
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocalized cancer photodynamic therapy approach based on core–shell electrospun nanofibers†
Sofia M.
Costa
 *a,
Leandro M. O.
Lourenço
b,
Ricardo C.
Calhelha
cd,
Isabel
Calejo
e,
Cristina C.
Barrias
efg,
Raul
Fangueiro
a and
Diana P.
Ferreira
*a
*a,
Leandro M. O.
Lourenço
b,
Ricardo C.
Calhelha
cd,
Isabel
Calejo
e,
Cristina C.
Barrias
efg,
Raul
Fangueiro
a and
Diana P.
Ferreira
*a
aCentre for Textile Science and Technology (2C2T), University of Minho, 4800-058 Guimarães, Portugal. E-mail: sofiamcosta@det.uminho.pt; diana.ferreira@det.uminho.pt
bLAQV-REQUIMTE, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
cCentro de Investigação de Montanha (CIMO), Instituto Politécnico de Bragança, Campus de Santa Apolónia, 5300-253 Bragança, Portugal
dLaboratório Associado para a Sustentabilidade e Tecnologia em Regiões de Montanha (SusTEC), Instituto Politécnico de Bragança, Campus de Santa Apolónia, 5300-253 Bragança, Portugal
ei3S – Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Portugal
fINEB – Instituto de Engenharia Biomédica, Universidade do Porto, Portugal
gICBAS – Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Portugal
First published on 18th July 2024
Abstract
Photodynamic therapy (PDT) has been considered a promising treatment for several types of cancer, including cervical cancer. Localized drug delivery systems (DDSs) based on nanofibers produced by electrospinning have emerged as a powerful platform to carry and deliver photosensitizers (PSs) onto or adjacent to the tumor site, thereby promoting higher therapeutic efficacy and reducing the side effects to healthy tissues associated with systemic administration. In this work, core–shell electrospun nanofibers were produced using biodegradable polymers, such as poly(vinyl alcohol) (PVA) and gelatin (Gel), to act as a localized DDS for the treatment of cervical cancer using PDT. The synthesized porphyrin (Por) was able to generate singlet oxygen (ΦΔ = 0.62) and displayed higher phototoxicity against tumor cells compared with healthy cells. The developed PVA–Gel membranes were fully characterized, revealing defect-free nanofibers with a core–shell structure. Different Por concentrations were added to the fibers’ core, and their presence and uniform distribution within the nanofibers were confirmed. The Por release profile from nanofibers showed an initial fast release stage, followed by continuous release for at least 9 days. The PVA–Gel + Por core–shell nanofibers exhibited a higher inhibition of cancer cell proliferation under light irradiation when compared to dark and a higher phototoxic effect against tumor cells compared with non-tumor cells. Overall, this study demonstrates the great potential of core–shell nanofibers to be used as localized DDSs of PSs for the treatment of cervical cancer.
1. Introduction
Currently, cervical cancer is ranked the fourth most common cancer among women, with 604![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 estimated new cases and 342000 deaths worldwide in 2020, according to the World Health Organization (WHO).1 The link between human papillomavirus (HPV) and cervical cancer is well established, although genetic and epigenetic alterations are also essential for the pathogenesis of cervical cancer. HPV strains are categorized based on their carcinogenicity as high-risk (HR), intermediate-risk (IR), and low-risk (LR), and the cases of persistent infection with HR-HPV types are responsible for up to 99.7% of cervical cancer cases. The treatment approach will depend on the stage and extent of cervical cancer progression, which may include surgery, radiotherapy, chemotherapy, or a combination of them.2,3 However, these conventional treatments have been associated with increased side effects and limited efficacy, which can impair the patient's quality of life.4
000 estimated new cases and 342000 deaths worldwide in 2020, according to the World Health Organization (WHO).1 The link between human papillomavirus (HPV) and cervical cancer is well established, although genetic and epigenetic alterations are also essential for the pathogenesis of cervical cancer. HPV strains are categorized based on their carcinogenicity as high-risk (HR), intermediate-risk (IR), and low-risk (LR), and the cases of persistent infection with HR-HPV types are responsible for up to 99.7% of cervical cancer cases. The treatment approach will depend on the stage and extent of cervical cancer progression, which may include surgery, radiotherapy, chemotherapy, or a combination of them.2,3 However, these conventional treatments have been associated with increased side effects and limited efficacy, which can impair the patient's quality of life.4
In an attempt to minimize the side effects and prevent cancer recurrence, alternative treatments have been explored, namely photodynamic therapy (PDT), which is characterized by being minimally invasive and localized.5,6 PDT involves the use of three components: a photoactive molecule, called the photosensitizer (PS), an external light source, and molecular oxygen (O2). After administration of the PSs and their absorption by cancer cells, the target area will be irradiated by light with a specific wavelength, and the PSs will be activated, resulting in the generation of reactive oxygen species (ROS), namely singlet oxygen (1O2). This will lead to the oxidation of different biomolecules and cause irreversible destruction of target tissues via cell death, vascular damage, and inflammation.7–10 Among all available PSs, porphyrin (Por) and its derivatives remain the most extensively used.11 These compounds have high purity, long triplet state lifetime, high 1O2 quantum yield (ΦΔ), minimal dark toxicity, and in some cases rapid clearance from the patient's body. Despite the great advantages of these PSs, they present some drawbacks when administered systemically that can affect their PDT application, such as high dose requirements to achieve a consistent uptake in tumors, which can lead to prolonged photosensitivity; low selectivity; low water solubility, which can result in aggregation under physiological conditions; difficulty in reaching certain depths in tissues; and the possibility of compound loss until targeting the localized tumor.12–15
Thus, the incorporation of PSs into localized drug delivery systems (DDSs) is a suitable approach to decrease the systemic toxicities of drugs.16 Localized DDSs implanted at the tumor site or close to it enable the direct delivery of PSs to the target area, thereby improving the treatment efficacy while decreasing the side effects on the surrounding healthy cells.17 Other advantages include the possibility of achieving a controlled and sustained PS release over time; PS protection from degradation before achieving the target; loading and release of water-insoluble molecules; prevention of PS aggregation; promotion of the molecules' rigidification (by diminishing the photoisomerization process), and other deactivation mechanisms; and one-time administration of the drug.12,13
Electrospinning has acquired increasing attention for the development of nanofibers to act as localized DDSs. Its simplicity of use, accuracy, cost-effectiveness, and scalability along with the ability to manipulate the nanofiber's composition and structure emphasize the versatility of this technique to adapt and respond to different requirements.18,19 Electrospun nanofibers stand out as fibrous nanoplatforms for the delivery of drugs in localized cancer treatment, due to their high surface-area-to-volume ratio, which is associated with high drug loading, more efficient drug encapsulation, wide selection of matrix materials and therapeutics, high porosity with interconnectivity, various possibilities for surface functionalization, which allows target-specific drug delivery, incorporation of several drugs simultaneously and control of the drug release profile. Moreover, their physical structure similar to the extracellular matrix (ECM) might be beneficial for cell–material interaction.16,18,20,21 Their small size, ultrathin, and flexible properties allow them to extravagate into confined space through small lesion cavities.22
Hence, incorporating PSs onto electrospun nanofibers is a suitable strategy to potentiate the effect of these molecules on tumor cells for a longer time, maintaining therapeutically effective drug concentrations in the target tissue.21,23 Their use as localized DDSs for PDT-assisted cervical cancer treatment is very promising due to the easy-accessible cervix through the vaginal canal, which allows the non-invasive implantation of DDSs directly or adjacent to the tumor site as well as their irradiation.3,24,25 Additionally, phototherapeutic nanofibers can function as theranostic platforms, enabling simultaneous diagnosis and therapy.13,26
