
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Converting commercial-grade silicone into a vitrimer using elemental sulfur†
Mahsaalsadat
Rokni
 ab,
Kun Woo
Park
ab,
Wing
Ho Leung
ac,
Zoran
Zujovic
ad and
Erin M.
Leitao
*ab
ab,
Kun Woo
Park
ab,
Wing
Ho Leung
ac,
Zoran
Zujovic
ad and
Erin M.
Leitao
*ab
aSchool of Chemical Sciences, The University of Auckland, 1010, New Zealand. E-mail: erin.leitao@auckland.ac.nz
bThe MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
cCentre for Advanced Materials Manufacturing and Design, Department of Mechanical and Mechatronics Engineering, The University of Auckland, 1023, New Zealand
dInstitute of General and Physical Chemistry, Studentski Trg 12/5, 1100, Belgrade, Republic of Serbia
First published on 15th May 2024
Abstract
The silicone industry is rapidly expanding due to the high demand for biocompatible materials exhibiting properties such as temperature and solvent resistence as well as flexibility. However, due to its heavily cross-linked structure, silicone is not repairable or easily recycled, resulting in a large quanitity of silicone waste. As a first step towards synthesising repairable commercial-grade silicone, we incorporated dynamic S–S cross-links through the addition of elemental sulfur. By partially intercepting the platinum-catalysed hydrosilylation process, that occurs during curing of commerical grade silicone, with a small amount of vulcanisation using sulfur, a vitrimeric material was formed. The success of integrating S–S cross-links was determined using SEM, elemental analysis, as well as SSNMR, FT-IR, and Raman spectroscopies. The physical and mechanical properties of the sulfur containing silicone was compared to the unmodified silicone through a comparsion of TGA, DSC, rheology, and contact angle measurements before and after the healing process. This research not only provides a sustainable pathway for the utilisation of sulfur, a byproduct of petroleum and gas refining, it also represents a significant advancement in the development of self-healing materials, aligning with the broader environmental and resource conservation goals in industrial applications.
Introduction
Polysiloxanes, commonly referred to as silicones, were first developed by Ladenburg in 1872 and have since evolved into a substantial industry, with the global silicone market valued at USD 18.5 billion in 2022 and projected to reach USD 27.0 billion by 2027.1,2 These polymers are characterised by an alternating silicon–oxygen (Si–O) backbone, where each silicon atom is typically bonded to two organic groups, most commonly methyl substituents, e.g., in polydimethylsiloxane (PDMS).3 The remarkable stability of silicone, across a wide temperature range, can be attributed to the strength and flexibility of the Si–O bond. For example, the comparatively long length of the Si–O bond (1.64 Å) and Si–C bond (1.87 Å) relative to the C–C (1.53 Å) bond, reduces the steric impact of functional groups on the chain and allows significant rotational freedom.4–6 This facilitates silicone's ability to adopt the lowest energy configurations at interfaces, resulting in surface tension values substantially lower than those of its organic polymer congeners.7 Moreover, the number of repeating units in the polymer chain and degree of cross-linking directly influence material properties, resulting in silicones that range from viscous oils to flexible resins.8 The diversity in the types of silicone reflect their broad range of applications across various fields, from construction to medical devices to consumer items.9 The properties of silicones are particularly beneficial in energy-efficient applications, such as window sealants, insulators for electrical wires, and adhesives in the production of solar cells. Their exceptional resistance to weather and environmental stability contribute to reduced maintenance and manufacturing costs, as well as increased efficiency and longevity for these applications.10 However, the process of preparing elemental silicon for use in creating silicone precursors is quite expensive, as it relies on a capital-intensive method known as carbothermal reduction. This process involves heating silica (SiO2) in the presence of carbon at temperatures exceeding 2000 °C, leading to the release of one mole of CO2 for each mole of silicon produced (Fig. 1).11–13 Furthermore, the growing use and disposal of silicone and silicone-containing materials, leads to significant resource waste. The non-melting and non-soluble cross-linked structures of silicones, while attractive for applications, make recycling them challenging. Currently, the most advanced recycling methods involve breaking silicones down into smaller molecules or monomers through chemical processes. This recycling approach is energy-intensive, has low production efficiency, and can lead to additional environmental pollution.14–16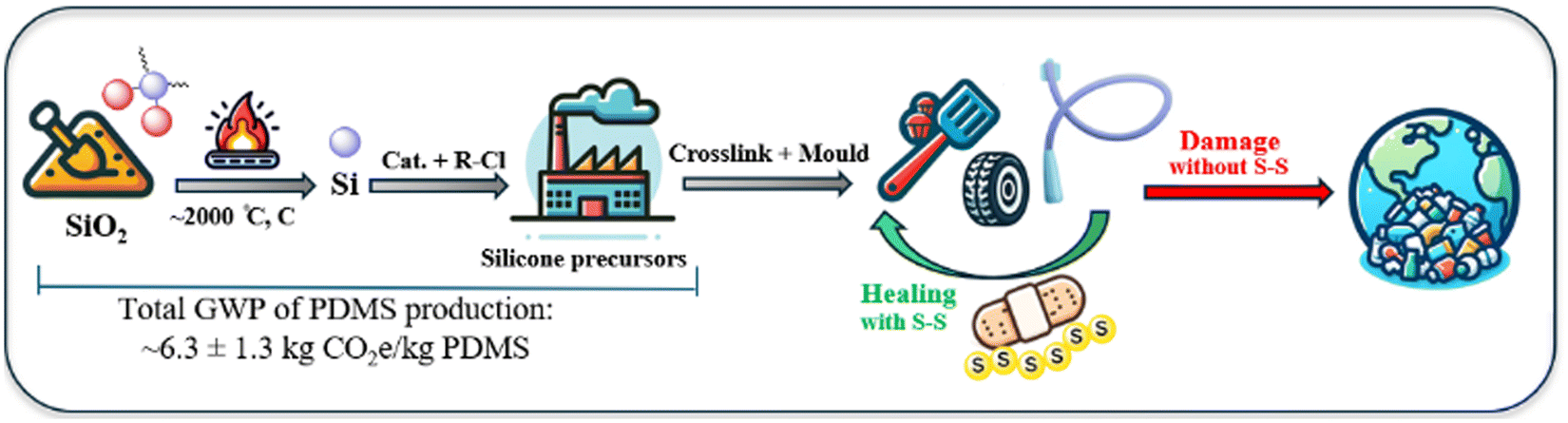 | ||
| Fig. 1 Simplified depiction of the energy-intensive production of silicone precursors and suggested strategy of using dynamic sulfur bonds for repairing and recycling them. | ||
Considering the challenges faced in the production, recycling, and waste management of silicones, there is an increasing need to develop repairable silicones. To this end, researchers have incorporated reversible non-covalent cross-links. Strategies such as Diels–Alder reactions,17 coordinating to ligands on metals,18 dynamic imine and aminal bonds,19 or taking inspiration from supramolecular chemistry are underway.20 Significantly, developing silicon vitrimer-based composites and functionalised boron nitride nanosheets represents progress in advancing repairable silicone technologies.21 In addition, combining disulfide bond-containing silanes with functionalised PDMS has allowed access to silicone which can self-repair under sunlight.22 This repairability can be tuned by the type of disulfide used, for example higher reactivity is achieved using aromatic disulfides.23,24 However, the use of disulfide bonds in silicone materials presents notable challenges, including the synthesis of the disulfide.
