
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas phase electronic structure of the DTDCTB small-molecule donor for vacuum-processed organic photovoltaics compared to its constituent building blocks†
Ambra
Guarnaccio
 *a,
Iulia Emilia
Brumboiu
*b,
Cesare
Grazioli
c,
Teng
Zhang
d,
Fredrik O. L.
Johansson
e,
Marcello
Coreno
f,
Monica
de Simone
c and
Carla
Puglia
e
*a,
Iulia Emilia
Brumboiu
*b,
Cesare
Grazioli
c,
Teng
Zhang
d,
Fredrik O. L.
Johansson
e,
Marcello
Coreno
f,
Monica
de Simone
c and
Carla
Puglia
e
aCNR – Istituto di Struttura della Materia (ISM), Tito Scalo Branch, C/da S. Loja, 85050 Tito Scalo (Pz), Potenza, Italy. E-mail: ambra.guarnaccio@cnr.it
bFaculty of Physics, Astronomy and Informatics, Nicolaus Copernicus University in Toruń, 87-100 Toruń, Poland. E-mail: iubr@umk.pl
cCNR – Istituto Officina dei Materiali (IOM), 34149 Trieste, Italy
dSchool of Integrated Circuits and Electronics, and Yangtze Delta Region Academy, Beijing Institute of Technology, 100081 Beijing, People's Republic of China
eProgram for Condensed Matter Physics of Energy Materials, Department of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden
fCNR – Istituto di Struttura della Materia (ISM), Trieste Branch, in Basovizza Area Science Park, 34149 Trieste, Italy
First published on 8th July 2024
Abstract
This study provides a comprehensive analysis of the electronic structure of the small-molecule (SM) donor DTDCTB in terms of its main molecular components, DPTA and BTD, with donor and acceptor characters, respectively. The occupied electronic states of gas phase DTDCTB and the two building block molecules are probed using photoelectron (PE) spectroscopy, while the unoccupied electronic states are probed using near-edge X-ray absorption fine structure (NEXAFS) spectroscopy. Jointly, density functional theory (DFT) calculations of the electronic structure and X-ray absorption spectra are used to assign the experimental peaks. We find that the frontier characteristic peaks of the gas phase building blocks can be identified in the spectra of the DTDCTB molecule. In particular, the highest occupied molecular orbital (HOMO) of DTDCTB, corresponding to the first peak in the outer photoelectron valence spectrum, is attributed mainly to the DPTA moiety (∼70%), with only a small contribution (∼30%) from BTD and from the dicyanovinylene (CN) terminal group. In contrast, the lowest unoccupied molecular orbital (LUMO), identified as the first spectral feature of the C and N K-edge NEXAFS spectra, originates almost entirely from the moieties with acceptor character, BTD and CN (∼81%), with a marginal contribution (∼19%) from DPTA. This study aims to elucidate how the electronic structure of DTDCTB, crucial for its technological functionalities in small-molecule organic photovoltaics (SMOPVs), is a result of the electronic characteristics of its constituent building blocks. The DTDCTB molecule precisely combines the electronic properties of its constituent donor and acceptor building blocks: the inclusion of thiophene in the donor moiety facilitates π-electron delocalization from the donor side to the acceptor side of DTDCTB, thus leading to the formation of mesomeric structures. Therefore, the D–A–A molecular architecture is confirmed to be a strategic solution to guarantee the efficient charge transfer among the two electron-donor and electron-withdrawing counterparts.
Introduction
Small-molecule (SM) donors possess the advantages of well-defined molecular weights, easy purifications, and small batch-to-batch variations in comparison with polymers. An additional advantage relates with the design of SM with push–pull architectures in which the resulting molecular system is made by two counterparts: one (or more) electron-rich moieties and one (or more) electron-donating groups. Within this push–pull architecture, the electron-donating and electron-withdrawing groups work synergistically to modulate the energy levels and electronic structure of the molecule, resulting in tailored optical and electronic properties. The electron-rich donor moieties facilitate the injection and transport of charge carriers, while the electron-deficient acceptor moieties enhance exciton dissociation and charge separation efficiency.1 Understanding the electronic structure of push–pull molecules is fundamental for optimizing their performance in various optoelectronic applications. In this study, we focus on elucidating the intramolecular charge transport through the mapping of the electronic structure of the push–pull SM donor 2-((7-(5-(Di-p-tolylamino)thiophen-2-yl)benzo[c][1,2,5]thiadiazol-4-yl)methylene)malononitrile (DTDCTB) in terms of its constituent building blocks.DTDCTB was first synthesized by Wong and co-workers2 for application as an electron donor in the active layer of small-molecule organic photovoltaics (SMOPVs). It has a donor–acceptor–acceptor (D–A–A) architecture which favors intramolecular charge transfer. DTDCTB is characterized by a highest occupied molecular orbital (HOMO) with high binding energy (∼5.30 eV3,4), contributing to a large open circuit voltage (VOC) in photovoltaic devices, where VOC is determined by the energy separation between the HOMO of the donor and the lowest unoccupied molecular orbital (LUMO) of the acceptor constituting the organic solar cell (OSC) active layer. Additionally, DTDCTB has a relatively low band gap (1.86 eV optical gap3) which results in a wide absorption in the near-infrared region with a high extinction coefficient.2,5 Coupled with the relatively high hole mobility in DTDCTB thin films, all these properties make DTDCTB an ideal active-layer material for SMOPVs. In contrast to typical organic photovoltaics (OPVs), where the donor is an organic polymer forming a bulk-heterojunction (BHJ) with the acceptor material, SMOPVs display a mixed heterojunction (MHJ) architecture, where the donor and acceptor domains do not form a truly biscontinuous interpenetrating network any longer.6 In this architecture and in combination with C60 or C70 fullerenes as electron acceptors, spectrally resolved transient absorption measurements showed that charge separation at the DTDCTB:fullerene interface takes place on the ∼100 fs time-scale,6 where the excitons in the DTDCTB film are rather short-lived (tens of ps life-time, compared to hundreds of ps for poly-3-hexylthiophene – P3HT).6 The first organic solar cells built using DTDCTB as the active donor material in combination with fullerenes and different fullerene derivatives as its acceptor counterpart, managed to achieve up to 5–6% power conversion efficiencies (PCEs) in single junction devices obtained by vacuum-deposition.2,7–10 It was shown that OPVs with a solution-processed active layer based on DTDCTB:C70 have similar PCEs.11 The properties of DTDCTB, as well as DTDCTB:fullerene interfaces have been studied computationally using different levels of theory,12–17 including exciton dynamics18,19 and recombination.20 Starting from DTDCTB, other similar SM donor materials have been synthesized by replacing either the donor moiety or one of the acceptor moieties with other similar groups to fine-tune molecular properties such as the band gap and HOMO energy level.3,21 This fine-tuning allowed to increase the PCE above 6.6%.3,21 Further improvements of PCEs and other OPV parameters have been obtained by using DTDCTB as an active material in tandem solar cells,22–24 in ternary devices,5,25 or in triple junction OPVs, reaching up to 11.1% PCE.24
This study presents novel experimental data and simulations on the electronic structures of DTDCTB and 1,2,3-benzothiadiazole (BTD), complemented by previous data on N,N-diphenyl-2-thiophenamine (DPTA).26 The optimized molecular structures of DTDCTB together with its DPTA and BTD building blocks are sketched, respectively, in Fig. 1a–c. As shown in Fig. 1a, ideally the DTDCTB molecule can be considered to be made up by three main building blocks: DPTA (i.e., ditolylaminothienyl), BTD (i.e, benzothiadiazole) and the CN (i.e., dicyanovinylene) moieties. The DPTA moiety represents the electron-donor portion, while the BTD and the CN moieties are the electron-withdrawing counterparts.
