
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light excited and temperature-responsive phosphorescent system in a phase-changing matrix†
Yingying
Hu
a,
Glib V.
Baryshnikov
 *c,
Xueru
Shan
a,
Weiyi
Zhang
b,
Sheng-yin
Zhao
*a,
Liangliang
Zhu
*d and
Hongwei
Wu
*ae
*c,
Xueru
Shan
a,
Weiyi
Zhang
b,
Sheng-yin
Zhao
*a,
Liangliang
Zhu
*d and
Hongwei
Wu
*ae
aState Key Laboratory for Modification of Chemical Fiber and Polymer Materials, Key Lab of Science and Technology of Eco-Textile, Ministry of Education, College of Chemistry and Chemical Engineering, Donghua University, Shanghai 201620, China. E-mail: syzhao8@dhu.edu.cn; wuhongwei@dhu.edu.cn
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China
cState Key Laboratory for Organic Electronics, Department of Science and Technology, Linköping University, SE-60174 Norrköping, Sweden. E-mail: glib.baryshnikov@liu.se
dState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200438, China. E-mail: zhuliangliang@fudan.edu.cn
eZheJiang YongFa Synthetic Leather Co. Ltd, Zhejiang 323000, China
First published on 21st May 2024
Abstract
Reversibly switching visible light excited phosphorescence emission by external stimuli is highly challenging. Herein, we report a series of tetrakis(arylthio)benzene derivatives with a D–A structure, exhibiting visible-light excited room-temperature phosphorescence. Significantly, the emission from their crystalline powder state responded to mechanical forces, attributed to alterations in molecular stacking changes, resulting in their phosphorescence color changes. Moreover, the monomer and aggregated phosphorescence transition could be reversibly switched by temperature when doping these molecules into a phase-changing matrix saturated fatty acid (FA) because the FA matrix is transformed between solid and fluid states under different temperatures. In addition, multi-color luminescent materials were also obtained by further introducing triphenylamine dye molecules. Finally, these doping systems exhibited excellent application potential in temperature indication and anti-counterfeiting. This successful design strategy provides a new idea for preparing reversible external stimuli-responsive phosphorescent materials.
Introduction
Organic room-temperature phosphorescence (RTP) materials have been widely applied in various fields such as organic light-emitting diodes,1–6 time-resolved bioimaging,7–14 dynamic anti-counterfeiting,15–19 and chemical sensing20–22 because of their excellent photophysical properties. In recent years, several rational strategies including supramolecular assembly,23–25 host–guest doping,26 covalent organic framework (COF) construction,27 and crystal engineering28 have been proposed to induce enhanced RTP emission. It is worth noting that RTP materials that are sensitive to external stimuli such as temperature,29 light,30,31 mechanical grinding,32,33 and humidity34 are a current research focus and a challenge. For example, Huang et al. developed a dual-emission phosphorescent polymer thermometer by doping two long-lived phosphorescent iridium(III) complexes into an acrylamide-based thermosensitive polymer, in which there was a conformational transition from an extended state to an aggregated state when the temperature was increased from 16 °C to 40 °C. The changes in the polymer microenvironment led to enhanced rigidity and decreased polarity, bringing a wavelength-ratiometric phosphorescence emission.35 However, the recent development of thermosensitive RTP materials including host–guest doped systems,26 copolymerization systems,23 and metal–organic frameworks (MOFs)36 mostly faced the daunting problem of slow responsiveness, single-color luminescence, and short excited wavelength.At present, the common construction strategies of thermosensitive systems possess the shortcomings of complicated preparation and poor repeatability, and it is urgent to seek a simpler and more efficient strategy.35 Phase-changing materials such as a novel thermo-responsive matrix, which could trigger a solid–liquid transition with increasing temperature, have been used to control the release of therapeutic drugs.37 FAs and fatty alcohols are the most common and widely used because of their excellent biocompatibility, natural availability, and low cost.37 Recently, Zhang et al. proposed a clever strategy to prepare thermosensitive fluorescent materials by doping fluorophores into a long-chain FA. These systems showed different fluorescence under room- and high-temperature because of the different existential states of fluorescent molecules in the high-crystallized and melting states of the FA.38 Our group also applied a similar strategy to regulate the reversible transition between the monomer and excimer emission of the fluorophores.39 The use of phase-changing materials to modulate the luminescence of fluorophores has been widely reported.40 However, few reports adopted this strategy for regulating the more complex and challenging phosphors.
As is known, the multi-sulfurated aromatic derivatives exhibited aggregation-induced phosphorescence,41 and their luminescence behavior could be controlled by their variable conformation and stacking, attributed to their flexible multiple C−S bond.42 Based on this, mechanical stimuli have often been used to regulate the solid-stated stacking of multi-sulfurated aromatic derivatives,32 further inducing different luminescence. Compared to the mechanical force, temperature as a non-invasive external stimulus provided a facile and promising way to control emission behavior.35 However, it is challenging to obtain the reversibly switching visible light excited phosphorescence in multi-sulfurated aromatic derivatives by heat. Herein, we proposed a strategy for developing a temperature-responsive controllable system of tetrakis(arylthio)benzene derivatives by employing the phase-changing material lauric acid (LA) as the matrix (Fig. 1). Notably, the different luminescence under room and high temperatures were also observed in the phase-changing doping system because of the different molecular existence states in the highly-crystallized and melting states of LA. Furthermore, the multi-color luminescent systems could be obtained by further adding other dye molecules, and they were used in anti-counterfeiting labels and temperature indicators.