Core–shell nanofibers can provide a more sustained and slower release than single ones, since the shell operates as a protection layer, hindering the diffusion of the drug. This will ensure an effective drug concentration on the target tissue for a long period while eliminating the need for repeat drug administration.27–29 Hence, the core can act as the carrier of bioactive molecules, and the shell as the barrier to prevent their degradation and premature release. Indeed, employing a co-axial needle also provides the possibility to use simultaneously two different polymeric solutions, allowing the combination of different polymers to achieve the desired application.28,30
The use of biodegradable polymers to produce these nanofibers is essential for effective drug release and to avoid surgical removal of the DDS at the end of its therapeutic lifetime.31 Poly(vinyl alcohol) (PVA), a hydrolyzed form of poly(vinyl acetate), is a synthetic biodegradable polymer, which is semi-crystalline, hydrophilic, biocompatible, presents good mechanical properties, and it can be electrospun into nanofibers in aqueous solution. DDSs composed only of synthetic polymers frequently exhibit appropriate mechanical properties but display minimal bioactivity due to their insufficient cellular recognition sites. Therefore, the combination with natural polymers, such as gelatin (Gel), is a promising approach to address this limitation.32,33 Gel is a natural polymer, formed by partial hydrolysis of collagen, which is available in animal bones, tendons, and skin. This protein is characterized by its biodegradability, biocompatibility, low immunogenicity, and cost-effectiveness. Moreover, it presents peptide domains for the recognition of integrin receptors in the cells. Despite presenting high biocompatibility it displays poor mechanical properties. Thus, the combination of the mechanical strength of synthetic polymers with the biological activity of natural ones may be a promising strategy for the development of DDS.33–37
Although the benefits of nanofibers as localized DDSs, and PDT as a cancer therapy are well established, the combination of both is still poorly explored, despite having enormous potential to improve the efficacy of cancer treatments. Some studies reported the use of PDT to treat cervical cancer,5,25,38–40 while others showed the potential of nanofibers as a DDS to treat it.22,27,41,42 Nevertheless, to the authors’ best knowledge, the combination of porphyrin-loaded electrospun core–shell nanofibers for the treatment of cervical cancer using PDT has not yet been reported. Thus, this study aimed to develop core–shell electrospun nanofibers loaded with a PS drug, specifically a porphyrin of TPPF16[S-CH2-COOH]4,43 to act as a localized DDS for PDT. In this case, PVA was used in the core, while Gel was used in the shell. Firstly, the singlet oxygen generation capacity and the photodynamic action of the synthesized Por were assessed and then incorporated into electrospun nanofibers. The viscosity and conductivity values of the polymeric solutions to be used in electrospinning were evaluated. All the developed membranes were characterized by Field Emission Scanning Electron Microscopy (FESEM), Transmission Electron Microscopy (TEM), Thermogravimetric Analysis (TGA), Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR-FTIR), Ground State Diffuse Reflectance (GSDR), and Confocal Laser Scanning Microscopy (CLSM). The drug Loading Capacity (LC) and Encapsulation Efficiency (EE) were also determined. Moreover, the drug release profile was evaluated as well as the cytotoxicity of the produced membranes in the dark and under irradiation against tumor and non-tumor cell lines.
2. Materials and methods
2.1. Synthesis of 5,10,15,20-tetrakis(4-carboxymethylthio-2,3,5,6-tetrafluorophenyl)porphyrin (TPPF16[S-CH2-COOH]4)
The synthesis of TPPF16[S-CH2-COOH]4 was performed from the already published experimental procedure43via nucleophilic substitution of the commercially available 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin (TPPF20) and thioglycolic acid under basic conditions in N,N-dimethylformamide (DMF). The reaction mixture was stirred for just 1 h at 50 °C, followed by their neutralization with citric acid, and their precipitation in chloroform. The obtained solid was filtrated and recrystallized using a mixture of methanol/chloroform. Then, the obtained purple compound was filtrated and dried in the vacuum system, being posteriorly identified as the desired product of TPPF16[S-CH2-COOH]4. The structural characterization of TPPF16[S-CH2-COOH]4 is provided in the ESI† (Fig. S1–S4).2.2. Singlet oxygen generation
The capability to generate singlet oxygen of the TPPF16[S-CH2-COOH]4 was evaluated by monitoring the photooxidation of 9,10-dimethylanthracene (9,10-DMA), a singlet oxygen quencher (Fig. S5, ESI†). DMF solutions of the PS, TPP (Abs420 ∼ 0.3)44 were aerobic irradiated in quartz cuvettes with monochromatic light (λ = 420 nm) in the presence of 9,10-DMA (∼135 μM). TPP (ΦΔ = 0.65 in DMF)44 was used as a reference. The kinetics of 9,10-DMA photooxidation photosensitized by TPPF16[S-CH2-COOH]4 and TPP were examined by following the depletion of their absorbance at 378 nm at each 30 s until 300 s, and the results were registered in a first-order plot. The kinetics of 9,10-DMA photooxidation in the absence of any compound in DMF was also assessed and no significant photodegradation was observed under the same irradiation conditions. The results are expressed as mean and standard deviation obtained from three independent experiments. Furthermore, the singlet oxygen quantum yields (ΦΔ) were determined using the equation indicated below: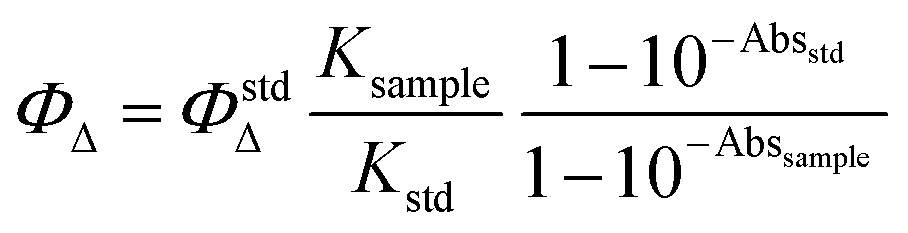 | (1) |
 is the singlet oxygen quantum yield of TPP, Ksample and Kstd are the photodecay constants of 9,10-DMA in the presence of the sample and reference, respectively; Abssample and Absstd are the absorbance of the sample and reference at ∼420 nm.
is the singlet oxygen quantum yield of TPP, Ksample and Kstd are the photodecay constants of 9,10-DMA in the presence of the sample and reference, respectively; Abssample and Absstd are the absorbance of the sample and reference at ∼420 nm.
2.3. In vitro photodynamic activity of the synthesized porphyrin
The human tumor cell line HeLa (cervical carcinoma) was purchased from DSMZ (Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen and Zellkulturen GmbH). The Vero (African green monkey kidney) cell line was purchased from the European Collection of Authenticated Cell Cultures (ECACC). The cells were routinely maintained as adherent cell cultures in RPMI-1640 medium containing 10% heat-inactivated FBS, 2 mM glutamine, and 1% antibiotics at 37 °C, in a humidified air incubator containing 5% CO2. It was plated at an appropriate density (1.0 × 104 cells per well) in 96-well plates and allowed to attach for 24 h. Cells were then treated for 24 h with various concentrations of the compounds (0.06–10 μM). Following this incubation period, the tested cells under irradiation were irradiated for 30 min. For light irradiation, a LED lamp (300–500 nm) was used with a fluence rate of 23–24 μW cm−2 measured with an ILT 2100 radiometer. The medium was changed and incubated again for 24 h. After this time, the adherent cells were fixed by adding cold 10% trichloroacetic acid (TCA; 100 μL) and incubated for 60 min at 4 °C. Plates were then washed with deionized water and subsequently dried; sulforhodamine B (SRB) solution (0.1% in 1% acetic acid, 100 μL) was then added to each plate well and incubated for 30 min at room temperature. Unbound SRB was removed by washing with 1% acetic acid. Plates were air-dried, the bound SRB was solubilized with 10 mM Tris (200 μL), and the absorbance was measured at 540 nm in an ELX800 Microplate Reader (Bio-Tek Instruments, Inc.; Winooski, USA). The results were expressed in compound concentration values that inhibited 50% of the net cell growth (GI50).2.4. Development of core–shell electrospun nanofibers
000, 88% hydrolyzed) and gelatin (Gel, from porcine skin Type A) were purchased from Polysciences Europe GmbH (Germany), and Merck-Sigma-Aldrich, respectively. Acetic acid glacial 99–100% a.r. was obtained from Chem-Lab NV.