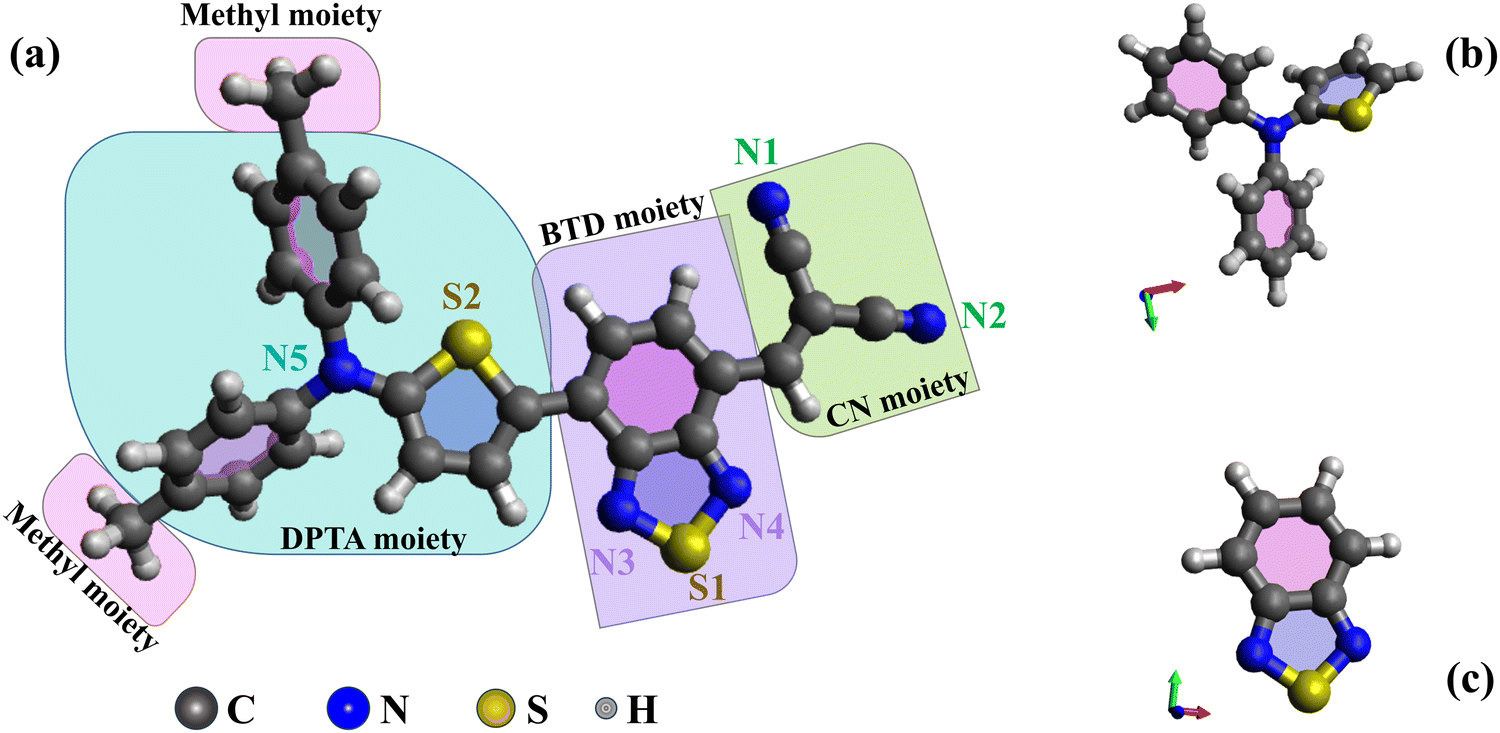 | ||
| Fig. 1 B3LYP/6-31G(d,p) optimized geometries of: (a) DTDCTB; (b) DPTA and (c) BTD molecules. | ||
In agreement with previous studies based on X-ray crystal structure analysis of DTDCTB,3 our geometry optimization confirms an almost coplanar conformation between the thiophene ring and the BTD ring which facilitates electronic coupling between the donor and acceptor units and enhances intramolecular charge transfer. The charge transport properties of the active materials significantly impact the performance of OPV cells. High carrier mobility ensures efficient exciton dissociation, facilitating charge transfer, and reducing the possibility of charge recombination.
The primary novelty of our work lies in the comparative analysis of these molecules to show if the D–A–A architecture of DTDCTB can be conceptualized as a structure composed of the distinct push–pull moieties, including the two main DPTA and BTD building blocks. With this aim in mind, we performed a comprehensive characterization of the gas phase electronic structure of DTDCTB focused on comparing it to the electronic structure of its building blocks, DPTA and BTD. We characterized the occupied electronic levels of all these molecules in gas phase by valence photoelectron spectroscopy (PES), as well as the unoccupied electronic structure by near-edge X-ray absorption fine structure (NEXAFS) spectroscopy. This allowed us to compare the electronic structure of the two main building blocks, DPTA and BTD, when they are isolated and when they are covalently bound in the D–A–A molecule to verify from the experimental point of view the charge transport ability of the push–pull architecture of DTDCTB.
Experimental details
Commercially available DTDCTB (C28H19N5S2, 99%) and 2,1,3-benzothiadiazole (BTD, C6H4N2S, 98%) from Merck and 2-(N,N-diphenylamino)thiophene (DPTA, C16H13NS, 97+%) from Georganics Ltd were used. All chemicals are crystalline solid powders that were sublimated in vacuum conditions by using a custom-built in situ resistively heated furnace (Knudsen cell).The purification process of DTDCTB involved placing a few milligrams of DTDCTB powder into the furnace. The temperature was slowly increased from room temperature to about 160 °C over a period of about 8 hours. Then the temperature was further increased until reaching the measurement temperature of approximately 170 °C following the removal of small amounts of impurities within the DTDCTB sample and at the same time preventing the molecular degradation caused by prolonged heating. The evaporation of DTDCTB was subject of a careful study, reported in a separated manuscript on the degradation of this technologically important molecule. Thanks to this study we were able to finally adjust the temperature to 172 °C for both PES and NEXAFS data acquisition, characterized by a modest but constant and reliable count rate.
Before starting the BTD measurements, a few milligrams of powder were put into an ex situ effusion cell and purged overnight at room temperature to allow the removal of the traces of impurities. Then the PES and NEXAFS measurements of BTD were performed by keeping the powder at room temperature. The experimental details of the sublimation and the characterization of the DPTA molecule are reported in detail in the paper by T. Zhang et al.26 For the DPTA molecule, a purging temperature of about 50 °C and a sublimation temperature of 61 °C were used.
Gas phase photoemission measurements were performed at the GasPhase beamline of Elettra Synchrotron in Trieste (Italy)27 by using, for the DTDCB and DPTA molecules, a Scienta SES200 electron analyzer28 mounted at the magic angle with respect to the electric vector of the linearly polarized incident light. For the BTD molecule, the photoemission measurements were acquired by using a VG-220i (150 mm) hemispherical electron energy analyzer mounted at the magic angle with respect to the electric vector of the linearly polarized incident light.29 For the calibration of the binding energy (BE) of the photoelectron (PE) spectra and of the photon energy of the NEXAFS spectra, a proper reference gas (see below) was introduced into the analysis chamber simultaneously with the sample, resulting in a pressure of about 10−5 mbar.
The experimental valence photoelectron spectra (VB) of DTDCTB, DPTA and BTD were taken with 100 eV photon energy, with a peak resolution of about 50 meV, 70 meV and 30 meV, respectively. The Ar 3p3/2 line, set to 15.76 eV,30 was used to calibrate the valence spectra.
All the NEXAFS spectra were normalized by the transmitted photon flux measured by a calibrated Si photodiode. The energy scales of the C 1s NEXAFS and N 1s NEXAFS spectra were calibrated by taking simultaneous spectra of the samples and of CO2 and N2 with the characteristic transitions at 290.77 eV (C 1s → π*, CO2)31 and at 401.10 eV32 (N 1s → π*, ν′ = 1), respectively.
The photon energy resolution of the C 1s NEXAFS spectra was about 120 meV for DTDCTB, 70 meV for DPTA and 50 meV for BTD. Moreover, the photon energy resolution of the N 1s NEXAFS spectra was around 110 meV for DTDCTB, 120 meV for DPTA and 40 meV for BTD.