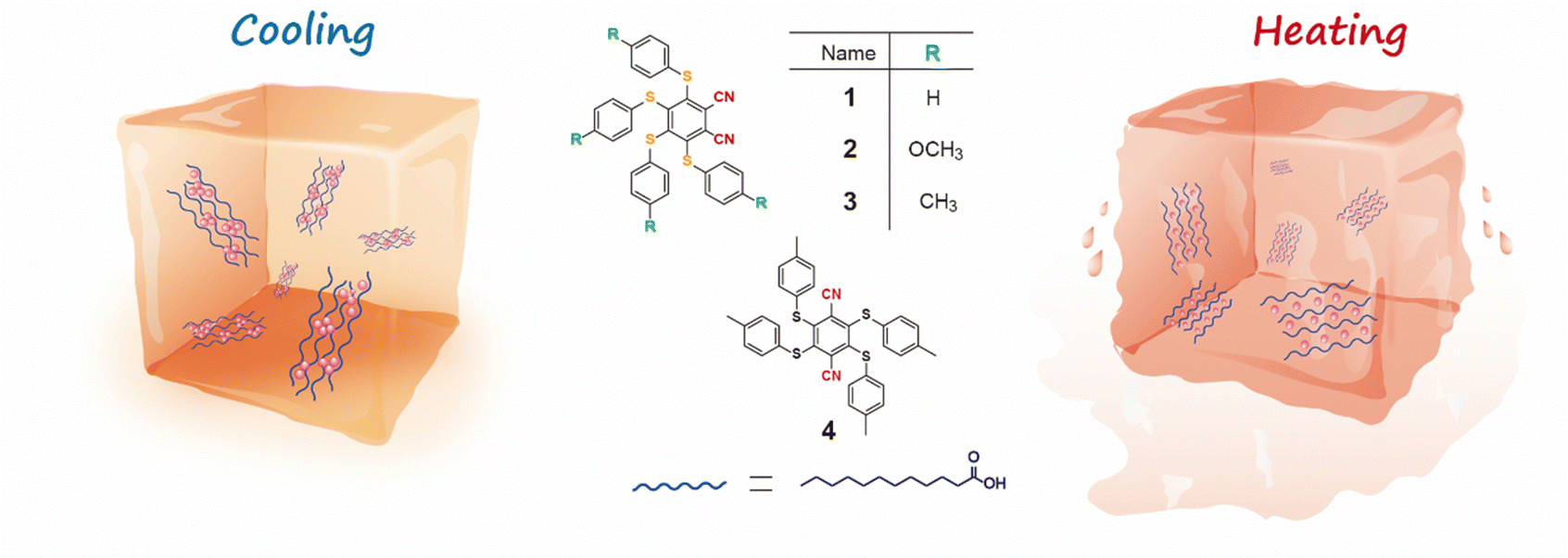 | ||
| Fig. 1 Schematic illustration of the regulation of phosphorescence emission behavior by temperature. | ||
Results and discussion
The target tetrakis(arylthio)benzene derivatives with D–A structure were obtained by one-step nucleophilic aromatic substitution reactions. Detailed synthesis procedures are placed in the ESI.† The purity of these compounds was confirmed using high-performance liquid chromatography (HPLC) (Fig. S1, ESI†). The UV-vis spectra of compounds 1–3 in dichloromethane (DCM) are shown in Fig. 2a, and they all exhibited similar absorption wavelengths. A broad low-energy absorption band that red-shifted to the visible region was attributed to their CT state (Fig. S2, ESI†). As the polarity of the solvent increased, the solution-stated emission gradually redshifted due to the strong CT effect of the D–A structure (Fig. 2b and Fig. S3, ESI†). Compared with other persulfurated benzene derivatives from previous reports,41 these compounds showed a more red-shifted emission, attributed to the stronger CT effect induced by the adjacent dicyano groups, which considerably enhanced the electron-withdrawing properties of the acceptor. These molecules showed weak luminescence in THF, and their luminescence quantum yields were 1.6% of 1, 0.9% of 2, 1.7% of 3, and 1.4% of 4, respectively. Comparatively, compounds 1 and 2 possessed a moderate luminescence in THF. However, when the H2O fraction reached up to 99% in the THF-H2O mixture, the emission intensity was enhanced (Fig. 2d and Fig. S4, ESI†), and their corresponding absorptions were also red-shifted (Fig. 2c and Fig. S4, ESI†), indicating the aggregation-induced emission (AIE) characteristic of these compounds. In addition, compound 3 with the adjacent dicyano groups, which further enhanced the electron-acceptor capability, possessed a stronger charge-transfer (CT) effect than compound 4 with separated dicyano groups, confirmed by the fact that compounds 3 possessed stronger redshifted absorption peaks than compound 4 in the solution state (Fig. S2, ESI†), which was a benefit to the visible-light excitation.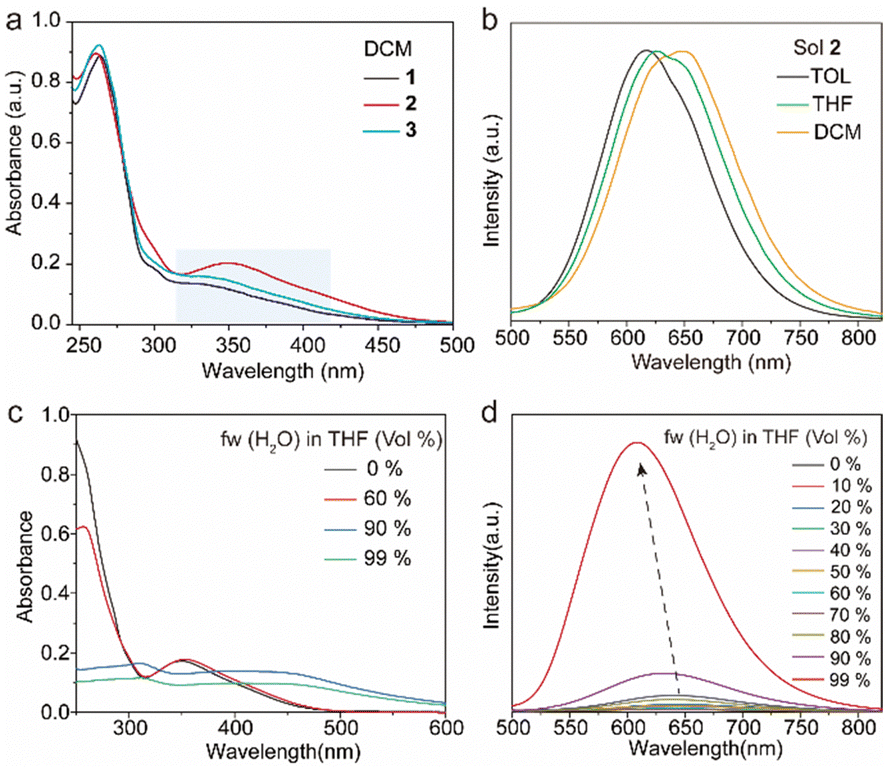 | ||
| Fig. 2 Photophysical properties of the tetrakis(arylthio)benzene derivatives in the solution state. (a) Absorption spectra of 1–3 in DCM. (b) Phosphorescence spectra of 2 in different solvents. (c) Absorption and (d) phosphorescence spectra of 2 in THF/H2O with different water fractions. All the measurements were performed under 420 nm excitation with a concentration of 10 μM. | ||
