
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of photoluminescent hydrogen-bonded frameworks based on pyromellitic diimide-tethered carboxylic acid hosts and multi-bonding solvent guests†
Raju Ram
Puniya‡
a,
Priyanka
Takhar‡
a,
Monika
Chhapoliya
a,
Rinki
Deka
b,
Dhruba Jyoti
Kalita
 b and
Devendra
Singh
*a
b and
Devendra
Singh
*a
aDepartment of Chemistry, Mohanlal Sukhadia University, Udaipur-313001, Rajasthan, India. E-mail: dsingh@mlsu.ac.in
bDepartment of Chemistry, Gauhati University, Guwahati-781014, Assam, India
First published on 4th September 2024
Abstract
The significance of hydrogen-bonding interactions in improving the chemical and physical properties of functional materials related to sustainable energy, gas absorption, catalysis, and pharmaceuticals has gained considerable research attention. In this report, some unprecedented hydrogen bond motifs between the –COOH group and the solvents capable of forming multiple hydrogen bonds with –COOH are studied. The effects of such diverse motifs on the construction of 3D supramolecular architectures of hydrogen-bonded host–guest frameworks and their optical properties are elucidated. For this purpose, structural studies on seven solvates, namely, 1a (1:2DMF), 1b (1:2pyridine), 1c (1:2quinoline), 2a (1:2DMF), 2b (1:2pyridine), 2c (1:2quinoline), and 2d (1:1quinoline:2piperidine), of two isomeric pyromellitic diimide hosts 1 and 2 were carried out. Single crystal X-ray diffraction (SCXRD) analyses revealed that solvates 1a, 2a, and 2b show 3D non-porous supramolecular host–guest networks, whereas solvates 1b, 1c, 2c, and 2d show 3D supramolecular host–guest channelled architectures accommodating guest solvent molecules within the cavities of different dimensions. Formation of different hydrogen bond motifs, either cyclic/ring (R) or discrete (D) or a combination of both, between the –COOH groups of isomeric hosts and identical guest molecules is analysed through density functional theory (DFT) calculations. Minor differences in the interaction energies of different motifs of isomeric hosts with the same guest suggest that the formation of either motif depends on the steric orientations of hosts and other weak host–guest interactions in the crystal lattices. Solid state fluorescence emission properties of solvates 1a, 2a, and 2b are found to be similar to their respective hosts, whereas those of solvates 1b, 1c, 2c, and 2d are different from their hosts. Along with the diversity of supramolecular synthons, frontier molecular orbital (FMO) analysis of hydrogen-bonded model structures explained well the different emission behaviours of solvates. Thermal analyses for the solvates are in good agreement for the association of the numbers of guest solvent molecules with both the isomeric hosts. Overall, this research is focused on establishing the phenomena for the formation of distinct hydrogen bond patterns between the two similar host–guest binding groups together with the effect of supramolecular states on the photophysical properties of such hydrogen-bonded complexes.
Introduction
Non-covalent intermolecular interactions play an important role in the organization of molecular assemblies for the development of new functional materials.1–4 Hydrogen bonds in combination with other weak interactions, such as π–π, CH–π, van der Waals and charge transfer interactions, facilitate the formation of extended molecular assemblies with novel properties.5–8 The significance of such weak interactions in the construction of fascinating supramolecular assemblies, such as hydrogen-bonded organic frameworks (HOFs), with potential applications in the fields of gas adsorption and separation,9–11 fluorescent sensing,12–14 catalysis,15–17 proton conduction,18,19 and biological relevance20,21 is well documented. HOFs are constructed entirely via intermolecular interactions between the molecular units and are considered superior crystalline porous materials than metal–organic frameworks (MOFs) and covalent-organic frameworks (COFs) owing to their unique features.22–25 Indeed, multi-component hydrogen-bonded host–guest frameworks, such as cocrystals, salts, solvates or hydrates, also offer advantages in terms of solubility, crystallinity, self-repairing ability, easy reproducibility, bioavailability, and mechanical properties over the distinctive crystalline materials.26–28 The derivatives of some multiple hydrogen bond donor/acceptor groups, such as carboxylic acid (–COOH),29 sulfonic acid (–SO3H),30 amine (–NH2),31 amidine (–CNHNH2),32 diaminotriazine (DAT),33 benzimidazolone,34 pyrazole,35 pyridine,36 and urea,37 have been reported to construct stable multi-component HOFs so far.The –COOH group, in conjunction with π–π and CH–π interactions of aromatic units, facilitates the formation of various supramolecular aggregates via dimeric, discrete, or charge-assisted interactions.38–41 An efficient study on these weak interactions provides scope to improve the properties and applications of active pharmaceutical ingredients,42 gas adsorbents,43 catalysts,44 sensors,45 receptors,46 and optical materials.47 For instance, slight variations in the host–guest hydrogen bond patterns in such multi-component systems lead to supramolecular isomerism, which results in the formation of polymorphs of particular scientific interest.48 The supramolecular features associated with the –COOH group connected to pyromellitic diimide (PMDI) and naphthalene diimide (NDI) building blocks make them attractive for such studies.49–52 In these systems, free rotation of cyclic imide ring around the C–N bond arranges hosts and guests in several orientations to facilitate the formation of dissimilar motifs between the –COOH group of host and same guest molecule containing multiple donor/acceptor hydrogen bonding sites. In this respect, minor differences in the hydrogen bond interactions between the N,N′-bis(benzoic acid)naphthalene diimide host and pyridine guest resulted in the formation of polymorphic solvates.53 A series of inclusion compounds of N,N′-bis(glycinyl)pyromellitic diimide host with aromatic hydrocarbon guests was also reported to exist via charge transfer and π–π interactions.54 In our recent study, the formation of two types of hydrogen bond motifs (D or R) between the identical interacting groups of two isomeric host PMDI molecules and some different guest solvent molecules, respectively, was explored using both experimental and theoretical investigations.55
Herein, with the objectives to rationalize hydrogen bonding patterns (as illustrated in Scheme 1) between the –COOH group and some multiple hydrogen bonds donor/acceptor groups, seven multi-component host–guest frameworks/solvates of PMDI carboxylic acid hosts (1 and 2) with different guest solvents are prepared (Scheme 2), and various types of intermolecular interactions responsible for the formation of their 3D supramolecular architectures are studied. In some of these supramolecular systems, the encapsulation of guests occurs through the creation of primary self-assembly of host molecules, whereas in other structures, the interaction of a host with a guest creates a secondary assembly for guest encapsulation. Unlike in our previous studies, where the D or R binding modes between a single –COOH group and a single guest molecule were analysed, some new and unexplored binding modes of a single –COOH group of host molecules with several guests, viz. DMF and pyridine, are analysed in this study. The photoluminescence properties of such systems are explored based on supramolecular interactions in the structures of solvates and FMO analyses of optimized model structures. It is believed that the results of this study will provide insight into the establishment of some newly exposed hydrogen bond motifs in the crystal lattice of solid-state materials.
 | ||
| Scheme 1 Illustration of the possible hydrogen bond motifs between (a) the –COOH group and DMF and (b) the –COOH group and pyridine in the structures of the solvates. | ||
 | ||
| Scheme 2 Structures of hosts 1 and 2 together with guest solvent molecules and host–guest complexes showing host–guest stoichiometry. | ||
Results and discussion
The host compounds N,N′-bis(butanoic acid)pyromellitic diimide (1) and N,N′-bis(2-methylpropanoic acid)pyromellitic diimide (2) are synthesized by acid anhydride-amine condensation reactions, and their solvates are prepared using the respective solvents for further studies. Except for the crystals of mixed solvate 2d grown from the quinoline solution of 2 in the presence of piperidine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 host–guest ratio, all other solvates are prepared by crystallizing host compounds 1 and 2 from the solutions of single solvent, viz. DMF, pyridine and quinoline solvents, respectively, in a 1:2 host–guest ratio (Scheme 2). The crystals of suitable diffraction quality could not be obtained for the mixed quinoline and piperidine solvate of host 1. All the solvates are characterized using various spectroscopic techniques, including FT-IR, NMR, solid state UV-visible and fluorescence emission, thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD) and single crystal X-ray diffraction (SCXRD). The formation of different types of hydrogen bond motifs in the solvates of hosts 1 and 2 with the same solvents (Scheme 1), namely, motifs III and IV (in DMF solvates 1a and 2a), and IX and X (in pyridine solvates 1b and 2b), are analysed by DFT calculations by optimizing formic acid–DMF and formic acid–pyridine molecules as model structures. In addition, the possibility of the formation of slightly different motifs between the –COOH group and the DMF/pyridine molecules (V, VI/XI, and XII), which are not observed in the solvates of DMF and pyridine, is also checked by applying optimized model structures. The purpose of using such model structures was to avoid the complexity of other interactions between the larger hosts so that the interactions between the –COOH group and choice of solvent molecules could be analysed clearly. The HOMO–LUMO band gaps of such model structures are also considered to understand the different emission behaviours of DMF and pyridine solvates of the same host. In this study, the guest quinoline molecules are found to attach with the –COOH groups of host molecules only via a similar D mode in the structures of quinoline solvates. Therefore, the formic acid-quinoline models are not considered to conduct theoretical studies. Various intermolecular interactions such as OH–O, OH–N, CH–O, CH–π, CO–π, O–π and short H–H contacts were found to be responsible for the formation of 3D host–guest supramolecular architectures of these solvates.
