
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence methods to probe mass transport and sensing in solid-state nanoporous membranes
H. Samet
Varol
 *a,
Dila
Kaya
bc,
Emma
Contini
a,
Chiara
Gualandi
a and
Damiano
Genovese
*a
*a,
Dila
Kaya
bc,
Emma
Contini
a,
Chiara
Gualandi
a and
Damiano
Genovese
*a
aDipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, 40126 Bologna, Italy. E-mail: samet.varol@unibo.it; damiano.genovese2@unibo.it
bGSI Helmholtzzentrum für Schwerionenforschung, 64291 Darmstadt, Germany
cDepartment of Chemistry, Istanbul Medeniyet University, 34700, Istanbul, Turkey
First published on 9th September 2024
Abstract
Single- and multi-nanoporous (1–100 nm pore size) solid-state membranes (SSNMs) receive significant attention in various fields, spanning from biosensing to water purification. Their finely tunable nanopore geometry and chemistry, combined with the large selection of materials that they can be made of, such as polymers, inorganic materials (e.g., silicon, silica, and alumina), and hydrogels, provide an excellent platform to control their mass transport and sensing capabilities for different cargoes from Å scale ions up to macromolecular biomaterials. The critical requirement to merge these nanoporous membranes’ advanced structural and chemical features with their applications is to find the most suitable analytical techniques that permit macro- and micro-scale and real-time probing of different nanopore activities. Luminescence-based detection of various physico-chemical processes in nanoporous membranes has recently received great attention: it permits rapid, non-invasive, and dynamic probing of nanoporous materials, yielding information on mass transport and sensing both (i) macroscopically, such as from an array of nanopores, and (ii) micro-nanoscopically, with ultra-high (e.g., single molecule) sensitivity and high resolution in time and space. Quantitative information arising from luminescence experiments on membrane-analyte interactions has uncovered the effects of nanoconfinement, membrane stability, and performance. This review article aims to provide the reader with a handbook of fluorescent methods–from the simplest to implement to the most advanced–helpful in studying different kinds of SSNMs for a specific application or function. To this end, we include examples from the literature published in the last ten years. At the end of our article, we also discuss limitations of the current state of fluorescence probing techniques and their future prospects.
 H. Samet Varol | H. Samet Varol received his PhD from the Molecular Spectroscopy Department of Max Planck Institute for Polymer Research (MPIP-Mainz, Germany) in 2017. Afterward, he worked as postdoc at MPIP-Mainz, the University of Zurich (Switzerland), and TU-Darmstadt (Germany). He is now working as an Alexander von Humboldt Postdoctoral Fellow at the Chemistry Department “Giacomo Ciamician” of the University of Bologna (Italy). His research mainly focuses on designing multi-functional nanoporous membranes and studying their function by spectroscopic, electrochemical, and microscopic methods. He also works on polymer-based nanocomposite mechanics and novel (super)antiwetting coatings. |
 Dila Kaya | Dila Kaya received her PhD degree in Physical Chemistry from Yildiz Technical University in Istanbul, Turkey. She is currently a post-doc in the Materials Research Department at the GSI Helmholtz Centre for Heavy Ion Research in Darmstadt, Germany. Her research mainly focuses on developing nanochannel-based sensors. Her work includes resistive-pulse sensing of DNA, fabricating nanopipettes for nanoparticle detection, studying gated ionic transport through single nanochannel membranes, and exploring various surface modification techniques. |
 Emma Contini | Emma Contini received her MSc degree in Photochemistry and Molecular Materials (2022) at the University of Bologna and is currently a PhD student in Industrial Chemistry at the University of Bologna. Her scientific interests mainly concern the development and characterization of mechanochromic polymeric materials and composites as sensors for early damage detection. |
 Chiara Gualandi | Chiara Gualandi received her PhD degree in Industrial Chemistry from the University of Bologna in 2010. She worked as post-doc at the Advanced Mechanics and Materials Interdepartmental Center for Industrial Research of the University of Bologna. She is currently an Associate Professor at the Chemistry Department “Giacomo Ciamician” (Bologna, Italy). Her research interests focus on stimuli-responsive and functional polymers, including mechano-responsive materials, photo-responsive materials, thermo-reversible polymeric systems, and shape-memory polymers. |
 Damiano Genovese | Damiano Genovese received his PhD in Chemistry from the University of Bologna in 2011, and after research periods in Harvard University (USA), Karlsruhe Institute of Technology (Germany) and Strasbourg (France) was appointed Associate Professor at the Chemistry Department “Giacomo Ciamician” (Bologna, Italy) in 2022. He was awarded the ENI medal for his “Debut in research” in 2013 and received the “Young Investigator Award” from the Italian group of photochemistry in 2018. His research group focuses on applying a broad range of luminescence-based techniques (including microscopy) to the study of hybrid nanomaterials. |
1. Introduction
Nature has countless examples to inspire materials scientists to design new and intelligent synthetic materials and/or advance their properties for specific applications. One of the most inspiring examples from nature is the biological ion channel activities in cell membranes, which play a vital role in living organisms based on their transport control mechanism for different matters.1,2 Inspired by the stimuli-responsive transport control mechanisms of biological channels, materials scientists have designed membranes featuring numerous tiny pores. These pores directly influence selectivity, rectification, and gating due to their high pore density. Such synthetic membranes carrying one or many nm size nanopores (when membrane thickness and diameter have similar sizes) or nanochannels (when the membrane thickness, thus, the pore channel length is larger than pore diameter) are called solid-state nanoporous membranes (SSNM).3,4 Unlike the biological ion channels, synthetic SSNMs are fabricated from abiotic materials, providing mechanical resistance, chemical functionality, and higher lifetime in severe conditions such as extreme pH and heat.3,5–7 Nanopores/Nanochannels of such SSNMs are mainly described as pores having a diameter <100 nm, and their small size, stimuli-responsive surface activities, and finely adjustable density on the membrane film make SSNM an ideal platform for various mass transport and sensing applications. Moreover, SSNMs could also find many applications in other fields, namely DNA/RNA sequencing, energy conversion and storage, water desalination and filtration, nanoreactors, dialysis, and nanoelectronics.4,8 However, many of these applications are based on their mass transport and sensing capability. Thus, our review article will include examples of membranes from the literature that are (or will be) used for mass transport and sensing applications. One significant factor towards applying such nanoporous materials for a specific application is their geometrical dimensionality which could be (i) 0-dimensional (0D) (e.g., cube, sphere, tube), (ii) membranes with 1D nanochannels (e.g., ion-track etched polymer membranes), (iii) single or multi-layered (2D) nanoporous membranes (e.g., mesoporous silica based membranes) or (iv) 3D nanoporous membranes (e.g., 3D-hydrogel membranes). There is a vast amount of important research presenting nanoporous membranes with different functions. However, for our review article, we kept the following three parameters to limit the content of the paper: (1) present studies on nanoporous membranes with pore size between 1–100 nm, where possible, and (2) present examples of membrane films in the form of >0D structure, such as film coating or free-standing film, rather than being 0D or similar geometries such as nanopipettes, nanoparticles, or nanotubes. (3) Selected examples published within the last ten years.In order to study the transport through solid-state nanoporous membranes analytically to understand their sensing and mass transport performance, the most well-known method is electrochemical characterization.4,9,10 In a typical steady-state electrochemical setup, an SSNM is sandwiched between the two halves of a container (cell) filled with electrolyte solution, and upon application of transmembrane voltage by a potentiostat, it is possible to record current–voltage curves (I–V curves). The changes in these curves are the direct indicator of the ion transport changes across the nanopores.4,11 Besides such steady-state electrochemical setup, electrochemical impedance spectroscopy (EIS) is also used to study the nanopore activities electrochemically, especially to estimate the membrane resistance with other parameters.12–14 While these ionic current-based probing of nanoporous materials offers excellent time response and sensitivity, it is often limited by characterizing single nanopore films. For the multiple nanopore membrane measurements, the signal-to-noise ratio decreases, and the current signals from individual pores become convoluted, making it challenging to discriminate small molecules or subtle differences between molecules. A strategy to overcome this limitation might be to connect each nanopore to an individual electrode and reservoir, though this presents significant engineering challenges.15 However, other sensing methods can leverage the molecular confinement provided by nanopores to yield additional information beyond ionic currents. Consequently, there has been significant interest in integrating nanopore platforms with various optical sensing strategies. The most used optical-sensing methods for probing the nanopore activities of SSNMs are (i) adsorption, (ii) fluorescent spectroscopy, (iii) plasmon resonance, or (iv) surface-enhanced Raman scattering. Of course, alternatives to the ionic-current-based probing of nanopores are not limited to optical probing. Further novel methods have been used to study nanoporous materials, such as field-effect transistors (FETs) and quantum tunneling.15 However, in this review, as the major method of optical nanopore probing techniques, we will focus only on fluorescence methods to study mass transport and sensing in nanoporous membranes.
Compared to other optical methods, fluorescence spectroscopy is a very fast and efficient method to collect data from nanoporous membranes; for instance, it is the only optical probing method to integrate spectra in short time scales, allowing the study of the translocation-diffusion dynamics of DNA or protein through nanopores of SSNMs.16–18 Depending on their working principle on probing luminescent spices, fluorescence methods have various sub-categories and classes. In this article, we classified the fluorescent methods used for solid-state nanoporous membranes into two main sub-categories: (1) macroscopic luminescence spectroscopy and microscopic luminescent imaging, and (2) single molecule fluorescence spectroscopy/microscopy. When considering fluorescence microscopy for nanopore imaging, the most known method is the Single Molecule Spectroscopy to probe nanopores. This is mainly because fluorescence spectroscopy is among the most widely used techniques for detecting single molecules in solution based on its ability to collect strong signals from a single fluorophore. Nanopores are exceptionally effective in being investigated by single-molecule fluorescence spectrometers since nanopores can confine analytes to a well-defined volume significantly smaller than a typical diffraction-limited probe volume, thereby substantially improving overall photon-collection efficiency. Additionally, solid-state nanopores offer the advantage of enabling the design of complex multilayer structures that can both enhance fluorescence and minimize background noise.15,19,20 However, even though the SSNM field focuses on probing nanopore activities by using fluorescent single-molecule detection, we still considered introducing macroscopic fluorescent spectroscopy and microscopy investigations. The first factor that motivated us is that single-molecule fluorescent spectroscopy/microscopy methods require special expertise and equipment. However, as provided in detail below, many fundamental nanopore activities of various SSNMs can be investigated using different macroscopic fluorescent techniques such as probing analyte cargoes permeating through nanoporous film in time21–25 or collecting data of translocation events in a high-density nanopore array rather than a single nanopore.9,10 Such macroscopic fluorescence spectroscopy/microscopic characterization of SSNMs provides averaged signals over large volumes, enabling high-throughput analysis and revealing detailed structural and morphological information with high spatial resolution. These methods are crucial for understanding bulk properties, ensemble effects, and spatial organization within the membranes. It is also important to understand the function of nanoporous membranes.
Therefore, this review shows recent scientific findings, including an analysis of the wide aspects of nanoporous membranes using fluorescence techniques. While introducing examples from the literature, we kept the connection between the following three factors as tight as possible: (1) structural information of the nanoporous membranes (2) the role of fluorescent methods in probing the nanopore activities, and (3) the application of the nanoporous membrane. In this way, we thought of providing the reader a handbook to combine their expertise with the other one or two remaining factors to progress in their research in materials science. As an outline, this review article is organized in the following structure: we first introduce the fluorescent methods to study the mass transport and sensing of various nanoporous materials (Fig. 1). In the subsequent section, we introduced the nanoporous materials and briefly explained their production methods, the importance of the nanopore activities, and their potential applications. Again, we distribute a large set of solid-state nanoporous membrane materials within three sub-categories: (a) membranes with 1D nanochannels, (b) 1D and 2D nanoporous membranes, and (c) 3D nanoporous membranes. For the rest of the manuscript, we mainly focus on different examples from the last 10 years, presenting how different fluorescent methods could help understand the various mass transport and sensing activities of different nanoporous membranes and how the fluorescently probed information could be beneficial in the application of a specific SSNM. In the final section, we present our vision for the importance of the future of fluorescence readout for investigating novel nanoporous membranes and their applications.
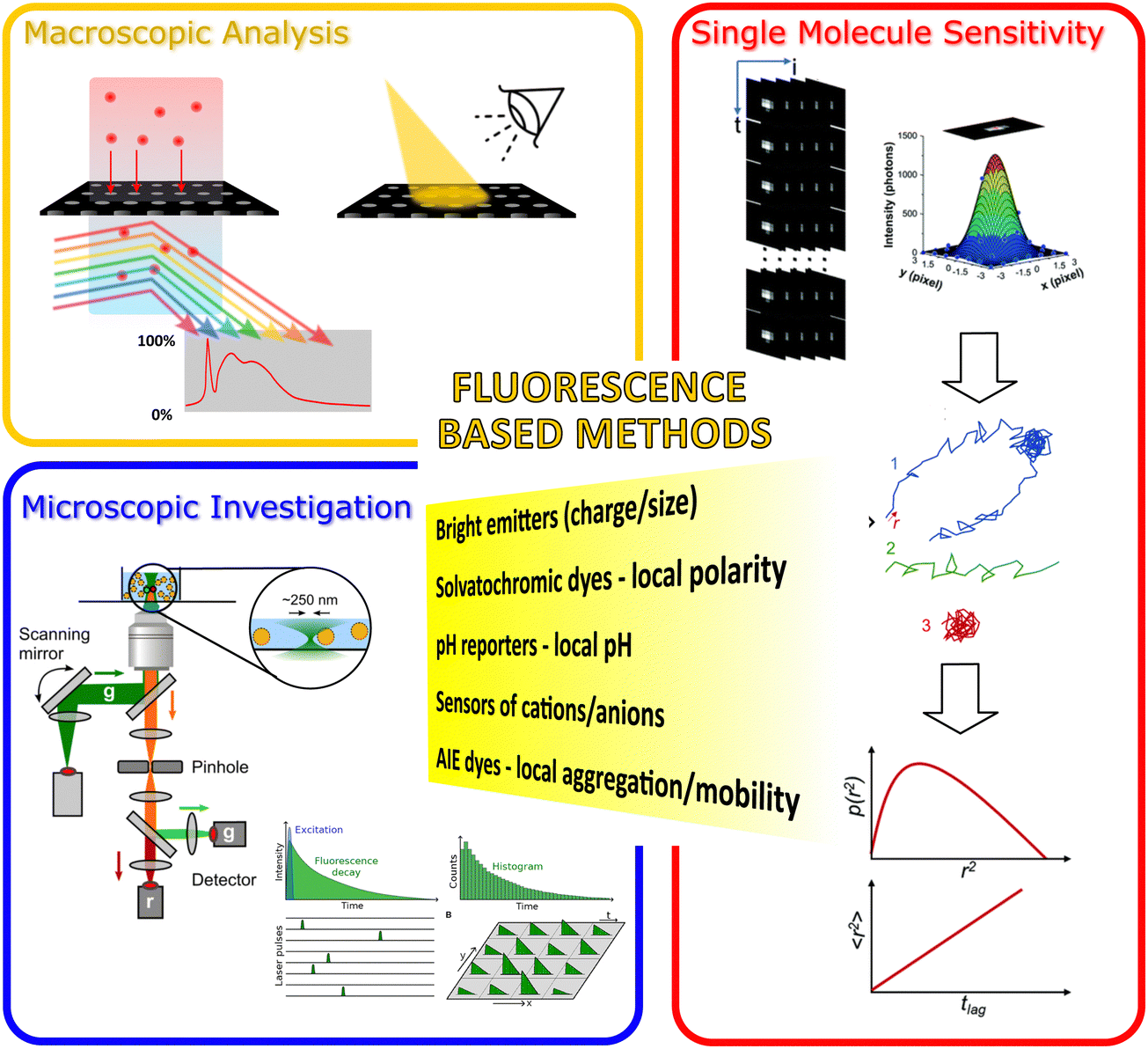 | ||
| Fig. 1 Illustration of the fluorescent methods included in our review article for probing the nanopore activities of various nanoporous materials. The figure's central text (in the yellow area) lists some significant functions of fluorescent methods that are critical to probe the nanopore activities. We categorized these luminescence-based methods into three main classes: (orange zone) Macroscopic fluorescence methods; (blue zone) investigation of the fluorescence emission from the nanoporous materials microscopically; and (red zone) high-resolution (e.g., single molecule sensitive fluorescent methods) fluorescent microscopy. Images in (blue zone) were reproduced from ref. 26 Copyright 2021, Elsevier and ref. 27 Copyright 2021, Frontiers. Images used in the red zone were reprinted with the permission from ref. 19 Copyright 2021, American Chemical Society. | ||
2. Luminescence-based methods
Photoluminescence (PL) is the widespread phenomenon whereby a molecule or a nanostructured species, upon absorption of a photon, emits another photon – typically at lower energy.The emission of a photon accompanies the relaxation from a high-energy excited state (which in photoluminescence is achieved upon absorption of a photon) to a lower energy state or to the ground state; when this transition occurs between states with same multiplicity, it is spin-allowed and therefore the kinetic constant of this process is high, in the range of 108–109 s−1. This kind of photoluminescence is named fluorescence and typically occurs in a time window of 0.1–100 ns. By contrast, phosphorescence concerns spin-forbidden radiative transitions that are, consequently, characterized by much smaller kinetic constants in the range of 106 s−1 or smaller. Phosphorescence typically occurs in a timeframe of μs up to seconds after the absorption of a photon.
During the time spent by a luminescent species at the excited state, a number of physico-chemical processes can occur that can change the properties of the excited state, thereby changing the luminescence in a more or less sensitive way. As inspiring examples–yet not as an exhaustive list–the excited state can be stabilized by reorganization of the solvent according to its polarity (solvatochromic dyes)28,29 or can be quenched by intramolecular motions that depend on the local viscosity experienced by the luminescent species (microviscosity probes such as molecular rotors and AIE–aggregation-induced emission–dyes).30,31 In addition, a species at the excited state is simply more energetic than the ground state; therefore, it may use this extra energy to undergo chemical reactions or reach metastable states (photocatalysis, photoisomerization, and photochemistry in general). Finally, the luminescent species can be subjected to acid–base or association–dissociation equilibria, where the different forms involved in these equilibria may have different luminescence properties, allowing the use of these species as chemical sensors for pH, ions, and other analytes. Quenching of luminescence can occur upon transient interaction with chemical species even in the absence of chemical association, a process called dynamic (or bimolecular) quenching: for example, owing to the spin-forbidden nature of the transition, phosphorescence is typically affected by the presence of dioxygen, which quenches emissive triplet states: a quantitative evaluation of the quenching process can then be used to measure the local concentration of O2.32
Finally, luminescence-based techniques can be extremely sensitive: an individual molecule can be subjected to a large number of absorption-emission cycles (up to 106 emitted photons for the best molecular emitters), laying the ground for the exceptional sensitivity of PL-based techniques, which even allows for monitoring the activity of a single molecule. For reaching the highest sensitivity, the absorption rate shall be pushed to the highest limit by using species with high molar extinction coefficient and light sources with high photon flux and density; furthermore, the species shall emit with high efficiency (high PL quantum yield), and the detecting arm of the instrument shall be characterized by high detection efficiency and low noise.
2.1. Macroscopic luminescence spectroscopy of porous membranes
Photoluminescence–bearing all information discussed above–can be investigated via bulk measurements of macroscopic systems (such as homogenous solutions in a cuvette or large areas of solid-state samples) that are taken as representative of the whole samples analyzed. Spectral properties of the emission of luminescent probes and their time decays can be investigated simply using a commercial fluorometer: if the fluorescent species is diffusing through the membrane, then the permeate solution may be flown to a cuvette, or the membrane may be accommodated in the instrument with a front-face geometry to study the emission building up in the membrane pores or surface.Suppose the membrane sample cannot be fruitfully accommodated in a commercial fluorometer. In that case, it is possible to bring excitation light and detect emission using optical fibers or build a dedicated optical path for excitation and luminescence detection.
In these configurations, spectral and time-dependent information on the emission of suitable luminescent probes is averaged in a region of space typically lying in the millimeter size range. Alternatively, luminescence can also be investigated as a function of the position in space, i.e., via imaging of PL properties, typically performed with micrometer (or sub-micron) resolution, as discussed in the following paragraph.
2.2. Microscopic luminescence imaging
The main and simplest PL property to image is the brightness (B), that is, the intensity resulting from the product of absorbance and emission probability coefficients (molar extinction coefficient (ε) and quantum yield (Φ), respectively) and the local concentration (c).B = ε![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Φ Φ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c | (1) |
Whether fluorescence or phosphorescence is involved, luminescence imaging methods are commonly called “fluorescence microscopies”. In a typical microscopy setup, the ratio between excitation and emission photons easily exceeds 106: an efficient separation of the huge amount of excitation photons is, therefore, vital to obtain a good signal/noise ratio (S/N) from the relatively few emitted photons. From this perspective, the “epifluorescence” geometry is advantageous over the transmittance geometry. Furthermore, suitable high-quality filters are necessary to reach the desired signal/background ratio and get the most from the sensitivity of fluorescence microscopy.33
Homogeneous illumination of the whole field of view and collection of the emitted photons via an imaging system (objective, tube lens, and imaging detector such as CCD or CMOS cameras) is called “wide-field fluorescence microscopy.” This relatively simple method can still reach the highest sensitivity, but it suffers from the PL emitted by species in optical planes that lay below or above the focal plane: for this reason, this technique is mostly indicated to image thin (<1 μm) samples.
The problem of poor contrast due to out-of-focus emission can be solved in different ways: laser-sheet microscopy and two-photon microscopy are two techniques of broad use to image thick, transparent samples, in particular in biomedical applications. TIRF (total internal reflection fluorescence) microscopy is a method that confines the excitation field to the very first layer (<200 nm) of the sample above a high refractive index interface, thus eliminating out-of-focus excitation and emission. Yet, the most widespread fluorescence microscopy technique that eliminates light from out-of-focus planes is undoubtedly confocal laser scanning microscopy (CLSM). The main novelties–and differences–of this technique compared to wide-field microscopy are that (i) the sample is not excited homogeneously in the whole field of view, yet the excitation is focused in a diffraction-limited focal spot, and that (ii) the emission is collected through a pinhole that–positioned in an image plane–cuts off most of the light emitted from out of focus planes.
In CLSM images are then reconstructed by scanning the diffraction-limited excitation spot throughout the sample. This allows the collection of other PL parameters than intensity: the emission spectrum and the emission decay can be measured in each excitation spot by implementing a CLSM with a spectral or a lifetime module, respectively. Furthermore, spectral information can be obtained more simply by separating the emission spectrum in 2 up-to 4 spectral regions that are independently analyzed by separate detectors.
The advantage of analyzing emission spectrum or decay over intensity is that, while intensity is a “relative” parameter (it provides information only relative to another measured intensity, in time or space), spectral and lifetime information are “absolute”, i.e., they contain information that can be directly given a physical meaning, thus yielding quantitative data in a straightforward fashion. As a general statement, fluorescence microscopy can yield quantitative data when a calibration curve is available and when accurate control on local concentration and volume can be achieved. Let us consider the example of a luminescent species (the donor) that is quenched by another luminescent species (the acceptor) via Förster resonance energy transfer (FRET): it is possible to evaluate the efficiency of FRET in three ways: (i) via the intensity of donor or acceptor emission, (ii) via the emission spectrum (ratio of donor and acceptor intensities) or (iii) via the lifetime of the donor emission. In the first case, in each pixel, the measured intensity depends on two variables: FRET efficiency and local concentration of the dyes: therefore, to assess whether, in a specific pixel, FRET is taking place, we have to know what the intensity would be in that very pixel in the absence of FRET. By contrast, it is sufficient to know the lifetime of the donor or the spectrum of donor and acceptor in the absence of FRET (from any instrument or any pixel, once for all) to estimate quantitatively the FRET efficiency in each pixel of a spectral or lifetime microscopy image. Emissive systems in which the spectral changes (i.e., the ratio between emission at different wavelengths) provide information are called “ratiometric”.34,35 Examples of this kind of system are FRET-based sensors, solvatochromic probes, and chemosensors where the emission spectrum changes upon interaction with the analyte. Similarly, the emission decay allows the mapping of an intrinsic property of the dyes–their emission lifetime–that is independent of concentration and, therefore, can be directly ascribed to the parameter to which the lifetime is sensitive, such as the presence of a chemical analyte or a physical parameter of the microenvironment such as viscosity, temperature or polarity.