The molecules 1–4 with AIE characteristics exhibited relatively bright and multicolor luminescence in the solid state, which could change from green to orange under 365 nm UV irradiation (Fig. 3a). To further explore the internal luminescent mechanism, we investigated their solid-stated photophysical properties. As shown in Fig. 3a, the maximum emission wavelengths of compounds 1–4 were between 500 and 550 nm and their luminescence quantum yields were 4.3% of 1, 11.2% of 2, 11.6% of 3, and 8.2% of 4, (Fig. 3b). In addition, their UV-vis absorption spectra in the solid state presented a more significant red-shift than that in the solution state (Fig. S2 and S5, ESI†), which are attributed to molecular aggregation inducing red-shifted absorption. Their solid-stated delayed and prompt luminescent peaks were highly overlapped (Fig. 3a, Fig. S6, and Table S1, ESI†). The lifetimes for 1–4 under ambient conditions were 67.4 μs, 11.0 μs, 45.8 μs, and 15.4 μs, respectively (Fig. 3d and Fig. S7, ESI†), confirming their phosphorescence nature as previously reported in the literature.43 Noteworthily, visible light could excite their phosphorescence, which was significant in reducing UV damage in practical applications.
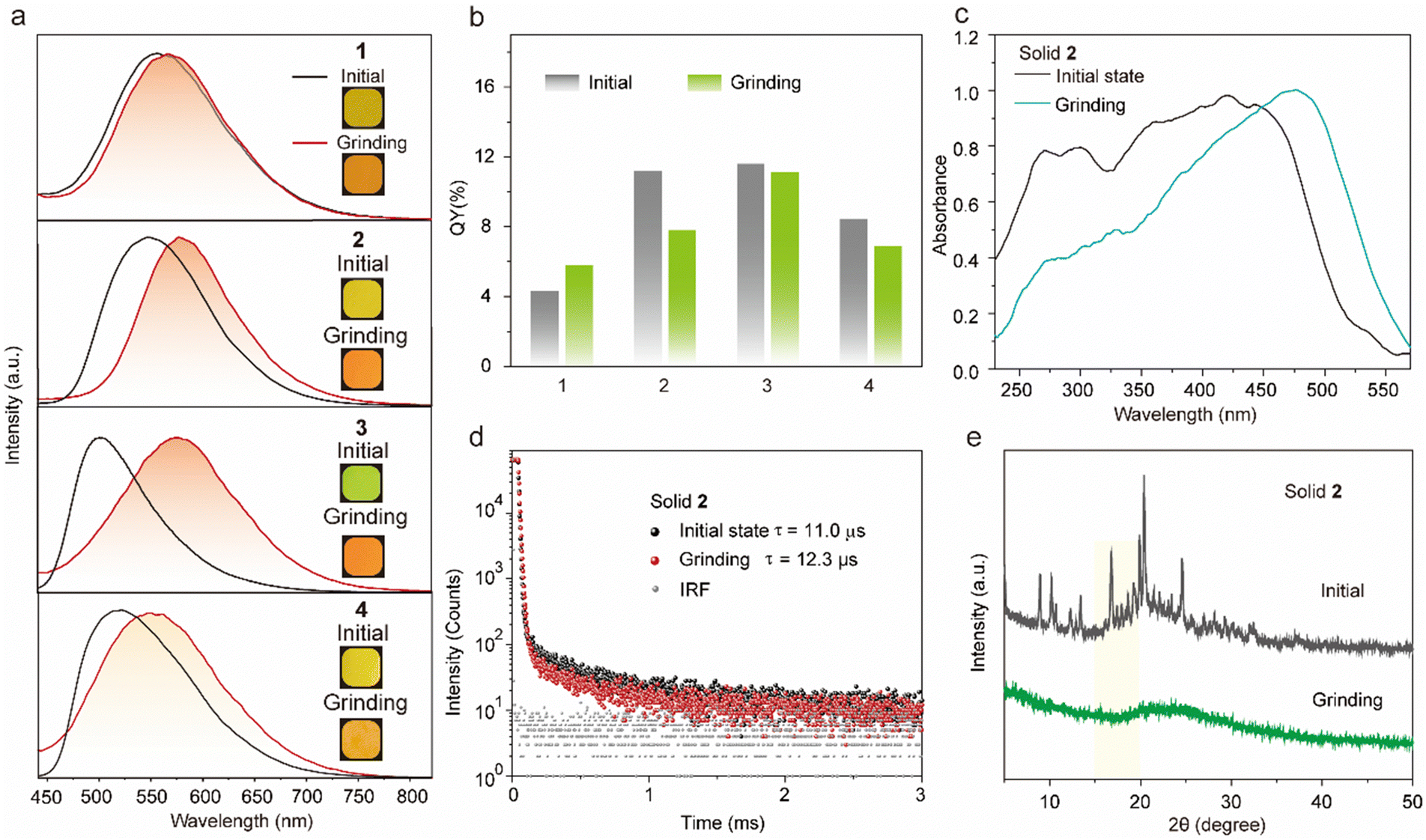 | ||
| Fig. 3 Photophysical properties of the tetrakis(arylthio)benzene derivatives in the solid powder state. (a) Normalized phosphorescence spectra (λex = 420 nm) of compounds 1–4 before and after grinding. (inset: photograph of solid powder 1–4 under 365 nm excitation). (b) Quantum yield of solid powders 1–4 before and after grinding. (c) Normalized absorption spectra, (d) luminescence lifetimes, and (e) XRD patterns of compound 2 before and after grinding. | ||
Certainly, the multi-sulfurated aromatic compounds are known for their diverse stacking in the solid state.42 Inspired by this, the feasibility of manipulating the stackings of compounds 1–4 through external stimuli to further regulate their luminescence behaviors was a topic that was worth researching deeply. Therefore, we first investigated their luminescence changes under mechanical stimuli. Encouragingly, compounds 2 and 3 powders showed high sensitivity to mechanical stimuli. As shown in Fig. 3a, the largest emission wavelength of compound 2 significantly redshifted from 548 nm to 582 nm under mechanical