:1:2 host–guest ratio, all other solvates are prepared by crystallizing host compounds 1 and 2 from the solutions of single solvent, viz. DMF, pyridine and quinoline solvents, respectively, in a 1:2 host–guest ratio (Scheme 2). The crystals of suitable diffraction quality could not be obtained for the mixed quinoline and piperidine solvate of host 1. All the solvates are characterized using various spectroscopic techniques, including FT-IR, NMR, solid state UV-visible and fluorescence emission, thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD) and single crystal X-ray diffraction (SCXRD). The formation of different types of hydrogen bond motifs in the solvates of hosts 1 and 2 with the same solvents (Scheme 1), namely, motifs III and IV (in DMF solvates 1a and 2a), and IX and X (in pyridine solvates 1b and 2b), are analysed by DFT calculations by optimizing formic acid–DMF and formic acid–pyridine molecules as model structures. In addition, the possibility of the formation of slightly different motifs between the –COOH group and the DMF/pyridine molecules (V, VI/XI, and XII), which are not observed in the solvates of DMF and pyridine, is also checked by applying optimized model structures. The purpose of using such model structures was to avoid the complexity of other interactions between the larger hosts so that the interactions between the –COOH group and choice of solvent molecules could be analysed clearly. The HOMO–LUMO band gaps of such model structures are also considered to understand the different emission behaviours of DMF and pyridine solvates of the same host. In this study, the guest quinoline molecules are found to attach with the –COOH groups of host molecules only via a similar D mode in the structures of quinoline solvates. Therefore, the formic acid-quinoline models are not considered to conduct theoretical studies. Various intermolecular interactions such as OH–O, OH–N, CH–O, CH–π, CO–π, O–π and short H–H contacts were found to be responsible for the formation of 3D host–guest supramolecular architectures of these solvates.
Analyses of crystal structures and DFT calculations
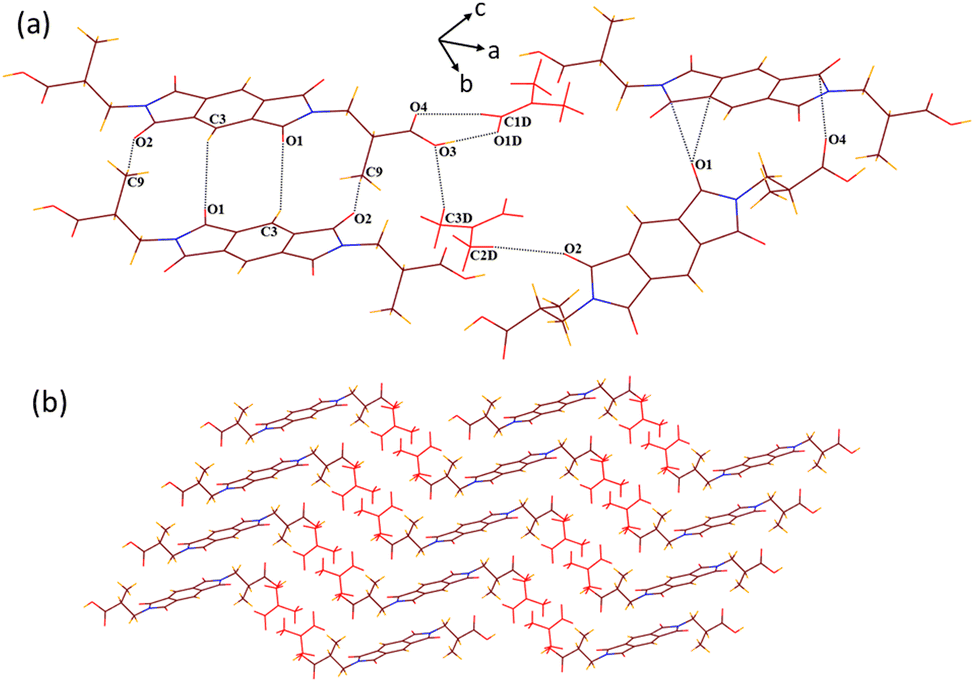
SCXRD structural analysis shows that solvate 1a crystallizes in a monoclinic P21/n space group and emerges as half of host molecule 1 along with a guest DMF molecule in its crystallographic asymmetric unit. In the crystal lattice of 1a, the guest DMF molecule interacts with the –COOH group of the host via discrete acceptor OH–O (O3–H3⋯O1D, dD⋯A 2.6 Å, ∠D–H⋯A 170°) and discrete donor CH–O (C2D–H2D3⋯O4, 3.53 Å, 168.3°) interactions (Fig. 1). It also interacts with the carbonyl oxygen of the cyclic imide ring of the host molecule via discrete donor C1D–H1D⋯O1 (3.36 Å, 173.9°) contact. Further, the host molecules are also found to interact with each other via C8–H8B⋯O3 (3.64 Å, 170°), O2⋯π (3.1 Å) and O4⋯π (3.0 Å) interactions, overall constructing a helical 3D host–guest network in which layers of single helix are arranged over each other, as viewed along the c axis. | ||
| Fig. 1 (a) Part of the crystal lattice of 1a showing intermolecular host–host and host–guest interactions. (b) A 3D supramolecular host–guest helical network of 1a (along the c axis). | ||
Solvate 2a crystallizes in the monoclinic P21/c space group. It appears as half of host molecule 2 lying on the inversion centre in its crystallographic asymmetric unit along with a guest DMF molecule. Contrary to the structure of 1a, in the structure of 2a, the guest DMF molecule interacts with the –COOH group of the host by the combination of acceptor OH–O (O3–H3⋯O1D, dD⋯A 2.58 Å, ∠D–H⋯A 170.9°) and donor CH–O (C1D–H1D⋯O4, 3.2 Å, 124.5°) interactions, making a cyclic R22(7) motif along with a discrete donor C3D–H3D3⋯O3 (3.64 Å, 161.9°) interaction (Fig. 2). It interacts similarly with the carbonyl oxygen of the cyclic imide ring of the host molecule via C2D–H2D1⋯O2 (3.6 Å, 159.2°) contact. The host molecules also interact with each other via C3–H3⋯O1 (3.27 Å, 128.5°) and C9–H9A⋯O2 (3.57 Å, 166.4°) interactions and stack over each other to create a 2D layered pattern along the b axis. These layers of host molecules further interact with each other via bifurcated O1⋯π (3.16 Å) and O4⋯π (3.029 Å) interactions to construct a 3D zigzag type of host–guest supramolecular architecture accommodating the layers of guest molecules between the layers of host molecules. In the structures of both DMF solvates 1a and 2a, no weak interactions among the guest molecules are observed in their crystal lattices.
 | ||
| Fig. 2 (a) Various weak interactions in the crystal lattice of 2a. (b) A 3D zigzag supramolecular architecture of solvate 2a (along the b axis). | ||
In the host–guest structures 1a and 2a, it is observed that the –COOH group of both the isomeric hosts interacts with two DMF molecules, making different types of hydrogen bond motifs. In the case of structure 1a, both the DMF molecules interact with the –COOH group via discrete C–H⋯O and O–H⋯O donor–acceptor interactions (motif III; Scheme 1), while in the case of structure 2a, one DMF molecule interacts with the –COOH group via a discrete donor C–H⋯O interaction and another DMF molecule forms a cyclic R22(7) hydrogen bond by the combination of C–H⋯O and O–H⋯O donor–acceptor interactions (motif IV). DFT calculations on analogous motifs constructed from formic acid and DMF are carried out to establish the existence of both types of motifs, as observed in the structures of solvates 1a and 2a. The optimized structures of formic acid and DMF motifs at the basis function B3LYP/6-31+G* are shown in Fig. 3.