 | ||
| Fig. 1 Co-axial accessory used to produce core–shell nanofibers. | ||
| Parameters | Values | |
|---|---|---|
| Feed rate | Core | 0.8 mL h−1 |
| Shell | 0.2 mL h−1 | |
| Applied voltage | 29 kV | |
| Distance between needle and collector | 250 mm | |
| Needle diameter | 27 G | |
| Deposition time | 2 h | |
| Humidity | 60–65% | |
| Temperature | 18–20 °C | |
2.5. Characterization of the polymeric solutions and electrospun nanofibers
 | (2) |
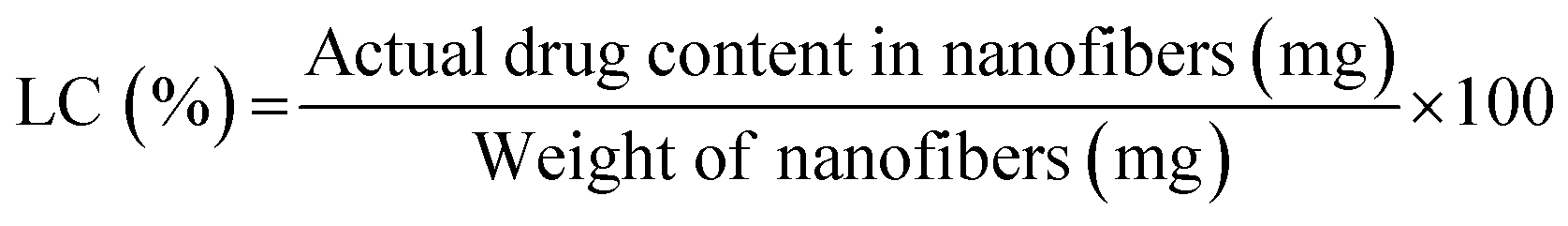

2.6. Drug release profile
The release profile of Por from core–shell nanofibers was monitored over 216 h by UV-Vis spectroscopy using a Shimadzu UV-1800 spectrophotometer. A section of each membrane (1 cm × 1 cm) was immersed in 4.5 mL of phosphate buffer saline (PBS) solution with a pH of 4.5, to mimic vaginal pH and tumor site.22,27,41 The samples were incubated in an orbital shaker at 37 °C, 120 rpm. At different time points (0, 1, 2, 4, 6, 8, 24, 48, 72, 96, 168, and 216 h), an aliquot was removed and evaluated by UV-Vis spectroscopy in the range of 300 to 650 nm. Three replicates of each sample were evaluated. The concentration of the Por released was calculated using a calibration curve, which was determined from absorption measurements of known Por concentrations in PBS pH 4.5.2.7. Evaluation of cytotoxicity of electrospun nanofibers in tumor and non-tumor cell lines
To evaluate the cytotoxicity of each electrospun nanofiber, a section (1 cm × 1 cm) was used according to the procedure described in Section 2.3. However, in this case, the cells were incubated with electrospun membranes for 24 h. Afterward, the membranes were removed before the cells’ irradiation. The results were expressed in the percentage of inhibition of each membrane in the dark and under light irradiation.3. Results and discussion
3.1. Singlet oxygen generation and cytotoxicity studies of porphyrin in tumor and non-tumor cell lines
Given that the 1O2 species is one of the ROS produced by this type of macrocycle upon irradiation and the main responsible species for damaging cells and inducing their death by PDT, the ability of the PS derivative in generating 1O2 was evaluated. Thus, the production of 1O2 was assessed using an indirect method based on measuring the absorption decay of the 9,10-dimethylanthracene (9,10-DMA) solution irradiated in the presence of the TPPF16[S-CH2-COOH]4 using TPP (ΦΔ = 0.65 in DMF)44 for comparison. The compounds were efficient 1O2 producers upon light irradiation, with TPP (ΦΔ = 0.65) being similar to TPPF16[S-CH2-COOH]4 (ΦΔ = 0.62).After confirming the ability of the synthesized Por TPPF16[S-CH2-COOH]4 to produce 1O2 species, its cytotoxicity in the dark and under irradiation was assessed against the tumor cell line HeLa (human cervical carcinoma) and the non-tumor cell line Vero (green monkey kidney fibroblast). The results are expressed in concentration values needed to inhibit 50% of cell growth (GI50), as shown in Table 2.
| Conditions | GI50 (μM) | |
|---|---|---|
| HeLa cells | Vero cells | |
| Dark | 6.29 ± 0.07 | 8.44 ± 0.51 |
| Light irradiation | 0.59 ± 0.03 | 4.09 ± 0.38 |
Ideally, a PS must exhibit high cytotoxicity upon light irradiation and absence or minimal toxicity in the dark. This will allow low levels of systemic toxicity until the irradiation of the target area. Additionally, it should be harmless to healthy cells.48,49 Considering the cytotoxicity results of Por in its free form against tumor HeLa cells, it was possible to observe that this compound presents some cytotoxicity under dark conditions (GI50 of 6.29 μM). Nevertheless, in the presence of light, the GI50 values were considerably lower (0.59 μM), which indicates that a very low concentration of Por is required to inhibit 50% of tumor cell growth. Thus, this PS exhibits much higher cytotoxicity when exposed to light than in the absence of light, demonstrating its photodynamic efficiency under light conditions against cancer cells.50 One of the desirable characteristics of any new PS is the lack or minimal toxicity for non-tumor cells.50 Therefore, the assessment of Por's cytotoxicity against non-tumoral cells was also performed both in the absence and presence of light. Por displayed GI50 values of 8.44% and 4.09% under dark and light conditions, respectively, which were superior to the ones determined for tumor cells, indicating the need for higher Por concentrations to inhibit 50% of non-tumor cell proliferation. This finding confirms a higher cytotoxicity against tumor cells compared to healthy ones. Moreover, Por exhibits about 11 times more phototoxicity compared to dark conditions against HeLa cells, whereas this difference was only 2 times more for Vero cells. Thus, the phototoxic effect of Por seems to be considerably reduced in healthy cells. For the production of electrospun nanofibers for PDT applications, the initial amount of Por incorporated was based on the GI50 values obtained against HeLa cells upon irradiation.
3.2. Development and characterization of PVA–Gel core–shell nanofibers
The developed core–shell nanofibers were characterized by their morphology and diameters, thermal stability, and chemical composition by FESEM, TEM, TGA, and ATR-FTIR, respectively (Fig. 2). | ||
| Fig. 2 (a) FESEM and (b) TEM images, and the respective diameter distribution histogram of PVA–Gel electrospun nanofibers. (c) TGA spectra and first-order derivate thermogravimetry (DTG) curves of the PVA powder, Gel powder, and PVA–Gel electrospun nanofibers, and (d) ATR-FTIR spectra of the PVA nanofibers, Gel nanofibers, and PVA–Gel nanofibers. | ||
As shown in Fig. 2a, uniform PVA–Gel nanofibers were successfully developed using the co-axial accessory, without practically any defects. The diameters of the developed nanofibers ranged from 55 to 229 nm, presenting an average of 125 nm. The core–shell structure was confirmed by TEM images (Fig. 2b), where the dark region represents the fiber's core, and the bright region indicates the shell.