friction. The similar phosphorescence lifetimes before and after grinding confirmed that the mechanical friction did not quench the triplet emission (Fig. 3d). In addition, compounds 3 and 4 possessed ortho- and para-positions of the cyano groups, respectively. The maximum wavelength of compound 4 was 565 nm after grinding in the solid state, while that of compound 3 was significantly red-shifted by about 82 nm (Fig. 3a). This phenomenon was attributed to the adjacent cyano groups that could more significantly enhance the CT effect and lead to a redshifted luminescence of compound 3.
The powder X-ray diffraction (XRD) was used to confirm the stacking change. As shown in Fig. 3e, the XRD peaks of compound 2 disappeared after grinding, indicating its crystal transition from the crystalline to amorphous form. Meanwhile, compounds 1 and 3 showed similar changes in XRD peaks and luminescent behaviors under mechanical stimuli (Fig. S8, ESI†).42,44,45 Then, as shown in Fig. S9 (ESI†), these small and numerous peaks at 600–800 cm−1 and 1000–1200 cm−1 in FTIR spectra were attributed to C–S and C–S–C bonds, respectively. Compared with the initial state, the tetrakis(arylthio)benzene derivative molecules after grinding exhibited delicate shifts in these infrared-absorption peaks, indicating that the C–S bond may be rotated during grinding, which may be caused by the molecular stacking changes.46 Theoretically, it is possible that the emission color may be changed by a few dimers generated by external stimuli. However, as shown in Fig. 3e and S8, the PXRD peaks of compounds 1–4 greatly diminished or even disappeared after grinding. Additionally, the crystals of compound 2 were obtained by slowly evaporating the mixed solvent of DCM and PE.47 As shown in Fig. 4a, the crystals of compound 2 exhibited a highly distorted structure and abundant intermolecular noncovalent interactions. At the same time, the central benzene ring distances between the two neighboring molecules were up to 7.369 Å (Fig. 4b). Hence, the irregular peripheral groups and long distance between the central benzene ring made it challenging to generate the tight dimerization even after grinding. In addition, the weakened molecular stacking after grinding considerably reduced luminescence intensity (Fig. 3b). The changes of the solid stated excitation spectra before and after grinding were similar to those in the UV-vis absorption spectra (Fig. 3c, Fig. S5, and S10, ESI†).