Computational details
The molecular structures of DTDCTB, BTD, and DPTA were optimized using the B3LYP functional33 and 6-31G(d,p) basis set34 as implemented in the Gaussian 16 quantum chemistry software.35 The valence electronic structure was determined for the optimized geometries, where the atomic orbital contributions to the valence spectrum were calculated using the c2 population analysis,36,37 as done previously.38,39 To facilitate the comparison to the experiment, a Gaussian broadening of 0.4 eV full width at half maximum (FWHM) was applied to the calculated data. The spectra were further shifted to align the calculated highest occupied molecular orbital (HOMO) peak to the first peak in the experimental PE spectra. The values of the shifts are given in the corresponding figures. The electron localization on the donor (DPTA) and acceptor (BTD, CN) subgroups of DTDCTB was analyzed for the HOMO and LUMO molecular orbitals using the VALET Python library for the visual analysis of electronic densities.40 This analysis allowed us to quantify the percentage by which each molecular subgroup contributes to the frontier orbitals and determine the charge transferred from the donor to the acceptors in a HOMO-to-LUMO transition.The C 1s and N 1s NEXAFS spectra were calculated within the transition potential DFT (TP-DFT) framework using the StoBe 2014 quantum chemistry software.41 In TP-DFT, the core-hole (CH) induced orbital relaxation is modeled by using a half-core-hole (HCH) localized at the core-excited site. The localized CH model has been shown to provide a good approximation for orbital relaxation,42 and TP-DFT calculated spectra are reasonably accurate, especially when it comes to excitation energies.43 With this model, a separate NEXAFS spectrum was calculated for each atom of the same species in the molecule (C or N). We used the DFT functional by Perdew, Burke, and Ernzerhof,44 in combination with the cc-pVTZ basis set for the S, C and N atoms,45,46 as well as the cc-pVDZ basis set for H.47 The core-excited atom was described using an IGLO-III triple zeta basis set,48 while the 1s orbitals of other atoms of the same species as the core-excited one were replaced by effective core potentials (ECPs) provided by the StoBe program package. The occupation of the 1s orbital of the core-excited atom was set to 0.5 and the transition dipole moments from this HCH state to the unoccupied molecular orbitals (MOs) were determined alongside the corresponding excitation energies. The spectrum was then shifted such that the calculated binding energy of the core-excited 1s orbital was equal to the calculated ionization energy (IE). The IE was calculated as the energy difference between the ground state and the core-ionized state (i.e. where the occupation of the orbital of interest was set to 0, ΔKohn–Sham). A Gaussian broadening of 0.5 eV FWHM was added to the calculated data to facilitate the comparison to the experiment. A total C 1s (N 1s) NEXAFS spectrum was calculated by summing up the individual C 1s (N 1s) calculated spectra. A further shift was applied to align the calculated position of the π* peak to the measured one. The values of the shifts for each molecule are given in the corresponding figures.
Results and discussion
DTDCTB electronic structure: the occupied states
The comparisons between the gas phase valence PE spectra calculated for the ground state of DTDCTB, DPTA and BTD are shown in Fig. 2. The calculated total density of states (DOS) allows us to predict the gas phase HOMO–LUMO gap (Eg(gas)) of the investigated molecules. The calculated HOMO and LUMO energy positions (shown in the inset table of Fig. 2) are reported together with the HOMO–LUMO gaps computed for DTDCTB (Eg(gas) 2.08 eV), DPTA (Eg(gas) 4.52 eV) and BTD (Eg(gas) 4.27 eV).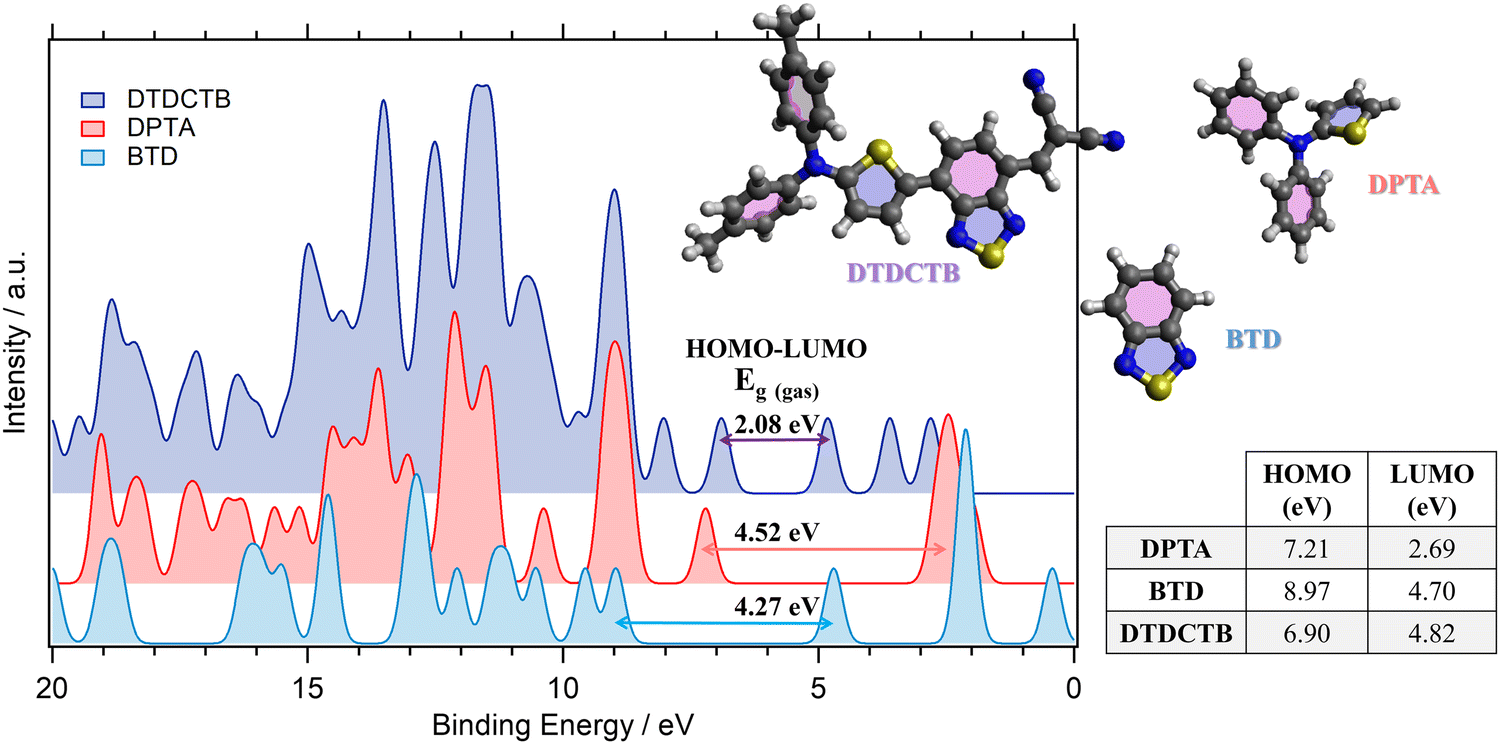 | ||
| Fig. 2 Comparison between the total calculated (B3LYP) valence photoelectron spectra and the resulting predicted HOMO–LUMO gaps of DTDCTB, DPTA and BTD molecules. The calculated valence spectra have been shifted by +1.67 eV (DTDCTB), +2.18 eV (DPTA) and +2.35 eV (BTD) to align each HOMO peak with the lower biding energy peak of the corresponding measured valence photoelectron spectrum (see Fig. 3). | ||
The energy gap reported in the literature for DTDCTB shows that the optical gap obtained from the extinction coefficient is 1.86 eV and the transport gap obtained from the HOMO–LUMO difference is 1.62 eV.2 As expected, the energy gap of DTDCTB obtained using the B3LYP functional is slightly higher49 (2.08 eV) than the reported experimental values.
The comparisons of the experimental PE valence spectrum with the calculated total DOS and PDOS of DTDCTB are shown in Fig. 3a–d where a very good agreement between the experimental and theoretical data can be observed. The partial contributions of electron-donor (DPTA and methyl moieties) and of electron-withdrawing (BTD and CN moieties) groups of the DTDCTB molecule are shown in Fig. 3a. A more detailed analysis related to the contributions from each C, N and S atomic orbitals (AOs) with s and p characters to the molecular orbitals (MOs) of DTDCTB is reported in Fig. 3b–d.