Fig. 2c shows the TGA spectra and the corresponding first-order derivate thermogravimetry (DTG) of PVA and Gel powders as well as PVA–Gel nanofibers. PVA powder exhibited three degradation stages: the first one at 30–200 °C corresponds to the loss of the absorbed water; the second step occurred at 200–400 °C with a maximum degradation temperature at 325 °C, and it is due to the loss of low molecular weight substances, such as residual acetate groups and non-conjugated polyenes; the third step was detected at 400–500 °C, which is related to the breakdown of the polymer backbone, and presented a maximum degradation peak at 429 °C. The weight losses of the first, second, and third stages were 5, 74, and 19%, respectively.51–53 Gel powder presented two mass loss steps. The first one occurred around 30–200 °C with a weight loss of 13%, which corresponds to the loss of water. In the second step, between 200 and 500 °C, a weight loss of 65% and a maximum degradation temperature of 328 °C were observed, which can be attributed to protein degradation.54–56 In the spectrum of PVA–Gel nanofibers, two main degradation stages were detected. The first step was observed between 200 and 400 °C, with a weight loss of 59% and a maximum degradation peak at 321 °C. This stage can be attributed to the degradation of both PVA and Gel polymers, since their degradation profile in this range of temperatures matches, thereby confirming the presence of PVA and Gel in the composition of nanofibers. Moreover, a second degradation step at 400–500 °C was found, which coincides with the decomposition of the main chain of PVA.
To evaluate the chemical composition of the developed nanofibers, ATR-FTIR analysis was performed (Fig. 2d). Electrospun nanofibers composed only by PVA as the core were used for comparison purposes as well as nanofibers composed only by Gel as the shell layer. PVA nanofibers displayed the characteristic peaks of this polymer: 3314 cm−1 (O–H stretching vibrations), 2941 and 2914 cm−1 (C–H stretching of the alkyl groups), 1732 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching from acetate group remaining in PVA), 1429 and 1329 cm−1 (C–H2 bending vibrations), 1252 cm−1 (C–C skeletal vibration), 1094 cm−1 (C–C and C–O groups stretching vibrations), 1024 cm−1 (C–O stretching vibration), and 847 cm−1 (C–C stretching vibrations and the O–H out-of-plane bending vibrations).57,58 Gel nanofibers exhibited a broad peak at 3281 cm−1, which corresponds to N–H and O–H stretching vibrations. The peaks observed at 1639, 1535, and 1244 cm−1, are attributed to CO stretching vibration in amide I, N–H bending in amide II and C–N stretching in amide III, respectively.54,59 Other typical peaks were observed at 2936 cm−1 (C–H stretching), 1449 cm−1 (aliphatic C–H bending), 1333 cm−1 (CH2 wagging of proline), and 1082 cm−1 (skeletal stretching vibrations).60 In the spectrum of PVA–Gel core–shell nanofibers, the characteristic peaks of both PVA and Gel were observed, confirming the existence of both polymers in electrospun nanofibers.32,33,61
O stretching from acetate group remaining in PVA), 1429 and 1329 cm−1 (C–H2 bending vibrations), 1252 cm−1 (C–C skeletal vibration), 1094 cm−1 (C–C and C–O groups stretching vibrations), 1024 cm−1 (C–O stretching vibration), and 847 cm−1 (C–C stretching vibrations and the O–H out-of-plane bending vibrations).57,58 Gel nanofibers exhibited a broad peak at 3281 cm−1, which corresponds to N–H and O–H stretching vibrations. The peaks observed at 1639, 1535, and 1244 cm−1, are attributed to CO stretching vibration in amide I, N–H bending in amide II and C–N stretching in amide III, respectively.54,59 Other typical peaks were observed at 2936 cm−1 (C–H stretching), 1449 cm−1 (aliphatic C–H bending), 1333 cm−1 (CH2 wagging of proline), and 1082 cm−1 (skeletal stretching vibrations).60 In the spectrum of PVA–Gel core–shell nanofibers, the characteristic peaks of both PVA and Gel were observed, confirming the existence of both polymers in electrospun nanofibers.32,33,61
3.3. Incorporation of porphyrin into the core of PVA–Gel core–shell nanofibers
After the development of PVA–Gel core–shell nanofibers, the Por was incorporated into the core of the fibers. Different Por concentrations were added to PVA solutions, considering the obtained GI50 values against HeLa cells of the compound when irradiated. In this way, 5.9 × 10−7, 1.18 × 10−5, 5.9 × 10−5, and 1.6 × 10−3 M of Por were used, which corresponds to the GI50 of the Por when irradiated, and 20×, 100×, and 2750× higher than that concentration, respectively. The viscosity and conductivity of Gel solutions, as well as PVA solutions containing different Por concentrations, were assessed. Moreover, to evaluate the morphology of nanofibers and to confirm the presence and distribution of the Por within the fibers, FESEM, GSDR, and CLSM analyses were performed (Fig. 3).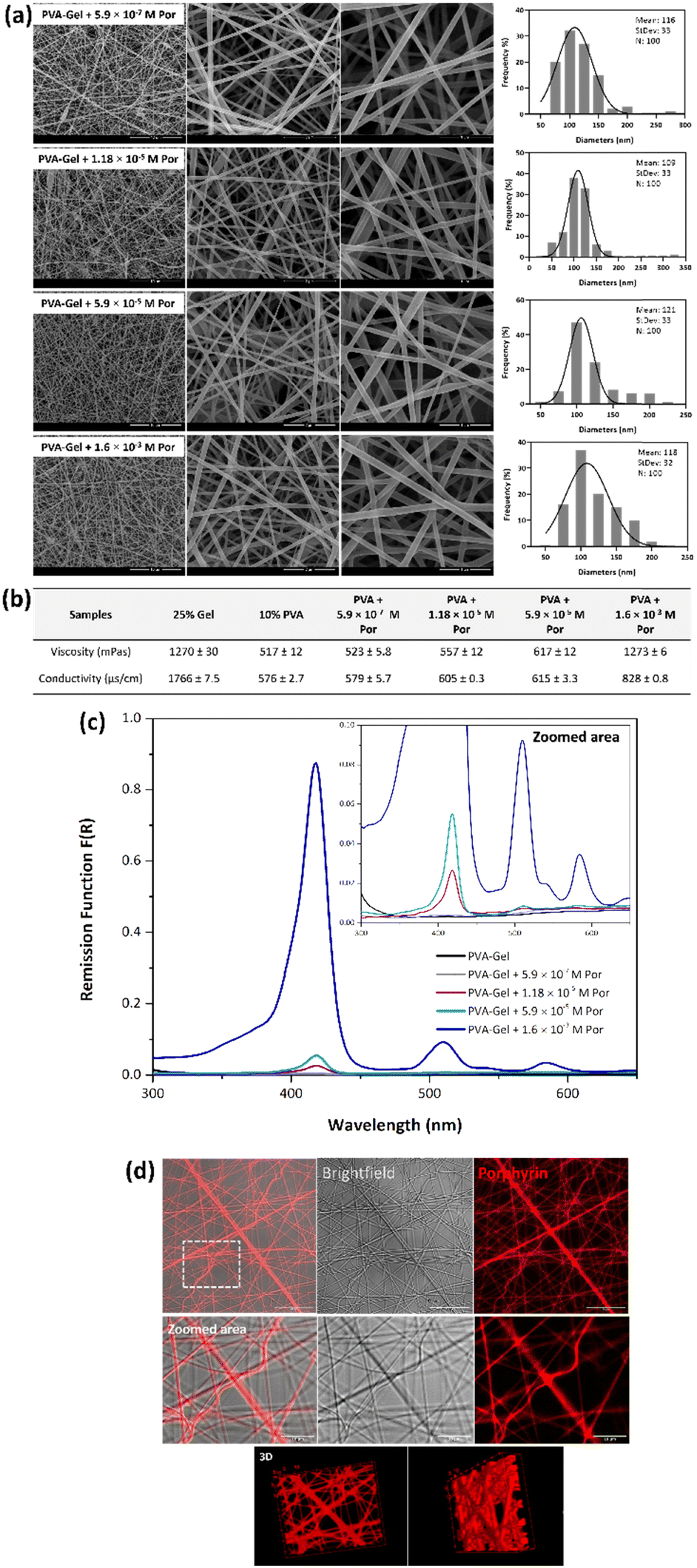 | ||
| Fig. 3 Images of (a) FESEM and the respective diameter distribution histograms of PVA–Gel nanofibers containing 5.9 × 10−7, 1.18 × 10−5, 5.9 × 10−5, and 1.6 × 10−3 M of Por, (b) viscosity and conductivity values of the different solutions; spectra of (c) GSDR of PVA–Gel without and with different Por concentrations; and images of (d) CLSM of PVA–Gel + Por nanofibers excited at 405 nm and detected at 500–699 nm. | ||
The incorporation of different Por concentrations into the core of PVA–Gel nanofibers did not affect the morphology of the fibers since defect-free nanofibers with smooth surfaces were obtained, as shown by FESEM images (Fig. 3a). Additionally, samples containing different amounts of Por showed a similar morphology between them. The average diameters of nanofibers containing 5.9 × 10−7, 1.18 × 10−5, 5.9 × 10−5, and 1.6 × 10−3 M of Por were 116, 109, 121, and 118 nm, respectively. Compared with PVA–Gel nanofibers, the incorporation of Por led to a very slight decrease in the fibers’ diameters. The addition of Por into the PVA solution promoted an increase in both viscosity and conductivity values, which increased according to PS concentration (Fig. 3b). The increase of conductivity values due to the increase of charge density with the incorporation of Por can result in stronger elongation forces in ejected jets, which promotes a decrease in fiber diameters.18