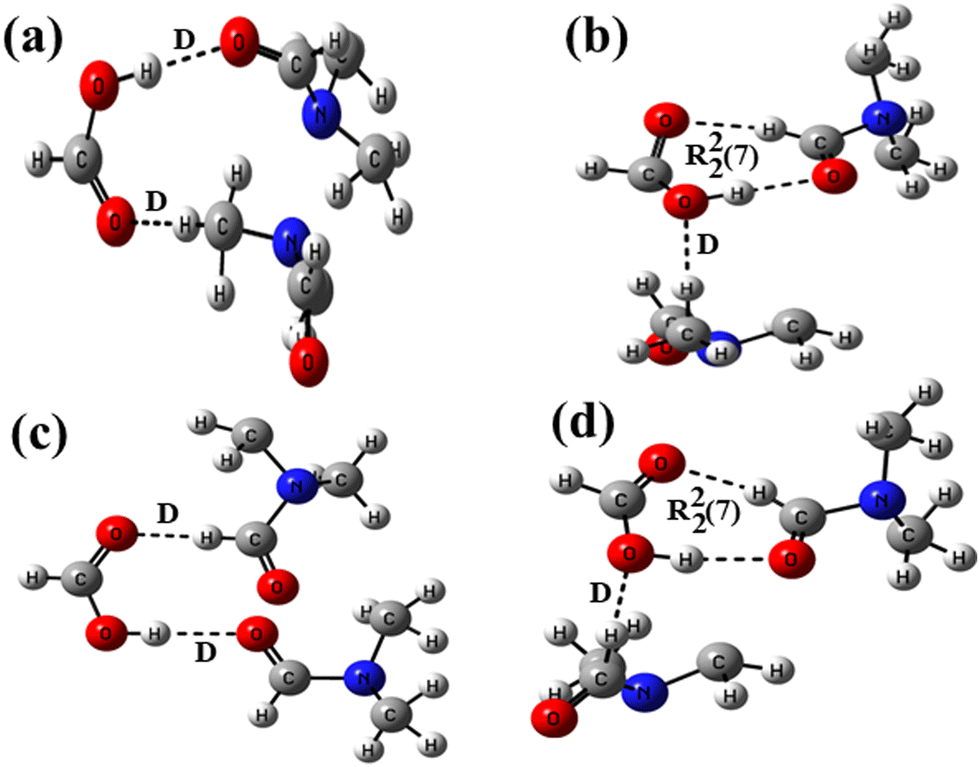 | ||
| Fig. 3 Optimized structures of formic acid–DMF at the B3LYP/6-31+G* level showing (a) two D motifs, (b) D and R22(7) motifs, (c) two D motifs and (d) D and R22(7) motifs. | ||
In structure 3a, the carbonyl oxygen and methyl hydrogen of two DMF molecules are allowed to form a discrete acceptor O–H⋯O interaction and a discrete donor C–H⋯O interaction with a formic acid molecule. In structure 3b, optimization of a formic acid and two DMF molecules facilitated the formation of a similar discrete donor C–H⋯O interaction along with two short range donor–acceptor C–H⋯O and O–H⋯O interactions. The interaction energies of 3a and 3b along with the relative stability of 3b calculated with different basis functions are shown in Table 1. It is evident from the inspections of crystal structures (1a and 2a) and optimized structures (3a and 3b) that the DMF molecules form two hydrogen bonds with the host in the case of the former, while three hydrogen bonds in the case of the latter; consequently, structure 3b was found to be more stable than 3a. The relative stability of 3b is found to be higher (4.0 eV) with lower-level basis function B3LYP/6-31+G*, lowered with higher-level basis functions such as 6-31++G* (3.99 eV) and 6-31+G** (3.78 eV), and is smaller (3.67 eV) with highly polarized and double diffused basis function AUG-cc-pVDZ. The formation of discrete and cyclic R22(7) motifs (I and II in Scheme 1) in an equal stoichiometric ratio between the –COOH groups of two isomeric hosts and the DMF molecule has recently been studied experimentally and theoretically.55 Our recently reported and current investigations confirm that the cyclic R22(7) is more stable than the D motif.
| Functional/basis sets | Interaction energies (Eint) | Relative stabilities | |
|---|---|---|---|
| 3a | 3b | Stability of 3b over 3a | |
| B3LYP/6-31+G* | −7.4430 | −11.4450 | 4.0020 |
| B3LYP/6-31++G* | −7.0742 | −11.0634 | 3.9892 |
| B3LYP/6-31+G** | −8.5083 | −12.2870 | 3.7787 |
| AUG-cc-pVDZ | −10.1790 | −13.8498 | 3.6708 |
| Functional/basis sets | Interaction energies (Eint) | Relative stabilities | |
|---|---|---|---|
| 3c | 3d | Stability of 3d over 3c | |
| B3LYP/6-31+G* | −6.5270 | −11.5357 | 5.0087 |
| B3LYP/6-31++G* | −6.5626 | −12.6610 | 6.0984 |
| B3LYP/6-31+G** | −7.9430 | −12.9110 | 4.9680 |
| AUG-cc-pVDZ | −9.6022 | −14.4675 | 4.8653 |
We have also designed two other motifs V and VI (as shown in Scheme 1), which are almost similar to motifs III and IV, respectively. In these two motifs, the position of one DMF molecule is changed, and it is allowed to create a discrete acceptor C–H⋯O interaction with the –COOH group via α-hydrogen instead of methyl-hydrogen. Because these motifs are not observed in the structures of DMF solvates, the possibility of their formation is also checked by DFT calculations. For this purpose, analogous structures 3c and 3d are constructed from formic acid and DMF (Fig. 3), and their interaction energies are calculated at different basis functions (Table 1). Similar to the aforementioned results, structure 3d is found to be more stable than structure 3c. The relative stability is found to be the highest (6.09 eV) in the case of basis function 6-31++G* and the lowest (4.86 eV) in the case of basis function AUG-cc-pVDZ. Moreover, to check the relative stabilities of motifs V and VI with motifs III and IV, the interaction energies of structures 3a and 3b are compared with those of 3c and 3d, respectively (Table S1; ESI†). It is observed that structure 3a, which is analogous to motif III and is observed in the structure of solvate 1a, is more stable than structure 3c, which is analogous to motif V and is not observed in the structures of DMF solvates. Unlikely, structure 3b is less stable than structure 3d, showing minor energy differences for each basis set. The stability of 3a over 3c is found in the range of 0.51–0.92 eV and the stability of 3d over 3b is found in the range of 0.62–1.59 eV, with various levels of functional calculations. The differences in the interaction energies of such alike motifs are negligible. Therefore, it can be concluded that various other weak interactions along with the interaction energies of dissimilar motifs play a crucial role in their formation. The significance of such small interaction energies of slightly different hydrogen bond motifs in the crystal lattice is documented well towards the improvement of chemical and physical properties of multi-component crystalline materials.56,57
Solvate 1b crystallizes in a triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group as a 1:2 host–guest complex containing half of the host molecule and one pyridine guest molecule in its crystallographic asymmetric unit. In the structure of 1b, all the six hydrogen bonding sites of the guest pyridine molecule make hydrogen bonds with the host molecule (Fig. 4). The guest pyridine molecule binds with the –COOH group of the host molecule via the cyclic R22(7) hydrogen bond motif, which is formed by the combination of acceptor–donor OH–N (O3–H3⋯N1P, dD⋯A 2.63 Å, ∠D–H⋯A 173.6°) and CH–O (C5P–H5P⋯O4, 3.61 Å, 124.7°) interactions. It also interacts with both the oxygen atoms of the –COOH group via two discrete CH–O (C1P–H1P⋯O3, 3.47 Å, 156.6° and C4P–H4P⋯O4, 3.64 Å, 121.6°) interactions. Moreover, the remaining two hydrogen bonding sites of pyridine engage in the formation of C3P–H3P⋯O1 (3.41 Å, 136.5°) and C2P–H2P⋯O2 (3.56 Å, 175.5°) bonds with the carbonyl oxygen atoms of the host molecule, making a 2D host–guest assembly. The host molecules interact with each other via short H–H (H3A⋯H7C, 2.35 Å) contacts to form a 3D channelled architecture, which is sustained by the repeated tetrameric assembly of host molecules as viewed along the b axis. The guest pyridine molecules reside inside the channels of 18.06 × 6.40 Å dimensions by interacting with each other via π–π (3.37 Å) contacts.
space group as a 1:2 host–guest complex containing half of the host molecule and one pyridine guest molecule in its crystallographic asymmetric unit. In the structure of 1b, all the six hydrogen bonding sites of the guest pyridine molecule make hydrogen bonds with the host molecule (Fig. 4). The guest pyridine molecule binds with the –COOH group of the host molecule via the cyclic R22(7) hydrogen bond motif, which is formed by the combination of acceptor–donor OH–N (O3–H3⋯N1P, dD⋯A 2.63 Å, ∠D–H⋯A 173.6°) and CH–O (C5P–H5P⋯O4, 3.61 Å, 124.7°) interactions. It also interacts with both the oxygen atoms of the –COOH group via two discrete CH–O (C1P–H1P⋯O3, 3.47 Å, 156.6° and C4P–H4P⋯O4, 3.64 Å, 121.6°) interactions. Moreover, the remaining two hydrogen bonding sites of pyridine engage in the formation of C3P–H3P⋯O1 (3.41 Å, 136.5°) and C2P–H2P⋯O2 (3.56 Å, 175.5°) bonds with the carbonyl oxygen atoms of the host molecule, making a 2D host–guest assembly. The host molecules interact with each other via short H–H (H3A⋯H7C, 2.35 Å) contacts to form a 3D channelled architecture, which is sustained by the repeated tetrameric assembly of host molecules as viewed along the b axis. The guest pyridine molecules reside inside the channels of 18.06 × 6.40 Å dimensions by interacting with each other via π–π (3.37 Å) contacts.