In this context, the direct incorporation of elemental sulfur (S8) offers a promising avenue. This approach utilises the inherent potential of elemental sulfur to form oligo- or polysulfide linkages, a technique that has been proven to effectively repair severe damage in materials, such as poly(sulfur-r-1,3-diisopropenylbenzene).25 By bypassing the need for disulfide silane synthesis, the direct use of elemental sulfur simplifies the production process and addresses scalability issues, allowing for easier pursuit of more efficient and sustainable repairable silicone materials. Furthermore, elemental sulfur, a by-product of petroleum and gas refining, is abundantly available. With production exceeding 70 million tonnes annually and consumption, to predominately make sulfuric acid, significantly lagging behind.26–28 This surplus, of >5 million tonnes per annum, along with sulfur's low cost, makes it a promising candidate for synthesizing new materials. Importantly, as polysulfides exhibit many of the same properties as polysiloxanes, including high strength and compressibility as well as resistance to solvents, temperature, and UV light, the material containing the dynamic S–S bonds will retain all desired properties of silicone.29,30
Expanding upon our recent research studying the inverse vulcanisation of sulfur31,32 with both linear and cyclic siloxane cross-linkers,33,34 we rationalised that the direct incorporation of sulfide cross-links into commercial silicone will produce silicones that are similar, but with the added benefit of repairability. To accomplish this, we aimed to partially intercept the commercial Pt-catalysed hydrosilylation cross-linking of silicone (top route, Fig. 2) with a minor amount of sulfur vulcanisation (bottom route, Fig. 2). Herein, the repairability of the sulfide cross-linked materials are assessed via a direct comparison of their properties relative to silicone synthesised by a related commercial process. Employing spectroscopic (FT-IR, Raman, SS NMR), thermal (TGA, DSC), physical (water contact angle), and mechanical (rheology) techniques, insights into the effects of integrating sulfur with silicone are provided. This advancement in silicone chemistry holds great potential for making silicone more sustainable and efficient, aligning with the broader objectives of reducing environmental impact and conserving resources in industrial applications.
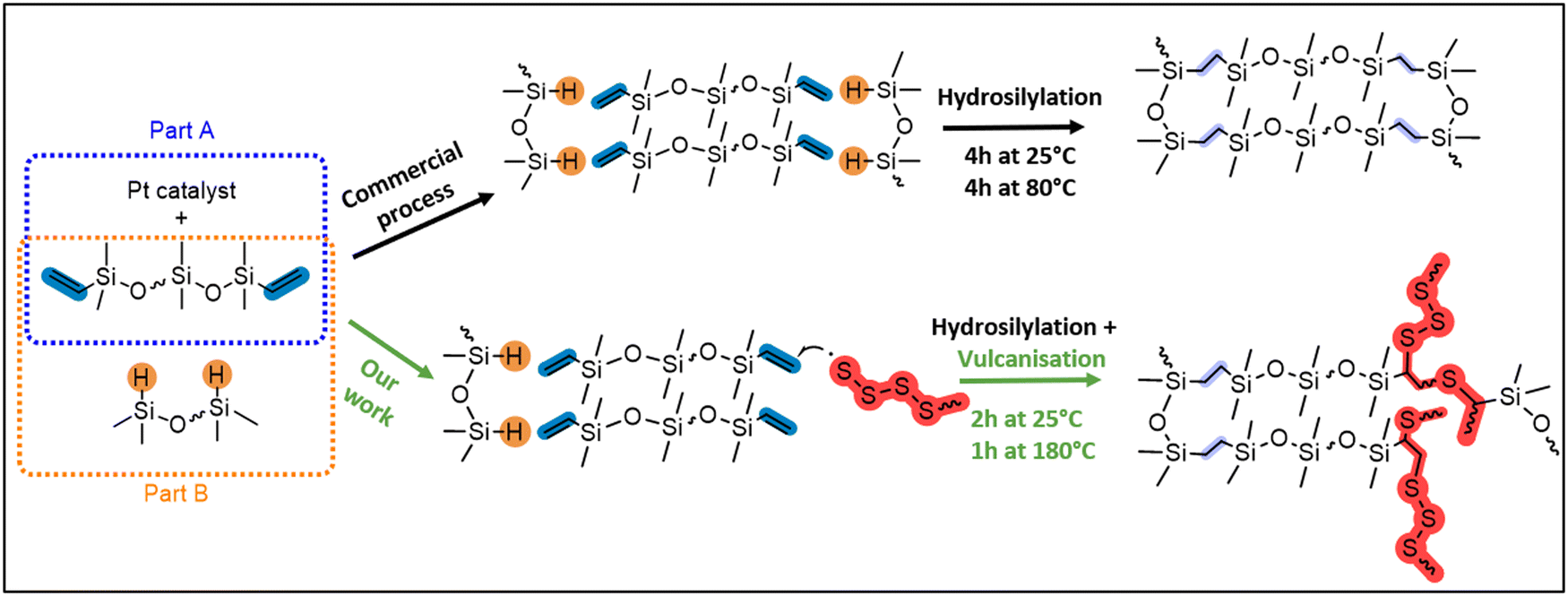 | ||
| Fig. 2 Commercial synthesis (top route) of polysiloxane from the part A and part B moulding kits and our modified synthesis (bottom route) using S8 to generate X-poly(siloxane-r-S) (X = wt% S) with thermally repairable properties. | ||
Experimental
Materials
Deuterated chloroform (CDCl3) was purchased from Cambridge Isotope Laboratories Inc., part A and part B high-modulus reprographic silicone (hPDMS) was purchased from Gelest, and sulfur (S8) powder was from Riedel-de Haen. All materials were used directly, as supplied.Instrumentation
For 1H NMR spectroscopy, solution-based samples were dissolved in CDCl3 and observed using a Bruker Avance 400 MHz spectrometer, with spectra recorded in parts per million (ppm). Tetramethylsilane (TMS) was used for calibration, and residual protium in NMR solvents provided a reference (7.26 ppm for CDCl3) and the data processing was carried out using MestReNova software. The SSNMR experiments were performed using a Bruker Avance 300 spectrometer operating at 300.13 MHz 1H frequency, 59.62 MHz 29Si frequency and 75.47 MHz 13C frequency. A multinuclear double-tuned Bruker probe with 7 mm zirconia rotors, retained with Kel-F end-caps was used. Recycle delays allow for complete relaxation of the longest components which was checked using corresponding T1 relaxation measurements. The spin rate was 7 kHz in all experiments. The spectra were analysed using Bruker TopSpin and MestReNova NMR processing software packages. The 29Si spectra were recorded using a single pulse excitation pulse sequence with 5.0 μs 90° pulse. The recycle delays were 500 s for 0-poly(siloxane-r-S), and 128 for 10-poly(siloxane-r-S) samples and numbers of scans were 124 and 160 respectively. The spectral width was 30 kHz. The 29Si chemical shift scale was referenced to a 29Si resonance in the tetramethylsilane spectrum (TMS, 0 ppm). The 1H spectra were recorded using a single pulse excitation pulse sequence with 6.0 μs 90° pulse. The recycle delays were 5 s and the numbers of scans were 24 for both samples. The spectral width was 25 kHz. The 1H chemical shift scale was referenced to a 1H resonance in the tetramethylsilane spectrum (TMS, 0 ppm). The 13C spectra were recorded using a single pulse excitation pulse sequence with 5.0 μs 90° pulse. The recycle delays were 20 s and numbers of scans were 3000 for both samples. The spectral width was 50 kHz. The 13C chemical shift scale was referenced to a 13C resonance in the tetramethylsilane spectrum (TMS, 0 ppm). Fourier-transform infrared spectroscopy (FT-IR) data were gathered using a Bruker Vertex70, between 400 cm−1 to 4000 cm−1 and Raman spectroscopy was conducted using a Nicolet is50 Raman Module instrument equipped with a 1064 nm laser. Rheological measurements were performed using a rheometer model MCR702, where the storage and loss modulus (G′ and G′′) were determined through a frequency sweep at 25 °C, 0.1% strain, and a range of 0.1–100 rad per second. A relaxation test was also conducted at 25 °C, 5% strain, for 1000 seconds. Differential Scanning Calorimetry (DSC) results were obtained using a TA Instrument Q1000 DSC, under nitrogen flow, with heating from −80 °C to 150 °C with heating rate of 10 °C min−1 and cooling rates of 5 °C min−1. Thermogravimetric analysis (TGA) was performed using a TA Instrument Q500, heating samples to 700 °C under nitrogen at a rate of 10 °C min−1. Scanning electron microscopy (SEM) analysis was updated to use a Hitachi SU-70 Schottky field emission scanning electron microscope, with specimens sputter-coated with platinum (Pt) for 60 seconds using a Hitachi E-1045 ion sputter. Water contact angle measurements were performed with a Biolin Scientific Theta Lite instrument.General curing and cross-linking procedure
To a vial a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of part A and part B (2 g each), were added. The mixture was stirred for 2 h at 25 °C to initiate the hydrosilylation process. For 0-poly(siloxane-r-S), the mixture was then poured into a mould to be cured at 180 °C for 5 min at which point it was a clear, transparent solid (yield = 98%). For the polymers containing sulfur, X-poly(siloxane-r-S), an appropriate amount of S8 (X = 5, 0.21 g, 6.56 mmol; X = 10, 0.44 g, 0.0137 mol; X = 15, 0.66 g, 0.0206 mol) was heated to 180 °C to induce the vulcanisation reaction. To the molten polysulfide, the 2 h cured polysiloxane was added. The reaction continued until a single phase formed (vitrification point) and the mixture began to become more viscous (90 min for 5 wt% and 10 wt% and 1 h for 15 wt%). This viscous solution was then poured into a preheated mould at 180 °C. At this temperature, the series of X-poly(siloxane-r-S) underwent final solidification over an additional 20 min in the oven. The higher wt% S samples were darker and more opaque, with 5-poly(siloxane-r-S) (yield = 94%) as a lighter yellow color than 10-poly(siloxane-r-S) (yield = 93%) which was lighter than the 15-poly(siloxane-r-S) (yield = 91%). After 24 h, the 15-poly(siloxane-r-S) sample showed signs of depolymerisation, as evidenced by the emergence of sulfur crystals and powder on its surface. This instability is attributed to the formation of weaker S–S bonds due to longer polysulfide chains within the polymer matrix when adding 15 wt% sulfur.