2.3. Single-molecule localization microscopy (SMLM) & fluorescence correlation spectroscopy (FCS)
One major limitation of fluorescence microscopy is the relatively low resolution, which is limited by diffraction to λ/2NA where λ is the wavelength of the light used for imaging and NA is the Numerical Aperture of the objective. In the last 30 years, several solutions have been proposed to overcome this limitation based on either instrumental variations or post-acquisition data analysis. These approaches (recently awarded with the Nobel Prize in Chemistry 2014) are collectively called “Super-Resolution Microscopies”.36There are two main possibilities to achieve images beyond the diffraction-limited resolution: using a special fluorescence microscope (“hardware-based” super-resolution techniques) or manipulating the photochemistry of the dyes to acquire super-resolved information (“sample-based” super-resolution techniques). The most powerful and successful hardware-based method is stimulated emission depletion microscopy (STED), a technique now embedded in several commercial fluorescence microscopes based on using a second laser beam with a peculiar doughnut-shape. This beam rapidly depletes the fluorescence via the process of stimulated emission: after such a pulse of depleting light, only emission from the very center of the excitation is left, that can be detected and assigned to a much smaller volume of origin than the initial (diffraction limited) excitation volume.
On the other hand, the majority of “sample-based” super-resolution techniques are grounded on the precise localization of a photon emitter: this family of techniques, called single-molecule localization microscopy (SMLM),37 relies on the fact that a single fluorescent species can be localized with much higher precision than the diffraction limit if its emission spot (called the point-spread function, PSF) does not overlap to the PSF of other single fluorescent species. In common samples, the density of fluorophores is much higher, and the PSFs of the various emitters do overlap. Therefore, to spatially isolate single emitters at a distance larger than the PSF, the emission of each species should be observed when the other emitters are in a non-emissive state, i.e., the PSFs of the emitters should be distributed in time, rather than in space. There are many ways to temporally isolate the emission of species: one possibility is to switch photoactivatable fluorophores between a fluorescent (on) state and a nonfluorescent (off) state (photoactivated localization microscopy, PALM) or to exploit the dynamic nature of transient binding (binding activated localization microscopy, BALM). This switching mechanism permits the detection of single molecules even in densely labeled samples, which can then be localized with high precision to yield a super-resolved image.
Under such conditions, the precision of localization of the center of emission within the PSF–and therefore of the real position of the single molecule–is only limited by the signal to noise (S/N) ratio: the higher the S/N (i.e., the flow of emitted photons vs. the detection noise), the higher the precision of localization and the resolution of the final reconstructed image. For this reason, all SMLM techniques require instrumental components and fluorophores that can guarantee the maximum S/N, reaching thus the highest sensitivity in fluorescence microscopy.
Two other techniques that are only feasible with instrumental components and fluorophores allowing single-molecule sensitivity are (i) single-molecule tracking (SMT) and (ii) fluorescence correlation spectroscopy (FCS).38 These techniques allow for the direct characterization of molecular motion in space and time, yielding information on diffusion properties, environmental viscosity, and confinement, and also on processes that impact the size or diffusion properties, such as molecular recognition, assembly, or disassembly. Both techniques can also be applied to nanosystems (dye-doped nanoparticles, QDs, fluorescent polymers etc.) that provide a much higher brightness and S/N ratio, thus paving the way to single particle analysis with standard instrumental components.
3. Solid state nanoporous membranes
The definition of nanoporous materials is comprehensive since it includes various natural, synthetic (also known as solid-state), and biological examples with different dimensions, chemistry, and structures, such as charcoal, biological tissues, thin films, multi-layered films, bulk polymeric porous structures, micro- or nano-particles, nanotubes, and many more. This review article does not include natural or biological nanoporous membranes; it focuses only on synthetic solid-state nanoporous materials (SSNMs). Moreover, among different SSNMs, we selected examples from different chemistry and functionality membrane films/coatings having, in general, high-density nanochannels or nanopores with different geometrical dimensions (1D, 2D, and 3D) where the mass transport takes place (Fig. 2). A second major factor in classifying the synthetic nanoporous films is their density of nanochannels/nanopores: single pore or high density of channels/pores (multi-pore). However, before describing the selected membranes’ structural features, fabrication, and applications, it is essential to provide some common structural properties that are valid for all the selected materials in this review. Primarily, based on IUPAC classification and their pore diameters, porous materials are divided into three major groups: microporous (<2 nm), mesoporous (>2 nm and <50 nm), and macroporous (>50 nm) materials.39,40 In our review article, we use the “nanoporous” term to define the nanopore size (diameter or opening) of all membrane films within the nanometer range of 1–100 nm. Therefore, various synthetic solid-state nanoporous membrane films with sub-nm (<1 nm) range nanopore size, such as single or multi-layer graphene, graphene-oxide membranes, Zeolite membranes, and metal–organic-framework (MOF) membranes, were excluded from our review article. Another crucial structural definition that needs to be clarified is the difference between “nanochannel” and “nanopore.” As also illustrated in Fig. 2a, especially for single or low-pore density nanoporous membranes, 1-dimensional (1D) “nanochannel” is used for channels whose diameter is significantly lower than the membrane thickness (t > d; in other words, high aspect ratio). On the other hand, the term “nanopores” is used for openings in 2D thin membrane structures having similar or lower pore diameter compared to the film thickness (t ≤ d; low aspect ratio) (Fig. 2b). In general, throughout our manuscript, we divided the nanoporous membrane films/coatings into the following three major classes: (1) nanoporous membranes with 1D nanochannels (Fig. 2a); (2) 1D and 2D nanoporous membranes (Fig. 2b); and (3) 3D membranes (Fig. 2c). We did not include almost any examples from 0D nanoporous materials such as nanoporous particles, nanotubes, etc. Below, we will briefly introduce these three classes of nanoporous membranes while explaining their typical structural properties, nanofabrication strategies for their production, and common applications.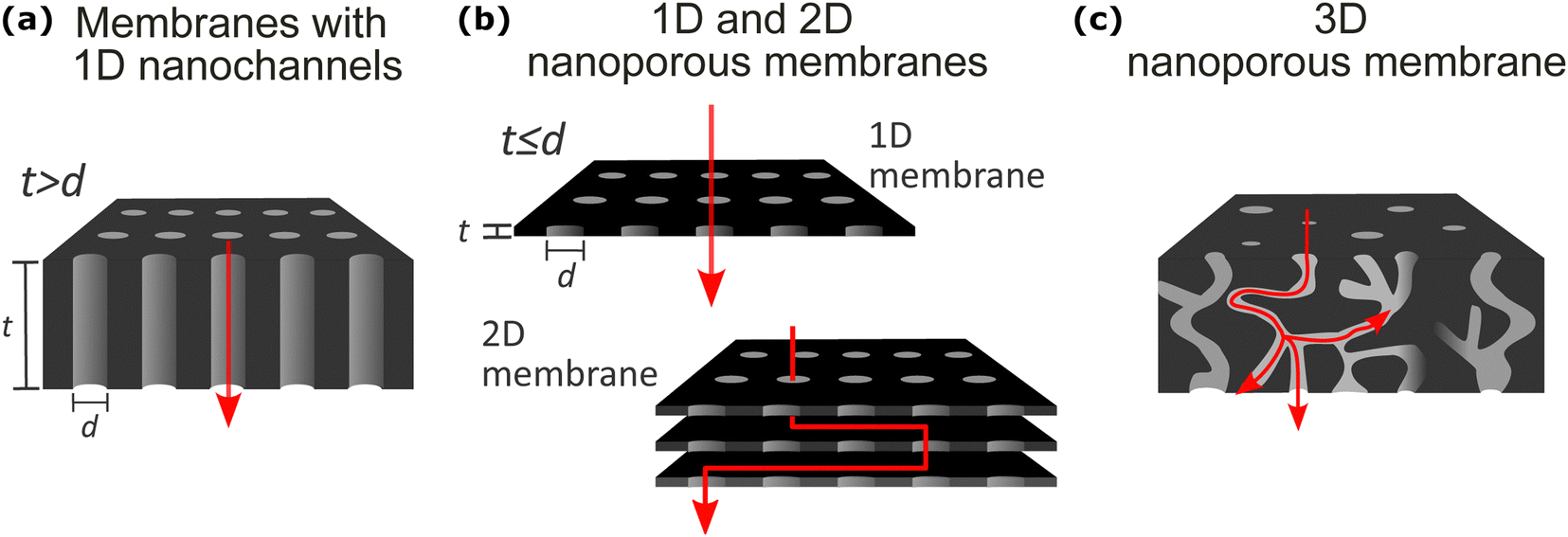 | ||
| Fig. 2 Illustrations of the solid-state membrane film structures included in our review: (a) 1D nanochannels with a longer channel length (or film thickness, t) than the diameter (d) of the nanopores; (b) 2D membrane thin films with 1D nanopores where d > t or the stacks of such 1D membranes (called 2D membranes); (c) 3D nanoporous membranes with irregularly connected nanopores. | ||
3.1. Membranes with 1D nanochannels
As “abiotic” analogues of biological channels, solid-state nanochannels provide a nanoconfined space where sensitive mass transport and detection of various targets can be achieved. The essential knowledge of the exact geometry, size, and surface properties of the nanochannels has enabled the design of nanochannel based ionic devices which are regularly employed as energy harvesters,41 chemical or biological sensors,42,43 separators,44 and iontronic devices such as nanofluidic transistors45 and diodes.46 These devices are preferred due to their high sensitivity while allowing the isolation and study of individual molecules.47 The interest in nanochannels stems from their unique chemical and physical properties compared to their bulk counterparts due to their spatial functions, such as size and surface charge distribution. These properties can be controlled and optimized by varying the size, shape, porosity, and charge carrying surface groups of the nanochannels for the desired application.Surface modification strategies for nanochannels are particularly significant for imparting specific properties, such as surface charge or wettability, as well as for introducing functional groups to the nanochannel surfaces. Various approaches can be employed to modify nanochannels, including covalent bonding of functionalities via coupling reactions, self-assembly of polyelectrolytes, atomic layer deposition, metallic deposition, and thiol chemistry.48,49 Unlike thermal evaporation (sputtering), which only covers the surface, all the above-mentioned techniques modify the entire nanochannel length. The modifications are routinely confirmed via electrochemical or fluorescent methods. In some cases, the modification of the surface of the nanoporous membrane may hinder its transport or sensing properties. In one example, it was found that the modification of AAO with a silane layer on the top surface caused adverse effects on the ionic transport properties of the membrane.50 The transport improved when the silane layer on the outer membrane surface was removed via air plasma treatment. In the case of metal deposition in polymer membranes, the top layer can easily be removed, for instance, by hand-polishing with fine-grain sandpaper.51 For large-size biomarkers as proteins, surface modification can be used to avoid the adsorption or aggregation of proteins onto polymer membrane surfaces for enhanced transport properties in sensing. It was shown that the self-assembly of PEG onto PET surface prevented the unwanted adsorption of protein aggregates onto the surface, ensuring the transient-state sensing through the nanochannels.52
Mass transport through nanochannels is usually detected and analyzed using conductivity measurements since the transport of an analyte through the nanoconfined space causes a measurable change in the ionic current due to volume exclusion, charge density, wettability etc.53 Any variation in the ionic current (e.g., the amplitude, duration, frequency etc.) can then be transformed into information concerning the analyte. On the other hand, electrical sensing can be complemented by optical detection methods to introduce the benefits of improved spatial resolution, specificity, simultaneous readouts, and regulated dwell times.53,54 Fluorescence is a useful tool in generating optical signals to capture information on the dynamic changes and translocation events within the nanochannels.
Here, we define one-dimensional (1D) nanostructures as nanochannels with high aspect ratios (i.e., length vs. diameter) (Fig. 2a) focusing on anodic aluminum oxide (AAO) and ion-track-etched polymer membranes. We provide insights into the fabrication processes and applications of 1D nanochannels.
Nanoporous AAO membranes are obtained in an acidic medium under an applied voltage. Larger diameters of nanochannels can be achieved by increasing the anodizing voltage, adjusting the pH of the acidic solution, prolonging the anodizing time, or reducing the concentration of the acidic electrolyte.55
The stages of AAO formation can be described as: (1) rapid anodic oxide formation at the metal/oxide interface, (2) current density increase as the pore nucleation starts until the maximum in which the oxide barrier layer is broken down, and disordered porous structure develops, (3) the disordered pores start to merge, and channels parallel to each other start to grow. (4) Steady stage where the thickness of the barrier layer is constant, and the porous structure grows in thickness through two competitive mechanisms: oxide formation and oxide dissolution due to the acid. An electric field assists both of these effects.56
Pretreatment processes, such as annealing and chemical cleaning, serve to eliminate internal stress in the metal and remove organic residues, respectively. Furthermore, a two-step anodization is usually carried out where the first step is employed to achieve an ordered surface on the Al metal to be used as a growth template in the second anodic oxidation.55 During this step, pre-patterning of the Al surface can be utilized to generate the desired channel morphologies. Additionally, further widening of the nanochannels can be achieved through acidic post-treatment.
AAO membranes are frequently employed as templates for designing and fabricating nanostructures with highly controllable features. Practical applications where AAO membranes are directly used include selective filtration for the separation of molecules,57 and drug delivery,58,59 as well as sensors for the electrochemical60–62 and optical detection63–65 of various analytes. The optical transparency of AAO membranes makes them suitable for sensing applications based on fluorescence detection.66 Single-molecule fluorescence can be used to investigate the mass transport mechanism of solutes through AAO nanochannels.67
Etched ion-track membranes are produced by swift heavy ion irradiation and their subsequent chemical etching. They are characterized by a narrow distribution of pore sizes and the unique advantage that all the relevant parameters can be tailored in an independent and controlled manner. The fabrication steps are,
(1) Irradiation of polymer foils with swift heavy-ions: each heavy ion projectile that passes through the polymer induces electronic excitation and ionization processes in a highly localized cylindrical zone along its trajectory.70 The ion fluence, typically between 1 and 1010 ions per cm2, is controlled by focusing or defocusing the ion beam. Single ion irradiation where only an individual heavy-ion impact with the polymer is possible, causing the production of a single ion-track at the center.
(2) Selective chemical etching of the ion-tracks to form tailored nanochannels: etching can be performed symmetrically or asymmetrically to control the nanochannel geometry. The nanochannel diameter is dependent on the etching time. Concentration or the composition of the chemical etchant, temperature of the etching solution, and pre-treatments directly influence the nanochannel size and geometry.71 Nanochannel length is determined by the thickness of the polymer, typically between 10–100 μm.
Ionic transport from track-etched single nanochannels provides valuable information for sensing purposes since their geometrical and electrical parameters can be adjusted and characterized independently. Ionic transport through PC, PET, and PI track-etched nanochannels has been investigated and regulated by external parameters such as pH, temperature, ion concentration.72–74 Asymmetry in the nanochannel geometry yields unique properties due to the broken symmetry in the nanochannel shape and the surface charge distribution. One effect is ion-current rectification, where the accumulation or depletion of ionic species within the nanochannel causes a diode-like ionic response, which can be implemented for sensing purposes. Additionally, ionic current gating can be performed on the electrically addressable nanochannels to further control the transmembrane current.5,45 Besides electrical signals generated by track-etched membrane systems, optical signals, particularly fluorescence intensity measurements and confocal imaging, have been extensively developed and utilized for both the characterization of nanochannel surfaces,75 and for separation76 and sensing purposes.77
3.2. 1D and 2D nanoporous membranes
As the second class of nanoporous films in this review, we present a few nm thick single (Fig. 2b-up) or multiple layers (Fig. 2b-bottom) 2D membrane films carrying single or multiple nanopores. As illustrated by red arrows in Fig. 2b, depending on the lamellar stack number (single or multiple) and pore density (e.g., single or multiple), the number of dimensions of mass transport changes. In single thin-layer films with one discrete nanopore or multipores, 1D mass transport (similar to membranes with 1D nanochannels) is possible (Fig. 2b-top). However, mass transport can also happen between layers for the multi-stack laminar nanosheets, thus creating 2D membrane nanopore activities (red arrow in Fig. 2b-bottom). In our article, among a large selection of 2D nanoporous solid-state membranes (single or multiple stacks and single- and multi-porous), we will focus on the following membranes mainly due to their tendency to be studied by various fluorescent sensing/investigation methods: mesoporous metal oxide films, covalent organic frameworks (COFs), and silicon-based thin membrane films.3.3. 3D nanoporous membranes
3D nanoporous membranes refer to matrices that display interconnected voids that weave throughout the material, allowing the passage of gases or liquids (Fig. 2c) with separation coefficients usually higher compared to 1D and 2D membranes. However, to obtain this interesting property, pore size and arrangement must be meticulously controlled to exploit size-controlled diffusion. Moreover, the complex interconnected design of these membranes provides a tortuous path for diffusing molecules, thus leading to different residence times arising based on physical and chemical pore-molecule chemical interactions.POPs encompass several subclasses distinguished by their monomer structures and polymerization methods. While most exhibit an amorphous 3D structure with irregular pore shapes and size distributions, covalent organic frameworks (COFs) are a notable exception. They are characterized by a crystalline structure with long-range order, enabling the formation of a highly regular 2D pore structure (Fig. 2b). Amorphous subclasses include hypercrosslinked polymers (HCPs),113 polymers of intrinsic microporosity (PIMs),114 conjugated microporous polymers (CMPs),115 and porous aromatic frameworks (PAFs).116
The versatile properties of POPs render them promising candidates for a wide range of scientific and industrial fields, such as heterogeneous catalysis,117 photocatalysis,118 CO2 capture and conversion,119,120 gas storage and separation,121 energy conversion and storage,122,123 chemical sensing,124 biomedical applications,125,126 and organic photovoltaic.127 POPs have gained valuable attention for their capability to host in their interconnected pores different kinds of guest molecules, such as metal ions,128 dyes,129 explosives,130 volatile organic compounds,131 radionuclides,132 and biological molecules.133 Guest molecule absorption is driven by specific interactions between POPs and guests, such as H-bonding, π–π interactions, electrostatic interactions, ionic interactions, hydrophobic interactions, and van der Waals's interactions. Due to their highly conjugated structures, most POPs are luminescent materials and work nicely as turn-off fluorescent sensors. In these cases, the fluorescence quenching in the presence of guest molecules can be driven by different mechanisms, including FRET, photoelectron transfer, absorption competition, and inner filter effects. For a detailed description of these systems, readers are invited to refer to recently published reviews.124,132
Depending on the embedded phase, these composite (or hybrid) membranes are classified as either mixed matrix membranes (MMMs) or nanocomposite membranes. In MMMs, the embedded fillers possess pores and contribute to mass transport through mechanisms like molecular sieving or diffusion.136
Nanocomposite membranes incorporate nanoparticles that enhance the composite's selectivity by improving separation properties or optimizing physico-chemical properties like hydrophilicity and porosity, overcoming the limitations of purely organic materials.137
In both cases, mass transport is governed not only by the individual properties of the components but also by the interactions between the matrix and the fillers. Therefore, having a well-dispersed filler and high compatibility between matrix and filler is mandatory to avoid aggregation of the filler and interfacial defects that could hinder performance.134
The potential applications of composite 3D nanoporous membranes are extensive and span numerous industries. These membranes can remove many contaminants in water purification, from heavy metals to emerging pollutants like pharmaceuticals.138–142 The chemical processing industry can leverage its capabilities to separate valuable products from complex mixtures and gas separation, leading to more efficient and sustainable production processes.143–146
Being 3D membranes, hydrogel properties depend immensely on their pore size.150,151 This parameter can be controlled during the synthesis by tailoring the crosslinking degree or by incorporating porogens, which are inert materials that can be subsequently removed to leave behind a defined pore structure within the hydrogel.152
However, determining pore size in hydrogels is an extremely challenging task: solid-state techniques, such as scanning electron microscopy, can easily provide this parameter; however, the result obtained highly depends on the technique employed for drying the hydrogel and does not account for the swelling-induced increase in pore size.153 To account for this, analysis performed in solutions, such as DLS (dynamic-light scattering) and FRAP (fluorescence recovery after photobleaching), are better suited despite being less straight-forward.153,154
The porosity of PEMs and the charged groups are critical parameters that govern their permeability and selectivity.155 PEMs can be fabricated with various pore sizes, from densely packed structures with nanometer-scale pores to highly porous networks with micron-sized channels.155
Dense PEMs with nm-scale pores exhibit high selectivity, allowing specific ions or molecules to pass while effectively rejecting others. This selectivity arises from the interplay of electrostatic interactions, steric hindrance, and size exclusion effects.156 This principle can be exploited for the production of ion exchange membranes in fuel cells, where PEMs selectively permit the transport of protons while rejecting bulk water molecules.157
High porosity PEMs, on the other hand, offer a compelling solution for applications demanding high permeability. Large pores within the membrane structure enhance the transport pathway for desired solutes. However, achieving high permeability often comes at the expense of selectivity.158
As for other membrane types, the selectivity of these membranes can be enhanced by the functionalization of pore walls with specific recognition groups, such as chelating agents159 and pH-responsive groups.160,161
4. Fluorescence methods used for probing nanopore activities of different nanoporous membranes
4.1. Bulk luminescence methods
In solid-state nanoporous materials, various analyte molecules with different luminescent activities have been monitored inside or in the vicinity of the nanopores, and variations in the fluorescence signal could be used as a direct indicator of various information on the nanopore activity, including nanoconfinement effects. The step was short, from striving for information on the behavior of nanopores to using fluorescence signals in various applications of nanoporous materials (first of all, sensing). The evolution of fluorescence based methods goes towards building up simpler methods and gaining higher sensitivity: since the early 2000s, fluorescence has been used primarily in probing the translocation of labeled biomolecules (e.g., intercalating dye such as YOYO-1 conjugated DNA) through solid-state nanopores.162,163 However, to capture the translocation information of biomolecules near the nanopores, a highly resolved and sensitive measurement was needed, which was obtained with fluorescence imaging techniques (Method-2, below). In some cases, detecting analytes from a single fluorophore sensitivity is desirable, which led to the use of fluorescence imaging techniques sensitive to single molecule detection with optimized signal/noise ratio, namely single-molecule localization microscopy (SMLM) and fluorescence correlation spectroscopy (FCS), techniques that will be discussed in Method-3. However, it is not always necessary to reach this level of sensitivity and resolution in space: in Method-1 we first want to introduce recent studies of nanoporous membrane examples carried out by spectroscopic (bulk) measurements such as steady-state fluorescence spectrometer.Nanoconfinement effects in nanopores have been explored by using dyes that are sensitive to local microviscosity and mobility (such as AIE luminogens), solvent polarity (solvatochromic dyes), or pH (fluorescent pH probes). AIE luminogens have mainly been applied to microscopy experiments and will then be reviewed in next chapter. To investigate solvation dynamics Braeuchle et al. measured the emission spectrum of a solvatochromic dye (nile blue) in ethanol confined to sol–gel glasses with 50 Å and 75 Å average pore size.169,170 Compared with the dynamics of the respective bulk solution, confinement reduces both the amplitude of the dynamic Stokes shift and the dynamics of the solvation process. This may be due to dye adsorbed at the inner pore surfaces, thus interacting with only a “half-space” of solvent molecules.
pH was instead locally probed via the use of pH-sensitive dyes: Andrieu-Brunsen and coworkers have recently explored the local interplay–inside 1D mesoporous silica films–between pH and the pKa of a fluorescent pH sensor, finding an apparent decrease of the pKa of the probe inside the pores, as compared to the same probe dissolved in water. This apparent pKa change may be explained either by an actual change of pKa of the dye itself or by a local increase of pH in nanoconfined pores (with respect to the bulk solution, where the pH is monitored).171 Furthermore, the same group found that the local mobility of water and of dissolved cations is much lower inside the nanopores compared to the bulk, which suggests that the activity of H3O+ ions is lower–and therefore the effective local pH higher–not (only) because of a lower concentration of protons, but (also) because of their lower activity.172
pH measurement inside nanoporous materials could be obtained in a faster and quantitative fashion using a ratiometric measurement: when the ratio between two emission bands changes as a function of pH, even an individual measurement of the emission spectrum (and thus of the ratio) is sufficient to provide a value of the local pH. This strategy was employed in 2019 by Stanzel et al., who grafted pH-sensitive (fluorescein isothiocyanate, FITC) and pH-insensitive (rhodamine B isothiocyanate, RITC) dyes inside the nanopores of the mesoporous silica films to monitor the pH-dependent emission spectrum (Fig. 3a).173 The presence of FRET between the dyes notably complicates this system, and authors only phenomenologically describe the observed pH dependence of the emission ratio, which is not simply sigmoidal. In fact, in the absence of FRET and of other parasite events such as aggregation, a simple and reproducible sigmoidal trend of this ratio should be expected as a function of pH, with the dynamic range of the sensor being ca. 2 pH units across the pKa of the pH-sensitive dye.