 | ||
| Fig. 4 (a) Intermolecular interactions of the host–guest assembly of 1b. (b) Formation of tetrameric assemblies of the hosts encapsulating the guests in the crystal lattice of 1b (along the b axis). (c) Space filling model of a 3D channel architecture after removal of guests inside the channels. | ||
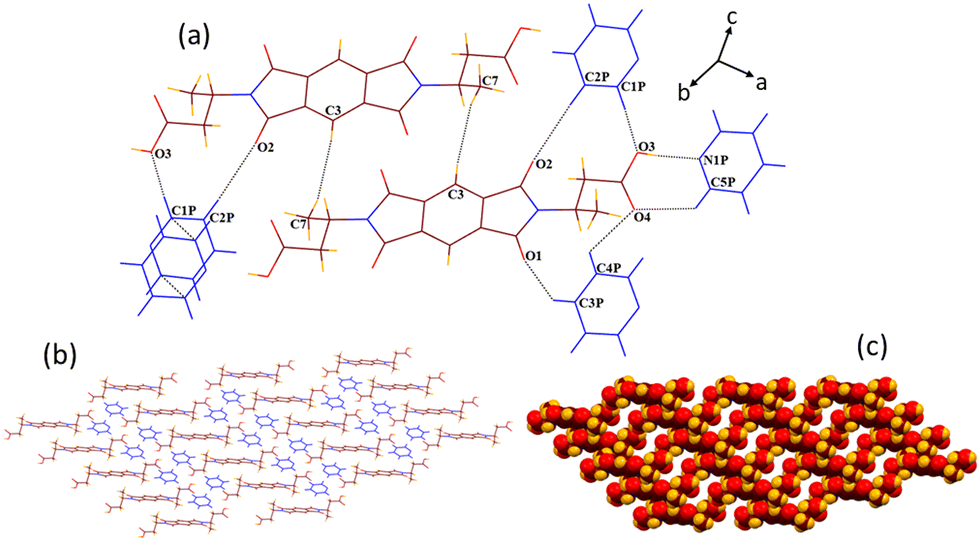
Solvate 2b crystallizes in the monoclinic P21/c space group. A crystallographic asymmetric unit of 2b includes half of the host molecule and one pyridine guest molecule. In the crystal lattice of 2b, all the six hydrogen bonding sites and the π-bonding site of the guest pyridine molecule interact with the host molecule (Fig. 5). Compared with the structure of 2a, guest pyridine similarly interacts with the host by the combination of O3–H3⋯N1P (dD⋯A 2.75 Å, ∠D–H⋯A 177.1°) and C1P–H1P⋯O4 (3.12 Å, 129.3°) interactions, making a cyclic R22(7) motif. In contrast to the structure of 2a, where guest pyridine was found to interact with both the oxygen atoms of the –COOH group via two discrete CH–O hydrogen bonds, the guest pyridine makes bifurcated CH–O (C3P–H3P⋯O4, 3.56 Å, 151.7° and C2P–H2P⋯O4, 3.46 Å, 138.6°) contacts with only one oxygen atom of the –COOH group in the structure of 2b. The formation of one more weak bond C5P–H5P⋯O1 (3.22 Å, 150.9°) also occurs between the guest and carbonyl oxygen of five membered cyclic imide rings of the host. The aromatic ring of the guest molecule interacts with the methylene group of the host via a weak C71–H71⋯π (2.87 Å) interaction. Apart from these host–guest interactions, host molecules also interact with each other via two types of CH–O (C81–H81⋯O1, 3.67 Å, 168.7° and C3–H3A⋯O2, 3.26 Å, 137.5°) and O–π (O3⋯π, 3.19 Å and O2⋯π, 3.0 Å) interactions. The guest molecules interact with each other via C4P–H4P⋯π (2.84 Å) and π–π (3.3 Å) contacts. These interactions result in the formation of a similar type of zigzag 3D supramolecular architecture (along the b axis), as observed in the case of DMF solvate 2a.
 | ||
| Fig. 5 (a) Various weak interactions in the crystal lattice of 2b. (b) Formation of a 3D zigzag host–guest structural arrangement in the supramolecular structure of 2b (along the b axis). | ||
The crystal structure analyses of solvates 1b and 2b also revealed the formation of distinct motifs between the –COOH groups of hosts and guest pyridine molecules with 1:3 stoichiometric ratios in both cases (motifs IX and X in Scheme 1). The analogous structures of such motifs between the formic acid and pyridine are constructed at the B3LYP/6-31+G* level of the basis function (Fig. 6). In these structures, one pyridine molecule is allowed to form a cyclic R22(7) hydrogen bond by the combination of an acceptor O–H⋯N interaction and a donor C–H⋯O interaction, while the other two pyridine molecules form two discrete donor C–H⋯O interactions with the –COOH group. In structure 6a, both the carbonyl and the hydroxyl oxygen atoms are involved in discretely binding two pyridine molecules, while in the case of structure 6b, only the carbonyl oxygen atom is involved in binding two pyridine molecules in a bifurcated manner. The interaction energies and the relative stabilities of these structures are calculated at different basis sets using the B3LYP functional (Table 2). Because structures 6a and 6b show almost similar types of binding patterns with four hydrogen bonds in each case, the differences in the interaction energies are also small on each basis set, demonstrating that structure 6b is more stable than structure 6a.
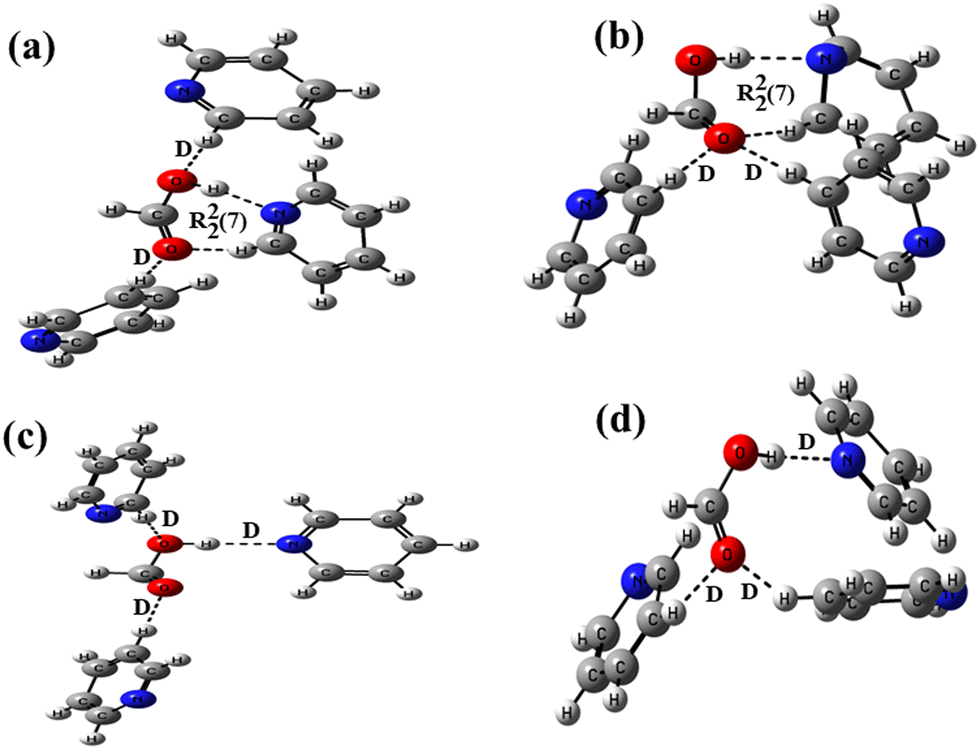 | ||
| Fig. 6 Optimized structures of formic acid–pyridine at the B3LYP/6-31+G* level making (a) two D and a R22(7) motifs, (b) two D and a R22(7) motifs, (c) three D motifs, and (d) three D motifs. | ||
| Functional/basis sets | Interaction energies (Eint) | Relative stabilities | |
|---|---|---|---|
| 6a | 6b | Stability of 6b over 6a | |
| B3LYP/6-31+G* | −12.4450 | −12.4942 | 0.0492 |
| B3LYP/6-31++G* | −12.4640 | −12.9875 | 0.5235 |
| B3LYP/6-31+G** | −13.2640 | −13.2695 | 0.0055 |
| AUG-cc-pVDZ | −13.6825 | −13.6892 | 0.0067 |
| Functional/basis sets | Interaction energies (Eint) | Relative stabilities | |
|---|---|---|---|
| 6c | 6d | Stability of 6d over 6c | |
| B3LYP/6-31+G* | −4.0820 | −6.9809 | 2.8989 |
| B3LYP/6-31++G* | −4.0093 | −7.2104 | 3.2011 |
| B3LYP/6-31+G** | −5.0489 | −7.9170 | 2.8681 |
| AUG-cc-pVDZ | −6.4721 | −9.3483 | 2.8762 |
The interaction energies of two probable motifs, XI and XII, which are not observed in pyridine solvates 1b and 2b, are also calculated to compare the stability of these motifs with motifs IX and X, respectively. In these motifs, three pyridine molecules are attached to the –COOH group via two discrete donor C–H⋯O and one discrete acceptor O–H⋯N bonds. The analogous structures between the formic acid and pyridine molecules are optimized for this purpose at B3LYP/6-31+G* level of basis function (6c and 6d depicted in Fig. 6), and their interaction energies and relative stabilities are calculated at different basis sets (Table 2). The results are similar to the trend observed for structures 6a and 6b, showing minor differences in the interaction energies between these structures. However, when the interaction energies of structures 6a and 6b are compared with 6c and 6d, respectively (Table S2, ESI†), the differences are noteworthy (∼5–8 eV) in determining the higher stability of structures 6a and 6b over 6c and 6d, respectively, which may be the reason for the formation of motifs IX and X in pyridine solvates 1b and 2b. The DFT calculations of the interaction energies of closely related motifs suggest that the formation of higher stable motifs is more feasible, and minor energy differences in the interaction energies may not impact alterations in hydrogen bond patterns, which are further guided by the presence of other weak interactions in the crystal lattices. The interaction energies of all the model structures between formic acid–DMF and formic acid–pyridine are also calculated at different basis sets of higher level B3LYP-D3 functional (Table S3, ESI†) and are found to follow a similar trend, as it was for different basis sets of functional B3LYP.