:1 ratio of part A and part B (2 g each), were added. The mixture was stirred for 2 h at 25 °C to initiate the hydrosilylation process. For 0-poly(siloxane-r-S), the mixture was then poured into a mould to be cured at 180 °C for 5 min at which point it was a clear, transparent solid (yield = 98%). For the polymers containing sulfur, X-poly(siloxane-r-S), an appropriate amount of S8 (X = 5, 0.21 g, 6.56 mmol; X = 10, 0.44 g, 0.0137 mol; X = 15, 0.66 g, 0.0206 mol) was heated to 180 °C to induce the vulcanisation reaction. To the molten polysulfide, the 2 h cured polysiloxane was added. The reaction continued until a single phase formed (vitrification point) and the mixture began to become more viscous (90 min for 5 wt% and 10 wt% and 1 h for 15 wt%). This viscous solution was then poured into a preheated mould at 180 °C. At this temperature, the series of X-poly(siloxane-r-S) underwent final solidification over an additional 20 min in the oven. The higher wt% S samples were darker and more opaque, with 5-poly(siloxane-r-S) (yield = 94%) as a lighter yellow color than 10-poly(siloxane-r-S) (yield = 93%) which was lighter than the 15-poly(siloxane-r-S) (yield = 91%). After 24 h, the 15-poly(siloxane-r-S) sample showed signs of depolymerisation, as evidenced by the emergence of sulfur crystals and powder on its surface. This instability is attributed to the formation of weaker S–S bonds due to longer polysulfide chains within the polymer matrix when adding 15 wt% sulfur.
Results and discussion
Synthesis of vitrimeric sulfide cross-linked commercial silicone
A series of oligosulfide cross-linked commercial silicones, X-poly(siloxane-r-S) (X = 5–15 wt% sulfur) were synthesised (Fig. 3). We used moulding kits for silicone elastomers provided by Gelest, which come in two parts. Part A contains telechelic (vinyl-terminated) polydimethylsiloxane (PDMSn=16) and a Pt catalyst, and part B contains the same telechelic PDMS as well as a polyhydromethylsiloxane-co-polydimethylsiloxane (PHMSn=4-co-PDMSn=12) cross-linker (Fig. S1–S8 and Table S1, ESI†). In the commercial process, the Pt catalyst facilitates hydrosilylation via the addition of Si–H bonds, pendent on the PHMS, across vinyl substituents (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) at the termini of PDMS. This occurs when the two parts are mixed at 25 °C for 4 h and they are either left to cure at 25 °C for a further 24 h or heated above 80 °C for 4 h (Fig. 2).35,36 In contrast, our process involved strategically modifying the polymer cross-linking, by deliberately allowing less hydrosilylation with the Pt catalyst, establishing only some ethylene (CH2CH2) bridges. In this case, 2 h after mixing part A and part B at 25 °C, we introduced the polysiloxane to sulfur that had been previously heated at 180 °C (5–15 wt%, Fig. 3). Both S8 and polysulfide are known to act as Pt catalyst poisons,37 thereby halting any further hydrosilylation and instead triggering the sulfur vulcanisation reaction to incorporate a low concentration of dynamic sulfide bridges. To create a control sample under the same conditions as our sulfur-modified samples, after mixing parts A and B at 25 °C for 2 hours, we cured the 0-poly(siloxanes-r-S) at 180 °C, resulting in solidification within 5 minutes (Fig. 3). This approach ensured the synthetic conditions were consistent, allowing for a direct comparison between sulfur modified and original siloxane polymers. The commercial conditions (4 h 25 °C followed by 4 h at 80 °C or 24 h at 25 °C) could not be applied to the sulfur containing materials as a higher temperature was required for the vulcanisation reaction to occur. In the sulfur incorporated samples, as the amount of sulfur increased from 5 to 15 wt% the polymers became darker (Fig. 3). The polymer containing the highest wt% sulfur, 15-poly(siloxane-r-S), showed signs of sulfur on the surface of the disc within 24 h at 25 °C (rightmost picture, Fig. 3), while the 5–10 wt% sulfur-containing polymers did not exhibit any depolymerisation over time (>1 year).
C) at the termini of PDMS. This occurs when the two parts are mixed at 25 °C for 4 h and they are either left to cure at 25 °C for a further 24 h or heated above 80 °C for 4 h (Fig. 2).35,36 In contrast, our process involved strategically modifying the polymer cross-linking, by deliberately allowing less hydrosilylation with the Pt catalyst, establishing only some ethylene (CH2CH2) bridges. In this case, 2 h after mixing part A and part B at 25 °C, we introduced the polysiloxane to sulfur that had been previously heated at 180 °C (5–15 wt%, Fig. 3). Both S8 and polysulfide are known to act as Pt catalyst poisons,37 thereby halting any further hydrosilylation and instead triggering the sulfur vulcanisation reaction to incorporate a low concentration of dynamic sulfide bridges. To create a control sample under the same conditions as our sulfur-modified samples, after mixing parts A and B at 25 °C for 2 hours, we cured the 0-poly(siloxanes-r-S) at 180 °C, resulting in solidification within 5 minutes (Fig. 3). This approach ensured the synthetic conditions were consistent, allowing for a direct comparison between sulfur modified and original siloxane polymers. The commercial conditions (4 h 25 °C followed by 4 h at 80 °C or 24 h at 25 °C) could not be applied to the sulfur containing materials as a higher temperature was required for the vulcanisation reaction to occur. In the sulfur incorporated samples, as the amount of sulfur increased from 5 to 15 wt% the polymers became darker (Fig. 3). The polymer containing the highest wt% sulfur, 15-poly(siloxane-r-S), showed signs of sulfur on the surface of the disc within 24 h at 25 °C (rightmost picture, Fig. 3), while the 5–10 wt% sulfur-containing polymers did not exhibit any depolymerisation over time (>1 year).