To confirm the presence of Por in nanofibers, the GSDR of the nanofibers with and without Por was performed (Fig. 3c). The electronic absorption spectrum of a typical Por compound is characterized by two distinct regions: the first one is related to the transition from the ground state to the second excited state S0 → S2, with the corresponding band called the Soret or B band, which is detected around 380–500 nm; the second one involves a weak transition to the first excited state S0 → S1, and the corresponding bands are called Q bands.13 The absorption spectrum of the Por under study exhibits a strong Soret band at 415 nm and moderate Q bands at 507 and 580 nm in DMF, as demonstrated by Lourenço et al.43 Regarding the nanofibers incorporated with this compound, it was possible to verify that in the samples containing 5.9 × 10−7 M of Por, no absorption bands were detected. Since this Por concentration is very low, it may not be sufficient to be detected by the equipment. On the other hand, in the spectra of nanofibers with 1.18 × 10−5 and 5.9 × 10−5 M of Por, a Soret band was detected with a maximum wavelength at 418 nm, and a Q-band was recorded at 512 and 511 nm, respectively. Moreover, in the spectrum of PVA–Gel + 1.6 × 10−3 M Por nanofibers, a Soret band, and two Q bands were detected. The maximum absorption of the Soret band was observed at 418 nm, while the first and the second Q bands were detected at 510 and 584 nm, respectively. Thus, it can be assumed that after the incorporation of 1.18 × 10−5, 5.9 × 10−5, and 1.6 × 10−3 M of Por into core–shell nanofibers, the PS's characteristic absorption bands appeared, with the obtained spectra being very similar to the spectrum of Por alone in solution,43 thereby confirming the presence of Por into nanofibers. Thus, it was possible to conclude that increasing the Por concentration led to an increase in the intensity of all bands.
Besides the presence of the Por, the distribution of these molecules within fibers was also evaluated by CLSM. Thus, PVA–Gel nanofibers containing the Por were excited at 405 nm and the emitted fluorescence was detected at 500–699 nm, considering the absorption and the emission spectra of the compound. No fluorescence was detected in nanofibers without Por (controls) when excited at 405 nm (Fig. S6, in the ESI†). On the other hand, as observed in Fig. 3d, red fluorescence was detected in Por-loaded PVA–Gel nanofibers when excited at 405 nm, which would be expected since these molecules emit fluorescence in the red spectral region upon excitation in the Soret band.43 Nevertheless, these results demonstrate the efficient incorporation of Por in PVA–Gel + Por nanofibers. Moreover, the continuous fluorescence along the fibers indicates a well-defined distribution of the Por, without any detectable aggregation, as demonstrated by both 2D and 3D reconstruction (Fig. 3d). Electrospun nanofibers are characterized by their high loading capacity (LC) and high encapsulation efficiency (EE), making them very attractive platforms for drug delivery. As an example, the LC and EE were determined for PVA–Gel + 1.6 × 10−3 M Por membranes, with the obtained values of 1.11% ± 0.216 and 91.4% ± 8.69, respectively, demonstrating the potential of the developed nanofibrous carriers for DDSs.62
Overall, GSDR and CLSM results demonstrate the presence and distribution of the PSs all over the fibers, confirming their successful incorporation into the electrospun nanofibers. Additionally, the Por encapsulation within the fibers did not affect their properties, namely their ability to emit fluorescence. The PSs' intrinsic fluorescence features can enable real-time therapeutic monitoring parameters as well as therapy response assessment, allowing for the planning and adjustment of subsequent therapeutic programs.63
3.4. Porphyrin release profile from core–shell nanofibers
To investigate the release profile of Por from core–shell nanofibers, the membranes were immersed in acidic conditions (in PBS at pH 4.5) to simulate the pH of vaginal fluid as well as the tumor site environment, and left under continuous stirring at 37 °C.22,27 PVA–Gel membranes loaded 1.6 × 10−3 M Por were used as an example and the PS's release was monitored by UV-Vis spectroscopy for 9 days at different time points (0, 1, 2, 4, 6, 8, 24, 48, 72, 96, 168, and 216 h). The absorption spectra of the release medium obtained using PVA–Gel and PVA–Gel + Por nanofibers as well as the drug release profile (curve of the concentration of released Por in the function of time) are represented in Fig. 4. | ||
| Fig. 4 Absorption spectra of the release medium obtained using (a) PVA–Gel nanofibers and (b) PVA–Gel + Por nanofibers and the respective zoomed area; (c) spectrum of the concentration of released Por over time from PVA–Gel + 1.6 × 10−3 M Por electrospun nanofibers to the PBS solution after 0, 1, 2, 4, 6, 8, 24, 48, 72, 96, 168, and 216 h. | ||
The spectrum corresponding to 0 h was measured immediately after the membranes were placed in contact with the PBS solution, thus no absorption band was observed. Regarding the spectra recorded for PVA–Gel nanofibers, no absorption bands were detected during the experiment (Fig. 4a). These results were expected since these membranes were not loaded with any Por. On the other hand, the spectra of nanofibers containing Por showed the appearance of the typical absorption bands of the compound (Soret and Q bands), indicating the release of Por from nanofibers over time (Fig. 4b).
Several key factors contribute to the drug release profile from nanofibers, including the physico-chemical properties of the drug, the structural characteristics of the polymer matrix, the release conditions, and the possible interactions between those factors.64 Drugs can be released from electrospun nanofibers by several mechanisms, such as desorption of drugs from the nanofiber's surface, diffusion through the channels and pores of the polymer matrix, and erosion or degradation of the polymer matrix.18 In a core–shell structure, the encapsulated molecules should pass through the matrix of both core and shell layers, providing a more sustained drug release.30 To better evaluate the release of Por, a zoom of the Soret band was performed. At 1 h, the presence of the Soret band and Q bands was already noticeable, indicating the existence of the compound in the medium. Moreover, an initial fast release of the Por was observed until 8 h. This release can be due to the desorption/dissolution of the drug deposited on the nanofiber's surface. In fact, during the electrospinning process, leakage of the PVA may have occurred, resulting in the exposure of some core parts on the surface of the fibers, which promotes the quick release of the Por.27 Hence, Por on the surface of nanofibers may diffuse into the release medium when they come into contact with it.