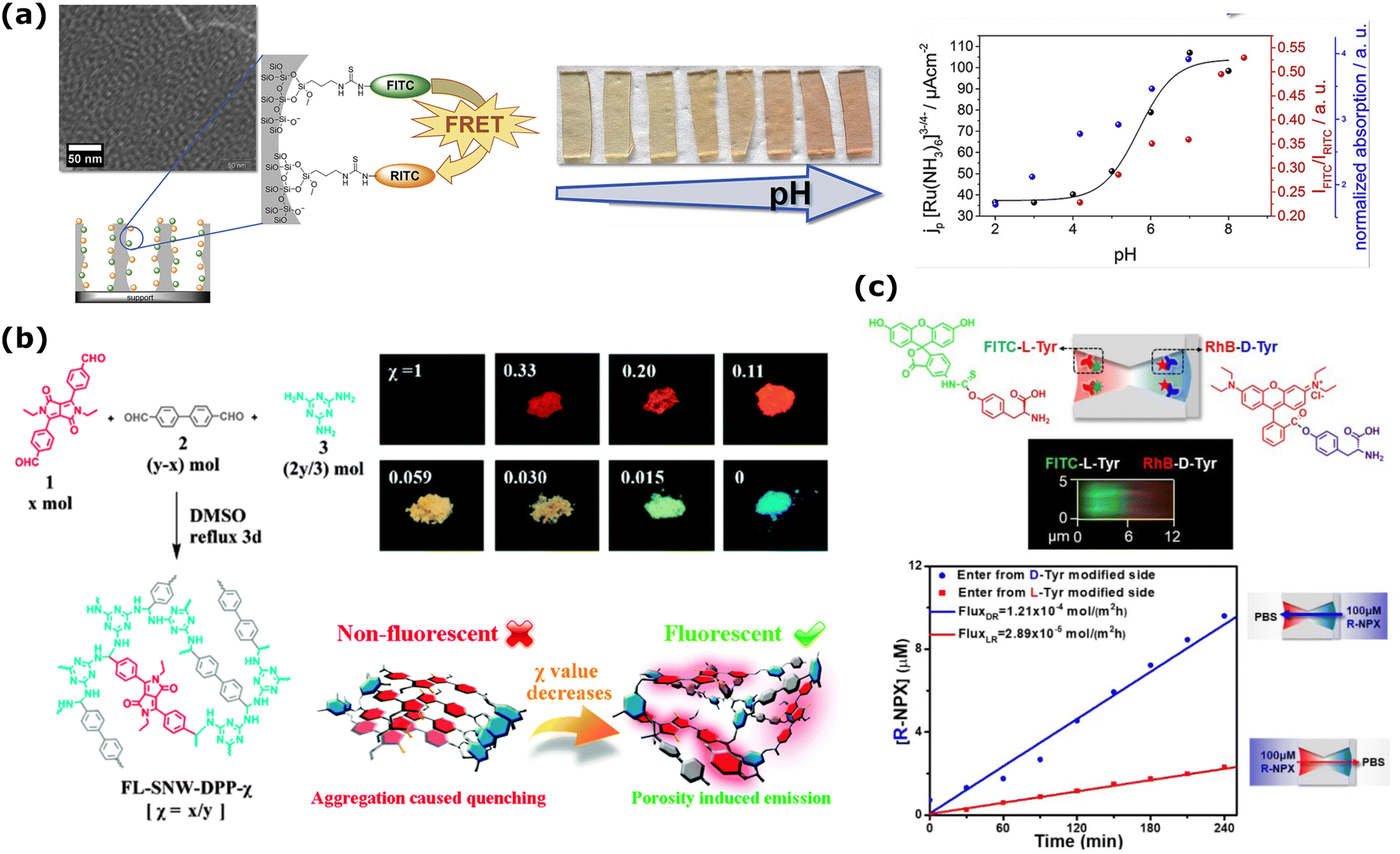 | ||
| Fig. 3 (a) FRET dye pair (FITC, green, donor and RITC, orange, acceptor) functionalized mesoporous silica film changes its fluorescence emission intensity (red spheres in the right graph) as a factor of changing pH. This change agrees with the pH-dependent transport properties of probe molecule [Ru(NH3)6]3−/4− from electrochemical investigations (black spheres in the right graph). Reprinted from ref. 173 with permission from Elsevier. Copyright 2019, Elsevier. (b) Mixing of 4,40-(2,5-diethyl-3,6-dioxo-2,3,5,6-tetrahydropyrrolo[3,4-c]pyrrole-1,4-diyl)dibenzaldehyde (1) with 4,40-biphenyldicarboxaldehyde (2) and melamine (3) to fabricate different FL-SNW-DPP-χ type POP membranes where χ is the molar ratio of (1). Solid-state photographic images of the samples with different χ taken after placing the samples under UV-lamp (λ = 365 nm). Schematic representation of “porosity induced emission” mechanism of the POPs. Adapted with the permission of ref. 174 Copyright 2015, Rocal Society of Chemistry. (c) (up) The schematic diagram of the asymmetrically modified nanochannel in PET and the fluorescence confocal image of labeled L-Tyr/D-Tyr channels. (bottom) The change of concentration with respect to time during the directional transport of R-NPX. Reprinted with permission from ref. 175 Copyright 2021, American Chemical Society. | ||
Track-etched membranes with 1D nanochannels are frequently employed as pH sensors based on electrochemical measurements after functionalization with pH-responsive groups.176,177 Additionally, track-etching an optically translucent polymer, such as polyethylene terephthalate (PET), permits the development of optical sensors. In 1D nanochannels, a similar approach to pH sensing was investigated by Espinoza et al., who fabricated track-etched PET membranes and used immobilized Fluorescein (Fluorescein isothiocyanate, FITC) or green fluorescent protein (GFP) as alternative chemosensors.77 They employed a grafting polymerization technique initially developed for PVDF and PP membranes, which they then extended to PET membranes with 1D hour-glass shaped nanochannels.178 Interestingly, authors found that FITC is not anymore pH-responsive when bound to the membrane, while GFP-immobilized track-etched PET membranes qualitatively exhibited a low emission at acidic pH and a high emission at alkaline pH, with repeatable fluorescence responses to pH changes. They also demonstrated the potential use of the developed sensor in biological environments by monitoring the pH of an E. coli cell culture in situ based on the emitted fluorescence intensity measurements.
Bulk fluorescence spectra can also be obtained by utilizing fluorophores as analyte labels or immobilized on 1D nanochannel walls. These optical readouts offer advantages in controlling channel properties for diverse functionalization paths and providing an alternative sensing route for various applications. Fluorescent sensors based on micro and nano-sized systems are used in several areas, including biotechnology and medicine.179 In a recent example, an optofluidic biosensor was developed based on an Anodic Alumina membrane (AAO) to provide a rapid and point-of-care alternative for the testing and diagnostics of the COVID-19 virus.66 After the fabrication of AAO nanochannel membranes with diameters of 20–200 nm, the probe DNA was immobilized on the nanochannel walls. The target was labeled with Cy3, and it was shown that the large surface area of the nanochannels increased the fluorescence enhancement compared to glass templates. A correlation was found between the fluorescence intensity and channel diameter, where the highest intensity was observed for the nanochannel with the largest diameter.
In the framework of 3D membranes, steady-state fluorescence emission of fluorescently labeled nanopores has been widely exploited in Porous Organic Polymers (POPs) for sensing purposes.124 Among numerous examples, an interesting work was published in 2015 to demonstrate that POP fluorescence can be tuned by dosing the fluorophore concentration and the system porosity (Fig. 3b).174 More specifically, the authors have covalently bonded 4-diketopyrrolo[3,4-c]pyrrole (DPP), a commercial dye that normally suffers from ‘‘aggregation-caused quenching’’ (ACQ), into the skeleton of POPs and demonstrated that the porosity contributes to spatially isolating the DPP dyes, efficiently canceling the ACQ effect. One of the synthesized POPs was used to detect both nitro-aromatics, through a turn-off mechanism driven by both energy and photoelectron transfer from the POP to the analyte, and electron-rich molecules, through a turn-on mechanism driven by photoelectron transfer from the analyte to the POP.174
The strategy of labeling nanopores with fluorescent dyes has also been extended to hydrogels. For instance, chitosan hydrogels have been modified by incorporating different amounts of NH2–Boron dipyrromethene (BODIPY) through Schiff base formation via reaction with terephthalaldehyde. The resulting fluorescent hydrogels showed an intense emission with a maximum at wavelength of 515 nm (λex = 380 nm). Fluorescence emission and lifetime measurements conducted using time-correlated single photon counting (TCSPC) revealed a significant decrease in emission upon exposure to Hg2+ and Hg+, while other metal ions did not induce notable changes. X-ray photoelectron spectroscopy (XPS) and FTIR confirmed that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds of the Schiff base serve as recognition sites for Hg2+/Hg+, forming a strong chelation.180 Fluorescent microgels based on poly(N-isopropylacrylamide) (PNIPAm) were synthesized by copolymerizing NIPAm with hyperbranched polysiloxane (HBPS) carrying vinyl groups. The emission observed from the resulting HBPS@PNIPAm microgels is attributed to HBPS, which exhibits a typical aggregation-induced emission (AIE) behavior, emitting intense light in aggregated states through the restriction of intramolecular motion. The characteristic thermo-responsiveness of PNIPAm was preserved in these new microgels, with a notable increase in emission intensity observed around 30–35 °C, corresponding to the volume phase transition temperature of PNIPAm. Furthermore, the emission of HBPS@PNIPAm was significantly quenched by a resonance energy transfer (RET) mechanism upon loading Luteolin, an antioxidant flavonoid, into the microgel. This phenomenon was effectively leveraged to monitor the release of Luteolin from the hydrogel by quantitatively tracking the increase in hydrogel emission intensity over time, indicative of drug leakage.181
N bonds of the Schiff base serve as recognition sites for Hg2+/Hg+, forming a strong chelation.180 Fluorescent microgels based on poly(N-isopropylacrylamide) (PNIPAm) were synthesized by copolymerizing NIPAm with hyperbranched polysiloxane (HBPS) carrying vinyl groups. The emission observed from the resulting HBPS@PNIPAm microgels is attributed to HBPS, which exhibits a typical aggregation-induced emission (AIE) behavior, emitting intense light in aggregated states through the restriction of intramolecular motion. The characteristic thermo-responsiveness of PNIPAm was preserved in these new microgels, with a notable increase in emission intensity observed around 30–35 °C, corresponding to the volume phase transition temperature of PNIPAm. Furthermore, the emission of HBPS@PNIPAm was significantly quenched by a resonance energy transfer (RET) mechanism upon loading Luteolin, an antioxidant flavonoid, into the microgel. This phenomenon was effectively leveraged to monitor the release of Luteolin from the hydrogel by quantitatively tracking the increase in hydrogel emission intensity over time, indicative of drug leakage.181
Direct detection of fluorescent analytes is used to quantify the different ionic cargo transport through the nanoporous membranes as a factor of increasing diffusion time. Differently from the Ca2+ ion binding methods (see below), this method is used to analyze the amount of fluorescent dye diffused through multipore nanoporous membranes rather than single pore films, and the analyte emission is studied after the analyte diffusion in the permeate container but not at the nanopore sites.182–188 In this technique, the ionic transport through multiporous membrane films was mainly quantified by UV-VIS spectroscopy since the analyte amount, and the absorbance signal intensity can be easily (linearly) correlated using Beer–Lambert's equation.184,186–189 However, in recent years, analyte diffusion dynamics through nanoporous membranes have also been extensively studied by fluorescent emission.24,25,190–193 Together with quantifying the analyte (e.g., ion) diffusion, using fluorescently emitting dyes also provides further advantages over UV-VIS measurements, such as microscopically resolving the dynamics of the analyte diffusion at the membrane/permeate phase interface.194,195
1D nanochannels can be integrated as abiotic alternatives to biological channels for sensing and separation purposes. For example, proteins are valuable biomarkers, as their concentrations can indicate specific diseases.196,197 In addition to conventional methods like ELISA or immunoassays, resistive pulse sensing offers a sensitive approach to protein detection. Zhang et al. utilized a tailored conical nanochannel (dbase: 1000 ± 80 nm, dtip: ∼5.6 nm) in PET for detecting HIV-1 protease.198 Fluorescence spectroscopy was employed to validate successful translocation events through the nanochannel. To achieve this, an enhanced fluorescence protein with similar dimensions to HIV-1 protease was introduced to the modified base side of the nanochannel under a voltage bias of 800 mV, driving the analyte towards the tip side. After three hours, the fluorescence intensity of the tip side compartment significantly increased, confirming analyte transport. Moreover, the research group of Haibing Li, has recently published several articles on selective transport through solid-state nanochannels.199–201 In a recent example, PET track-etched membrane with hourglass-shaped nanochannels were asymmetrically modified with L-tyrosine (L-Tyr)/D-tyrosine (D-Tyr) for the enantiomeric separation of a chiral drug, naproxen (NPX) (Fig. 3c).175 The smallest part in the middle of the nanochannel was 10 nm, while both large openings were ∼500 nm. Here, an important undertaking was to increase the enantioselectivity of the nanochannels with asymmetric modification prior to the mass transport of the racemic NPX sample. For this part, qualitative confirmation of the modification was realized through confocal imaging using two different fluorescent groups, fluorescein isothiocyanate (FITC) and rhodamine B (RhB), for the labeling of L-Tyr and D-Tyr, respectively (Fig. 3c-up). The directional mass transport of S/R-NPX through modified channels, solely driven by a concentration gradient, revealed that the transport rate of S-NPX was over 5 times greater than R-NPX (in the direction from L-Tyr-modified side to the other). Additionally, the active transport of S/R-NPX through asymmetrically modified nanochannels without any applied voltage was demonstrated by the quantitative evaluation based on the changing intensity of the fluorescence emission. The change in concentration with time is shown in Fig. 3c-bottom for the directional transport of R-NPX. The same research group also conducted selective transportation and enantiomeric separation of S/R-NPX using conical nanochannels within PET membranes.202 This was achieved by employing self-assembled layers of β-cyclodextrin for selective interaction, with quantification relying on the fluorescence intensity of the transported analyte. Additionally, direct fluorescent analyte detection is routinely used to determine the surface charge density of multiporous 1D nanochannels.21,198,203,204 Here, the measurement is based on electro-osmotic flow of phenol and monitored by the change in the fluorescence intensity of the permeate half-cell.
In another study, polyimide (PI) nanochannels were employed as synthetic biomimetic systems to understand the protein transport process through nuclear pore complexes.204 Bovine serum albumin (BSA) was selected as the model analyte. Given BSA's chiral selectivity towards cysteine enantiomers, the nanochannels were functionalized with L-cysteine (L-Cys) and D-cysteine (D-Cys). BSA was transported through the nanochannels via electro-phoresis and electro-osmosis under an applied voltage. Mass transport rates were quantified through fluorescence intensity analysis of the permeate half-cell. The selectivity coefficient, defined as the ratio of the BSA flux through L-Cys and D-Cys modified channels, was found to be 2.7. This chiral effect, indicating preferential protein translocation, was also observed in resistive-pulse sensing experiments using modified single nanochannels.
One known example of detecting time-dependent diffusion of differently charged ionic fluorescent analytes through 2D nanoporous membranes is to show the high anion selectivity of Janus membranes prepared via the phase separation of two block copolymers.24 The asymmetric geometry of these Janus membranes, and thus, their high anion selectivity, enables very effective osmotic energy conversion with high power densities (ca. 2.04 W m−2) after mixing natural seawater and river water. A 2D nanoporous composite membrane consisting of a graphene oxide layer sandwiched between two ultrathin layers of polydopamine was also recently used for osmotic energy conversion.25 Thanks to its sufficient cation selectivity, this layered 2D membrane has an enhanced surface charge density guiding a power density generation of 3.4 W m−2 under the river and seawater. The high cationic selectivity of this membrane was determined by using an ionic diffusion setup and quantifying the higher permeability rate of rhodamine 6G (as cationic dye) than that detected from sodium fluorescein (anionic dye).
The diffusion of fluorescent analytes through nanoporous 3D polyelectrolyte membranes (PEMs) has been extensively investigated by Wen L. and coworkers.22,205,206 In a series of papers they have prepared different types of PEMs, including polyether sulfone/sulfonated polyether sulfone (PES/SPES) membranes,205 sulfonated poly(ether ether ketone)/poly(ether sulfone) (SPEEK/PES) membranes,206 and poly [3-cyanomethyl-1-vinylimidazolium bis (trifluoromethane sulfonyl) imide]/poly (acrylic acid) (PCMVImTf2N/PAA).22 These membranes, produced through potentially scalable processes generating interconnected pores with diameters ranging from 10 to 100 nm, have been engineered for salinity gradient power generation. Depending on the specific ionic polymers, the authors developed membranes selective to the transport of either positive ions206,207 or negative ones.22 To deduce information about the ion transport properties of the membranes, they tested the membrane permeability towards two oppositely charged fluorescent dyes, namely sulforhodamine (anionic (−) dye) and rhodamine 6G (cationic (+) dye), by quantifying the diffusion of charged compounds through the membranes via their emission intensity. The authors also applied the same experimental methodology to hydrogels intended for salinity gradient power generation.23,208
On the other hand, pushing these two microscopy techniques to their sensitivity limits can lead to advanced microscopy methods single molecule localization microscopy (SMLM), fluorescence correlation spectroscopy (FCS), or stimulated emission depletion microscopy (STED), techniques that will be discussed in the following Method section.
4.1.2.1. Aggregation-induced emission (AIE). Confocal microscopy stands out as a convenient method to increase optical contrast, minimizing out-of-focus emission. Contrast can also be enhanced by specific processes based on the photochemical properties of the luminescent dyes: fluorogenic and luminogenic dyes are molecules that are initially non-emissive and only become emissive upon specific conditions or transformations, allowing to monitor with high contrast when and where such conditions are met. Among luminogenic dyes, those based on aggregation-induced emission (AIE) have been intensively studied over the past decade.209–211 Aggregation-induced emission luminogens (known as AIEgens) are nearly non-emissive as individual molecules due to nonradiative decay pathways through intramolecular motion. When these molecules aggregate, this motion is restricted, resulting in strong fluorescence.
Fluorescence and ionic current measurements can yield a sensor with a dual-signal output for detection purposes. Such a device was developed based on a functionalized PET membrane combined with AIE luminogens, allowing in situ, non-invasive monitoring of H2O2 released from living cells.212 Cylindrical nanochannels on PET were modified in a two-step approach with hydroxyl groups, as shown in the schematics in Fig. 4a, to serve as capture probes for tyrosine-containing TPE (tetraphenylethylene) derivative AIE luminogen (TT). In the presence of H2O2 released from living cells, and added horseradish peroxidase (HRP) enzyme, the luminogen formed linkages in the nanochannels, effectively blocking them. SEM images of the functionalized nanochannels showed that before and after adding H2O2 (10 μM), the average diameters of the nanochannels were 27 ± 3 nm and 7 ± 1 nm, respectively. The LSCM observations in Fig. 4a (bottom) show that the fluorescence intensity was highest for the combination of TT + HRP + H2O2. Consequently, a dual-signal could be used for the quantitative H2O2 detection, where the ionic current exhibited an “off” state with the reduced current flow because of the partially blocked nanochannel, while a heightened fluorescence was observed due to the linkage of the luminogens.
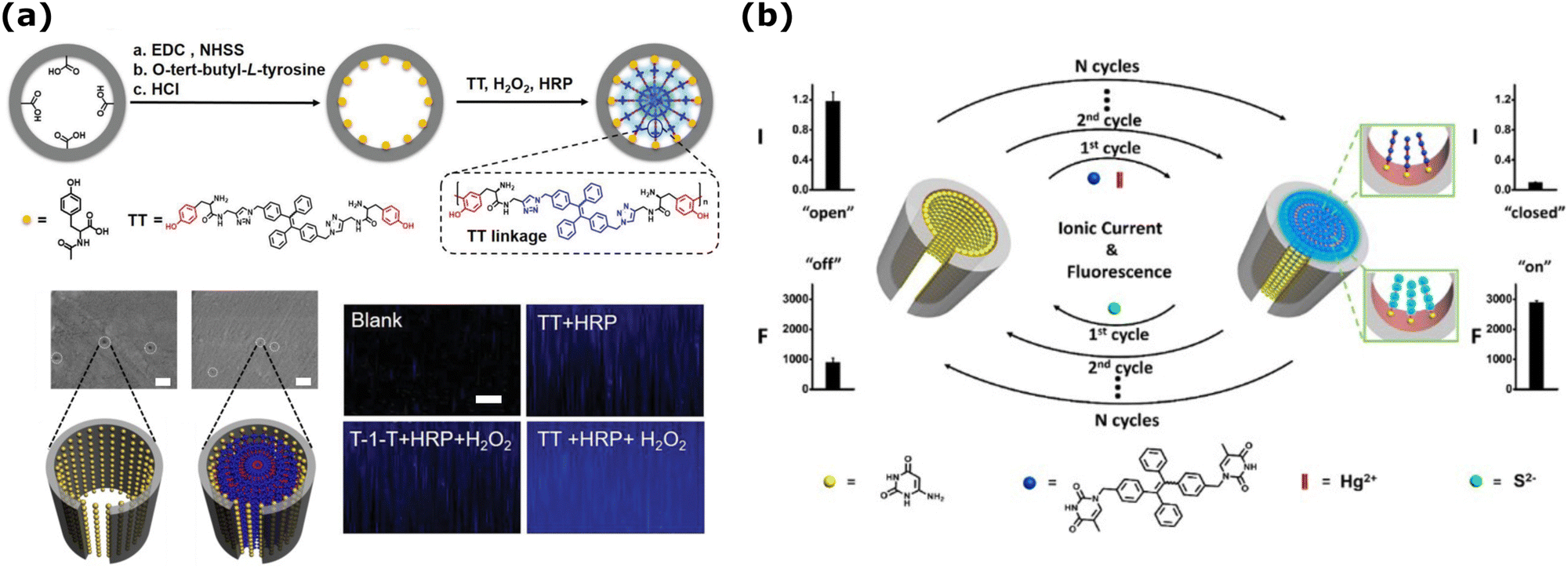 | ||
| Fig. 4 (a) (Top) Schematics of the functionalized nanochannels for H2O2 analysis using HRP and AIEgen (TT) oligomerization reaction. (middle) SEM images of the functionalized nanochannels before and after adding H2O2 (10 μM). Scale bars are 100 nm. (Bottom) CLSM observation of the nanochannel membrane in different conditions. The membrane thickness is ≈12 μm. Adapted with the permission of ref. 212 Copyright 2020, Wiley. (b) Schematics of the electro-optical detection in PET nanochannel membrane and its reversibility modulated by Hg2+ and S2−. Reprinted with permission from ref. 213 Copyright 2016, American Chemical Society. | ||
Although 1D nanochannel sensors based on electrical responses are widely employed, some issues persist. Monitoring conductance changes through nanochannels is influenced by environmental factors. To improve accuracy, combining ionic current signals with optical methods, i.e., fluorescence, allows direct observation of whether current changes are due to actual blockages. Electro-optical methods in 1D nanochannel systems have so far been implemented for the detection of analytes such as ions214,215 and small molecules.216,217 A novel dual-signal-output nanochannel detection system was developed by Xu et al. that combines fluorescence and ion current signals to detect Hg2+ ions by the use of S2− ions (Fig. 4b).213 Here, PET nanochannel was functionalized with the capture probe (6-aminouracil), yielding an on state for ionic current and an off state for the fluorescence readout. When Hg2+ and AIE molecules (TPE-2E)(TPE-2T) were introduced into the nanochannel, long complexes were formed on the interior channel wall, blocking the ionic current but introducing aggregation-induced fluorescence emission. S2− ions were chosen as the recovery agent and unblocked the nanochannels. Importantly, repeated plug–unplug processes and successive calibration curves, showcased reversible switching properties, alternatively switching between on and off states for both signals (Fig. 4b).