 | ||
| Fig. 3 Moulded discs of X-poly(siloxane-r-S). From left to right: X = 0, 5, 10, 15 wt% S and 15 wt% S after 24 h at 25 °C. | ||
Spectroscopy
Once cured and moulded, we investigated any chemical bond alterations by FT-IR spectroscopy. Initially, part A exhibited peaks at 1596 cm−1 and 912 cm−1 (CC stretching from Si–CHCH2 functional group), while part B showed a distinctive peak at 2158 cm−1 (Si–H stretch)38 (Fig. 4). During the synthesis of 10-poly(siloxane-r-S), continuous measurements taken at 30-min intervals showed a consistent decrease in the intensity of the CC and Si–H stretches (Fig. 4, top). This decrease signifies the consumption of the vinyl groups and Si–H bonds as the vulcanisation and hydrosilylation reactions progressed, indicating that the sulfur chains were successfully incorporated into the copolymer. However, it should be noted that unfortunately, direct evidence by FT-IR spectroscopy of the formation of a C–S bond (C–S stretch at 1159 cm−1) was not possible due to its low concentration within the material and peak overlap with the Si–CH3 stretches at 1260 cm−1 and Si–O–Si in the region of 1000–1130 cm−1, typical for polysiloxanes.39
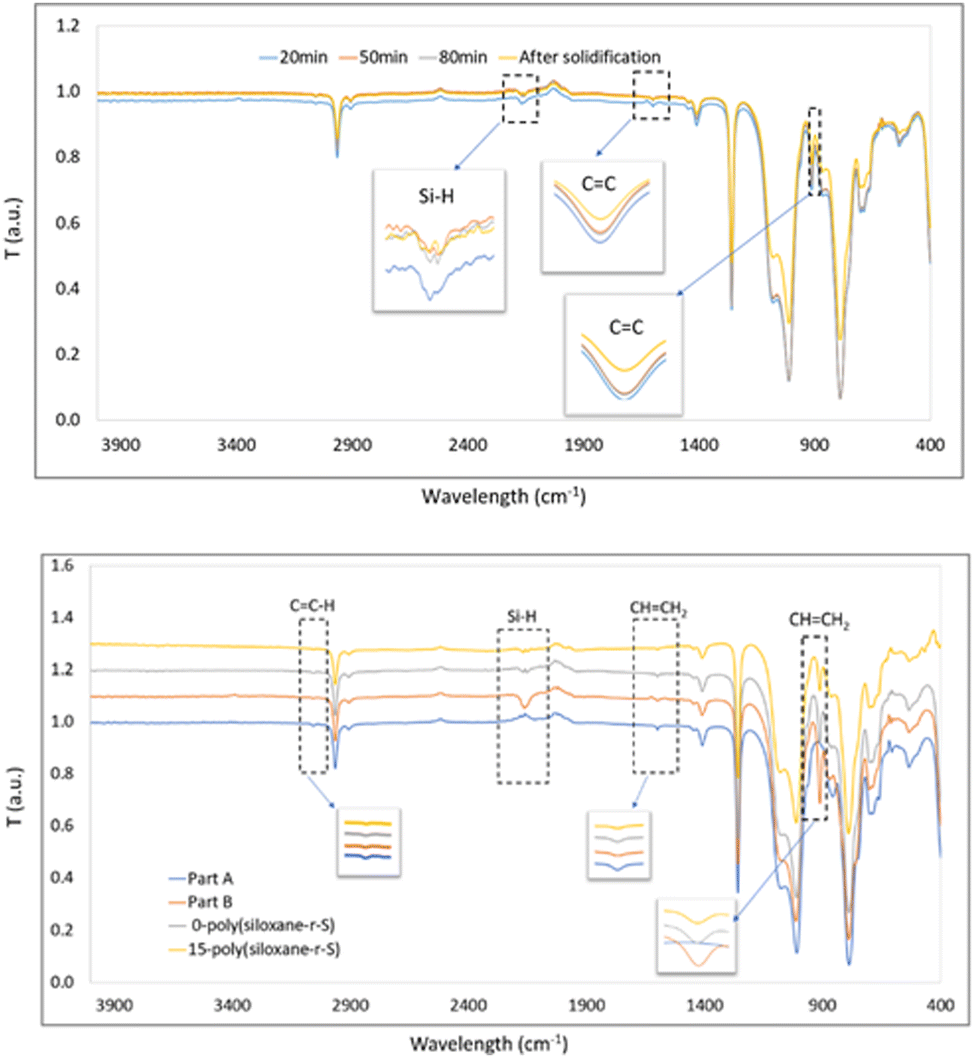 | ||
| Fig. 4 Top: FT-IR spectra of (a) 10-poly(siloxane-r-S) after reacting with sulfur for 20 min (blue), 50 min (orange), 80 min (grey) and after solidification (yellow), and bottom: part A (blue), part B (orange), and 0-poly(siloxane-r-S) after stirring for 2 h (grey) and 15-poly(siloxane-r-S) after solidification at 180 °C (yellow). | ||
In order to prove that C–S bonds were formed during the synthesis of X-poly(siloxane-r-S) (X > 0, wt% S), we turned to solid state (SS) NMR spectroscopy (Fig. 5; Fig. S9–S11, ESI†). Using SS 13C single pulse excitation, weak signals were observed at 48.9 and 52.4 ppm, corresponding to CH–S and CH2–S, respectively (Fig. 5, top). Additionally, SS 1H NMR spectroscopy confirmed the presence of CH–S and CH2–S signals with broad signals observed in the 2.0–3.5 ppm range (Fig. 5, bottom). The presence of unreacted vinyl groups and Si–H bonds were detected using both SS 13C and SS 1H NMR analyses (Fig. 5), corroborating what was observed by FT-IR spectroscopy. Notably, the Si–H signal (δH = 4.6 ppm) was substantially weaker in the spectrum for 0-poly(siloxane-r-S) compared to the spectrum for 10-poly(siloxane-r-S). This difference in signal strength confirms that the hydrosilylation reaction was suppressed as there was a shift towards vulcanisation upon sulfur integration.
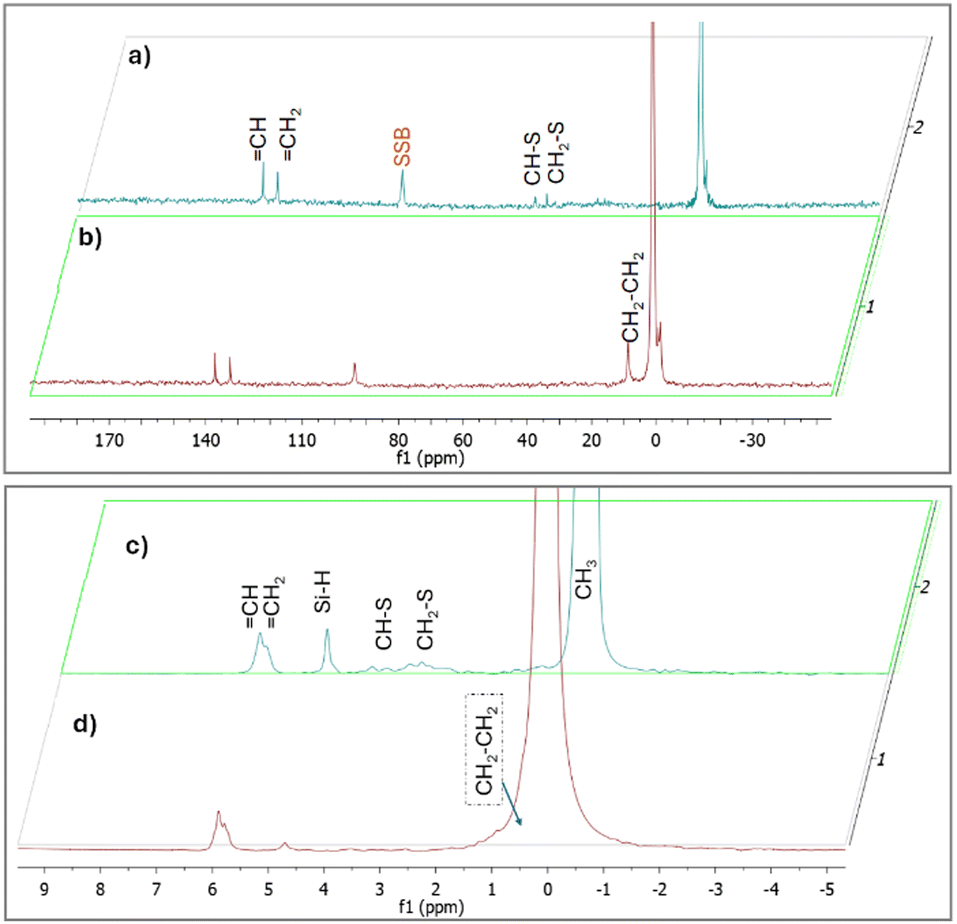 | ||
| Fig. 5 SSNMR spectra of X-poly(siloxane-r-S) (X = wt% S): 13C NMR (top) of (a) X = 10 (blue) and (b) X = 0 (red), and 1H NMR (bottom) of (c) X = 10 (blue) and (d) X = 0 (red). [note: SSB = spinning side band] | ||
Raman spectroscopy provided further insight into the composition of our synthesised sulfide cross-linked polysiloxanes, particularly in terms of sulfur integration. Notably, in 0-poly(siloxane-r-S), 5-poly(siloxane-r-S) and 10-poly(siloxane-r-S), the characteristic elemental sulfur peaks at 217 and 150 cm−1 (red, Fig. 6) were missing, suggesting the absence (X = 0, blue) or successful integration of S–S bonds into the polysiloxane (X = 5, purple; X = 10 green).40 Supporting our visual observations, 15-poly(siloxane-r-S) clearly displayed the typical spectral signature of elemental sulfur (yellow, Fig. 6). Our Raman spectroscopy results thus reflect the successful incorporation of S–S bonds within the polymer matrix and the formation of C–S bonds through the vulcanisation reaction.