Afterward, Por was gradually released over time. From 24 to 216 h, a continuous increase in the bands’ intensity, and consequently, an increase in the Por concentration in the release medium was observed. As the release medium occupies the interfibrous pores over time, the polymer matrix swells, allowing more access to the core layer and further Por diffusion may occur.64 At the same time, this result can be related to the degradation of the core–shell structure, which allowed the molecules to diffuse from the core of the nanofibers to the solution. These findings are consistent with the work of Zandi et al., who demonstrated that the Gel layer functioned as a physical barrier, providing a more sustained drug release since more time was required for water molecules to penetrate the polymer matrix, thereby delaying the molecule's diffusion from the core to the release medium.30
A plot of the released Por concentration over time was also performed (Fig. 4c), where it was possible to observe an initial fast drug release profile until 8 h, specifically during the first 2 h, followed by a sustained and gradual release. In fact, from 24 to 216 h, a slower release was detected, which may be related to the presence of the outer layer, which can delay the Por diffusion.65 Overall, these results demonstrate a burst release of the Por during the first hours, followed by a sustained release at least for 9 days, confirming the ability of these electrospun membranes to act as a sustained and continuous DDS, namely for cervical cancer.
3.5. Cytotoxicity studies of electrospun nanofibers in tumor and non-tumor cell lines
The cytotoxicity of the synthesized Por TPPF16[S-CH2-COOH]4 was already assessed under dark and light conditions (Table 2) against cancer cells and healthy ones. To evaluate the ability of these molecules to act as a PS after their incorporation into nanofibrous membranes, cytotoxicity studies of PVA–Gel and PVA–Gel + Por membranes were performed in the same tumor and non-tumor cell lines used for the evaluation of Por's photodynamic effect in its free form. For this, cells were incubated with electrospun nanofibers for 24 h, followed by the membranes’ removal. Then, the cells were irradiated for 30 min with a LED lamp (300–500 nm). The percentage of inhibition of each membrane was calculated in the dark and under light irradiation. The results are shown in Table 3.| Samples | Inhibition percentage (%) | |||
|---|---|---|---|---|
| HeLa cells | Vero cells | |||
| Dark | Light irradiation | Dark | Light irradiation | |
| PVA–Gel | 6.32 ± 0.23 | 7.33 ± 1.08 | 2.11 ± 0.17 | 2.82 ± 0.24 |
| PVA–Gel + 5.9 × 10−7 M Por | 7.28 ± 0.09 | 33.87 ± 2.78 | 3.14 ± 0.23 | 7.44 ± 0.83 |
| PVA–Gel + 1.18 × 10−5 M Por | 29.76 ± 2.87 | 75.29 ± 5.32 | 4.91 ± 0.2 | 10.04 ± 0.84 |
| PVA–Gel + 5.9 × 10−5 M Por | 47.56 ± 4.1 | 89.76 ± 6.44 | 6.18 ± 0.29 | 15.21 ± 1.51 |
| PVA–Gel + 1.6 × 10−3 M Por | 53.76 ± 3.29 | 97.65 ± 8.56 | 8.19 ± 0.94 | 20.74 ± 1.44 |
The percentage of inhibition of each membrane on cells refers to the capacity of the electrospun membranes to inhibit cell proliferation. Therefore, the highest the percentage of inhibition, the lowest the cell proliferation, which means that the cells are being hindered from proliferating normally.
By the results shown in Table 3, PVA–Gel nanofibers presented a very low percentage of inhibition both in the dark (6.32%) and under irradiation (7.33%) against HeLa cells, demonstrating minimal cytotoxicity. Nevertheless, the reduction in cell viability was less than 30%, which is the threshold level for cytotoxicity according to International Standard.49,66 Moreover, the inhibition was very similar under both conditions, which indicates no specificity under irradiation. This low toxicity could be associated with the presence of residual acetic acid in electrospun membranes since this solvent was used to dissolve Gel polymer (shell layer).
Por incorporation promoted an increase in the percentage of inhibition of PVA–Gel membranes under the dark and under light conditions against tumor cells, and this increase is proportional to Por concentration. Nevertheless, the percentage of inhibition was always higher under light irradiation than in the dark, showing the ability of these electrospun membranes to inhibit the HeLa cell's growth when they are irradiated. Although the photodynamic action of single Por was already confirmed (Table 2), these results demonstrate that this molecule is still phototoxic even after its incorporation into nanofiber mats. The addition of 5.9 × 10−7 M Por promoted a strong increase in the percentage of inhibition under light conditions in comparison with the dark. Thus, using the GI50 value of 5.9 × 10−7 M Por (Table 2), it was already possible to inhibit cancer cell proliferation by 33.87% under light conditions. Therefore, this result demonstrates the higher ability of PVA–Gel + 5.9 × 10−7 M Por membranes to inhibit cell proliferation after radiation exposure, confirming the necessity of light for drug activation.13 Thus, a photodynamic effect could be achieved by adding a minimal amount of Por to the membranes, which can reduce dark toxicity and help avoid skin photosensitivity.67
The difference between the percentage of inhibition in the dark and under irradiation using PVA–Gel + 5.9 × 10−7 M Por membranes was 26.59%, which increased to around 42–46% with the incorporation of 1.18 × 10−5, 5.9 × 10−5 and 1.6 × 10−3 M Por. PVA–Gel + 1.18 × 10−5 M Por membranes (20× more of Por than the GI50 calculated concentration) were able to inhibit 75% of cancer cellular growth, while the membranes with the highest Por concentration (1.6 × 10−3 M) promoted an inhibition of almost 100% of cell proliferation under light conditions. It is important to note that in this experiment, electrospun nanofibers were put in contact with cells for 24 h followed by their removal, which means that the observed phototoxic effect is due to the released molecules after that period. According to the in vitro drug release tests, a continuous release of Por from nanofibers was detected at least until 216 h, suggesting that these fibrous membranes can be applied for long-lasting release and thereby avoiding repeatable drug administration.
PVA–Gel nanofibers showed very low cytotoxicity and similar inhibition values in the absence (2.11%) and presence (2.82%) of light against non-tumor cells, demonstrating no specificity to light. Although the inhibition of cell proliferation increased with the incorporation and concentration of Por for both conditions, these values were consistently lower than those observed with HeLa cells. Additionally, the difference between the values obtained for light irradiation and darkness (4.3, 5.13, 9.03, and 12.55%, according to Por concentration) was considerably lower compared to the ones obtained for tumor cells. These findings suggest the selective photocytotoxic effect in the tumor cell line.
Overall, the photodynamic effect of Por molecules was not affected by their incorporation into nanofibrous membranes, showing the potential of these substrates to carry the photoactive molecules, that can destroy cancer cells in the presence of light. These fibrous therapeutic platforms also seem to have a selective photocytotoxic effect in tumor cell lines (HeLa cells), highlighting the suitability and potentiality of this PDT approach to treat tumor cells while maintaining their safety for healthy ones.