The primary structure of 2D nanoporous COFs with the strong π–π interactions can lead to rapid quenching due to the thermal decay of photoexcitation in photocarrier systems, resulting in decreased fluorescence with an increasing number of layers.95 However, Wang et al., showed that post-processing and activation of COFs could induce desired geometries to enhance or decrease emission collected from these COFs films with different interlayer spacing and nanopore sizes around 4 nm (Fig. 5a(i)).218 They demonstrated that the interlayer distance (3.7 Å) of the stacked 2D – COF thin membranes with high fluorescence emission decreases significantly (to 3.4 Å) after the effective solvent removal at cryogenic temperatures (Fig. 5a(ii)). Such decreased interlayer distance changes the aggregate-induced emission (AIE) in phosphorescence and makes the sample non-emissive (Fig. 5a(ii and iii)). Time-resolved emission decays (TCSPC) of these COF thin films at 77 K and 298 K showed that upon an increase in interlayer spacing, there is also a decrease in the intramolecular rotation barriers playing a role in the phosphorescence.
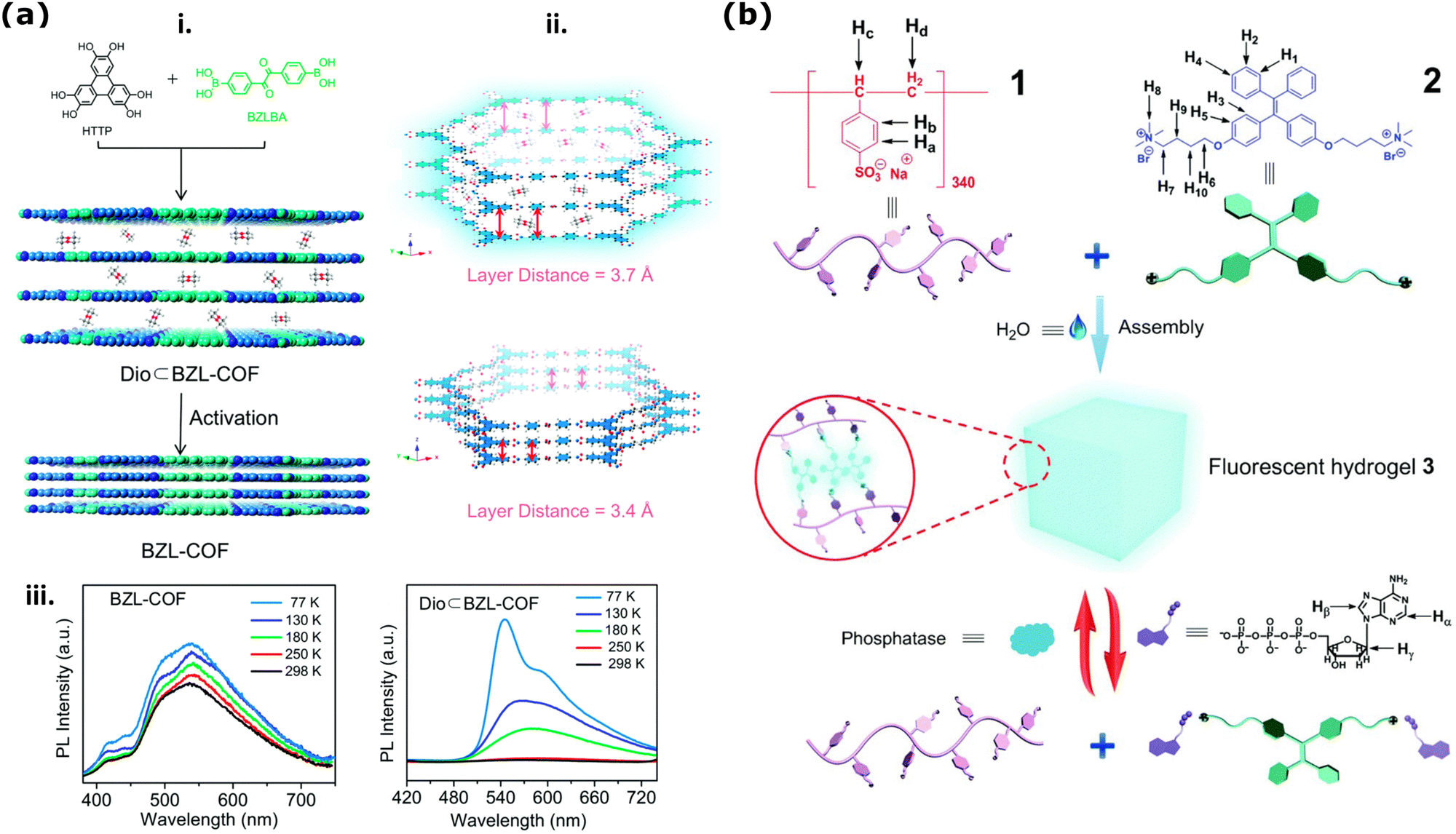 | ||
| Fig. 5 (a) Illustration of COF stacking arrangements and its relation to optical fluorescence emission. (i) Schematics presenting COF activation via decreasing the interlayer distance and influencing the AIE and ACQ (ii) As a factor of decreasing interlayer distance (iii) and the temperature detected phosphorescence emissions increase. Reproduced from ref. 218 with permission from the Royal Society of Chemistry. (b) Chemical structures poly(sodium p-syrenesulfonate) hydrogel (1) and ammino-derivative of TPE employed for the synthesis of the hydrogel (2) and the cartoon representation of the formation and bioresponsive reversible properties of the supramolecular fluorescent hydrogel in the presence of ATP (3). Reproduced from ref. 219 with permission from the Royal Society of Chemistry. | ||
Covalent triazine frameworks (CTFs) are one of the main classes of porous organic polymers together with COFs. CTFs, with their triazine ring units, offer stable and adjustable porosity, ease of architectural modification, and robust performance under acidic or alkaline conditions.220 Recently Tang et al. proposed incorporating AIEgen blocks into 2D fluorescent covalent triazine framework (F-CTF) nanosheets to create a highly luminescent material for detecting and removing nitrofurans (NFs) from water.221 The F-CTFs demonstrated high adsorption capacity, rapid adsorption rates for NFs, and 99% efficiency of NF removal after equilibrium.
In 3D-polymeric based materials and membranes, the use of AIE dyes for qualitative (e.g., fluorescent emission “on”/“off”)222 and quantitative (e.g., gradually changing fluorescence lifetime)223 sensing is a fairly well-established technique to sense changes in the matrix, such as strain and aging. These dyes can be easily incorporated into polymeric based materials by simple dispersion or by covalently bonding the dye to the polymeric chains. In particular, the covalent incorporation of AIE dyes into polymeric hydrogels is of great interest since it could provide valuable insight into the gelation process without having to perform destructive analysis, such as rheological tests.222–224 During hydrogel gelation the mobility of the chains becomes limited due to the formation of either covalent or non-covalent bonds and such RIM effect leads to an increase in the emission of AIE dyes present in the hydrogel. This theoretically allows us to follow the gelation-induced decrease in mobility by following the emission intensity in real-time.224 For a detailed description of these systems, readers are invited to refer to recently published reviews.225,226 Moreover, when dealing with stimuli-responsive hydrogels, other processes such as degradation and variation in the chemical surrounding, temperature, and pH can induce contraction or expansion in the hydrogel network, thus leading to changes in the emission intensity of AIE when introduced in the network.227,228 Concerning the study of mass transport, several studies report using AIE-containing hydrogels for real-time studies on the drug release process.219,229,230 In this context, two different approaches have been employed: (1) the use of drugs labeled with AIE dyes or (2) the use of stimuli-responsive hydrogels functionalized with AIE dyes. In the first approach, when the AIE-functionalized drug is embedded in the hydrogel, the mobility of the dye is limited, and thus, its emission intensity is enhanced; conversely, when the drug is released, its mobility is higher, thus leading to a decrease in emission intensity.229 This approach was employed by Wang et al.229 to study the release of non-conjugated luminescent polymers (NCLPs) presenting both AIE and antibacterial properties from a sodium alginate (SA) hydrogel. By first developing a calibration curve correlating the concentration of NCLPs in the hydrogel and emission intensity, they determined its cumulative release by collecting the hydrogel's emission spectra after different incubation times in a PBS buffer. Moreover, by analyzing the hydrogel under a fluorescence microscope, they could also visualize the release through the after mentioned decrease in emission intensity. Conversely, in the second approach, the hydrogel is functionalized with an AIE dye that displays different emission intensities when specific molecules are present or absent, thus making it possible to detect analytes entering the pores.219 This approach was employed by Wang et al.219 for developing a bio-responsive hydrogel that can signal the presence of adenosine triphosphate (ATP). In the study, they developed a poly(sodium p-syrenesulfonate) hydrogel crosslinked via electrostatic interaction with an ammino-derivative of TPE (Fig. 5b(1) and (2)). Due to the strong electrostatic interaction between the polymer and the TPE-derivative the obtained hydrogel was found to be a strong emitter as schematized in Fig. 5b(3). When ATP was added, the phosphate groups of the biomolecule were found to disrupt the hydrogel by interacting with the chromophore, thus leading to both a sol–gel transition and a decrease in the emission intensity (Fig. 5b(3)).
4.1.2.2. Widefield/epifluorescence microscopy. Traditional widefield/epifluorescence microscopy is a widely employed imaging technique that excels in its simplicity and versatility. Illuminating the entire field of view with a broad light source allows for observing fluorescently labeled molecules as they traverse through nanoporous membranes. Widefield microscopy offers rapid imaging capabilities and is particularly suitable for capturing large-scale molecular transport events collected, for instance, in situ, from the membrane films and their surrounding environment. However, as explained above, it may suffer reduced contrast and resolution due to out-of-focus fluorescence signals from regions above and below the focal plane.
One main use of widefield fluorescence imaging of nanoporous membranes is the fluorescent indicator dye sensing using Ca2+ ion binding to an indicator dye. In general, as the common step of the fluorescent indicator dye sensing methods, a nanoporous membrane is placed between two containers, and the transport of Ca2+ ions or other fluorescent analytes from one container to the other container is triggered by: (i) electromigration based on the applied voltage difference, (ii) diffusion based on the concentration difference and (iii) filtration based on the applied external pressure.231 For the Ca2+ ion – indicator dye-binding technique, via an applied electric field, Ca2+ ions start traveling from one container to the second container where the Ca2+ ion indicator dyes such as Fluo-8,232,233 Rhod-2,232 are present. During the transport of Ca2+ ions through the nanopores, Ca2+ ions interact with the indicator dyes and emit fluorescence signals. The emitted fluorescence signal is proportional to the Ca2+ flux at the nanopores. Ca2+ binding to Fluo-8 dye was used, in part, to quantify the ionic transport through hydrophobic 1D nanochannels in AAO prepared by silanization modification.50 The ion transport process was found to be influenced by metal ions as well as proton and hydroxide ions. For the demonstration of this finding, Ca2+ ions were driven through the hydrophobic nanochannels under an applied electric field. As a result of the applied negative voltage, Ca2+ ions were transported to the other side of the membrane, where they combined with the Fluo-8, generating a fluorescence response. Moreover, this response increased by 2-fold when the bias voltage was elevated from −1 V to −2 V. Among the recent studies for 2D nanoporous membranes, this technique is mainly used to study the biomolecule (DNA, RNA, folded proteins) translocation at nanopore(s) inside a 2D film (e.g., SiNx) and when a biomolecule translocates at the nanopores then this effect the Ca2+ flux and thus the detected fluorescent emission intensity.232–235
Epifluorescence imaging is routinely integrated as the fluorescent method of choice in 1D sensor and detector design for biomolecules. Direct visualization of transport processes has the potential to pave the way for the development of porous structures with tailored characteristics. Track-etched polypropylene (PP) membranes with ∼500 nm-diameter nanochannels were incorporated for the fluorescence imaging of individual quantum dots (CdSeS/ZnS, core/shell nanocrystals) with diameters of ∼10 nm.236 Here, the authors employed an epiluminescence microscope to capture images of adsorbed quantum dots following ultrafiltration. Their observations revealed that the adsorbed quantum dots could be imaged down to a depth of 10 μm with a decrease in the number of quantum dots as the focal depth increased. 1D nanochannels and their widefield fluorescence imaging are employed to detect other analytes such as ions. Recent examples have demonstrated selective detection of copper ions (Cu2+) through fluorescence quenching. In such a nanofluidic device utilizing AAO nanochannels (pore diameter is 200 nm), Cu2+ detection was achieved with a detection limit as low as 4 fM.237 Here, the synergistic effect of a high-porosity metal organic framework (ZIF-8) and fluorescent quantum dots (CdSe QD) was incorporated in the design of the AAO membrane. After the ZIF-QD composite was loaded on the AAO nanochannel membrane, it was possible to visualize the surface via an inverted fluorescence microscope. The Cu2+ was enriched on the AAO surface by applying 5 V potential, which caused the fluorescence quenching with respect to the ion concentration. Another robust sensor application for Cu2+ was based on conical track-etched nanochannels in PET membranes.214 The nanochannels were covalently functionalized with peptide-like sequence coupled with a fluorophore, 5,6-corboxyfluorescein. The formation of the peptide-metal complex, which serves as the primary mechanism for fluorescence quenching, was found to be pH dependent. A pH of 6.5 was explicitly selected for detection, ensuring selectivity towards Cu2+ ions. The decrease in fluorescence intensity exhibited a linear dependency within the range of 0–60 μM. Furthermore, the sensor could be reused up to seven times without losing its sensing capabilities. These examples highlight that 1D nanochannels offer a rapid and highly sensitive sensor platform for both qualitative and quantitative detection of copper ions in solution.
8-Hydroxyquinoline-functionalized 2D covalent organic framework (COF-HQ) membrane is an interesting example of a fluorescent pH-reporting (sensing) material (Fig. 6a).238 For their work in 2018, Wang and colleagues synthesized these novel COF-HQ through a Schiff base reaction, and the characteristic pore diameter and the spacing between the COF layers were reported as 1.4 nm and 3.62 Å, as presented in Fig. 6a (top-left). In aqueous solution and steady-state fluorescence investigations, they found that the fluorescence intensity of COF-HQ increased as a factor of higher environmental pH (Fig. 6a (top-right)). As an optical chemical sensor, COF-HQ demonstrated the ability to differentiate various acidic solutions within the pH range of 1 to 5, exhibiting rapid response times and straightforward operation. As presented in Fig. 6a-bottom, incorporating COF-HQ into a mixed matrix membrane (MMM) showcased its potential for practical applications and processability in composite film forms that can qualitatively (eye-visible color variations) report changing pH. The color of the COF-HQ-loaded composite membrane transitioned to black with increasing acidity within just one minute, providing a fast and visually recognizable detection method. pH-dependent fluorescence quenching was quantified by epifluorescence images of the free-standing films in contact with different acidic solutions. It was attributed to the porous nature of COF-HQ, facilitating the penetration and diffusion of protons, thereby disrupting the p-electron conjugation of HQ grafted within the COF-HQ structure. Besides pH-detection, epifluorescence microscopy helps study molecular selectivity and permeability inside various 1D and 2D nanoporous membranes. Another known example of fluorescence microscopy is studying the time-dependent molecular permselectivity through 2D silica nanoporous membranes prepared by electrochemically assisted self-assembly (known as EASA)239 and Stoeber solution growth methods.240 These two methods yield structures with uniform nanopore sizes typically ranging from 2 to 3 nm, and high pore density that could reach values up to 7.5 × 1012 cm−2. Therefore, such 1D nanoporous silica membranes are particularly well-suited for separating small molecules. For instance, in 2015, PMMA-assisted transfer combined with Stöber-solution growth approach was used to fabricate free-standing ultrathin (10–120 nm thickness) multiporous (ca. 2.3 nm pore size) 2D silica nanoporous membrane films and used for precise and rapid molecular sieving monitored by time-dependent epifluorescent microscopy imaging.195 Time-dependent molecular transport through the ultrathin nanoporous films was investigated by a microfluidic device functioning with fluorescence microscopy under electrochemical stimuli and detection. In the end, they reported that the cationic Ru(bpy)32+ (where bpy = 2,2′-bipyridine, diameter 1.3 nm) and Ru(dpp)32+ (where dpp = 4,7-diphenyl-1,10-phenanthroline, diameter 2.0 nm) could be effectively separated based on their molecular sizes. Furthermore, compounds bearing different charge states, such as the anionic fluorescein anion (FL2−), neutral rhodamine B, and cationic Ru(bpy)32+, are separated according to electrostatic interactions, which can be modulated by varying the ionic strength of the solutions.
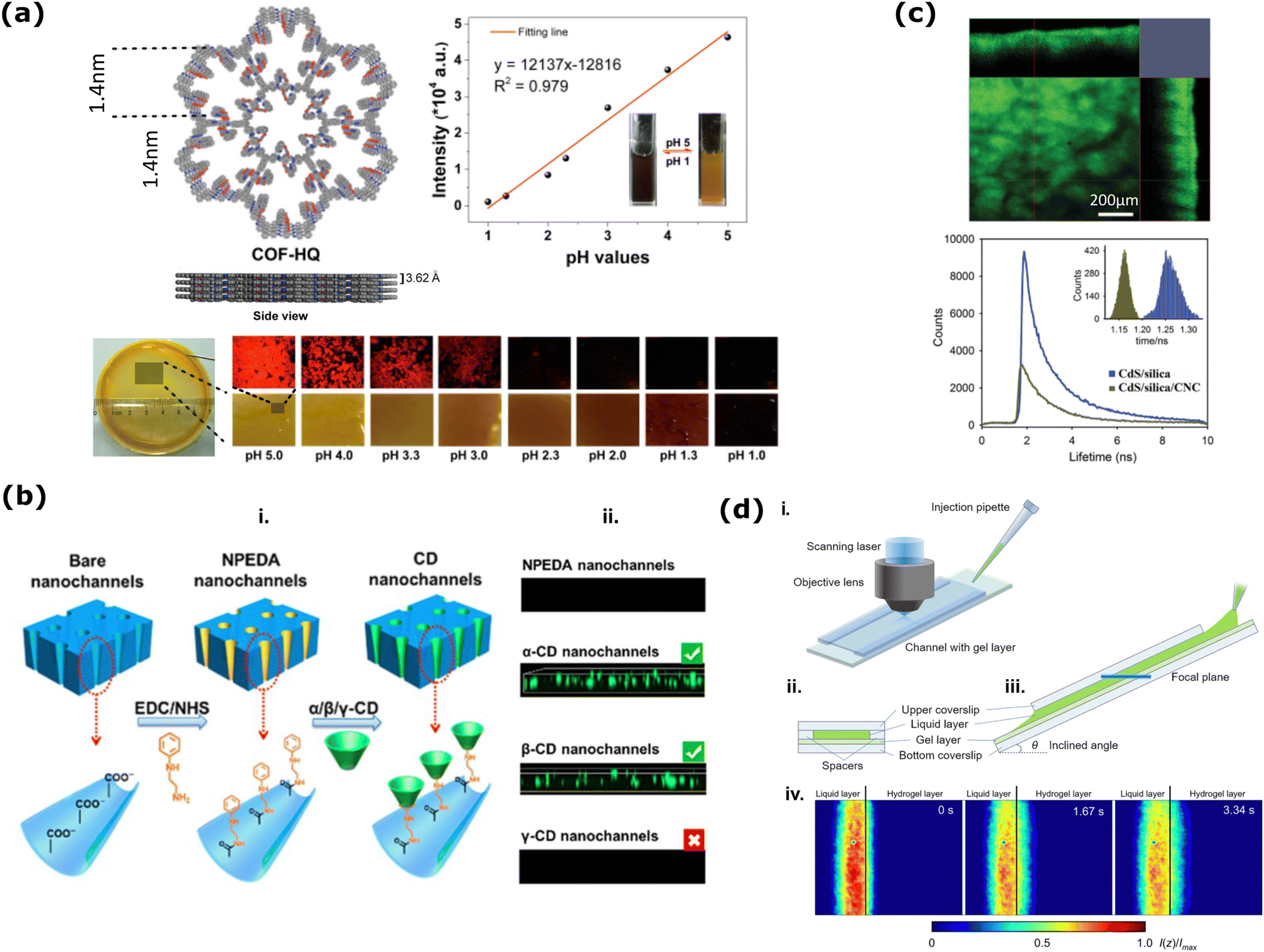 | ||
| Fig. 6 (a) (top-left) Top and side view schemes of 8-hydroxyquinoline-functionalized covalent organic framework (COF-HQ). (top-right) The relation between probed environmental pH and the fluorescence intensity. (bottom) Naked eye visible pH-dependent color changes of the free-standing COF-HQ based matrix membrane. Reprinted with permission from ref. 238 Copyright 2018, American Chemical Society. (b) (i) Schematics of functionalization of the CD-self-assembled nanochannel membranes, (ii) the fluorescence confocal microscope images of nanochannel membranes before and after NPEAD modification and CD self-assembly. Reprinted with permission from ref. 202 Copyright 2020, American Chemical Society. (c) (top) CLSM z-stack image of iridescent, luminescent chiral nematic CdS QD loaded mesoporous silica film presenting the film's (large area) surface and (top and right) cross-section. (bottom) Fluorescent lifetime decays of the CdS/mesoporous films that were fabricated differently and the inset presents corresponding histograms of pixel-by-pixel lifetime distributions of these films. Adapted with the permission from ref. 241 Copyright 2013, Wiley. (d) (i) Bird view of the setup employed for transient molecular transport measurement using inclined microscopic observation; (ii) cross-sectional view of the channel containing the gel layer; (iii) side view of the channel with the observed focal plane indicated with a blue line; (iv) transient permeation of uranine from the upper liquid layer to the lower hydrogel layer. The colored bar shows the normalized fluorescence intensities. Reprinted with permission from ref. 242 Copyright 2019, Plos One. | ||
4.1.2.3. Laser scanning confocal microscopy (LSCM). Fluorescence microscopy and confocal imaging techniques are powerful approaches to visualize and quantify mass transport phenomena within 3D nanoporous structures.243 However, in the contest of this family of materials -up to now-this type of characterization techniques has been mainly employed on the structural characterization of hydrogel membranes, with a particular interest in the visualization of the 3D network,244–246 phase separation in mixed systems247–249 and sol–gel transition250,251 rather than mass transport-related processes. These topics have been thoroughly discussed in two recent review articles by Saini et al.252 and Zhong et al.253 Nonetheless, the visualization of analyte diffusion is of great interest since it could provide useful insight into the design of 3D nanoporous membranes. In fact, by tracking the emission of luminescent analytes in space and in real-time, it is possible to gain valuable insights into factors like pore connectivity, diffusion coefficients, and potential transport barriers within the membrane.243
Compared to widefield/epifluorescence imaging, laser scanning confocal microscopy (LSCM) presents a powerful solution for three-dimensional imaging with enhanced spatial resolution. As mentioned in detail previously (in Section 2), by employing a pinhole to collect light from a single focal plane selectively, confocal microscopy eliminates out-of-focus fluorescence, resulting in improved contrast and clarity in imaging. This capability enables resolving activities of various functional nanomaterials or nanocoatings with surrounding or in-contact media from a nm level.254,255 For nanoporous membranes, LSCM helps to precisely visualize molecular transport dynamics within specific regions of nanoporous membranes, offering insights into pore structure, connectivity, and diffusion processes. Additionally, confocal microscopy facilitates the acquisition of optical sections and three-dimensional reconstructions, providing a comprehensive view of mass transport phenomena at the nanoscale.