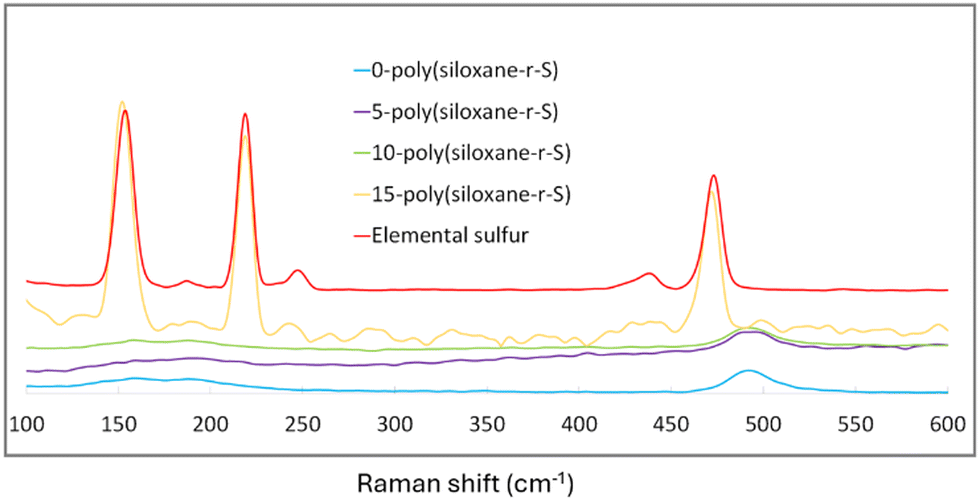 | ||
| Fig. 6 Raman spectra of X-poly(siloxane-r-S) (X = wt% S); X = 0 (blue), 5 (purple), 10 (green), 15 (yellow), and S8 (red). | ||
Imaging
Magnified photos (left) and scanning electron microscopy (SEM) images (right) of 5-poly(siloxane-r-S) and 10-poly(siloxane-r-S) samples confirmed that their morphology closely resembled the 0-poly(siloxane-r-S), displaying smooth and relatively homogeneous surfaces, indicating a uniform integration of sulfur chains within the polymer matrix (Fig. 7a–c). However, both magnified images and SEM analysis of the 15-poly(siloxane-r-S) revealed a different surface morphology, showing visible sulfur formation (Fig. 7d). This finding was consistent with the observed depolymerisation after 24 h. | ||
| Fig. 7 Magnified (9x zoom) photos (left) and SEM images (right) to show surface features of X-poly(siloxane-r-S) (X = wt% S); X = (a) 0 (b) 5 (c) 10 (d) 15. | ||
Thermal properties
Thermogravimetric analysis (TGA) provides information on the decomposition behaviour of X-poly-(siloxane-r-S) (X = wt% S). Elemental sulfur shows a characteristic initial weight loss at about 230 °C (red, Fig. 8). As expected, this particular onset is reflected in the 15-poly(siloxane-r-S) sample (yellow). For 5-poly(siloxane-r-S) (purple) and 10-poly(siloxane-r-S) (green), the onset of weight loss occurs later, at 305 °C for 5-poly(siloxane-r-S) and 304 °C for 10-poly(siloxane-r-S), due to the cleavage of C–S bonds.31,41 The sample with higher wt% S, namely 15-poly(siloxane-r-S), also displays a slightly lower degradation temperature at 284 °C suggesting the presence of C–S bonds in the cross-linked polymer, along with the additional early weight loss at 223 °C attributed to the depolymerisation of S–S bonds. In fact, the TGA profile of the 15 wt% S sample is much closer to that of the 5 and 10 wt% S samples, than to elemental sulfur, signifying significantly more incorporation of sulfur into the polysiloxane structure than the surface techniques, Raman and SEM, suggest. In contrast, weight loss of 0-poly(siloxane-r-S) (blue, Fig. 8) is determined to be around 352 °C followed by three mass losses at 464 °C, 531 °C and 606 °C and it maintains roughly 60% of its weight at 700 °C, demonstrating its relative thermal stability (Fig. S12–S15, ESI†). The samples with some sulfur content demonstrate a more noticeable decrease in remaining weight as the temperature increases. Specifically, the 5-poly(siloxane-r-S) sample retains just under 50% of its weight at 700 °C, while the 10-poly(siloxane-r-S) and 15-poly(siloxane-r-S) samples show residuals of about 40% and 30%, respectively. These observations suggest that the incorporation of sulfur reduces the thermal stability of polysiloxane.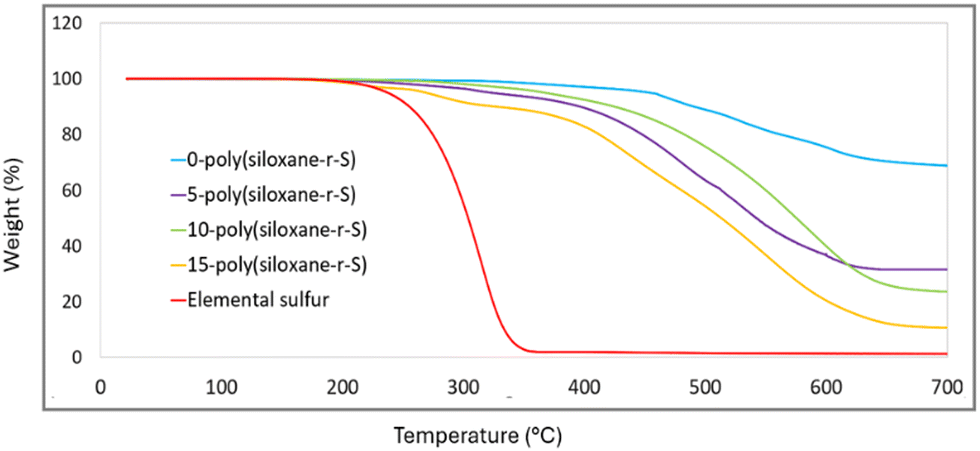 | ||
| Fig. 8 Comparative TGA thermograms of X-poly(siloxane-r-S) (X = wt% S); X = 0 (blue), 5 (purple), 10 (green), 15 (yellow), and S8 (red). | ||
Further insight into the thermal transitions of our series of synthesised polymers, X-poly(siloxane-r-S) (X = 0–15 wt% S), was possible using differential scanning calorimetry (DSC). The DSC thermograms revealed varied thermal behaviours indicative of the different compositions. For example, elemental sulfur exhibits a distinct endothermic peak at approximately 117.36 °C, typical of its melting transition (red, Fig. 9).42 This peak was not observed in 5-poly(siloxane-r-S) (purple, Fig. 9) or 10-poly(siloxane-r-S) (green), which suggests that elemental sulfur is either absent or present in negligible amounts within these materials. However, 15-poly(siloxane-r-S) clearly showed this endothermic peak (yellow), indicative of a significant level of depolymerised sulfur. Unsurprisingly, this peak disappeared in the second heating cycle as the sulfur was reincorporated into the material upon heating (Fig. S16, ESI†). With respect to the glass transition temperature (Tg), PDMS has a notoriously low Tg, between −150 °C and −123 °C,43,44 which is not observed in the 0-poly(siloxane-r-S) as the lower limit of our instrument is −100 °C. Similarly, for the other samples which have 5–15 wt% incorporation of S, no Tg was observed under our temperature range. This suggests that these materials likely retain low-temperature flexibility and mobility, characteristic of 0-poly(siloxane-r-S), even after 15 wt% sulfur incorporation.