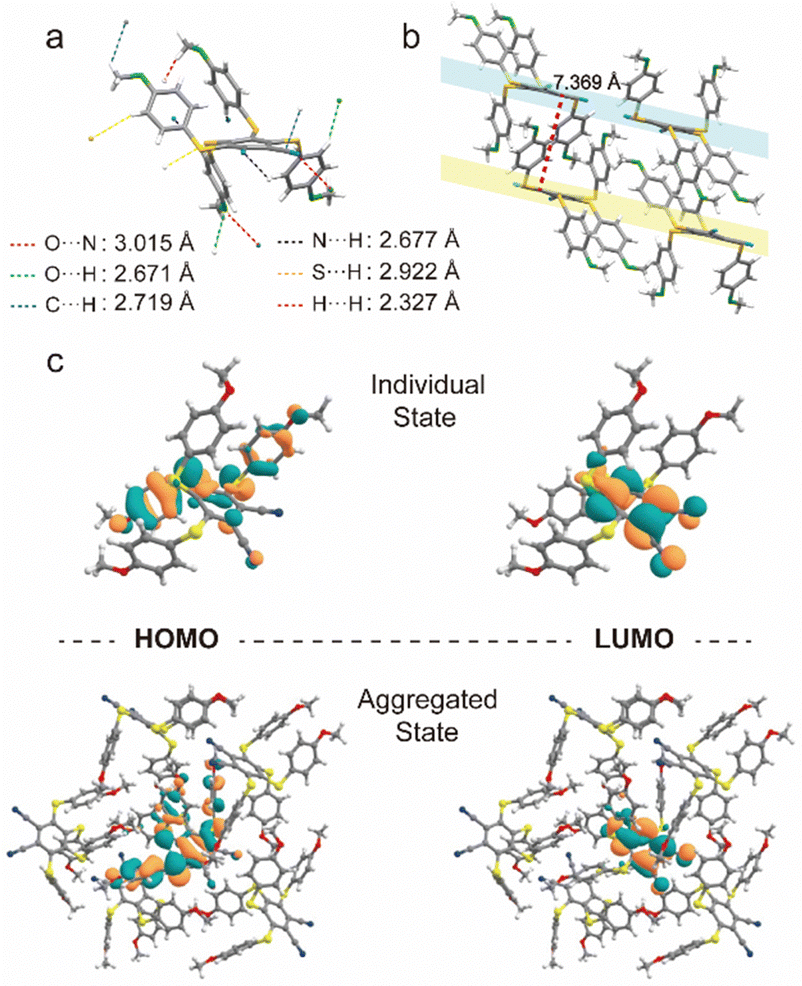 | ||
| Fig. 4 (a) The molecular geometry and (b) intermolecular interactions of compound 2. (c) HOMO and LUMO orbit of T1 in individual and aggregated states. | ||
To deeply elucidate the luminous mechanism regarding the molecular level, we performed the theoretical calculations using ONIOM simulation with Grimme's empirical dispersion correction, and the structure of compound 2 for calculation was extracted from an X-ray crystal structure containing 6 molecules where one molecule was fully surrounded by other ones. We optimized the target (internal) molecule in a T1 state at high-level B3LYP/6-311G(d,p),3–6 while the surrounding molecule was optimized by molecular mechanics method employing the UFF force field. These calculations simulate the behaviour of compound 2 in a crystal state (before grinding). Then, we optimized individual molecule 2 without surrounding using the same B3LYP/6-311G(d,p) method as for the target molecule in the ONIOM method. The more calculation details are provided in the ESI.† As for individual and aggregated states, their electronic configurations of HOMO and LUMO in the T1 state were similar, corresponding to the CT nature from the S atom of side phenyl substituents to the central benzene ring (Fig. 4c). It is widely known that multi-sulfurated aromatic derivatives sustained numerous minima on the T1 potential energy surface with a relatively flat potential in the broad range of the central ring deformations. Based on the crystal structure of compound 2, its monomolecular ONIOM optimization (as a part of the crystal packing fragment) of the T1 state exhibited a slightly distorted geometry (the central ring curvature: 21°) with predicted phosphorescence wavelength of 640 nm (Table S2, ESI†). After grinding, crystal powder 2 could generate more free molecules with strong T1 state distortion, and much stronger central ring distortion resulting in a red-shifted emission. Meanwhile, the predicted phosphorescence and distorted central cores of individual molecule 2 were 696 nm and 26°, respectively. The predicated simulations of 640 nm in the crystal and 696 nm for the individual molecules were consistent with experimentally observed values of 548 nm of the initial state and 582 nm after grinding. These calculations elucidated the structure of the T1 state in the highly dispersed state after grinding. Meanwhile, the crystal structure of compound 2 was completely disrupted, in good agreement with the XRD change, resulting in diminished molecular interactions with the surrounding molecules. Significantly, the tight crystal was absent after grinding, enabling molecule 2 to easily relax after the T1 population without confinement by surrounding molecules.
Considering that the emission of these molecules can be regulated by environmental change, we utilized the temperature-responsive phase-changing LA matrix with a melting point of 44–46 °C to control the microenvironment.38 At a temperature below its melting point, the highly crystalline solid-stated LA interacts sufficiently with the luminescent molecules, creating an environment with strong intermolecular interactions. However, when the temperature exceeds the melting threshold of LA, the intermolecular interactions weaken, resulting in a “soft” environment. Hence, it is possible to realize a temperature-induced luminescent molecular transition from monomer to aggregate states within LA.