Solvate 1c crystallizes in a triclinic P space group showing half of the host molecule 1 and one guest quinoline molecule in its crystallographic asymmetric unit. In the crystal lattice of 1c, guest quinoline interacts with the –COOH group of the host molecule only via discrete O3–H3⋯N1Q (dD⋯A 2.65 Å, ∠D–H⋯A 173.9°) interaction (Fig. 7). Further, it makes contact with the carbonyl oxygens of the host molecule via C3Q–H3Q⋯O1 (3.3 Å, 122.1°) and C6Q–H6Q⋯O2 (3.58 Å, 162.9°) interactions and also makes weak π–π (3.26 Å) contact with the aromatic ring of the host molecule. The host molecules interact with each other via C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯π (3.14 Å) and weak H3A–H7C (2.34 Å) contacts. No interactions among the guest molecules are observed in the structure of 1c. Similar to pyridine solvate 1b, the 3D supramolecular network of 1c consists of tetrameric assemblies of host molecules encapsulating the guest molecules inside the channels of 19.7 × 6.6 Å dimension, as viewed along the a-axis.
O⋯π (3.14 Å) and weak H3A–H7C (2.34 Å) contacts. No interactions among the guest molecules are observed in the structure of 1c. Similar to pyridine solvate 1b, the 3D supramolecular network of 1c consists of tetrameric assemblies of host molecules encapsulating the guest molecules inside the channels of 19.7 × 6.6 Å dimension, as viewed along the a-axis.
 | ||
| Fig. 7 (a) Part of the crystal structure of 1c showing different types of weak interactions. (b) Formation of tetrameric assemblies of hosts encapsulating guests in a 3D hydrogen-bonded structure of 1c (along the a-axis). (c) Space filling model after removal of guest quinoline molecules inside the channels. | ||
Solvate 2c crystallizes in the triclinic P space group, and the asymmetric unit of this has one molecule of host 2 and two symmetry non-equivalent guest quinoline molecules. In the crystal lattice of 2c, both sets of symmetry independent guest quinoline molecules make discrete OH–N (O7–H7⋯N1Q, dD⋯A 2.67 Å, ∠D–H⋯A 154.4° and O5–H5⋯N2Q, 2.62 Å, 144.3°) contacts with both the –COOH groups of host molecule (Fig. 8). Apart from that, the first set of quinoline molecules (shown as blue) interact with the carbonyl oxygen of host molecule via C7Q–H7Q⋯O4 (3.29 Å, 144.3°) interaction, while the second set of quinoline molecules (shown in red) form two more discrete donor CH–O (C10Q–H10Q⋯O8, 3.47 Å, 145.6° and C12Q–H12Q⋯O8 Å, 3.25, 160°) contacts with the –COOH group of the host along with the same type of CH–O (C15Q–H15Q⋯O1, 3.37 Å, 156.4°) contact, as formed by the first set of guest molecules with the host. The host molecules interact with each other via C3–H3⋯O6 (3.46 Å, 157.2°) and CO⋯π (C10–O4⋯π, 3.17 Å) interactions. Overall, a 3D host–guest channelled architecture is formed along the b axis by the combination of the host and second type of guest quinoline molecules. The first type of guest molecules resides inside these channels of approximately 14.8 × 8.8 Å dimensions. Even though the second type of guest molecules form π–π interactions with the host (3.391 Å), no weak interactions among the guest molecules are observed in the supramolecular structure of 2c.
 | ||
| Fig. 8 (a) Various weak interactions between the host and two types of symmetry independent guest molecules in the structure of 2c. (b) Formation of a 3D supramolecular channel architecture by the combination of host and one type of symmetry independent guest molecule (red) (along the b axis). (c) Space filling model after removal of another type of guest molecules (blue) inside the channels. | ||
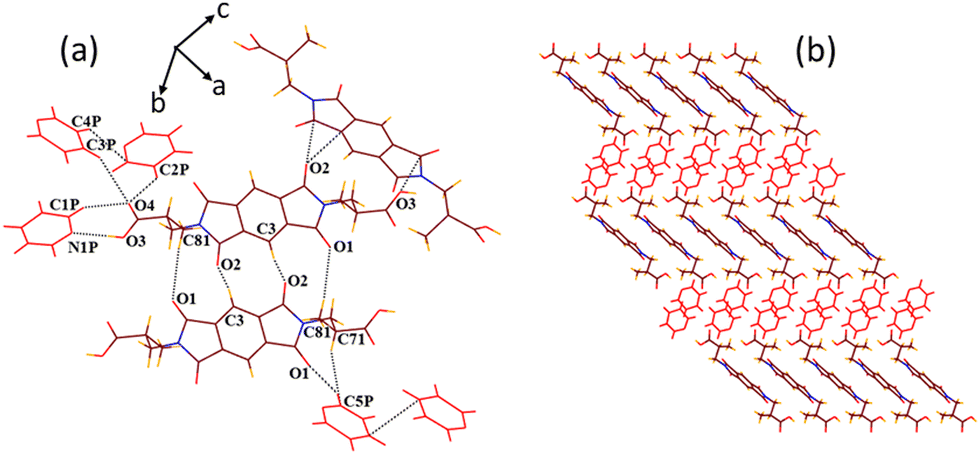
Solvate 2d crystallizes in the triclinic P space group, including half molecule of host 2 lying on the inversion centre with half guest quinoline and one guest piperidine molecule in its crystallographic asymmetric unit. Because the host and guest quinoline molecules are found disordered in the structure of 2d, a better description of hydrogen bonding is not possible. However, the guest piperidine molecules are found to interact with the –COOH group of the host via two discrete CH–O (C2P–H2P2⋯O31, dD⋯A 2.72 Å, ∠D–H⋯A 169.5° and C2P–H2P1⋯O41, 2.67 Å, 157.3°) interactions (Fig. 9). Moreover, two methylene hydrogens of guest piperidine interact with the carbonyl oxygen of the host via bifurcated CH–O (C1P–H1P1⋯O1, 3.59 Å, 170.8° and C4P–H4P1⋯O1, 3.61 Å, 159°) interactions. A 2D host–guest assembly thus formed by the combination of host and guest piperidine is further converted to a 3D supramolecular channelled architecture via C3–H3⋯O2 (3.28 Å, 127°) host–host interactions. The guest quinoline molecules interact with the hosts only via strong π–π (3.38 Å) contacts and reside inside the channels of approximately 9.4 × 19.5 dimensions, as viewed along the b axis. No interactions among both types of guest molecules are observed in the crystal lattice of 2d.
 | ||
| Fig. 9 (a) Intermolecular interactions in the host–guest assembly of 2d. (b) Formation of 3D supramolecular channel network by the combination of hosts and guest piperidine molecules accommodating guest quinoline molecules (along the b axis). (c) Space filling model after the removal of guest quinoline molecules inside the channels. | ||
Fluorescence emission, PXRD and thermal analyses
Aromatic imides are a class of important π-conjugated organic luminescent materials used in optoelectronics due to their rigid structures, excellent photochemical and thermal stabilities, high fluorescence quantum yields and good electron transmission abilities. In particular, aromatic cyclic imides, such as PMDI and NDI, exhibited remarkable contributions in a wide range of applications, including organic light-emitting diodes,58 organic field-effect transistors,59 organic solar cells,60 chemosensors,61 and bioprobes.62 Such types of π-conjugated small organic emitters have been reported to exhibit emissions in the blue spectral region.In view of their potential applications, the solid state fluorescence emission properties of both the host compounds along with their solvates are recorded. All these materials are found to show emissions in the blue spectral region (Fig. 10). Upon excitation at 340 nm, host 1 and its DMF solvate 1a show similar emission patterns with an emission peak at 455 nm along with two shoulder peaks at 400 and 485 nm, respectively. However, major differences are observed between the emission spectra of host 1 and its pyridine and quinoline solvates (1b and 1c). Solvates 1b and 1c show highly intense emission peaks at 410 and 450 nm, respectively. It should be noted that along with the intensity of emission peaks, the emission of pyridine solvate 1b appears to be blue shifted at ∼40–45 nm in comparison to the emissions of host 1 and its other solvates (Fig. 10a).