LSCM was conducted to explore the chiral selectivity of bovine serum albumin (BSA) for the cysteine-modified PI nanochannels (1D). Following the transport of fluorescein isothiocyanate (FITC)-labeled BSA through modified nanochannels for 10 minutes, LSCM revealed intense fluorescent signals throughout the porous membrane, indicating the presence of BSA adsorbed on the nanopore surfaces for L-Cys modified nanochannel while the D-Cys modified nanopores exhibited weaker fluorescence.204
An important part of 1D nanochannel based systems is the functionalization for improved sensitivity and selectivity. Fluorescence and confocal imaging are employed to confirm various functionalization routes by detecting changes in the fluorescent properties of the functionalized nanochannels.178,215,256 For instance, asymmetric modification of polymer nanochannels was visualized (qualitatively) by confocal imaging.21 Fluorescein isothiocyanate (FITC) and Rhodamine B (RhB), were used to label L-Tyr and D-Tyr, respectively, which led to two distinct regions in the image, confirming the asymmetric surface modification (recall: Fig. 3c). The same group also used similar confocal imaging to visualize cyclodextrin (CD) self-assembled nanochannel membranes for chiral separation of drug enantiomers.202 Through EDC chemistry, they first covalently functionalized the surface of the conical nanochannels with N-phenylethylenediamine (NPEDA) (Fig. 6b(i)). On this layer, α-, β-, and γ-CD were self-assembled. Based on the confocal images in Fig. 6b(ii), it was shown that γ-CD could not self-assemble on the NPEDA layer due to its large cavity. In the end, it was concluded α-CD was incapable of chiral identification and only the β-CD-self-assembled nanochannel membrane could effectively regulate chiral transport. In another example, fluorescence properties of coordination polymers were utilized for qualitative visualization of the functionalization of the AAO surface to be used in a high-performance Janus Device for sensing purposes.75 While AAO itself is not fluorescent, the assembly of coordination polymers on the nanochannel surface (both on the open surface and inside the nanochannels) enabled accurate observation (emission) through scanning confocal microscopy.
In a further example of ionic detection, a dual-signal nanosensor was developed for reusable Hg2+ sensing based on FITC and hydrazine functionalized 35 nm AAO nanochannels.257 Here, the change in the current response was due to the surface charge-reversal of the nanochannel, and the fluorescence intensity enhancement (studied by confocal z-stack imaging) was caused by Hg2+-induced ring opening mechanism. To assess the sensing abilities of the device, a fluorescence gating ratio was defined as ((F − F0)/F0, 518 nm) where F0 and F are the fluorescence intensity at 518 nm before and after the introduction of Hg2+. For 10 μM Hg2+, this ratio was 5.7. Furthermore, the sensor could be restored to its initial state by N2H4 treatment and reused multiple times. The ionic flow through 1D nanochannels can be controlled by applying various gates, such as pH, electric field, light, temperature, or ions. Through functionalization routes, solid-state nanochannels can switch between “on” and “off” states in the presence or absence of the applied gate, similar to biological nanochannels. Such a device engineered featuring conical nanochannels in PI modified with the functional probe multi-amino conjugated rhodamine B (NH2-RhB) for ATP-activated ionic transport gating.258 This platform utilized a dual-signal approach involving ionic current and fluorescence measurements. Ionic current measurements demonstrated that the modified nanochannels could selectively respond to ATP with a detection limit in the femtomolar range. Additionally, the reversibility of the target-probe interaction was achieved by immersing the membrane in a pH = 4.4 solution. This on-and-off behavior was qualitatively visualized through confocal fluorescence imaging, where, in response to ATP, the nanochannel membrane transitioned from a non-fluorescent to a fluorescent state, indicating good reversibility for gated ionic transport.
Especially for the membranes with 1D nanochannels, research has largely focused on modifying functional groups on the inner walls of the nanopores, while the role of the outer surface has been overlooked.259–261 However, recent experimental studies have shown that probes attached only on the outer surface of the membranes (not inside the pores) can independently modulate ionic current, offering significant advantages for in situ target detection. This is especially valid for targets larger than the nanochannel diameter that cannot pass through such as proteins, cells or viruses.261 For instance, in 2023, Qiao et al. used the free space on the outer surface of 1D nanochannel membranes to detect protein conformations.262 They developed a platform combining nanochannel arrays and protein origami structures to detect unfolded proteins through both electrical and optical signals. The detection mechanism relied on the specific interaction between maleimide groups of the probes on the outer surface of the membranes and cysteine thiols in the protein. This selective interaction results in changes in ion current and fluorescence intensity of the nanochannel array quantified from the LSCM images. This approach enables discrimination between folded and unfolded protein states based on the exposure of individual cysteine residues.
One particular application of LSCM imaging and various steady-state fluorescent spectroscopy measurements is to investigate the liquid and gas phase TNT detection performance of novel luminescent chiral nematic CdS QD filled mesoporous silica film sensors (Fig. 6c).241 Steady-state fluorescence emission results showed that these emissive films exhibit luminescence quenching upon exposure to TNT solution or vapor, indicating their potential as selective sensory materials for the trace detection of TNT explosives. The authors used further stead-state fluorescence spectral measurements to demonstrate CdS QDs’ survival during different calcination processes during film fabrication. LSCM z-stack images of the films before (not shown here) and after their calcination (Fig. 6c-top) qualitatively showed the good dispersion of the luminescent CdS QDs in these films. Luminescence lifetime decay profiles of the composite films before (CdS/silica/CNC) and after (CdS/silica) their calcination (Fig. 6c-bottom) change significantly. The prolonged lifetime of the CdS/mesoporous silica structures (after calcination) can be attributed to the enhanced porosity of the materials, which may facilitate air diffusion into the silica pores during calcination. At the same time, the CdS QDs may undergo annealing, creating a passivated surface through oxidation.240,263 Consequently, a gradual reduction in surface carrier recombination processes occurs, thereby extending the luminescence lifetime. Similar to the above-described work used for detecting the molecular permeation at 2D nanoporous silica membranes by time-dependent epifluorescence imaging, also in 2021, Zhou et al. used LSCM images to understand the ionic selectivity at mesoporous silica/macroporous alumina (MS/AAO) frame-work-based nanofluidic heterostructure membrane.264 The presence of a mesoporous silica layer rich in silanol groups and having a high specific surface area confers several advantages to the heterostructure membrane. It achieves a low inner resistance of about 7 KΩ, exceptional ion selectivity, and efficient osmotic energy conversion. In the end, when exposed to a mixture of artificial seawater and river water, the membrane exhibits a high power density of up to 4.50 W m−2, surpassing conventional 2D nanofluidic membranes by 20 times and outperforming other 3D porous membranes by around 30%. CLSM was used to demonstrate the ionic selectivity of the MS/AAO heterostructure membrane quantitatively by collecting confocal images from fluorescent dyes with different charges upon permeation through the membrane from a perpendicular direction setup. They realized a higher fluorescence signal from the cationic dye throughout the heterostructure membrane compared to the signal collected from anionic fluorescence. In this way, they demonstrated the precise cation selectivity of the heterostructured membrane.
Regarding 3D membranes, the possibility of confocal microscopy to create depth profiles is particularly valuable for studying mass transport, allowing researchers to not only observe the presence of fluorescent molecules within the pores but also quantify their spatial distribution throughout the 3D structure.243 Among numerous examples,228,265–267 an interesting work was published in 2019 to report a new technique for high time-resolved depth measurement of molecular concentration distribution in a permeable hydrogel film (Fig. 6d).242 Researchers have developed a novel technique for measuring the depth profile of molecules permeating through a hydrogel film. This method, called inclined confocal microscopy, utilizes a modified confocal microscope equipped with laser-induced fluorescence (LIF) (Fig. 6d(i–iii)). By tilting the focal plane, researchers can achieve simultaneous depth scanning within the microscopic field of view (Fig. 6d(iii and iv)). This enables real-time, non-invasive measurement of concentration distribution throughout the hydrogel with micrometer-scale depth resolution (Fig. 6d(iv)). The study demonstrates the effectiveness of this technique by monitoring the permeation of uranine, a fluorescent molecule, from a liquid layer into a hydrogel. The results reveal high time resolution (16.7 milliseconds) and a depth measurement interval of approximately 1 micrometer (Fig. 6d(iv)). The researchers were also able to determine the concentration of uranine with an accuracy of 1.3 nanomoles per liter. This approach offers valuable insights into the dynamics of passive transport driven by concentration gradients within permeable materials. In fact, by analyzing the concentration profiles, the researchers determined the mass transport coefficient, a key parameter describing the rate of passive transport driven by the concentration gradient.
Besides its widespread impact on biomedical research, fluorescence lifetime imaging microscopy (FLIM) coordinated with confocal microscopy is also a powerful tool for studying various luminescent material functions by combining the spatial resolution of confocal microscopy with the temporal resolution of FLIM.268–270 In many sensing devices, photoluminescence quantum yield (QY) is considered a critical parameter for evaluating their efficiency. However, measuring the quantum yield of luminescent molecules and nanocrystals in mesoporous 2D membranes poses significant challenges, especially when spatially resolved quantitative luminescence imagining (e.g., high-resolution FLIM images) is required. In 2019, Ozelci et al. proposed a new Drexhage-type experiment, utilizing a silver-coated millimeter-sized sphere to modify the local density of states, offering a sophisticated solution to this problem.271 Their FLIM image mapping detected local lifetime changes of the fluorophores inside the mesoporous film from a few ten micrometer resolutions. They also demonstrate that the QY results collected with this novel imaging method agree with those collected from the same dye in a solution at a different pH. Their novel FLIM imaging approach bridges the gap between single fluorophore studies and ensemble measurements. It facilitates spatially resolved fluorescence measurements of ultralow emitter concentrations in thin sensor films or membranes, which function as optically active elements and reporters.
4.1.2.4. Total internal reflection fluorescence (TIRF) microscopy. Finally, TIRF allows precisely confine the excitation energy (and thus the emissive molecules) to an extremely thin layer above an interface with a large variation of refractive index, for example glass/water interface. The principle of total internal reflection fluorescence (TIRF) microscopy involves directing an excitation laser light from a medium with a higher refractive index to one with a lower refractive index at a larger angle than the critical angle. This results in total internal reflection at the interface and, thus, generates an exponentially decaying electromagnetic field near the interface, which is called the evanescent field. Consequently, single molecules located far from this confined region (>200 nm) are not excited, thereby reducing background light and enhancing signal specificity.
TIRF method, owing to its ability to deplete emission from other layers than the ∼200 nm thick layer excited by the evanescent field, can help imaging nanoporous structures with single molecule sensitivity (i.e., with high S/N), even when coupled with electrochemical setup.272,273 For instance, the high refractive index (n ≈ 2.06) of 1D nanoporous SiNx membranes was exploited to overcome its high transparency, due to the minimal thickness, and to specifically visualize processes occurring within the membrane. In 2010, Soni et al. introduced asymmetric fluids on the cis (n ≈ 1.33) and trans (n ≈ 1.41) sides, facilitating total internal reflection at the 20–30 nm thick SiNx membrane film interface (Fig. 7a).274 They synchronized ion current signals with fluorescent signals to monitor, for the first time, the translocation behaviors of single DNA biomolecules through 4 nm SiNx pore. This method could also be used to visualize various FRET tags to collect positional information of molecules inside nanopores, and to image multiple pores at a time. Recently, an innovative attempt has been made to efficiently couple solid-state multi array nanopores with zero-mode waveguides (ZMW).275,276 In this approach, researchers first employed a molecular-layer deposition method to create a cost-effective porous membrane. Subsequently, a waveguide array was fabricated on the porous membrane at the wafer scale. This porous membrane, containing serpentine confined pores, was used to electrophoretically capture DNA at picogram levels.163 Upon applying a bias potential, the YOYO-DNA molecules trapped within the confined spaces of the porous ZMW were selectively excited. Inspired by this work of Bohn with ZMW, in 2017, Lu et al. were able to study adsorption, desorption, and heterogeneous single redox events of single resorufin molecules inside ca. 3 nm size ordered nanopores of 1D ultra thin (<100 nm) mesoporous silica film coated on the top of ITO electrode by using TIRF (Fig. 7b).277 These narrow silica nanopores could host redox molecules (resorufin, see molecule in Fig. 7b-bottom) inside and restrict their diffusional motions. Single molecule analysis from the collected TIRF data helped them to precisely calculate the rate constants of the adsorption and desorption processes at the nanopores. From fluorescent resorufin to non-fluorescent dihydroresorufin redox events were demonstrated by quantifying the change surface population of single molecules upon the applied potential to the system.
 | ||
| Fig. 7 (a) (Right) The cartoon on the left presents the TIRF method used to investigate single-molecule detection from 1D thin SiNx nanoporous film (orange color) sandwiched between two aqueous fluids. Schematic illustration on the left-top presents the flow cell setup to mobilize the ATTO647N fluorophore conjugated DNA molecules towards the nanopores by the applied voltage and image them at this nanoconfinement. (Right) Images of the SiN membrane where individual DNA molecules were trapped. Reprinted from ref. 274 with the permission of AIP Publishing. (b) (Top) Scheme presenting the setup used for single-molecule electrochemistry detection from a porous 1D nanoporous silica-coated ITO electrode. (Bottom) Irreversible reduction of Resazurin (weakly fluorescent) to Resofuring (highly fluorescent) and the redox reaction between resorufin and Dihydroresorufin (non-fluorescent), which is reversible. Reprinted with permission from ref. 277 Copyright 2017, American Chemical Society. | ||
4.2. Method-3: super-resolution and single-molecule methods (STED, single-molecule localization microscopy, SMLM & fluorescence correlation spectroscopy, FCS)
Especially in recent years, fluorescent single-molecule spectroscopy (SMS) has been developed among different microscopic methods to directly monitor individual molecules’ behaviors (dyes or analytes) in confined spaces. This advancement enables spatial and temporal visualization, providing quantitative insights into heterogeneous molecular dynamics within diverse nanoconfinements in complex porous materials.278 Overall, three distinct SMS techniques–i.e., techniques that require the highest sensitivity achievable in fluorescence microscopy–are utilized to study the mass transport of fluorescent dye molecules.The first technique involves directly analyzing a series of fluorescence microscopy images, or movies, of diluted fluorescent species. In these images, the fluorescence of individual species appears as diffraction-limited patterns that can be tracked and analyzed. Fluorescence of individual molecules can be tracked, but it requires sufficient accumulation of photons in each position; therefore, only slow motions of single molecules are compatible (e.g., slow diffusion in a membrane or through a pore); on the contrary, highly fluorescent species such as QDs or dye-doped nanostructures containing hundreds of dyes can be tracked more efficiently, even during Brownian motion in a low viscosity solvent. This method, known as single molecule tracking (SMT), or more generally as nano tracking analysis (NTA), does not require model assumptions and allows for the direct characterization of molecular motion in space and time, yielding information on diffusion properties, environmental viscosity and confinement, and molecular assembly or disassembly. SMT provides detailed observations of various diffusion behaviors by analyzing the reconstructed trajectories. The second technique is fluorescence correlation spectroscopy (FCS). FCS employs a microscopy apparatus to observe the diffusion of individual molecules as they enter and exit a precisely defined small detection volume. Diffusion coefficients can be extracted by statistically analyzing the temporal fluctuations of detected fluorescence intensity I(t). A third set of methods based on single-molecule imaging is the single-molecule localization microscopy (SMLM). This family of techniques enables the determination of individual molecule positions with nanometer precision, allowing for the reconstruction of images with lateral resolutions down to 10 nm and axial resolutions down to 20 nm. This method relies on the fact that the localization of single fluorescent species can be determined with high precision if their point-spread functions (PSF) do not overlap; therefore, the emitters should be spatially isolated at a distance larger than the PSF, a condition that can be obtained distributing the emitters in time. There are many ways to temporally isolate the emission of species: one possibility is to switch photoactivatable fluorophores between a fluorescent (on) state and a nonfluorescent (off) state (photoactivated localization microscopy, PALM) or to exploit the dynamic nature of transient binding (binding activated localization microscopy, BALM). This switching mechanism permits the detection of single molecules even in densely labeled samples, facilitating high-resolution imaging.
Another method overcoming the resolution limit imposed by diffraction is STED microscopy, which employs a targeted depletion laser to selectively switch off fluorescence in specific regions around the focal point. This process sharpens the effective point spread function, thereby achieving nanoscale imaging. One very interesting use of STED imaging was performed to explain the electrochemical performance of 1D conical nanochannels (Fig. 8a).279,280 Such conical nanochannels in polymer membranes are known to have unique electrochemical behaviors, caused by the asymmetry in their geometry and fixed surface charges. Especially when the diameter of the nanochannel is comparable to the electrical double layer formed on the channel walls, the nanochannel will preferentially incorporate ions of the opposite charge and reject ions with the same charge.281 The asymmetry in the nanochannel geometry also lowers electrical resistance compared to symmetrical nanochannels, and a so-called “capture zone” with a higher electric field at the tip side of the nanochannel is formed.282 To prolong the translocation times of analytes within conical PET nanochannels, alumina (Al2O3) was deposited onto the nanochannel walls using atomic layer deposition (ALD) (Fig. 8a).280 The obtained nanochannel was used as a separation platform for single-strand (ss) and double-strand (ds) DNA as well as a resistive-pulse sensor. It was demonstrated that 22-base ss-DNA exhibited a higher affinity for the alumina layer compared to ds- DNA, resulting in translocation times increased by one to three orders of magnitude, as shown in Fig. 8b- left. Stimulated emission depletion (STED) imaging was conducted on the alumina-deposited membrane to elucidate qualitatively the mechanism behind the extended translocation time and resistive-pulse sensing of ss-DNA. Based on the super-resolution image presented in Fig. 8b-right and the 100 nm depletion ring in the image, it was suggested that ss-DNA initially adsorbs to the alumina-coated nanochannel surface before migrating into the conical nanochannel through the tip side.
 | ||
| Fig. 8 (a) Schematic illustration of uniform ALD coating on a single conical nanochannel and SEM images of the tip and base sides. The scale bar is 100 nm. Reprinted with permission from ref. 279 Copyright 2015, American Chemical Society. (b) (Left) Resistive pulse signals of a 1:1 mixture of 22 b ss-DNA and ds-DNA after 5 min and 10 min. (Right) STED super-resolution microcopy image at 30 min. Reprinted from ref. 280 with the permission of AIP Publishing. | ||
Membrane separation applications incorporating nanochannels are challenging since nanoconfined solutes behave differently than bulk, and a better understanding of the mechanism for the solute-nanochannel interactions is needed if new devices are to be developed. In an advanced chemical separation device for the dehydration of renewable biofuels, Rashidi et al. explored with FCS the diffusion of Rhodamine B through AAO membranes containing cylindrical (1D) channels of 10 nm and 20 nm diameters, and with solvent compositions ranging from pure ethanol to 33% water-ethanol mixtures (Fig. 9a).283 The study utilized single-molecule detection and Fluorescence Correlation Spectroscopy (FCS) methods to quantitatively investigate the rates and mechanisms of RhB diffusion as functions of AAO channel size, and solvent composition. With the experimental setup shown in Fig. 9a(i), the translational motions of individual RhB molecules at a depth of 5 μm could be detected. The representative time transients for 2 nM RhB in Fig. 9a(ii) yielded that fluorescence bursts were evident in all cases, but bursts in bulk solution were too fast to observe. Due to the slower RhB diffusion within AAO nanochannels, the bursts were more distinct and longer. This visualization of single-molecule events was crucial to understanding the dye diffusion mechanism. Autocorrelations derived from fluorescence time transient data (Fig. 9a(iii)) allowed the assignment of two different dye diffusion mechanisms. The dashed lines were the fitted data, while the dotted lines depicted the 95% confidence intervals. Two distinct diffusion mechanisms were unveiled: fast and slow 1D diffusion, both notably slower than RhB diffusion in bulk ethanol. While fast and slow diffusion increased with channel size, a complex diffusion process was proposed for RhB mass transport under varying solvent compositions. Fast diffusion decreased with increasing water content due to hindered diffusion caused by heightened hydrodynamic drag and electrostatic interactions. Conversely, slow diffusion increased with water fraction, attributed to the reversible adsorption of RhB dye to pore surfaces via electrostatic interactions or hydrogen bonding, potentially moderated in mixtures with higher water content.