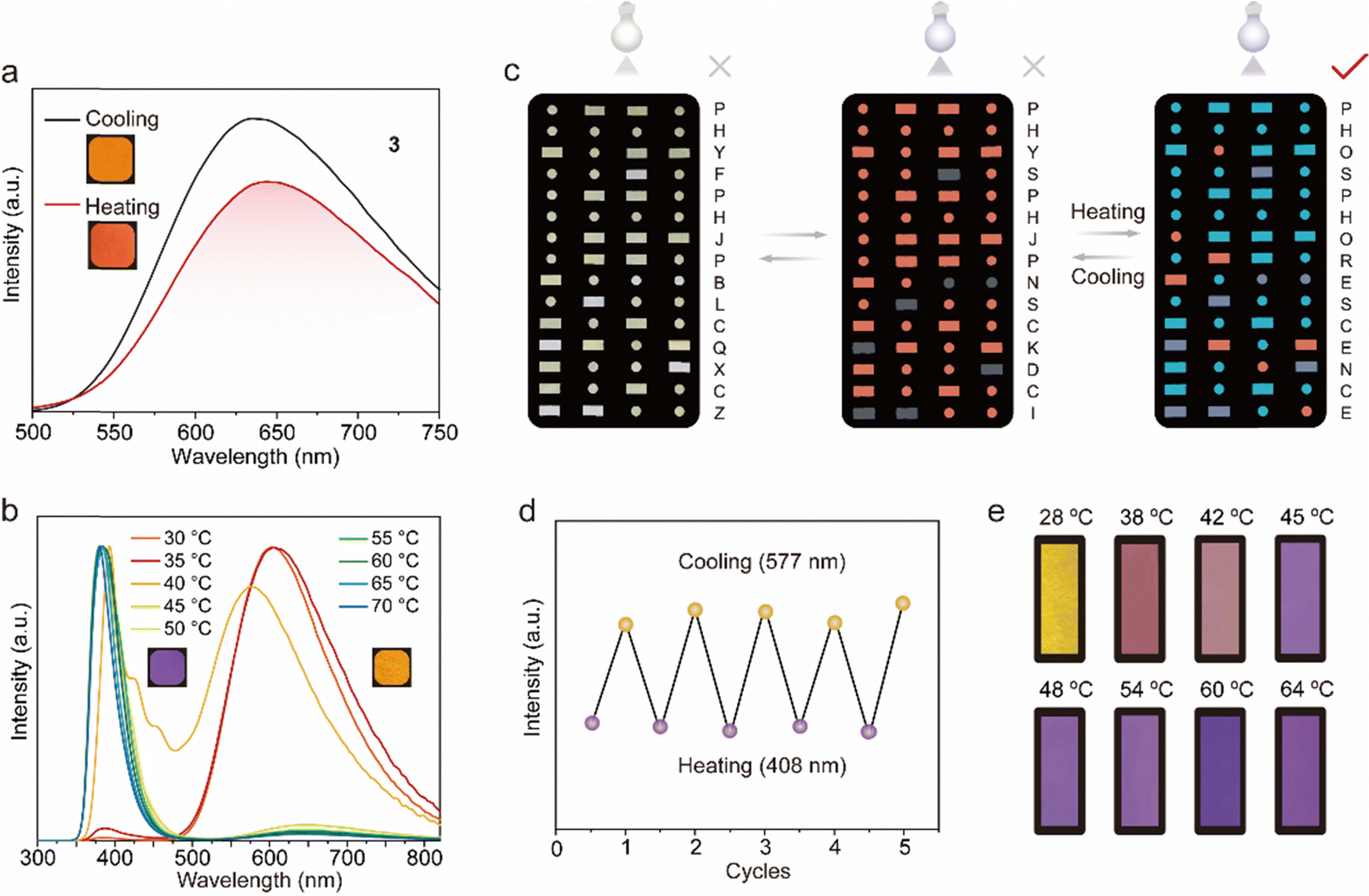
As shown in Fig. 5a, 3@LA (compound 3 with doping ratios about 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixed with LA) possessed a visible luminescent color, which was chosen for the following study. At room temperature, the 3@LA within a confined environment exhibited a maximum emission peak at 625 nm. As the temperature increased to 60 °C, the intermolecular interactions became weaker, and compound 3 was in a “loose” environment, resulting in a “monomer state”. At the same time, the maximum emission peak of 3@LA was located at 654 nm, which was red-shifted by about 29 nm compared with that at room temperature. This phenomenon was similar to the mechanical stimuli responsive phosphorescence behavior of compound 3, confirming that the transition of the dual-state phosphorescence emission can be realized by controlling the temperature. To demonstrate the universality of this construction strategy, we prepared other doped systems. Upon increasing the temperature, 2@LA showed similar redshifts to that of 3@LA, mainly because the methyl and methoxyl groups could induce more intermolecular interactions, while these interactions at different temperatures varied considerably (Fig. S11, ESI†). However, as for 1@LA and 4@LA, their temperature-responsive phosphorescence behaviors were weaker in the range of 30 °C to 60 °C. (Fig. S11, ESI†).
:1 mixed with LA) possessed a visible luminescent color, which was chosen for the following study. At room temperature, the 3@LA within a confined environment exhibited a maximum emission peak at 625 nm. As the temperature increased to 60 °C, the intermolecular interactions became weaker, and compound 3 was in a “loose” environment, resulting in a “monomer state”. At the same time, the maximum emission peak of 3@LA was located at 654 nm, which was red-shifted by about 29 nm compared with that at room temperature. This phenomenon was similar to the mechanical stimuli responsive phosphorescence behavior of compound 3, confirming that the transition of the dual-state phosphorescence emission can be realized by controlling the temperature. To demonstrate the universality of this construction strategy, we prepared other doped systems. Upon increasing the temperature, 2@LA showed similar redshifts to that of 3@LA, mainly because the methyl and methoxyl groups could induce more intermolecular interactions, while these interactions at different temperatures varied considerably (Fig. S11, ESI†). However, as for 1@LA and 4@LA, their temperature-responsive phosphorescence behaviors were weaker in the range of 30 °C to 60 °C. (Fig. S11, ESI†).