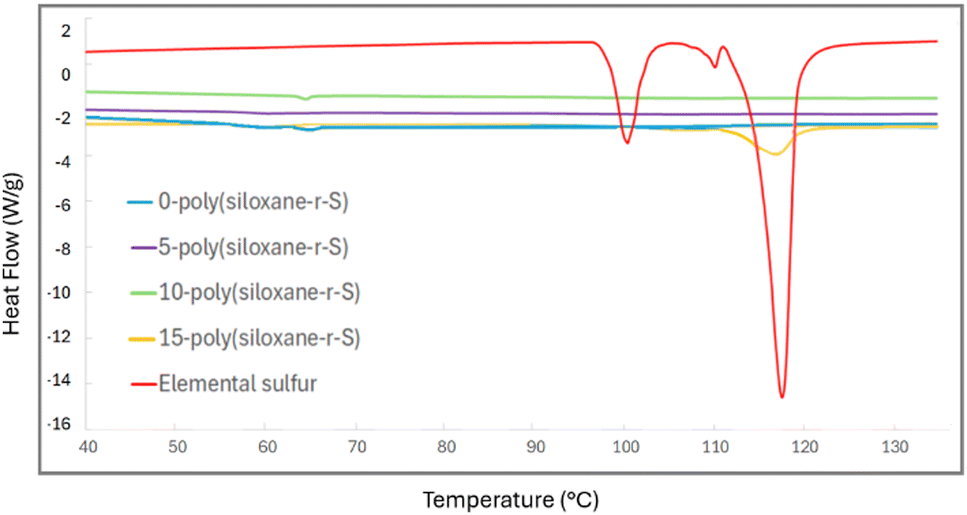 | ||
| Fig. 9 Comparative DSC thermograms of X-poly(siloxane-r-S) (X = wt% S); X =0 (blue), 5 (purple), 10 (green), and 15 (yellow) and S8 (red). | ||
 | ||
| Fig. 10 Thermally repairable properties of polysiloxane samples with varying wt% S (X) and their corresponding SEM images, shown before (left) and after (right) the healing process for X-poly(siloxane-r-S); X = (a) 0 (b) 5 (c) 10. | ||
Physical properties
Contact angle measurements were carried out to assess the surface wettability of the samples to determine if the incorporation of 5–15 wt% S has an effect on the material's hydrophobicity. Narrower contact angles are observed on materials with increased wettability (hydrophilic, <90°) and wider angles are observed on more hydrophobic surfaces (>90°). The unmodified polysiloxane, 0-poly(siloxane-r-S), exhibited a contact angle of 105.91°, while similar contact angles of 105.90° and 104.90° were measured for 5-poly(siloxane-r-S) and 10-poly(siloxane-r-S), respectively. Considering the margin of experimental error, it can be concluded that the materials exhibit comparable hydrophobic properties. However, for the sample containing 15 wt% S, 15-poly(siloxane-r-S), the presence of the surface sulfur resulted in a contact angle of 111.4°, showing even higher hydrophobicity. This difference is attributed to the hydrophobic presence of surface elemental sulfur which increases roughness making it less wettable.45 An assessment of the relative solubilities of X-poly(siloxane-r-S) (X = 0, 5, 10, 15 wt%) revealed limited solubility and swelling behaviours in common solvents (Fig. S17 and Table S3, ESI†) for both control and sulfur-containing samples, with only DMF showing partial solubility due to its ability to catalyse the degradation of oligosulfides.46 The similar solubilities suggest the minor impact that sulfur has on the solvent resistance of polysiloxane.Thermal repair properties
After confirming the integration of sulfide bonds into the cross-linked polysiloxanes, we investigated their self-healing potential. We explored this property in X-poly(siloxane-r-S), where X = 0, 5, 10 wt% S. In our healing experiments, each material was intentionally cut with a knife (Fig. 10, top left). To facilitate the self-healing process, the damaged samples were subjected to 120 °C alongside applied physical pressure through a heat press. Irrespective of healing time, the unmodified silicone, 0-poly(siloxane-r-S), remained damaged (Fig. 10a). On the other hand, a remarkable transformation was observed in the sulfur-incorporated samples. After 30 h and 24 h healing time, for 5 and 10 wt% S, respectively (Fig. 10b and c), the two sulfur containing samples were completely repaired. These observations highlight the critical influence of the dynamic S–S bonds on the self-healing capability of silicone, with increased sulfur content resulting in faster healing times. In order to investigate the presence of any subtle changes in the microstructure and material properties after healing, FT-IR spectroscopy, SEM, water contact angles, and rheology of the samples before and after healing, were measured. FT-IR spectroscopy conducted on the 5-poly(siloxane-r-S) sample post-healing revealed no noticeable change in the overall spectra (Fig. S18, ESI†), suggesting that the healing process did not alter any major functional groups in the polymer. The SEM results (Fig. 10, bottom) demonstrated that the surface properties remained consistent with their initial state. Healed 5-poly(siloxane-r-S) gave a contact angle of 106.2°, while 10-poly(siloxane-r-S) gave a contact angle of 105.7°. Both values are similar to the values obtained prior to healing, indicating the preservation of surface wettability and hydrophobicity during the healing step.Rheological analysis was conducted on 0-poly(siloxane-r-S) and 10-poly(siloxane-r-S), focusing on their storage modulus (G′) and loss modulus (G′′) (Fig. 11). This evaluation included examining their initial states and responses to healing. As mentioned previously, it was observed that 0-poly(siloxane-r-S) exhibited no visible signs of healing or improvement after 36 h hot pressing (Fig. 10a). However, when comparing the storage modulus (G′) before (solid orange line) and after (dotted orange line) the unsucsessful healing attempt, the G′ observed in the damaged sample subjected to thermal healing was higher than that of the initial sample in undamaged state (Fig. 11). This slight increase in G′ could be due to a minor amount of further hydrosilylation occuring in this sample during the thermal treatment. On the other hand, the sulfur-modified sample, 10-poly(siloxane-r-S) (solid blue line), exhibited a slightly lower G′ across the frequency sweep after healing (dotted blue line, Fig. 11). In this case, the trend is attributed to the presence of cleaved and unrepaired Si–O, Si–C, and C–C bonds within the sample. During the healing process, sulfur aids in the creation of new S–S cross-links, facilitating the recovery of the material's elasticity that was diminished due to damage. This includes elasticity lost because of the presence of cleaved and not repaired Si–O, Si–C, and C–C bonds. While the formation of S–S cross-links helps in regaining elasticity, the remaining cleaved and unrepaired bonds lead to an increase in bond mobility. This increased mobility is likely responsible for the observed slight reduction in the storage modulus.