 | ||
| Fig. 10 Solid state fluorescence emission spectra of (a) host 1 and its solvates 1a, 1b and 1c and (b) host 2 and its solvates 2a, 2b, 2c and 2d. | ||
Similarly, upon excitation at 340 nm, the emission spectra of host 2 and its DMF solvate 2a also appear to resemble each other, with the spectra of host 1 and its DMF solvate 1a, respectively. The emission spectrum of pyridine solvate 2b is also similar to the host and its DMF solvates except showing a comparatively more intense blue-shifted peak at 400 nm. Similar to the quinoline solvate of host 1, strong emission peaks for quinoline solvate 2c and mixed quinoline/piperidine solvate 2d of host 2 are also obtained in the 445–455 nm region (Fig. 10b). To directly compare the variations in emission wavelengths of these samples, normalised emission spectra of both the hosts and their solvates, along with the photographs of emissive materials in the ground state, are shown in Fig. S5 and S6 (ESI†), respectively. Because the emission spectra of all the solvates are recorded in the solid state, the relative quantum yields for these systems could not be achieved.
It is quite obvious from the structural investigations of all solvates that the 3D supramolecular architectures of pyridine and quinoline solvates, namely, 1b, 1c, 2c and 2d, are moderately different from other solvates showing intense emissions. In the structures of these solvates, the size of guest molecules as well as minor differences in the weak interactions changes the packing pattern, and the formation of porous architectures occurs containing channels and cavities of different dimensions accommodating the guest molecules inside them. The structures of other DMF and pyridine solvates, namely, 1a, 2a and 2b, show tight-packed 3D layered architectures of hosts and guests with no channels and cavities. Therefore, the differences in the emission spectra are probably due to changes in the host–guest supramolecular arrangements in the solid crystalline phases. It has been demonstrated that the emissive properties of materials can be tuned from lower to higher by increasing the porosity of supramolecular aggregates.63 Moreover, extending the conjugated backbone of aromatic imides or adding a π-conjugated guest by cooperative donor–acceptor interactions is an efficient way to enhance luminescence efficiency. It is also evident from the crystal structure inspections that the photoluminescence of the solvates with aromatic guests is found to increase 2–3 fold due to the aggregation-induced emission enhancement (AIEE), which is attributed to the strong π–π interactions between the electron deficient PMDI hosts and electron rich aromatic guests. It has been established that strong π–π stacking in a solid state can deactivate the non-radiative decay pathway, leading to a longer lifetime of luminescent materials, and is the main cause for enhanced emission behaviour.64 Such photoluminescence enhancements originate from the emissive exciplex or the excited electronic intermolecular charge transfer state formed by the hosts and aromatic guest molecules in the crystals.65 The photoluminescence enhancement of a metal NDI framework is revealed by the formation of an exciplex electronic charge transfer state between the framework and electron rich naphthalene guest molecules.66 It has already been reported that the photoluminescence of electron deficient NDI hosts can be enhanced by the interaction with π-electron rich aromatic systems through a charge-transfer excitation mechanism.67
Notably, the emission spectra of pyridine solvates 1b and 2b appear to be blue shifted (∼40–50 nm) compared with the other solvates of hosts 1 and 2, e.g., DMF solvates 1a and 2a, respectively. Alternatively, the energies of the FMOs of the formic acid–DMF and formic acid–pyridine model structures are considered to justify the blue-shifted luminescence of solvates 1b and 2b. The theoretical band gaps between the LUMO and HOMO of model structures 3a and 6a as well as 3b and 6b, respectively, are calculated and compared with each other (Fig. 11). The higher band gap (2.58 eV) between the LUMO and HOMO of structure 6a than the band gap (1.29 eV) between the LUMO and HOMO of structure 3a suggested a lower emission wavelength for solvate 1b than for solvate 1a (Fig. 11a). Similarly, the higher band gap (1.46 eV) between the LUMO and HOMO of structure 6b than the band gap (0.75 eV) between the LUMO and HOMO of structure 3b suggested a lower emission wavelength for solvate 2b than for solvate 2a (Fig. 11b). According to the FMO theory, a higher band gap between the LUMO and HOMO implies a higher energy requirement for electronic transition, suggesting that the material emits light at shorter wavelength.68 The guest-dependent emission variations in the crystals of multimolecular hydrogen-bonded systems via guest–host donor–acceptor interactions have also been reported by Hisaeda's group.67
 | ||
| Fig. 11 Optimized Frontier molecular orbitals and orbital energies (in eV) of structures (a) 3a and 6a and (b) 3b and 6bvia DFT at the B3LYP/6-31+G* level. | ||
The PXRD patterns of all host–guest crystalline materials are recorded along with their host compounds to check the phase purity of bulk samples. The differences in the PXRD peaks of the individual host and its various complexes are clearly visible due to the presence of different guest solvent molecules in the crystal lattice of each host–guest complex (Fig. 12). The peaks obtained in the experimental PXRD patterns of all the complexes are found to correlate well with the peaks of their simulated patterns from SCXRD (Fig. S7–S13, ESI†). However, some small deviations among the theoretical and experimental peaks are attributed to the loss of solvent molecules from the solvates, leading to the loss of crystallinity.39,51
 | ||
| Fig. 12 PXRD patterns of (a) host 1 and its solvates 1a, 1b and 1c and (b) host 2 and its solvates 2a, 2b, 2c and 2d. | ||
To examine the stabilities and host–guest stoichiometries, TGA and DSC analyses of all the host–guest complexes are carried out (Fig. S14–S20, ESI†). The TGA curve of solvate 1a shows that it loses both the guest DMF molecules in one step (exp. 25.15% weight) in the temperature range of 50–100 °C (calcd 27.3%). A sharp endothermic peak appears at 85 °C for the loss of solvent molecules in its DSC profile. Solvate 1b also loses two guest pyridine molecules in a single step between 55 and 115 °C, showing a sharp endotherm in DSC at 110 °C (exp. 29.6%, calcd 28.9%). Solvate 1c similarly loses two guest quinoline molecules in the 115–165 °C temperature range, for which an endothermic peak appears at 152 °C in the DSC profile (exp. 40.13%, calcd 39.93%).
Solvate 2a loses two guest DMF molecules between 55 and 90 °C in one step, showing a sharp peak at 85 °C in the DSC curve (exp. 28.08%, calcd 27.3%). Solvate 2b starts losing guest molecules in one step as the temperature increases above the room temperature and ends at 110 °C, showing a sharp endothermic peak at 70 °C (exp. 22.9%, calcd 28.9%). Solvate 2c loses two quinolines in the 110–180 °C temperature range, showing two endothermic peaks at 120 and 155 °C for successive loss of two symmetry non-equivalent guest quinolines due to their different binding tendencies with the host (exp. 42.3%, calcd 39.93%). A single step weight loss for two piperidine and one quinoline guest molecule in the temperature range of 80–130 °C is observed for solvate 2d in the TGA curve with an endothermic peak at 125 °C in the DSC curve (exp. 40.13%, calcd 45%).
Conclusions
In this work, the attachment of multiple solvent molecules with the –COOH group of two isomeric pyromellitic diimide host molecules showing diverse interactions is assessed. The role of various intermolecular interactions in the construction of 3D supramolecular host–guest frameworks is discussed along with their varying photoluminescence behaviour. In the crystal lattices of DMF solvates 1a and 2a, both DMF guests are attached to the –COOH groups of hosts via two D motifs and via one R motif and one D motif, respectively. In pyridine solvates 1b and 2b, three pyridine molecules are also attached with –COOH groups of hosts having different combinations of hydrogen bonds, viz., via two dissimilar types of D and one R and via two similar types of D and one R motifs, respectively. In quinoline solvates 1c and 2c, quinoline molecules are attached with the –COOH groups via a similar D motif, while no interaction of guest quinoline with the host is observed in the case of mixed solvate 2d. Both the experimental and theoretical estimations are carried out to analyse the formation of some new and distinct hydrogen bond motifs between the two similar host–guest binding entities. The possibility of the formation of some other closely related motifs between the identical host and guest with the same number of donor/acceptor groups, as observed in the structures of solvates, is also supported by the results of the DFT calculations. The variations in the photophysical properties of hydrogen-bonded frameworks are closely associated with the energy differences between the HOMO and LUMO of the designed model synthons. It is concluded that minor differences in the interaction energies may not restrict the formation of such diverse motifs between the same binding entities, and the formation of either motif depends on the geometric alignment of the host and other host–guest weak interactions in the crystal lattices. However, major differences in the interaction energies of hydrogen bond patterns may result in desired molecular arrangements, leading to changes in physicochemical properties and potential applications of multi-component hydrogen-bonded solids, such as pharmaceuticals, agrochemicals, biomedicals, and optical materials.Author contributions
R. R. Puniya, P. Takhar and M. Chhapoliya performed the experiments and analysed the data. R. Deka and D. J. Kalita performed DFT calculations. D. Singh wrote the manuscript and approved the final version with the contributions of all authors.Data availability
The data supporting this article are available in the ESI.† Crystallographic data for this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2324804–2324810.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the University Grant Commission (UGC), New Delhi, India for financial support and RUSA 2.0 for providing instrument facilities to the Department of Chemistry, MLSU, Udaipur, India.References
- D. Venkataraman, S. Lee, J. Zhang and J. S. Moore, An organic solid with wide channels based on hydrogen bonding between macrocycles, Nature, 1994, 371, 591–593 CrossRef CAS.
- M. E. Davis, Ordered porous materials for emerging applications, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Multifunctional porous hydrogen-bonded organic framework materials, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- T.-U. Yoon, S. B. Baek, D. Kim, E.-J. Kim, W.-G. Lee, B. K. Singh, M. S. Lah, Y.-S. Bae and K. S. Kim, Efficient separation of C2 hydrocarbons in a permanently porous hydrogen-bonded organic framework, Chem. Commun., 2018, 54, 9360–9363 RSC.
- M. Simard, D. Su and J. D. Wuest, Use of hydrogen bonds to control molecular aggregation. Self-assembly of three-dimensional networks with large chambers, J. Am. Chem. Soc., 1991, 113, 4696–4698 CrossRef CAS.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, Origin of attraction and directionality of the π/π interaction: model chemistry calculations of benzene dimer interaction, J. Am. Chem. Soc., 2002, 124, 104–112 CrossRef CAS PubMed.
- I. Hisaki, S. Nakagawa, N. Ikenaka, Y. Imamura, M. Katouda, M. Tashiro, H. Tsuchida, T. Ogoshi, H. Sato, N. Tohnai and M. Miyata, A series of layered assemblies of hydrogen-bonded, hexagonal networks of C3-symmetric π-conjugated molecules: a potential motif of porous organic materials, J. Am. Chem. Soc., 2016, 138, 6617–6628 CrossRef CAS PubMed.
- P. Wei, X. He, Z. Zheng, D. He, Q. Li, J. Gong, J. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, M. Liu and B. Z. Tang, Robust supramolecular nano-tunnels built from molecular bricks, Angew. Chem., Int. Ed., 2021, 60, 7148–7154 CrossRef CAS PubMed.
- W. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa, N. R. Champness and M. Schröder, Exceptional thermal stability in a supramolecular organic framework: porosity and gas storage, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS.
- S. Yu, G.-L. Xing, L.-H. Chen, T. Ben and B.-L. Su, Crystalline porous organic salts: from micropore to hierarchical pores, Adv. Mater., 2020, 32, 2003270 CrossRef CAS.
- J. Liang, S. Xing, P. Brandt, A. Nuhnen, C. Schlüsener, Y. Sun and C. Janiak, A chemically stable cucurbit[6]uril-based hydrogen-bonded organic framework for potential SO2/CO2 separation, J. Mater. Chem. A, 2020, 8, 19799–19804 RSC.
- H. Wang, Z. Bao, H. Wu, R.-B. Lin, W. Zhou, T.-L. Hu, B. Li, J. C.-G. Zhao and B. Chen, Two solvent-induced porous hydrogen-bonded organic frameworks: solvent effects on structures and functionalities, Chem. Commun., 2017, 53, 11150–11153 RSC.
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling, M. Gu, C. Ma, L. Gu, Z. An and W. Huang, Hydrogen-bonded organic aromatic frameworks for ultralong phosphorescence by intralayer π–π interactions, Angew. Chem., Int. Ed., 2018, 57, 4005–4009 CrossRef CAS PubMed.
- B. Wang, R. He, L.-H. Xie, Z.-J. Lin, X. Zhang, J. Wang, H. Huang, Z. Zhang, K. S. Schanze, J. Zhang, S. Xiang and B. Chen, Microporous hydrogen-bonded organic framework for highly efficient turn-up fluorescent sensing of aniline, J. Am. Chem. Soc., 2020, 142, 12478–12485 CrossRef CAS PubMed.
- Y. Tang, M. Yuan, B. Jiang, Y. Xiao, Y. Fu, S. Chen, Z. Deng, Q. Pan, C. Tian and H. Fu, Inorganic acid-derived hydrogen-bonded organic frameworks to form nitrogen-rich carbon nitrides for photocatalytic hydrogen evolution, J. Mater. Chem. A, 2017, 5, 21979–21985 RSC.
- B. Han, H. Wang, C. Wang, H. Wu, W. Zhou, B. Chen and J. Jiang, Postsynthetic metalation of a robust hydrogen-bonded organic framework for heterogeneous catalysis, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed.
- F. Q. Liu, J. W. Liu, Z. Gao, L. Wang, X.-Z. Fu, L. X. Yang, Y. Tao, W. H. Yin and F. Luo, Constructing bimetal-complex based hydrogen-bonded framework for highly efficient electrocatalytic water splitting, Appl. Catal., B, 2019, 258, 117973 CrossRef CAS.
- A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Hydrogen-bonded organic frameworks (HOFs): a new class of porous crystalline proton-conducting materials, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS PubMed.
- Y. Wang, M. Zhang, Q. Yang, J. Yin, D. Liu, Y. Shang, Z. Kang, R. Wang, D. Sun and J. Jiang, Single-crystal-to-single-crystal transformation and proton conductivity of three hydrogen-bonded organic frameworks, Chem. Commun., 2020, 56, 15529–15532 RSC.
- W. Liang, F. Carraro, M. B. Solomon, S. G. Bell, H. Amenitsch, C. J. Sumby, N. G. White, P. Falcaro and C. J. Doonan, Enzyme encapsulation in a porous hydrogen-bonded organic framework, J. Am. Chem. Soc., 2019, 141, 14298–14305 CrossRef CAS PubMed.
- X.-T. He, Y.-H. Luo, D.-L. Hong, F.-H. Chen, Z.-Y. Zheng, C. Wang, J.-Y. Wang, C. Chen and B.-W. Sun, Atomically thin nanoribbons by exfoliation of hydrogen-bonded organic frameworks for drug delivery, ACS Appl. Nano Mater., 2019, 2, 2437–2445 CrossRef CAS.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Designing hydrogen-bonded organic frameworks (HOFs) with permanent porosity, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS PubMed.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, Hydrogen-bonded organic frameworks as a tunable platform for functional materials, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- Y. He, S. Xiang and B. Chen, A microporous hydrogen-bonded organic framework for highly selective C2H2/C2H4 separation at ambient temperature, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- M. R. di Nunzio, I. Hisaki and A. Douhal, HOFs under light: relevance to photon-based science and applications, J. Photochem. Photobiol., C, 2021, 47, 100418 CrossRef CAS.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Pharmaceutical cocrystals: along the path to improved medicines, Chem. Commun., 2016, 52, 640–655 RSC.
- F. Hu, C. Liu, M. Wu, J. Pang, F. Jiang, D. Yuan and M. Hong, An ultrastable and easily regenerated hydrogen-bonded organic molecular framework with permanent porosity, Angew. Chem., Int. Ed., 2017, 56, 2101–2104 CrossRef CAS PubMed.
- N. A. Mir, R. Dubey and G. R. Desiraju, Strategy and methodology in the synthesis of multicomponent molecular solids: the quest for higher cocrystals, Acc. Chem. Res., 2019, 52, 2210–2220 CrossRef CAS PubMed.
- O. Ermer, Five-Fold Diamond Structure of Adamantane-1,3,5,7-Tetracarboxylic Acid, J. Am. Chem. Soc., 1988, 110, 3747–3754 CrossRef CAS.
- V. A. Russell, M. C. Etter and M. D. Ward, Layered materials by molecular design: structural enforcement by hydrogen bonding in guanidinium alkane- and arenesulfonates, J. Am. Chem. Soc., 1994, 116, 1941–1952 CrossRef CAS.
- A. Comotti, S. Bracco, A. Yamamoto, M. Beretta, T. Hirukawa, N. Tohnai, M. Miyata and P. Sozzani, Engineering switchable rotors in molecular crystals with open porosity, J. Am. Chem. Soc., 2014, 136, 618–621 CrossRef CAS PubMed.
- G. Xing, I. Bassanetti, S. Bracco, M. Negroni, C. Bezuidenhout, T. Ben, P. Sozzani and A. Comotti, A double helix of opposite charges to form channels with unique CO2 selectivity and dynamics, Chem. Sci., 2019, 10, 730–736 RSC.
- K. E. Maly, E. Gagnon, T. Maris and J. D. Wuest, Engineering hydrogen-bonded molecular crystals built from derivatives of hexaphenylbenzene and related compounds, J. Am. Chem. Soc., 2007, 129, 4306–4322 CrossRef CAS PubMed.
- M. Mastalerz and I. M. Oppel, Rational construction of an extrinsic porous molecular crystal with an extraordinary high specific surface area, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. S. Miljanić, Thermally robust and porous noncovalent organic framework with high affinity for fluorocarbons and CFCs, Nat. Commun., 2014, 5, 5131 CrossRef CAS PubMed.
- W. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa, N. R. Champness and M. Schröder, Exceptional thermal stability in a supramolecular organic framework: porosity and gas storage, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS PubMed.
- H. Zhou, Q. Ye, X. Wu, J. Song, C. M. Cho, Y. Zong, B. Z. Tang, T. S. A. Hor, E. K. L. Yeow and J. Xu, A thermally stable and reversible microporous hydrogen-bonded organic framework: aggregation induced emission and metal ion-sensing properties, J. Mater. Chem. C, 2015, 3, 11874–11880 RSC.
- D. Singh, P. K. Bhattacharyya and J. B. Baruah, Structural studies on solvates of cyclic imide tethered carboxylic acids with pyridine and quinoline, Cryst. Growth Des., 2010, 10, 348–356 CrossRef CAS.
- D. Singh and J. B. Baruah, Metal(II) complexes derived from conformation flexible cyclic imide tethered carboxylic acids: syntheses, supramolecular structures, and molecular properties, Cryst. Growth Des., 2012, 12, 2109–2121 CrossRef CAS.
- D. Singh and J. B. Baruah, Molecular complex from two different binuclear copper 1,4,5,8-naphthalenetetracarboxylate complexes, Inorg. Chim. Acta, 2012, 390, 37–40 CrossRef CAS.
- D. Singh and J. B. Baruah, Varieties in symmetry non-equivalent structural arrangements in solvates of 2-(3-methylene-1,3,7-trioxo-6-(2-carboxy-phenyl)-3,5,6,7-tetrahydro-1H-pyrrolo[3,4-f]isoindol-2-yl)benzoic acid, J. Mol. Struct., 2009, 937, 75–80 CrossRef CAS.
- G. Bolla and A. Nangia, Pharmaceutical cocrystals: walking the talk, Chem. Commun., 2016, 52, 8342–8360 RSC.
- X.-Z. Luo, X.-J. Jia, J.-H. Deng, J.-L. Zhong, H.-J. Liu, K.-J. Wang and D.-C. Zhong, A microporous hydrogen-bonded organic framework: exceptional stability and highly selective adsorption of gas and liquid, J. Am. Chem. Soc., 2013, 135, 11684–11687 CrossRef CAS PubMed.
- W. Gong, D. Chu, H. Jiang, X. Chen, Y. Cui and Y. Liu, Permanent porous hydrogen-bonded frameworks with two types of Brønsted acid sites for heterogeneous asymmetric catalysis, Nat. Commun., 2019, 10, 600 CrossRef CAS PubMed.
- Z. Sun, Y. Li, L. Chen, X. Jing and Z. Xie, Fluorescent hydrogen-bonded organic framework for sensing of aromatic compounds, Cryst. Growth Des., 2015, 15, 542–545 CrossRef CAS.
- P. Li, Y. He, J. Guang, L. Weng, J. C.-G. Zhao, S. Xiang and B. Chen, A homochiral microporous hydrogen-bonded organic framework for highly enantioselective separation of secondary alcohols, J. Am. Chem. Soc., 2014, 136, 547–549 CrossRef CAS PubMed.
- G. Xia, Z. Jiang, S. Shen, K. Liang, Q. Shao, Z. Cong and H. Wang, Reversible specific vapoluminescence behavior in pure organic crystals through hydrogen-bonding docking strategy, Adv. Opt. Mater., 2019, 7, 1801549 CrossRef.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, Supramolecular polymorphism in one-dimensional self-assembly by kinetic pathway control, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- G. D. Pantoş, P. Pengo and J. K. M. Sanders, Hydrogen-bonded helical organic nanotubes, Angew. Chem., Int. Ed., 2007, 46, 194–197 CrossRef PubMed.
- D. Singh and J. B. Baruah, Solvation controlling reaction paths and gel-formation in imide derivatives, Tetrahedron Lett., 2008, 49, 4374–4377 CrossRef CAS.
- D. Singh and J. B. Baruah, Solid state assemblies of cyclic imides tethered hydroxy benzoic acids with pyridine and quinoline: toward the formation of channels and cavities, Cryst. Growth Des., 2012, 12, 3169–3180 CrossRef CAS.
- D. Singh and J. B. Baruah, Structural study on solvates of dopamine-based cyclic imide derivatives, Cryst. Growth Des., 2011, 11, 768–777 CrossRef CAS.
- D. Singh and J. B. Baruah, Different solvates of two isomeric dicarboxylic acids with pyridine and quinoline, CrystEngComm, 2009, 11, 2688–2694 RSC.
- N. Barooah, R. J. Sarma and J. B. Baruah, Solid-state hydrogen bonded assembly of N,N′-bis(glycinyl)-pyromellitic diimide with aromatic guests, CrystEngComm, 2006, 8, 608–615 RSC.
- R. R. Puniya, P. Takhar, T. Kalita, D. J. Kalita and D. Singh, Design and Development of Hydrogen Bonded Molecular Assemblies Based on Pyromellitic Diimide Tethered Carboxylic Acids as Optical Materials, Mol. Syst. Des. Eng., 2023, 8, 929–941 RSC.
- M. Igarashi, T. Nozawa, T. Matsumoto, F. Yagihashi, T. Kikuchi and K. Sato, Parallel-stacked aromatic molecules in hydrogen-bonded inorganic frameworks, Nat. Commun., 2021, 12, 7025 CrossRef CAS PubMed.
- Y. Liu, L. Wang, L. Zhao, Y. Zhang, Z.-T. Li and F. Huang, Multiple hydrogen bonding driven supramolecular architectures and their biomedical applications, Chem. Soc. Rev., 2024, 53, 1592–1623 RSC.
- H. F. Higginbotham, P. Pander, R. Rybakiewicz, M. K. Etherington, S. Maniam, M. Zagorska, A. Pron, A. P. Monkman and P. Data, Triphenylamine disubstituted naphthalene diimide: elucidation of excited states involved in TADF and application in near-infrared organic light emitting diodes, J. Mater. Chem. C, 2018, 6, 8219–8225 RSC.
- H. Ran, X. Duan, R. Zheng, F. Xie, L. Chen, Z. Zhao, R. Han, Z. Lei and J.-Y. Hu, Two isomeric azulene-decorated naphthodithiophene diimide-based triads: molecular orbital distribution controls polarity change of OFETs through connection position, ACS Appl. Mater. Interfaces, 2020, 12, 23225–23235 CrossRef CAS PubMed.
- M. A. Jameel, T. C.-J. Yang, G. J. Wilson, R. A. Evans, A. Gupta and S. J. Langford, Naphthalene diimide-based electron transport materials for perovskite solar cells, J. Mater. Chem. A, 2021, 9, 27170–27192 RSC.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Luminescent chemodosimeters for bioimaging, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- B. Zhang, C. Ge, J. Yao, Y. Liu, H. Xie and J. Fang, Selective selenol fluorescent probes: design, synthesis, structural determinants, and biological applications, J. Am. Chem. Soc., 2015, 137, 757–769 CrossRef CAS PubMed.
- C. M. Carbonaro, S. V. Thakkar, R. Ludmerczki, C. Olla, A. Pinna, D. Loche, L. Malfatti, F. Cesare Marincola and M. F. Casula, How porosity affects the emission of fluorescent carbon dot-silica porous composites, Microporous Mesoporous Mater., 2020, 305, 110302 CrossRef CAS.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, The influence of the molecular packing on the room temperature phosphorescence of purely organic luminogens, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- T. Ono, A. Taema, A. Goto and Y. Hisaeda, Switching of monomer fluorescence, charge-transfer fluorescence, and room-temperature phosphorescence induced by aromatic guest inclusion in a supramolecular host, Chem. – Eur. J., 2018, 66, 17487–17496 CrossRef PubMed.
- J.-J. Liu, Y.-B. Shan, C.-R. Fan, M.-J. Lin, C.-C. Huang and W.-X. Dai, Encapsulating naphthalene in an electron-deficient MOF to enhance fluorescence for organic amines sensing, Inorg. Chem., 2016, 55, 3680–3684 CrossRef CAS PubMed.
- T. Ono, M. Sugimoto and Y. Hisaeda, Multicomponent molecular puzzles for photofunction design: emission color variation in Lewis acid–base pair crystals coupled with guest-to-host charge transfer excitation, J. Am. Chem. Soc., 2015, 137, 9519–9522 CrossRef CAS PubMed.
- J. Yu, X. Yong, Z. Tang, B. Yang and S. Lu, Theoretical understanding of structure–property relationships in luminescence of carbon dots, J. Phys. Chem. Lett., 2021, 12, 7671–7687 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2324804–2324810. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ma00634h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |