 (dd, J = 8.8, 1.6 Hz, 1H), 7.87 (s, 1H), 7.79 (d, J = 9.9 Hz, 1H), 7.54 (dd, J = 8.8, 0.9 Hz, 1H), 7.11 (d, J = 10.0 Hz, 1H), 4.53 (dd, J = 4.5, 2.4 Hz, 2H), 3.85 (dt, J = 12.6, 1.1 Hz, 2H), 3.24 (dd, J = 12.4, 2.6 Hz, 2H), 2.59 (s, 3H), 2.03–1.88 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 155.7, 142.7, 140.3, 137.0, 128.9, 128.5, 125.9, 124.9, 122.1, 121.1, 117.8, 109.9, 109.3, 73.8, 51.3, 27.6, 10.3. HRMS (ESI†) m/z calcd. for C20H20N6O [M + H]+ 361.1777, found 361.1763.(2S,6S)-4-(3-Bromoimidazo[1,2-b]pyridazin-6-yl)-2,6-dimethylmorpholine (S11)
Off-white solid (236 mg, 35%). 1H NMR (500 MHz, chloroform-d) δ 7.68 (d, J = 9.9 Hz, 1H), 7.53 (s, 1H), 6.79 (d, J = 9.9 Hz, 1H), 4.27–4.06 (m, 2H), 3.65 (dd, J = 12.8, 3.4 Hz, 2H), 3.27 (dd, J = 12.8, 6.3 Hz, 2H), 1.30 (d, J = 6.4 Hz, 6H); 13C NMR (125 MHz, chloroform-d) δ 155.4, 136.8, 132.0, 126.0, 109.4, 100.3, 66.0, 50.7, 17.9. HRMS (ESI†) m/z calcd. for C12H16BrN4O [M + H]+ 311.0507, found 311.0507.(2R,6R)-2,6-Dimethyl-4-(3-(3-methyl-1H-indazol-5-yl)imidazo[1,2-b]pyridazin-6-yl)morpholine (35)
Synthesized using substrate S11. Brown solid (131 mg, 37%). 1H NMR (500 MHz, methanol-d4) δ 8.66 (s, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.84 (s, 1H), 7.72 (d, J = 9.8 Hz, 1H), 7.49 (d, J = 8.7 Hz, 1H), 7.07 (d, J = 9.8 Hz, 1H), 4.15 (tt, J = 6.4, 3.4 Hz, 2H), 3.62 (dd, J = 12.8, 3.4 Hz, 2H), 3.26 (dd, J = 12.7, 6.4 Hz, 2H), 2.54 (d, J = 5.5 Hz, 3H), 1.27 (d, J = 6.4 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 155.2, 142.5, 140.2, 136.9, 128.8, 128.2, 125.5, 124.8, 122.0, 121.0, 117.2, 109.9, 109.7, 66.0, 50.6, 16.8, 10.4. HRMS (ESI†) m/z calcd. for C20H22N6O [M + H]+ 363.1933, found 363.1931.Cell viability assays
The myeloma cell lines used in this study were MPC-11 and H929. MPC-11 cells were maintained in DMEM with 10% FBS and H292 cells were maintained in RPMI with 10% heat-inactivated FBS. All cells were purchased from ATCC and maintained in culture below 10 passages. All cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. 4–5 × 103 cells were seeded in 96 well plates and incubated for 24 hours. Cells were then treated with various concentrations of indicated compounds for 72 hours. After the indicated period of treatment, the CellTiter-Blue cell viability assay reagent (Promega) was added based on the manufacturer’s recommendations and incubated for 3 hours. The fluorescence (λex/em = 560/590 nm) of each well was quantified via a Biotek Cytation 5 multi-mode reader. Experiments were performed in triplicate, with data reported as the mean and standard deviation of 3 data points. Readings from treated groups were normalized to cells treated with DMSO.Evaluation of pTAK1 levels in MPC-11 cells via western blot