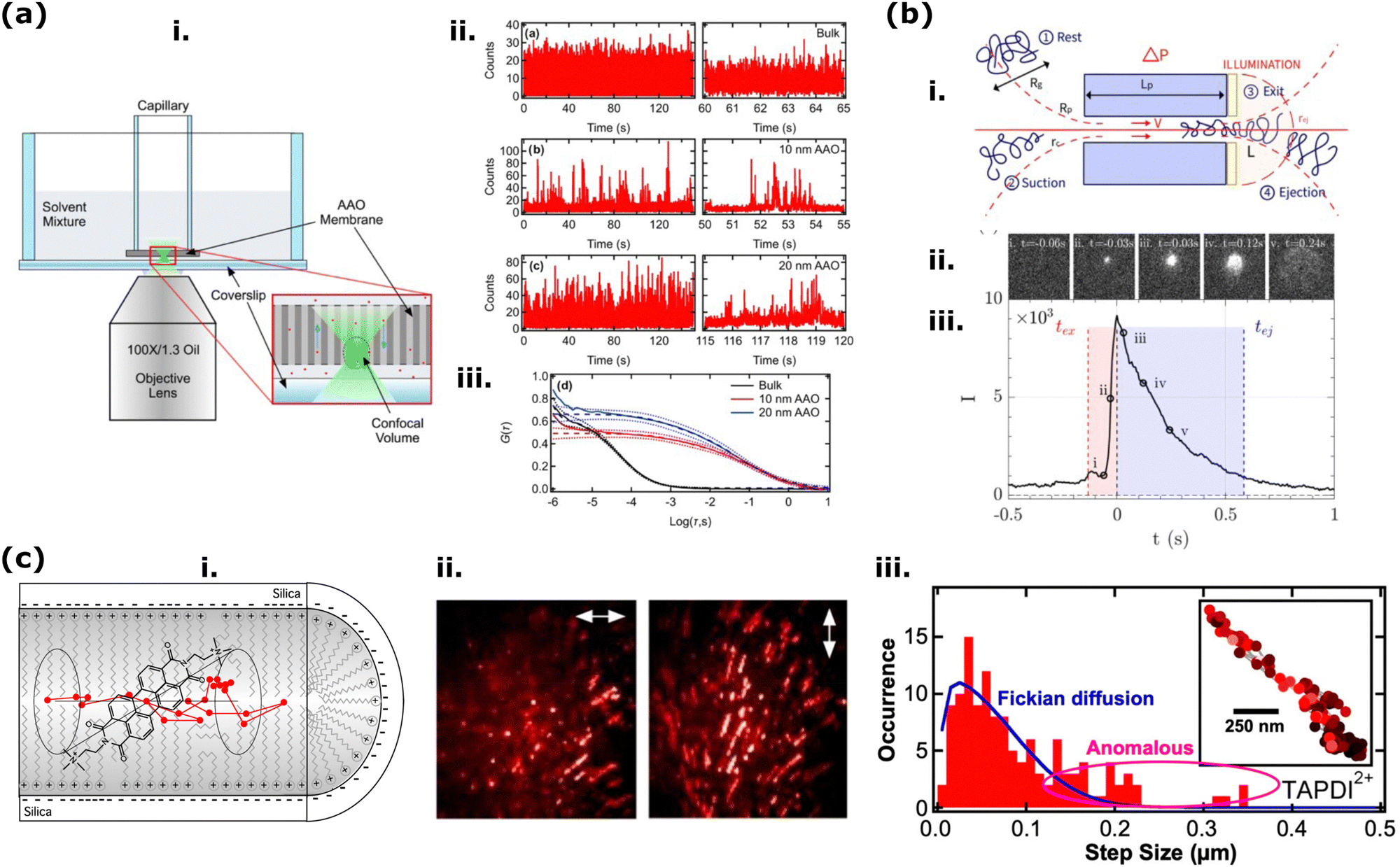 | ||
| Fig. 9 (a) (i) Experimental FCS setup with the expanded view of the AAO membrane. (ii) Representative time transients for RhB in a bulk ethanol solution, ethanol-filled 10 nm AAO nanochannels, and ethanol-filled 20 nm AAO nanochannel, top to bottom, respectively. (iii) Autocorrelation data (solid lines) obtained from the same series of samples based on averages from multiple replicate measurements. Reprinted with permission from ref. 283 Copyright 2023, American Chemical Society. (b) (i) Schematic explaining the translocation mechanism of polymers (also applied for different DNA) through 1D nanochannel. Rg stands for the gyration radius of the polymer coil. (iii) Mean event quantified after averaging all segmented events tracked by single-molecule fluorescence microscopy images (ii). The red region (tex) and blue region (tej) correspond to the time of images captured while the polymer coil exits the nanopore and the coil moves outside the illumination region, respectively. Reprodiced from ref. 284 with permission from the Royal Society of Chemistry. (c) (i) Illustration of a cationic PDI molecule inside a CTAB-loaded silica mesopore. (ii) Simultaneously collected Z-projection images of the PDI molecules at two orthogonal polarizations (double-ended white arrows with 3 μm length). (iii) Detected single-molecule trajectory presenting the 1D motion (inset) and related step-size histograms (red bars). The solid blue line represents the fit to a 1D Fickian diffusion model, accounting for finite localization precision. Populations that did not conform to this model are labeled as “Anomalous.” Reprinted with permission from ref. 285 Copyright 2020, American Chemical Society. | ||
1D Nanochannels are also widely used to detect oligonucleotides such as DNA, RNA by measuring the changes in the ionic currents. In a recent example, a promising system was proposed for detecting single molecules based on optical methods (Fig. 9b).284 Track-etched membranes with varying nanochannel diameters and lengths were incorporated in a chamber where controlled pressure was applied to transport various natural and synthetic polymer analytes, including labeled DNA and fluorescent polymers. The device was placed on an inverted fluorescence microscope to visualize the translocation events directly. Image processing and analysis were important in detecting and assessing a series of simultaneous events (Fig. 9b(ii and iii)). With the tools described in this work, it was possible to characterize the molecular transport by experimentally visualizing and measuring the translocation events. Among other findings, it was shown that it is possible to detect and discriminate two different DNA populations and even quantify their volume fractions.
Especially in recent years, to understand the mass transport and diffusion dynamics from a molecular level inside 1D and 2D nanoporous membranes, single molecule tracking (SMT) has been used very effectively.19,286–288 Among such mesoporous materials, surfactant (cetyltrimethylammonium bromide (CTAB))-filled silica mesopores have the smallest diameters (≈3.7 nm)289 and these pores behave as chemically heterogeneous environments: hydrophobic micelle cores, hydrophilic micelle-silica interface, and solid silica matrix (Fig. 9c(i)).285 In such heterogeneous nanoconfinement, upon solvent(s) addition, the system pushes the fluorescent molecules to mobilize. In the end, single-molecule tracking (SMT) and single-molecule emission polarization (SMEP) methods, allowing polarized emission detection of single molecules in the nanopores, are used to understand the relation between orientational dynamics of the single molecules and their shape-selective separations.286,290,291 Recently, Kumarasinghe and others285 quantitatively investigated the diffusion and distribution dynamics of charged and uncharged perylenediimide (PDI) fluorescent probes in such CTAB-filled silica mesopores using SMT and single molecule emission polarization (SMEP) (Fig. 9c). As presented in their SMEP images, both charged and uncharged PDI molecules in extremely narrow CTAB-templated nanopores exhibited one-dimensional diffusion trajectories and strong emission polarization parallel to the diffusion direction (Fig. 9c(ii)). This behavior indicates the orientational confinement of the dyes within the cylindrical CTAB-filled nanopores, as illustrated in Fig. 9c(i). Charged PDI dyes showed smaller diffusion coefficients and less orientational confinement than uncharged PDI dyes, suggesting that the charged dyes diffused closer to the micelle-silica interface. Interestingly, the distribution of their frame-to-frame step sizes (Fig. 9c(iii)) indicated contributions from anomalous diffusion, potentially based on the chemical and structural heterogeneity of the silica mesopores.
Fluorescence correlation spectroscopy (FCS) has also been receiving great interest for studying the nanopore activities in 1D and 2D nanoporous membranes especially due to its powerful output to quantify diffusion coefficients of dyes in nanoconfinement and their evolution in time.288,292–294 In 2015, Park et al. gained quantitative insights into the distributions of charged and uncharged PDI fluorescent molecules in silica mesopores filled with an uncharged surfactant template (Pluronic F127) by examining the dimensionality of single-molecule diffusion by FCS.292 Based on the reported FCS data, they found that a significant population of the charged molecules exhibited two-dimensional diffusion trajectories (recall: Fig. 2b-bottom), in contrast to the uncharged molecules, which displayed one-dimensional trajectories (recall: Fig. 2b-top). This difference suggests that charged molecules have easier access to hydrophilic interfacial regions and pass through defects in the silica matrix more frequently.
Single-molecule localization spectroscopy in 3D membranes (Fig. 10) is a novel approach, but it cannot be broadly used due to some limitations. One major limitation is the complexity of determining free molecule trajectories in complex three-dimensional space. This is mainly related to the impossibility of differentiating between Brownian diffusion and other factors, such as active transport or variation in the transport kinetics caused by intermolecular interactions.295 Recently, Xiang et al. developed a fascinating methodology called single-molecule displacement mapping (SMdM) to partially overcome this limitation. This is done by analyzing the local statistics of a molecule's instantaneous displacements. By statistically evaluating how far and in which direction a molecule moves over short time intervals, with SMdM it is possible to build a map of its diffusion behavior across the cellular landscape. This allows scientists to not only observe movement but also quantify how easily a molecule can move within different and complex environments, allowing them to determine the nanoscale displacement of freely diffusing single molecules in time windows of roughly 1 ms.295 With this technique, they were first able to determine the diffusion of an average-size protein inside a mammalian cell and then to study in-gel diffusion of a fluorophore (Cy3B) and a model protein (BSA) labeled with the same (Cy3B) in poly(acrylamide-co-sodium acrylate) hydrogels (Fig. 10a). In the latter study, to evaluate the relationship between diffusion and hydrogel mesh size, they tuned the hydrogel's mesh size by varying the sodium acrylate fraction and the ionic strength of the solution (Fig. 10a-left).296 By doing so, they were able to alter the hydrogels expansion ratio, and to demonstrate that for the same diffusing molecule, diffusion is progressively hindered when the mesh size is reduced and that this effect becomes more prominent when the diffusing molecule's dimensions are increased (Fig. 10a-right). Moreover, they were also able to prove that a decrease in mesh size and an increase in solution viscosity, despite having a similar effect in limiting molecular diffusivity, are not correlated. This proves that both mechanisms can be employed to modify diffusion rate in a controlled manner.296 Al Sulaiman et al. employed this strategy to detect small DNA fragments in a composite quartz nanopipette filled with chemically crosslinked PVA hydrogels (Fig. 10b(i)). Fig. 10b(ii) reports the wide variety of chemically modified hydrogels where R represents one of the three functional groups employed: anionic carboxylate functional group on PVA-CO− (green), hydrophobic phenyl functional group on PVA-Phe (orange), and cationic ammonium functional group on PVA-NH+ (red).297 By fine tuning pore size (11–30 nm) and pore functionality of this hydrogel functionalized nanopipette (HFN) and the application of an electrical potential, the researchers were able to extract reversibly single 10 kbp YOYO-1 labeled DNA fragments and to visualize their presence by fluorescence microscopy. As reported in Fig. 10b(iii), a single DNA molecule is trapped at the HFN tip upon applying a positive voltage bias. The molecule remains at the tip for over 50 s, but fluorescence intensity decreases due to photobleaching within this time scale. Once a single 10 kbp molecule is trapped at the tip at t = 0 s, the application of a negative voltage bias forces the DNA molecule out of the HFN, evident by the motion of the molecule at t = 0.1 s and the disappearance of fluorescence at 5 s.
 | ||
| Fig. 10 (a) (Left) Schematic representation of hydrogel synthesis and of the diffusion process studied; (right) Normalized count distributions of the SMdM-measured 400 μs single-molecule displacements for Cy3B diffusing in PBS (dash line) and in hydrogels with different expansion factors (solid lines), with respective maximum likelihood estimation results shown in the legend. Adapted with permission from ref. 298 Copyright 2023, American Chemical Society. (b) (i) Schematic representation of the hydrogel functionalized nanopipette (HFN) sensor showing a 3D cross-linked nanoporous mesh at the tip of the nanopipette, (ii) simplified chemical structure of the photo-cross-linked PVA-based hydrogels, (iii) optical tracking of individual YOYO-1 labeled 10 kbp dsDNA molecules at the HFN tip (scale bars show 2 μm). Reprinted with permission from ref. 297 Copyright 2018, American Chemical Society. | ||
5. Summary, limitations, and prospects
Especially in the last decade, Solid-state nanoporous membranes (SSNMs) have become increasingly important due to their critical roles in various applications such as energy conversion, water management, drug delivery, sensing, separation, and novel batteries. SSNMs provide unique opportunities for selective transport of various cargoes from ions to macromolecular biomolecules (e.g., DNA, RNA) and precise sensing at the nanoscale, which are essential for improving the efficiency and functionality of these membranes. Despite significant advancements, there is still ample room for improvement in engineering these nanoporous membranes for future applications. For instance, free-standing thin films with single nanopore/nanochannel helped us understand the nanopore's potential from a fundamental point of view, such as its ionic permselectivity, rectification, and many more. A current challenge to apply this knowledge in engineering systems is the fabrication of arrays of smart nanopores, with ultraprecision8 and cost-effective. Another key challenge is expanding the nanopore response functionality (against various stimuli) and gaining a deeper understanding of structure–property relationships of SSNMs (e.g., nanopore geometry).4,8,15,299 In recent years, significant nanopore functionalization efforts have been made to expand the nanopore response beyond their charge responsive selectivity, such as (de)protonation of the functional groups upon changing pH. For instance, multifunctional (also zwitterionic) polydopamine has recently been electrodeposited inside and on the top of SSNMs to help the nanopores sort the pH-dependent ionic transport.300,301 However, the multifunctionality of polydopamine can also make the functionalized nanoporous membranes improve their adhesion, catalytic photothermal, and even fluorescence quenching responses with zwitterionic response.302–304As introduced in our paper, among other nanopore optical-probing methods, especially during the last ten years, fluorescence spectroscopy and microscopy have become very popular tools for studying mass transport and sensing performance in SSNMs. Fluorescent readouts can provide information on molecular interactions and dynamics from (i) individual molecules within the nanopores (microscale), as well as from (ii) an array of nanopores or different media in contact with nanoporous membranes (macroscale). For instance, to date, fluorescence readout provided crucial information about localized pH at nanoconfinement, asymmetric functionalization of nanopores, and the nanoporous materials’ structural changes, such as their interlayer stacking or porosity; fluorescence readout also yielded time-dependent, in situ and non-invasive monitoring of various nanopore interaction with ions, molecules, and macromolecules; and allowed to quantitatively sense chemical species in different physical forms, such as gas and liquid at the nanoporous membranes, including explosive TNT gas detection; it allowed understanding the fundamental mechanism of different cargo permeation through nanoporous membranes such as mixture of solvents, ions, protons, DNA, RNA and many more; it yielded–at a single molecule level–dynamic data of macromolecular translocation at the nanopores, and of interaction between various macromolecules (e.g., labeled DNA, proteins) and 3D polymeric matrix. Of course, this review does not aim to be exhaustive of the examples of nanoporous materials that can be studied by fluorescent methods, nor of the fluorescent methods that can provide information about nanoporous materials. This review can certainly be complemented by other reviews focusing on more specific fluorescent probing methods, such as single molecule detection of nanoconfinement,19,20,288 or on fluorescence probing methods applied to specific nanoporous materials such as COFs, POPs, Mxene, and others.10,288,305–307 In this article, we tried to give an overall idea of how to apply various fluorescent spectroscopy and microscopy techniques to understand the function of different high-dimension (>0D) nanoporous membranes.
Above-sorted recent advancements in fluorescent probing of nanoporous membranes have opened new avenues; however, the marriage of fluorescence methods and nanoporous materials should progress towards new research areas, for example: (i) dynamics of nanoconfinement: by designing fluorescent probes with variable size to ref. 19 provide insights into how nanoconfinement affects transport and reaction processes; (ii) nanopore functionalization kinetics: how functional groups are incorporated into the nanopores and how they interact with target molecules over time. (iii) Machine learning-assisted analysis: machine learning techniques can be applied to analyze complex fluorescence data, enhancing the interpretation and understanding of nanopore behaviors and interactions.15 This can lead to more accurate models and predictions of membrane performance. (iv) Combining methods: integrating fluorescence with other analytical techniques, such as vibrational spectroscopy (e.g., Raman spectroscopy, ATR-FTIR), electrochemiluminescence, X-ray fluorescence to study various complex nanoporous systems.10,308–313 (v) Multiplexed and enhanced detection: combining electrical detection with fluorescence can increase specificity, adding the chemical information intrinsically present in the fluorescent probe. Additionally, using plasmonic devices can localize light at the nanoscale, enhancing the fluorescence signals and improving detection sensitivity.15 (vi) New fluorogenic probes: designing fluorogenic probes that correlate with essential chemical transformations, such as water splitting, oxygen reduction, carbon dioxide reduction, and small alcohol formation, is a promising area of research.314,315 These probes can provide real-time monitoring of chemical reactions within nanopores. (vii) Investigation of key functional sites: many crucial functions of materials like metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) occur at their nanoscale pores and interlayer spacing. Applying single-molecule fluorescent techniques to study these sites can reveal critical insights into their functional mechanisms.
These prospects in fluorescence probing for nanoporous membrane investigations will strongly impact the field of materials science, enhancing the understanding of the complex physico-chemical processes occurring in the nanopores during mass transport and chemical activity. Furthermore, future realization of these perspectives can boost the interaction and cooperation between researchers and engineers across multiple disciplines, including chemistry, physics, biology and engineering, with the final goal of bringing the exceptional functionalities of SSNMs to the application and to the market, spanning across diverse sectors from water treatment, to chemical sensing, to clinical and biological detection assays and to high-end materials for theranostics and for energy storage and conversion.
Author contributions
This review was conceptualized by HSV and DG. All the co-authors contributed to writing the original draft, editing and reviewing the manuscript.Data availability
In this review article, no primary research results, software or code was included. No new data were generated and/or analyzed for this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
HSV gratefully acknowledge Alexander von Humboldt Foundation for financial support. DG gratefully acknowledges funding by the European Union – NextGenerationEU under the National Recovery and Resilience Plan (PNRR) – Mission 4 Education and research – Component 2 From research to business – Investment 1.1 Notice Prin 2022 – DD N. 104 del 2/2/2022, from title “REDOX TRIGGERED AGGREGATION INDUCED EMISSION FOR HIGH END APPLICATIONS”, proposal code 2022ETBCER – CUP J53D23008650001.References
- B. Corry and S. H. Chung, Cell. Mol. Life Sci., 2006, 63, 301–315 CrossRef CAS PubMed.
- S. S. Patel, B. J. Belmont, J. M. Sante and M. F. Rexach, Cell, 2007, 129, 83–96 CrossRef CAS PubMed.
- M. Tagliazucchi and I. Szleifer, Mater. Today, 2015, 18, 131–142 CrossRef CAS.
- G. Laucirica, Y. Toum Terrones, V. Cayón, M. L. Cortez, M. E. Toimil-Molares, C. Trautmann, W. Marmisollé and O. Azzaroni, TrAC, Trends Anal. Chem., 2021, 144, 116425 CrossRef CAS.
- G. Pérez-Mitta, A. G. Albesa, C. Trautmann, M. E. Toimil-Molares and O. Azzaroni, Chem. Sci., 2017, 8, 890–913 RSC.
- C. Dekker, Nat. Nanotechnol., 2007, 2, 209–215 CrossRef CAS PubMed.
- L. Jiang and X. Hou, ACS Nano, 2009, 3, 3339–3342 CrossRef PubMed.
- H. Liu, Q. Zhou, W. Wang, F. Fang and J. Zhang, Small, 2023, 19, 2205680 CrossRef CAS PubMed.
- W. Lu, R. Hu, X. Tong, D. Yu and Q. Zhao, Small Struct., 2020, 1, 2000003 CrossRef.
- J. P. Fried, Y. Wu, R. D. Tilley and J. J. Gooding, Nano Lett., 2022, 22, 869–880 CrossRef CAS PubMed.
- G. Laucirica, V. M. Cayón, Y. Toum Terrones, M. L. Cortez, M. E. Toimil-Molares, C. Trautmann, W. A. Marmisollé and O. Azzaroni, Nanoscale, 2020, 12, 6002–6011 RSC.
- M. Liu, J. Li and C. S. Tan, Biosensors, 2023, 13, 598 CrossRef CAS PubMed.
- Y. Lu, D. Zhang, Q. Zhang, Y. Huang, S. Luo, Y. Yao, S. Li and Q. Liu, Biosens. Bioelectron., 2016, 79, 251–257 CrossRef CAS PubMed.
- N. Li, S. Yu, C. C. Harrell and C. R. Martin, Anal. Chem., 2004, 76, 2025–2030 CrossRef CAS PubMed.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS.
- P. Ponzellini, X. Zambrana-Puyalto, N. Maccaferri, L. Lanzanò, F. De Angelis and D. Garoli, Nanoscale, 2018, 10, 17362–17369 RSC.
- R. Wang, T. Gilboa, J. Song, D. Huttner, M. W. Grinstaff and A. Meller, ACS Nano, 2018, 12, 11648–11656 CrossRef CAS PubMed.
- D. Punj, M. Mivelle, S. B. Moparthi, T. S. Van Zanten, H. Rigneault, N. F. Van Hulst, M. F. García-Parajó and J. Wenger, Nat. Nanotechnol., 2013, 8, 512–516 CrossRef CAS PubMed.
- B. Dong, N. Mansour, T. X. Huang, W. Huang and N. Fang, Chem. Soc. Rev., 2021, 50, 6483–6506 RSC.
- B. N. Miles, A. P. Ivanov, K. A. Wilson, F. Dogan, D. Japrung and J. B. Edel, Chem. Soc. Rev., 2013, 42, 15–28 RSC.
- S. Zhang, M. Cheng, M. K. Dhinakaran, Y. Sun and H. Li, ACS Nano, 2021, 15, 13148–13154 CrossRef CAS PubMed.
- Y. Hu, Y. Teng, Y. Sun, P. Liu, L. Fu, L. Yang, X. Y. Kong, Q. Zhao, L. Jiang and L. Wen, Nano Energy, 2022, 97, 107170 CrossRef CAS.
- W. Chen, Q. Wang, J. Chen, Q. Zhang, X. Zhao, Y. Qian, C. Zhu, L. Yang, Y. Zhao, X. Y. Kong, B. Lu, L. Jiang and L. Wen, Nano Lett., 2020, 20, 5705–5713 CrossRef CAS PubMed.
- Z. Zhang, X. Sui, P. Li, G. Xie, X. Y. Kong, K. Xiao, L. Gao, L. Wen and L. Jiang, J. Am. Chem. Soc., 2017, 139, 8905–8914 CrossRef CAS PubMed.
- J. Hao, Y. Ning, Y. Hou, S. Ma, C. Lin, J. Zhao, C. Li and X. Sui, J. Colloid Interface Sci., 2023, 630, 795–803 CrossRef CAS PubMed.
- J. Hohlbein, Food Struct., 2021, 30, 100236 CrossRef CAS.
- L. M. Hirvonen and K. Suhling, Front. Phys., 2020, 8, 161 CrossRef.
- J. Valanciunaite, E. Kempf, H. Seki, D. I. Danylchuk, N. Peyriéras, Y. Niko and A. S. Klymchenko, Anal. Chem., 2020, 92, 6512–6520 CrossRef CAS PubMed.
- F. Palomba, D. Genovese, L. Petrizza, E. Rampazzo, N. Zaccheroni and L. Prodi, Sci. Rep., 2018, 8, 1–8 CAS.
- A. Kyrychenko, Methods Appl. Fluoresc., 2015, 3, 042003 CrossRef PubMed.
- M. Paez-Perez and M. K. Kuimova, Angew. Chem., Int. Ed., 2024, 63, e202311233 CrossRef CAS PubMed.
- T. V. Esipova, M. J. P. Barrett, E. Erlebach, A. E. Masunov, B. Weber and S. A. Vinogradov, Cell Metab., 2019, 29, 736–744 CrossRef CAS PubMed.
- U. Kubitscheck, Fluorescence Microscopy: From Principles to Biological Applications, Wiley-VCH, 2013 Search PubMed.
- P. Wu, X. Hou, J. J. Xu and H. Y. Chen, Nanoscale, 2016, 8, 8427–8442 RSC.
- L. Y. Liu, Y. Zhao, N. Zhang, K. N. Wang, M. Tian, Q. Pan and W. Lin, Anal. Chem., 2021, 93, 1612–1619 CrossRef CAS PubMed.
- L. Schermelleh, A. Ferrand, T. Huser, C. Eggeling, M. Sauer, O. Biehlmaier and G. P. C. Drummen, Nat. Cell Biol., 2019, 21, 72–84 CrossRef CAS PubMed.
- M. Lelek, M. T. Gyparaki, G. Beliu, F. Schueder, J. Griffié, S. Manley, R. Jungmann, M. Sauer, M. Lakadamyali and C. Zimmer, Nat. Rev., 2021, 1, 1–27 Search PubMed.
- J. Ries and P. Schwille, Phys. Chem. Chem. Phys., 2008, 10, 3487–3497 RSC.
- X. Y. Yang, L. H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B. L. Su, Chem. Soc. Rev., 2017, 46, 481–558 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- G. Laucirica, A. G. Albesa, M. E. Toimil-Molares, C. Trautmann, W. A. Marmisollé and O. Azzaroni, Nano Energy, 2020, 71, 104612 CrossRef CAS.
- Y. Toum Terrones, G. Laucirica, V. M. Cayón, G. E. Fenoy, M. L. Cortez, M. E. Toimil-Molares, C. Trautmann, W. A. Mamisollé and O. Azzaroni, Chem. Commun., 2022, 58, 10166–10169 RSC.