4. Conclusions
Photoresponsive core–shell nanofibrous membranes were successfully developed via co-axial electrospinning to be used as possible localized DDSs in PDT for cancer. Biodegradable polymers, PVA and Gel, were applied to produce the membranes, and different concentrations of synthesized Por were incorporated into the core of nanofibers. The synthesized Por was shown to be a promising candidate to act as a PS for PDT since it was able to generate 1O2 and exhibited much higher cytotoxicity under light irradiation in comparison to dark conditions against tumor cells. Additionally, it displayed higher phototoxicity toward tumor cells than toward healthy ones. The incorporation of Por into nanofibers did not affect their morphology, and these molecules were well distributed all over the fibers. These fibrous nanoplatforms exhibited the high LC and EE of Por. Por was continuously released from nanofibers for at least 9 days, the initial stage characterized by a fast release, followed by a slower and sustained release, which could be due to the use of a core–shell structure. Finally, PVA–Gel nanofibers with different Por concentrations showed a photodynamic effect, since their percentage of inhibition of HeLa cell proliferation was higher under light irradiation than in the dark, confirming the ability of these electrospun membranes to inhibit the growth of cancer cells when exposed to radiation. Furthermore, a selective photocytotoxic effect seems to occur in tumor cell lines compared to non-tumor. Thus, this study shows the great potential of nanofibers incorporated with PSs to act as localized DDS for cancer PDT, particularly for cervical cancer, allowing a continuous release of Por. The production of these electrospun mats can be upscaled to the industrial level, which is advantageous for these applications.Author contributions
Conceptualisation, S. M. C. and D. P. F.; methodology, S. M. C.; validation, L. M. O. L., R. C. C., I. C., and D. P. F.; investigation, S. M. C., R. C. C., I. C., and C. B.; resources, L. M. O. L., and D. P. F.; writing – original draft S. M. C.; writing – review and editing, L. M. O. L., R. C., I. C., C. B., R. F. and D. P. F.; supervision: R. F., and D. P. F.; funding acquisition, L. M. O. L., and D. P. F. All authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks to the University of Minho, University of Aveiro, and FCT/MCTES (Fundação para a Ciência e a Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) for the financial support to 2C2T – Centre for Textile Science and Technology under the projects UID/CTM/00264/2020 on its components Base (https://doi.org/10.54499/UIDB/00264/2020) and programmatic (https://doi.org/10.54499/UIDP/00264/2020), LAQV-REQUIMTE (UIDB/50006/2020 and UIDP/50006/2020), and the project EndoSWITCH (PTDC/BTM-ORG/5154/2020) funded by FCT/MCTES through national funds. The authors are also thankful to Project DRI/India/0447/2020 (https://doi.org/10.54499/DRI/India/0447/2020). The authors are also grateful to the Foundation for Science and Technology (FCT, Portugal) for financial support through national funds FCT/MCTES (PIDDAC) to CIMO (UIDB/00690/2020 and UIDP/00690/2020) and SusTEC (LA/P/0007/2020); the contract of R. C. Calhelha through the institutional scientific employment program-contract. Sofia M. Costa is thankful to FCT PhD Scholarship (SFRH/BD/147517/2019) and Diana P. Ferreira to CEECIND/02803/2017.References
- World Health Organization (WHO), Cervical cancer, https://www.who.int/news-room/fact-sheets/detail/cervical-cancer, (accessed 1 June 2023).
- C. A. Burmeister, S. F. Khan, G. Schäfer, N. Mbatani, T. Adams, J. Moodley and S. Prince, Tumour Virus Res., 2022, 13, 200238 CrossRef CAS.
- S. Ghosh, P. Jayaram, S. P. Kabekkodu and K. Satyamoorthy, Eur. J. Pharmacol., 2022, 917, 174751 CrossRef CAS.
- E. P. Chizenga, R. Chandran and H. Abrahamse, Oncotarget, 2019, 10, 4380–4396 CrossRef.
- N. Hodgkinson, C. A. Kruger, M. Mokwena and H. Abrahamse, J. Photochem. Photobiol., B, 2017, 177, 32–38 CrossRef CAS.
- Y.-Y. Zhao, L. Zhang, Z. Chen, B.-Y. Zheng, M. Ke, X. Li and J.-D. Huang, J. Am. Chem. Soc., 2021, 143, 13980–13989 CrossRef CAS.
- B. Pucelik, A. Sułek, A. Drozd, G. Stochel, M. M. Pereira, S. M. A. Pinto, L. G. Arnaut and J. M. Dąbrowski, Int. J. Mol. Sci., 2020, 21, 2786 CrossRef CAS PubMed.
- F. Adnane, E. El-Zayat and H. M. Fahmy, Tissue Cell, 2022, 77, 101856 CrossRef CAS.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef.
- X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- M. Yang, Z. Özdemir, H. Kim, S. Nah, E. Andris, X. Li, Z. Wimmer and J. Yoon, Adv. Healthcare Mater., 2022, 11, 2200529 CrossRef CAS.
- D. P. Ferreira, D. S. Conceição, F. Fernandes, T. Sousa, R. C. Calhelha, I. C. F. R. Ferreira, P. F. Santos and L. F. Vieira Ferreira, J. Phys. Chem. B, 2016, 120, 1212–1220 CrossRef CAS PubMed.
- D. P. Ferreira, D. S. Conceicao, R. C. Calhelha, T. Sousa, R. Socoteanu, I. C. F. R. Ferreira and L. F. Vieira Ferreira, Carbohydr. Polym., 2016, 151, 160–171 CrossRef CAS PubMed.
- X. Feng, Y. Shi, L. Xie, K. Zhang, X. Wang, Q. Liu and P. Wang, J. Med. Chem., 2018, 61, 7189–7201 CrossRef CAS PubMed.
- X. Li, D. Lee, J.-D. Huang and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 9885–9890 CrossRef CAS.
- A. Erickson, P. A. Chiarelli, J. Huang, S. L. Levengood and M. Zhang, Nanoscale Horiz., 2022, 7, 1279–1298 RSC.
- Y. Fu, X. Li, Z. Ren, C. Mao and G. Han, Small, 2018, 14, 1801183 CrossRef.
- S. M. Costa, R. Fangueiro and D. P. Ferreira, Macromol. Biosci., 2022, 22, 2100512 CrossRef CAS.
- A. S. Ribeiro, S. M. Costa, D. P. Ferreira, R. C. Calhelha, L. Barros, D. Stojković, M. Soković, I. C. F. R. Ferreira and R. Fangueiro, React. Funct. Polym., 2021, 159, 104808 CrossRef CAS.
- D. Alves, J. C. Araújo, R. Fangueiro and D. P. Ferreira, Molecules, 2023, 28, 3053 CrossRef CAS.
- R. CeCe, L. Jining, M. Islam, J. G. Korvink and B. Sharma, Adv. Eng. Mater., 2023, 2301297 Search PubMed.
- U. Aggarwal, A. K. Goyal and G. Rath, Mater. Sci. Eng., C, 2017, 75, 125–132 CrossRef CAS.
- N. Aminu, S. Ilyasu, M. Al-Kassim Hassan, F. S. Kurfi, A. I. Jatau, S.-Y. Chan and D. Alfred-Ugbenbo, J. Drug Delivery Sci. Technol., 2023, 90, 105128 CrossRef CAS.
- I. Major and C. McConville, Drug Delivery Transl. Res., 2017, 7, 817–828 CrossRef CAS.
- M. S. Afanasiev, A. D. Dushkin, T. G. Grishacheva, S. S. Afanasiev and A. V. Karaulov Academician, Photodiagn. Photodyn. Ther., 2022, 37, 102620 CrossRef CAS PubMed.
- X. Li, S. Yu, Y. Lee, T. Guo, N. Kwon, D. Lee, S. C. Yeom, Y. Cho, G. Kim, J.-D. Huang, S. Choi, K. T. Nam and J. Yoon, J. Am. Chem. Soc., 2019, 141, 1366–1372 CrossRef CAS PubMed.
- E. Yan, J. Jiang, X. Yang, L. Fan, Y. Wang, Q. An, Z. Zhang, B. Lu, D. Wang and D. Zhang, J. Drug Delivery Sci. Technol., 2020, 55, 101455 CrossRef CAS.
- S. Tabakoglu, D. Kołbuk and P. Sajkiewicz, Biomater. Sci., 2023, 11, 37–61 RSC.
- G. Kabay, A. E. Meydan, T. Eom, B. S. Shim, M. Mutlu and G. Kaleli-Can, Int. J. Pharm., 2023, 630, 122442 CrossRef CAS.
- N. Zandi, R. Lotfi, E. Tamjid, M. A. Shokrgozar and A. Simchi, Mater. Sci. Eng., C, 2020, 108, 110432 CrossRef CAS.
- N. Goonoo, A. Bhaw-Luximon and D. Jhurry, J. Biomed. Nanotechnol., 2014, 10, 2173–2199 CrossRef CAS.