0.5–1 × 106 MPC-11 cells were seeded in 6-well plates. After 24 h, the cells were treated with compounds 26, 3, and takinib for 48 h. The cells were then lysed with RIPA lysis buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate) with protease inhibitor cocktail (Roche) and 1 mM phenylmethylsulfonyl fluoride (Themo-Fisher Scientific). The cells were centrifuged and the supernatant was collected into new 1.5 mL Eppendorf tubes. The Pierce Rapid Gold BCA Protein Assay Kit (Thermo-Fisher) was used to quantify total protein. Next, the proteins were separated using 10% SDS polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was probed with pTAK1 (CST #9339), TAK1 (CST, #4505), and β-actin (CST #8457) antibodies overnight at 4 °C. After overnight incubation, the membrane was washed and further incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies at 24 °C for 2 h. A SuperSignal West Pico PLUS chemiluminescent substrate (Thermo-Fisher) was used for signal detection on an Azure 300 imaging system (Azure Biosystems). All antibodies were purchased from Cell Signaling Technology.Author contributions
HOS designed and supervised the project. DA and ND synthesized the compounds. DA performed the kinase assays. ALK and JL performed cell culture. NRB performed computational docking. All authors were involved in the writing of the manuscript.Conflicts of interest
HOS is a co-founder of KinaRx Inc., a company developing kinase inhibitors for potential oncology applications.Acknowledgements
We thank The Paula and Rodger Riney Foundation and the Purdue Institute for Cancer Research for funding.References
- S. V. Rajkumar, Am. J. Hematol., 2020, 95, 548–567 CrossRef CAS.
- A. Palumbo, K. Anderson and S. G. Battista, Clin. Cancer Res., 2016, 22, 5419–5427 CrossRef.
- K. L. Wu and P. Sonneveld, Clin. Lymphoma Myeloma, 2005, 6, 96–101 CrossRef PubMed.
- D. E. Reece, Hematology Am. Soc. Hematol. Educ. Program, 2005, 1, 353–359 CrossRef PubMed.
- M. S. Raab, K. Podar, I. Breitkreutz, P. G. Richardson and K. C. Anderson, Int. J. Mol. Sci., 2022, 23, 1649 CrossRef.
- S. V. Rajkumar and R. A. Kyle, Mayo Clin. Proc., 2005, 80, 1371–1382 CrossRef PubMed.
- N. Giuliani, V. Rizzoli and G. D. Roodman, Blood, 2006, 108, 3992–3996 CrossRef CAS PubMed.
- T. Hideshima, P. Richardson, D. Chauhan, V. J. Palombella, P. J. Elliott, J. Adams and K. C. Anderson, Cancer Res., 2001, 61, 3071–3076 CAS.
- S. Aashaq, A. Batool and K. I. Andrabi, Apoptosis, 2019, 24, 3–20 CrossRef CAS PubMed.
- M. Nishimura, M.-S. Shin, P. Singhirunnusorn, S. Suzuki, M. Kawanishi, K. Koizumi, I. Saiki and H. Sakurai, Mol. Cell. Biol., 2009, 29, 5529–5539 CrossRef CAS.
- B. Skaug, X. Jiang and Z. J. Chen, Annu. Rev. Biochem., 2009, 78, 769–796 CrossRef CAS PubMed.
- A. A. Ajibade, H. Y. Wang and R. F. Wang, Trends Immunol., 2013, 34, 307–316 CrossRef CAS PubMed.
- A. Adhikari, M. Xu and Z. J. Chen, Oncogene, 2007, 26, 3214–3226 CrossRef CAS PubMed.
- H. Sakurai, Trends Pharmacol. Sci., 2012, 33, 522–530 CrossRef CAS PubMed.
- J. Li, C. Liang, Z. K. Zhang, X. Pan, S. Peng, W. S. Lee, A. Lu, Z. Lin, G. Zhang, W. N. Leung and B. T. Zhang, Cell Discovery, 2017, 3, 17023 CrossRef CAS.
- R. Niesvizky, D. Siegel and J. Michaeli, Med. J. Aust., 2019, 210, 375–380 CrossRef.
- K. J. Gordon and G. C. Blobe, Biochim. Biophys. Acta, 2008, 1782, 197–228 CrossRef CAS PubMed.
- D. E. Joshua, C. Bryant, C. Dix, J. Gibson and J. Ho, Med. J. Aust., 2019, 210, 375–380 CrossRef.
- K. Podar, G. Tonon, M. Sattler, Y.-T. Tai, S. Legouill, H. Yasui, K. Ishitsuka, S. Kumar, R. Kumar, L. N. Pandite, T. Hideshima, D. Chauhan and K. C. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19478–19483 CrossRef CAS.
- J. Teramachi, H. Tenshin, M. Hiasa, A. Oda, A. Bat-Erdene, T. Harada, S. Nakamura, M. Ashtar, S. Shimizu, M. Iwasa, K. Sogabe, M. Oura, S. Fujii, K. Kagawa, H. Miki, I. Endo, T. Haneji, T. Matsumoto and M. Abe, Haematologica, 2021, 106, 1401–1413 CrossRef CAS PubMed.
- T. Harada, M. Hiasa, J. Teramachi and M. Abe, Cancers, 2021, 13, 4441 CrossRef CAS PubMed.
- J. Totzke, D. Gurbani, R. Raphemot, P. F. Hughes, K. Bodoor, D. A. Carlson, D. R. Loiselle, A. K. Bera, L. S. Eibschutz, M. M. Perkins, A. L. Eubanks, P. L. Campbell, D. A. Fox, K. D. Westover, T. A. J. Haystead and E. R. Derbyshire, Cell Chem. Biol., 2017, 24, 1029–1039 CrossRef CAS.
- A. Fauster, M. Rebsamen, K. V. M. Huber, J. W. Bigenzahn, A. Stukalov, C. H. Lardeau, S. Scorzoni, M. Bruckner, M. Gridling, K. Parapatics, J. Colinge, K. L. Bennett, S. Kubicek, S. Krautwald, A. Linkermann and G. Superti-Furga, Cell Death Dis., 2015, 6, e1767 CrossRef CAS PubMed.
- A. Garrido, G. Vera, P. O. Delaye and C. Enguehard-Gueiffier, Eur. J. Med. Chem., 2021, 226, 113867 CrossRef CAS PubMed.
- M. Al-Ghorbani, B. A. Begum, M. S. Zabiulla and S. Ara Khanum, J. Chem. Pharm. Res., 2015, 7, 281–301 CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- H. Zegzouti, M. Zdanovskaia, K. Hsiao and S. A. Goueli, Assay Drug Dev. Technol., 2009, 7, 560–572 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- L. M. Lima and E. J. Barreiro, Curr. Med. Chem., 2005, 12, 23–49 CrossRef CAS.
- A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7–10 CrossRef CAS.
- X. Chen, S. Hussain, S. Parveen, S. Zhang, Y. Yang and C. Zhu, Curr. Med. Chem., 2012, 19, 3578–3604 CrossRef CAS PubMed.
- A. Ovung and J. Bhattacharyya, Biophys. Rev., 2021, 13, 259–272 CrossRef CAS PubMed.
- E. Lenci, L. Calugi and A. Trabocchi, ACS Chem. Neurosci., 2021, 12, 378–390 CrossRef CAS PubMed.
- A. Kumari and R. K. Singh, Bioorg. Chem., 2020, 96, 103578 CrossRef CAS.
- A. P. Kourounakis, D. Xanthopoulos and A. Tzara, Med. Res. Rev., 2020, 40, 709–752 CrossRef CAS.
- C. Ouyang, L. Nie, M. Gu, A. Wu, X. Han, X. Wang, J. Shao and Z. Xia, J. Biol. Chem., 2014, 289, 24226–24237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00415e |
| This journal is © The Royal Society of Chemistry 2024 |