 | ||
| Fig. 5 (a) Emission spectra of 3@LA at 30 °C and 60 °C under 420 nm excitation. (b) Emission spectra of 3–6@LA under different temperatures. (c) The information encryption with the Morse code. (d) The thermostability of 3–6@LA. (e) The luminescence photograph of 3–6@LA under different temperatures. | ||
Since multi-color luminescence can convey richer and more comprehensive information, two new systems (3–5@LA and 3–6@LA) were further constructed by doping two classical triphenylamine (TPA) dyes into 3@LA. As can be seen from Fig. 5b, the maximum emission peak of 3–6@LA at 30 °C located at 605 nm, similar to the luminescence of compound 3. When the temperature was increased to 60 °C, the maximum emission peak of this doped system shifted to 383 nm, identical to that of compound 6, accompanied by the luminescence color change from red to purple. Similar changes in luminescence from red to green also occurred in 3–5@LA when the temperature was increased from 30 °C to 60 °C (Fig. S12a and b, ESI†). These thermochromism phenomena were attributed to the ACQ characteristic of triphenylamine (TPA) dyes in the aggregated state, which could show brighter luminescence than compound 3 in the dispersed state. These doping systems exhibited different color changes at varied temperatures, and therefore they were suitable for temperature indication and anti-counterfeiting. As shown in Fig. 5c, 3 and 3–5@LA were used for advanced information encryption because the luminescent color 3–5@LA in the crystalline solid state was approximately the same as that in the solid powder 3. The molten 3–5@LA was first applied to a part of the message in the Morse code, and the chloroform solution of compound 3 was then applied to the rest of the Morse code and allowed to dry. At temperatures below the melting point of LA, compounds 3 and 3–5@LA exhibited an identical color under 365 nm UV light. The correct message was well hidden (the wrong message: PHYSPHJPNSCKDCI). However, when the temperature surpassed its melting point, the color of the region corresponding to compound 3 remained unchanged, while that of the area covering 3–5@LA transitioned to green. This alteration in coloration unveiled the correct information (PHOSPHORESCENCE), enabling the decryption of the accurate information. It should be emphasized that this process was very reproducible (Fig. 5d). As can be seen in Fig. 5e, 3–6@LA was heated to a molten state, then the filter paper with the appropriate shape was immersed into it to sufficiently moisten it to make a temperature-indicating filter paper. The temperature-indicating filter paper showed different luminescent colors at different temperatures. Therefore, shown to be a promising candidate for luminescent thermometers.
Conclusions
In conclusion, a series of D–A-type tetrakis(arylthio)benzene derivatives with aggregation-induced phosphorescence characteristics were obtained by introducing different substituents in the periphery of the polysulfur aromatic core. Significantly, the adjacent dicyano groups, which further enhanced the electron-acceptor capability, induced more bathochromic phosphorescence emission. Their solid-state powders were sensitive to mechanical stimuli, and the phosphorescence emission significantly red-shifted upon grinding. Moreover, the monomer and the aggregated phosphorescence transition could be reversibly switched by temperature when doping these molecules into the phase-changing matrix LA, which transformed between the highly crystallized and melting states by temperature. Additionally, the multi-color luminescent systems could be obtained by adding other dye molecules, which were used in anticounterfeit labels and temperature indicators. This successful design provides a new idea for developing more temperature-responsive phosphorescent materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Fundamental Research Funds for the Central Universities (No. 2232022A-03; 2232022G-04), the National Natural Science Foundation of China (No. 22375036 and No. 21975046), the Natural Science Foundation of Shanghai (No. 21ZR1402000), International Cooperation Fund of Science and Technology Commission of Shanghai Municipality (No. 21130750100). G. B. thanks for support to the Swedish Research Council (starting grant No. 2020-04600), and the Swedish Government Strategic Research Area in Materials Science on Advanced Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009-00971). The quantum-chemical calculations were performed with computational resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS 2023/5–77 and SNIC 2022-3-34) at the National Supercomputer Centre (NSC) at Linköping University partially funded by the Swedish Research Council through grant agreement no. 2022-06725.References
- X. Li, S. Shen, C. Zhang, M. Liu, J. Lu and L. Zhu, Sci. China: Chem., 2021, 64, 534–546 CrossRef CAS.
- H. Chen, Y. Deng, X. Zhu, L. Wang, L. Lv, X. Wu, Z. Li, Q. Shi, A. Peng, Q. Peng, Z. Shuai, Z. Zhao, H. Chen and H. Huang, Chem. Mater., 2020, 32, 4038–4044 CrossRef CAS.
- G. Zhan, Z. Liu, Z. Bian and C. Huang, Front. Chem., 2019, 7, 305 CrossRef CAS PubMed.
- A. Krishna, V. Darshan, C. H. Suresh, K. N. Narayanan Unni and R. L. Varma, J. Photochem. Photobio. A, 2018, 360, 249–254 CrossRef CAS.
- Y. He, N. Cheng, X. Xu, J. Fu and J.-A. Wang, Org. Electron., 2019, 64, 247–251 CrossRef CAS.
- M. Ji and X. Ma, Ind. Chem. Mater., 2023, 57, 10854–10858 Search PubMed.
- Y. Li, G. V. Baryshnikov, F. Siddique, P. Wei, H. Wu and T. Yi, Angew. Chem., Int. Ed., 2022, 61, e202213051 CrossRef CAS PubMed.
- S. Zhao, L. Chen, Y. Yang and X. Liu, J. Mol. Struct., 2022, 1269, 133855 CrossRef CAS.
- Q. Feng, Z. Xie and M. Zheng, Sens. Actuators, B, 2022, 351, 130976 CrossRef CAS.
- J. Wang, M. Zhang, S. Han, L. Zhu and X. Jia, J. Mater. Chem. C, 2022, 10, 15565–15572 RSC.
- F. Lin, H. Wang, Y. Cao, R. Yu, G. Liang, H. Huang, Y. Mu, Z. Yang and Z. Chi, Adv. Mater., 2022, 34, 2108333 CrossRef CAS PubMed.
- Q. Dang, Y. Jiang, J. Wang, J. Wang, Q. Zhang, M. Zhang, S. Luo, Y. Xie, K. Pu, Q. Li and Z. Li, Adv. Mater., 2020, 32, 2006752 CrossRef PubMed.