- V. M. Merkle, L. Zeng, M. J. Slepian and X. Wu, Biopolymers, 2014, 101, 336–346 CrossRef CAS PubMed.
- M. Sengor, A. Ozgun, G. Corapcioglu, M. Ipekoglu, B. Garipcan, N. Ersoy and S. Altintas, J. Appl. Polym. Sci., 2018, 135, 46582 CrossRef.
- I. Zulkiflee and M. B. Fauzi, Biomedicines, 2021, 9, 979 CrossRef CAS.
- V. M. Merkle, D. Martin, M. Hutchinson, P. L. Tran, A. Behrens, S. Hossainy, J. Sheriff, D. Bluestein, X. Wu and M. J. Slepian, ACS Appl. Mater. Interfaces, 2015, 7, 8302–8312 CrossRef CAS PubMed.
- M. Santoro, A. M. Tatara and A. G. Mikos, J. Controlled Release, 2014, 190, 210–218 CrossRef CAS.
- V. Merkle, L. Zeng, W. Teng, M. Slepian and X. Wu, Polymer, 2013, 54, 6003–6007 CrossRef CAS.
- N. M. Inada, H. H. Buzzá, M. F. M. Leite, C. Kurachi, J. R. Trujillo, C. A. de Castro, F. M. Carbinatto, W. Lombardi and V. S. Bagnato, Pharmaceuticals, 2019, 12, 107 CrossRef CAS.
- Y. Tang, Y. Su, Y. Xu, Y. Zhang, Y. Shen, L. Qin, L. Zhang, L. Cao, Y. Zhou, T. Zhang and M. Zhang, Photodiagn. Photodyn. Ther., 2022, 39, 102884 CrossRef CAS.
- A. Unanyan, L. Pivazyan, J. Davydova, K. Murvatova, A. Khrapkova, R. Movsisyan, A. Ishchenko and A. Ishchenko, Photodiagn. Photodyn. Ther., 2021, 36, 102530 CrossRef.
- S. Zong, X. Wang, Y. Yang, W. Wu, H. Li, Y. Ma, W. Lin, T. Sun, Y. Huang, Z. Xie, Y. Yue, S. Liu and X. Jing, Eur. J. Pharm. Biopharm., 2015, 93, 127–135 CrossRef CAS.
- Y.-P. Chen, Y.-W. Liu, D. Lee, J. T. Qiu, T.-Y. Lee and S.-J. Liu, Int. J. Nanomed., 2019, 14, 421–429 CrossRef CAS PubMed.
- L. M. O. Lourenço, J. Resende, B. A. Iglesias, K. Castro, S. Nakagaki, M. J. Lima, A. F. da Cunha, M. G. P. M. S. Neves, J. A. S. Cavaleiro and J. P. C. Tomé, J. Porphyrins Phthalocyanines, 2014, 18, 967–974 CrossRef.
- J. M. D. Calmeiro, S. R. D. Gamelas, A. T. P. C. Gomes, M. A. F. Faustino, M. G. P. M. S. Neves, A. Almeida, J. P. C. Tomé and L. M. O. Lourenço, Dyes Pigm., 2020, 181, 108476 CrossRef CAS.
- T. Eren Böncü and N. Ozdemir, Beilstein J. Nanotechnol., 2022, 13, 245–254 CrossRef.
- A. Razzaq, Z. U. Khan, A. Saeed, K. A. Shah, N. U. Khan, B. Menaa, H. Iqbal and F. Menaa, Pharmaceutics, 2021, 13, 349 CrossRef CAS.
- L.-F. Zhu, Y. Zheng, J. Fan, Y. Yao, Z. Ahmad and M.-W. Chang, Eur. J. Pharm. Sci., 2019, 137, 105002 CrossRef CAS.
- E. Lima, O. Ferreira, J. F. Silva, A. O. Santos, R. E. Boto, J. R. Fernandes, P. Almeida, S. M. Silvestre and L. V. Reis, Dyes Pigm., 2020, 174, 108024 CrossRef CAS.
- E. Lima, A. G. Barroso, M. A. Sousa, O. Ferreira, R. E. Boto, J. R. Fernandes, P. Almeida, S. M. Silvestre, A. O. Santos and L. V. Reis, Eur. J. Med. Chem., 2022, 229, 114071 CrossRef CAS PubMed.
- F. Mandim, V. C. Graça, R. C. Calhelha, I. L. F. Machado, L. F. V. Ferreira, I. C. F. R. Ferreira and P. F. Santos, Molecules, 2019, 24, 863 CrossRef.
- G.-M. Kim, A. S. Asran, G. H. Michler, P. Simon and J.-S. Kim, Bioinspiration Biomimetics, 2008, 3, 46003 CrossRef PubMed.
- M. T. Taghizadeh, N. Yeganeh and M. Rezaei, J. Appl. Polym. Sci., 2015, 132, 42117 CrossRef.
- I. Restrepo, C. Medina, V. Meruane, A. Akbari-Fakhrabadi, P. Flores and S. Rodríguez-Llamazares, Polim.: Cienc. Tecnol., 2018, 28, 169–177 CrossRef.
- A. S. Ribeiro, S. M. Costa, D. P. Ferreira, H. Abidi and R. Fangueiro, Key Eng. Mater., 2021, 893, 45–55 Search PubMed.
- D. Kotatha, M. Hirata, M. Ogino, S. Uchida, M. Ishikawa, T. Furuike and H. Tamura, J. Nanotechnol., 2019, 2019, 2501039 Search PubMed.
- R. Longo, M. Catauro, A. Sorrentino and L. Guadagno, J. Therm. Anal. Calorim., 2022, 147, 5391–5399 CrossRef CAS.
- M. A. Teixeira, J. C. Antunes, C. L. Seabra, A. Fertuzinhos, S. D. Tohidi, S. Reis, M. T. P. Amorim, D. P. Ferreira and H. P. Felgueiras, Biomater. Adv., 2022, 137, 212830 CrossRef CAS PubMed.
- H. S. Mansur, C. M. Sadahira, A. N. Souza and A. A. P. Mansur, Mater. Sci. Eng., C, 2008, 28, 539–548 CrossRef CAS.
- M. Alishahi, M. Khorram, Q. Asgari, F. Davani, F. Goudarzi, A. Emami, A. Arastehfar and K. Zomorodian, Int. J. Biol. Macromol., 2020, 163, 288–297 CrossRef CAS PubMed.
- V. Mosayebi, M. Fathi, M. Shahedi, N. Soltanizadeh and Z. Emam-Djomeh, Food Biosci., 2022, 47, 101759 CrossRef CAS.
- T.-Y. Kuo, C.-F. Jhang, C.-M. Lin, T.-Y. Hsien and H.-J. Hsieh, Open Phys., 2017, 15, 1004–1014 CrossRef CAS.
- A. Farboudi, K. Mahboobnia, F. Chogan, M. Karimi, A. Askari, S. Banihashem, S. Davaran and M. Irani, Int. J. Biol. Macromol., 2020, 150, 178–188 CrossRef CAS PubMed.
- P. Sarbadhikary, B. P. George and H. Abrahamse, Theranostics, 2021, 11, 9054–9088 CrossRef CAS.
- M. K. Gaydhane, C. S. Sharma and S. Majumdar, RSC Adv., 2023, 13, 7312–7328 RSC.
- W. Li, N. Cicek, D. B. Levin, S. Logsetty and S. Liu, J. Biomater. Sci., Polym. Ed., 2020, 31, 394–406 CrossRef CAS PubMed.
- International Standard Is, Biological Evaluation of Medical Devices. Part 5. Tests for in Vitro Cytotoxicity, 3rd edn, 2009. ISO-10993-5.
- M. Mušković, R. Pokrajac and N. Malatesti, Pharmaceuticals, 2023, 16, 613 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00647j |
| This journal is © The Royal Society of Chemistry 2024 |