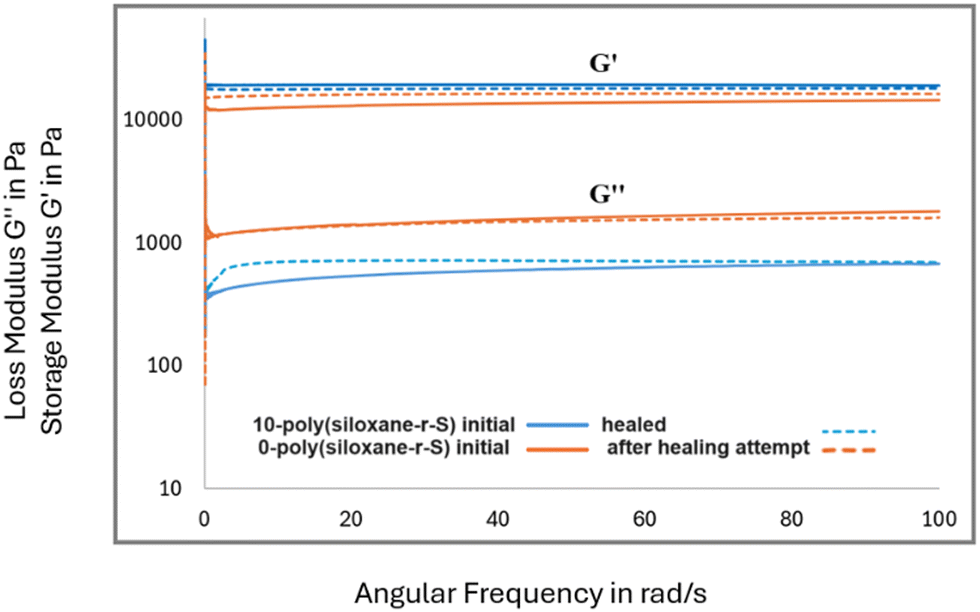 | ||
| Fig. 11 Storage modulus (G′) and loss modulus (G′′) measurements of 0-poly(siloxane-r-S) (orange) and 0-poly(siloxane-r-S) (blue) initial (solid line) and after healing attempt (dotted line). | ||
Analysis of the loss modulus (G′′) shows a higher overall G′′ for the unmodified silicone in comparison to the sulfur-modified variant, suggesting that the bond mobility is higher in the unmodified material. The unmodified silicone, 0-poly(siloxane-r-S) maintains the same loss modulus post-healing attempt (dotted orange line), with a slightly lower G′′ at some frequencies, likely due to the additional hydrosilylation during the 36 h of the thermal treatment. Conversely, the sulfur-modified silicone, 10-poly(siloxane-r-S), displays a slight increase in G′′ across the frequency sweep after healing (dotted blue line) relative to the initial sample (solid blue line, Fig. 11). This observation supports the notion that the polymer network is cleaved and the unhealed sites, including Si–O, Si–C and C–C bonds, achieve a minor enhanced mobility following damage, thereby leading to an increased loss modulus. Overall, the elasticity properties before and after healing appear comparable, largely due to the formation of additional S–S cross-links, which effectively maintains the elasticity of the material.
The relaxation modulus graph presents a noticeable slope for 0-poly(siloxane-r-S), whereas the 10-poly(siloxane-r-S) sample, both in its initial and healed states, shows a relatively flatter profile. This behavior is indicative of a higher cross-link density in the sulfur-modified silicone compared to the control sample. Such an analysis suggests that the inclusion of sulfur cross-links contributes to a denser network within the polymer. This supports the observed lower loss modulus and slightly higher storage modulus in the sulfur modified sample compared to the unmodified silicone (Fig. S19, ESI†).
Conclusions
In this study, we have demonstrated the feasibility of incorporating sulfide cross-links, derived from the petroleum waste by-product elemental sulfur, into commercial-grade silicone. These dynamic bonds enhance the repairability and recyclability of silicone without significantly changing their desirable properties. Our results highlight the potential of sulfur-containing polysiloxanes to significantly extend the lifespan of silicone-based products through improved self-healing capabilities. Additionally, the properties introduced by integrating polysulfides into these materials suggest alternative pathways to less energy-intensive degradation processes, aligning with environmental sustainability goals by reducing the carbon footprint associated with the disposal or recycling of silicone products. Future work in this area will include investigating different chain lengths of telechetic (vinyl-terminated) and vinyl-functionalised PDMS, eliminating all ethylene cross-links formed through hydrosilylation (see Fig. S20, ESI† for part A + 10 wt% S). Through this approach, the potential for a completely circular material which can be degraded back into its original components, S8 and PDMS, or precursors thereof, will be realised.Alongside this, further optimisation using existing reactive extrusion protocols and equipment, commonly used for mixing and curing polysiloxanes, could yield improved outcomes. This breakthrough enhances the functional performance of silicone-based products and paves the way for more sustainable manufacturing and end-of-life management practices. As we move forward, the insights gained from this study will continue to guide our exploration into the development of sulfur-containing polysiloxanes, including investigating their environmental stability (i.e., resistance to hydrolysis, pH, microbes, UV, and ozone exposure). We aim to not only broaden the possible applications for these materials but also to refine and scale up the production processes for both cross-linked and linear silicone variants. Achieving commercial viability while retaining the observed laboratory-scale benefits will be key to making a substantial impact on the sustainability and functionality of silicone materials.
Author contributions
M. R. performed the synthesis, characterisation and tested the properties of the polysiloxanes (e.g., FTIR, NMR, Raman, SEM, TGA, DSC, thermal repair, contact angle) and wrote the initial draft of the manuscript. K. W. P. performed preliminary studies on the synthesis of the cured polymers, demonstrating proof of concept. Rheological studies were supported by W. H. L., including interpretation and review & editing. SSNMR studies were performed by Z. Z., including interpretation and review & editing. Project conceptualization, methodology, supervision, and review & editing of manuscript was contributed by E. M. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the School of Chemical Sciences at the University of Auckland for financial and infrastructure support. ZZ is grateful for financial support of the Ministry of Science, Technological Development and Innovation of the Republic of Serbia (Contract No 451-03-55/2024-673 03/20051). We are grateful to Albina Avzalova for assistance with Raman spectroscopy, Daniel Clyde for assistance with DSC and TGA measurements, Stephen Cawley for assistance with rheology, Hoa Nguyen for assistance with EA, and Dr Yuan Tao for assistance SEM. Each technician has significantly contributed to our research, and we appreciate their expertise.References
- A. Ladenburg, Ueber die Reductionsproducte des Kieselsäureäthers und deren Derivate, Justus Liebigs Ann. Chem., 1872, 164(2), 300–332 CrossRef.
- MarketsandMarkets [Internet], [cited 2024 Mar 5], Silicone Market Size, Industry Share Growth Forecast, Global Trends Report, [Latest], Available from: https://www.marketsandmarkets.com/Market-Reports/silicone-market-709.html.
- B. Rupasinghe and J. C. Furgal, Degradation of silicone-based materials as a driving force for recyclability, Polym. Int., 2022, 71(5), 521–531 CrossRef CAS.
- Zeigler J. M. and Fearon F. W. G., Silicon-based polymer science: a comprehensive resource, American Chemical Society, Washington, DC, 1990. p. 801 (Advances in chemistry series) Search PubMed.
- B. Beagley, J. J. Monaghan and T. G. Hewitt, Electron-diffraction studies of tetramethylsilane and hexamethyldisilane, and discussion of the lengths of Si-C bonds, J. Mol. Struct., 1971, 8(4), 401–411 CrossRef CAS.
- İ. Yilgör and J. E. McGrath,Polysiloxane containing copolymers: A survey of recent developments. in Polysiloxane Copolymers/Anionic Polymerization [Internet]. Springer-Verlag, Berlin/Heidelberg, 1988. pp. 1–86. https://link.springer.com/10.1007/BFb0025274. (Advances in Polymer Science; vol. 86) Search PubMed.
- A. J. O’Lenick, Silicones—Basic chemistry and selected applications, J Surfactants Deterg., 2000, 3(2), 229–236 CrossRef.
- S. Pawlenko, Organosilicon Chemistry [Internet]. Walter de Gruyter, Berlin/Boston, GERMANY, GmbH, 1986 [cited 2024 Mar 5]. Available from: https://ebookcentral.proquest.com/lib/auckland/detail.action?docID=3042274 Search PubMed.
- W. Noll, Chemistry and Technology of Silicones, Elsevier, 2012. p. 716 Search PubMed.
- Global Silicones Council [Internet], [cited 2024 Apr 30], Energy, Available from: https://globalsilicones.org/explore-silicones/benefits-uses/energy/.
- B. Abolpour and R. Shamsoddini, Mechanism of reaction of silica and carbon for producing silicon carbide, Prog. React. Kinet. Mech., 2020, 45, 146867831989141 CrossRef.
- J. C. Furgal and C. U. Lenora, Green routes to silicon-based materials and their environmental implications, Phys. Sci. Rev., 2020, 5(1), 20190024 Search PubMed.