- M. Cui, P. Dai, J. Ding, M. Li, R. Sun, X. Jiang, M. Wu, X. Pang, M. Liu, Q. Zhao, B. Song and Y. He, Angew. Chem., Int. Ed., 2022, 61, e202200172 CrossRef CAS PubMed.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed.
- C. Qian, Z. Ma, B. Yang, X. Li, J. Sun, Z. Li, H. Jiang, M. Chen, X. Jia and Z. Ma, J. Mater. Chem. C, 2021, 9, 14294–14302 RSC.
- Z. Yuan, L. Zou, D. Chang and X. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 52059–52069 CrossRef CAS PubMed.
- D. Chávez, C. R. Garcia, J. Oliva and L. A. Diaz-Torres, Ceram. Int., 2021, 47, 10–41 CrossRef.
- X. Wang, H. Ma, M. Gu, C. Lin, N. Gan, Z. Xie, H. Wang, L. Bian, L. Fu, S. Cai, Z. Chi, W. Yao, Z. An, H. Shi and W. Huang, Chem. Mater., 2019, 31, 5584–5591 CrossRef CAS.
- Y. Li, G. V. Baryshnikov, C. Xu, H. Ågren, L. Zhu, T. Yi, Y. Zhao and H. Wu, Angew. Chem., Int. Ed., 2021, 60, 23842–23848 CrossRef CAS PubMed.
- D. Lee, J. Jung, D. Bilby, M. S. Kwon, J. Yun and J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 2993–2997 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- J. Wei, M. Zhu, T. Du, J. Li, P. Dai, C. Liu, J. Duan, S. Liu, X. Zhou, S. Zhang, L. Guo, H. Wang, Y. Ma, W. Huang and Q. Zhao, Nat. Commun., 2023, 14, 4839 CrossRef CAS PubMed.
- X. Ma, C. Xu, J. Wang and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 10854–10858 CrossRef CAS PubMed.
- L. Ma and X. Ma, Sci. China: Chem., 2022, 66, 304–314 CrossRef.
- H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS PubMed.
- E. Hamzehpoor, C. Ruchlin, Y. Tao, C.-H. Liu, H. M. Titi and D. F. Perepichka, Nat. Chem., 2022, 15, 83–90 CrossRef PubMed.
- Z. Chen, X. Chen, D. Ma, Z. Mao, J. Zhao and Z. Chi, J. Am. Chem. Soc., 2023, 145, 16748–16759 CrossRef CAS PubMed.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS PubMed.
- Q. Huang, X. Mei, Z. Xie, D. Wu, S. Yang, W. Gong, Z. Chi, Z. Lin and Q. Ling, J. Mater. Chem. C, 2019, 7, 2530–2534 RSC.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS.
- B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC.
- X. Jia and L. Zhu, Acc. Chem. Res., 2023, 56, 655–666 CrossRef CAS PubMed.
- D. Li, J. Yang, M. Fang, B. Z. Tang and Z. Li, Sci. Adv., 2022, 8, eabl8392 CrossRef CAS PubMed.
- Z. Chen, K. Y. Zhang, X. Tong, Y. Liu, C. Hu, S. Liu, Q. Yu, Q. Zhao and W. Huang, Adv. Funct. Mater., 2016, 26, 4386–4396 CrossRef CAS.
- D. Li, X. Yang and D. Yan, ACS Appl. Mater. Interfaces, 2018, 10, 34377–34384 CrossRef CAS PubMed.
- J. Qiu, D. Huo and Y. Xia, Adv. Mater., 2020, 32, 2000660 CrossRef CAS PubMed.
- J. Du, L. Sheng, Y. Xu, Q. Chen, C. Gu, M. Li and S. X. A. Zhang, Adv. Mater., 2021, 33, 2008055 CrossRef CAS PubMed.
- X. Shan, W. Chi, H. Jiang, Z. Luo, C. Qian, H. Wu and Y. Zhao, Angew. Chem., Int. Ed., 2022, 62, e202215652 CrossRef PubMed.
- K. Xue, C. Wang, J. Wang, S. Lv, B. Hao, C. Zhu and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 14147–14157 CrossRef CAS PubMed.
- J. Gu, B. Yue, G. V. Baryshnikov, Z. Li, M. Zhang, S. Shen, H. Ågren and L. Zhu, Research, 2021, 2021, 11 Search PubMed.
- H. Wu, G. V. Baryshnikov, A. Kuklin, B. F. Minaev, B. Wu, L. Gu, L. Zhu, H. Ågren and Y. Zhao, ACS Appl. Mater. Interfaces, 2020, 13, 1314–1322 CrossRef PubMed.
- M. Villa, M. Roy, G. Bergamini, M. Gingras and P. Ceroni, Dalton Trans., 2019, 48, 3815–3818 RSC.
- Z.-A. Xia, X. Zhang, C. Xi, Q. Bai, H. Liu, S.-T. Zhang and B. Yang, CrystEngComm, 2023, 25, 5802–5809 RSC.
- V. Vendrell-Criado, V. Lhiaubet-Vallet, M. Yamaji, M. C. Cuquerella and M. A. Miranda, Org. Biomol. Chem., 2016, 14, 4110–4115 RSC.
- D. A. Zimmerman, J. L. Koenig and H. Ishida, Polymer, 1999, 40, 4723–4731 CrossRef CAS.
- Deposition numbers 2343600 (for compound 2) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2343600. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ma00318g |
| This journal is © The Royal Society of Chemistry 2024 |