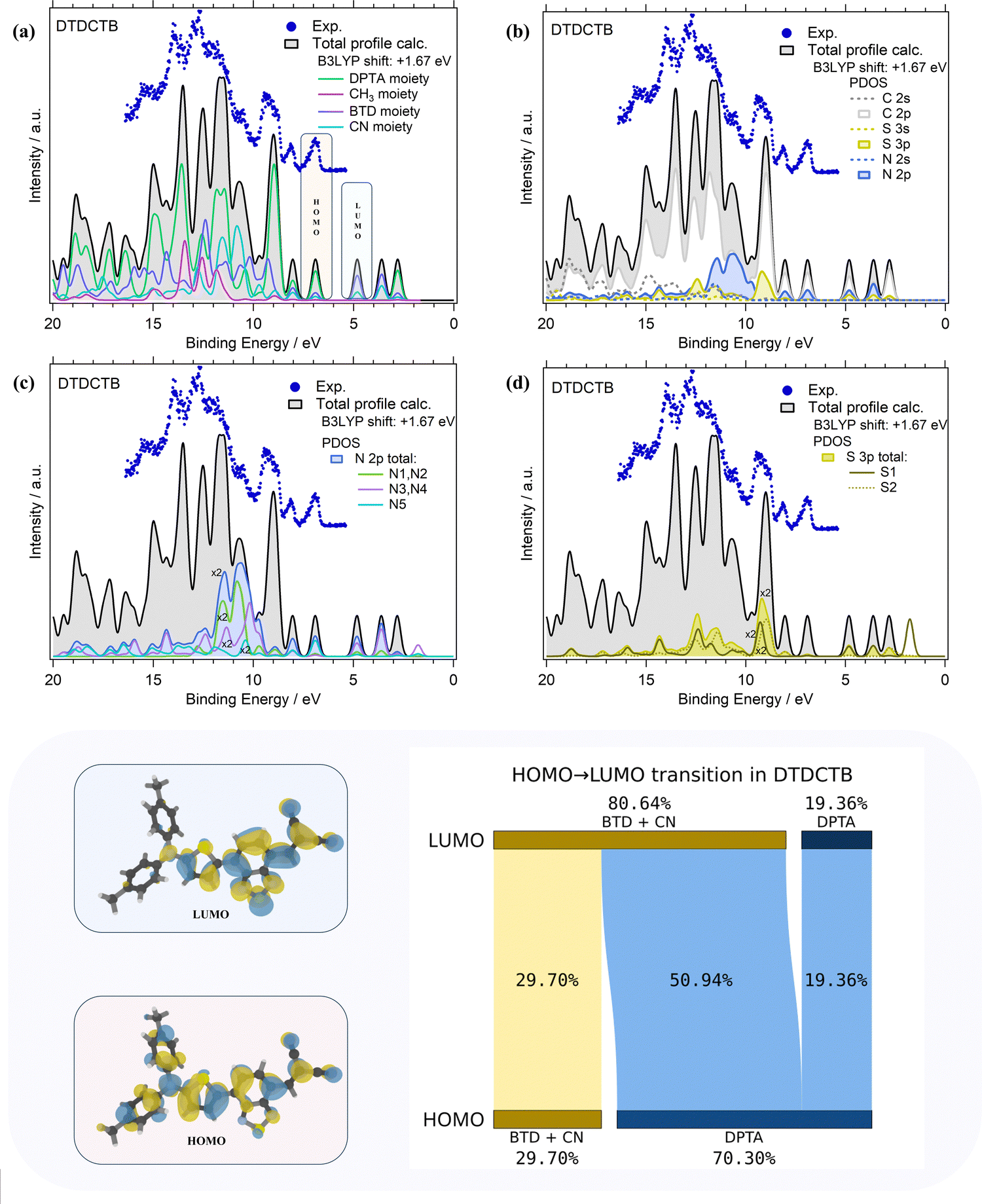 | ||
| Fig. 3 Comparison of the experimental PE valence spectrum (hv 100 eV, dotted blue line) and the total calculated (B3LYP) density of states (DOS, solid black line) of the DTDCTB molecule are shown together with the PDOS (solid colored lines) related to: (a) the partial contributions of DPTA, CN, BTD and METHYL moieties belonging to the DTDCTB molecule; (b) C 2s, C 2p, S 3s, S 3p, N 2s, N 2p orbitals; (c) N 2p orbital with the disentangled contributions from the N1, N2, N3, N4 and N5 atoms; (d) S 3p orbital with the disentangled contributions from the S1 and S2 atoms. The calculated valence spectrum of DTDCTB has been shifted by +1.67 eV to match the corresponding experimental spectrum. Bottom panel: the HOMO and LUMO molecular orbital sketches and the numerical analysis of the fragment decomposition of the HOMO and LUMO orbitals of DTDCTB over its donor and acceptor moieties. | ||
From the experimental valence PE spectrum (Fig. 3), the HOMO of DTDCTB is found as an asymmetric peak at 6.96 eV with a FWHM of about 0.30 eV. The calculated PDOS in Fig. 3a indicates that the main contribution to the HOMO (as well as to the two adjacent features at higher BEs) comes from the electron-donor DPTA moiety and only a small part comes from the BTD and CN electron-accepting groups. To further quantify the contribution to HOMO and LUMO coming from the different fragments of the DTDCTB molecule, we performed a numerical analysis of the HOMO and LUMO using VALET (see bottom panel of Fig. 3). From this analysis, we obtained a ∼70% contribution to HOMO from the electron-donating DPTA subgroup, while only ∼30% is attributed to the BTD and CN moieties. It is interesting to note that even if the CH3 is an electron-donating group, its first intense contributions only appear at 11–15 eV. The same quantitative VALET analysis reported in the bottom panel of Fig. 3 predicts instead that the electron-withdrawing groups (BTD and CN moieties) contribute to the LUMO by ∼81%, while only about 19% is attributed to DPTA. This analysis clearly shows a high electron density localized mostly on the electron-acceptor part of the DTDCTB molecule demonstrating how the extended π-electron delocalization favors the intramolecular charge transfer for the HOMO-to-LUMO transition from the donor to the acceptor counterparts through the quinoidal D–A–A molecular architecture.
Furthermore, the theory reveals that the HOMO of DTDCTB has a significant contribution from the C 2p and N 2p AOs (see also the HOMO sketch in the bottom panel of Fig. 3). These C 2p and N 2p orbitals appear to also give the major contributions together with the S 3p AO to the LUMO. This can be also observed in the DTDCTB LUMO sketch in the bottom panel of Fig. 3 in which the electronic density appears to increase around the S2 atom compared with the HOMO for which the electronic density is shifted more on the donor moiety and the S1 atom is more involved. The features in the PE valence spectrum of DTDCTB in the BE range between 10 eV and 15 eV are due to the contributions of the N 2p of the N1 and N2 atoms belonging to the CN groups (Fig. 3c). Among the N heteroatoms, the N5 atom of the electron-donor DPTA moiety gives the greater contribution to the HOMO N 2p PDOS (Fig. 3c). Instead, the largest contribution from the N atoms to the LUMO molecular orbital comes from the N3, N4 atoms and the second largest contribution comes from the N1, N2 atoms which are the heteroatoms involved, respectively, in the BTD and CN electron-withdrawing moieties. These nitrogen atoms (N1, N2 and N3, N4) give similar contributions to the LUMO+1 feature. For what concerns the S 3p PDOS, the contribution of S1 and S2 heteroatoms (Fig. 3d) to the HOMO is quite negligible; instead, their contribution becomes strong to the LUMO and LUMO+1 features (where S1 predominates) and to the LUMO+2 intensity (where S2 predominates).
The valence PE spectra of DPTA and BTD along with the calculated different atomic contributions are shown in Fig. 4a and b. In the experimental valence PE spectrum (Fig. 4a) of DPTA, the HOMO is found as a mostly symmetric peak centered at 7.13 eV with a FWHM of about 0.25 eV.