- A. W. Weimer, K. J. Nilsen, G. A. Cochran and R. P. Roach, Kinetics of carbothermal reduction synthesis of beta silicon carbide, AIChE J., 1993, 39(3), 493–503 CrossRef CAS.
- P. Döhlert, J. Pfrommer and S. Enthaler, Recycling Concept for End-of-Life Silicones: Boron Trifluoride Diethyl Etherate as Depolymerization Reagent to Produce Difluorodimethylsilane as Useful Commodity, ACS Sustainable Chem. Eng., 2015, 3(1), 163–169 CrossRef.
- D. J. Krug, M. Z. Asuncion and R. M. Laine, Facile Approach to Recycling Highly Cross-Linked Thermoset Silicone Resins under Ambient Conditions, ACS Omega, 2019, 4(2), 3782–3789 CrossRef CAS PubMed.
- M. E. Noman, M. K. Bayo, Y. Chen and M. A. Brook, Selective oxidative devulcanization of hydrosilylation-cured silicone elastomers: enhanced circularity, Polym. Chem., 2024, 13, 1310–1317 RSC.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, A self-healing, re-moldable and biocompatible crosslinked polysiloxane elastomer, J. Mater. Chem. B, 2016, 4(5), 982–989 RSC.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin and Y. Sun, et al., A highly stretchable autonomous self-healing elastomer, Nat. Chem, 2016, 8(6), 618–624 CrossRef CAS PubMed.
- H. Deng, J. Ye, Z. Zu, Z. Lin, H. Huang and L. Zhang, et al., Repairable, reprocessable and recyclable rigid silicone material enabled by dual dynamic covalent bonds crosslinking side-chain, Chem. Eng. J., 2023, 465, 143038 CrossRef CAS.
- A. Zhang, L. Yang, Y. Lin, L. Yan, H. Lu and L. Wang, Self-healing supramolecular elastomers based on the multi-hydrogen bonding of low-molecular polydimethylsiloxanes: Synthesis and characterization, J. Appl. Polym. Sci., 2013, 129(5), 2435–2442 Search PubMed.
- C. Yue, L. Zhao, L. Guan, X. Zhang, C. Qu and D. Wang, et al., Vitrimeric silicone composite with high thermal conductivity and high repairing efficiency as thermal interface materials, J. Colloid Interface Sci., 2022, 620, 273–283 CrossRef CAS PubMed.
- H. P. Xiang, M. Z. Rong and M. Q. Zhang, A facile method for imparting sunlight driven catalyst-free self-healability and recyclability to commercial silicone elastomer, Polymer, 2017, 108, 339–347 Search PubMed.
- L. Zhao, Y. Yin, B. Jiang, Z. Guo, C. Qu and Y. Huang, Fast room-temperature self-healing siloxane elastomer for healable stretchable electronics, J. Colloid Interface Sci., 2020, 573, 105–114 Search PubMed.
- X. Wu, J. Li, G. Li, L. Ling, G. Zhang and R. Sun, et al., Heat-triggered poly(siloxane-urethane)s based on disulfide bonds for self-healing application, J. Appl. Polym. Sci., 2018, 135(31), 46532 Search PubMed.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass and R. A. Norwood, et al., Dynamic Covalent Polymers via Inverse Vulcanization of Elemental Sulfur for Healable Infrared Optical Materials, ACS Macro Lett., 2015, 4(9), 862–866 CrossRef CAS PubMed.
- P. D. Bartlett, Elemental Sulfur: Chemistry and Physics, (Meyer, Beat, ed.). J. Chem. Educ., 1966;43(12):A1096 Search PubMed.
- R. Javadli and A. De Klerk, Desulfurization of heavy oil, Appl. Petrochem. Res., 2012, 1(1–4), 3–19 CrossRef CAS.
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Polymerizations with elemental sulfur: A novel route to high sulfur content polymers for sustainability, energy and defense, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS.
- B. A. Dogadkin, The mechanism of vulcanization and the action of accelerators, J. Polym. Sci., 1958, 30(121), 351–361 CrossRef CAS.
- N. Bacon, Vulcanization of Rubber, J. Phys. Chem., 1928, 32(6), 801–828 CrossRef CAS.
- M. J. H. Worthington, R. L. Kucera, I. S. Albuquerque, C. T. Gibson, A. Sibley and A. D. Slattery, et al., Laying Waste to Mercury: Inexpensive Sorbents Made from Sulfur and Recycled Cooking Oils, Chem. A Eur J., 2017, 23(64), 16219–16230 Search PubMed.
- The use of elemental sulfur as an alternative feedstock for polymeric materials | Nature Chemistry [Internet], [cited 2024 Mar 11], Available from: https://www.nature.com/articles/nchem.1624.
- K. W. Park, E. A. Tafili, F. Fan, Z. Zujovic and E. M. Leitao, Synthesis and characterization of polysulfides formed by the inverse vulcanisation of cyclosiloxanes with sulfur, Polym. Chem., 2022, 13(32), 4717–4726 RSC.
- K. W. Park, Z. Zujovic and E. M. Leitao, Synthesis and Characterization of Disiloxane Cross-Linked Polysulfides, Macromolecules, 2022, 55(6), 2280–2289 CrossRef CAS.
- PP2-RG07-Gelest®-hPDMS-TDS - Gelest, Inc, [Internet], [cited 2024 Mar 5], Available from: https://www.gelest.com/themencode-pdf-viewer/?file=https://www.gelest.com/wp-content/uploads/PP2-RG07-Gelest%C2%AE-hPDMS-TDS.pdf.
- C. Sturgess, C. J. Tuck, I. A. Ashcroft and R. D. Wildman, 3D reactive inkjet printing of polydimethylsiloxane, J. Mater. Chem. C, 2017, 5(37), 9733–9743 RSC.
- J. Stieghorst and T. Doll, Rheological behavior of PDMS silicone rubber for 3D printing of medical implants, Addit. Manuf., 2018, 24, 217–223 CAS.
- B. Arkles and G. Larson, Silicon Comp., 2013, 178–179 Search PubMed.
- P. Launer and B. Arkles, Infrared Analysis of Organsilicon Compounds: Spectra-Structure Correlations. in Reprinted from Silicone Compounds Register and Review. 2008. p. 223–6 Search PubMed.
- J. L. Dodd, C. Lima, D. Costa-Milan, R. A. Neale, B. Saunders and B. Zhang, et al., Raman analysis of inverse vulcanised polymers, Polym. Chem., 2023, 14(12), 1369–1386 RSC.
- A. D. Tikoalu, N. A. Lundquist and J. M. Chalker, Mercury Sorbents Made By Inverse Vulcanization of Sustainable Triglycerides: The Plant Oil Structure Influences the Rate of Mercury Removal from Water, Adv. Sustainable Syst., 2020, 4(3), 1900111 CrossRef CAS.
- B. Zhang, H. Gao, P. Yan, S. Petcher and T. Hasell, Inverse vulcanization below the melting point of sulfur, Mater. Chem. Front., 2020, 4(2), 669–675 RSC.
- A. Volkov in Polydimethylsiloxane (PDMS). ed. Drioli E., Giorno L.Encyclopedia of Membranes [Internet], Springer, Berlin, Heidelberg, 2015, [cited 2024 Mar 5]. p. 1–2. Available from: https://doi.org/10.1007/978-3-642-40872-4_1400-1 Search PubMed.
- R. G. Jones, W. Ando and J. Chojnowski, Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications, Springer, Netherlands, Dordrecht, 2000, [cited 2024 Mar 5]. Available from: https://link.springer.com/10.1007/978-94-011-3939-7 Search PubMed.
- G. W. Walker, C. P. Walters and P. E. Richardson, Hydrophobic effects of sulfur and xanthate on metal and mineral surfaces, Int. J. Min. Proc., 1986, 18(1), 119–137 CrossRef CAS.
- P. Yan, W. Zhao, S. J. Tonkin, J. M. Chalker, T. L. Schiller and T. Hasell, Stretchable and Durable Inverse Vulcanized Polymers with Chemical and Thermal Recycling, Chem. Mater., 2022, 34(3), 1167–1178 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00297k |
| This journal is © The Royal Society of Chemistry 2024 |