- D. Kaya, A. Dinler, N. San and K. Kececi, Electrochim. Acta, 2016, 202, 157–165 CrossRef CAS.
- D. Y. Butylskii, N. D. Pismenskaya, P. Y. Apel, K. G. Sabbatovskiy and V. V. Nikonenko, J. Membr. Sci., 2021, 635, 119449 CrossRef CAS.
- G. Laucirica, Y. Toum Terrones, M. F. P. Wagner, V. M. Cayón, M. L. Cortez, M. E. Toimil-Molares, C. Trautmann, W. Marmisollé and O. Azzaroni, Nanoscale, 2023, 15, 1782–1793 RSC.
- G. Laucirica, Y. Toum Terrones, V. M. Cayón, M. L. Cortez, M. E. Toimil-Molares, C. Trautmann, W. A. Marmisollé and O. Azzaroni, Nanoscale, 2020, 12, 18390–18399 RSC.
- M. Nazari, A. Davoodabadi, D. Huang, T. Luo and H. Ghasemi, ACS Nano, 2020, 14, 16348–16391 CrossRef CAS PubMed.
- M. Tagliazucchi and I. Szleifer, Chemically modified nanopores and nanochannels, Elsevier, 2017 Search PubMed.
- Y. T. Terrones, V. M. Cayón, G. Laucirica, M. L. Cortez, M. E. Toimil-Molares, C. Trautmann, W. A. Marmisollé and O. Azzaroni, Miniaturized Biosensing Devices: Fabrication and Applications, Springer, Singapore, 2022, pp. 57–81 Search PubMed.
- X. L. Ding, Z. Q. Wu, Z. Q. Li and X. H. Xia, Chin. J. Chem., 2021, 39, 1511–1516 CrossRef CAS.
- J. Vetter and D. Dobrev, Nucl. Instrum. Methods Phys. Res., Sect. B, 1999, 156, 177–182 CrossRef CAS.
- N. Meyer, N. Arroyo, J. M. Janot, M. Lepoitevin, A. Stevenson, I. A. Nemeir, V. Perrier, D. Bougard, M. Belondrade, D. Cot, J. Bentin, F. Picaud, J. Torrent and S. Balme, ACS Sens., 2021, 6, 3733–3743 CrossRef CAS PubMed.
- G. Lu, N. Lin, Z. Chen, W. Jiang, J. J. Hu, F. Xia and X. Lou, Chin. J. Chem., 2023, 41, 1374–1384 CrossRef CAS.
- T. Gilboa and A. Meller, Analyst, 2015, 140, 4733–4747 RSC.
- S. Liu, J. Tian and W. Zhang, Nanotechnology, 2021, 32, 222001 CrossRef CAS PubMed.
- A. Ruiz-Clavijo, O. Caballero-Calero and M. Martín-González, Nanoscale, 2021, 13, 2227–2265 RSC.
- S. Zhang, Y. Wang, Y. Tan, J. Zhu, K. Liu and J. Zhu, Mater. Res. Express, 2016, 3, 074004 CrossRef.
- S. Manzoor, M. W. Ashraf, S. Tayyaba and M. K. Hossain, CMES - Comput. Model. Eng. Sci., 2021, 135, 1007–1052 Search PubMed.
- P. Kapruwan, J. Ferré-Borrull and L. F. Marsal, Adv. Mater. Interfaces, 2020, 7, 2001133 CrossRef CAS.
- S. K. Podgolin, D. I. Petukhov, S. G. Dorofeev and A. A. Eliseev, Talanta, 2020, 219, 121248 CrossRef CAS PubMed.
- L. Shi, C. Mu, T. Gao, T. Chen, S. Hei, J. Yang and G. Li, Chem. Commun., 2018, 54, 11391–11394 RSC.
- P. Gao, L. Hu, N. Liu, Z. Yang, X. Lou, T. Zhai, H. Li and F. Xia, Adv. Mater., 2016, 28, 460–465 CrossRef CAS PubMed.
- A. S. Peinetti, R. J. Lake, W. Cong, L. Cooper, Y. Wu, Y. Ma, G. T. Pawel, M. E. Toimil-Molares, C. Trautmann, L. Rong, B. Mariñas, O. Azzaroni and Y. Lu, Sci. Adv., 2021, 7, 1–12 Search PubMed.
- L. Pla, S. Santiago-Felipe, M. A. Tormo-Mas, J. Pemán, F. Sancenón, E. Aznar and R. Martínez-Máñez, Sens. Actuators, B, 2020, 320, 128281 CrossRef CAS.
- L. Pol, C. Eckstein, L. K. Acosta, E. Xifré-Pérez, J. Ferré-Borrull and L. F. Marsal, Nanomaterials, 2019, 9, 478 CrossRef CAS PubMed.
- M. Makela, Z. Lin and P. T. Lin, IEEE Sens. J., 2021, 21, 22645–22650 CAS.
- T. Ito, J. Phys. Chem. B, 2023, 127, 5733–5741 CrossRef CAS PubMed.
- P. Sornmek, D. Phromyothin, J. Supadech, K. Tantisantisom and T. Boonkoom, Phys. Chem. Chem. Phys., 2022, 24, 24866–24872 RSC.
- Z. S. Siwy, Adv. Funct. Mater., 2006, 16, 735–746 CrossRef CAS.
- M. E. Toimil-Molares, Beilstein J. Nanotechnol., 2012, 3, 860–883 CrossRef PubMed.
- P. Y. Apel, I. V. Blonskaya, O. L. Orelovitch, P. Ramirez and B. A. Sartowska, Nanotechnology, 2011, 22, 175302 CrossRef PubMed.
- Z. Siwy, P. Apel, D. Dobrev, R. Neumann, R. Spohr, C. Trautmann and K. Voss, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 208, 143–148 CrossRef CAS.
- B. Yameen, M. Ali, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Small, 2009, 5, 1287–1291 CrossRef CAS PubMed.
- X. Hou, Y. Liu, H. Dong, F. Yang, N. Li and L. Jiang, Adv. Mater., 2010, 22, 2440–2443 CrossRef CAS PubMed.
- D. Zhang and X. Zhang, Nano Lett., 2022, 22, 32 Search PubMed.
- S. Zhang, J. Zhou and H. Li, Angew. Chem., 2022, 134, e202204012 CrossRef.
- S. Soto Espinoza, C. Aguiar, F. Richieri and M. Grasselli, React. Funct. Polym., 2019, 135, 1–7 CrossRef CAS.
- E. Topoglidis, Coatings, 2020, 10, 668 CrossRef CAS.
- M. Mierzwa, E. Lamouroux, A. Walcarius and M. Etienne, Electroanalysis, 2018, 30, 1241–1258 CrossRef CAS.
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098 RSC.
- P. Innocenzi and L. Malfatti, Chem. Soc. Rev., 2013, 42, 4198–4216 RSC.
- A. B. Farimani, K. Min and N. R. Aluru, ACS Nano, 2014, 8, 7914–7922 CrossRef CAS PubMed.
- H. L. Qian, C. X. Yang and X. P. Yan, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- L. Guo, L. Yang, M. Li, L. Kuang, Y. Song and L. Wang, Coord. Chem. Rev., 2021, 440, 213957 CrossRef CAS.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- X. Hu, Y. Long, M. Fan, M. Yuan, H. Zhao, J. Ma and Z. Dong, Appl. Catal., B, 2019, 244, 25–35 CrossRef CAS.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- Z. Mu, Y. Zhu, B. Li, A. Dong, B. Wang and X. Feng, J. Am. Chem. Soc., 2022, 144, 5145–5154 CrossRef CAS PubMed.
- S. L. Cai, Y. B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S. R. Zheng, W. G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 RSC.
- D. B. Shinde, G. Sheng, X. Li, M. Ostwal, A. H. Emwas, K. W. Huang and Z. Lai, J. Am. Chem. Soc., 2018, 140, 14342–14349 CrossRef CAS PubMed.
- K. Dey, M. Pal, K. C. Rout, S. S. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- Z. Li, Y. Zhang, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 6613–6616 RSC.
- L. K. Beagle, Q. Fang, L. D. Tran, L. A. Baldwin, C. Muratore, J. Lou and N. R. Glavin, Mater. Today, 2021, 51, 427–448 CrossRef CAS.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- Q. Gao, X. Li, G. H. Ning, K. Leng, B. Tian, C. Liu, W. Tang, H. Sen Xu and K. P. Loh, Chem. Commun., 2018, 54, 2349–2352 RSC.
- H. Q. Yin, F. Yin and X. B. Yin, Chem. Sci., 2019, 10, 11103–11109 RSC.
- H. L. Qian, C. Dai, C. X. Yang and X. P. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 24999–25005 CrossRef CAS PubMed.
- L. Zhang, Y. Zhou, M. Jia, Z. Liu, J. Cheng and H. Deng, Matter, 2020, 2, 1049–1063 CrossRef.
- C. Montoro, D. Rodríguez-San-Miguel, E. Polo, R. Escudero-Cid, M. L. Ruiz-González, J. A. R. Navarro, P. Ocón and F. Zamora, J. Am. Chem. Soc., 2017, 139, 10079–10086 CrossRef CAS PubMed.
- I. Vlassiouk, P. Y. Apel, S. N. Dmitriev, K. Healy and Z. S. Siwy, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21039–21044 CrossRef CAS PubMed.
- H. S. Varol, C. Förster and A. Andrieu-Brunsen, Adv. Mater. Interfaces, 2023, 10, 2201902 CrossRef CAS.
- C. C. Striemer, T. R. Gaborski, J. L. McGrath and P. M. Fauchet, Nature, 2007, 445, 749–753 CrossRef CAS PubMed.
- J. Li, D. Stein, C. McMullan, D. Branton, M. J. Aziz and J. A. Golovchenko, Nature, 2001, 412, 166–169 CrossRef CAS PubMed.
- H. D. Tong, H. V. Jansen, V. J. Gadgil, C. G. Bostan, E. Berenschot, C. J. M. Van Rijn and M. Elwenspoek, Nano Lett., 2004, 4, 283–287 CrossRef CAS.
- M. M. Deshmukh, D. C. Ralph, M. Thomas and J. Silcox, Appl. Phys. Lett., 1999, 75, 1631–1633 CrossRef CAS.
- A. J. Storm, J. H. Chen, X. S. Ling, H. W. Zandbergen and C. Dekker, Nat. Mater., 2003, 2, 537–540 CrossRef CAS PubMed.
- M. G. Mohamed, A. F. M. El-Mahdy, M. G. Kotp and S. W. Kuo, Mater. Adv., 2022, 3, 707–733 RSC.
- Y. Song, C. Zhu and S. Ma, EnergyChem, 2022, 4, 100079 CrossRef CAS.
- W. Song, Y. Zhang, C. H. Tran, H. K. Choi, D. G. Yu and I. Kim, Prog. Polym. Sci., 2023, 142, 101691 CrossRef CAS.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- N. B. Mc Keown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- J. S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- W. Wang, M. Zhou and D. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC.
- J. Liang, Y. B. Huang and R. Cao, Coord. Chem. Rev., 2019, 378, 32–65 CrossRef CAS.
- S. Lu, Q. Liu, R. Han, M. Guo, J. Shi, C. Song, N. Ji, X. Lu and D. Ma, J. Environ. Sci., 2021, 105, 184–203 CrossRef CAS PubMed.
- X. Liu, C. F. Liu, S. Xu, T. Cheng, S. Wang, W. Y. Lai and W. Huang, Chem. Soc. Rev., 2022, 51, 3181–3225 RSC.
- X. Liu, C. F. Liu, W. Y. Lai and W. Huang, Adv. Mater. Technol., 2020, 5, 2000154 CrossRef CAS.
- S. Wang, H. Li, H. Huang, X. Cao, X. Chen and D. Cao, Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- N. Singh, S. Son, J. An, I. Kim, M. Choi, N. Kong, W. Tao and J. S. Kim, Chem. Soc. Rev., 2021, 50, 12883–12896 RSC.
- Y. Zhu, P. Xu, X. Zhang and D. Wu, Chem. Soc. Rev., 2022, 51, 1377–1414 RSC.
- H. Bildirir, V. G. Gregoriou, A. Avgeropoulos, U. Scherf and C. L. Chochos, Mater. Horiz., 2017, 4, 546–556 RSC.
- L. Huang, R. Liu, J. Yang, Q. Shuai, B. Yuliarto, Y. V. Kaneti and Y. Yamauchi, Chem. Eng. J., 2021, 408, 127991 CrossRef CAS.
- A. Waheed, N. Baig, N. Ullah and W. Falath, J. Environ. Manage., 2021, 287, 112360 CrossRef CAS PubMed.
- J. H. Song and D. W. Kang, Coord. Chem. Rev., 2023, 492, 215279 CrossRef CAS.
- S. Lu, Q. Liu, R. Han, M. Guo, J. Shi, C. Song, N. Ji, X. Lu and D. Ma, J. Environ. Sci., 2021, 105, 184–203 CrossRef CAS PubMed.
- E. A. Gendy, D. T. Oyekunle, J. Ali, J. Ifthikar, A. El-Motaleb Mosad Ramadan and Z. Chen, J. Environ. Radioact., 2021, 238–239, 106710 CrossRef CAS PubMed.
- S. Wang, H. Li, H. Huang, X. Cao, X. Chen and D. Cao, Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- E. Drioli and L. Giorno, Encyclopedia of Membranes, Springer Berlin, Heidelberg, 2016 Search PubMed.
- Y. Cheng, Y. Ying, S. Japip, S.-D. Jiang, T.-S. Chung, S. Zhang, D. Zhao, Y. Cheng, Y. Ying, S. Japip, S. Jiang, T. Chung, S. Zhang and D. Zhao, Adv. Mater., 2018, 30, 1802401 CrossRef PubMed.
- M. Ahmadi, S. Janakiram, Z. Dai, L. Ansaloni and L. Deng, Membranes, 2018, 8, 50 CrossRef PubMed.
- S. Janakiram, M. Ahmadi, Z. Dai, L. Ansaloni and L. Deng, Membranes, 2018, 8, 24 CrossRef PubMed.
- J. Yin and B. Deng, J. Membr. Sci., 2015, 479, 256–275 CrossRef CAS.
- J. Yin, G. Zhu and B. Deng, Desalination, 2016, 379, 93–101 CrossRef CAS.
- S. Zinadini, A. A. Zinatizadeh, M. Rahimi, V. Vatanpour and H. Zangeneh, J. Membr. Sci., 2014, 453, 292–301 CrossRef CAS.
- J. Mendret, M. Hatat-Fraile, M. Rivallin and S. Brosillon, Sep. Purif. Technol., 2013, 111, 9–19 CrossRef CAS.
- P. Gao, Z. Liu, M. Tai, D. D. Sun and W. Ng, Appl. Catal., B, 2013, 138–139, 17–25 CrossRef CAS.
- J. Hu, H. Cai, H. Ren, Y. Wei, Z. Xu, H. Liu and Y. Hu, Ind. Eng. Chem. Res., 2010, 49, 12605–12612 CrossRef CAS.
- X. Bi, Y. Zhang, F. Zhang, S. Zhang, Z. Wang and J. Jin, ACS Appl. Mater. Interfaces, 2020, 12, 49101–49110 CrossRef CAS PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2014, 14, 48–55 CrossRef PubMed.
- T. Yang, Y. Xiao and T. S. Chung, Energy Environ. Sci., 2011, 4, 4171–4180 RSC.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- F. L. Buchholz and A. T. Graham, Modern superabsorbent polymer technology, Wiley-VCH, 1998 Search PubMed.
- L. Brannon-Peppas and R. S. Harland, Absorbent Polymer Technology, Elsevier, New York, 1990 Search PubMed.
- F. Ganji, S. Vasheghani-Farahani and E. Vasheghani-Farahani, Polym. J., 2010, 19, 375–398 CAS.
- W. R. Gombotz and S. F. Wee, Adv. Drug Delivery Rev., 1998, 31, 267–285 CrossRef CAS PubMed.
- M. V. Badiger, M. E. McNeill and N. B. Graham, Biomaterials, 1993, 14, 1059–1063 CrossRef CAS PubMed.
- C. S. A. de Lima, T. S. Balogh, J. P. R. O. Varca, G. H. C. Varca, A. B. Lugão, L. A. Camacho-Cruz, E. Bucio and S. S. Kadlubowski, Pharmaceutics, 2020, 12, 970 CrossRef CAS PubMed.
- J. Karvinen, T. O. Ihalainen, M. T. Calejo, I. Jönkkäri and M. Kellomäki, Mater. Sci. Eng., C, 2019, 94, 1056–1066 CrossRef CAS PubMed.
- R. V. Klitzing and B. Tieke, Adv. Polym. Sci., 2004, 165, 177–210 CrossRef.
- R. M. DuChanois, R. Epsztein, J. A. Trivedi and M. Elimelech, J. Membr. Sci., 2019, 581, 413–420 CrossRef CAS.
- K. Hooshyari, B. Amini Horri, H. Abdoli, M. Fallah Vostakola, P. Kakavand and P. Salarizadeh, Energies, 2021, 14, 5440 CrossRef CAS.
- W. Zhang, Q. Zhao and J. Yuan, Angew. Chem., Int. Ed., 2018, 57, 6754–6773 CrossRef CAS PubMed.
- M. A. Mokhter, S. Lakard, C. Magnenet, M. Euvrard and B. Lakard, Environ. Technol., 2017, 38, 2476–2485 CrossRef CAS PubMed.
- H. Kuroki, C. Islam, I. Tokarev, H. Hu, G. Liu and S. Minko, ACS Appl. Mater. Interfaces, 2015, 7, 10401–10406 CrossRef CAS PubMed.
- Y. Si, L. Wang, X. Wang, N. Tang, J. Yu and B. Ding, Adv. Mater., 2017, 29, 1700339 CrossRef PubMed.
- A. Zrehen, D. Huttner and A. Meller, ACS Nano, 2019, 13, 14388–14398 CrossRef CAS PubMed.
- V. Jadhav, D. P. Hoogerheide, J. Korlach and M. Wanunu, Nano Lett., 2019, 19, 921–929 CrossRef PubMed.
- G. Rateni, P. Dario and F. Cavallo, Sensors, 2017, 17, 1453 CrossRef PubMed.
- L. Zhang, Z. Tian, H. Bachman, P. Zhang and T. J. Huang, ACS Nano, 2020, 14, 3159–3169 CrossRef CAS PubMed.
- H. J. S. de Oliveira, P. L. de Almeida, B. A. Sampaio, J. P. A. Fernandes, O. D. Pessoa-Neto, E. A. de Lima and L. F. de Almeida, Sens. Actuators, B, 2017, 238, 1084–1091 CrossRef.
- S. Sanllorente, L. Rubio, M. C. Ortiz and L. A. Sarabia, Sens. Actuators, B, 2019, 285, 240–247 CrossRef CAS.
- C. P. Bacon, Y. Mattley and R. DeFrece, Rev. Sci. Instrum., 2004, 75, 1–16 CrossRef CAS.
- R. Baumann, C. Ferrante, F. W. Deeg and C. Bräuchle, J. Chem. Phys., 2001, 114, 5781–5791 CrossRef CAS.
- N. R. Aluru, F. Aydin, M. Z. Bazant, D. Blankschtein, A. H. Brozena, J. P. de Souza, M. Elimelech, S. Faucher, J. T. Fourkas, V. B. Koman, M. Kuehne, H. J. Kulik, H. K. Li, Y. Li, Z. Li, A. Majumdar, J. Martis, R. P. Misra, A. Noy, T. A. Pham, H. Qu, A. Rayabharam, M. A. Reed, C. L. Ritt, E. Schwegler, Z. Siwy, M. S. Strano, Y. H. Wang, Y. C. Yao, C. Zhan and Z. Zhang, Chem. Rev., 2023, 123, 2737–2831 CrossRef CAS PubMed.
- R. Brilmayer, M. Brodrecht, C. Kaiser, H. Breitzke, B. Kumari, J. Wachtveitl, G. Buntkowsky and A. Andrieu-Brunsen, ChemNanoMat, 2020, 6, 1843–1853 CrossRef CAS.
- S. Schneider, M. Brodrecht, H. Breitzke, T. Wissel, G. Buntkowsky, H. S. Varol, R. Brilmayer, A. Andrieu-Brunsen and M. Vogel, J. Chem. Phys., 2022, 157, 034503 CrossRef CAS PubMed.
- M. Stanzel, R. Brilmayer, M. Langhans, T. Meckel and A. Andrieu-Brunsen, Microporous Mesoporous Mater., 2019, 282, 29–37 CrossRef CAS.
- Y. Li, S. Bi, F. Liu, S. Wu, J. Hu, L. Wang, H. Liu and Y. Hu, J. Mater. Chem. C, 2015, 3, 6876–6881 RSC.
- S. Zhang, M. Cheng, M. K. Dhinakaran, Y. Sun and H. Li, ACS Nano, 2021, 15, 13148–13154 CrossRef CAS PubMed.
- G. Pérez-Mitta, W. A. Marmisolle, L. Burr, E. Toimil-Molares, C. Trautmann, O. Azzaroni, G. Pérez-Mitta, W. A. Marmisollé, O. Azzaroni, L. Burr, M. E. Toimil-Molares and C. Trautmann, Small, 2018, 14, 1703144 CrossRef PubMed.
- G. Pérez-Mitta, J. S. Tuninetti, W. Knoll, C. Trautmann, M. E. Toimil-Molares and O. Azzaroni, J. Am. Chem. Soc., 2015, 137, 6011–6017 CrossRef PubMed.
- S. L. Soto Espinoza, C. R. Arbeitman, M. C. Clochard and M. Grasselli, Radiat. Phys. Chem., 2014, 94, 72–75 CrossRef CAS.
- M. I. Khan, K. Mukherjee, R. Shoukat and H. Dong, Microsyst. Technol., 2017, 23, 4391–4404 CrossRef CAS.
- C. Li, L. Duan and X. Cheng, Carbohydr. Polym., 2022, 288, 119417 CrossRef CAS PubMed.
- L. Zhu, Q. Song and H. Ma, J. Macromol. Sci., Part A: Pure Appl. Chem., 2023, 60, 18–28 CrossRef CAS.
- S. K. Dalvie and R. E. Baltus, J. Membr. Sci., 1992, 71, 241–255 CrossRef.
- K. Froehlich, M. Ali, P. Ramirez, J. Cervera, V. García-Morales, M. Erdmann and W. Ensinger, Electrochim. Acta, 2021, 367, 137263 CrossRef CAS.
- I. Duznovic, M. Diefenbach, M. Ali, T. Stein, M. Biesalski and W. Ensinger, J. Membr. Sci., 2019, 591, 117344 CrossRef CAS.
- Q. Yang, X. Lin and B. Su, Anal. Chem., 2016, 88, 10252–10258 CrossRef CAS PubMed.
- Q. H. Nguyen, M. Ali, S. Nasir and W. Ensinger, Nanotechnology, 2015, 26, 485502 CrossRef PubMed.
- P. Wang, M. Wang, F. Liu, S. Ding, X. Wang, G. Du, J. Liu, P. Apel, P. Kluth, C. Trautmann and Y. Wang, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- K. Froehlich, M. Ali, P. Ramirez, J. Cervera, V. García-Morales, M. Erdmann and W. Ensinger, Electrochim. Acta, 2021, 367, 137263 CrossRef CAS.
- Q. Yang, X. Lin and B. Su, Anal. Chem., 2016, 88, 10252–10258 CrossRef CAS PubMed.
- X. Huang, Z. Zhang, X. Y. Kong, Y. Sun, C. Zhu, P. Liu, J. Pang, L. Jiang and L. Wen, Nano Energy, 2019, 59, 354–362 CrossRef CAS.