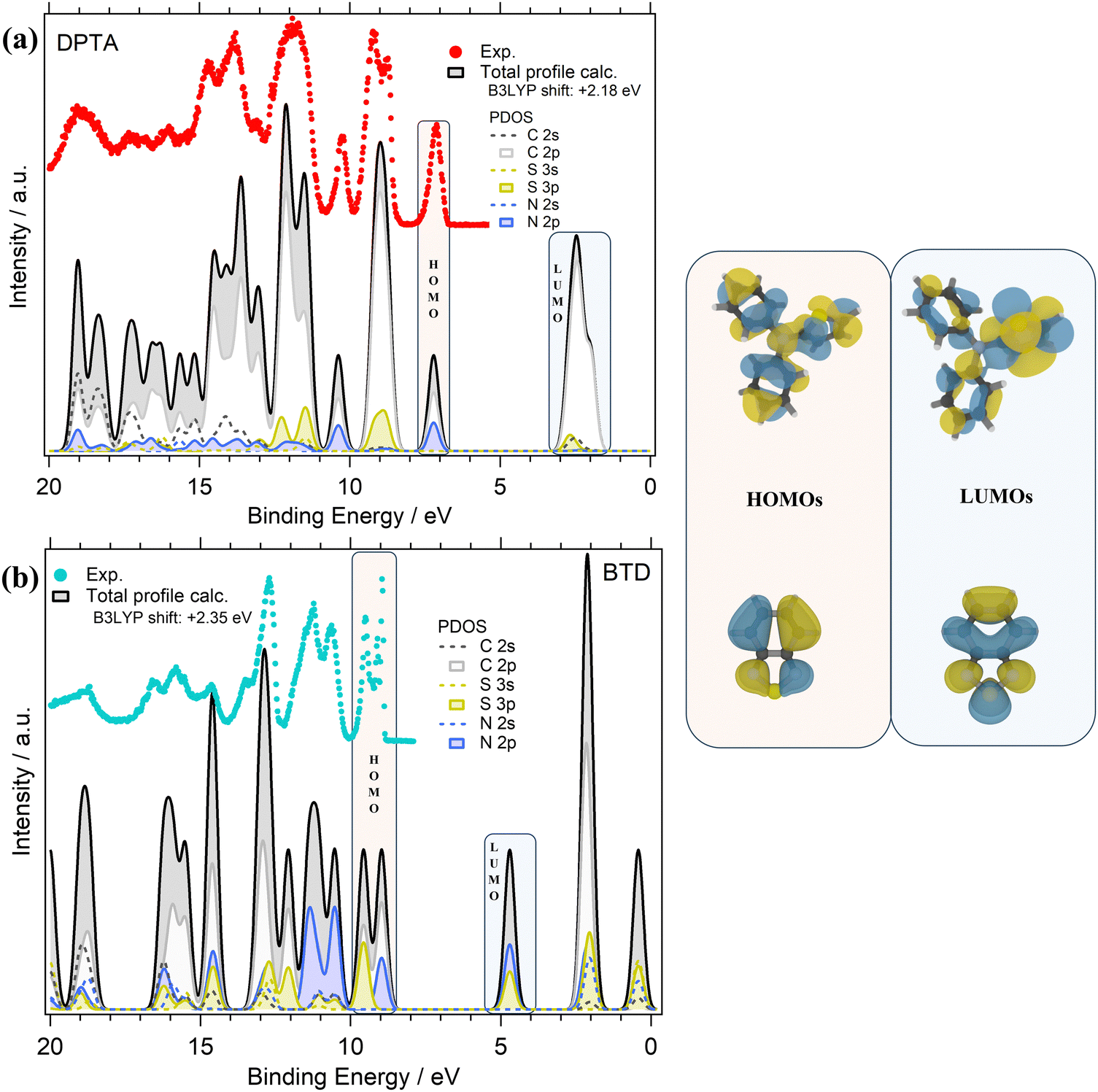 | ||
| Fig. 4 Comparison of the experimental PE spectrum (hv 100 eV, dotted red line for DPTA and light blue line for BTD) and the total calculated (B3LYP) DOS (solid black lines) of: (a) DPTA and (b) BTD molecules. For each molecule, the calculated PDOS (solid colored lines) related to: C 2s, C 2p, S 3s, S 3p, N 2s, and N 2p orbitals are shown. The calculated valence peaks have been shifted by: +2.18 eV (DPTA) and +2.35 eV (BTD) to match the corresponding experimental spectra. Right panel: the HOMO and LUMO molecular orbital sketches. | ||
Like DTDCTB, also for DPTA the theory predicts that the HOMO is mainly derived from the C 2p and N 2p atomic orbitals. For more details about AOs contribution to the valence electronic structure of DPTA the readers can refer to our previous study of this molecule.26 At higher BEs, the feature centered at about 9 eV originates mainly from C 2p and S 3p AOs. Like the HOMO, the third main feature at about 10.2 eV shows once more a contribution from the C 2p and N 2p AOs. In the inner valence energy region (11–20 eV), the PE peaks are due to contributions from C 2p, but also S 3p, N 2p, and C 2s.
Considering the theoretical data, the LUMO originates mainly from C 2p states with a small but non-negligible contribution also from the S 3p and C 2s AOs. Considering the LUMO sketch of DPTA (see the right panel of Fig. 4) it seems that the LUMO electron density has a great tendency to shift toward the thiophene ring favoring the opening of the “channel” toward the electron-accepting moieties of the whole DTDCTB molecule thanks to the quinoidal character of the conjugated backbone of this D–A–A molecular architecture.
The experimental valence PE spectrum (Fig. 4b) of BTD shows the HOMO as a sharp double peak with two maxima at 9.49 eV (FWHM 0.18 eV) and 8.95 eV (FWHM 0.65 eV). The calculated total DOS and PDOS of BTD is shown in Fig. 4b considering the contribution from each C, N and S AOs with s and p characters to the to the MO of BTD. The theory predicts that the main contributions to the HOMO come from the C 2p, S 3p and N 2p overlapping within this double peak. The second doublet at higher BEs (11.31 eV FWHM 0.56 eV and 10.68 eV FWHM 0.38 eV) in the valence spectrum shows an important contribution coming from the N 2p states. In the range 12–20 eV instead, mixed contributions from C 2p, S 3p, N 2p, N 2s and C 2s are evidenced. In the LUMO feature, the prevalent contributions come from N 2p and S 3p, as also shown by the LUMO sketch (Fig. 4b).
DTDCTB electronic structure: the empty states
C K-edge NEXAFS measurements were performed for the isolated DPTA and BTD building block molecules (respectively, in Fig. 5b and c), to identify the electronic structure modifications induced when they are covalently bound in the donor–acceptor–acceptor DTDCTB molecule (Fig. 5a).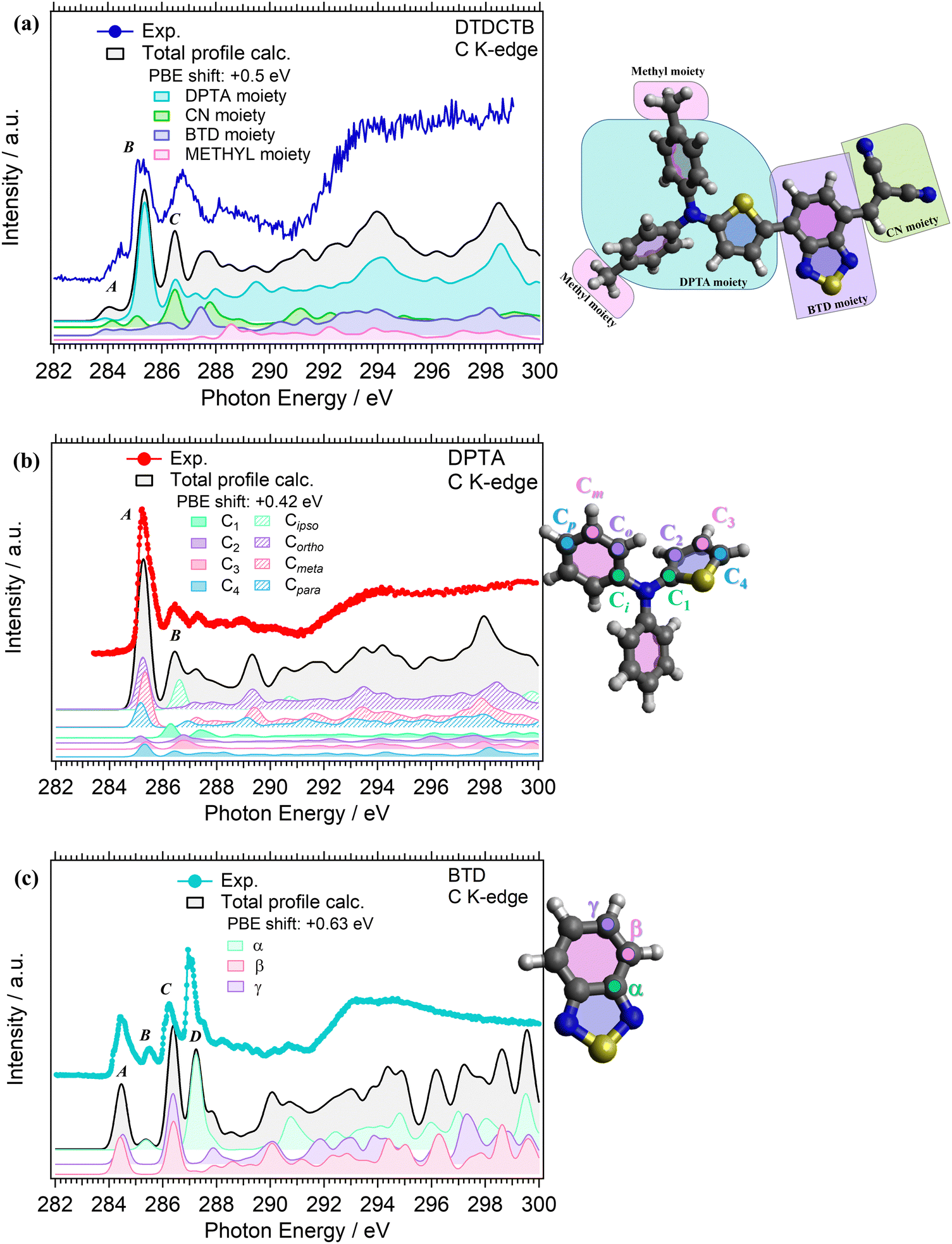 | ||
| Fig. 5 Experimental (dotted colored lines) and calculated (PBE/cc-pVTZ, solid black lines) NEXAFS C K-edge of (a) DTDCTB, (b) DPTA and (c) BTD molecules. (a) The total calculated profile for DTDCTB is shown together with the partial contributions related to the: DPTA, CN, BTD and METHYL moieties belonging to the DTDCTB molecule. The calculated total and the partial profiles have been shifted by +0.5 eV to match the experimental spectrum. (b) and (c) The total calculated profiles are shown together with the partial contributions related to each inequivalent carbon belonging to the molecules. The calculated total and the partial profiles have been shifted by +0.42 eV (DPTA) and +0.63 eV (BTD) to match the corresponding experimental spectra. | ||
The experimental C K-edge NEXAFS spectrum of DTDCTB (Fig. 5a), evidences a low-intensity feature, peak A (at 283.8–284.7 eV), before the more intense peak B (at 285.3 eV, FWHM 0.4 eV). These two A and B features are confirmed by the calculated total C K-edge profile (Fig. 5a). The calculated partial contributions related to the DPTA, BTD and CN moieties of DTDCTB clearly reveal that the main contribution to the intensive peak B comes from the DPTA electron-donor moiety (light blue band in Fig. 5a), while the main contribution to the feature at the absorption threshold (peak A) originates from the BTD and CN electron-withdrawing moieties (respectively, green and purple bands in Fig. 5a). The third intensity (peak C at 286.0–287.7 eV) is mostly due to the contributions from the DPTA, CN and BTD moieties. The first important contribution from the methyl moiety starts at photon energies higher than 287.0 eV, as shown from the pink area in Fig. 5a.
Looking at the experimental C K-edge NEXAFS spectrum of the isolated DPTA molecule (Fig. 5b) peak A, resulting from C 1s to LUMO transitions, is centered roughly at the same photon energy (285.2 eV FWHM 0.44 eV) as peak B in the DTDCTB molecule. The theory assigns the NEXAFS peaks to contributions from the chemically inequivalent atoms in the DPTA molecule. Referring to the central nitrogen atom, the C atoms of the DPTA phenyl rings are labeled as C ipso (Ci), C ortho (Co), C meta (Cm) and C para (Cp), while the atoms belonging to the thiophene ring are indicated as C1, C2, C3, and C4 (see the sketch in Fig. 5b). Peak A of DPTA gets mainly contributions from the C atoms which are not directly bonded to the central N (i.e., Co, Cm, Cp and C2, C3, C4). Conversely, the C atoms directly bonded to the central N atom (i.e., Ci and C1) provide strong contributions to peak B at higher photon energies.
The experimental C K-edge spectrum of the isolated BTD building block (Fig. 5c) shows four main features. The first one is peak A (at 284.5 eV FWHM 0.4 eV) which is located at 0.7–0.8 eV lower photon energy compared to features A of DPTA and B of DTDCTB, respectively. To analyze the contributions of the chemically inequivalent atoms to the NEXAFS peaks, we label the carbon atoms as α, β and γ depending on the distance to the nitrogen atoms going, respectively, from the adjacent to the farthest (more about this in the ESI†). The theory (solid black line in Fig. 5c) predicts for peak A of BTD unique contributions coming from β and γ C atoms, i.e. not directly bonded to the N atoms. Instead, at 285.5 eV (FWHM 0.2 eV), the feature B is attributed only to the α C atom of the BTD building block. The same description of peaks A and B applies, respectively, to the third (peak C at 286.2 eV FWHM 0.5 eV) and fourth (peak D at 287.0 eV FWHM 0.2 eV) C K-edge features. For the former peak, the theory predicts contributions coming from the β and γ C atoms, while the latter originates totally from the α C atom of the BTD building block.
NEXAFS measurements were performed at the N K-edge for investigating the electronic structure of the DPTA and BTD building blocks when they are isolated (respectively, in Fig. 6b and c) and when they are covalently bound to the whole donor–acceptor–acceptor DTDCTB molecule (Fig. 6a).
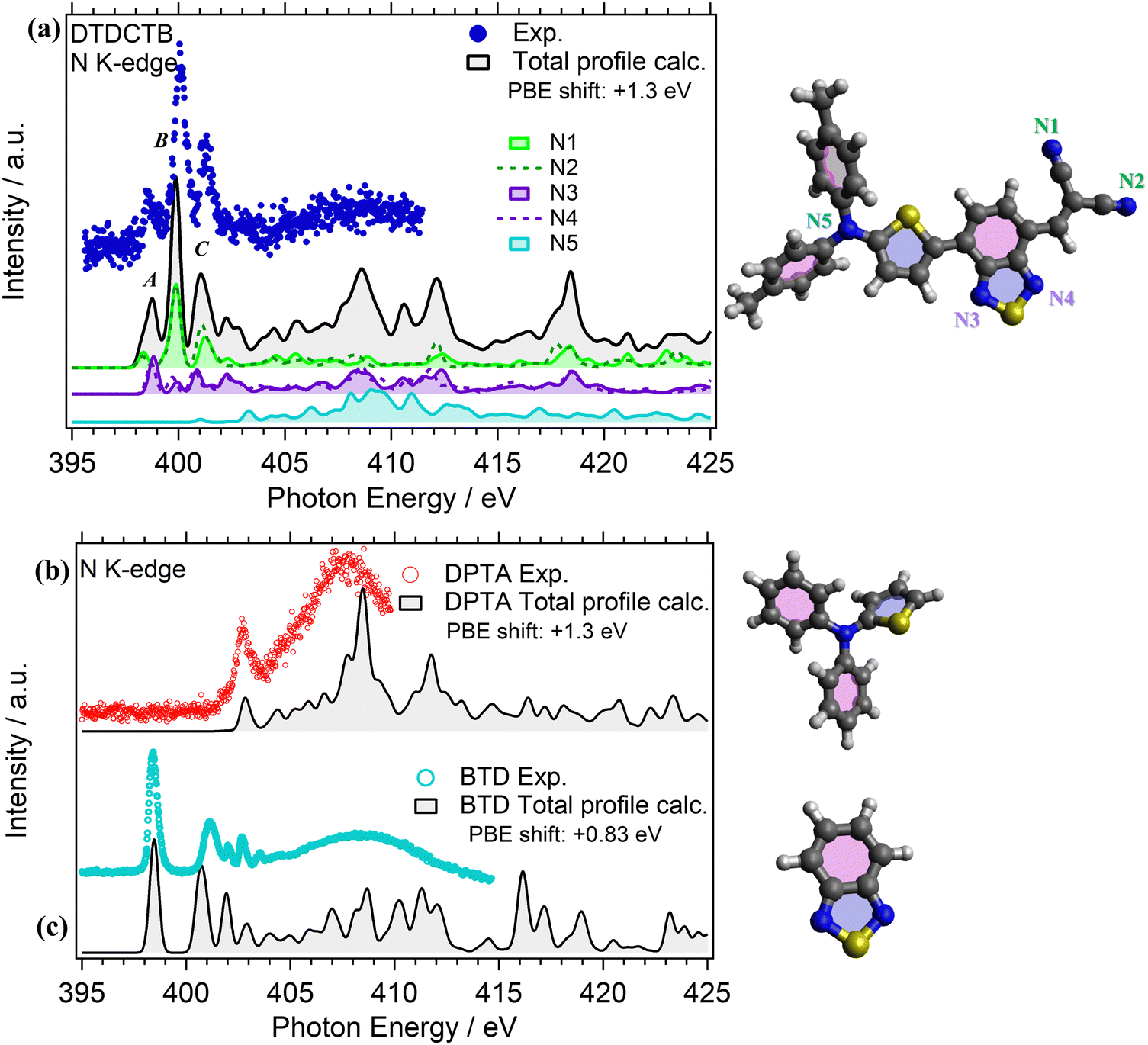 | ||
| Fig. 6 (a) Experimental (dotted colored lines) and calculated (StoBe, PBE/cc-pVTZ+GENA4, 0.5 eV FWHM, solid black lines) NEXAFS N K-edge spectra of the DTDCTB molecule. The calculated total profile is compared with the partial contributions related to the inequivalent N1, N2, N3, N4 and N5 nitrogen atoms as indicated in the sketched molecule. The calculated total and the partial intensities have been shifted by +1.3 eV to match the experimental spectrum. Experimental and calculated (StoBe, PBE/cc-pVTZ+GENA4, 0.5 eV FWHM) NEXAFS N K-edge spectra of the (b) DPTA and (c) BTD molecules. The experimental spectra are compared with the corresponding calculated total N K-edge profiles. The calculated total profiles have been shifted by: +1.3 eV (DPTA) and +0.83 eV (BTD) to match the experimental spectra. | ||
The experimental N K-edge NEXAFS spectrum of DTDCTB (Fig. 6a) consists of three main features: peak A (398.40 eV FWHM 0.20 eV), corresponding to transitions from N 1s to LUMO, peak B (399.90 eV FWHM 0.30 eV) and peak C (401.10 eV FWHM 0.40 eV) that are well reproduced by the theory (Fig. 6a) and attributed to π* resonances. Furthermore, the theory allows us to disentangle the contributions of the five inequivalent N atoms (i.e., N1, N2, N3, N4, N5) as reported in detail in Fig. 6a. As expected, all the N atoms of the moieties with electron-acceptor character contribute to peak A: the N1 and N2 atoms of the CN moiety and the N3 and N4 atoms of the BTD moiety of the whole DTDCTB molecule with a slight dominance of the N3 and N4 atoms. Moreover, to peak B we can identify the same contributions, but with a prevalence of the N1 and N2 atoms. The first contribution but still not dominant to the N K-edge coming from the N5 atom of the DPTA moiety is predicted in peak C of DTDCTB together with the contributions of all the other N atoms. In total agreement with the published results of the N K-edge NEXAFS spectrum of DPTA26 (Fig. 6b), the LUMO+4 (located at 403 eV) is the first unoccupied state of the molecule with significant contributions from the central N atom.
Comparing the N K-edge spectrum of the isolated electron-withdrawing BTD building block (Fig. 6c), the peak corresponding to transitions into the LUMO is centered at 398.4 eV (FWHM 0.3 eV). This corresponds exactly to the first π* peak in the DTDCTB NEXAFS spectrum, attributed to the N3 and N4 atoms by our TP-DFT calculations. Starting from about 404 eV of the BTD N K-edge spectrum, the intensity is attributed to σ* resonances instead.
Conclusions
Gas phase DTDCTB and its DPTA and BTD building blocks have been characterized by valence photoelectron spectroscopy to probe their occupied electronic structure and NEXAFS experiments to probe their empty density of states at the C and N K-edges. DFT calculations have been applied for a detailed understanding of the molecular electronic structures and to disentangle the atomic orbital contributions to the valence electronic structure of DTDCTB. By disentangling the electronic structure of DTDCTB and examining the contributions of its building blocks (i.e., the donor and the acceptors moieties), we contributed to elucidate the underlying principles governing the electronic properties of the DTDCTB small-molecule donor and provide insights for the rational design of novel push–pull materials with enhanced functionality and performances. We found that the frontier states of DTDCTB can be attributed mainly to one moiety or the other. From the comparison of the DTDCTB PE spectrum to the spectra of BTD and DPTA, the DTDCTB HOMO peak is mainly assigned to the DPTA moiety. This is confirmed by our DFT calculations, where the decomposition into contributions from AOs from the different moieties shows that DPTA is mainly responsible for the HOMO, with smaller contributions from BTD and CN. The occupied states at higher BEs have a more mixed character, but some of the strong spectral features can still be attributed to one moiety or the other. In particular, the DPTA building block is mainly responsible for the strong peak at ∼9 eV BE, while the signature of the CN groups can be identified in the small feature at 10.5 eV BE.When it comes to the unoccupied states, they were probed using C and N K-edge NEXAFS spectroscopy. In the C K-edge NEXAFS spectrum of DTDCTB, we identified a pre-edge feature located at the same photon energy as the π* peak measured for BTD. This assignment was confirmed by our TP-DFT calculations, which attributed this feature to transitions from the C 1s core orbitals localized on the BTD moiety or on the CN terminal groups to the DTDCTB LUMO. A similar assignment was obtained for the first π* peak in the DTDCTB N K-edge NEXAFS spectrum, which also sharply corresponds in photon energy to the measured gas phase BTD π* peak.
The study clearly shows that the electronic structure of the molecular building blocks is preserved when covalently bonded together and has a very important role in the electronic properties of the resulting DTDCTB molecular system. It can be concluded that the DTDCTB molecule precisely combines the electronic properties of its constituent donor and acceptor building blocks, demonstrating that the inclusion of thiophene in donor moiety facilitates π-electron delocalization and results an enhanced quinoidal character to the conjugated backbone. Thus, the adoption of the ditolylaminothienyl donor moiety promotes efficient intramolecular charge transfer from the donor side to the acceptor side of DTDCTB, thus leading to the formation of mesomeric structures like D–A ↔ D = A−. Therefore, the D–A–A molecular architecture is confirmed to be a strategic solution to guarantee the efficient charge transfer among the two electron-donor and electron-withdrawing counterparts.
The contribution of the donor DPTA moiety to the HOMO is still preserved in the donor–acceptor–acceptor molecular architecture of DTDCTB: this together with the increased density of state at the LUMO due to the molecular building blocks of acceptor character would guarantee an effective charge transfer of the excited electrons. In future studies of more applied character, it will be interesting to check these properties with dynamic studies and compare the results of DTDCTB when it is combined with other acceptor molecules in more complex systems.
Data availability
Data for this article are available at the open science framework (OSF) repository at https://osf.io/jzqyu/?view_only=b76d81b880a74faaaaf729596b7e6541.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Author A. G. acknowledges Consiglio Nazionale delle Ricerche (CNR) – Unità Relazioni Europee e Internazionali for funding part of this research through the Program “Mobilità di Breve Durata – STM (Short Term Mobility) – Call 2020”. T. Z. thanks the support by the National Natural Science Foundation of China (NSFC No. 62271048). The calculations were carried out on resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC) in Linköping, Sweden, partially funded by the Swedish Research Council through grant agreement No. 2022-06725 and No. 2018-05973. The Authors thank the Carl Trygger Foundation (CTS 22:2225) for financial support and for making available the VG-Scienta SES200 photoelectron analyzer at the Gas Phase beamline, Elettra, Italy. The Authors acknowledge the EU CERIC-ERIC Consortium for granting access to experimental facilities and financial support. The Authors thank the staff at GaPh beamline, at Elettra for all the help provided during the beamtimes.References
- C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416–10434 RSC.
- L.-Y. Lin, Y.-H. Chen, Z.-Y. Huang, H.-W. Lin, S.-H. Chou, F. Lin, C.-W. Chen, Y.-H. Liu and K.-T. Wong, J. Am. Chem. Soc., 2011, 133, 15822–15825 CrossRef CAS PubMed.
- Y.-H. Chen, L.-Y. Lin, C.-W. Lu, F. Lin, Z.-Y. Huang, H.-W. Lin, P.-H. Wang, Y.-H. Liu, K.-T. Wong, J. Wen, D. J. Miller and S. B. Darling, J. Am. Chem. Soc., 2012, 134, 13616–13623 CrossRef CAS PubMed.
- J. Yoo, K. Jung, J. Jeong, G. Hyun, H. Lee and Y. Yi, Appl. Surf. Sci., 2017, 402, 41–46 CrossRef CAS.
- H.-S. Shim, C.-K. Moon, J. Kim, C.-K. Wang, B. Sim, F. Lin, K.-T. Wong, Y. Seo and J.-J. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 1214–1219 CrossRef CAS.
- A. Y. Chang, Y.-H. Chen, H.-W. Lin, L.-Y. Lin, K.-T. Wong and R. D. Schaller, J. Am. Chem. Soc., 2013, 135, 8790–8793 CrossRef CAS PubMed.
- J. Kim, H.-S. Shim, H. Lee, M.-S. Choi, J.-J. Kim and Y. Seo, J. Phys. Chem. C, 2014, 118, 11559–11565 CrossRef CAS.
- S. Madduri, V. S. Katta, S. S. K. Raavi and S. G. Singh, Mater. Lett., 2021, 300, 130159 CrossRef CAS.
- S. Madduri, V. G. Kodange, S. S. K. Raavi and S. G. Singh, 2021 IEEE 48th Photovoltaic Specialists Conference (PVSC), 2021, 0732–0736.
- H.-W. Lin, Y.-H. Chen, Z.-Y. Huang, C.-W. Chen, L.-Y. Lin, F. Lin and K.-T. Wong, Org. Electron., 2012, 13, 1722–1728 CrossRef CAS.
- H.-W. Lin, J.-H. Chang, W.-C. Huang, Y.-T. Lin, L.-Y. Lin, F. Lin, K.-T. Wong, H.-F. Wang, R.-M. Ho and H.-F. Meng, J. Mater. Chem. A, 2014, 2, 3709–3714 RSC.
- D. Wang, X. Zhang, W. Ding, X. Zhao and Z. Geng, Comput. Theor. Chem., 2014, 1029, 68–78 CrossRef CAS.
- H. Lee, Y. Yi, S. W. Cho and W. K. Choi, Synth. Met., 2014, 194, 118–125 CrossRef CAS.
- X. Shen, G. Han and Y. Yi, Phys. Chem. Chem. Phys., 2016, 18, 15955–15963 RSC.
- X.-X. Shen, G.-C. Han and Y.-P. Yi, Chin. Chem. Lett., 2016, 27, 1453–1463 CrossRef CAS.
- G. Han, X. Shen and Y. Yi, Adv. Mater. Interfaces, 2015, 2, 1500329 CrossRef.
- H. Ting, Y. Chen, L. Lin, S. Chou, Y. Liu, H. Lin and K. Wong, ChemSusChem, 2014, 7, 457–465 CrossRef CAS.
- S. Jiang, J. Zheng, Y. Yi, Y. Xie, F. Yuan and Z. Lan, J. Phys. Chem. C, 2017, 121, 27263–27273 CrossRef CAS.
- S. Jiang, Y. Xie and Z. Lan, Chem. Phys., 2018, 515, 603–613 CrossRef CAS.
- X. Shen, G. Han, D. Fan, Y. Xie and Y. Yi, J. Phys. Chem. C, 2015, 119, 11320–11326 CrossRef CAS.
- N. Kim, S. Park, M.-H. Lee, J. Lee, C. Lee and S. C. Yoon, J. Nanosci. Nanotechnol., 2016, 16, 2916–2921 CrossRef CAS.
- H. Shim, F. Lin, J. Kim, B. Sim, T. Kim, C. Moon, C. Wang, Y. Seo, K. Wong and J. Kim, Adv. Energy Mater., 2015, 5, 1500228 CrossRef.
- D. Cheyns, M. Kim, B. Verreet and B. P. Rand, Appl. Phys. Lett., 2014, 104, 093302 CrossRef.
- X. Che, X. Xiao, J. D. Zimmerman, D. Fan and S. R. Forrest, Adv. Energy Mater., 2014, 4, 1400568 CrossRef.
- L. Ye, H.-H. Xu, H. Yu, W.-Y. Xu, H. Li, H. Wang, N. Zhao and J.-B. Xu, J. Phys. Chem. C, 2014, 118, 20094–20099 CrossRef CAS.
- T. Zhang, I. E. Brumboiu, C. Grazioli, A. Guarnaccio, M. Coreno, M. de Simone, A. Santagata, H. Rensmo, B. Brena, V. Lanzilotto and C. Puglia, J. Phys. Chem. C, 2018, 122, 17706–17717 CrossRef CAS.
- R. R. Blyth, R. Delaunay, M. Zitnik, J. Krempasky, R. Krempaska, J. Slezak, K. C. Prince, R. Richter, M. Vondracek, R. Camilloni, L. Avaldi, M. Coreno, G. Stefani, C. Furlani, M. de Simone, S. Stranges and M.-Y. Adam, J. Electron Spectrosc. Relat. Phenom., 1999, 101, 959–964 CrossRef.
- N. Mårtensson, P. Baltzer, P. A. Brühwiler, J.-O. Forsell, A. Nilsson, A. Stenborg and B. Wannberg, J. Electron Spectrosc. Relat. Phenom., 1994, 70, 117–128 CrossRef.
- R. M. Pinto, A. A. Dias, M. Coreno, M. de Simone, B. M. Giuliano, J. P. Santos and M. L. Costa, Chem. Phys. Lett., 2011, 516, 149–153 CrossRef CAS.
- H.-H. Perkampus and D. W. Turner, Molecular Photoelectron Spectroscopy, Unter Mitarbeit von C. Baker and A. D. Becker und, Wiley-Interscience, JohnWiley & Sons Ltd., London, New York, Sydney, Toronto 1970, vol. 386 Seiten.Preis: p. 140 sh., Berichte der Bunsengesellschaft für physikalische Chemie, 1971, vol. 75, 181 Search PubMed.
- M. Tronc, G. C. King and F. H. Read, J. Phys. B: At. Mol. Phys., 2001, 12, 137 CrossRef.
- M. Tronc, G. C. King and F. H. Read, J. Phys. B: At. Mol. Phys., 1999, 13, 999 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- P. Ros and G. C. A. Schuit, Theor. Chim. Acta, 1966, 4, 1–12 CrossRef CAS.
- T. Zhang, I. E. Brumboiu, V. Lanzilotto, J. Lüder, C. Grazioli, E. Giangrisostomi, R. Ovsyannikov, Y. Sassa, I. Bidermane, M. Stupar, M. de Simone, M. Coreno, B. Ressel, M. Pedio, P. Rudolf, B. Brena and C. Puglia, J. Phys. Chem. C, 2017, 121, 26372–26378 CrossRef CAS.
- I. E. Brumboiu, R. Totani, M. de Simone, M. Coreno, C. Grazioli, L. Lozzi, H. C. Herper, B. Sanyal, O. Eriksson, C. Puglia and B. Brena, J. Phys. Chem., 2014, 118, 927–932 CrossRef CAS PubMed.
- T. B. Masood, S. S. Thygesen, M. Linares, A. I. Abrikosov, V. Natarajan and I. Hotz, Comput. Graph. Forum, 2021, 40, 287–298 CrossRef.
- K. Hermann, L. G. M. Pettersson, M. E. Casida, C. Daul, A. Goursot, A. Koester, E. Proynov, A. St-Amant, D. R. Salahub, V. Carravetta, H. Duarte, C. Friedrich, N. Godbout, M. Gruber, J. Guan, C. Jamorski, M. Leboeuf, M. Leetmaa, M. Nyberg, S. Patchkovskii, L. Pedocchi, F. Sim, L. Triguero and A. Vela, StoBe-deMon version 3.3, 2014 Search PubMed.
- I. E. Brumboiu and T. Fransson, J. Chem. Phys., 2022, 156, 214109 CrossRef CAS.
- T. Fransson, I. E. Brumboiu, M. L. Vidal, P. Norman, S. Coriani and A. Dreuw, J. Chem. Theory Comput., 2021, 17, 1618–1637 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- U. Fleischer, W. Kutzelnigg, H.-H. Limbach, G. J. Martin, M. L. Martin, M. Schindler, W. Kutzelnigg, U. Fleischer and M. Schindler, NMR: Basic Princ. Prog., 1990, 23, 165–262 Search PubMed.
- A. J. Garza and G. E. Scuseria, J. Phys. Chem. Lett., 2016, 7, 4165–4170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00515e |
| This journal is © The Royal Society of Chemistry 2024 |