- Y. Hu, Y. Teng, Y. Sun, P. Liu, L. Fu, L. Yang, X. Y. Kong, Q. Zhao, L. Jiang and L. Wen, Nano Energy, 2022, 97, 107170 CrossRef CAS.
- W. Chen, Q. Wang, J. Chen, Q. Zhang, X. Zhao, Y. Qian, C. Zhu, L. Yang, Y. Zhao, X. Y. Kong, B. Lu, L. Jiang and L. Wen, Nano Lett., 2020, 20, 5705–5713 CrossRef CAS PubMed.
- C. Lin, J. Hao, J. Zhao, Y. Hou, S. Ma and X. Sui, J. Colloid Interface Sci., 2024, 654, 840–847 CrossRef CAS PubMed.
- P. Gao, D. Wang, C. Che, Q. Ma, X. Wu, Y. Chen, H. Xu, X. Li, Y. Lin, D. Ding, X. Lou and F. Xia, Nat. Protoc., 2021, 16, 4201–4226 CrossRef CAS PubMed.
- X. Lin, Q. Yang, L. Ding and B. Su, ACS Nano, 2015, 9, 11266–11277 CrossRef CAS PubMed.
- D. Maltseva, S. Chatterjee, C. C. Yu, M. Brzezinski, Y. Nagata, G. Gonella, A. C. Murthy, J. C. Stachowiak, N. L. Fawzi, S. H. Parekh and M. Bonn, Nat. Chem., 2023, 15, 1146–1154 CrossRef CAS PubMed.
- D. Mozhdehi, K. M. Luginbuhl, J. R. Simon, M. Dzuricky, R. Berger, H. S. Varol, F. C. Huang, K. L. Buehne, N. R. Mayne, I. Weitzhandler, M. Bonn, S. H. Parekh and A. Chilkoti, Nat. Chem., 2018, 10, 496–505 CrossRef CAS PubMed.
- Y. Zhang, X. Chen, C. Wang, G. M. Roozbahani, H. C. Chang and X. Guan, Biosens. Bioelectron., 2020, 165, 112289 CrossRef CAS PubMed.
- F. Zhu, W. Wang, F. Zhang, M. K. Dhinakaran, Y. Wang, R. Wang, J. Cheng, M. E. Toimil-Molares, C. Trautmann and H. Li, Chem. Commun., 2021, 57, 215–218 RSC.
- Y. Wang, S. Zhang, H. Yan, J. Quan, L. Yang, X. Chen, M. E. Toimil-Molares, C. Trautmann and H. Li, Anal. Chem., 2021, 93, 6145–6150 CrossRef CAS PubMed.
- S. Zhang, J. Zhou and H. Li, Angew. Chem., 2022, 134, e202204012 CrossRef.
- S. Zhang, X. Chen, L. Sun and H. Li, ACS Appl. Nano Mater., 2020, 3, 4351–4356 CrossRef CAS.
- Y. Li, L. Tu, X. Ma, H. Chen, Y. Fan, Q. Zhou and Y. Sun, ACS Sens., 2021, 6, 808–814 CrossRef CAS PubMed.
- F. Zhang, Y. Sun, D. Tian and H. Li, Angew. Chem., 2017, 129, 7292–7296 CrossRef.
- X. Huang, Z. Zhang, X. Y. Kong, Y. Sun, C. Zhu, P. Liu, J. Pang, L. Jiang and L. Wen, Nano Energy, 2019, 59, 354–362 CrossRef CAS.
- Y. Sun, Y. Wu, Y. Hu, C. Zhu, H. Guo, X. Y. Kong, E. Luo, L. Jiang and L. Wen, NPG Asia Mater., 2021, 13, 1–10 CrossRef.
- X. Huang, Z. Zhang, X. Y. Kong, Y. Sun, C. Zhu, P. Liu, J. Pang, L. Jiang and L. Wen, Nano Energy, 2019, 59, 354–362 CrossRef CAS.
- W. Chen, Q. Zhang, Y. Qian, W. Xin, D. Hao, X. Zhao, C. Zhu, X. Y. Kong, B. Lu, L. Jiang and L. Wen, ACS Cent. Sci., 2020, 6, 2097–2104 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- X. Lou, Y. Song, R. Liu, Y. Cheng, J. Dai, Q. Chen, P. Gao, Z. Zhao and F. Xia, Small Methods, 2020, 4, 1900432 CrossRef CAS.
- X. Xu, R. Hou, P. Gao, M. Miao, X. Lou, B. Liu and F. Xia, Anal. Chem., 2016, 88, 2386–2391 CrossRef CAS PubMed.
- L. K. Müller, I. Duznovic, D. Tietze, W. Weber, M. Ali, V. Stein, W. Ensinger and A. A. Tietze, Chem. – Eur. J., 2020, 26, 8511–8517 CrossRef PubMed.
- H. Chen, L. Xu, W. Tuo, X. Chen, J. Huang, X. Zhang and Y. Sun, Anal. Chem., 2020, 92, 4131–4136 CrossRef CAS PubMed.
- X. Xu, W. Zhao, P. Gao, H. Li, G. Feng, Z. Zhao and X. Lou, NPG Asia Mater., 2016, 8, e234 CrossRef CAS.
- X. Lou, Y. Song, R. Liu, Y. Cheng, J. Dai, Q. Chen, P. Gao, Z. Zhao and F. Xia, Small Methods, 2020, 4, 1900432 CrossRef CAS.
- S. Wang, L. Ma, Q. Wang, P. Shao, D. Ma, S. Yuan, P. Lei, P. Li, X. Feng and B. Wang, J. Mater. Chem. C, 2018, 6, 5369–5374 RSC.
- H. Wang, X. Ji, Y. Li, Z. Li, G. Tang and F. Huang, J. Mater. Chem. B, 2018, 6, 2728–2733 RSC.
- N. Kundachira Subramani, H. Siddaramaiah and J. Hee, Lee, Polymer-based advanced functional composites for optoelectronic and energy applications, Elsevier, 2021 Search PubMed.
- Y. Tang, H. Huang, W. Xue, Y. Chang, Y. Li, X. Guo and C. Zhong, Chem. Eng. J., 2020, 384, 123382 CrossRef CAS.
- V. A. Dini, A. Gradone, M. Villa, M. Gingras, M. L. Focarete, P. Ceroni, C. Gualandi and G. Bergamini, Chem. Commun., 2023, 59, 1465–1468 RSC.
- V. A. Dini, D. Genovese, C. Micheletti, N. Zaccheroni, A. Pucci and C. Gualandi, Aggregate, 2023, 4, e373 CrossRef CAS.
- Z. Wang, J. Nie, W. Qin, Q. Hu and B. Z. Tang, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- Z. Li, X. Ji, H. Xie and B. Z. Tang, Adv. Mater., 2021, 33, 2100021 CrossRef CAS PubMed.
- W. Lu, S. Wei, H. Shi, X. Le, G. Yin and T. Chen, Aggregate, 2021, 2, e37 CrossRef CAS.
- J. Tavakoli, E. Laisak, M. Gao and Y. Tang, Mater. Sci. Eng., C, 2019, 104, 109951 CrossRef CAS PubMed.
- L. Chen, H. Jiang, M. Zheng, Z. Li, N. Li, S. Zhao and X. Liu, J. Hazard. Mater., 2022, 434, 128868 CrossRef CAS PubMed.
- Y. Wang, J. Zhao, Z. Dong, C. Wang, H. Meng, Y. Li and H. Jin, Mater. Today Chem., 2021, 21, 100537 CrossRef CAS.
- S. S. Liow, Q. Dou, D. Kai, Z. Li, S. Sugiarto, C. Y. Y. Yu, R. T. K. Kwok, X. Chen, Y. L. Wu, S. T. Ong, A. Kizhakeyil, N. K. Verma, B. Z. Tang and X. J. Loh, Small, 2017, 13, 1603404 CrossRef PubMed.
- Z. S. Siwy, M. L. Bruening and S. Howorka, Chem. Soc. Rev., 2023, 52, 1983–1994 RSC.
- B. N. Anderson, O. N. Assad, T. Gilboa, A. H. Squires, D. Bar and A. Meller, ACS Nano, 2014, 8, 11836–11845 CrossRef CAS PubMed.
- A. Ivankin, R. Y. Henley, J. Larkin, S. Carson, M. L. Toscano and M. Wanunu, ACS Nano, 2014, 8, 10774–10781 CrossRef CAS PubMed.
- J. P. Fried, J. L. Swett, B. Paulose Nadappuram, A. Fedosyuk, P. Miguel Sousa, D. P. Briggs, A. P. Ivanov, J. B. Edel, J. A. Mol, J. R. Yates, J. P. Fried, J. L. Swett, B. P. Nadappuram, A. Fedosyuk, A. P. Ivanov, J. B. Edel, P. M. Sousa, J. R. Yates, D. P. Briggs and J. A. Mol, Small, 2021, 17, 2102543 CrossRef CAS PubMed.
- A. Zrehen, T. Gilboa and A. Meller, Nanoscale, 2017, 9, 16437–16445 RSC.
- S. I. Kulik, I. Y. Eremchev, P. Y. Apel, D. L. Zagorski and A. V. Naumova, J. Appl. Spectrosc., 2018, 85, 916–922 CrossRef CAS.
- H. Gao, R. Sun, L. He, Z. J. Qian, C. Zhou, P. Hong, S. Sun, R. Mo and C. Li, ACS Appl. Mater. Interfaces, 2020, 12, 4849–4858 CrossRef CAS PubMed.
- L. Chen, L. He, F. Ma, W. Liu, Y. Wang, M. A. Silver, L. Chen, L. Zhu, D. Gui, J. Diwu, Z. Chai and S. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 15364–15368 CrossRef CAS PubMed.
- A. Walcarius, E. Sibottier, M. Etienne and J. Ghanbaja, Nat. Mater., 2007, 6, 602–608 CrossRef CAS PubMed.
- Z. Teng, G. Zheng, Y. Dou, W. Li, C. Y. Mou, X. Zhang, A. M. Asiri and D. Zhao, Angew. Chem., Int. Ed., 2012, 51, 2173–2177 CrossRef CAS PubMed.
- T. D. Nguyen, W. Y. Hamad and M. J. Maclachlan, Adv. Funct. Mater., 2014, 24, 777–783 CrossRef CAS.
- K. Kikuchi, S. Shigeta and T. Ishikawa, PLoS One, 2019, 14, e0214504 CrossRef CAS PubMed.
- J. C. Thiele, D. A. Helmerich, N. Oleksiievets, R. Tsukanov, E. Butkevich, M. Sauer, O. Nevskyi and J. Enderlein, ACS Nano, 2020, 14, 14190–14200 CrossRef CAS PubMed.
- H. Bodugoz-Senturk, C. E. Macias, J. H. Kung and O. K. Muratoglu, Biomaterials, 2009, 30, 589–596 CrossRef CAS PubMed.
- H. Bodugoz-Senturk, J. Choi, E. Oral, J. H. Kung, C. E. Macias, G. Braithwaite and O. K. Muratoglu, Biomaterials, 2008, 29, 141–149 CrossRef CAS PubMed.
- F. Fergg, F. J. Keil and H. Quader, Colloid Polym. Sci., 2001, 279, 61–67 CrossRef CAS.
- C. S. Wang, G. Natale, N. Virgilio and M. C. Heuzey, Food Hydrocolloids, 2016, 60, 374–383 CrossRef CAS.
- A. B. Norton, R. D. Hancocks, F. Spyropoulos and L. M. Grover, Food Hydrocolloids, 2016, 53, 93–97 CrossRef CAS.
- F. Fergg, F. J. Keil and H. Quader, Colloid Polym. Sci., 2001, 279, 61–67 CrossRef CAS.
- N. Sereni, A. Enache, G. Sudre, A. Montembault, C. Rochas, P. Durand, M. H. Perrard, G. Bozga, J. P. Puaux, T. Delair and L. David, Langmuir, 2017, 33, 12697–12707 CrossRef CAS PubMed.
- R. Le Dizès Castell, E. Mirzahossein, M. Grzelka, S. Jabbari-Farouji, D. Bonn and N. Shahidzadeh, J. Phys. Chem. Lett., 2024, 15, 628–635 CrossRef PubMed.
- A. Saini and L. Kisley, J. Appl. Phys., 2019, 126, 81101 CrossRef.
- J. Zhong, T. Zhao and M. Liu, NPG Asia Mater., 2022, 14, 1–23 CrossRef.
- H. S. Varol and S. Seeger, Langmuir, 2022, 38, 231–243 CrossRef CAS PubMed.
- P. Papadopoulos, X. Deng, L. Mammen, D.-M. Drotlef, G. Battagliarin, C. Li, K. Müllen, K. Landfester, A. Del Campo, H.-J. Rgen Butt and D. Vollmer, Langmuir, 2020, 28, 8392–8398 CrossRef PubMed.
- I. Duznovic, A. Gräwe, W. Weber, L. K. Müller, M. Ali, W. Ensinger, A. Tietze and V. Stein, Small, 2021, 17, 2101066 CrossRef CAS PubMed.
- D. Zhang, C. Wang, C. Wu and X. Zhang, Nanomaterials, 2022, 12, 1468 CrossRef CAS PubMed.
- Y. Bai, M. Xiang, Z. Zhou, Z. Mao and F. Zhang, Sens. Actuators, B, 2024, 402, 135100 CrossRef CAS.
- X. Zhang, Y. Dai, J. Sun, J. Shen, M. Lin and F. Xia, Anal. Chem., 2024, 96, 2277–2285 CrossRef CAS PubMed.
- Y. Dai, Y. Zhang, Q. Ma, M. Lin, X. Zhang and F. Xia, Anal. Chem., 2022, 94, 17343–17348 CrossRef CAS PubMed.
- L. Dai, W. Q. Zhang, D. Ding, C. Luo, L. Jiang, Y. Huang and F. Xia, ACS Nano, 2024, 18, 7677–7687 CrossRef CAS PubMed.
- Y. Qiao, J. J. Hu, Y. Hu, C. Duan, W. Jiang, Q. Ma, Y. Hong, W. H. Huang, F. Xia and X. Lou, Angew. Chem., Int. Ed., 2023, 62, e202309671 CrossRef CAS PubMed.
- S. Jun, J. Lee and E. Jang, ACS Nano, 2013, 7, 1472–1477 CrossRef CAS PubMed.
- S. Zhou, L. Xie, L. Zhang, L. Wen, J. Tang, J. Zeng, T. Liu, D. Peng, M. Yan, B. Qiu, Q. Liang, K. Liang, L. Jiang and B. Kong, ACS Appl. Mater. Interfaces, 2021, 13, 8782–8793 CrossRef CAS PubMed.
- L. Chen, Y. Zhang, R. Li, H. Zhang, M. Zhang and H. Zhang, Water Res., 2020, 185, 116240 CrossRef CAS PubMed.
- K. A. Leonidakis, P. Bhattacharya, J. Patterson, B. E. Vos, G. H. Koenderink, J. Vermant, D. Lambrechts, M. Roeffaers and H. Van Oosterwyck, Acta Biomater., 2017, 47, 25–39 CrossRef CAS PubMed.
- A. Matlschweiger, P. Fuks, G. Carta and R. Hahn, J. Chromatogr. A, 2019, 1585, 121–130 CrossRef CAS PubMed.
- V. Caponetti, A. Mavridi-printezi, M. Cingolani, E. Rampazzo, D. Genovese, L. Prodi, D. Fabbri and M. Montalti, Polymers, 2021, 13, 1588 CrossRef CAS PubMed.
- X. Liu, D. Lin, W. Becker, J. Niu, B. Yu, L. Liu and J. Qu, J. Innovative Opt. Health Sci., 2019, 12, 12 Search PubMed.
- K. Tao, B. Xue, S. Frere, I. Slutsky, Y. Cao, W. Wang and E. Gazit, Chem. Mater., 2017, 29, 4454–4460 CrossRef CAS PubMed.
- E. Özelci, B. Rühle, F. Weigert, B. Lubotzky, G. Kewes, U. Resch-Genger and O. Benson, J. Phys. Chem. C, 2019, 123, 20468–20475 CrossRef.
- S. M. Lu, Y. Y. Peng, Y. L. Ying and Y. T. Long, Anal. Chem., 2020, 92, 5621–5644 CrossRef CAS PubMed.
- R. Hao, Z. Peng and B. Zhang, ACS Omega, 2020, 5, 89–97 CrossRef CAS PubMed.
- G. V. Soni, A. Singer, Z. Yu, Y. Sun, B. McNally and A. Meller, Rev. Sci. Instrum., 2010, 81, 014301 CrossRef PubMed.
- L. P. Zaino, D. A. Grismer, D. Han, G. M. Crouch and P. W. Bohn, Faraday Discuss., 2015, 184, 101–115 RSC.
- J. Zhao, L. P. Zaino and P. W. Bohn, Faraday Discuss., 2013, 164, 57–69 RSC.
- J. Lu, Y. Fan, M. D. Howard, J. C. Vaughan and B. Zhang, J. Am. Chem. Soc., 2017, 139, 2964–2971 CrossRef CAS PubMed.
- J. J. Gooding and K. Gaus, Angew. Chem., Int. Ed., 2016, 55, 11354–11366 CrossRef CAS PubMed.
- C. Wang, Q. Fu, X. Wang, D. Kong, Q. Sheng, Y. Wang, Q. Chen and J. Xue, Anal. Chem., 2015, 87, 8227–8233 CrossRef CAS PubMed.
- A. Egatz-Gomez, C. Wang, F. Klacsmann, Z. Pan, S. Marczak, Y. Wang, G. Sun, S. Senapati and H. C. Chang, Biomicrofluidics, 2016, 10, 032902 CrossRef PubMed.
- Z. Siwy, E. Heins, C. C. Harrell, P. Kohli and C. R. Martin, J. Am. Chem. Soc., 2004, 126, 10850–10851 CrossRef CAS PubMed.
- D. Kaya and K. Kececi, J. Electrochem. Soc., 2020, 167, 037543 CrossRef CAS.
- H. Rashidi, K. J. Howard, T. Ito and D. A. Higgins, J. Phys. Chem. C, 2023, 127, 411–420 CrossRef CAS.
- C. de Blois, M. Engel, M. A. Rejou, B. Molcrette, A. Favier and F. Montel, Nanoscale, 2023, 16, 138–151 RSC.
- R. Kumarasinghe, T. Ito and D. A. Higgins, Anal. Chem., 2020, 92, 1416–1423 CrossRef CAS PubMed.
- D. A. Higgins, S. C. Park, K. H. Tran-Ba and T. Ito, Annu. Rev. Anal. Chem., 2015, 8, 193–216 CrossRef CAS PubMed.
- T. Ito and D. A. Higgins, Chem. Rec., 2021, 21, 1417–1429 CrossRef CAS PubMed.
- T. Ito, J. Phys. Chem. B, 2023, 127, 5733–5741 CrossRef CAS PubMed.
- J. Cho and Y. Ishida, Adv. Mater., 2017, 29, 1605974 CrossRef PubMed.
- R. Pramanik, T. Ito and D. A. Higgins, J. Phys. Chem. C, 2013, 117, 15438–15446 CrossRef CAS.
- D. A. Higgins, K. H. Tran-Ba and T. Ito, J. Phys. Chem. Lett., 2013, 4, 3095–3103 CrossRef CAS.
- S. C. Park, T. Ito and D. A. Higgins, J. Phys. Chem. C, 2015, 119, 26101–26110 CrossRef CAS.
- A. Y. Jee, N. Yanai and S. Granick, J. Phys. Chem. Lett., 2018, 9, 6399–6403 CrossRef CAS PubMed.
- X. Zambrana-Puyalto, P. Ponzellini, N. MacCaferri and D. Garoli, Phys. Rev. Appl., 2020, 14, 054065 CrossRef CAS.
- L. Xiang, K. Chen, R. Yan, W. Li and K. Xu, Nat. Methods, 2020, 17, 524–530 CrossRef CAS PubMed.
- H. H. Park, A. A. Choi and K. Xu, J. Phys. Chem. B, 2023, 127, 3333–3339 CrossRef CAS PubMed.
- D. Al Sulaiman, P. Cadinu, A. P. Ivanov, J. B. Edel and S. Ladame, Nano Lett., 2018, 18, 6084–6093 CrossRef CAS PubMed.
- H. H. Park, A. A. Choi and K. Xu, J. Phys. Chem. B, 2023, 127, 3333–3339 CrossRef CAS PubMed.
- R. Pardehkhorram and A. Andrieu-Brunsen, Chem. Commun., 2022, 58, 5188–5204 RSC.
- J. Yu, T. Marchesi D’alvise, I. Harley, A. Krysztofik, I. Lieberwirth, P. Pula, P. W. Majewski, B. Graczykowski, J. Hunger, K. Landfester, L. Kuan, R. Shi, C. V. Synatschke, T. Weil, J. Yu, T. Marchesi D’alvise, S. L. Kuan, R. Shi, C. V. Synatschke, T. Weil, I. Harley, I. Lieberwirth and K. Landfester, Adv. Mater., 2024, 2401137 CrossRef CAS PubMed.
- H. S. Varol, T. Herberger, M. Kirsch, J. Mikolei, L. Veith, V. Kannan-Sampathkumar, R. D. Brand, C. V. Synatschke, T. Weil and A. Andrieu-Brunsen, Chem. Mater., 2023, 35, 9192–9207 CrossRef CAS PubMed.
- M. E. Lynge, R. Van Der Westen, A. Postma and B. Städler, Nanoscale, 2011, 3, 4916–4928 RSC.
- B. Stöckle, D. Y. W. Ng, C. Meier, T. Paust, F. Bischoff, T. Diemant, R. J. Behm, K. E. Gottschalk, U. Ziener and T. Weil, Macromol. Symp., 2014, 346, 73–81 CrossRef.
- V. Ball, Nanotechnol. Rev., 2024, 13, 20230216 CrossRef CAS.
- S. Xu and Q. Zhang, Mater. Today Energy, 2021, 20 Search PubMed.
- D. Chen, C. Liu, J. Tang, L. Luo and G. Yu, Polym. Chem., 2019, 10, 1168–1181 RSC.
- Y. Li, S. Huang, S. Peng, H. Jia, J. Pang, B. Ibarlucea, C. Hou, Y. Cao, W. Zhou, H. Liu, G. Cuniberti, Y. Li, J. Pang, C. Hou, W. Zhou, H. Liu, S. Huang, G. Cuniberti, S. Peng, H. Jia and Y. Cao, Small, 2023, 19, 2206126 CrossRef CAS PubMed.
- Y. Alqaheem and A. A. Alomair, Membranes, 2020, 10, 33 CrossRef CAS PubMed.
- G. Valenti, E. Rampazzo, S. Kesarkar, D. Genovese, A. Fiorani, A. Zanut, F. Palomba, M. Marcaccio, F. Paolucci and L. Prodi, Coord. Chem. Rev., 2018, 367, 65–81 CrossRef CAS.
- P. Nikolaou, G. Valenti and F. Paolucci, Electrochim. Acta, 2021, 388, 138586 CrossRef CAS.
- H. S. Varol, A. Srivastava, S. Kumar, M. Bonn, F. Meng and S. H. Parekh, Polymer, 2020, 200, 122529 CrossRef CAS.
- H. S. Varol, F. Meng, B. Hosseinkhani, C. Malm, D. Bonn, M. Bonn, A. Zaccone and S. H. Parekh, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3170–E3177 CrossRef CAS PubMed.
- H. S. Varol, M. A. Sánchez, H. Lu, J. E. Baio, C. Malm, N. Encinas, M. R. B. Mermet-Guyennet, N. Martzel, D. Bonn, M. Bonn, T. Weidner, E. H. G. Backus and S. H. Parekh, Macromolecules, 2015, 48, 7929–7937 CrossRef CAS.
- M. Vendrell, D. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS PubMed.
- S. Singha, Y. W. Jun, S. Sarkar and K. H. Ahn, Acc. Chem. Res., 2019, 52, 2571–2581 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |